

Abstract
Background
Hypertrophic cardiomyopathy (HCM) is a complex disease partly explained by the effects of individual gene variants on sarcomeric protein biomechanics. At the cellular level, HCM mutations most commonly enhance force production, leading to higher energy demands. Despite significant advances in elucidating sarcomeric structure-function relationships, there is still much to be learned about the mechanisms that link altered cardiac energetics to HCM phenotypes. In this work, we test the hypothesis that changes in cardiac energetics represent a common pathophysiologic pathway in HCM.
Methods
We performed a comprehensive multi-omics profile of the molecular (transcripts, metabolites, and complex lipids), ultrastructural, and functional components of HCM energetics using myocardial samples from 27 HCM patients and 13 normal controls (donor hearts).
Results
Integrated omics analysis revealed alterations in a wide array of biochemical pathways with major dysregulation in fatty acid metabolism, reduction of acylcarnitines, and accumulation of free fatty acids. HCM hearts showed evidence of global energetic decompensation manifested by a decrease in high energy phosphate metabolites [ATP, ADP, and phosphocreatine (PCr)] and a reduction in mitochondrial genes involved in creatine kinase and ATP synthesis. Accompanying these metabolic derangements, electron microscopy showed an increased fraction of severely damaged mitochondria with reduced cristae density, coinciding with reduced citrate synthase (CS) activity and mitochondrial oxidative respiration. These mitochondrial abnormalities were associated with elevated reactive oxygen species (ROS) and reduced antioxidant defenses. However, despite significant mitochondrial injury, HCM hearts failed to upregulate mitophagic clearance.
Conclusions
Overall, our findings suggest that perturbed metabolic signaling and mitochondrial dysfunction are common pathogenic mechanisms in patients with HCM. These results highlight potential new drug targets for attenuation of the clinical disease through improving metabolic function and reducing mitochondrial injury.
Keywords: hypertrophic cardiomyopathy, altered metabolism, mitochondrial abnormalities, reactive oxygen species, mitophagy
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common heritable cardiovascular disease, affecting 1 in 500 individuals, and a leading cause of heart failure and sudden death1–4. HCM is characterized by often asymmetric thickening of the left ventricle (LV), fibrosis and reduced diastolic function. Systolic function is usually hyperdynamic initially, but can progress to systolic dysfunction and clinical heart failure5. Additional clinical phenotypes include LV outflow tract (LVOT) obstruction and arrhythmia6, 7. To date, no treatment has been shown to slow disease progression, although mavacamten has shown promise in reducing LV hypertrophy8. Advances in genetics over the past two decades have uncovered over 1,000 mutations associated with HCM most often in sarcomeric genes such as cardiac myosin-binding protein-C (MYBPC3) and β-cardiac myosin (MYH7)3, 9. Biochemical and biophysical studies of isolated human cardiac myosin suggest that different HCM mutations can have heterogeneous effects on actin gliding velocity, intrinsic force, and ATPase activity3, 10, 11. At the cellular level, most HCM mutations increase total force production and ATP utilization, resulting in higher myocardial energy demand12. Inefficient energy utilization for force generation has been suggested as a primary biophysical consequence of HCM sarcomeric mutations, leading to energetic stress and adverse remodeling12–15,16. The normal heart has an extreme degree of metabolic flexibility, utilizing various substrates to meet changes in energy demand4. Alterations in cardiac substrate utilization have been associated with compromised energy supply in HCM17–21 including lipid metabolism and mitochondrial oxidative phosphorylation21. Of note, patients with mutations in mitochondrial DNA often develop an HCM phenotype, indicating that primary energetic alterations can also lead to HCM pathology22, 23. Early electron micrographic studies demonstrated mitochondrial fission and disruption of cristae before their role in cardiac remodeling was fully understood24–26. Impairment of mitochondrial function and morphology have more recently been described in murine models20, 27 and in HCM patients19. Despite evidence linking energy metabolism and HCM, the molecular mechanisms involved in the initiation and progression of HCM disease are still largely unknown as are the role of alterations in mitochondrial structure and function.
We performed multi-omics profiling of human myocardial tissue including metabolome, lipidome and transcriptome; ultrastructural tissue analysis using transmission electron microscopy (TEM); and mitochondrial functional studies. We uncovered a distinct metabolic profile in HCM, accompanied by markedly abnormal mitochondrial structure, respiratory dysfunction, and failure to upregulate mitophagic clearance, a normal mechanism for maintenance of mitochondrial integrity. These diffuse metabolic alterations could help identify new treatments targeted on rescuing progressive metabolic dysfunction and mitochondrial damage at an earlier stage of disease.
METHODS
The data that support the findings of the present study are available from the corresponding author upon reasonable request.
Study cohort and myocardial sample collection
HCM patients.
27 patients who underwent septal myectomy for clinical indications Inclusion criteria: normal or hyperdynamic LV function (ejection fraction≥55%) with a gradient across the LVOT (See Table 1 and Supplemental Materials online for details).
Table 1.
Characteristics of 42 subjects included in the cohort.
| Control | Disease | ||
|---|---|---|---|
| Group | Donor | Mitral Stenosis | HCM |
| NO. of patient samples | 13 | 2 | 27 |
| Female, n (%) | 9 (69) | 2 (100) | 6 (22) |
| Age, year | 50 ± 10 | 49 ± 6 | 54 ± 14 |
| Positive Family Hx, n (%) | 8 (30) | ||
|
| |||
|
Mutations
*
| |||
| MYBPC3, n (%) | 7 (28) | ||
| MYH7, n (%) | 3 (12) |
||
| Others**,***, n (%) | 2 (8) | ||
| VUS or no mutation, n (%) | 13 (52) | ||
|
| |||
|
Medication
| |||
| β-blocker, n (%) | 11 (41) | ||
| Calcium blocker, n (%) | 19 (71) | ||
| Statin, n (%) | 14 (52) | ||
|
| |||
|
Echocardiogram
| |||
| LVEF | 58 ± 6 | 62 ± 6 | 67 ± 7 |
| IVSd, cm | 1.8 ± 0.5 | ||
| LVPWd, cm | 1.4 ± 0.4 | ||
| IVSd/LVPWd ratio | 1.4 ± 0.6 | ||
| LVOT peak grad 40-60 mmHg, n (%) | 2 (8)# | ||
| LVOT peak grad 60-90 mmHg, n (%) | 4 (15) | ||
| LVOT peak grad > 90 mmHg, n (%) | 20 (75) | ||
| LV Mass (ASE Method), g/m2; (LV mass index) | 264 ± 100; (134 ± 45)## | ||
|
| |||
|
Magnetic resonance imaging (MRI)
| |||
| Delayed Enhancement (range) | 1.5 ± 1.7 (0 - 4) | ||
| LV Mass, g/m2; (LV mass index) | 187 ± 43; (94 ± 18)$ | ||
Continuous variables are expressed as mean ± SD and categorical variables as number (percentage); normal LV mass index28 in men, 70.0 ± 8.9 g/m2 and in women, 60.7 ± 8.1 g/m2.
Genetic testing was performed on 25 patients.
one patient had two mutations.
mutations in TNNT, CSRP, and SCN.
in one patient with a resting gradient of 33 mmHg, exercise gradient was not available.
left ventricular mass on 24 patients calculated from echo data.
left ventricular mass on 9 patients calculated from MRI data.
HCM, hypertrophic cardiomyopathy; MYBPC, myosin binding protein C; MYH, myosin heavy chain; VUS, variants of unknown significance; LVEF, left ventricular ejection fraction; IVSd, interventricular septum diameter in diastole; LVPWd, left ventricular posterior wall thickness in diastole; LVOT peak grad, left ventricular outflow tract peak gradient; ASE, American Society of Echocardiography.
Controls.
Myocardial tissue was obtained from 13 donor hearts with no cardiac history, and 2 patients with primary mitral stenosis without significant insufficiency. The Stanford Institutional Review Board approved the study. All subjects provided written informed consent.
An overview of all techniques and data analysis is summarized in Figure 1A. A detailed method section including a list of software and reagents (Table I and Table II in the Supplement) is available in the Supplemental Materials online.
Figure 1. Study design and molecular profiles in HCM.
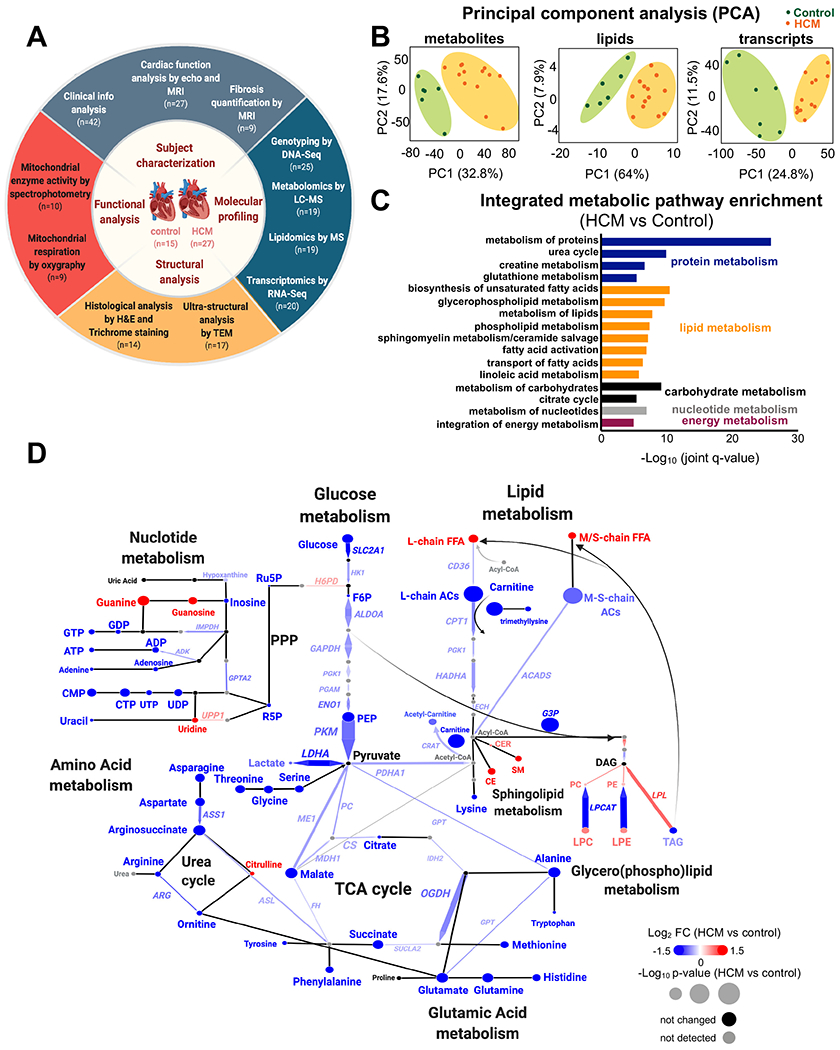
(A) Summary of the experimental design workflow. (B) Metabolomic, lipidomic, and transcriptomic profiling was performed on left ventricle tissue from HCM patients and donor controls. Principal component analysis (PCA) plots of all 6,189 metabolite features(left), 728 lipids (middle), and 48,167 transcripts (right) clearly separate the profiles of HCM from controls. (C) Top metabolic pathways enriched in HCM identified by IMPaLA, integrating metabolites, lipids, and transcripts (FDR<0.05). Metabolic pathways were categorized as follows: proteins, blue; lipids, orange; carbohydrates, black; nucleotides, gray and energy metabolism (purple). (D) Integrated network analysis combining all omic datasets using Shiny Genes and Metabolites (GAM) platform. Resulting networks were annotated and plotted in Cytoscape. Each node (circle) represents a metabolite or lipid and each edge connecting nodes represents an enzyme-encoding transcript based on biochemical relationships. The arrowhead determines the principal direction of the biochemical reaction. The size of each node and of connecting line reflects their p value; the color represents the relative change (blue, decrease; red, increase; black, not significant; and gray, not detected). n=6 control and n=13 HCM samples for metabolites/lipids; n=7 control and n=13 HCM for transcripts. FFA, free fatty acids; M/S-chain, medium/short-chain; L-chain, long chain; AC, acylcarnitine; G3P, glyceraldehyde 3-phosphate; PPP, pentose phosphate pathway; Ru5P, ribulose-5-phosphate; R5P, ribose 5-phosphate; CER, ceramide; CE, cholesterol ester; PC, phosphatidylcholine; PE, phosphatidylethanolamine; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; DAG, diacylglycerol; and TAG, triacylglycerol.
