

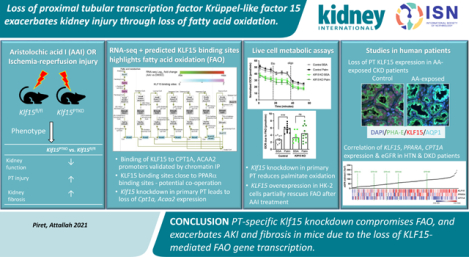
Abstract
Loss of fatty acid β-oxidation (FAO) in the proximal tubule is a critical mediator of acute kidney injury and eventual fibrosis. However, transcriptional mediators of FAO in proximal tubule injury remain understudied. Krüppel-like factor 15 (KLF15), a highly enriched zinc-finger transcription factor in the proximal tubule, was significantly reduced in proximal tubule cells after aristolochic acid I (AAI) treatment, a proximal tubule-specific injury model. Proximal tubule specific knockout of Klf15 exacerbated proximal tubule injury and kidney function decline compared to control mice during the active phase of AAI treatment, and after ischemia-reperfusion injury. Furthermore, along with worsening proximal tubule injury and kidney function decline, knockout mice exhibited increased kidney fibrosis as compared to control mice during the remodeling phase after AAI treatment. RNA-sequencing of kidney cortex demonstrated increased transcripts involved in immune system and integrin signaling pathways and decreased transcripts encompassing metabolic pathways, specifically FAO, and PPARα signaling, in knockout versus control mice after AAI treatment. In silico and experimental chromatin immunoprecipitation studies collectively demonstrated that KLF15 occupied the promoter region of key FAO genes, CPT1A and ACAA2, in close proximity to transcription factor PPARα binding sites. While the loss of Klf15 reduced the expression of Cpt1a and Acaa2 and led to compromised FAO, induction of KLF15 partially rescued loss of FAO in AAI-treated cells. Klf15, Ppara, Cpt1a, and Acaa2 expression was also decreased in other mouse kidney injury models. Tubulointerstitial KLF15 independently correlated with eGFR, PPARA and CPT1A appearance in expression arrays from human kidney biopsies. Thus, proximal tubule-specific loss of Klf15 exacerbates acute kidney injury and fibrosis, likely due to loss of interaction with PPARα leading to loss of FAO gene transcription.
Keywords: proximal tubule, KLF15, fatty acid oxidation, PPARα, AKI, kidney fibrosis
Graphical abstract
INTRODUCTION
The proximal tubule (PT) is a major target for different insults that lead to acute kidney injury (AKI), such as ischemia and toxic effects of drugs and chemicals. Injured PT cells undergo significant changes in morphology, cell cycle, intracellular and extracellular signaling, and metabolic pathways.1 Thus, damaged PT cells dedifferentiate, and initially undergo cell cycle arrest, predominantly at the G2/M checkpoint.2 This cell cycle arrest may allow repair of DNA damage caused by reactive oxygen species secondary to mitochondrial damage (e.g. after ischemia) or directly by DNA-damaging toxins (e.g. cisplatin, aristolochic acid (AAI)). Sustained cell cycle arrest is associated with a switch to secretion of pro-fibrotic signaling molecules, such as transforming growth factor-beta (TGF-β), and Wnt and Hedgehog family members.3, 4 Such signaling molecules induce resident fibroblasts to proliferate and differentiate to myofibroblasts, beginning the transition to a fibrotic injury. PT cells also undergo metabolic reprograming, with severe downregulation of fatty acid β-oxidation (FAO), and limited compensation by anerobic glycolysis.5 Uninjured PT cells use FAO for the majority of their ATP production, and inhibition of this process using the carnitine palmitoyl transferase (CPT1) inhibitor etomoxir alone can induce dedifferentiation and apoptosis of tubular cells in vitro, and worsen folic acid-induced kidney injury in vivo.5 Conversely, upregulation of FAO either by overexpressing Ppargc1a or by using a peroxisome proliferator activated receptor alpha (PPARα) agonist, attenuated kidney injury.5 However, PPARα agonists have so far not translated to clinical use, suggesting that other transcription factors may also contribute to the regulation of PT FAO in the setting of kidney injury.
Krüppel-Like Factor 15 (KLF15) is a member of the Krüppel-like subfamily of zinc finger transcription factors, involved in a diverse range of cellular processes.6 Although KLF15 is highly enriched in the kidney, it is required in multiple epithelial cell types for cellular differentiation and for control of cellular metabolism (e.g. in adipocytes, hepatocytes, skeletal myocytes, and cardiomyocytes).6, 7 In cardiomyocytes, which also use FAO for the majority of their cellular ATP production, Klf15 knockout led to reduced palmitate utilization due to dysregulation of several FAO genes, and KLF15 was shown to interact with p300 to promote open chromatin structure, and with PPARα to actively transcribe FAO genes.8, 9 KLF15 also plays a role in kidney interstitial fibrosis and glomerulosclerosis. In 5/6 nephrectomized rats, KLF15 expression was downregulated in podocytes, and in fibroblasts by TGFβ1 via ERK and JNK/MAPK signaling pathways.10, 11 Nephrectomized rats fed a low protein diet had improvement of kidney pathology, which correlated with increased KLF15 expression, whilst global Klf15−/− mice had significant glomerulosclerosis after uninephrectomy compared to control mice.10 We recently showed that stromal specific loss of Klf15 exacerbated myofibroblast proliferation and interstitial fibrosis, in unilateral ureteric obstruction (UUO) and angiotensin II overload models, due to activation of canonical Wnt/β-catenin signaling.12 KLF15 is also critical for podocyte differentiation, and in its absence, podocytes are more susceptible to injury.13 Given the protective effects of podocyte and stromal KLF15 in kidney injury14, 15, and the potential role of KLF15 in regulation of FAO, we hypothesized that loss of PT-specific KLF15 exacerbates PT injury and subsequent fibrosis.
