

Abstract
The heat shock response (HSR) is a highly conserved cellular pathway that is responsible for stress relief and the refolding of denatured proteins [1]. When a host cell is exposed to conditions such as heat shock, ischemia, or toxic substances, heat shock factor- 1 (HSF- 1), a transcription factor, activates the genes that encode for the heat shock proteins (Hsps), which are a family of proteins that work alongside other chaperones to relieve stress and refold proteins that have been denatured [2]. Along with the refolding of denatured proteins, Hsps facilitate the removal of misfolded proteins by escorting them to degradation pathways, thereby preventing the accumulation of misfolded proteins [3]. Research has indicated that many pathological conditions, such as diabetes, cancer, neuropathy, cardiovascular disease, and aging have a negative impact on HSR function and are commonly associated with misfolded protein aggregation [4], [5]. Studies indicate an interplay between mitochondrial homeostasis and HSF-1 levels can impact stress resistance, proteostasis, and malignant cell growth, which further support the role of Hsps in pathological and metabolic functions [6]. On the other hand, Hsp activation by specific small molecules can induce the heat shock response, which can afford neuroprotection and other benefits [7]. This review will focus on the modulation of Hsps and the HSR as therapeutic options to treat these conditions.
Graphical Abstract
Introduction to the Heat Shock Response
The heat shock response (HSR) is an evolutionary conserved process that induces molecular chaperone levels in response to cellular stress [8]. The HSR is activated via several stressors such as an increase in temperature, oxidative stress, glucose depletion, and/or the overexpression of misfolded proteins [9]–[11]. Small molecules such as Hsp90 inhibitors, proteasome inhibitors, amino acid analogs, and ribosome biogenesis inhibitors also induce chaperone levels through the HSR [8], [12]. Upon induction of the HSR, there is a robust expression of molecular chaperones and other cell-protective pathways that prevent the misfolding and aggregation of cellular proteins, and promote recovery from stress-induced damage [13]. Molecular chaperones protect proteins from the deleterious effects of stressors by stabilizing and refolding protein-folding intermediates or facilitating their degradation [14]. The heat shock proteins (Hsps), 70 kDa and 90 kDa are the focus of this review and are highly conserved ATP-dependent molecular chaperones that fold and activate proteins. Along with other families of Hsps, they are regulated by several mechanisms, including heat shock factor one (HSF-1), which plays an essential role in their activation [15]. This topic is of great interest in medical research as small molecule modulators of the heat shock response have been developed for the treatment of several diseases and disorders. Specific examples of small molecule activators and inhibitors will be succinctly discussed in this review.
Heat Shock Transcription Factors
Heat shock transcription factors (HSFs) are a family of DNA-binding proteins that regulate gene expression on a transcriptional level [16]. They exhibit a complex winged helix-loop-helix structure, manifest DNA-binding selectivity, can be regulated by a number of post-translational modifications (PTMs), and play various roles in response to cellular stress [16][17]. There are six HSFs encoded by the human genome; HSF-1, HSF-2, HSF-4, HSF-5, HSFX and HSFY [18]. The two most studied HSF’s are HSF-1 and HSF-2, both of which play a functional role in the HSR [8], [16]. HSF-2 is expressed early in development and serves as an activator of chaperone genes at febrile temperatures [19], [20]. HSF-2 has been linked to tumor suppression and activation [19]. HSF-1 is essential to the HSR and is considered the master regulator of the heat shock response. For example, mouse embryonic fibroblasts failed to upregulate chaperone genes upon heat shock in the absence of HSF-1, suggesting that HSF-1 is essential for regulation of the HSR in mammals [21], [22].
Heat Shock Factor-1 (HSF-1)
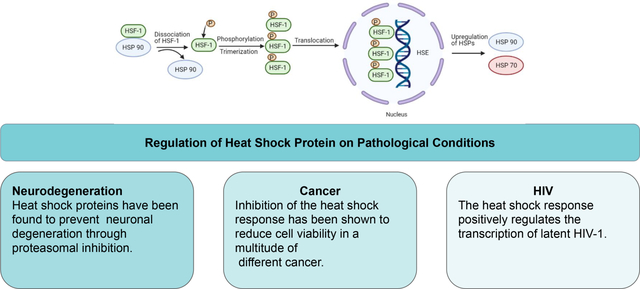
HSF-1 is described as the master regulator of protein expression for genes that encode for the quality-control machinery [8], [18]. Under normal cellular conditions, inactive HSF-1 monomers reside in the cytoplasm in a complex with HSP40, 70, 90, and the cytosolic chaperonin TCP1 ring complex (TRiC) [16]. Upon activation by a stressor, HSF-1 undergoes a multistep activation cycle, whereby it dissociates from the Hsp complex, trimerizes, and then translocates to the nucleus wherein the HSF-1 trimer binds DNA to initiate transcriptional activation [23][24]. As a result of the nuclear localization signal, HSF can be found at increased levels in the nucleus once activated [16]. Although it is currently unclear how HSF-1 senses stress and converts into an active form, it is likely that different stressors lead to alternative mechanisms of activation [16], [25].
PTMs that promote DNA binding and transcriptional activation also increase HSF-1 activity [25]. The heat-shock binding elements are identified by the 5′-nGAAn-3′ nucleotide sequence to which activated HSF-1 binds to alter transcription [16]. HSF-1 is edited by inhibitory PTMs and p23, which modulate DNA dissociation, HSF-1 deactivation, and the degradation of HSF to complete the cycle [16], [26].
The transformation of HSF from its monomeric to multimeric form occurs through an oligomerization process that is not fully understood, although it has been shown that elevated temperatures can cause intrinsic structural changes in HSF-1 that directly facilitate oligomerization [16]. There is further evidence that HSF-1 oligomerization is regulated by PTMs such as acetylation, phosphorylation and sumoylation [16], [27]. Even though the multimeric form of HSF-1 has been classically referred to as “the active form,” it should be noted that oligomerization is not sufficient for transcriptional activation of HSF-1 target genes [28].
HSF-1 steady state levels are controlled by acetyltransferase p300, which acetylates Lys80, Lys208, and Lys298 to promote HSF-1 stability and prevent degradation [16]. Lys80 inhibits the binding of HSF-1 to DNA [28]. Deacetylase sirtuin 1 (SIRT1) deacetylated HSF-1 and resulted in the silencing of p300 (in HeLa cells) and led to increased proteasomal degradation, which reduced HSF-1 levels [16].
Phosphorylation events have also been shown to regulate the degradation of HSF-1. Specifically, phosphorylation of Ser303 and Ser307 by glycogen synthase kinase 3 beta (GSK3β), casein kinase 2 (CK2), and the mitogen-activated protein (MAP) kinases MEK1 and/or ERK1 promote HSF-1 degradation, and consequently reduce HSF-1 activity [29], [30]. Under metabolic stress in the presence of cancer, the 5’-AMP-activated protein kinase (AMPK) has also been shown to inactivate HSF-1 through phosphorylation [31].
Heat Shock Factor-2 (HSF-2)
Although HSF-2 is not considered a master regulator of the HSR, HSF-2 plays a fundamental role in regulation of the HSR. Both HSF-1 and HSF-2 bind to the Hsp70 promoter in response to cellular stress, but HSF-1 is required to achieve maximum promoter occupancy, suggesting HSF-1 influences the DNA binding activity of HSF-2 [7]. Furthermore, HSF-2 is able to modulate HSF-1 expression, which demonstrates HSF-1 is required for transcriptional regulation of the HSR [31]. However, HSF-2 does not compensate for HSF-1 knockout during the HSR, further implicating HSF-1 as the master regulator of the HSR [32]–[34].
Medical Relevance of Heat Shock Transcription Factors
HSF-1 is reduced in neurodegenerative diseases, which results in increased stress, misfolded proteins, and cell death [35], [36]. P300 is attuned in aging mice and potentially contributes to the lower levels of HSF-1. The exact mechanism for the reduction in HSF levels is unknown, but it is proposed that decreased SIRT1 levels lead to a decrease in HSF-1, resulting in increased Lys80 acetylation-dependent proteasomal degradation [16]. Increased expression of HDAC1 in aging cells contributes to HSF-1 inhibition by recruiting the histone acetyltransferase, GCN5 (KAT2A), through involvement of the p23 cochaperone [16], [37]. Although impaired HSF-1 activation does not cause neurodegenerative diseases, it did increase the levels of misfolded and aggregated proteins by decreasing chaperone expression and inhibiting the protein quality control system, resulting in neuronal dysfunction, neuronal cell death and disease progression [38], [39]. Studies have shown that enhancement of the protein folding capacity via elevated expression of HSF-1 can exhibit therapeutic potential [40].
HSF-1 and HSF-2 activity is altered in cancer [40]. HSF-1-knockout in mice has been shown to decrease tumorigenesis caused by RAS and loss of p53 [16], [40], [41]. In cancer, HSF-1 promotes cellular adaptation and survival under stressful conditions that are encountered by the cell [42], [43]. Although increased levels of HSF-1 do not cause cancer initiation, cancer cells are more dependent upon HSF-1 than normal cells, which results in increased levels of HSF-1 in cancer [16]. High HSF-1 levels are the result of increased HSF-1 translation or a decrease in HSF-1 degradation [43], [44]. PTMs also alter HSF-1 activity in cancer [45]. For example, inhibition of GSK3β leads to a decrease in Ser303 and Ser307 phosphorylation, resulting in increased HSF-1 levels [46]. Increased HSF-1 levels promote cell survival by increasing MAPK signaling, which has been linked to tumor growth and proliferation [38]. In addition, key oncological primers such as p53, AKT, RAF1, BCR–ABL1 fusion, cyclin-dependent kinase 4 (CDK4), Cyclin D, ERBB2, hormone receptors and hypoxia-inducible factor 1α (HIF1α) are also dependent upon chaperones that are regulated by HSF-1, highlighting a clear link between HSF-1 and oncogenesis [16]. HSF-2 has also been linked to cancer, however, it appears as though HSF-2 acts as a tumor suppressor [47]. A reduction in HSF-2 levels leads to increased invasion by cancer organoids, suggesting that low levels of HSF-2 can promote metastasis [48].
The Role of the Heat Shock Response in the Nervous System and Neuropathies
The nervous system is an extremely crucial and complex system that is responsible for transmitting and integrating information from various parts of the body. It consists of two major parts; the central nervous system (CNS) and the peripheral nervous system (PNS). The brain and spinal cord comprise the CNS and are imperative for sending and receiving signals to and from the rest of the body. The PNS is the system that links the CNS to other bodily systems and allows for their regulation via the CNS. This process is accomplished via afferent neurons that carry signals from the sensory stimuli to the CNS, while efferent neurons carry signals away from the CNS to initiate a response [49]. Diseases and/or dysfunctions of these nerves can result in a variety of neuropathies, which include neurodegenerative diseases, stroke, and a handful of other conditions [50]. The division between the CNS and PNS plays a major role in categorizing the various types of neuropathies. The proposed mechanisms and the functions regulated by Hsps in neuropathies are described below.
Neurodegeneration
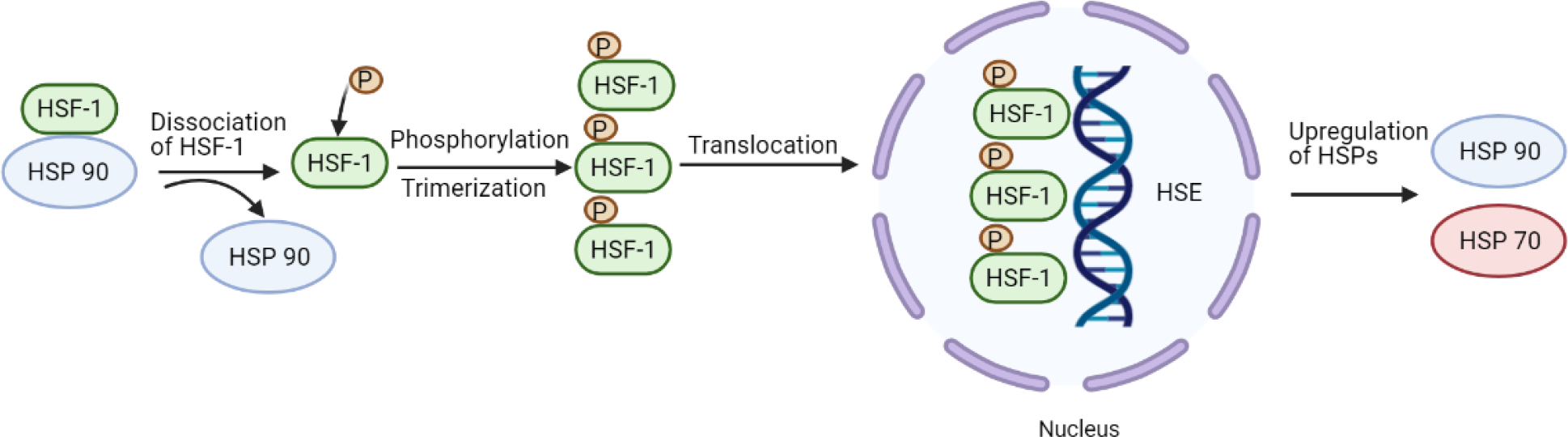
Certain neuronal subtypes are especially vulnerable to protein misfolding and aggregation in neurodegenerative diseases. This may be the result of a multitude of factors, including an age-related decline in the systems that maintain proteostasis (protein homeostasis), an inability of the proteostasis network to identify pathogenic aggregates, or a combination of these factors. Thus, chaperones are critical elements of the proteostasis network as they function to facilitate the proper folding of nascent polypeptides and also recognize and promote the disaggregation of malformed proteins [51]. For this reason, the HSR is believed to be neuroprotective due to the Hsp’s ability to regulate and disaggregate misfolded proteins that trigger central and peripheral neuropathies via pathogenic aggregation [52]. Specifically, in Alzheimer’s Disease (AD), the most common form of dementia, there is a strong connection between neuropathy and the pathogenic aggregation of multiple proteins. Aggregation of tau protein, a protein involved in the stabilization of microtubules in AD, is associated with the formation of neurofibrillary lesions and cell death. When tau becomes abnormally hyperphosphorylated, the microtubules disassemble, and the untethered tau protein aggregates into helical filaments [53][54]. A number of protein kinases have been implicated in the hyperphosphorylation of tau, including stress-activated protein (SAP) kinases, mainly SAP kinase-3 and SAP kinase-4 [55].