Statistical Analysis
Data are shown as mean ± standard deviation (SD) or standard error (SE). Student’s t-test and Wilcoxon-Mann-Whitney U test were used for two-group comparisons of all non-omics data. P<0.05 was considered significant. For omics data, our statistical methods are described in the respective methods sections. Sample sizes are indicated in Figure 1A and figure legends. Pearson’s correlation coefficient and linear regression analyses were performed using pandas and scipy.
RESULTS
Cohort characteristics
LV septal myectomy samples were obtained from 27 patients with HCM: 22% women; age 54±14 years (mean±SD); and ejection fraction 67±7% (Table 1). Genetic testing was performed in 25 subjects. Twelve (48%) had a known or likely pathogenic HCM variants. Seven were identified in MYBPC3; three in MYH7; and one each in CSRP3, TNNT2, and SCN5A. Thirteen patients had one or more variants of unknown significance or no mutation detected. All patients had increased LV mass by both echocardiography and magnetic resonance imaging (MRI). Thirteen donor hearts were used as controls: 69% women; age, 50±10 years; and ejection fraction, 58±6%. Two additional patients, with mitral stenosis undergoing valve repair, were used as controls for mitochondrial respiration assays only: all women; age, 49±6 years; and ejection fraction, 62±6%. Clinical features of the study cohort are summarized in Table 1. Detailed clinical characteristics on HCM patients and controls are described in Table III and Table IV in the Supplement. Sample workflow and assay details are described in Table V in the Supplement.
Level of fibrosis was measured using gadolinium-enhanced cardiac MRI in 9 patients and was rated as mild (<4%) (Figure IA and Figure IB in the Supplement). However, given the limited resolution of MRI for detection of focal and interstitial fibrosis29, we also performed histological analysis using hematoxylin/eosin and Masson Trichrome. HCM samples showed marked myocyte hypertrophy and increased interstitial collagen (%fibrosis HCM=11 vs. control=3.5) (Figure IC and Figure ID in the Supplement). We then analyzed the correlation between the degree of tissue fibrosis with the expression of fibrotic genes (ECM organization gene-set ; GO-0030198) and found only few genes were positively correlated (Excel I in the Supplement). Altogether, these data suggest that there is a mild/moderate level of fibrosis in septal myectomy samples.
Distinct molecular profiles discriminate HCM from controls
A multi-omics approach was used to characterize molecular composition of HCM (n=13) and control (n=6-7) myocardium as well as gaining insights into regulatory mechanisms. Metabolite and lipid profiling was performed using untargeted and targeted mass spectrometry and gene expression by RNA sequencing. A large proportion of metabolites and lipids were significantly altered (FDR<0.05; 40% (2,456/6,189) and 73% (530/728), respectively) (Figure II, Excel II, and Excel III in the Supplement), while 10% (5,033) of a total of 48,167 genes were differentially expressed in HCM (FDR<0.05) (Figure III in the Supplement). Principal component analysis (PCA) illustrates a clear separation of HCM from controls across omic datasets (Figure 1B).
Integrative omics reveals impaired energy metabolism in HCM
Integrated Molecular Pathway Level Analysis (IMPaLA)30 was used to identify dysregulated pathways. Pathway enrichment revealed marked alterations in all metabolic pathways including lipids, amino acids, carbohydrates, and nucleotides suggesting a global dysregulation in energy metabolism (Figure 1C). A summary of the major metabolic changes was plotted on a metabolic network map (Figure 1D), demonstrating concordant alterations in the transcription of key metabolic enzymes and their associated metabolites or lipids.
In lipid metabolism, we observed a significant increase in free fatty acid (FFA) concentration (Figure 1D, Figure 2A, Figure IVA in the Supplement), despite reduced expression of the genes involved in their transport across the plasma membrane (SLC27A4, FABP4, and CD36) (Figure 2B, Figure IVB in the Supplement). After entering the cells, FFA are converted to acyl-CoAs and then acylcarnitine (AC) to enable entry into the mitochondria and ß-oxidization4. HCM myocardium showed a decrease in the abundance of all ACs, with long-chain ACs (>C14) the most impacted (Figure 1D, Figure 2C). This suggests a defect in conversion of FFA to AC, consistent with the marked reduction in free carnitine along with decreased trimethyllysine, the precursor of free carnitine (Figure 1D, Figure 2D). Consistent with this, mitochondrial carnitine acetyltransferase (CRAT), which catalyzes the conversion of acyl-CoA to Acetyl-carnitine using free carnitine, was reduced (Figure 1D). Additionally, genes associated with transporting ACs across the mitochondrial membrane (SLC25A, and SLC22A5) and carnitine palmitoyltransferase I (CPT1) which facilitate their transport were reduced in HCM (Figure 1D, Figure 2B, Figure IVB and Figure IVC in the Supplement). Importantly, the expression of genes encoding key enzymes involved in fatty acid β-oxidation were all reduced including: ACADVL, responsible for the first step of long-chain fatty acid oxidation; ACSL1, converting long-chain fatty acids to their active acyl-CoAs; HADHA and HADHB, catalyzing the last three steps of long-chain fatty acid oxidation; and ECH1, the final step of fatty acid oxidation to produce acetyl CoA (Figure 1D, Figure 2B). Besides entering mitochondria for oxidation, acyl-CoAs can also enter the de novo sphingolipid synthesis pathway to form ceramide (CER) and sphingomyelin (SM) which were both elevated in HCM (Figure 1D, Figure 2E). Similarly, there were significant increases in several phospholipids and lysophospholipids including cholesterol esters (CE), phosphatidylcholines (PC), phosphatidylethanolamines (PE), lysophosphatidylcholines (LPC) and lysophosphatidylethanolamines (LPE) (Figure 1D, Figure IVD–F in the Supplement). Despite the normal heart’s preference for lipids as an energy substrate, several studies have shown pathological effects of lipid overload in the heart, especially CER31. Triacylglycerol (TAG), the main source of lipid storage, however, was lower in HCM, with no changes in diacylglycerol (DAG), suggesting that overall energy storage (likely as lipid droplets) was lower (Figure 1D, Figure IVG in the Supplement). Consistent with reduced TAG levels, lipoprotein lipase (LPL), which hydrolyzes TAG for fatty acid generation, was significantly increased, along with downregulation of DGAT1, which forms TAG from DAG for energy storage (Figure 1D, Figure IVH in the Supplement).
Figure 2. Dysregulated metabolism in HCM.
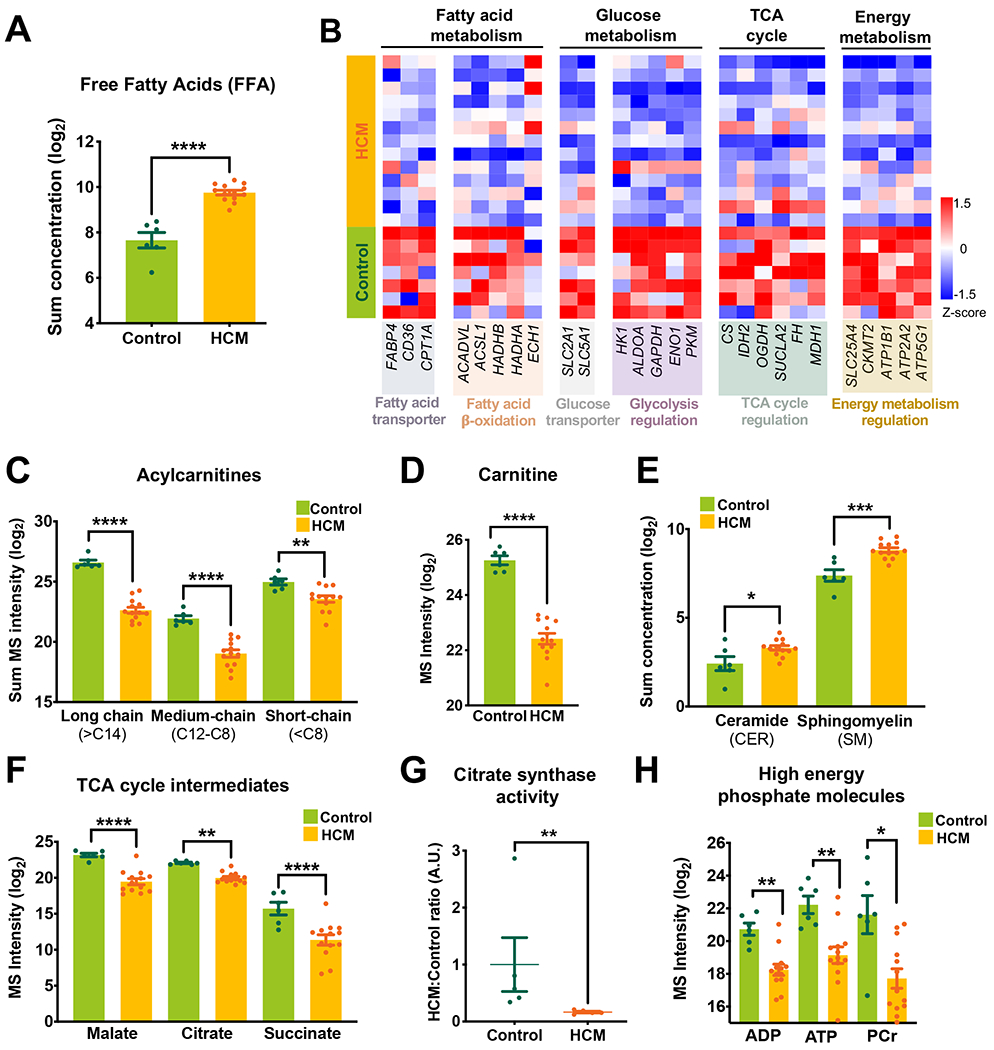
(A) Total concentration (log2 scale) of free fatty acids (FFA) in HCM as measured by the targeted lipidomics platform. (B) Heatmap shows overall changes in genes regulating myocardial energetics. Shown are expression of genes encoding major fatty acid transporters, β-oxidation enzymes, a glucose transporter, and enzymes involved in regulation of glycolysis, TCA cycle, and mitochondrial energy metabolism. (C-F) Levels of acylcarnitines (total concentration, log2 scale), carnitine, sphingolipids (total concentration), and TCA cycle metabolites measured by untargeted metabolomics. (G) Citrate synthase (CS) activity in HCM vs control (spectrophotometry analysis). (H) High energy phosphate metabolites measured by untargeted metabolomics. Error bars represent mean ± SEM. n=6 control and n=13 HCM in A, C-F, and H; n=7 control and n=13 HCM in B; n=5 control and n=5 HCM in G. Between-group comparisons performed using Whitney U test in A, C, E, and G. Two-sided Welch’s t-test or Wald test with Benjamini-Hochberg’s FDR correction method used in B, D, F and H. *p<0.05, **p<0.01, ***p<0.005, or ****p<0.001.