METHODS
Generation and genotyping of mice
All animal studies conducted were approved by the Stony Brook University Animal Institute Committee approved all animal studies. NIH Guide for the Care and Use of Laboratory Animals was followed strictly. Klf15fl/fl (C57BL/6) mice were generated by inserting LoxP sites flanking exon 2 of the Klf15 gene as previously reported.16 Klf15fl/fl mice were crossed with C57BL/6 mice expressing Cre recombinase under the control of the Pepck promoter17 to generate Pepck-Cre Klf15fl/fl (Klf15PTKO) mice. This Pepck promoter is mutagenized to minimize liver expression,17 and previous studies have shown absence of knockdown of floxed genes in liver using this promoter.18 After two generations of breeding, Klf15fl/fl (control) and Klf15PTKO mice were used in the experimental groups. Genotyping by tail preparation and PCR were performed at two weeks of age as previously described.15 Mice transgenic for the Gt(ROSA)26Sor(CAG-tdTomato) allele (JAX stock number 007905) were crossed with Pepck-Cre mice to generate reporter mice. Tg26 mice aged 10 weeks were used as a model of human immunodeficiency virus (HIV)-associated nephropathy, as described previously.14 All mouse experiments were undertaken at a similar time of day to negate any effects of circadian rhythm on kidney function or metabolism.
Aristolochic acid I (AAI), ischemia-reperfusion injury (IRI), and unilateral ureteric obstruction (UUO) models
Klf15fl/fl and Klf15PTKO littermates either underwent DMSO (vehicle) or AAI treatment with modification as previously reported19 starting at 10 weeks of age. DMSO or 3mg/kg AAI dissolved in DMSO was administered by intraperitoneal injection every 3 days for 2 weeks followed by euthanasia after either 3 days (day 15 after first injection) to study the active phase of the disease, or 2 weeks (day 27 after first injection) to allow fibrosis development and remodeling.
Simultaneous uninephrectomy (UNx) of the right kidney with mild IRI of the left kidney was performed in separate groups of control and Klf15PTKO mice aged ~24 weeks.20 This protocol reduces variability compared to bilateral IRI, whilst still resulting in functional changes, in contrast to unilateral IRI. Under anesthesia, the blood vessels of the right kidney were ligated, the kidney removed, and one half fixed for histological analysis with the other half preserved for RNA analysis. During the same surgery, the blood vessels of the left kidney were exposed and clamped using a vascular clamp (ROBOZ, RS-5459) for 24 minutes. After releasing the clamp, the kidney was returned to the retroperitoneum. Successful ischemia and reperfusion were confirmed visually in all mice. Blood samples were collected at days −1, +1, +4 and +7 relative to the surgery, and mice were euthanized on day 7.
UUO was performed in C57Bl/6 mice as previously reported, and mice were euthanized after 3 days.12 Under terminal anesthesia, blood was collected by cardiac puncture, followed by cardiovascular perfusion with PBS, and collection of kidneys for histological, RNA and protein analyses.
Assessment of kidney function
Serum urea nitrogen was measured by colorimetric detection method (Arbor Assays) as per manufacturer’s protocol. Serum creatinine was measured by the Isotope Dilution LC-MS/MS at the University of Alabama at Birmingham O’Brien Core Center.
Real-time PCR
Total RNA was extracted from tissue samples using the RNeasy kit (Qiagen) or from cultured cells using TRIzol (GIBCO Life Technology). After DNase treatment, first-strand cDNA was prepared from RNA (1.5μg) using the SuperScript VILO cDNA Synthesis Kit and Master Mix (Life Technologies). cDNA was diluted and amplified in triplicate using Powerup SYBR Green qPCR Master Mix on an ABI Quantstudio 3 Real-Time PCR instrument (Applied Biosystems). Primers for mouse (m)Klf15, mPpara, mCpt1a, and mAcaa2 were designed using NCBI/Primer-BLAST (Supplementary Table 1). Quantstudio software was used to determine CT values using the second derivative method. Data were normalized to a reference gene (mActb) and presented as fold increase compared with RNA isolated from the control group using the 2−ΔΔCT method.21
Histopathological analysis
Kidneys were fixed in 10% phosphate-buffered formalin for 24 hours, processed, embedded in paraffin, and sectioned at 3μm thickness. Sections were stained with Periodic Acid Schiff (PAS) (Sigma-Aldrich) or Hematoxylin and Eosin (H&E) according to standard protocols and mounted in permanent mounting medium. Slides were viewed using a Nikon Eclipse 90i microscope and DS-Fi1 camera. Histological scoring for tubular damage, interstitial fibrosis and inflammation was performed using a semiquantitative scale from 0 to 3 (zero: none; mild (1): ≤25%; moderate (2): >25–50%; and severe (3): >50%) by a histopathologist (MPR), who was blinded to genotype and treatment.
Immunofluorescence
Immunofluorescence was performed by deparaffinizing and rehydrating the sections, followed by heat induced epitope retrieval in citrate buffer pH6. After blocking, sections were incubated with the following primary antibodies: rabbit anti-Col1α1 (ab34710; Abcam), mouse anti-α-SMA (D21H3; Cell Signaling), rabbit monoclonal anti-aristolactam (AL)-DNA adducts (Thomas Rosenquist, SBU)22, rabbit anti-Ki67 (Biocare Medical), rabbit anti Aquaporin-1 (Alpha Diagnostics), rabbit anti-cytokeratin-20 (ab97511; Abcam), rabbit anti-KLF15 (Genscript), mouse anti-Aquaporin-1 (ab9566; Abcam), rat anti-GR1 (MCA2387; BioRad) or rat anti-CD68 (137001; BioLegend) at 4°C overnight. After washing, sections were incubated with unlabeled secondary goat anti-rabbit (111-005-144; Jackson Immunoresearch), or rabbit anti-rat (312-005-003; Jackson Immunoresearch), followed by fluorescently labeled tertiary donkey anti-goat-568 (A11057; Life Technologies), donkey anti-mouse-647 (A31571; Life Technologies), or donkey anti-rabbit-568 (A10042; Life Technologies); or goat anti-mouse-488 (A11029; Life Technologies) antibodies. After counterstaining with fluorescein-labeled Lotus lectin (Vector Labs) or Phaseolus vulgaris Erythroagglutinin (PHA-E) (Vector Labs) and/or Hoechst (Invitrogen), slides were mounted in ProLong Gold Antifade mounting media (Invitrogen) and photographed under a Nikon Eclipse i90 microscope and DS-Qi1Mc camera. Quantifications of intensity, area stained, and cell numbers were performed using ImageJ 1.52p software.