There is evidence that Hsps may affect the production of misfolded proteins, and subsequent aggregates, by regulation of their respective SAP kinase activities [56], [57]. Specific Hsps have the potential to regulate aggregation prone intermediates, and therefore can influence the stability of SAP kinase pathways [58]. However, other evidence implicates Hsps in the prevention and dysregulation of misfolded protein aggregates. For example, the aggregation of tau was associated with a decrease in the chaperone activity of Hsp70, its co-chaperones, and other Hsps. Specifically, Hsp70 provided a critical function in the degradation of hyperphosphorylated tau by assisting its ubiquitination with CHIP, an E3 ubiquitin ligase responsible for interacting with Hsp70 and Hsp90 to promote protein degradation [59][60]. Under normal conditions, ubiquitin, a cellular protein that marks misfolded proteins for proteasomal degradation, allows the ubiquitin-proteasome system to hydrolyze damaged proteins into small peptides or individual amino acids. Hsp70 regulates the ubiquitination process in conjunction with CHIP when tau causes microtubule destabilization [61]. Evidence indicates that knockdown of Hsp70 and Hsp90 by RNAi is associated with an increase in aggregated tau and microtubule damage [58]. Therefore, these data suggest that AD and other tau-related neuropathies can be regulated by direct modulation of Hsp90 and Hsp70 levels. The molecular chaperones’ abilities to regulate the proteasome degradation of misfolded tau and to prevent unbound tau from aggregating, leads to increased soluble tau for binding to microtubules. Research indicates a reduction in intracellular Hsps contribute to neurodegeneration, as there was a strong association between Hsp70 activity, its co-chaperones, and other Hsps and tau aggregation [61]. Furthermore, lower levels of Hsp90 in the serum and the frontal cortex of the brain have been observed in patients with AD, suggesting Hsp activation could present a potential therapy for AD [62].
A chronic inflammatory response also arises in AD brain tissue and involves the increased secretion of proinflammatory cytokines, and reactive oxygen species (ROS), which manifest a cytotoxic effect on neurons. In addition, toll-like receptor 4 (TLR4) and NF-κB activities are increased in the AD brain [63]. TLR4 belongs to the pattern recognition receptor family, and its activation leads to an intracellular signaling pathway involving NF-κB, an inflammatory cytokine that is responsible for activating the innate immune system [61]. However, activation of TLR4 by protein aggregation causes the overproduction of proinflammatory cytokines and ROS, which leads to neuronal damage in AD [64]. Hsp70 inhibits the TLR4 activation-induced stimulation of proinflammatory factors. This is due to intracellular Hsp70, which mediates TLR4 ubiquitination and inhibits the NF-κB cascade that is triggered upon TLR4 activation. Therefore, the overexpression of Hsp70 results in a decreased rate of apoptosis in neurons through the prevention of oxidative stress that is associated with such inflammatory responses [65][66 ].
The roles played by Hsps in the inflammatory response are also associated with exosomes. High levels of Hsp70 are expressed in exosomes, which appear to increase natural killer (NK) cells as well as lymphocytes that are responsible for the termination of infected cells, migration and cytotoxicity. Multiple myeloma (MM) exosomes overexpress membrane-bound Hsp70 and have been found to induce dendritic cell maturation and stimulate a type 1 CD4C and CD8C T-cell response via the induction of NK cell-mediated immunity in clinical samples of patients with MM. This data suggests that Hsp70 displayed on the surface of MM exosomes is a trigger for TLR2 activation in NK cells, and could provide another opportunity to regulate neurological or oncological disease pathways [67].
Ischemic Brain Injury
Traumatic brain injury (TBI) is a significant health condition and a leading cause of mortality and long- term neurological disability, which appears to be increasing in prevalence worldwide [68]. TBI is determined by the damage of cognitive, physical and psychosocial functions, but this is also dependent upon the severity of the initial injury [69]. It is understood that the etiology of TBI is complex and consists of primary and secondary injury mechanisms. Primary injury is defined as the direct result of the mechanical effects of initial impact that cause contusion, laceration, ischemia, diffuse swelling and/or intracranial hemorrhage, which may result in neurological impairment [70]. Meanwhile, secondary injuries are thought to result in brain edema, neurological deficits and cognitive dysfunction following TBI via inflammation, oxidative stress, neurotransmitter release, and mitochondrial dysfunction [71]. Secondary injuries differ from primary injuries because they are likely preventable and can be modified via treatment [72]. The pathological reactions that follow TBI and contribute to secondary injury can also lead to neuronal cell death, including apoptosis and necroptosis [73].
TBI is an underappreciated stroke risk factor as trauma to the head and neck may increase risk through vascular dissection, microvascular injury, or abnormal coagulation [74]Studies indicate the median survival after stroke is 1.8 years, compared with 5.7 years for matched non-stroke samples [75]. Stroke is a syndrome in which two types exist; ischemic (85% of cases) or hemorrhagic (15%) [75]. Hemorrhagic stroke is due to bleeding or blood vessel leakage in the CNS; whereas, ischemic stroke is caused by a deficient blood and oxygen supply to the brain [76]Due to its prevalence, ischemic stroke will be the focus of this section. Different regions of the brain provide varying capacities to modulate ischemic cell damage, with white matter being more elastic than gray. In addition, certain populations of neurons are more vulnerable to ischemia; for example, in the hippocampus, rodent CA1 pyramidal neurons were highly susceptible to ischemia, whereas dentate granule neurons were more resistant [76]. However, evidence indicates Hsps function in response to ischemia. Inducible Hsp70 in neurons of transgenic mice produced significantly less neuronal damage in postischemic brain tissue than in the wild-type controls, implicating Hsp70 is beneficial to neuronal survival following ischemia [76], [77]. Furthermore, it has been shown that Hsp70 was able to decrease postischemic brain injury via modulation of the anti-inflammatory response in rodents [78]. Previous studies have fused Hsp70 to the transactivator of transcription (TAT) domain of the human immunodeficiency virus (HIV) and shown that the infusion of TAT-Hsp70 minimized microglial activation three days after stroke, indicating the regulation of post-ischemic inflammation is a key factor for neuroprotection [79]. These data support the hypothesis that through proteasomal inhibition, Hsp70 prevents neuronal degeneration via regulation of the pro-inflammatory NF-κB pathway [80].
Hsp modulators in HIV and Latency
Human immunodeficiency virus (HIV) is a complex virus that is known to attack the host’s immune cells with no cure currently existing [81]. HIV can become transcriptionally inactive in resting memory CD4+ T-cells, which have great longevity, and in turn produces a latent viral reservoir that remains undetectable by the immune system [82]. Studies indicate that HIV latency is promoted by multiple members of the NF-κB family [83]. Specifically, p50, a transcription factor of the NF-κB/Rel family, was found to play an active role in maintaining HIV-1 latency [84]. Since the NF-κB pathway is activated by innate and intrinsic immune responses and Hsp90 functions in pathways that regulate cellular homeostasis when exposed to external stressors, it has been suggested that Hsp90 is associated with HIV-1 reactivation from latency [85]–[87]. Furthermore, HIV-1 infection results in increased expression of Hsp90 in mononuclear cells and T-cells implicating HIV reverse transcriptase as a client of Hsp90 [88]. Inhibition of Hsp90 and Cdc37 demonstrated an ability to regulate innate immune response following exposure to HIV-1 DNA [89]. Furthermore, Cdc37 is an Hsp90 co-chaperone essential for the function of several kinases [90]. Studies have demonstrated that AUY922, an Hsp90 inhibitor, is able to displace Cdc37 from the Hsp90 complex, and inhibit HIV reactivation [91]. Since the HSR has been shown to positively regulate the transcription of latent HIV, it represents a potential therapeutic target to treat HIV [92].
Role of the Mitochondria in Heat Shock Response
Mitochondria are energy-generating, membrane-bound organelles present in almost all eukaryotic cells. The main function of the mitochondria is to organize cellular energy production; these organelles are critical to organism survival and are also responsible for regulating cell death [93]. Mitochondrial positioning is modulated by processes of fission and fusion as well as biogenesis and autophagy to ensure that healthy mitochondria carry out their essential functions [94], [95]. Mitochondrial dysfunction is associated with many metabolic and age-related disorders, neurodegenerative diseases, and ischemic injury in both the heart and brain [96].
Studies indicate that the mitochondrial electron transport chain (ETC) regulates the HSR and the proteostasis networks that control biogenesis, folding, trafficking, and the degradation of proteins inside and outside of the cell [94]. Mitochondrial HSF is heavily integrated in the heat shock response [97]. For example, loss of function mutations in hsf-1 severely reduced the ability to induce stress response genes linked with HSR and the mitochondrial unfolded protein response (UPRmt), suggesting that mitochondrial signaling can prevent repression of the HSR and maintain proteostasis and somatic health despite suboptimal environmental conditions [98].
The mitochondrial chaperonin, heat shock protein 60 (mtHsp60), consists of both Hsp60 and heat shock protein 10 subunits, and forms a barrel-shaped complex that is responsible for the folding of relatively small, soluble proteins. Together, Hsp10 and Hsp60 shift the distribution of Bcl-2, a regulator of apoptosis, to an apoptosis-resistant state and regulate the cross-membrane electrochemical gradient of the mitochondria in the presence of chemotherapeutic drug treated cells. This provides an association between Hsp60/Hsp10 and cardiac defense within the context of cardiotoxicity [99]. Overexpression of Hsp60, Hsp10, or both in cultured cardiomyocytes sheltered cardiac cells from apoptosis induced by simulated ischemia [100]. Furthermore, when an inducible cardiac-specific Hsp60 knockout mouse (Hsp60CKO) model was generated, the removal of Hsp60 in adult mouse cardiomyocytes resulted in heart failure due to impaired mitochondrial proteostasis [101].
Mitochondria and the Heat Shock Response in Neuroprotection
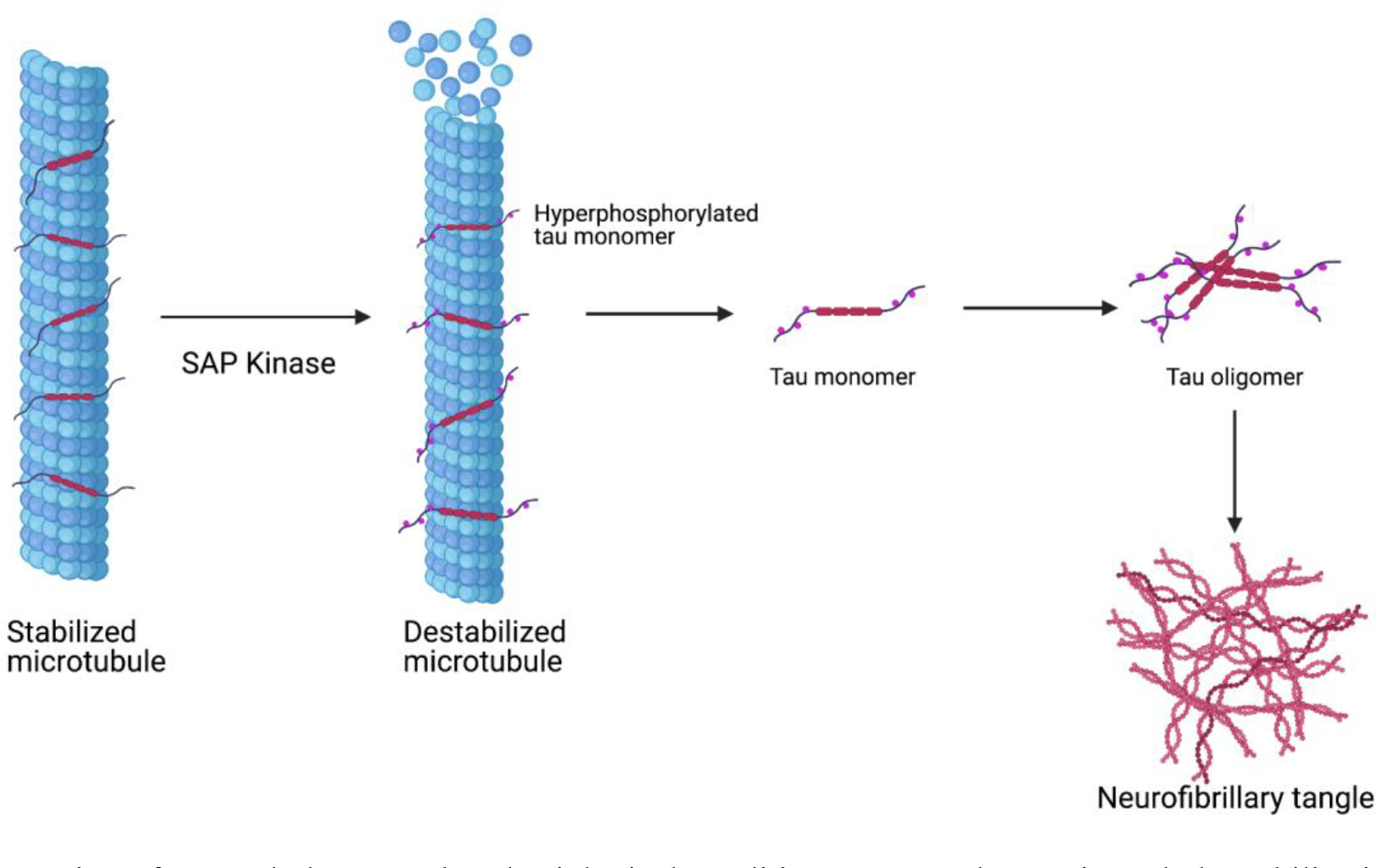
Impaired mitochondrial function plays a critical role in the development of Alzheimer’s Disease (AD) pathology, however augmentation with Hsp70 resulted in the recovery of cell viability and reduced apoptosis, mitochondrial fragmentation, expression of NF-κB and reactive oxygen species (ROS) levels [102]. Oxidative stress is a tissue damaging complication associated with neurodegeneration. However, research supports neuroprotective roles for Hsp60 and Hsp70 in the brain as the HSR and UPRmt have been shown to counter mitochondrial oxidative stress and defend against protein aggregation [103]. For instance, studies indicate that metformin, an anti-diabetic medication, prevented the accumulation of aggregated proteins in the cell via the upregulation of Hsp60 and Hsp70, which catalyzed the clearance of oxidized proteins, and ultimately enhanced mitochondrial function [104].