In addition to changes in lipids, carbohydrate metabolism was significantly decreased in HCM as demonstrated by downregulation of GLUT1 (SLC2A1) and SGLT1 (SLC5A1, sodium-glucose linked transporter), reduced levels of glucose, glycolysis intermediates (fructose 6-phosphate, phosphoenolpyruvic acid) and their associated enzymes (HK1, ALDOA, GAPDH, ENO1, PKM), as well as a decrease in pentose phosphate pathway metabolites (ribose-5-phosphate|ribulose 5-phosphate) (Figure 1D, Figure 2B, Figure VA–C in the Supplement). Pyruvate dehydrogenase (PDHA1), which converts pyruvate to acetyl-CoA, a primary link between glycolysis and the TCA cycle, was reduced (Figure 1D, Figure VD in the Supplement). Pyruvate can also enter the TCA cycle by forming malate via malic enzyme 1 (ME1) and pyruvate carboxylase (PC)32, both of which were decreased in HCM (Figure 1D, Figure VD in the Supplement). Notably, TCA cycle intermediates (malate, citrate, succinate) and their associated genes (CS, IDH2, OGDH, SUCLA2, FH, and MDH1) were all decreased (Figure 1D, Figure 2B, Figure 2F). In line with these observations, citrate synthase activity was reduced (Figure 2G). In addition, most amino acids (16/20) as well as key intermediates of nucleotide biosynthesis (NAD, ATP, GDP, CMP) were also decreased, suggesting a global energetic decompensation (Figure 1D, Figure VE–G in the Supplement).
Consistent with the notion of the energetic imbalance in HCM, there was a significant reduction in high energy phosphate molecules (ATP, ADP, PCr) along with reduced expression of genes in the phosphocreatine–creatine kinase (PCr-CK) system including ADP/ATP translocase 1 (SLC25A4), mitochondrial creatine kinase II (CKMT2) and several subunits of ATP synthase machinery (Figure 2B, Figure 2H). These perturbations could compromise energy transfer between mitochondria and the contractile apparatus, exhausting myocardial energy reserve and contributing to the development of contractile dysfunction.
Together these results suggest that HCM pathophysiology induces metabolic alterations that limit fatty acid oxidation and impair entry of several anaplerotic substrates into the TCA cycle. These changes, combined with a decrease in the PCr-CK and ATP synthesis, suggest a globally reduced capacity for mitochondrial oxidative metabolism, ultimately reducing cardiac energy generation in HCM.
HCM is associated with impaired mitochondrial ultrastructure and function
We next sought to characterize the extent to which the above metabolic alterations were associated with alterations in mitochondrial morphology, dynamics and function. Consistent with increased energetic stress, TEM showed a profound disruption in mitochondrial ultrastructure, in which a subset of interfibrillar mitochondria (IFM) were swollen with disorganized and reduced cristae density (Figure 3A). Quantification of cristae density confirmed a marked increase in the percent of mitochondria with severely damaged cristae (Figure 3B–D, Figure VI in the Supplement). To evaluate mitochondrial morphology at a higher resolution, we used electron tomography to reconstruct a 3D volume and found a high variance in cristae density between individual mitochondria in the same sample (Figure 3E, e–e”), consistent with heterogeneous mitochondrial injury. Importantly, these morphological changes were correlated with downregulation of key genes regulating mitochondrial membrane organization, respiratory chain complex assembly, and cristae formation (Figure 3F).
Figure 3. Mitochondrial damage in HCM.
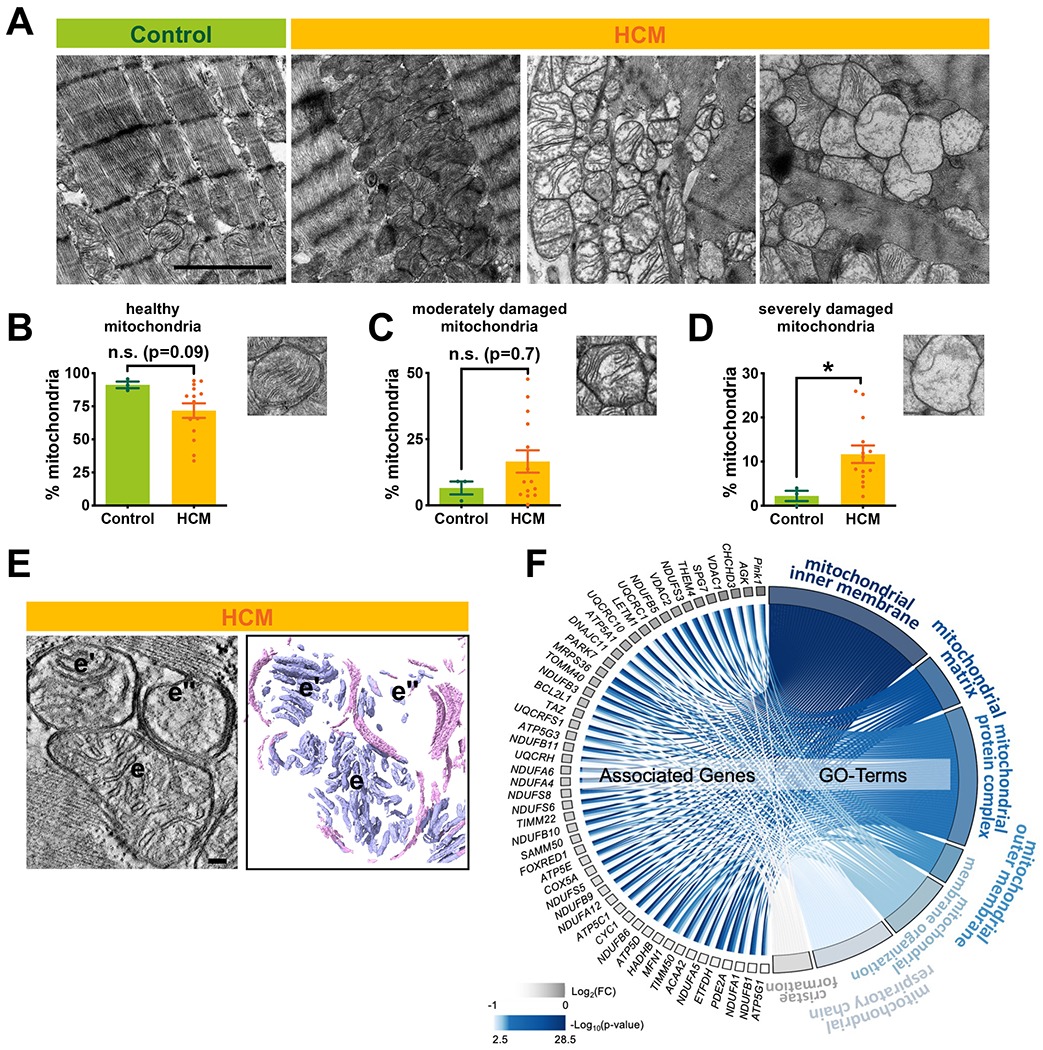
(A) Representative electron micrographs of myocardial tissue from donor and HCM. Each image represents one individual subject (scale bar=2µm). (B-D) Quantitative measurements of interfibrillar mitochondrial cristae density using ImageJ (~20 randomly selected images from each sample). (E) A slice through reconstructed 3D volume and corresponding segmented 3D volumes showing heterogeneous mitochondrial morphology within a single HCM sample (scale bar=100nm). (e-e”) Mitochondrial membranes are color coded in pink and cristae are shown in blue. (F) Chord plot representation of 51 differentially expressed genes (HCM vs control, FDR<0.05) from 7 enriched pathways generated by GOplot. The color map represents fold change of genes (log2 scale) and p value of go terms (−log10 scale). Error bars represent mean ± SEM. n=3 control and n=14 HCM in B-D; n=7 control and n=13 HCM in F. Between-group comparisons performed using Whitney U test. *p<0.05 or n.s., not significant (p>0.05).
In addition to disruption of cristae architecture, we also found increased mitochondrial number and a trend towards a decrease in mitochondrial size (Figure VII in the Supplement), suggestive of mitochondrial fission, a response to pathologic stress33. However, expression of key regulators of both mitochondrial fission (Drp1, phosphorylated-Drp1) and fusion (Mfn1/2, Opa1) showed no evidence of activation of these mediators (Figure VIIIA–F in the Supplement). Transcript levels of regulators of mitochondrial biogenesis (PPARGC1, PPARA, NRF1) as well as protein level of PGC1-a were all unchanged (Figure VIIIG and Figure VIIIH in the Supplement). Taken together, these results show that HCM is associated with substantial mitochondrial ultrastructural remodeling, characterized by a reduction in cristae density, but without significant activation of the fission/fusion and biogenesis processes, at least at the clinical time point at which we made this assessment.