Manual Microdissection of PT Segments
Kidneys from mice euthanized 24 hours after a single DMSO or 3mg/kg AAI injection were sliced and tubule microdissection was performed in ice-cold dissection solution (135mM NaCl, 1mM Na2HPO4, 1.2mM Na2SO4, 1.2mM MgSO4, 5mM KCl, 2mM CaCl2, 5.5mM glucose, 5mM HEPES, 0.1% BSA; pH 7.4) using a Leica MZ12.5 stereomicroscope equipped with Leica MC170 HD microscope camera and ThermaZone cooling device. Five to ten S2/S3 PT tubules were pooled together from each kidney and washed with PBS in a separate petri dish. RNA was extracted using PicoPure RNA kit (Applied Biosystems).
RNA Sequencing and Enrichment Analyses
Total RNA was isolated from kidney cortex of Klf15fl/fl and Klf15PTKO mice treated with DMSO, or AAI in the active and remodeling phases (n=3 per group) using the RNeasy mini kit (Qiagen, Hilden, Germany). Following RNA isolation, and DNase digestion, total RNA integrity was checked using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA concentrations were measured using the NanoDrop system (Thermo Fisher Scientific, Inc., Waltham, MA). Preparation of RNA sample library and RNA-seq were performed by the Genomics Core Laboratory at Weill Cornell Medicine. cDNA was prepared using TruSeq Stranded Total RNA Sample Preparation kit (Illumina, San Diego, CA), according to the manufacturer’s instructions. The normalized cDNA libraries were pooled and sequenced on Illumina HiSeq4000 sequencer with single-end 50 cycles. The raw sequencing reads in BCL format were processed through bcl2fastq 2.19 (Illumina) for FASTQ conversion and demultiplexing. Differentially expressed genes (DEGs) were determined via the BioJupies platform23 using Characteristic Direction (CD)24 or limma methods. Significantly differentially expressed genes were defined as those with >1.5-fold or <0.7-fold change, and BH adjusted p-value of <0.05. Heat maps were generated using Morpheus software (https://software.broadinstitute.org/morpheus) and genes were clustered using the one minus Pearson’s correlation method. Pathway and transcription factor enrichment analyses of the DEGs were performed using Enrichr,25, 26 and matching against the geneset libraries of KEGG 2019 (mouse) pathways, WikiPathways 2019 (mouse), and the NCI-Nature Pathway Interaction Database (2016), and Gene Expression Omnibus (GEO) datasets, mined through the Enrichr “TF perturbation followed by expression” library, for transcription factor enrichment analysis.
Promoter Analyses
Potential transcriptional regulators of Klf15 were identified using the ChIP-X Enrichment Analysis (ChEA) dataset27 accessed via Enrichr25, 26, and cross-matching with those expressed in the PT using available single cell RNA-Seq data, and those binding within +/− 3kb of the transcription start site (TSS) of Klf15. To visualize the locations of the binding sites relative to a region of open chromatin around the Klf15 TSS, we utilized the R Bioconductor package Gviz. The ATAC-seq BigWig coverage file from mouse p2 nephron (GSE12480428) was used along with bed files of KLF4 (ref29), c-MYC (ref29), and CEBPβ (GSE2782630) binding sites, which had first been converted to mm10 coordinates using the UCSC liftover tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Predicted transcriptional targets of KLF15 were determined as reported previously.14 Predicted PPAR binding sites were obtained from the PPARgene database (www.ppargene.org)31 and experimentally determined PPARα binding sites were obtained from previously published ChIP-Seq data for PPARα in WT and Ppara knockout mouse livers (GSE113157)32 and mapped against the mouse p2 nephron ATAC-seq data as above. Binding site locations were mapped between equivalent mouse and human locations using the UCSC liftover tool.
Chromatin Immunoprecipitation Assay
HEK293 cells were transiently transfected with V5 empty vector (EV) control or KLF15-V5 vector using Viafect reagent (Promega), and 24 hours later were treated with DMSO or 20μM AAI for a further 24 hours. The ChIP assay was performed using the SimpleChIP Enzymatic Chromatin IP Kit from Cell Signaling Technology (#9003) as per the manufacturer’s protocol. Briefly, one confluent 15cm plate of the treated HEK293 cells for each condition was used for crosslinking with 1% formaldehyde for 10 minutes followed by the addition of 1/10 volume of 1.25 M glycine to quench unreacted formaldehyde. Cells were lysed using a series of lysis buffers as per the manufacturer’s protocol. Chromatin extracted from the lysed cells was digested with Micrococcal Nuclease and sonicated using a Sonic Dismembrator 550 sonicator (Fisher Scientific) with microtip to generate chromatin fragments of 150–1000bp. Immunoprecipitation of chromatin was carried out in triplicate using rabbit anti-V5 (ab15828; Abcam) antibody or normal rabbit IgG (#2729; Cell Signaling Technology) control for nonspecific IgG binding. After incubation of chromatin with antibody at 4°C overnight, protein G–coupled magnetic beads were added and incubated for 2 hours followed by washing and elution of immunoprecipitated chromatin complexes from the beads. DNA-protein crosslinks were reversed by incubation at 65°C for 16 hours with NaCl and Proteinase K, followed by addition of RNAase A. Purified DNA was used for the analysis of predicted KLF15 binding sites in the CPT1A and ACAA2 proximal promoter regions by real-time PCR on an ABI QuantStudio 3 RT-PCR system using PowerUp SYBR Green Master Mix. PCR primers for the KLF15 binding sites in the human CPT1A and ACAA2 promoter region are listed in Supplementary Table 2. The relative amplification of the promoter sequence of each site was calculated using the 2−ΔΔCT method, and normalization was performed against the 1:50 diluted input of DNA.