AD pathogenesis is believed to be driven by the production and deposition of β-amyloid peptide (Aβ) and the aggregation of tau. In fact, the extracellular deposits of Aβ plaques and neurofibrillary tangles represent the two hallmarks required for AD diagnosis [105]. The aggregation of Aβ is thought to prevent and disrupt both synaptic and cellular function, including the blockage of nuclear-encoded proteins from the mitochondria (such as components of the ETC) and impair other aspects of mitochondrial function. As a result, neuronal dysfunction and degeneration often occur [106]. There is extensive evidence to suggest that mitochondria may mediate, drive, or contribute to a variety of AD pathologies and that Aβ precursors affect various aspects of mitochondrial function. Such consequences may result in reduced mitochondria distribution throughout dendrites, causing energy depletion in the neuron [107]. Ultrastructural examination has revealed a significant decrease in the mitochondria of vulnerable neurons in AD, thus increasing mitochondrial degradation products and mitochondrial dysfunction. ROS generation often follows mitochondrial disruption and leads to the release of apoptosis-inducing molecules [108]. However, the overexpression of Hsp60 improved the function of ETC complexes III and IV, when subjected to ischemia and reoxygenation. Hsp60 and Hsp90 overexpression together were sufficient to prevent inactivation of ATP regulating enzymes, such as pyruvate dehydrogenase and ɑ-ketoglutarate dehydrogenase [109]. These data indicate a correlation between protection by Hsp overexpression and decreased ROS production. Furthermore, decreased mortalin expression has been found in AD patient brains and AD mouse models. Mortalin is a mitochondrial chaperone protein and member of the Hsp70 family, and the inhibition of mortalin also induces mitochondrial fragmentation and dysfunction [110].
Introduction to Heat Shock and Cancer
Due to their unique tumor environment, many cancer cells are “addicted” to the heat shock proteins, and inhibition of the HSR could be an effective tool for cancer treatment. Hsp90 and Hsp70 are molecular chaperones required for the heat shock response and are critical to a cell’s ability to respond to cellular stress. Hsp70 inhibits apoptosis through attenuation of cytosolic calcium levels and stabilization of lysosomes within cancer cells [111][112]. Inhibition of Hsp70 has been shown to reduce cell viability in multiple myeloma, pleural mesothelioma and breast cancer, while enhancing the efficacy of other treatments such as BRAF inhibition [113][114]–[116]. Likewise, cancer cells often overexpress Hsp90, and this protein maintains many oncogenic proteins such as the mutant c-SRC tyrosine kinase. c-SRC requires constant stabilization by Hsp90 to maintain its kinase activity. Mutated tumor-suppressor protein p53, which is the most common mutation found in human cancers, is also dependent upon Hsp90 for stabilization, and mutants of this protein do not function as tumor suppressors [117]. Hsp90 inhibitors have been shown to act as anti-cancer agents and induce the degradation of oncogenic Hsp90 client proteins in a wide array of tumors [118][119 ]. Hsp90 inhibition has also been shown to work in conjunction with other cancer therapies. Hsp90 inhibition damages DNA repair mechanisms, sensitizing cancer cells to other treatment pathways such as the inhibition of poly (ADP-ribose) polymerase. It also serves to upregulate the interferon response genes, which enhance the ability of T-cells to kill cancer cells [120][121]. A host of small-molecules have been developed to inhibit these two proteins, with varying degrees of success.
First Generation Hsp90 Inhibitors
Molecules derived from geldanamycin and its semi-synthetic derivatives are known as “first-generation” inhibitors, and inhibit Hsp90 function by competing with ATP for binding to the N-terminal nucleotide-binding pocket [122]. Geldanamycin also produces reactive oxygen species, which can induce oxidative-stress and apoptosis in addition to geldanamycins’ ability to inhibit Hsp90 [123]. However, this process also leads to the increased expression of both Hsp70 and Hsp90, which exhibit resistance to treatment [124]. These problems, along with geldanamycin’s poor solubility and hepatotoxicity, necessitated the creation of new derivatives such as tanespimycin and alvespimycin [125].
Tanespimycin, or 17-AAG, manifests lower toxicity than geldanamycin, better stability, and improved anti-cancer activity at nanomolecular concentrations [126]. Unfortunately, tanespimycin faced similar problems as its predecessor; facilitating the expression of Hsp70 and Hsp90 while maintaining a level of unacceptable toxicity in clinical trials [126][127]. Tanespimycin also suffered from poor solubility [124].
Alvespimycin, or 17-DMAG, fared slightly better, as it was designed to enhance solubility over 17-AAG. When compared to tanespimycin, alvespimycin exhibits stronger anti-tumor activity, improved solubility, good oral bioavailability, and greater selectivity for the Hsp90 heteroprotein complexes present in cancer [124]. Unfortunately, alvespimycin still retains the ability to induce Hsp70 and Hsp90 expression, which is contradictory [128].
Geldanamycin derivatives IPI-493 and IPI-504, also underwent clinical evaluation as Hsp90 inhibitors [128], [129]. IPI-493 demonstrated strong antitumor potential against gastrointestinal tumors, and was shown to possess a longer half-life in-vivo and better potency when compared to Tanespimycin [130]. However, research on IPI-493 was halted in favor of the more active and clinically feasible candidate, IPI-504 [131][132 ]. IPI-504 demonstrated anti-proliferative activity against pancreatic cancer cells and also blocked the unfolded protein response in multiple myeloma [133]. Disappointingly, it has also demonstrated low impact and unacceptable toxicity in patients with castration resistant prostate cancer [134]–[139].
While developing small molecule inhibitors, it is important to address target selectivity and document potential off-target effects [140]. Hsp90-family members have multiple functions in proteostasis and the Hsp90 family includes two cytoplasmic/nuclear isoforms, Hsp90β and Hsp90α, which are constitutively expressed and stress-expressed, respectively [141]. Therefore, inhibition of Hsp90 can inhibit all four of these isoforms and impact the cellular environment [142]. Non-selective targeting of Hsp90 increases the risk of undesired effects, whereas pan-inhibition of all four isoforms can also produce adverse events due to the high levels of Hsp90 isoforms and the large number of client substrates [143]. Pan-Hsp90 inhibitors, such as 17-AAG, inhibit all four isoforms of Hsp90 [144]. More recently, isoform-selective inhibitors were developed and utilized to identify Hsp90α as the key isoform that promotes opioid antinociception and signaling in the brain [145]. In addition, compound KUNB31, which exhibits selectivity for Hsp90β prevents Hsp90 induction and represents a significant advantage over the use of pan-Hsp90 inhibitors [146].
Second Generation N-Terminal Inhibitors
Geldanamycin and its derivatives face significant problems; namely, their dependence upon NAD(P)H:quinone oxidoreductase activity in vivo, significant hepatotoxicity, difficult synthesis, and their ability to induce the expression of Hsp70 and Hsp90 [138], [139], [147], [148]. As a result, a series of second-generation of Hsp90 inhibitors has been developed. Note that the inhibitors focused upon in this section will be those that inhibit Hsp90 via competition with ATP at the Hsp90 N-terminal domain.
AUY922 and STA9090 are second generation inhibitors that demonstrated limited success in clinical trials and exhibited less toxicity. Specifically, AUY922 did not exhibit the extent of hepatotoxicity that was observed with first generation Hsp90 inhibitors, but did induce reversible ocular toxicity, diarrhea, and the upregulation of Hsp70 and Hsp27 [148]–[152]. AUY922 demonstrated potent in vitro and in vivo anti-tumor activity against a wide range of cancers [153]. Likewise, this compound was shown to synergize with other treatment methods such as radiation therapy, cytarabine treatment, treatment with the histone deacetylase inhibitor PXD101, and treatment with the MEK inhibitor, trametinib [154], [155]. In addition, it appeared to overcome resistance to the TNF-related apoptosis-inducing ligand in one trial [155]–[162]. AUY922 is currently in the second phase of clinical development.
Likewise, STA9090 and ganetespib displayed greater efficacy than the first generation inhibitor tanespimycin in terms of tumor reduction and the degradation of Hsp90-dependent substrates [154], [157], [158], [163]. Ganetespib alone or in combination with other drugs, has shown in vivo and in vitro efficacy against a number of different cancers, and displayed less hepatotoxicity than the first generation drugs [164]. Unfortunately, ganetespib induced the upregulation of Hsp70, Hsp27, Hsp90, and HSF-2 and caused undesired adverse events [165], [166].
A number of other second-generation drugs are currently being evaluated. XL888 is a small molecule inhibitor that has been shown to reduce Hsp90 levels and lead to tumor regression [167]. However, XL888’s in vivo efficacy is still uncertain [168]–[170]. Another second-generation inhibitor, PU-3, is based on the purine scaffold and allows for the creation of easily synthesizable and orally bioavailable analogs [171], [172]. A number of derivatives have been prepared based on the purine scaffold, such as BIIB021, PU-H71, and MPC3100. BIIB021 has demonstrated in vivo inhibition of Hsp90 in advanced solid tumors and induced an autophagic response [173]. Likewise, research demonstrated PU-H71 to induce a complete response in triple-negative breast cancer models and the induction of apoptosis through both endoplasmic reticulum and mitochondrial pathways [174]. Research with MPC3100 established safe dosing concentrations, but in vivo effectiveness remains unknown [175]. Clinical trials with PU-H71 have identified some dose-limiting toxicities, but more comprehensive trials have yet to be conducted [176]. The Hsp90 inhibitor, AT13387, has also been shown to reduce tumor size in clinical trials without the hepatotoxic effects of earlier Hsp90 inhibitors. However, like other Hsp90 inhibitors targeting the N-Terminus, it too upregulated the HSR [126].
Hsp70 Inhibitors
Hsp70 inhibitors possess the potential to augment small molecule Hsp90 inhibition by countering the effects of Hsp70 upregulation that is induced via the heat shock response. Examination of 17-AAG against colon carcinoma cells determined that upon treatment with the Hsp70 inhibitor VER-155008, 17-AAG exhibited increased efficacy. 0.5 μM of 17-AAG reduced cell survival by 15%, whereas 10 or 20 μM of VER-155008 reduced cell survival by 4% and 8%, respectively. However, when 0.5 μM of 17-AAG was given in combination with 10 μM of VER-155008, cell survival was reduced by 70%. When VER-155008 was increased to 20 μM, cell death increased to 92% [177]. A similar combinatorial effect was observed in glioblastoma cells upon treatment with 17-AAG and the small-molecule kinase inhibitor D11, which mediates Hsp70 down-regulation. This combination led to a significant decrease in Hsp70 and Hsp27 levels, and enhanced cytotoxicity as compared to either compound alone [178]. Combination therapy with VER-155008 and the Hsp90 inhibitor STA9090 also synergized to manifest an efficacy greater than either monotherapy alone [179]. Likewise, combination of the Hsp70 inhibitor intravenous immunoglobulin (IVIgG), and the small molecule Hsp90 inhibitor AUY922, reduced the viability of OPM-2, U266, and RPMI 8226 cells [180].
While Hsp70 inhibitors can be used to mitigate the effects of upregulated Hsp70, targeting Hsp70 can also act as an effective standalone anti-cancer strategy. The small molecule inhibitor JG-98, which disrupts the Hsp70-Bag3 complex has been shown to regulate cancer cell survival and metastasis and, has demonstrated activity against a panel of ovarian, cervical, breast, skin, and bone marrow cancer cells with EC50 values varying from ~0.3 μmol/L to 4 μmol/L [163]. Hsp70/Bag-3 disruption with JG-98 represents a novel method of Hsp-related cancer treatment. Interestingly, JG-98 was found to increase DNA damage and induce the unfolded protein response, while simultaneously downregulating the heat shock response. The Hsp90 inhibitors discussed in this article do the exact opposite [181]. Dual-targeting of Hsp70 in invasive bladder cancer by MAL3–101, which inhibits Hsp70 ATPase activity by blocking Hsp40 co-chaperone interactions, and the Hsp70 inhibitor VER155008 reduced cell viability and depleted levels of growth-related kinases, but did not upregulate the HSR [182][183].
C-Terminal Hsp90 Inhibitors
A novel method to evade the heat shock response and concomitant upregulation of Hsp70 and Hsp90 is via specific targeting of the Hsp90 C-terminal binding pocket (rather than the N-terminal ATP binding pocket). Hsp90 C-terminal inhibitors exercise allosteric control over the N-terminal binding site [184]. The antimicrobial drug novobiocin and its analogs bind to this pocket and interfere with client protein maturation and the binding of co-chaperones [185]. Fungal mycotoxin, penisuloxazin A, was identified as an Hsp90 C-terminus inhibitor that binds to cysteine residues C572/C598 of CT-Hsp90, which inhibits Hsp90 chaperone activity and induces apoptosis and growth inhibition both in vivo and in vitro. Analogs of this compound, PEN-A and HDN-1, also exhibit Hsp90 inhibitory activity. All three of these molecules disrupt Hsp90 dimerization, but appear to affect Hsp90 client proteins differently. Likewise, the small molecule chaetocin interacts directly with the C-terminal binding pocket of Hsp90α and significantly inhibits the phosphorylation levels of a wide array of Hsp90 client proteins such as Akt, c-Raf, C-ABL, BCR-ABL, AML1-ETO, and SUV39H1. The novobiocin derivatives KU-32, KU-174, and KU-596 also demonstrate modulating effects on Hsp90 via C-terminal binding. KU-174 derivatives manifest anti-cancer activity without induction of the heat shock response and demonstrate good inhibitory activity against an array of cancers in the NCI 60-cell line panel assay [186][187 ].