We next sought to determine whether these mitochondrial morphologic abnormalities resulted in mitochondrial dysfunction, and thus could contribute to HCM pathogenesis. Consistent with our findings of reduced ATP and ADP content, we found decreased oxidative phosphorylation capacity through complex V (ATP synthase) measured by Oroboros oximeter (Figure 4A, Figure 4B, Figure IXA–C in the Supplement). Activity of complexes II and V and transcript levels of several mitochondrial complex components were also significantly reduced (Figure 4C, Figure IXD in the Supplement). Additionally, UCP2, which uncouples oxygen consumption from ATP synthesis and thus decreases the cellular energy state34, was significantly upregulated (Figure 4D). To ascertain whether reduced ATP levels, in the setting of increased metabolic demand in HCM, activates the AMPK pathway (sensor of low cellular ATP levels35) we measured the catalytic subunit, α-AMPK, and its activated form, pAMPK (Thr172), and found significantly increased pAMPK (Figure 4E–G). Taken together, these data suggest that despite increased metabolic demand and activation of AMPK in HCM hearts, decreased energy supply due to mitochondrial dysfunction contributes to energetic deprivation.
Figure 4. Mitochondrial respiratory function and capacity in HCM.
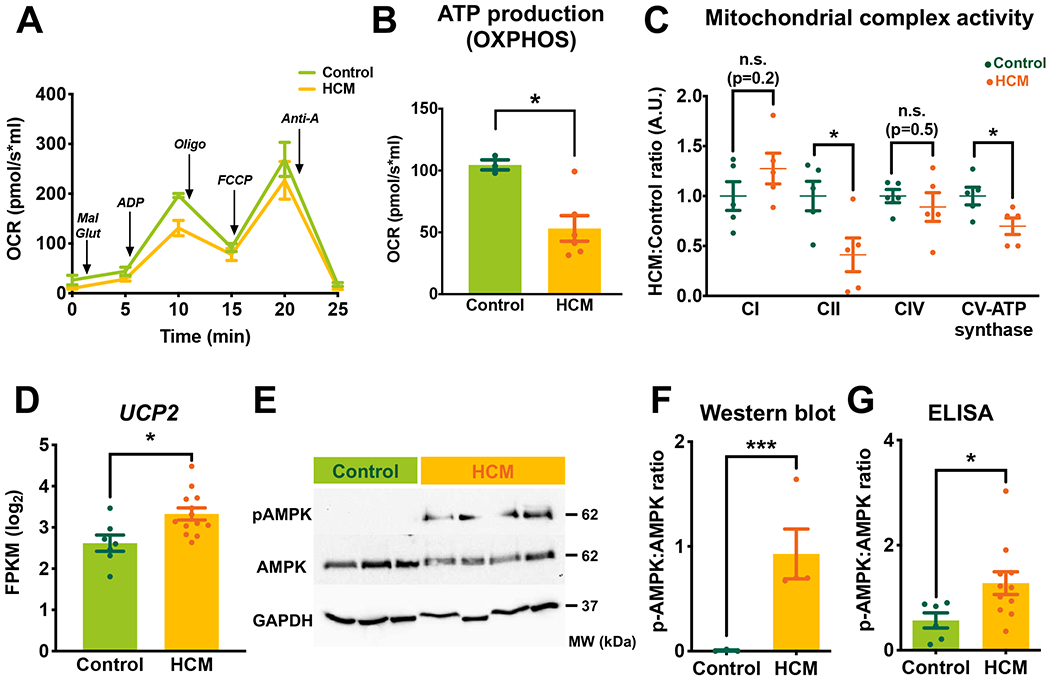
(A) Average oxygen consumption rate (OCR) showing respiratory activity measured by Oroboros oximeter. Arrows indicate the sequential additions of mitochondrial substrates to assess respiratory states in freshly isolated myocardial tissue. (B) Analysis of ATP production capacity through oxidative phosphorylation (OXPHOS). (C) Mitochondrial complex activities in HCM vs control (spectrophotometry analysis). (D) The transcript level of UCP2 (log2 scale) measured by RNA-Seq. (E and F) Representative western blot and quantification of phosphorylated- and total AMPK. GAPDH was used as a loading control. (G) ELISA measurements of pAMPK to total AMPK. Error bars represent mean ± SEM. n=3 control and n=9 HCM in A and B; n=5 control and n=5 HCM in C; n=7 control and n=13 HCM in D; n=3 control and n=5 HCM in E and F; n=6 control and n=11 HCM in G. Between-group comparisons performed using Whitney U test in B, C, F, and G. Wald test with Benjamini-Hochberg’s FDR correction method used in D. *p<0.05, ***p<0.005, or n.s, not significant (p>0.05). Mal, malate; Glut, glutamate; Oligo, oligomycin A; FCCP, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone; Anti-A, antimycin A; CI-CV, mitochondrial complex I-V; UCP, uncoupling protein.
Mitochondrial damage is associated with increased Reactive Oxygen Species (ROS)
To investigate mechanisms for HCM mitochondrial structural damage, we investigated the deleterious effects of ROS on mitochondrial lipid membranes and mitochondrial DNA (mtDNA). 4-hydroxynonenal (4-HNE)-modified protein (an end-degradation product of phospholipid peroxidation36) was significantly elevated (Figure 5A, Figure XA in the Supplement). We also found a decrease in various cardiolipin (CL) species including LO3, L2O2, and L3O as well as a trend toward decreased L4, measured by untargeted LC-MS lipidomics (Figure 5B, Figure XB, Figure XC and Excel IV in the Supplement). Cardiolipins are essential for the optimal function of numerous mitochondrial enzymes and are particularly susceptible to ROS-induced oxidation due to their unsaturated fatty acyl composition and proximity to ROS-generating centers in the mitochondria37, 38. Similarly, mtDNA is susceptible to elevated ROS due to the lack of protective histones, inefficient DNA repair mechanisms, and similar proximity to ROS production sites39. Mitochondrial-to-genomic DNA ratios were reduced in HCM (Figure 5C) accompanied by reduced expression of genes associated with mitochondrial DNA integrity and mitochondrial transcription and translation (Figure XD, Figure XE in the Supplement). Interestingly, reduced mtDNA copy number was correlated with decreased cardiolipin species and downregulation of genes associated with cristae formation/maintenance (DNAJ11, TIMM50, SAMM50, C19orf70, TAZ) suggesting that the magnitude of the mtDNA damage coincided with impaired mitochondrial integrity (Figure XF, Figure XG in the Supplement).
Figure 5. Increased oxidative damage in HCM.
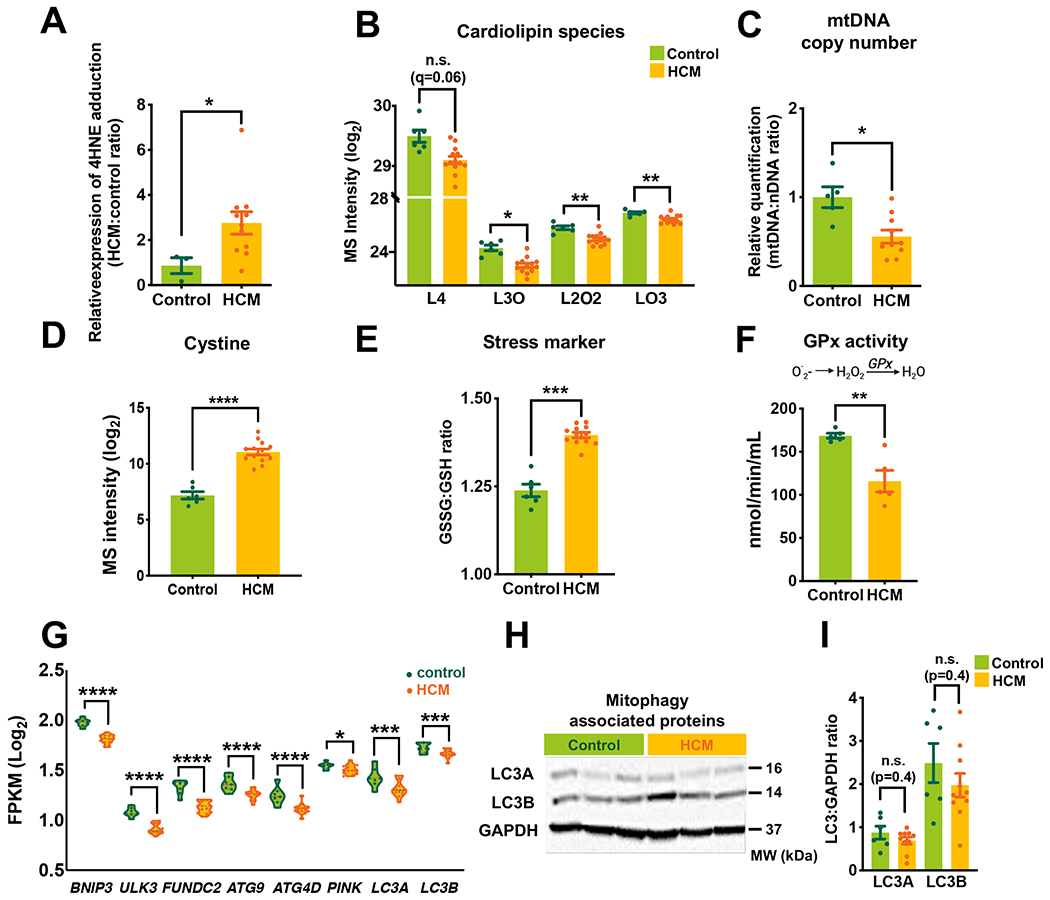
(A) 4-hydroxynonenal (4HNE) immunoreactivity indicative of oxidative stress measured by western blot. (B) Major cardiolipin species were measured by mass spec. (C) Quantification of mitochondrial DNA (mtDNA) to nuclear DNA (nDNA) using qPCR. (D) Cystine level measured by mass spec (log2 scale). (E) The ratio of oxidized glutathione (GSSG) to glutathione (GSH) (calculated from mass spec data). (F) The enzymatic activity of glutathione peroxidase (spectrophotometry analysis). (G) Violin plot showing mitophagy associated genes. (H and I) Representative Western blots and quantitative measurements of LC3A and B. GAPDH was used as a loading control. Violin plot was reserved to show RNA-Seq data. Error bars represent mean ± SEM. n=3 control and n=8 HCM in A; n=6 control and n=13 HCM in B, D, and E; n=5 control and n=10 HCM in C; n=5 control and n=5 HCM in F; n=7 control and n=13 HCM in G. n=6 control and n=10 HCM in H and I. Between-group comparisons performed using Whitney U test in A, C, E, F, and I. Two-sided Welch’s t-test and Benjamini-Hochberg’s FDR correction method used in B and D. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 or n.s, not significant (p>0.05). L4, tetralinoleoyl (18:2)4; L3O, trilinoleoyl-oleoyl (18:2)3(18:1)1; L2O2, dilinoleoyl-dioleoyl (18:2)2(18:1)2; LO3, linoleoyl-trioleoyl (18:2)1(18:1)3; O.2, superoxide radical; H2O2, hydrogen peroxide; GPx, glutathione peroxidase; LC3, Microtubule-associated proteins 1A/1B-light chain 3.