Stable overexpression of KLF15 in vitro
Human kidney (HK-2) cells with stable overexpression of KLF15 were generated using Genecopoeia Lentiviral expression vector EX-U0762-Lv216 (FLAG-tagged KLF15 ORF), with EX-NEG-Lv216 as the control vector. Lentiviral particles were produced by transfecting HEK293 cells with a combination of lentiviral expression plasmid, pCD/NL-BH ΔΔΔ packaging plasmid, and VSV-G-encoding pLTR-G plasmid as previously reported.14 For HK-2 cell infection, viral supernatants were supplemented with 8μg/ml polybrene and incubated with cells for 24 hours. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, 100units/ml penicillin and 100μg/ml streptomycin, and cells with stable integration of the vectors were selected with 1μg/ml puromycin for >3 weeks prior to use in all studies. Real-time PCR was performed to confirm KLF15 overexpression.
Primary PT cell cultures
Mouse primary PT cells were isolated from kidney cortex of Klf15fl/fl mice immediately following cardiovascular perfusion with PBS. Minced cortex was incubated in 2mg/ml collagenase A solution at 37°C for 1 hour with gentle shaking. Digested tissue was passed through 2 100μm strainers, followed by centrifugation and resuspension in PBS, centrifugation and resuspension in 5% fetal bovine serum/PBS, and finally centrifugation and resuspension in complete PT cell media (DMEM:F12 with 10μg/L EGF, 5pM T3, 3.5mg/L ascorbic acid, 25μg/L prostaglandin E1, 25μg/L hydrocortisone, 1X insulin transferrin selenium (ITS) supplement, 100units/ml penicillin, and 100μg/ml streptomycin).33 Cells were infected with adenovirus expressing GFP (control) (Vector Biolabs #1060) or Cre recombinase and GFP (Vector Biolabs #1700) with multiplicity of infection=50 overnight, to induce recombination between the LoxP sites and delete Klf15. Cells were harvested for RNA or subjected to live-cell metabolic assays 48 hours after removal of the adenovirus.
Live Cell Metabolic Assays
A Seahorse Bioscience XFe96 extracellular flux analyzer was used to measure oxygen consumption rate (OCR) in by cells cultured in an XF96 cell culture microplate. EV and KLF15-OE HK2 cells were seeded at 4×104 cells per well, and primary PT cells at 2×104 per well. After 24 hours, growth medium was removed, cells washed with PBS, and serum-free Seahorse DMEM pH7.4 supplemented with 1mM glucose was added. HK2 cells were treated with DMSO or 20μM AAI for 24 hours, followed by removal of the medium and replacement with fresh medium of the same composition. After incubation for 45–60 minutes in a CO2-free incubator, either BSA (control) or palmitate-conjugated BSA (180μM) was added and OCR measured initially, and after addition of 40μM (HK2 cells) or 150μM (primary PT cells) etomoxir (inhibitor of CPT1) and 1μM (HK2) or 1.5μM (primary PT) oligomycin. OCR data were subsequently normalized for the number of cells per well using CyQuant reagent (Life Technologies).
Human AA-exposed Patient Samples
Human renal cortex and matched tumors from renal pelvis and/or ureter were obtained from patients at National Taiwan University Hospital who underwent unilateral nephrectomy due to a diagnosis of upper tract urothelial cancer. Clinical data, along with molecular evidence for AA exposure (AL-DNA adducts in renal cortex) and its effect (TP53 A>T mutations in urothelial tumors) in these samples were published previously as part of a larger dataset.34 Five-micron thick sections of formalin-fixed, paraffin-embedded samples of renal cortex were used for immunostaining in the present study.
Statistical Analysis
All mouse data were assessed for normality, and then parametric or non-parametric tests were employed for data analysis, as appropriate. The unpaired two-tailed t test was used to compare data between two groups and one-way ANOVA with Sidak’s correction for multiple testing to compare data between more than two groups. For data sets that we could not assume normality, nonparametric statistical tests were performed using the Mann-Whitney test to compare data between the two groups and the Kruskal-Wallis test with Dunn’s post-hoc test to compare data between more than two groups. The exact test used for each experiment is noted in the figure legends. Data are expressed as mean±SEM. Statistical significance was considered when p<0.05. All statistical analysis was performed using GraphPad Prism 8. For analysis of human data, GFR, and gene expression levels were extracted from previously reported tubulointerstitial microarrays from healthy donor nephrectomy and kidney disease patients deposited in Nephroseq.35 Pearson r correlation coefficients were calculated using GraphPad Prism 9. A multivariate linear regression model was used to calculate effect sizes of gene expression on GFR, using SAS v9.4 software.
RESULTS
Proximal tubular KLF15 is reduced after AAI treatment
The expression of KLF15 has previously been reported to be decreased in podocytes and fibroblasts after injury, and in the PT after UUO.10–12 To explore the potential role of PT-specific KLF15 after DNA damage during AKI and kidney fibrosis, we used the naturally occurring and clinically relevant PT-specific toxin, AAI,36 to induce multiple AKIs during 2 weeks (active phase, AKI) followed by a 2 week period after stopping AAI treatment (remodeling phase, fibrosis). AAI treatment resulted in the expected kidney injury, including loss of PT, inflammation, and protein casts as compared to vehicle (DMSO)-treated mice (Figure 1a). Aristolactam (AL)-DNA adducts, which are pathognomonic for AAI intoxication, were found in both Lotus lectin (LL) positive (brush border of fully differentiated PT) and negative (likely de-differentiated PT) tubular structures (Figure 1b, S1a). In addition, the amount of Lotus lectin staining was reduced indicating loss of differentiated PT (Figure 1b, S1b). In the remodeling phase, interstitial collagen I (COL1α1) and alpha-smooth muscle actin (α-SMA) were increased, indicating fibrosis, compared to DMSO-treated mice (Figure 1c, d, S1c, d). PT-specific KLF15 expression, as defined by LL positive tubules with KLF15 expression, was significantly decreased in the kidneys of mice with AAI in both the active and remodeling phases as compared to vehicle-treated mice (Figure 1e, S1f, g). Furthermore, Klf15 mRNA expression was reduced in the active and remodeling phases as compared to vehicle-treated mice (Figure S1e). Analysis of PT-expressed transcription factors binding in a region of open chromatin close to the Klf15 transcription start site revealed several candidates that may provide a mechanistic explanation for downregulation of Klf15 expression in injured PT (Figure S1h, i). In particular, c-Myc, which was induced after AAI, and is also upregulated in PT segments after IRI37 and UUO38 may be a driver of decreased PT Klf15 expression across different types of injury. These data indicate that the PT-specific toxin, AAI, reduced PT-specific KLF15 expression in the active and remodeling phases of kidney injury.