Small Molecule Activators of the Heat Shock Response
While inhibition of HSPs can be useful to combat cancer, activation of the chaperone network can mitigate damage resulting from the accumulation of misfolded proteins that result from neurodegenerative disorders or ischemic brain injuries. Consequently, the development of small molecule activators of the heat shock response can provide a new treatment option for difficult to treat neurological conditions. For example, Bimoclomol binds the HSF-1 complex and enhances HSF1’s ability to bind DNA, which ultimately leads to increased activation of the HSR [188]. Interestingly, geldamycin and its derivatives can also serve as small molecule heat shock activators. These molecules have been demonstrated to inhibit Hsp90 in cancer, with the unfortunate side effect of increasing Hsp70 and Hsp90 expression. In normal tissue, the administration of geldamycin can stimulate the heat shock response. As such, geldamycin and its derivatives have shown potential as neuroprotective agents, but face a number of problems, such as a limited ability to cross the blood-brain barrier and the degradation of essential growth factor receptors and intracellular signaling molecules. KU-32, a derivative of novobiocin, stimulates the function of Hsp90 through binding to the C-terminal domain, which causes the N-terminal domain to enter a partially closed intermediate phase that increases ATPase activity. This function enables KU-32 to effectively manage neuropathies by activation of the HSR, which results in a reduction of misfolded proteins in cells [189]. KU-32 induces the heat shock response, but at significantly lower concentrations than what is needed to induce the degradation of client proteins, resulting in a large therapeutic window [190][191]. KU-596 also manifests excellent neuroprotective activity through induction of the heat shock response and its ability to increase HSP levels via binding to the C-terminal domain of Hsp90 [192]. KU-596 has been shown to reverse clinical measures of diabetic peripheral neuropathy in diabetic animal models, decrease oxidative stress in diabetic sensory neurons, and has led to improvements in mitochondrial maximal respiratory capacity [193]. KU-596 is currently undergoing phase II clinical trials for the treatment of neuropathy [194],[195]. Other small molecules such as protein translation inhibitors like puromycin, amino acid analogs that prevent proper folding such as canvanine and azetidine, as well as proteasome and protease inhibitors like MG132 can also act as HSR activators. These molecules are defined by their ability to disrupt various targets within a cell’s proteostasis network [196].
Conclusion
The HSR serves a critical function to relieve stress and refold denatured proteins in both normal and stressed cells. Specifically, the Hsps within the HSR are responsible for the folding, activation, and assembly of target proteins as ubiquitous molecular chaperones. Hsp overexpression and inhibition can impact apoptosis regulation within the cellular environment and is therefore targeted within a wide variety of therapies against pathological conditions. In this review, the function of Hsps within peripheral and central neuropathies as well as cancer and the clinical relevance of small molecule inhibitors and activators to these pathologies were examined. Expanding upon our understanding of the interplay between Hsps and each specific pathology will allow for small molecule inhibition and activation of the HSR to become a more compelling therapeutic target in the future.
Figure 1.
The heat shock response. HSF-1 dissociates from HSP90. HSF-1 then undergoes phosphorylation and trimerization. It is then translocated to the nucleus where it interacts with heat shock elements (HSE) resulting in the upregulation of heat shock proteins. Created with BioRender.com.
Figure 2.
Progression of tau pathology. Under physiological conditions, tau regulates microtubule stabilization. In neuropathy, the hyperphosphorylation of tau impacts microtubule affinity. Soluble tau aggregates into pathological tau oligomers, ultimately forming pathological insoluble neurofibrillary tangles. Created with BioRender.com
Figure 3.

Heat shock proteins are closely involved with the ubiquitin-proteasome pathway. Ubiquitin-proteasome degradation requires coordinated reactions by three enzymes (E1, E2, and E3). Poly-ubiquitinated proteins are recognized by the proteasome and degraded. Created with BioRender.com
Figure 4.
Mitochondrial dysfunction and oxidative stress are codependent and the basis of dysregulation in neurodegenerative disease. Increased production of ROS and transcriptional dysregulation are all consequences of mitochondrial dysfunction and oxidative stress that contributes to the apoptosis of neurons. Created with BioRender.com
Figure 5.
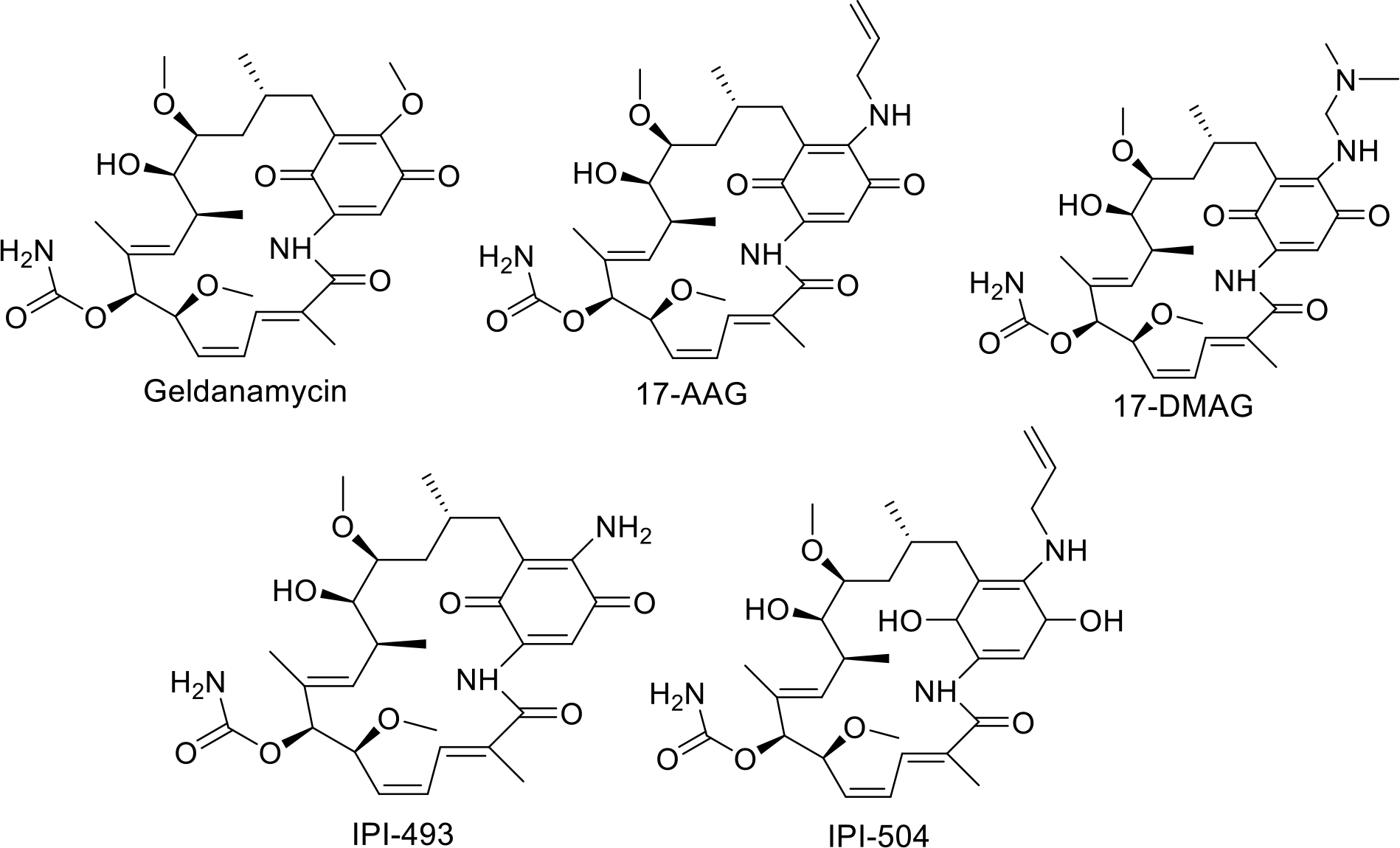
First generation HSP90 inhibitors
Figure 6.
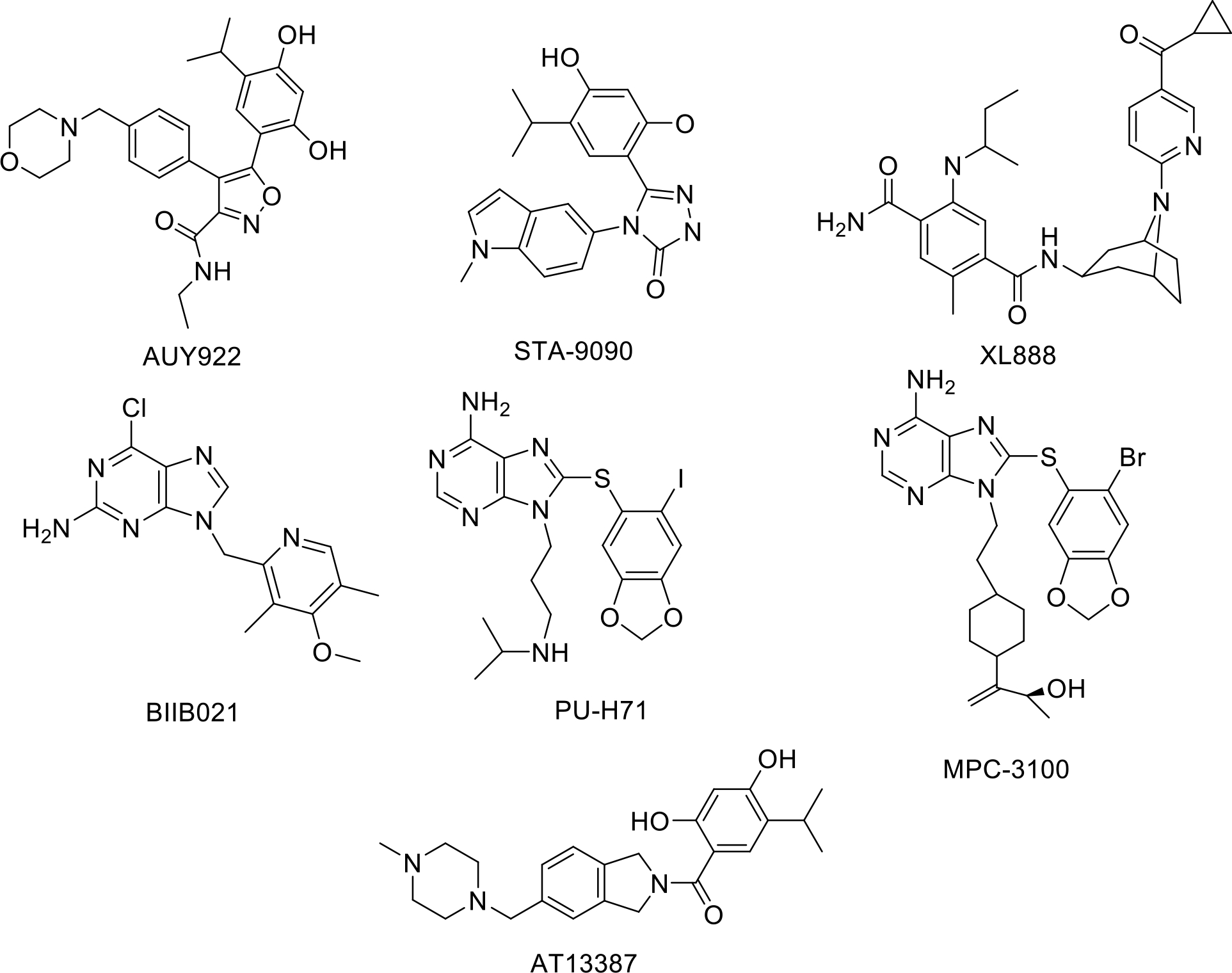
Second generation HSP90 N-Terminal inhibitors
Figure 7.
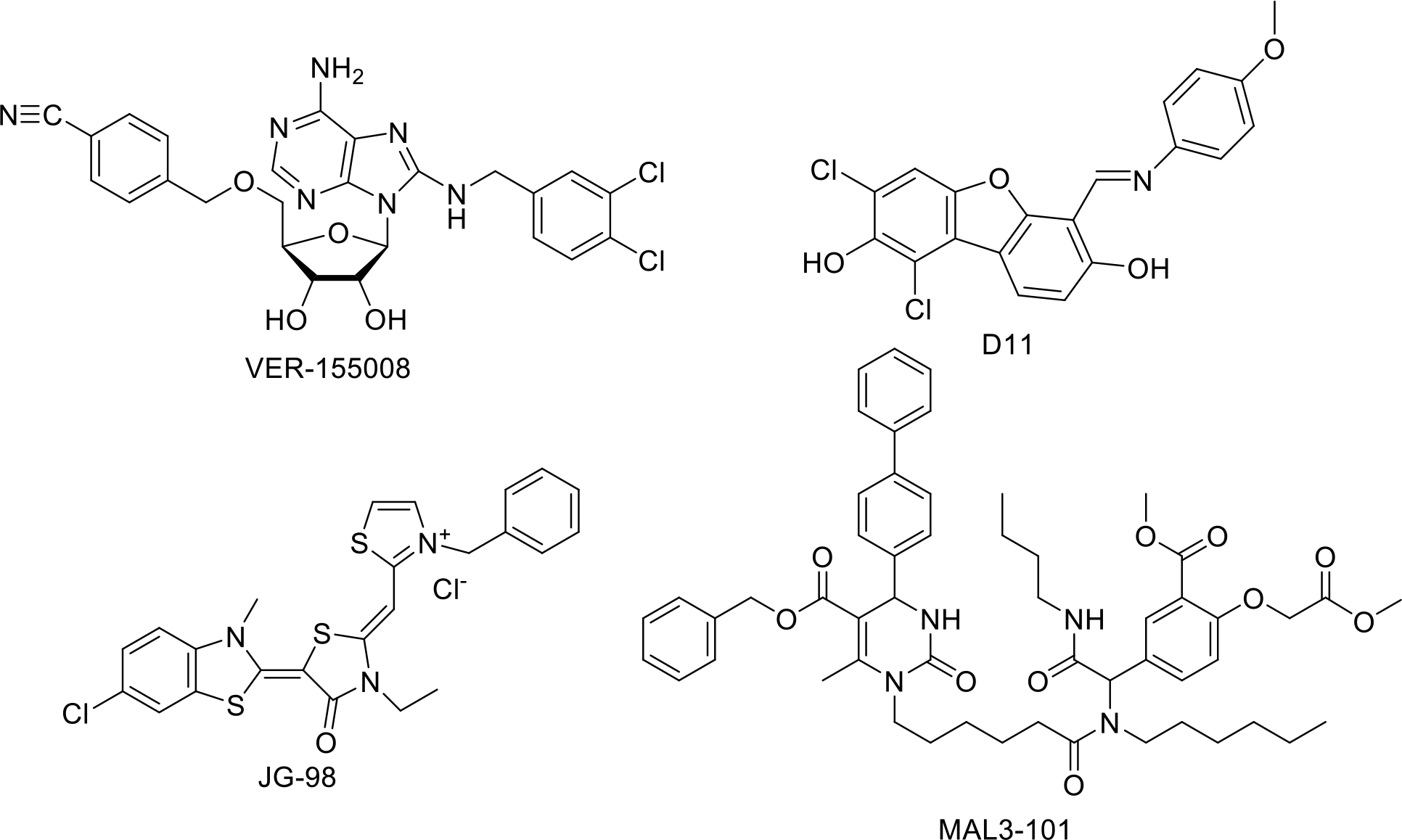
HSP70 inhibitors
Figure 8.