In healthy myocardium, oxidative stress is tightly counterbalanced by reducing agents, coordinated by transcriptional regulation of enzymatic antioxidant systems40. Metabolomic data showed high oxidative stress in HCM, indicated by an elevated level of oxidized cysteine (cystine), increased oxidized glutathione (GSSG) to reduced glutathione (GSH) ratio and a reduced level of glutathione, a major antioxidant (Figure 5D, Figure 5E, Figure XH in the Supplement). Levels of other key antioxidants including superoxide dismutase (SOD), catalase (CAT), glutathione reductase (GSR), and glutathione peroxidase (GPX) were all reduced (Figure XI in the Supplement). Confirming the functional effect of these transcriptional changes, enzymatic activity of glutathione peroxidase (GPx1), which reduces hydrogen peroxide and lipid peroxides41, was significantly decreased (Figure 5F). These results suggest that increased oxidative stress, due to a combination of elevated ROS and limited antioxidant activity, could contribute to the mitochondrial pathology we observed in HCM.
Impaired mitochondrial quality control/mitophagy
Mitophagy is the principal mechanism for clearing damaged mitochondria and maintenance of myocardial mitochondrial integrity42. The abundance of damaged mitochondria in HCM suggests that mitophagic pathways might be either dysfunctionally downregulated or overwhelmed by the degree of mitochondrial damage. We observed a significant downregulation of several genes associated with mitophagy including BNIP3, FUNDC2, ATG9, PINK1, and MAP1LC3 (Figure 5G). LC3 protein expression, which has a central role in autophagosome formation43, was also not increased (Figure 5H, Figure 5I), suggesting a failure to upregulate mitophagy. Similarly, we found no significant difference in Parkin and P62/SQSTM1 expression, which play central roles in autophagic clearance44 (Figure XI in the Supplement). Taken together, these data suggest that failure to upregulate or impairment of mitochondrial quality control could be in part responsible for the accumulation of aberrant mitochondria in HCM.
Genotype-phenotype correlation in HCM
Mechanisms by which myocyte-specific mutations, especially those in genes encoding sarcomere proteins, affect the HCM phenotype remain unknown. Additionally, in many HCM patients a disease-causing mutation cannot be identified45. To determine the impact of genotype on the phenotypic expression of HCM, we compared key molecular features in patients with or without sarcomeric gene mutations. Unsupervised hierarchical clustering of differentially expressed genes and metabolites revealed no cluster based on genotype (Figure XIIA and Figure XIIB in the Supplement). Levels of FFA, AC (>C14) and carnitine, major altered lipids in HCM, were also similar between the two groups (Figure XIIC–E in the Supplement). Additionally, we found no difference in the percentage of severely damaged mitochondria or oxygen consumption based on patient genotype (Figure XIIF and XIIG in the Supplement). Thus, there were no correlations between the presence of a known mutation and severity of the HCM phenotype, indicating that the HCM metabolomic profile at the time of sampling was relatively homogeneous.
DISCUSSION
Despite several clinical and experimental studies showing metabolic remodeling and inflexibility in hypertrophic and failing hearts46–48, there are few comprehensive studies in human cardiomyopathies, especially in HCM21, 49. Here, we present an in-depth multi-omics characterization of human HCM myocardium combined with analysis of mitochondrial structure and function. This integrative omics analysis reveals potential molecular mechanisms underlying HCM pathophysiology, including major metabolic derangements across all main biochemical pathways (summarized in Figure 6). These are associated with reduced mitochondrial respiration and accumulation of damaged mitochondria due to a combination of increased oxidative stress, reduced antioxidant defenses, and failure to upregulate mitophagic clearance.
Figure 6. Mitochondrial alterations in HCM.
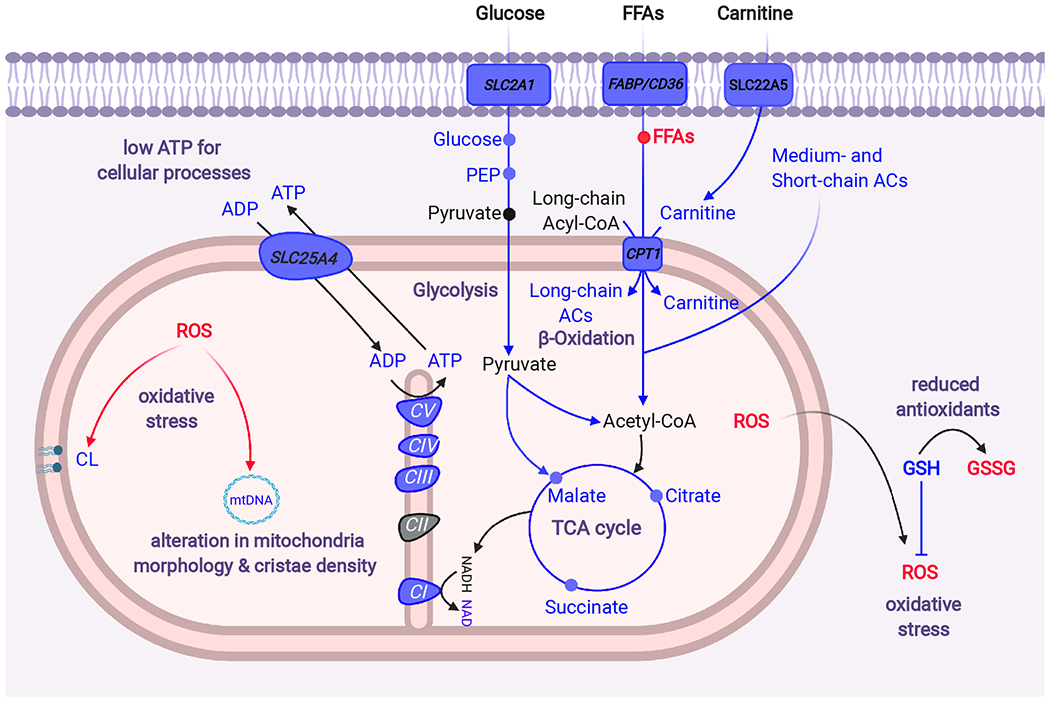
HCM hearts exhibit impaired fatty acid oxidation and reduced glucose metabolism. Along with these metabolic changes, increased energy demands in hypercontractile HCM hearts enhance ROS production, which together with insufficient antioxidant contents causes damage to diverse mitochondrial sites including mtDNA, cardiolipins, cristae and mitochondrial respiratory complexes. These mitochondrial abnormalities lead to reduced mitochondrial respiration and high energy phosphate molecules ultimately causing myocardial energy deprivation. Italics represents changes at the transcript level while metabolites and enzymatic activities are shown in regular font. Color indicates relative changes in the respective biomolecules: blue, decrease; red, increase. FABP, fatty acid binding protein; CD36, cluster of differentiation 36; CPT1, carnitine palmitoyl transferase type 1; PEP, phosphoenolpyruvate; CoA, Coenzyme A; CI-V, Complex I-V; mtDNA, mitochondrial DNA; TCA, tricarboxylic acid; ACs, acylcarnitines; ROS, reactive oxygen species; GSH, reduced glutathione; GSSG, oxidized GSH; SCLC2A1, SLC25A4 and SLC22A5 genes encode glucose transporter1 (GLUT1), the mitochondrial ADP/ATP translocator (ANT1), and carnitine transporter respectively.
Lipids are the preferred substrate for energy production in the normal heart50. HCM tissues manifested a substantial accumulation of free fatty acids, whereas the concentration of acylcarnitines and free carnitine were markedly reduced. Our findings are consistent with recent proteomic studies showing downregulation of fatty acid β-oxidation in HCM patients21, 45. Although the underlying mechanism linking these changes to alterations in sarcomere function remains unclear, the increased level of free fatty acids, along with reduced expression of their transporters (SLC27A4, FABP4, and CD36), suggest that the diminished abundance of acylcarnitines is most likely not due to increased fatty acid uptake or retention46, but rather to the inability of the heart to increase its capacity to oxidize fatty acids. In support of this hypothesis, we found reduced expression of major enzymes regulating different steps of fatty acid β-oxidation (ACADVL, ACSL1, PGK1, HADHA, ECH1) while the PGC1-PPARA pathway, a major regulator of mitochondrial fatty acid oxidation51, was not upregulated. Additionally, HCM hearts accumulated toxic lipid intermediates including ceramides. Circulating and myocardial ceramides have been previously shown to be elevated in heart failure and, in some instances, correlated to severity and adverse events52–54.
In addition to metabolic alterations that limit fatty acid oxidation in HCM, we found reduced levels of glucose and glycolytic metabolites (F6P and PEP) as well as anaplerotic substrates (e.g. aspartate, glutamate, tyrosine, and phenylalanine) that fuel the TCA cycle, ultimately limiting energy provision. Supporting these metabolomic data, the majority of genes catalyzing the TCA cycle (CS, IDH2, OGDH, SUCLA2, FH, and MDH1) were downregulated, leading to reduced levels of malate, citrate, and succinate. These alterations impact downstream energy production through high energy phosphates, with downregulation of key genes involved in mitochondrial energy metabolism including ANT1, CKM2, many subunits of the ETC complex and ATP synthase. These findings are consistent with previous studies suggesting that both HCM and acquired forms of cardiomyopathy are associated with overall reduced cardiac bioenergetics10, 11, 15, 18, 45, 55–58. Imaging assessment of myocardial nucleotide content using phosphorus-NMR (31P-NMR) in animal models and humans with HCM56, 59–61 detected significant reductions in the PCr/ATP ratio, suggesting limited myocardial energy regenerative capacity. Additionally, both asymptomatic (mutation carriers) and symptomatic HCM patients showed reduced cardiac efficiency using 11C-acetate PET and NMR imaging62. Two recent studies21, 45 used proteomics analysis to demonstrate that several metabolic pathways related to energy metabolism including oxidative phosphorylation, glycolysis, and β-oxidation were downregulated in HCM. Other evidence supporting an energy deficit in HCM comes from studies of cardiac hypertrophy in inherited syndromes including Barth, Sengers, MELAS and MERRF, in which mitochondrial energy production is impaired63, 64. In addition, patients with mutations in mitochondrial DNA often develop an HCM-like phenotype22, 65. Further evidence supporting the role of altered energetics in HCM is based on mutations of the AMPK gene in families with combined HCM and Wolff-Parkinson-White syndrome66–68. A small randomized study of HCM patients treated with the metabolic modulator perhexiline, which shifts myocardial substrate utilization from fatty acids to lactate, showed improvements in exercise capacity and New York Heart Association (NYHA) functional class69. Thus, impaired energy metabolism could be an important mechanism of functional deterioration in HCM and a potential target for therapeutic intervention.