Figure 1. AAI toxicity results in PT injury, fibrosis, and reduced KLF15 expression.
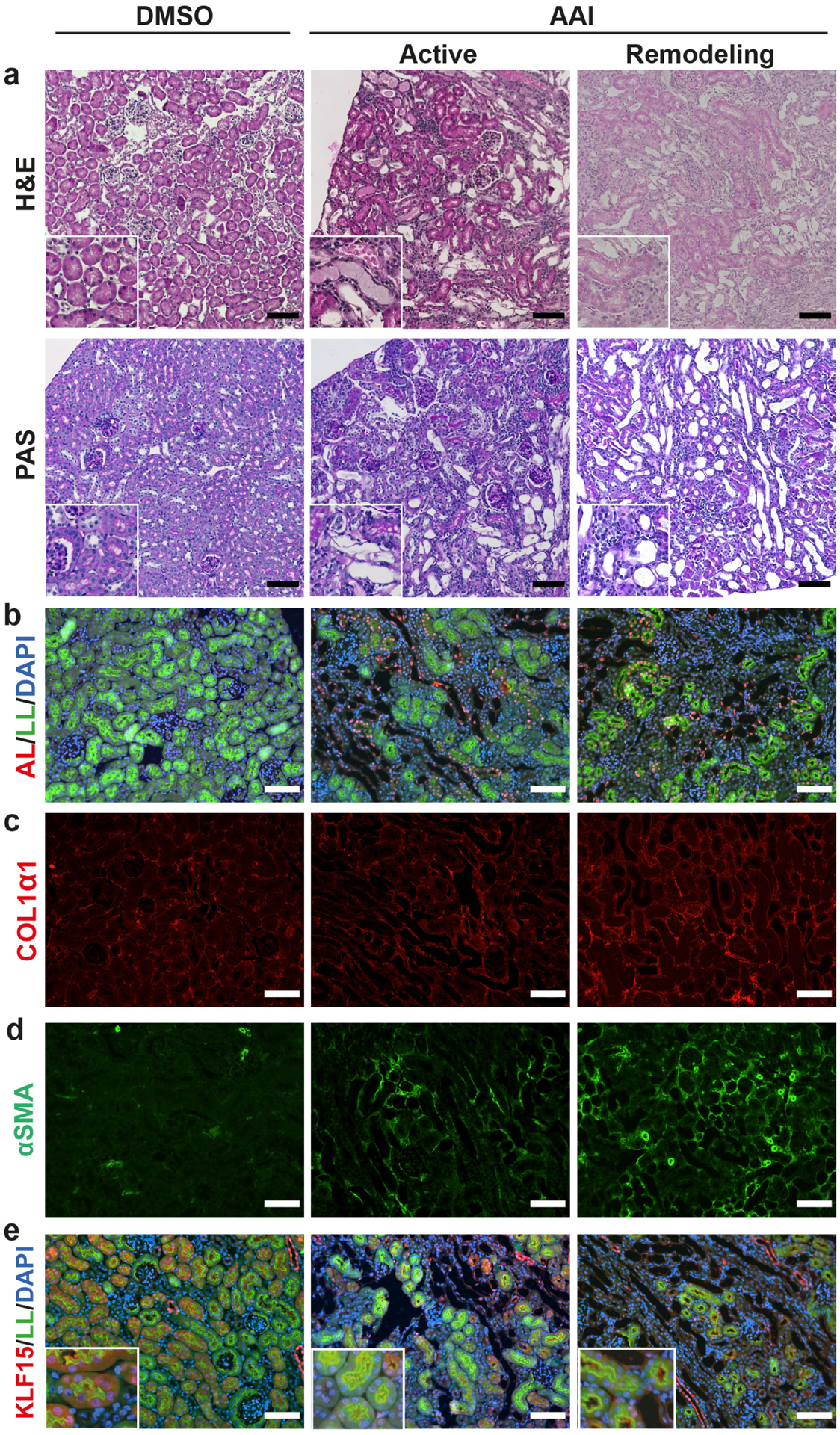
Wildtype mice were administered DMSO or AAI for 2 weeks, and examined 3 days later (active phase) or 2 weeks later (remodeling phase). (a) Histological analysis using Hematoxylin and Eosin (H&E), and periodic acid-Schiff (PAS) stains. (b-e) Immunofluorescent staining for: aristolactam (AL)-DNA adducts, with counterstaining for Lotus lectin (LL) and DAPI (b), COL1α1 (c), α-SMA (d), and KLF15 with counterstaining for LL and DAPI (e). Representative images provided from each group (n=6 per group). Scale bars = 100μm, insets are 150μm × 150μm.