HSP90 C-Terminal inhibitors
Figure 9.
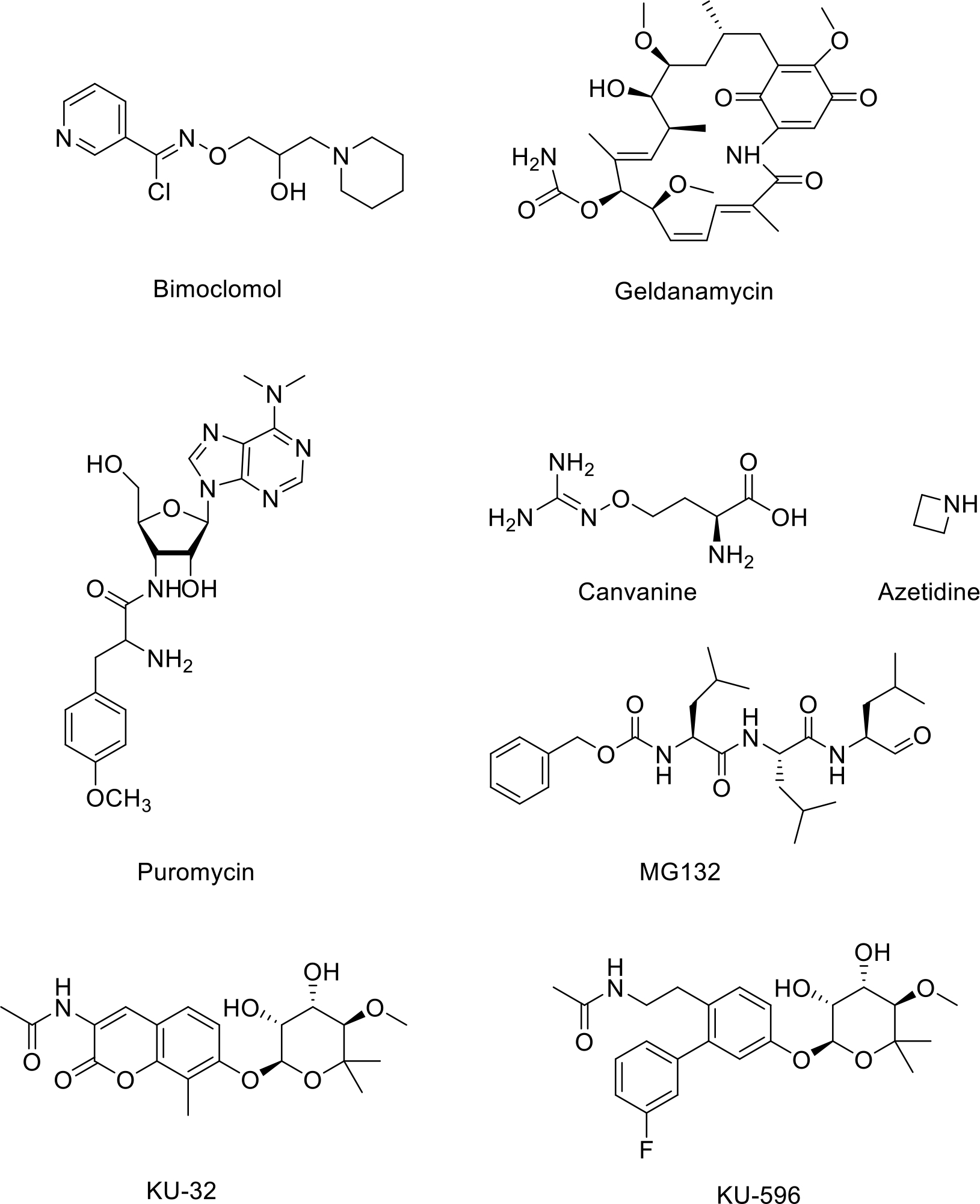
Small Molecule Heat Shock Response Activators
Mitochondrial signaling promotes the heat shock response and maintains proteostasis
Small molecule activators mitigate damage from neurodegeneration or brain injury
Heat shock protein inhibition exhibits anti-proliferative activity against cancer cells
Isoform-selective heat shock protein inhibitors decrease the risk of undesired effects
Footnotes
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
KU-596 is a small molecule therapeutic that was developed by the Blagg Laboratory and then licensed to Reata Therapeutics, whom is evaluating the drug in clinical trials for neuropathy.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Craig EA and Schlesinger MJ, “The Heat Shock Response,” Critical Rev Biochem, vol. 18, no. 3, pp. 239–280, 2008, doi: 10.3109/10409238509085135. [DOI] [PubMed] [Google Scholar]
- [2].Burdon RH, “Heat shock and the heat shock proteins,” Biochem J, vol. 240, no. 2, pp. 313–324, 1986, doi: 10.1042/bj2400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lecker SH, Goldberg AL, and Mitch WE, “Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States,” J Am Soc Nephrol, vol. 17, no. 7, pp. 1807–1819, 2006, doi: 10.1681/asn.2006010083. [DOI] [PubMed] [Google Scholar]
- [4].Pourhamidi K, Dahlin LB, Boman K, and Rolandsson O, “Heat shock protein 27 is associated with better nerve function and fewer signs of neuropathy,” Diabetologia, vol. 54, no. 12, pp. 3143–3149, 2011, doi: 10.1007/s00125-011-2303-5. [DOI] [PubMed] [Google Scholar]
- [5].Fargnoli J, Kunisada T, Fornace AJ, Schneider EL, and Holbrook NJ, “Decreased expression of heat shock protein 70 mRNA and protein after heat treatment in cells of aged rats,” Proc National Acad Sci, vol. 87, no. 2, pp. 846–850, 1990, doi: 10.1073/pnas.87.2.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chou S-D, Prince T, Gong J, and Calderwood SK, “mTOR Is Essential for the Proteotoxic Stress Response, HSF1 Activation and Heat Shock Protein Synthesis,” Plos One, vol. 7, no. 6, p. e39679, 2012, doi: 10.1371/journal.pone.0039679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].DeFranco DB, Ho L, Falke E, and Callaway CW, “Small molecule activators of the heat shock response and neuroprotection from stroke,” Curr Atheroscler Rep, vol. 6, no. 4, pp. 295–300, 2004, doi: 10.1007/s11883-004-0061-2. [DOI] [PubMed] [Google Scholar]
- [8].Pincus D, “HSF1 and Molecular Chaperones in Biology and Cancer,” Adv Exp Med Biol, vol. 1243, pp. 41–50, 2020, doi: 10.1007/978-3-030-40204-4_3. [DOI] [PubMed] [Google Scholar]
- [9].Hahn J-S and Thiele DJ, “Activation of the Saccharomyces cerevisiae Heat Shock Transcription Factor Under Glucose Starvation Conditions by Snf1 Protein Kinase*,” J Biol Chem, vol. 279, no. 7, pp. 5169–5176, 2004, doi: 10.1074/jbc.m311005200. [DOI] [PubMed] [Google Scholar]
- [10].Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, and Drummond DA, “Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast,” Proc National Acad Sci, vol. 108, no. 2, pp. 680–685, 2011, doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].ÅKERFELT M, TROUILLET D, MEZGER V, and SISTONEN L, “Heat Shock Factors at a Crossroad between Stress and Development,” Ann Ny Acad Sci, vol. 1113, no. 1, pp. 15–27, 2007, doi: 10.1196/annals.1391.005. [DOI] [PubMed] [Google Scholar]
- [12].Kim D, Kim S-H, and Li GC, “Proteasome Inhibitors MG132 and Lactacystin Hyperphosphorylate HSF1 and Induce hsp70 and hsp27 Expression,” Biochem Bioph Res Co, vol. 254, no. 1, pp. 264–268, 1999, doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- [13].Morimoto RI, “The Heat Shock Response: Systems Biology of Proteotoxic Stress in Aging and Disease,” Cold Spring Harb Sym, vol. 76, no. 0, pp. 91–99, 2011, doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- [14].Morimoto RI, Kline MP, Bimston DN, and Cotto JJ, “The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones.,” Essays Biochem, vol. 32, pp. 17–29, 1997. [PubMed] [Google Scholar]
- [15].Voellmy R and Boellmann F, “Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks,” Adv Exp Med Biol, vol. 594, pp. 89–99, 2007, doi: 10.1007/978-0-387-39975-1_9. [DOI] [PubMed] [Google Scholar]
- [16].Gomez-Pastor R, Burchfiel ET, and Thiele DJ, “Regulation of heat shock transcription factors and their roles in physiology and disease,” Nat Rev Mol Cell Bio, vol. 19, no. 1, pp. 4–19, 2018, doi: 10.1038/nrm.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Littlefield O and Nelson HCM, “A new use for the ‘wing’ of the ‘winged’ helix-turn-helix motif in the HSF–DNA cocrystal,” Nat Struct Biol, vol. 6, no. 5, pp. 464–470, 1999, doi: 10.1038/8269. [DOI] [PubMed] [Google Scholar]
- [18].Åkerfelt M, Morimoto RI, and Sistonen L, “Heat shock factors: integrators of cell stress, development and lifespan,” Nat Rev Mol Cell Bio, vol. 11, no. 8, pp. 545–555, 2010, doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shinkawa T et al. , “Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation,” Mol Biol Cell, vol. 22, no. 19, pp. 3571–3583, 2011, doi: 10.1091/mbc.e11-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fujimoto M and Nakai A, “The heat shock factor family and adaptation to proteotoxic stress,” Febs J, vol. 277, no. 20, pp. 4112–4125, 2010, doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- [21].McMillan DR, Xiao X, Shao L, Graves K, and Benjamin IJ, “Targeted Disruption of Heat Shock Transcription Factor 1 Abolishes Thermotolerance and Protection against Heat-inducible Apoptosis*,” J Biol Chem, vol. 273, no. 13, pp. 7523–7528, 1998, doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- [22].Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT, “Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation,” Mol Cell, vol. 62, no. 1, pp. 63–78, 2016, doi: 10.1016/j.molcel.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anckar J and Sistonen L, “Regulation of HSF1 Function in the Heat Stress Response: Implications in Aging and Disease,” Annu Rev Biochem, vol. 80, no. 1, pp. 1089–1115, 2011, doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- [24].Mercier PA, Winegarden NA, and Westwood JT, “Human heat shock factor 1 is predominantly a nuclear protein before and after heat stress.,” J Cell Sci, vol. 112 ( Pt 16), pp. 2765–74, 1999. [DOI] [PubMed] [Google Scholar]
- [25].Morimoto RI, “Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators,” Gene Dev, vol. 12, no. 24, pp. 3788–3796, 1998, doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- [26].Hentze N, Breton LL, Wiesner J, Kempf G, and Mayer MP, “Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1,” Elife, vol. 5, p. e11576, 2016, doi: 10.7554/elife.11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jurivich DA, Pachetti C, Qiu L, and Welk JF, “Salicylate Triggers Heat Shock Factor Differently than Heat (* ),” J Biol Chem, vol. 270, no. 41, pp. 24489–24495, 1995, doi: 10.1074/jbc.270.41.24489. [DOI] [PubMed] [Google Scholar]
- [28].Raychaudhuri S et al. , “Interplay of Acetyltransferase EP300 and the Proteasome System in Regulating Heat Shock Transcription Factor 1,” Cell, vol. 156, no. 5, pp. 975–985, 2014, doi: 10.1016/j.cell.2014.01.055. [DOI] [PubMed] [Google Scholar]
- [29].Jin X, Moskophidis D, and Mivechi NF, “Heat Shock Transcription Factor 1 Is a Key Determinant of HCC Development by Regulating Hepatic Steatosis and Metabolic Syndrome,” Cell Metab, vol. 14, no. 1, pp. 91–103, 2011, doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dai S et al. , “Suppression of the HSF1- mediated proteotoxic stress response by the metabolic stress sensor AMPK,” Embo J, vol. 34, no. 3, pp. 275–293, 2015, doi: 10.15252/embj.201489062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Östling P, Björk JK, Roos-Mattjus P, Mezger V, and Sistonen L, “Heat Shock Factor 2 (HSF2) Contributes to Inducible Expression of hsp Genes through Interplay with HSF1*,” J Biol Chem, vol. 282, no. 10, pp. 7077–7086, 2007, doi: 10.1074/jbc.m607556200. [DOI] [PubMed] [Google Scholar]
- [32].Li Q, Xiao H, and Isobe K, “Histone Acetyltransferase Activities of cAMP-Regulated Enhancer-Binding Protein and p300 in Tissues of Fetal, Young, and Old Mice,” Journals Gerontology Ser, vol. 57, no. 3, pp. B93–B98, 2002, doi: 10.1093/gerona/57.3.b93. [DOI] [PubMed] [Google Scholar]
- [33].Yang J, Oza J, Bridges K, Chen KY, and Liu AY-C, “Neural differentiation and the attenuated heat shock response,” Brain Res, vol. 1203, pp. 39–50, 2008, doi: 10.1016/j.brainres.2008.01.082. [DOI] [PubMed] [Google Scholar]
- [34].Liu DJ, Hammer D, Komlos D, Chen KY, Firestein BL, and Liu AY-C, “SIRT1 Knockdown Promotes Neural Differentiation and Attenuates the Heat Shock Response,” J Cell Physiol, vol. 229, no. 9, pp. 1224–1235, 2014, doi: 10.1002/jcp.24556. [DOI] [PubMed] [Google Scholar]
- [35].Zelin E, Zhang Y, Toogun OA, Zhong S, and Freeman BC, “The p23 Molecular Chaperone and GCN5 Acetylase Jointly Modulate Protein-DNA Dynamics and Open Chromatin Status,” Mol Cell, vol. 48, no. 3, pp. 459–470, 2012, doi: 10.1016/j.molcel.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zelin E and Freeman BC, “Lysine Deacetylases Regulate the Heat Shock Response Including the Age-Associated Impairment of HSF1,” J Mol Biol, vol. 427, no. 7, pp. 1644–1654, 2015, doi: 10.1016/j.jmb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Neef DW, Jaeger AM, and Thiele DJ, “Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases,” Nat Rev Drug Discov, vol. 10, no. 12, pp. 930–944, 2011, doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Björk JK et al. , “Heat-shock factor 2 is a suppressor of prostate cancer invasion,” Oncogene, vol. 35, no. 14, pp. 1770–1784, 2016, doi: 10.1038/onc.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mendillo ML et al. , “HSF1 Drives a Transcriptional Program Distinct from Heat Shock to Support Highly Malignant Human Cancers,” Cell, vol. 150, no. 3, pp. 549–562, 2012, doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dai C, Whitesell L, Rogers AB, and Lindquist S, “Heat Shock Factor 1 Is a Powerful Multifaceted Modifier of Carcinogenesis,” Cell, vol. 130, no. 6, pp. 1005–1018, 2007, doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luo J, Solimini NL, and Elledge SJ, “Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction,” Cell, vol. 136, no. 5, pp. 823–837, 2009, doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao YH et al. , “Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth,” Oncogene, vol. 28, no. 42, pp. 3689–3701, 2009, doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- [43].Kourtis N et al. , “FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification,” Nat Cell Biol, vol. 17, no. 3, pp. 322–332, 2015, doi: 10.1038/ncb3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gomez-Pastor R et al. , “Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease,” Nat Commun, vol. 8, no. 1, p. 14405, 2017, doi: 10.1038/ncomms14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dai C et al. , “Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis,” J Clin Invest, vol. 122, no. 10, pp. 3742–3754, 2012, doi: 10.1172/jci62727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dai C and Sampson SB, “HSF1: Guardian of Proteostasis in Cancer,” Trends Cell Biol, vol. 26, no. 1, pp. 17–28, 2016, doi: 10.1016/j.tcb.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Silverthorn DU, Human Physiology: An Integrated Approach, vol. 8. Pearson Education, 2019. [Google Scholar]
- [48].de Tommaso M, Arendt-Nielsen L, Defrin R, Kunz M, Pickering G, and Valeriani M, “Pain in Neurodegenerative Disease: Current Knowledge and Future Perspectives,” Behav Neurol, vol. 2016, pp. 1–14, 2016, doi: 10.1155/2016/7576292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Houry W, “Chaperone-Assisted Protein Folding in the Cell Cytoplasm,” Curr Protein Pept Sc, vol. 2, no. 3, pp. 227–244, 2001, doi: 10.2174/1389203013381134. [DOI] [PubMed] [Google Scholar]