Increased energy demand in the hypercontractile HCM heart enhances ROS production as by-products of oxidative phosphorylation4. Elevated ROS and insufficient antioxidant protection expose mitochondria to increased oxidative stress leading to mitochondrial damage70. ROS react with DNA, proteins and lipids (especially mitochondrial membranes), inactivating ETC complexes and mitochondrial proteins and impairing mitochondrial respiration21, 71. We found increased oxidative stress markers, including 4HNE, GSSG, and cystine in HCM hearts, while transcript levels of several major antioxidants (GPX1, GSR, SOD, CAT) and enzymatic activity of GPx were significantly diminished. HCM myocardium demonstrates a reduction in several cardiolipin species, shown to impair mitochondrial respiratory activity by destabilizing inner mitochondrial membrane integrity, feed-forwarding additional electron leakage, and further increasing ROS formation72. Similar to previous studies in hypertrophied and failing hearts73–75, mtDNA was reduced in HCM, along with downregulation of mitochondrial genes encoding ribosomal, tRNA, and ETC subunits. Finally, our systematic analysis of mitochondrial morphology using quantitative electron microscopy revealed a significant subset of severely damaged mitochondria with reduced cristae density. Cristae loss may be explained by the reduced expression of several genes associated with cristae formation and maintenance of cristae junctions. CHCHD3, a member of cristae organizing proteins (MICOS) and genes encoding DNAJC11 and SAMM50 which interact with the MICOS complex76 for proper cristae formation77 were all significantly downregulated.
Mitochondrial structural alterations seen in HCM were associated with reduced mitochondrial oxidative phosphorylation capacity and decreased respiratory complex activity, confirming mitochondrial functional impairment. Previous studies have reported mitochondrial dysfunction and abnormal morphology in HCM19, 20, 27. In pigs, there were signs of swollen mitochondria with disrupted cristae, mtDNA depletion, and reduced mitochondrial complex activity78. In a single case study, an end-stage heart failure patient with HCM who underwent heart transplant showed lowered respiratory capacity in mitochondria isolated from the septum compared to other regions of the heart79. Interestingly, patients with end-stage heart failure who underwent left ventricular assist device placement showed an overall increase in substrate oxidation and improved mitochondrial function after mechanical unloading, suggesting reversibility of these defects80. Consistent with this result, we found broad heterogeneity in the degree of cristae damage within the same patient sample, suggesting that a subset of mitochondria in HCM myocardium may still have normal respiratory function but may be insufficient to meet the high energy demands of the hypercontractile HCM heart.
Activation of the mitophagy pathway to eliminate aberrant mitochondria with reduced respiratory capacity helps to prevent the cytotoxic impact of ROS, maintaining cellular homeostasis and improving mitochondrial function11, 81–84. Despite the presence of damaged mitochondria, we found downregulation of several genes associated with mitophagy including BNIP3, FUNDC2, ATG9, PINK1, and MAP1LC3. LC3 protein expression was also not changed, suggesting a failure to upregulate mitochondrial quality control pathways in the presence of significant mitochondrial damage. Similar to our results, autophagy was found to be defective in mice-carrying MYBPC3 mutations and in a small group of HCM patients85. Activation of autophagy, was shown to attenuate cardiomyopathy in these mice85. We speculate that the presence of so many aberrant mitochondria in HCM could be due to failure of mitophagic mitochondrial clearance. Although accumulation of autophagic vacuoles caused by defective proteolytic systems has also been associated with HCM pathology82, 86, the lack of LC3 upregulation combined with the absence of accumulated autophagic vesicles in our EM data suggest that autophagy is not activated, at least at this stage of the disease. Altogether, impaired mitochondrial quality control is likely to contribute to the accumulation of damaged mitochondria in HCM, ultimately leading to progressive mitochondrial damage, a vicious cycle in the progression of the HCM phenotype.
A recent study by Coats et al.21, using label-free proteomics to characterize HCM tissue, corroborates some of our findings, including alterations in proteins associated with energy metabolism and mitochondrial dysfunction. In line with our data, they showed reduced β-oxidation and downregulation of ACSL1, ACADVL, and HADHB. Similar to our data, there were decreased levels of mitochondrial respiratory complex proteins despite no change in citrate synthase, implying similar mitochondrial number. Coates et al. hypothesized that mtDNA depletion or perturbation in HCM could result in alteration of the electron transport chain. In support of their hypothesis, we found reduced mtDNA, previously reported in pigs with HCM78 as well as in patients with hypertrophy due to congenital heart disease74. Although we did not measure the protein expression of citrate synthase, its enzymatic activity was reduced in HCM, confirming limited energy metabolism. Besides validation of some of our data by Coats et al., our integrated omics approach provides a more in-depth analysis of metabolic changes in HCM. Importantly, by combining omics data with mitochondrial morphological and functional assessments our study provides mechanistic insights into cellular processes such as the role of oxidative stress and reduced antioxidant defenses on the observed metabolic alterations and reduced metabolic reserve. We believe our approach of using combinational analysis from different modalities is a robust way to understand the multiple perturbations in a disease as complex as HCM.
Overall, our findings suggest that altered metabolic signaling and mitochondrial dysfunction are common pathogenic mechanisms among HCM patients undergoing myectomy surgery, a time when their global cardiac function is still hyperdynamic, and far from end-stage heart failure. Using an unbiased approach, we observed similar molecular changes in the HCM cohort independent of whether a sarcomeric gene mutation was present or not. Consistent with this finding, two recent studies45, 87 showed similar proteomic and proteoform profiles in HCM patients regardless of their individual gene mutation, suggesting that secondary mechanisms (e.g. cellular and mitochondrial stress) lead, through common pathways, to the HCM clinical phenotype. HCM mutations in sarcomeric proteins primarily cause diverse alterations in biophysical properties of the cardiomyocyte, which ultimately perpetuate the hypertrophic response in the heart. The molecular and histologic alterations (e.g. alterations in cardiac energetics) that occur in response to these primary changes are influenced by a combination of genetic, epigenetic, and environmental factors88. Therefore, whereas the initial alterations are divergent, they converge into common pathways that lead to altered metabolism, which we have identified as a major component of the HCM phenotype88.
LIMITATIONS
This study represents the most comprehensive multi-omics and functional characterization of HCM in human myocardium. However, there are several limitations associated with a study of this kind. First, the samples were obtained from HCM patients undergoing surgical myectomy, only providing us with a narrow temporal window into the disease. Thus, we cannot extrapolate our findings to earlier or later stages of the HCM disease process. All of our patients were undergoing myectomy for symptomatic LVOT obstruction, so they represent a sub-class (hypertrophic obstructive cardiomyopathy), albeit a large one, of all HCM patients. Importantly, the vast majority of our patients had hyperdynamic left ventricular function, and the remainder had preserved function, thus these samples were from hearts that had not yet developed many of the secondary and tertiary changes associated with the transition from hypercontractility to heart failure. It is therefore even more notable that we found such a high degree of metabolic and mitochondrial alterations in patients who were at this stage in their disease. Second, global metabolomic and transcriptomic studies of human heart tissue, especially in HCM, are influenced by multiple factors in addition to disease stage, including heterogeneity within the myocardium (septum, apex, free wall), genetic diversity between patients, secondary remodeling mechanisms, medical comorbidities, and drug therapy. It is possible that the septum represents a more advanced stage of HCM pathophysiology compared to the LV free wall in these patients. Third, metabolic measurements preclude definitive conclusions on metabolic flux changes. However, when combined with expected changes in expression of their regulatory enzymes, provide insights into likely mechanisms explaining the metabolic alterations. Future tracing experiments with isotopically-labeled metabolites are warranted to confirm metabolic flux dysregulation in HCM. Nevertheless, our extensive metabolic profiling provides a global map of altered metabolic pathways. Fourth, our sample size limited our ability to perform correlation analysis between any individual sarcomeric mutation and HCM phenotype, warranting further exploration in a larger cohort. Fifth, most of the control samples were obtained from unused donor hearts, and thus were from individuals who suffered catastrophic neurological injury, suggesting the potential for metabolic alterations in the controls. However, all donor hearts had normal function at the time of harvesting. Both control and HCM hearts were subjected to cardioplegia, although the duration was longer for the donor controls; importantly none were longer than 4 hours. Finally, we cannot eliminate the effect of regional heterogeneity on our analysis. Different regions of the left ventricle were assessed in our groups as follow: LV septal tissue in HCM, interventricular septum and LV apex in donors, and LV free wall in mitral stenosis patients.
CONCLUSION
In summary, our study demonstrates the power of using an unbiased integrated multi-omic analysis to determine the role of global alterations in cardiac metabolism and energetics as a pathophysiologic mechanism in HCM. The correlation of diffuse metabolomic and lipidomic alterations with changes in transcription of key enzymes regulating those pathways strengthens this evidence and provides mechanistic insights. Accompanying these metabolic derangements, we found increased oxidative damage leading to dramatic mitochondrial structural and functional defects, exacerbated by a failure to upregulate mitochondrial quality control, and thereby increasing energetic compromise. The degree to which these alterations are present at this earlier stage suggests that developing new strategies directed toward improving metabolic and mitochondrial function could be promising approaches to slow down the progression of HCM.
Supplementary Material
Clinical Perspective.
What is new?
We leveraged a novel integrated multi-omics platform to explore the role of altered cardiac metabolism as a common pathway leading to the pathophysiological phenotypes in patients with hypertrophic cardiomyopathy (HCM).
We describe widespread alterations across a broad array of metabolic pathways and in mitochondrial structure and function at a relatively early stage in the progression of HCM.
These findings provide a crucial connection between two central aspects of HCM disease: biophysical properties of the cardiomyocyte and cellular metabolism.
What are the clinical implications?
This degree of metabolic derangement has not been previously suspected in patients with HCM, especially at this stage of disease, when contractile function is still hyperdynamic.
Our integrated multi-omics approach should be applicable to diverse forms of cardiovascular disease for discovery of both mechanism and drug targets.
These findings highlight the potential for development of new HCM therapeutics targeting metabolic alterations, in addition to therapies targeting the sarcomere.