PT-specific loss of Klf15 exacerbates kidney injury in the active phase after AAI treatment
To define the pathophysiologic function of KLF15 suppression in mice in response to AAI, we generated PT-specific Klf15 knockdown (Klf15PTKO) mice by crossing Klf15fl/fl mice with Pepck-Cre mice. TdTomato reporter mice showed that Pepck was expressed in the PT, with variable expression in the S1 segment and more consistent expression in the S2/S3 segments (Figure S2A), consistent with single cell RNA-seq data for Pepck expression.38 Klf15PTKO mice were viable, fertile, and phenotypically indistinguishable from Klf15fl/fl mice. Klf15PTKO mice had reductions of ~60% in PT-specific KLF15 protein expression and kidney cortical Klf15 mRNA expression (Figure S2B–D), which was similar to the reduction in KLF15 expression in the active phase of injury in control mice (Figure S1f), and therefore physiologically relevant, and consistent with transcription factor biology whereby even relatively small changes in expression can have profound biological effects.39
Klf15fl/fl and Klf15PTKO littermates were administered 3mg/kg AAI every 3 days for 2 weeks and euthanized 3 days after the last injection to assess the active phase of injury. Serum creatinine and urea nitrogen were significantly increased in Klf15PTKO mice but not in Klf15fl/fl mice with AAI versus DMSO, and serum urea nitrogen was significantly increased in Klf15PTKO versus Klf15fl/fl after AAI (Figure 2a, b). Histological analysis using H&E and PAS staining showed that AAI induced PT loss, interstitial inflammation and protein casts (Figure 2c). PT loss and protein casts were more extensive in Klf15PTKO versus Klf15fl/fl mice in this active phase. PT loss was further assessed by staining for cytokeratin-20 indicating the presence of injured PT, and LL showing that mature PT were lost in Klf15fl/fl mice, and this was significantly exacerbated in Klf15PTKO mice (Figure 2d). Immunofluorescence for aquaporin 1 (AQP1) also showed extensive loss of polarity of PT, indicated by dense intracellular staining in AAI-treated mice instead of the usual apical membrane staining in DMSO-treated mice, which was exacerbated in Klf15PTKO mice as compared to Klf15fl/fl mice (Figure 2d). Collectively, these data suggest that PT-specific loss of Klf15 exacerbated AKI after AAI treatment.
Figure 2. Loss of PT-specific Klf15 exacerbates tubular injury in the active phase of injury.
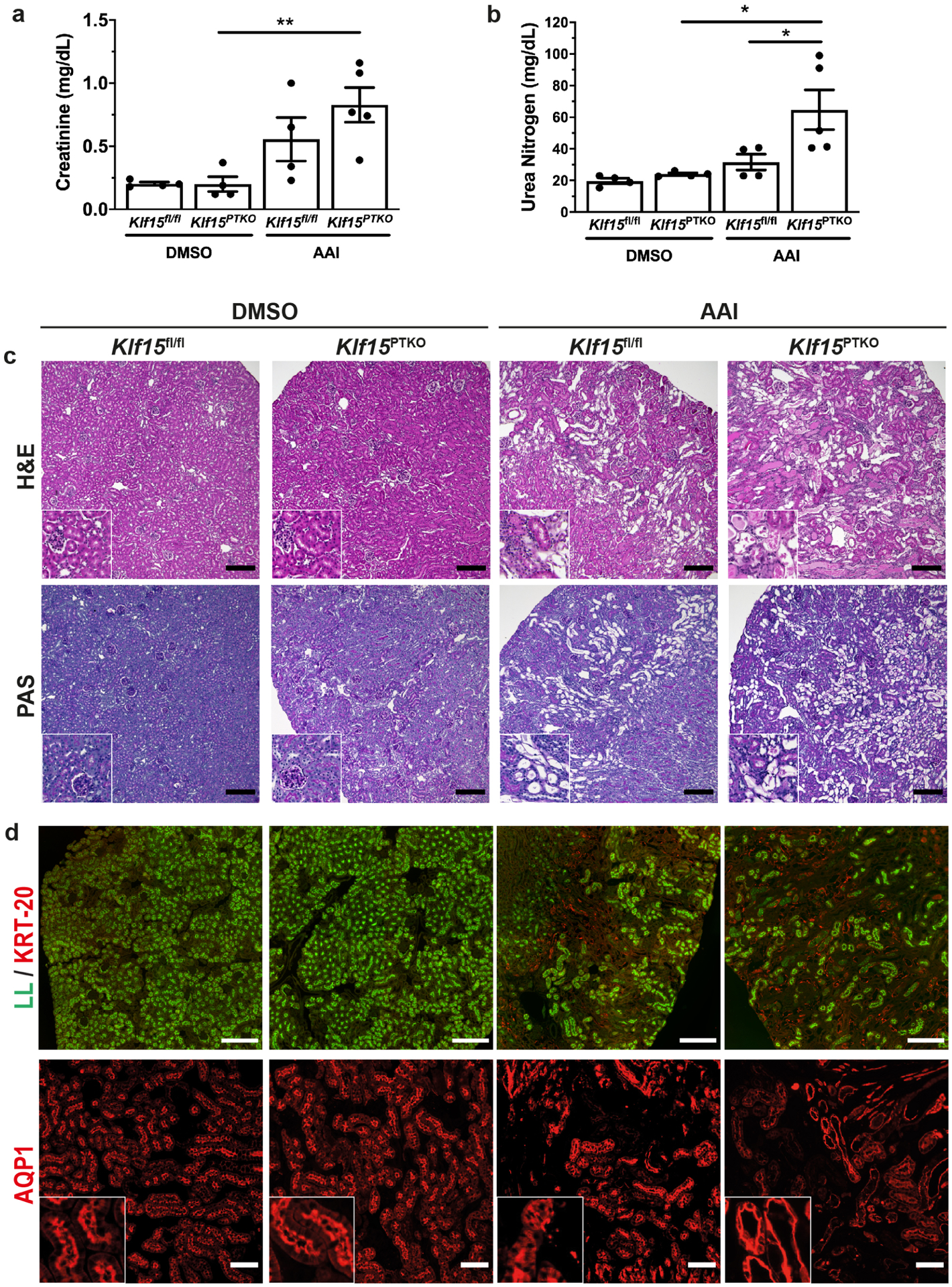
Klf15PTKO and Klf15fl/fl mice were treated with DMSO or AAI 2 weeks and examined 3 days after the last injection. (a,b) Serum creatinine (a) and urea nitrogen (b) concentrations. *p<0.05, **p<0.01; one-way ANOVA with Sidak’s correction. (c) Histological analysis using Hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) stains. Scale bars = 100μm. (d) Examination of PT by cytokeratin-20 immunofluorescence with Lotus lectin (LL) staining and AQP1 immunofluorescence. Representative images provided from each group (n=6 per group). Scale bars = 250μm for LL, 100μm for AQP1; insets are 100μm × 100μm.