- [50].Brehme M et al. , “A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease,” Cell Reports, vol. 9, no. 3, pp. 1135–1150, 2014, doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Medeiros R, Baglietto- Vargas D, and LaFerla FM, “The Role of Tau in Alzheimer’s Disease and Related Disorders,” Cns Neurosci Ther, vol. 17, no. 5, pp. 514–524, 2011, doi: 10.1111/j.1755-5949.2010.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dou F et al. , “Chaperones increase association of tau protein with microtubules,” Proc National Acad Sci, vol. 100, no. 2, pp. 721–726, 2003, doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tolnay and Probst, “REVIEW: tau protein pathology in Alzheimer’s disease and related disorders,” Neuropath Appl Neuro, vol. 25, no. 3, pp. 171–187, 1999, doi: 10.1046/j.1365-2990.1999.00182.x. [DOI] [PubMed] [Google Scholar]
- [54].Richter-Landsberg C, Wyttenbach A, and Arrigo AP, “Heat Shock Proteins in Neural Cells,” pp. 81–99, 2009, doi: 10.1007/978-0-387-39954-6_7. [DOI] [Google Scholar]
- [55].Harper SJ and Wilkie N, “MAPKs: new targets for neurodegeneration,” Expert Opin Ther Tar, vol. 7, no. 2, pp. 187–200, 2005, doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- [56].Zhang H-T, Zeng L-F, He Q-Y, Tao WA, Zha Z-G, and Hu C-D, “The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5,” Biochimica Et Biophysica Acta Bba - Mol Cell Res, vol. 1863, no. 2, pp. 335–346, 2016, doi: 10.1016/j.bbamcr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang P et al. , “Ubiquitin ligase CHIP regulates OTUD3 stability and suppresses tumour metastasis in lung cancer,” Cell Death Differ, vol. 27, no. 11, pp. 3177–3195, 2020, doi: 10.1038/s41418-020-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Franklin TB, Krueger-Naug AM, Clarke DB, Arrigo A-P, and Currie RW, “The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system,” Int J Hyperther, vol. 21, no. 5, pp. 379–392, 2009, doi: 10.1080/02656730500069955. [DOI] [PubMed] [Google Scholar]
- [59].Leak RK, “Heat shock proteins in neurodegenerative disorders and aging,” J Cell Commun Signal, vol. 8, no. 4, pp. 293–310, 2014, doi: 10.1007/s12079-014-0243-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dou F et al. , “Chaperones increase association of tau protein with microtubules,” Proc National Acad Sci, vol. 100, no. 2, pp. 721–726, 2003, doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Garbuz DG, Zatsepina OG, and Evgen’ev MB, “The Major Human Stress Protein Hsp70 as a Factor of Protein Homeostasis and a Cytokine-Like Regulator,” Mol Biol+, vol. 53, no. 2, pp. 176–191, 2019, doi: 10.1134/s0026893319020055. [DOI] [PubMed] [Google Scholar]
- [62].Molteni M, Gemma S, and Rossetti C, “The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation,” Mediat Inflamm, vol. 2016, pp. 1–9, 2016, doi: 10.1155/2016/6978936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jin J-J, Kim H-D, Maxwell JA, Li L, and Fukuchi K, “Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease,” J Neuroinflamm, vol. 5, no. 1, p. 23, 2008, doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Calabrese V, Stella AMG, Butterfield DA, and Scapagnini G, “Redox Regulation in Neurodegeneration and Longevity: Role of the Heme Oxygenase and HSP70 Systems in Brain Stress Tolerance,” Antioxid Redox Sign, vol. 6, no. 5, pp. 895–913, 2004, doi: 10.1089/ars.2004.6.895. [DOI] [PubMed] [Google Scholar]
- [65].Vulpis E et al. , “Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis,” Oncoimmunology, vol. 6, no. 3, pp. 00–00, 2017, doi: 10.1080/2162402x.2017.1279372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li X et al. , “Quercetin induces mitochondrial biogenesis in experimental traumatic brain injury via the PGC-1α signaling pathway,” American journal of translational research, vol. 8, no. 8, pp. 3558–3566, August. 2016, [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/27648146 [PMC free article] [PubMed] [Google Scholar]
- [67].Stein SC, Georgoff P, Meghan S, Mizra K, and Sonnad SS, “150 Years of Treating Severe Traumatic Brain Injury: A Systematic Review of Progress in Mortality,” J Neurotraum, vol. 27, no. 7, pp. 1343–1353, 2010, doi: 10.1089/neu.2009.1206. [DOI] [PubMed] [Google Scholar]
- [68].Zhang M-H, Zhou X-M, Cui J-Z, Wang K-J, Feng Y, and Zhang H-A, “Neuroprotective effects of dexmedetomidine on traumatic brain injury: Involvement of neuronal apoptosis and HSP70 expression,” Mol Med Rep, vol. 17, no. 6, pp. 8079–8086, 2018, doi: 10.3892/mmr.2018.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mckee AC and Daneshvar DH, “Chapter 4 The neuropathology of traumatic brain injury,” Handb Clin Neurology, vol. 127, pp. 45–66, 2015, doi: 10.1016/b978-0-444-52892-6.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kelly S et al. , “Targeting Expression of hsp70i to Discrete Neuronal Populations Using the Lmo-1 Promoter: Assessment of the Neuroprotective Effects of hsp70i In Vivo and In Vitro,” J Cereb Blood Flow Metabolism, vol. 21, no. 8, pp. 972–981, 2001, doi: 10.1097/00004647-200108000-00010. [DOI] [PubMed] [Google Scholar]
- [71].Zhou W and Yuan J, “Necroptosis in health and diseases,” Semin Cell Dev Biol, vol. 35, pp. 14–23, 2014, doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- [72].Burke JF, Stulc JL, Skolarus LE, Sears ED, Zahuranec DB, and Morgenstern LB, “Traumatic brain injury may be an independent risk factor for stroke,” Neurology, vol. 81, no. 1, pp. 33–39, 2013, doi: 10.1212/wnl.0b013e318297eecf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Fang MC et al. , “Long-term survival after ischemic stroke in patients with atrial fibrillation,” Neurology, vol. 82, no. 12, pp. 1033–1037, 2014, doi: 10.1212/wnl.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Musuka TD, Wilton SB, Traboulsi M, and Hill MD, “Diagnosis and management of acute ischemic stroke: speed is critical,” Can Med Assoc J, vol. 187, no. 12, p. cmaj.140355, 2015, doi: 10.1503/cmaj.140355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Woodruff TM, Thundyil J, Tang S-C, Sobey CG, Taylor SM, and Arumugam TV, “Pathophysiology, treatment, and animal and cellular models of human ischemic stroke,” Mol Neurodegener, vol. 6, no. 1, p. 11, 2011, doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kelly S et al. , “Targeting Expression of hsp70i to Discrete Neuronal Populations Using the Lmo-1 Promoter: Assessment of the Neuroprotective Effects of hsp70i In Vivo and In Vitro,” J Cereb Blood Flow Metabolism, vol. 21, no. 8, pp. 972–981, 2001, doi: 10.1097/00004647-200108000-00010. [DOI] [PubMed] [Google Scholar]
- [77].Doeppner TR et al. , “TAT-Hsp70-Mediated Neuroprotection and Increased Survival of Neuronal Precursor Cells after Focal Cerebral Ischemia in Mice,” J Cereb Blood Flow Metabolism, vol. 29, no. 6, pp. 1187–1196, 2009, doi: 10.1038/jcbfm.2009.44. [DOI] [PubMed] [Google Scholar]
- [78].Doeppner TR, Kaltwasser B, Fengyan J, Hermann DM, and Bähr M, “TAT-Hsp70 Induces Neuroprotection Against Stroke Via Anti-Inflammatory Actions Providing Appropriate Cellular Microenvironment for Transplantation of Neural Precursor Cells,” J Cereb Blood Flow Metabolism, vol. 33, no. 11, pp. 1778–1788, 2013, doi: 10.1038/jcbfm.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Osellame LD, Blacker TS, and Duchen MR, “Cellular and molecular mechanisms of mitochondrial function,” Best Pract Res Cl En, vol. 26, no. 6, pp. 711–723, 2012, doi: 10.1016/j.beem.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Züchner S et al. , “Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A,” Nat Genet, vol. 36, no. 5, pp. 449–451, 2004, doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- [81].Bailon L, Mothe B, Berman L, and Brander C, “Novel Approaches Towards a Functional Cure of HIV/AIDS,” Drugs, vol. 80, no. 9, pp. 859–868, 2020, doi: 10.1007/s40265-020-01322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chan JK and Greene WC, “NF-κB/Rel: agonist and antagonist roles in HIV-1 latency,” Curr Opin Hiv Aids, vol. 6, no. 1, pp. 12–18, 2011, doi: 10.1097/coh.0b013e32834124fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Reuse S et al. , “Synergistic Activation of HIV-1 Expression by Deacetylase Inhibitors and Prostratin: Implications for Treatment of Latent Infection,” Plos One, vol. 4, no. 6, p. e6093, 2009, doi: 10.1371/journal.pone.0006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Williams SA, Chen L, Kwon H, Ruiz- Jarabo CM, Verdin E, and Greene WC, “NF- κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation,” Embo J, vol. 25, no. 1, pp. 139–149, 2006, doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Vallabhapurapu S and Karin M, “Regulation and Function of NF-κB Transcription Factors in the Immune System,” Immunology, vol. 27, no. 1, pp. 693–733, 2009, doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- [86].Pearl LH, Prodromou C, and Workman P, “The Hsp90 molecular chaperone: an open and shut case for treatment,” Biochem J, vol. 410, no. 3, pp. 439–453, 2008, doi: 10.1042/bj20071640. [DOI] [PubMed] [Google Scholar]
- [87].Roesch F et al. , “Hyperthermia Stimulates HIV-1 Replication,” Plos Pathog, vol. 8, no. 7, p. e1002792, 2012, doi: 10.1371/journal.ppat.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kim H, Choi M-S, Inn K-S, and Kim B-J, “Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner,” Sci Rep-uk, vol. 6, no. 1, p. 28896, 2016, doi: 10.1038/srep28896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lee MN et al. , “Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach,” Nat Immunol, vol. 14, no. 2, pp. 179–185, 2013, doi: 10.1038/ni.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Li J, Soroka J, and Buchner J, “The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones,” Biochimica Et Biophysica Acta Bba - Mol Cell Res, vol. 1823, no. 3, pp. 624–635, 2012, doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- [91].Anderson I et al. , “Heat shock protein 90 controls HIV-1 reactivation from latency,” Proc National Acad Sci, vol. 111, no. 15, pp. E1528–E1537, 2014, doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Pan X-Y et al. , “Heat Shock Factor 1 Mediates Latent HIV Reactivation,” Sci Rep-uk, vol. 6, no. 1, p. 26294, 2016, doi: 10.1038/srep26294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kanungo S, Morton J, Neelakantan M, Ching K, Saeedian J, and Goldstein A, “Mitochondrial disorders,” Ann Transl Medicine, vol. 6, no. 24, pp. 475–475, 2018, doi: 10.21037/atm.2018.12.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Labbadia J, Brielmann RM, Neto MF, Lin Y-F, Haynes CM, and Morimoto RI, “Mitochondrial Stress Restores the Heat Shock Response and Prevents Proteostasis Collapse during Aging,” Cell Reports, vol. 21, no. 6, pp. 1481–1494, 2017, doi: 10.1016/j.celrep.2017.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bouchecareilh M and Balch WE, “Proteostasis,” Proc Am Thorac Soc, vol. 8, no. 2, pp. 189–195, 2011, doi: 10.1513/pats.201008-055ms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Koike N, Hatano Y, and Ushimaru T, “Heat shock transcriptional factor mediates mitochondrial unfolded protein response,” Curr Genet, vol. 64, no. 4, pp. 907–917, 2018, doi: 10.1007/s00294-018-0809-9. [DOI] [PubMed] [Google Scholar]
- [97].Shan Y-X, Liu T-J, Su H-F, Samsamshariat A, Mestril R, and Wang PH, “Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells,” J Mol Cell Cardiol, vol. 35, no. 9, pp. 1135–1143, 2003, doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- [98].Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, and Dillmann WH, “Combined and Individual Mitochondrial HSP60 and HSP10 Expression in Cardiac Myocytes Protects Mitochondrial Function and Prevents Apoptotic Cell Deaths Induced by Simulated Ischemia-Reoxygenation,” Circulation, vol. 103, no. 13, pp. 1787–1792, 2001, doi: 10.1161/01.cir.103.13.1787. [DOI] [PubMed] [Google Scholar]
- [99].Fan F et al. , “Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure,” Cell Death Differ, vol. 27, no. 2, pp. 587–600, 2020, doi: 10.1038/s41418-019-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Li H, Yang J, Wang Y, Liu Q, Cheng J, and Wang F, “Neuroprotective effects of increasing levels of HSP70 against neuroinflammation in Parkinson’s disease model by inhibition of NF-κB and STAT3,” Life Sci, vol. 234, p. 116747, 2019, doi: 10.1016/j.lfs.2019.116747. [DOI] [PubMed] [Google Scholar]