ACKNOWLEDGMENTS
We are indebted to the HCM patients seen at the Stanford Hospital for their participation in this study. We would like to thank Dr. Hyun Tae Hwang for his guidance and training on Oroboros oximetry. Also, we like to thank Dr. Anson Lee who helped us to obtain myocardial samples. The Stanford EM microscopy core facility provided resources to prepare and analyze EM samples with special thanks to Ruth Yamawaki. Serial EM sample preparation and imaging was supported by ARRA Award Number 1S10RR026780-01 from the National Center for Research Resources (NCRR). Novogene provided services for RNA sequencing. Mitochondrial enzymatic activity assessment was performed by UAB Bioanalytical Redox Biology Core with the support from DRC (NIH P30DK079626), and NORC (NIDDK DK056336). BioRender was used to generate schematic figures.
SOURCE OF FUNDING
This work was supported by an RM1 grant from the NIGMS/National Institutes of Health RM1GM131981 to DB and JAS, AHA Collaborative Investigator Award 17CSA33590101 to DB and the Stanford Translational and Clinical Innovation Award to DB and JAS. Mass spectrometry analysis was supported by NIH grant 2RM1HG00773506 to MS. SR and KK were supported by a T32 Post-Doctoral Fellowship (HL094274). RAW was supported in part by NIBIB (2T32EB009035).
Non-standard Abbreviations and Acronyms
- HCM
Hypertrophic Cardiomyopathy
- ROS
Reactive Oxygen Species
- LV
Left Ventricle
- LVOT
LV Outflow Tract
- TEM
Transmission Electron Microscopy
- FFA
Free Fatty Acid
- AC
Acylcarnitine
- CER
Ceramide
- TAG
Triacylglycerol
- DAG
Diacylglycerol
- PCr
Phosphocreatine
- PCr-CK
Phosphocreatine–Creatine Kinase
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1.Maron BJ and Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–55. [DOI] [PubMed] [Google Scholar]
- 2.Semsarian C, Ingles J, Maron MS and Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. [DOI] [PubMed] [Google Scholar]
- 3.Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch. 2019;471:701–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritterhoff J and Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovascular Research. 2017;113:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marian AJ and Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKenna WJ and Behr ER. Hypertrophic cardiomyopathy: management, risk stratification, and prevention of sudden death. Heart. 2002;87:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotkar KD, Said SM, Dearani JA and Schaff HV. Hypertrophic obstructive cardiomyopathy: the Mayo Clinic experience. Ann Cardiothorac Surg. 2017;6:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, Kubanek M, Wojakowski W, Jensen MK, Gimeno-Blanes J, Afshar K, Myers J, Hegde SM, Solomon SD, Sehnert AJ, Zhang D, Li W, Bhattacharya M, Edelberg JM, Waldman CB, Lester SJ, Wang A, Ho CY, Jacoby D, Bartunek J, Bondue A, Van Craenenbroeck E, Kubanek M, Zemanek D, Jensen M, Mogensen J, Thune JJ, Charron P, Hagege A, Lairez O, Trochu J-N, Axthelm C, Duengen H-D, Frey N, Mitrovic V, Preusch M, Schulz-Menger J, Seidler T, Arad M, Halabi M, Katz A, Monakier D, Paz O, Viskin S, Zwas D, Olivotto I, Brunner-La Rocca HP, Michels M, Dudek D, Oko-Sarnowska Z, Oreziak A, Wojakowski W, Cardim N, Pereira H, Barriales-Villa R, García Pavia P, Gimeno Blanes J, Hidalgo Urbano R, Rincón Diaz LM, Elliott P, Yousef Z, Abraham T, Afshar K, Alvarez P, Bach R, Becker R, Choudhury L, Fermin D, Jacoby D, Jefferies J, Kramer C, Lakdawala N, Lester S, Marian A, Masri A, Maurer M, Nagueh S, Owens A, Owens D, Rader F, Saberi S, Sherrid M, Shirani J, Symanski J, Turer A, Wang A, Wever-Pinzon O, Wheeler M, Wong T and Yamani M. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2020;396:759–769. [DOI] [PubMed] [Google Scholar]
- 9.Marian AJ and Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adhikari AS, Trivedi DV, Sarkar SS, Song D, Kooiker KB, Bernstein D, Spudich JA and Ruppel KM. beta-Cardiac myosin hypertrophic cardiomyopathy mutations release sequestered heads and increase enzymatic activity. Nat Commun. 2019;10:2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adhikari AS, Kooiker KB, Sarkar SS, Liu C, Bernstein D, Spudich JA and Ruppel KM. Early-Onset Hypertrophic Cardiomyopathy Mutations Significantly Increase the Velocity, Force, and Actin-Activated ATPase Activity of Human beta-Cardiac Myosin. Cell Rep. 2016;17:2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashrafian H, Redwood C, Blair E and Watkins H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003;19:263–8. [DOI] [PubMed] [Google Scholar]
- 13.Belus A, Piroddi N, Scellini B, Tesi C, D’Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I and Poggesi C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008;586:3639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C and Poggesi C. Mechanical and energetic consequences of HCM-causing mutations. J Cardiovasc Transl Res. 2009;2:441–51. [DOI] [PubMed] [Google Scholar]
- 15.Witjas-Paalberends ER, Güçlü A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA, et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res. 2014;103:248–57. [DOI] [PubMed] [Google Scholar]
- 16.Timmer SA, Germans T, Brouwer WP, Lubberink M, van der Velden J, Wilde AA, Christiaans I, Lammertsma AA, Knaapen P and van Rossum AC. Carriers of the hypertrophic cardiomyopathy MYBPC3 mutation are characterized by reduced myocardial efficiency in the absence of hypertrophy and microvascular dysfunction. Eur J Heart Fail. 2011;13:1283–9. [DOI] [PubMed] [Google Scholar]
- 17.Rosello-Lleti E, Tarazon E, Barderas MG, Ortega A, Otero M, Molina-Navarro MM, Lago F, Gonzalez-Juanatey JR, Salvador A, Portoles M and Rivera M. Heart mitochondrial proteome study elucidates changes in cardiac energy metabolism and antioxidant PRDX3 in human dilated cardiomyopathy. PLoS One. 2014;9:e112971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG and Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unno K, Isobe S, Izawa H, Cheng XW, Kobayashi M, Hirashiki A, Yamada T, Harada K, Ohshima S, Noda A, Nagata K, Kato K, Yokota M and Murohara T. Relation of functional and morphological changes in mitochondria to myocardial contractile and relaxation reserves in asymptomatic to mildly symptomatic patients with hypertrophic cardiomyopathy. Eur Heart J. 2009;30:1853–62. [DOI] [PubMed] [Google Scholar]
- 20.Lucas DT, Aryal P, Szweda LI, Koch WJ and Leinwand LA. Alterations in mitochondrial function in a mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2003;284:H575–83. [DOI] [PubMed] [Google Scholar]
- 21.Coats CJ, Heywood WE, Virasami A, Ashrafi N, Syrris P, Dos Remedios C, Treibel TA, Moon JC, Lopes LR, McGregor CGA, et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ Genom Precis Med. 2018;11:e001974. [DOI] [PubMed] [Google Scholar]
- 22.Elliott P and McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–91. [DOI] [PubMed] [Google Scholar]
- 23.Okajima Y, Tanabe Y, Takayanagi M and Aotsuka H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Heart. 1998;80:292–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mudhar HS, Wagner BE and Suvarna SK. Electron microscopy of myocardial tissue. A nine year review. Journal of Clinical Pathology. 2001;54:321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grasso M, Diegoli M, Brega A, Campana C, Tavazzi L and Arbustini E. The mitochondrial DNA mutation T12297C affects a highly conserved nucleotide of tRNA(Leu(CUN)) and is associated with dilated cardiomyopathy. Eur J Hum Genet. 2001;9:311–5. [DOI] [PubMed] [Google Scholar]
- 26.Robbins RC, Bernstein D, Berry GJ, VanMeurs KP, Frankel LR and Reitz BA. Cardiac transplantation for hypertrophic cardiomyopathy associated with Sengers syndrome. Ann Thorac Surg. 1995;60:1425–7. [DOI] [PubMed] [Google Scholar]
- 27.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J and Leinwand LA. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizukoshi K, Takeuchi M, Nagata Y, Addetia K, Lang RM, Akashi YJ and Otsuji Y. Normal Values of Left Ventricular Mass Index Assessed by Transthoracic Three-Dimensional Echocardiography. J Am Soc Echocardiogr. 2016;29:51–61. [DOI] [PubMed] [Google Scholar]
- 29.Ricciardi MJ, Wu E, Davidson CJ, Choi KM, Klocke FJ, Bonow RO, Judd RM and Kim RJ. Visualization of discrete microinfarction after percutaneous coronary intervention associated with mild creatine kinase-MB elevation. Circulation. 2001;103:2780–3. [DOI] [PubMed] [Google Scholar]
- 30.Kamburov A, Cavill R, Ebbels TM, Herwig R and Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27:2917–8. [DOI] [PubMed] [Google Scholar]
- 31.Drosatos K and Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013;10:109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell RR, 3rd and Taegtmeyer H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am J Physiol. 1991;261:H1756–62. [DOI] [PubMed] [Google Scholar]
- 33.Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, Hu D-Q, Reddy S, Sandoval E, Stotland A, Gottlieb RA and Bernstein D. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circulation research. 2018;122:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian XY, Ma S, Tse G, Wong WT and Huang Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front Physiol. 2018;9:1060–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gianazza E, Brioschi M, Fernandez AM and Banfi C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019;23:101119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paradies G, Paradies V, Ruggiero FM and Petrosillo G. Cardiolipin and mitochondrial function in health and disease. Antioxid Redox Signal. 2014;20:1925–53. [DOI] [PubMed] [Google Scholar]
- 38.Paradies G, Paradies V, Ruggiero FM and Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X and Chen Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. Journal of Translational Medicine. 2017;15:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D, Knight AR, Taylor EL, Oettrich J, Ruskovska T, et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid Redox Signal. 2015;23:1144–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Touyz RM and Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5–14. [DOI] [PubMed] [Google Scholar]
- 42.Saito T and Sadoshima J. Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circulation Research. 2015;116:1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanida I, Ueno T and Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. [DOI] [PubMed] [Google Scholar]
- 44.Tong M and Sadoshima J. Mitochondrial autophagy in cardiomyopathy. Current opinion in genetics & development. 2016;38:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuldt M, Pei J, Harakalova M, Dorsch LM, Schlossarek S, Mokry M, Knol JC, Pham TV, Schelfhorst T, Piersma SR, et al. Proteomic and Functional Studies Reveal Detyrosinated Tubulin as Treatment Target in Sarcomere Mutation-Induced Hypertrophic Cardiomyopathy. Circ Heart Fail. 2021;14:e007022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP and Hill BG. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.An Raghow R. ‘Omics’ Perspective on Cardiomyopathies and Heart Failure. Trends Mol Med. 2016;22:813–827. [DOI] [PubMed] [Google Scholar]