PT-specific loss of Klf15 exacerbates kidney injury in the remodeling phase after AAI treatment
Mice were administered 3mg/kg AAI every 3 days for 2 weeks and euthanized 2 weeks after the last injection to assess the remodeling (fibrotic) phase of injury. Serum creatinine was significantly increased in both Klf15fl/fl and Klf15PTKO mice in this remodeling phase after AAI treatment, with a further significant increase in Klf15PTKO versus Klf15fl/fl mice (Figure 3a). Serum urea nitrogen was significantly increased in Klf15PTKO mice with AAI versus DMSO, and in Klf15PTKO versus Klf15fl/fl with AAI treatment (Figure 3b). Histological analysis showed more extensive tubule loss, and inflammation (GR1+ and CD68+ cells per high power field) in Klf15PTKO versus Klf15fl/fl mice treated with AAI (Figure 3c, Figure S3a, b, Table 1). Immunofluorescent staining for α-SMA and COL1α1 showed myofibroblast proliferation (Ki67/α-SMA staining), and deposition of α-SMA and COL1α1 in both Klf15fl/fl and Klf15PTKO mice after AAI, which was more extensive in Klf15PTKO versus Klf15fl/fl mice (Figure 3d, Figure S3c–e). We confirmed this occurred at the transcript level with the increase in Col1a1 mRNA expression in AAI-treated Klf15PTKO as compared to AAI-treated Klf15fl/fl mice (Figure S3f). Finally, cytokeratin-20 and LL staining showed that PT were not recovered in AAI-treated mice in the remodeling phase, and Klf15PTKO mice had fewer mature PT remaining than Klf15fl/fl mice (Figure 3e). Thus, loss of PT-specific Klf15 exacerbates AKI and eventual kidney fibrosis after treatment with a PT-specific injury model with AAI.
Figure 3. Loss of PT-specific Klf15 exacerbates tubulointerstitial fibrosis.
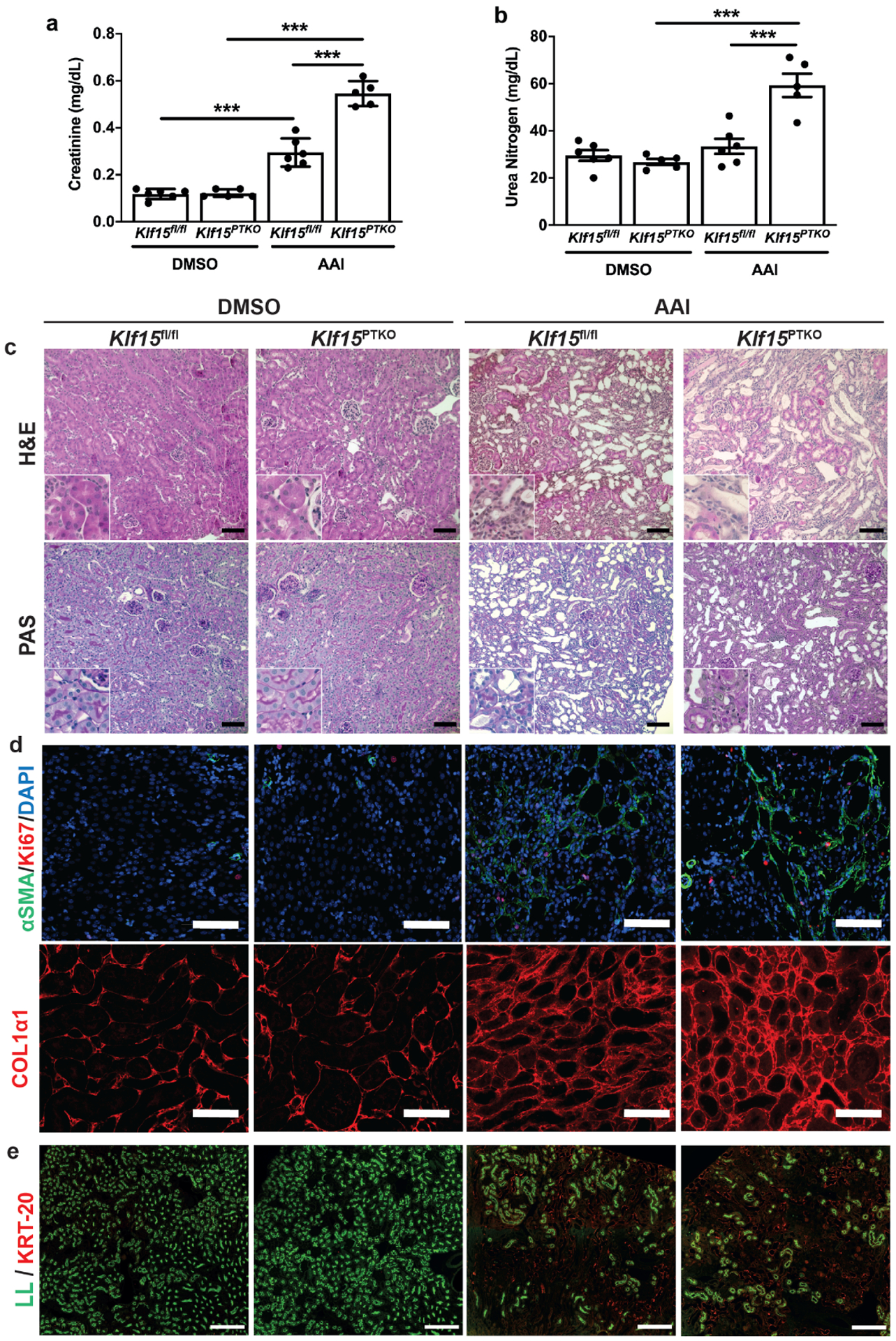
Klf15PTKO and Klf15fl/fl mice were treated with DMSO or AAI 2 weeks and examined 2 weeks after the last injection. (a,b) Serum creatinine (a) and urea nitrogen (b) concentrations. ***p<0.001; one-way ANOVA with Sidak’s correction. (c) Histological analysis using Hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) staining. Scale bars = 100μm. (d) Immunofluorescent staining for α-SMA with Ki67 and DAPI, and COL1α1. Scale bars = 100μm. (e) Examination of PT by cytokeratin-20 immunofluorescence with Lotus lectin staining. Representative images provided from each group (n=6 per group). Scale bars = 250μm.