- [101].Min S-W, Sohn PD, Cho S-H, Swanson RA, and Gan L, “Sirtuins in neurodegenerative diseases: an update on potential mechanisms,” Front Aging Neurosci, vol. 5, p. 53, 2013, doi: 10.3389/fnagi.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Docrat TF, Nagiah S, Naicker N, Baijnath S, Singh S, and Chuturgoon AA, “The protective effect of metformin on mitochondrial dysfunction and endoplasmic reticulum stress in diabetic mice brain,” Eur J Pharmacol, vol. 875, p. 173059, 2020, doi: 10.1016/j.ejphar.2020.173059. [DOI] [PubMed] [Google Scholar]
- [103].Murphy MP and H. L. III, “Alzheimer’s Disease and the Amyloid-β Peptide,” J Alzheimer’s Dis, vol. 19, no. 1, pp. 311–323, 2010, doi: 10.3233/jad-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Pagani L and Eckert A, “Amyloid-Beta Interaction with Mitochondria,” Int J Alzheimer’s Dis, vol. 2011, p. 925050, 2011, doi: 10.4061/2011/925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Swerdlow RH, “Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease,” J Alzheimer’s Dis, vol. Preprint, no. Preprint, pp. 1–14, 2017, doi: 10.3233/jad-170585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Hirai K et al. , “Mitochondrial Abnormalities in Alzheimer’s Disease,” J Neurosci, vol. 21, no. 9, pp. 3017–3023, 2001, doi: 10.1523/jneurosci.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Veereshwarayya V, Kumar P, Rosen KM, Mestril R, and Querfurth HW, “Differential Effects of Mitochondrial Heat Shock Protein 60 and Related Molecular Chaperones to Prevent Intracellular β-Amyloid-induced Inhibition of Complex IV and Limit Apoptosis*,” J Biol Chem, vol. 281, no. 40, pp. 29468–29478, 2006, doi: 10.1074/jbc.m602533200. [DOI] [PubMed] [Google Scholar]
- [108].Park SJ et al. , “Down-regulation of Mortalin Exacerbates Aβ-mediated Mitochondrial Fragmentation and Dysfunction* * This work was supported by Korean Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea Grant A092042 and the Korea-UK Collaborative Alzheimer’s Disease Research Project, Ministry of Health & Welfare, Republic of Korea Grant A120196.,” J Biol Chem, vol. 289, no. 4, pp. 2195–2204, 2014, doi: 10.1074/jbc.m113.492587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dudeja V et al. , “Heat Shock Protein 70 Inhibits Apoptosis in Cancer Cells Through Simultaneous and Independent Mechanisms,” Gastroenterology, vol. 136, no. 5, pp. 1772–1782, 2009, doi: 10.1053/j.gastro.2009.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Jäättelä M, “Escaping Cell Death: Survival Proteins in Cancer,” Exp Cell Res, vol. 248, no. 1, pp. 30–43, 1999, doi: 10.1006/excr.1999.4455. [DOI] [PubMed] [Google Scholar]
- [111].Sakai K et al. , “Functional inhibition of heat shock protein 70 by VER- 155008 suppresses pleural mesothelioma cell proliferation via an autophagy mechanism,” Thorac Cancer, vol. 12, no. 4, pp. 491–503, 2021, doi: 10.1111/1759-7714.13784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yu B, Yang H, Zhang X, and Li H, “Visualizing and Quantifying the Effect of the Inhibition of HSP70 on Breast Cancer Cells Based on Laser Scanning Microscopy,” Technol Cancer Res T, vol. 17, p. 1533033818785274, 2018, doi: 10.1177/1533033818785274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Budina-Kolomets A et al. , “HSP70 Inhibition Limits FAK-Dependent Invasion and Enhances the Response to Melanoma Treatment with BRAF Inhibitors,” Cancer Res, vol. 76, no. 9, pp. 2720–2730, 2016, doi: 10.1158/0008-5472.can-15-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Workman P, “Altered states: selectively drugging the Hsp90 cancer chaperone,” Trends Mol Med, vol. 10, no. 2, pp. 47–51, 2004, doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [115].Nimmanapalli R, Gerbino E, Dalton WS, Gandhi V, and Alsina M, “HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma,” Brit J Haematol, vol. 142, no. 4, pp. 551–561, 2008, doi: 10.1111/j.1365-2141.2008.07217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Whitesell L and Lindquist SL, “HSP90 and the chaperoning of cancer,” Nat Rev Cancer, vol. 5, no. 10, pp. 761–772, 2005, doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- [117].Kamal A et al. , “A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors,” Nature, vol. 425, no. 6956, pp. 407–410, 2003, doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- [118].Gabbasov R et al. , “Targeted blockade of HSP90 impairs DNA-damage response proteins and increases the sensitivity of ovarian carcinoma cells to PARP inhibition,” Cancer Biol Ther, vol. 20, no. 7, pp. 1–11, 2019, doi: 10.1080/15384047.2019.1595279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mbofung RM et al. , “HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes,” Nat Commun, vol. 8, no. 1, p. 451, 2017, doi: 10.1038/s41467-017-00449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bedin M, Gaben A, Saucier C, and Mester J, “Geldanamycin, an inhibitor of the chaperone activity of HSP90, induces MAPK- independent cell cycle arrest,” Int J Cancer, vol. 109, no. 5, pp. 643–652, 2004, doi: 10.1002/ijc.20010. [DOI] [PubMed] [Google Scholar]
- [121].Zhang M-H et al. , “HSP90 protects apoptotic cleavage of vimentin in geldanamycin-induced apoptosis,” Mol Cell Biochem, vol. 281, no. 1–2, pp. 111–121, 2006, doi: 10.1007/s11010-006-0638-x. [DOI] [PubMed] [Google Scholar]
- [122].McCollum AK et al. , “P-Glycoprotein–Mediated Resistance to Hsp90-Directed Therapy Is Eclipsed by the Heat Shock Response,” Cancer Res, vol. 68, no. 18, pp. 7419–7427, 2008, doi: 10.1158/0008-5472.can-07-5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Supko JG, Hickman RL, Grever MR, and Malspeis L, “Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent,” Cancer Chemoth Pharm, vol. 36, no. 4, pp. 305–315, 1995, doi: 10.1007/bf00689048. [DOI] [PubMed] [Google Scholar]
- [124].Gorska M et al. , “Geldanamycin and its derivatives as Hsp90 inhibitors,” Front Biosci, vol. 17, no. 7, p. 2269, 2012, doi: 10.2741/4050. [DOI] [PubMed] [Google Scholar]
- [125].Saif MW et al. , “Open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of intravenously administered CNF1010 (17-(allylamino)-17-demethoxygeldanamycin [17-AAG]) in patients with solid tumors,” Cancer Chemoth Pharm, vol. 71, no. 5, pp. 1345–1355, 2013, doi: 10.1007/s00280-013-2134-9. [DOI] [PubMed] [Google Scholar]
- [126].Schaefer S, Svenstrup TH, and Guerra B, “The small-molecule kinase inhibitor D11 counteracts 17-AAG-mediated up-regulation of HSP70 in brain cancer cells,” Plos One, vol. 12, no. 5, p. e0177706, 2017, doi: 10.1371/journal.pone.0177706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Smith V, Sausville EA, Camalier RF, Fiebig H-H, and Burger AM, “Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models,” Cancer Chemoth Pharm, vol. 56, no. 2, pp. 126–137, 2005, doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- [128].Floris G et al. , “The Novel HSP90 Inhibitor, IPI-493, Is Highly Effective in Human Gastrointestinal Stromal Tumor Xenografts Carrying Heterogeneous KIT Mutations,” Clin Cancer Res, vol. 17, no. 17, pp. 5604–5614, 2011, doi: 10.1158/1078-0432.ccr-11-0562. [DOI] [PubMed] [Google Scholar]
- [129].Lee J et al. , “153 POSTER IPI-493, a potent, orally bioavailable Hsp90 inhibitor of the ansamycin class,” European J Cancer Suppl, vol. 6, no. 12, p. 49, 2008, doi: 10.1016/s1359-6349(08)72085-8. [DOI] [Google Scholar]
- [130].Somu P and Paul S, “Heat Shock Protein 90 in Human Diseases and Disorders,” Heat Shock Proteins, pp. 159–182, 2019, doi: 10.1007/978-3-030-23158-3_8. [DOI] [Google Scholar]
- [131].Song D et al. , “Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer,” Mol Cancer Ther, vol. 7, no. 10, pp. 3275–3284, 2008, doi: 10.1158/1535-7163.mct-08-0508. [DOI] [PubMed] [Google Scholar]
- [132].Patterson J, Palombella VJ, Fritz C, and Normant E, “IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells,” Cancer Chemoth Pharm, vol. 61, no. 6, pp. 923–932, 2008, doi: 10.1007/s00280-007-0546-0. [DOI] [PubMed] [Google Scholar]
- [133].Oh WK et al. , “Multicenter Phase II Trial of the Heat Shock Protein 90 Inhibitor, Retaspimycin Hydrochloride (IPI-504), in Patients With Castration-resistant Prostate Cancer,” Urology, vol. 78, no. 3, pp. 626–630, 2011, doi: 10.1016/j.urology.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Sessa C et al. , “First-in-Human Phase I Dose-Escalation Study of the HSP90 Inhibitor AUY922 in Patients with Advanced Solid Tumors,” Clin Cancer Res, vol. 19, no. 13, pp. 3671–3680, 2013, doi: 10.1158/1078-0432.ccr-12-3404. [DOI] [PubMed] [Google Scholar]
- [135].Roman D et al. , “Ocular toxicity of AUY922 in pigmented and albino rats,” Toxicol Appl Pharm, vol. 309, pp. 55–62, 2016, doi: 10.1016/j.taap.2016.08.025. [DOI] [PubMed] [Google Scholar]
- [136].Johnson ML et al. , “Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer With Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors,” J Clin Oncol, vol. 33, no. 15, pp. 1666–1673, 2015, doi: 10.1200/jco.2014.59.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Bendell JC et al. , “A Phase 2 Study of the Hsp90 Inhibitor AUY922 as Treatment for Patients with Refractory Gastrointestinal Stromal Tumors,” Cancer Invest, vol. 34, no. 6, pp. 1–6, 2016, doi: 10.1080/07357907.2016.1193746. [DOI] [PubMed] [Google Scholar]
- [138].Felip E et al. , “Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non–Small Cell Lung Cancer,” J Thorac Oncol, vol. 13, no. 4, pp. 576–584, 2018, doi: 10.1016/j.jtho.2017.11.131. [DOI] [PubMed] [Google Scholar]
- [139].Jensen MR et al. , “NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models,” Breast Cancer Res, vol. 10, no. 2, p. R33, 2008, doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Stothert AR et al. , “Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma,” Sci Rep-uk, vol. 7, no. 1, p. 17951, 2017, doi: 10.1038/s41598-017-18344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Mak OW, Chand R, Reynisson J, and Leung IKH, “Identification of Isoform-Selective Ligands for the Middle Domain of Heat Shock Protein 90 (Hsp90),” Int J Mol Sci, vol. 20, no. 21, p. 5333, 2019, doi: 10.3390/ijms20215333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, and Picard D, “Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development,” Cell Stress Chaperones, vol. 23, no. 4, pp. 467–482, 2018, doi: 10.1007/s12192-018-0877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Li J and Buchner J, “Structure, Function and Regulation of the Hsp90 Machinery,” Biomed J, vol. 36, no. 3, pp. 106–117, 2013, doi: 10.4103/2319-4170.113230. [DOI] [PubMed] [Google Scholar]
- [144].Lei W et al. , “Heat-shock protein 90 (Hsp90) promotes opioid-induced anti-nociception by an ERK mitogen-activated protein kinase (MAPK) mechanism in mouse brain,” J Biol Chem, vol. 292, no. 25, pp. 10414–10428, 2017, doi: 10.1074/jbc.m116.769489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Lei W, Duron DI, Stine C, Mishra S, Blagg BSJ, and Streicher JM, “The Alpha Isoform of Heat Shock Protein 90 and the Co-chaperones p23 and Cdc37 Promote Opioid Anti-nociception in the Brain,” Front Mol Neurosci, vol. 12, p. 294, 2019, doi: 10.3389/fnmol.2019.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Khandelwal A et al. , “Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor,” Nat Commun, vol. 9, no. 1, p. 425, 2018, doi: 10.1038/s41467-017-02013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Gaspar N et al. , “Mechanistic Evaluation of the Novel HSP90 Inhibitor NVP-AUY922 in Adult and Pediatric Glioblastoma,” Mol Cancer Ther, vol. 9, no. 5, pp. 1219–1233, 2010, doi: 10.1158/1535-7163.mct-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Okui T et al. , “Antitumor effect of novel HSP90 inhibitor NVP-AUY922 against oral squamous cell carcinoma.,” Anticancer Res, vol. 31, no. 4, pp. 1197–204, 2011. [PubMed] [Google Scholar]
- [149].Gandhi N et al. , “Novel Hsp90 inhibitor NVP-AUY922 radiosensitizes prostate cancer cells,” Cancer Biol Ther, vol. 14, no. 4, pp. 347–356, 2013, doi: 10.4161/cbt.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Walsby EJ, Lazenby M, Pepper CJ, Knapper S, and Burnett AK, “The HSP90 inhibitor NVP- AUY922- AG inhibits the PI3K and IKK signalling pathways and synergizes with cytarabine in acute myeloid leukaemia cells,” Brit J Haematol, vol. 161, no. 1, pp. 57–67, 2013, doi: 10.1111/bjh.12215. [DOI] [PubMed] [Google Scholar]