- 48.Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE and Wang TJ. Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ Res. 2016;118:1659–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pal N, Acharjee A, Ament Z, Dent T, Yavari A, Mahmod M, Ariga R, West J, Steeples V, Cassar M, et al. Metabolic Profiling of Aortic Stenosis and Hypertrophic Cardiomyopathy Identifies Mechanistic Contrasts in Substrate Utilisation. bioRxiv. 2019:715680. [DOI] [PubMed] [Google Scholar]
- 50.Maekawa K, Hirayama A, Iwata Y, Tajima Y, Nishimaki-Mogami T, Sugawara S, Ueno N, Abe H, Ishikawa M, Murayama M, et al. Global metabolomic analysis of heart tissue in a hamster model for dilated cardiomyopathy. J Mol Cell Cardiol. 2013;59:76–85. [DOI] [PubMed] [Google Scholar]
- 51.Cheng C-F, Ku H-C and Lin H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. International journal of molecular sciences. 2018;19:3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight. 2017;2:e82922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu J, Pan W, Shi R, Yang T, Li Y, Yu G, Bai Y, Schuchman EH, He X and Zhang G. Ceramide Is Upregulated and Associated With Mortality in Patients With Chronic Heart Failure. Canadian Journal of Cardiology. 2015;31:357–363. [DOI] [PubMed] [Google Scholar]
- 55.Jung WI, Hoess T, Bunse M, Widmaier S, Sieverding L, Breuer J, Apitz J, Schmidt O, van Erckelens F, Dietze GJ et al. Differences in cardiac energetics between patients with familial and nonfamilial hypertrophic cardiomyopathy. Circulation. 2000;101:E121. [DOI] [PubMed] [Google Scholar]
- 56.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Östman-Smith I, Clarke K and Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. Journal of the American College of Cardiology. 2003;41:1776–1782. [DOI] [PubMed] [Google Scholar]
- 57.Finck BN, Lehman JJ, Barger PM and Kelly DP. Regulatory networks controlling mitochondrial energy production in the developing, hypertrophied, and diabetic heart. Cold Spring Harb Symp Quant Biol. 2002;67:371–82. [DOI] [PubMed] [Google Scholar]
- 58.Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP and Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–7. [DOI] [PubMed] [Google Scholar]
- 59.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG and Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. The Journal of clinical investigation. 1998;101:1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Javadpour MM, Tardiff JC, Pinz I and Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. The Journal of Clinical Investigation. 2003;112:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He H, Javadpour MM, Latif F, Tardiff JC and Ingwall JS. R-92L and R-92W Mutations in Cardiac Troponin T Lead to Distinct Energetic Phenotypes in Intact Mouse Hearts. Biophysical Journal. 2007;93:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Güçlü A, Knaapen P, Harms HJ, Parbhudayal RY, Michels M, Lammertsma AA, Rossum ACv, Germans T and Velden Jvd. Disease Stage–Dependent Changes in Cardiac Contractile Performance and Oxygen Utilization Underlie Reduced Myocardial Efficiency in Human Inherited Hypertrophic Cardiomyopathy. Circulation: Cardiovascular Imaging. 2017;10:e005604. [DOI] [PubMed] [Google Scholar]
- 63.Frey N, Luedde M and Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nature Reviews Cardiology. 2012;9:91–100. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz ML, Cox GF, Lin AE, Korson MS, Perez-Atayde A, Lacro RV and Lipshultz SE. Clinical approach to genetic cardiomyopathy in children. Circulation. 1996;94:2021–38. [DOI] [PubMed] [Google Scholar]
- 65.Obayashi T, Hattori K, Sugiyama S, Tanaka M, Tanaka T, Itoyama S, Deguchi H, Kawamura K, Koga Y, Toshima H and et al. Point mutations in mitochondrial DNA in patients with hypertrophic cardiomyopathy. Am Heart J. 1992;124:1263–9. [DOI] [PubMed] [Google Scholar]
- 66.Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I and Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–20. [DOI] [PubMed] [Google Scholar]
- 67.Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–31. [DOI] [PubMed] [Google Scholar]
- 68.Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG and Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562–9. [DOI] [PubMed] [Google Scholar]
- 70.Addabbo F, Montagnani M and Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dai D-F, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF and Rabinovitch PS. Mitochondrial Targeted Antioxidant Peptide Ameliorates Hypertensive Cardiomyopathy. Journal of the American College of Cardiology. 2011;58:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saini-Chohan HK, Holmes MG, Chicco AJ, Taylor WA, Moore RL, McCune SA, Hickson-Bick DL, Hatch GM and Sparagna GC. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res. 2009;50:1600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K and Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–90. [DOI] [PubMed] [Google Scholar]
- 74.Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P and Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail. 2011;4:707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N and Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35. [DOI] [PubMed] [Google Scholar]
- 76.Guarani V, McNeill EM, Paulo JA, Huttlin EL, Fröhlich F, Gygi SP, Van Vactor D and Harper JW. QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. eLife. 2015;4:e06265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ioakeimidis F, Ott C, Kozjak-Pavlovic V, Violitzi F, Rinotas V, Makrinou E, Eliopoulos E, Fasseas C, Kollias G and Douni E. A Splicing Mutation in the Novel Mitochondrial Protein DNAJC11 Causes Motor Neuron Pathology Associated with Cristae Disorganization, and Lymphoid Abnormalities in Mice. PLOS ONE. 2014;9:e104237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin CS, Sun YL and Liu CY. Structural and biochemical evidence of mitochondrial depletion in pigs with hypertrophic cardiomyopathy. Res Vet Sci. 2003;74:219–26. [DOI] [PubMed] [Google Scholar]
- 79.Cordero-Reyes AM, Youker K, Hamilton DJ, Torre-Amione G, Marian AJ and Nagueh SF. Molecular, cellular, and functional characterization of myocardial regions in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2012;5:419–22. [DOI] [PubMed] [Google Scholar]
- 80.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, et al. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. 2014;7:266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carchman EH, Rao J, Loughran PA, Rosengart MR and Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T and Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol. 2012;107:235. [DOI] [PubMed] [Google Scholar]
- 83.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. [DOI] [PubMed] [Google Scholar]
- 84.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh SR, Zech ATL, Geertz B, Reischmann-Düsener S, Osinska H, Prondzynski M, Krämer E, Meng Q, Redwood C, van der Velden J, Robbins J, Schlossarek S and Carrier L. Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circulation Heart failure. 2017;10:e004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Song L, Su M, Wang S, Zou Y, Wang X, Wang Y, Cui H, Zhao P, Hui R and Wang J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. Journal of cellular and molecular medicine. 2014;18:2266–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tucholski T, Cai W, Gregorich ZR, Bayne EF, Mitchell SD, McIlwain SJ, de Lange WJ, Wrobbel M, Karp H, Hite Z, et al. Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proceedings of the National Academy of Sciences. 2020;117:24691–24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marian AJ and Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitchell C, Rahko PS, Blauwet LA, Canaday B, Finstuen JA, Foster MC, Horton K, Ogunyankin KO, Palma RA and Velazquez EJ. Guidelines for Performing a Comprehensive Transthoracic Echocardiographic Examination in Adults: Recommendations from the American Society of Echocardiography. J Am Soc Echocardiogr. 2019;32:1–64. [DOI] [PubMed] [Google Scholar]
- 90.Foppa M, Duncan BB and Rohde LE. Echocardiography-based left ventricular mass estimation. How should we define hypertrophy? Cardiovasc Ultrasound. 2005;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K, Li M and Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sosa JM, Huber DE, Welk B and Fraser HL. Development and application of MIPAR™: a novel software package for two- and three-dimensional microstructural characterization. Integrating Materials and Manufacturing Innovation. 2014;3:123–140. [Google Scholar]
- 93.Contrepois K, Jiang L and Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry. Mol Cell Proteomics. 2015;14:1684–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Contrepois K, Mahmoudi S, Ubhi BK, Papsdorf K, Hornburg D, Brunet A and Snyder M. Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma. Sci Rep. 2018;8:17747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walter W, Sánchez-Cabo F and Ricote M. GOplot: an R package for visually combining expression data with functional analysis: Fig. 1. Bioinformatics. 2015;31:2912–2914. [DOI] [PubMed] [Google Scholar]
- 96.Kamburov A, Cavill R, Ebbels TM, Herwig R and Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27:2917–8. [DOI] [PubMed] [Google Scholar]
- 97.Sergushichev AA, Loboda AA, Jha AK, Vincent EE, Driggers EM, Jones RG, Pearce EJ and Artyomov MN. GAM: a web-service for integrated transcriptional and metabolic network analysis. Nucleic Acids Res. 2016;44:W194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Picard M, White K and Turnbull DM. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J Appl Physiol (1985). 2013;114:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kremer JR, Mastronarde DN and McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–6. [DOI] [PubMed] [Google Scholar]
- 100.Mastronarde DN and Held SR. Automated tilt series alignment and tomographic reconstruction in IMOD. J Struct Biol. 2017;197:102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen M, Dai W, Sun SY, Jonasch D, He CY, Schmid MF, Chiu W and Ludtke SJ. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat Methods. 2017;14:983–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH and Ferrin TE. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Makrecka-Kuka M, Krumschnabel G and Gnaiger E. High-Resolution Respirometry for Simultaneous Measurement of Oxygen and Hydrogen Peroxide Fluxes in Permeabilized Cells, Tissue Homogenate and Isolated Mitochondria. Biomolecules. 2015;5:1319–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ and Rodenburg RJ. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–34. [DOI] [PubMed] [Google Scholar]
- 105.Ragan CI, Wilson MT, Darley-Usmar VM, and Lowe PN Mitochondria, A Practical Approach IRL Press at Oxford University Press, Oxford; 1987. [Google Scholar]
- 106.Horstman LL and Racker E. Partial resolution of the enzyme catalyzing oxidative phosphorylation. XXII. Interaction between mitochondrial adenosine triphosphatase inhibitor and mitochondrial adenosine triphosphatase. J Biol Chem. 1970;245:1336–44. [PubMed] [Google Scholar]
- 107.Feniouk BA, Suzuki T and Yoshida M. Regulatory interplay between proton motive force, ADP, phosphate, and subunit epsilon in bacterial ATP synthase. J Biol Chem. 2007;282:764–72. [DOI] [PubMed] [Google Scholar]
- 108.Shepherd D and Garland PB. The kinetic properties of citrate synthase from rat liver mitochondria. Biochem J. 1969;114:597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.