- [151].Kim SH et al. , “Novel Heat Shock Protein 90 Inhibitor NVP-AUY922 Synergizes With the Histone Deacetylase Inhibitor PXD101 in Induction of Death of Anaplastic Thyroid Carcinoma Cells,” J Clin Endocrinol Metabolism, vol. 100, no. 2, pp. E253–E261, 2015, doi: 10.1210/jc.2014-3101. [DOI] [PubMed] [Google Scholar]
- [152].Park K-S et al. , “The HSP90 inhibitor, NVP-AUY922, sensitizes KRAS-mutant non-small cell lung cancer with intrinsic resistance to MEK inhibitor, trametinib,” Cancer Lett, vol. 372, no. 1, pp. 75–81, 2016, doi: 10.1016/j.canlet.2015.12.015. [DOI] [PubMed] [Google Scholar]
- [153].Lee D-H, Sung KS, Bartlett DL, Kwon YT, and Lee YJ, “HSP90 inhibitor NVP-AUY922 enhances TRAIL-induced apoptosis by suppressing the JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells,” Cell Signal, vol. 27, no. 2, pp. 293–305, 2015, doi: 10.1016/j.cellsig.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Shimamura T et al. , “Ganetespib (STA-9090), a Nongeldanamycin HSP90 Inhibitor, Has Potent Antitumor Activity in In Vitro and In Vivo Models of Non–Small Cell Lung Cancer,” Clin Cancer Res, vol. 18, no. 18, pp. 4973–4985, 2012, doi: 10.1158/1078-0432.ccr-11-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lai C-H et al. , “HSP-90 inhibitor ganetespib is synergistic with doxorubicin in small cell lung cancer,” Oncogene, vol. 33, no. 40, pp. 4867–4876, 2014, doi: 10.1038/onc.2013.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Wang Y, Trepel JB, Neckers LM, and Giaccone G, “STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer.,” Curr Opin Investigational Drugs Lond Engl 2000, vol. 11, no. 12, pp. 1466–76, 2010. [PubMed] [Google Scholar]
- [157].Goyal L et al. , “A phase 2 clinical trial of the heat shock protein 90 (HSP 90) inhibitor ganetespib in patients with refractory advanced esophagogastric cancer,” Invest New Drug, vol. 38, no. 5, pp. 1533–1539, 2020, doi: 10.1007/s10637-019-00889-y. [DOI] [PubMed] [Google Scholar]
- [158].Subramaniam DS et al. , “A Phase Ib/II Study of Ganetespib With Doxorubicin in Advanced Solid Tumors Including Relapsed-Refractory Small Cell Lung Cancer,” Frontiers Oncol, vol. 8, p. 64, 2018, doi: 10.3389/fonc.2018.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lin S-F et al. , “Efficacy of a HSP90 inhibitor, ganetespib, in preclinical thyroid cancer models,” Oncotarget, vol. 5, no. 0, pp. 41294–41304, 2014, doi: 10.18632/oncotarget.17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Nagaraju GP, Mezina A, Shaib WL, Landry J, and El-Rayes BF, “Targeting the Janus-activated kinase-2-STAT3 signalling pathway in pancreatic cancer using the HSP90 inhibitor ganetespib,” Eur J Cancer, vol. 52, pp. 109–119, 2016, doi: 10.1016/j.ejca.2015.10.057. [DOI] [PubMed] [Google Scholar]
- [161].Socinski MA et al. , “A Multicenter Phase II Study of Ganetespib Monotherapy in Patients with Genotypically Defined Advanced Non–Small Cell Lung Cancer,” Clin Cancer Res, vol. 19, no. 11, pp. 3068–3077, 2013, doi: 10.1158/1078-0432.ccr-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Goldman JW et al. , “A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies,” Bmc Cancer, vol. 13, no. 1, p. 152, 2013, doi: 10.1186/1471-2407-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Prince T et al. , “Dual targeting of HSP70 does not induce the heat shock response and synergistically reduces cell viability in muscle invasive bladder cancer,” Oncotarget, vol. 9, no. 66, pp. 32702–32717, 2018, doi: 10.18632/oncotarget.26021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Bussenius J et al. , “Discovery of XL888: A novel tropane-derived small molecule inhibitor of HSP90,” Bioorg Med Chem Lett, vol. 22, no. 17, pp. 5396–5404, 2012, doi: 10.1016/j.bmcl.2012.07.052. [DOI] [PubMed] [Google Scholar]
- [165].Eroglu Z et al. , “Combined BRAF and HSP90 inhibition in patients with unresectable BRAF V600E mutant melanoma,” Clin Cancer Res, vol. 24, no. 22, p. clincanres.0565.2018, 2018, doi: 10.1158/1078-0432.ccr-18-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Azimi A et al. , “Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors,” Mol Syst Biol, vol. 14, no. 3, p. e7858, 2018, doi: 10.15252/msb.20177858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Chiosis G, Lucas B, Shtil A, Huezo H, and Rosen N, “Development of a Purine-Scaffold Novel Class of Hsp90 Binders that Inhibit the Proliferation of Cancer Cells and Induce the Degradation of Her2 Tyrosine Kinase,” Bioorgan Med Chem, vol. 10, no. 11, pp. 3555–3564, 2002, doi: 10.1016/s0968-0896(02)00253-5. [DOI] [PubMed] [Google Scholar]
- [168].Dickson MA et al. , “Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors,” Ann Oncol, vol. 24, no. 1, pp. 252–257, 2013, doi: 10.1093/annonc/mds275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Hong D et al. , “Phase I Study of BIIB028, a Selective Heat Shock Protein 90 Inhibitor, in Patients with Refractory Metastatic or Locally Advanced Solid Tumors,” Clin Cancer Res, vol. 19, no. 17, pp. 4824–4831, 2013, doi: 10.1158/1078-0432.ccr-13-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].HE W et al. , “Hsp90 inhibitor, BIIB021, induces apoptosis and autophagy by regulating mTOR-Ulk1 pathway in imatinib-sensitive and -resistant chronic myeloid leukemia cells,” Int J Oncol, vol. 48, no. 4, pp. 1710–1720, 2016, doi: 10.3892/ijo.2016.3382. [DOI] [PubMed] [Google Scholar]
- [171].Caldas-Lopes E et al. , “Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models,” Proc National Acad Sci, vol. 106, no. 20, pp. 8368–8373, 2009, doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kale Ş, Korcum AF, Dündar E, and Erin N, “HSP90 inhibitor PU-H71 increases radiosensitivity of breast cancer cells metastasized to visceral organs and alters the levels of inflammatory mediators,” Naunyn-schmiedeberg’s Archives Pharmacol, vol. 393, no. 2, pp. 253–262, 2020, doi: 10.1007/s00210-019-01725-z. [DOI] [PubMed] [Google Scholar]
- [173].Yu MK et al. , “MPC-3100, a fully synthetic, orally bioavailable Hsp90 inhibitor, in cancer patients.,” J Clin Oncol, vol. 28, no. 15_suppl, pp. e13112–e13112, 2010, doi: 10.1200/jco.2010.28.15_suppl.e13112. [DOI] [Google Scholar]
- [174].Speranza G et al. , “First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile,” Invest New Drug, vol. 36, no. 2, pp. 230–239, 2018, doi: 10.1007/s10637-017-0495-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Shapiro GI et al. , “First-in-Human Phase I Dose Escalation Study of a Second-Generation Non-Ansamycin HSP90 Inhibitor, AT13387, in Patients with Advanced Solid Tumors,” Clin Cancer Res, vol. 21, no. 1, pp. 87–97, 2015, doi: 10.1158/1078-0432.ccr-14-0979. [DOI] [PubMed] [Google Scholar]
- [176].Massey AJ et al. , “A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells,” Cancer Chemoth Pharm, vol. 66, no. 3, pp. 535–545, 2010, doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- [177].Cavanaugh A, Juengst B, Sheridan K, Danella JF, and Williams H, “Combined inhibition of heat shock proteins 90 and 70 leads to simultaneous degradation of the oncogenic signaling proteins involved in muscle invasive bladder cancer,” Oncotarget, vol. 6, no. 37, pp. 39821–39838, 2015, doi: 10.18632/oncotarget.5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Jones RJ et al. , “Intravenous Immunoglobulin G Suppresses Heat Shock Protein (HSP)-70 Expression and Enhances the Activity of HSP90 and Proteasome Inhibitors,” Front Immunol, vol. 11, p. 1816, 2020, doi: 10.3389/fimmu.2020.01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Li X et al. , “Validation of the Hsp70–Bag3 Protein–Protein Interaction as a Potential Therapeutic Target in Cancer,” Mol Cancer Ther, vol. 14, no. 3, pp. 642–648, 2015, doi: 10.1158/1535-7163.mct-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Yaglom JA et al. , “Cancer cell responses to Hsp70 inhibitor JG-98: Comparison with Hsp90 inhibitors and finding synergistic drug combinations,” Sci Rep-uk, vol. 8, no. 1, p. 3010, 2018, doi: 10.1038/s41598-017-14900-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Koren J and Blagg BSJ, “HSF1 and Molecular Chaperones in Biology and Cancer,” Adv Exp Med Biol, vol. 1243, pp. 135–146, 2020, doi: 10.1007/978-3-030-40204-4_9. [DOI] [PubMed] [Google Scholar]
- [182].Prince T et al. , “Dual targeting of HSP70 does not induce the heat shock response and synergistically reduces cell viability in muscle invasive bladder cancer,” Oncotarget, vol. 9, no. 66, pp. 32702–32717, 2018, doi: 10.18632/oncotarget.26021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Marcu MG, Chadli A, Bouhouche I, Catelli M, and Neckers LM, “The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-binding Domain in the Carboxyl Terminus of the Chaperone*,” J Biol Chem, vol. 275, no. 47, pp. 37181–37186, 2000, doi: 10.1074/jbc.m003701200. [DOI] [PubMed] [Google Scholar]
- [184].Dai J et al. , “Fungal mycotoxin penisuloxazin A, a novel C-terminal Hsp90 inhibitor and characteristics of its analogues on Hsp90 function related to binding sites,” Biochem Pharmacol, vol. 182, p. 114218, 2020, doi: 10.1016/j.bcp.2020.114218. [DOI] [PubMed] [Google Scholar]
- [185].Lian B, Lin Q, Tang W, Qi X, and Li J, “SUV39H1 is a New Client Protein of Hsp90 Degradated by Chaetocin as a Novel C-Terminal Inhibitor of Hsp90,” Biomol Ther, vol. 29, no. 1, pp. 73–82, 2021, doi: 10.4062/biomolther.2020.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Eskew JD et al. , “Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells,” Bmc Cancer, vol. 11, no. 1, p. 468, 2011, doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Zhao H, Anyika M, Girgis A, and Blagg BSJ, “Novologues containing a benzamide side chain manifest anti-proliferative activity against two breast cancer cell lines,” Bioorg Med Chem Lett, vol. 24, no. 15, pp. 3633–3637, 2014, doi: 10.1016/j.bmcl.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Hargitai J et al. , “Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1,” Biochem Bioph Res Co, vol. 307, no. 3, pp. 689–695, 2003, doi: 10.1016/s0006-291x(03)01254-3. [DOI] [PubMed] [Google Scholar]
- [189].Chatterjee BK et al. , “Stimulation of heat shock protein 90 chaperone function through binding of a novobiocin analog KU-32,” J Biol Chem, vol. 294, no. 16, pp. 6450–6467, 2019, doi: 10.1074/jbc.ra118.002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Zhang L, Zhao H, Blagg BSJ, and Dobrowsky RT, “C-Terminal Heat Shock Protein 90 Inhibitor Decreases Hyperglycemia-induced Oxidative Stress and Improves Mitochondrial Bioenergetics in Sensory Neurons,” J Proteome Res, vol. 11, no. 4, pp. 2581–2593, 2012, doi: 10.1021/pr300056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Li C, Ma J, Zhao H, Blagg BSJ, and Dobrowsky RT, “Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-Jun,” Asn Neuro, vol. 4, no. 7, p. e00102, 2012, doi: 10.1042/20120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ma J, Pan P, Anyika M, Blagg BSJ, and Dobrowsky RT, “Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons,” Acs Chem Neurosci, vol. 6, no. 9, pp. 1637–1648, 2015, doi: 10.1021/acschemneuro.5b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].You Z, Zhang Z, Blagg BSJ, and Dobrowsky RT, “KU-596 decreases mitochondrial superoxide and improves bioenergetics following downregulation of manganese superoxide dismutase in diabetic sensory neurons,” Exp Neurol, vol. 313, pp. 88–97, 2019, doi: 10.1016/j.expneurol.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Birar VC, Gelis I, Zuo A, and Blagg BSJ, “Synthesis of paramagnetic ligands that target the C-terminal binding site of Hsp90,” Bioorg Med Chem Lett, vol. 30, no. 16, p. 127303, 2020, doi: 10.1016/j.bmcl.2020.127303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].DeFranco DB, Ho L, Falke E, and Callaway CW, “Small molecule activators of the heat shock response and neuroprotection from stroke,” Curr Atheroscler Rep, vol. 6, no. 4, pp. 295–300, 2004, doi: 10.1007/s11883-004-0061-2. [DOI] [PubMed] [Google Scholar]
- [196].West JD, Wang Y, and Morano KA, “Small Molecule Activators of the Heat Shock Response: Chemical Properties, Molecular Targets, and Therapeutic Promise,” Chem Res Toxicol, vol. 25, no. 10, pp. 2036–2053, 2012, doi: 10.1021/tx300264x. [DOI] [PMC free article] [PubMed] [Google Scholar]