Table 1.
Histological scoring of kidney injury in DMSO and AAI-treated Klf15fl/fl and Klf15PTKO mice
| Fibrosis | Inflammation | Tubular injury | |
|---|---|---|---|
| Klf15fl/fl + DMSO | 0.0 | 0.0 | 0.0 |
| Klf15PTKO + DMSO | 0.0 | 0.0 | 0.0 |
| Klf15fl/fl + AAI | 0.50±0.24 | 1.3±0.23$$ | 1.8±0.28$$$ |
| Klf15PTKO + AAI | 1.0±0.0$ | 2.4±0.24$$$,** | 2.8±0.20$$$,* |
Fibrosis, inflammation and tubular injury score: 0 (none), 1 (<10%), 2 (10–25%), 3 (25–50%) n= 4–6 mice per group
All data are expressed as mean ± SEM; $p<0.05, $$p<0.01, $$$p<0.001 versus same genotype with DMSO; *p<0.05, **p<0.01 versus AAI-treated Klf15fl/fl mice, one-way ANOVA with Sidak’s correction for multiple testing.
Loss of PT Klf15 exacerbates ischemia-reperfusion injury (IRI)
To determine whether the exacerbatory effect of loss of Klf15 was specific for AAI-induced injury or applicable to other models of injury, we utilized a uninephrectomy (UNx) + IRI protocol in control and Klf15PTKO mice. Simultaneous UNx of the right kidney and IRI of the left kidney was undertaken, with the uninephrectomized kidney used as the contralateral (uninjured) control tissue. Serum creatinine and urea nitrogen levels increased in both control and Klf15PTKO mice at day 1 after IRI; however, whilst control mice recovered back to baseline levels, Klf15PTKO mice still had significantly elevated serum creatinine and urea nitrogen levels at day 7 (Figure S4a, b). Histological analysis showed only small foci of injury in control mice, whilst Klf15PTKO mice had much more extensive injury, such as tubular dilation, loss of PT brush border, inflammation, and occasional protein casts (Figure S4c). PT injury was confirmed by immunofluorescence for cytokeratin-20 and costaining for LL, showing significantly more PT injury in Klf15PTKO mice compared to control mice (Figure S4d).
Loss of PT Klf15 activates pathways specific to immune responses and integrin signaling, and suppresses metabolic pathways
We sought to determine the changes that occur at the transcriptome level mediated by KLF15 both at baseline, and in the setting of AAI-induced injury, by undertaking RNA-seq of mRNA from kidney cortex of Klf15fl/fl and Klf15PTKO mice treated with DMSO or AAI in the active and remodeling phases. A combined total of 3,042 DEGs were identified between pairwise comparisons of genotypes or treatments (Figure 4a, Supplementary Data file). AAI-treated Klf15PTKO mice versus AAI-treated Klf15fl/fl mice revealed significant increases in immune system and integrin pathways. Interestingly, complement pathways were increased in the active phase, whereas other immune pathways such as macrophage- and phagocytic pathways were not significantly different until the remodeling phase, indicating a bi-phasic activation of different immune components in Klf15PTKO mice compared to Klf15fl/fl mice (Figure 4b). Several of the integrin and focal adhesion pathways were already upregulated without injury (DMSO-treated mice), and became more significantly different in the remodeling phase, rather than the active phase, suggesting that Klf15PTKO mice may be predisposed to developing fibrosis, leading to increased exacerbation of injury and fibrosis in Klf15PTKO mice over time after AKI. Pathways that were decreased in Klf15PTKO mice versus Klf15fl/fl mice were predominantly related to cellular metabolism, and could be classified into amino acid, carbohydrate, and fatty acid (FA) metabolic pathways (Figure 4c). In contrast to the upregulated pathways, these metabolic pathways were much more significantly downregulated in the active phase compared to the remodeling phase, suggesting that downregulation of these metabolic pathways may be a driving force for the later exacerbations in inflammation and/or fibrosis. Interestingly, PPAR signaling pathways were significantly downregulated in Klf15PTKO mice without injury (DMSO), suggesting that compromised FAO may predispose Klf15PTKO mice to worse injury (Figure 4c). Circadian rhythm genes (Per1-3, Bhlhe40) were also slightly downregulated in Klf15PTKO mice without injury; however these differences were not detected in the active or remodeling phases, suggesting that disrupted circadian rhythm is unlikely to be a major contributor to the exacerbation of injury in Klf15PTKO mice. Analysis of PT transporters showed that there were no differences between Klf15fl/fl and Klf15PTKO either at baseline or after AAI (Figure S5a, b). Interestingly, S1 PT transporters were much less downregulated after AAI compared to S2/S3 transporters, consistent with AAI predominantly affecting the S2/S3 PT segments36. To determine potential transcription factors that may regulate the DEGs, we undertook transcription factor enrichment analysis by matching the DEGs against GEO datasets, using the “Transcription factor perturbation followed by expression” library in Enrichr. Analysis of the genes that were upregulated between Klf15PTKO and Klf15fl/fl mice showed that transcription factors predicted to drive differential gene expression in the remodeling phase were also predicted in the absence of injury (Figure 4d). Upregulated genes in Klf15PTKO mice matched multiple genesets, including genes upregulated by over-expression of GATA6, which is associated with cell death, and genes upregulated by loss of GLIS2, which results in nephronophthisis with fibrosis and increased hedgehog signaling.40, 41 Transcription factors accounting for downregulated genes were also frequently enriched without even injury, suggesting compromised kidney function as a result of loss of KLF15. For example, genes downregulated in Klf15PTKO mice matched with gene sets downregulated in GLIS2 knockout mice, downregulated in mice with knockout of HNF4α which is a critical transcription factor in PT development,42 and downregulated in mice with loss of PPARα (Figure 4e).
Figure 4. RNA-Seq reveals increased immune activity and cell adhesion, and decreased cellular metabolism, in Klf15PTKO mice.
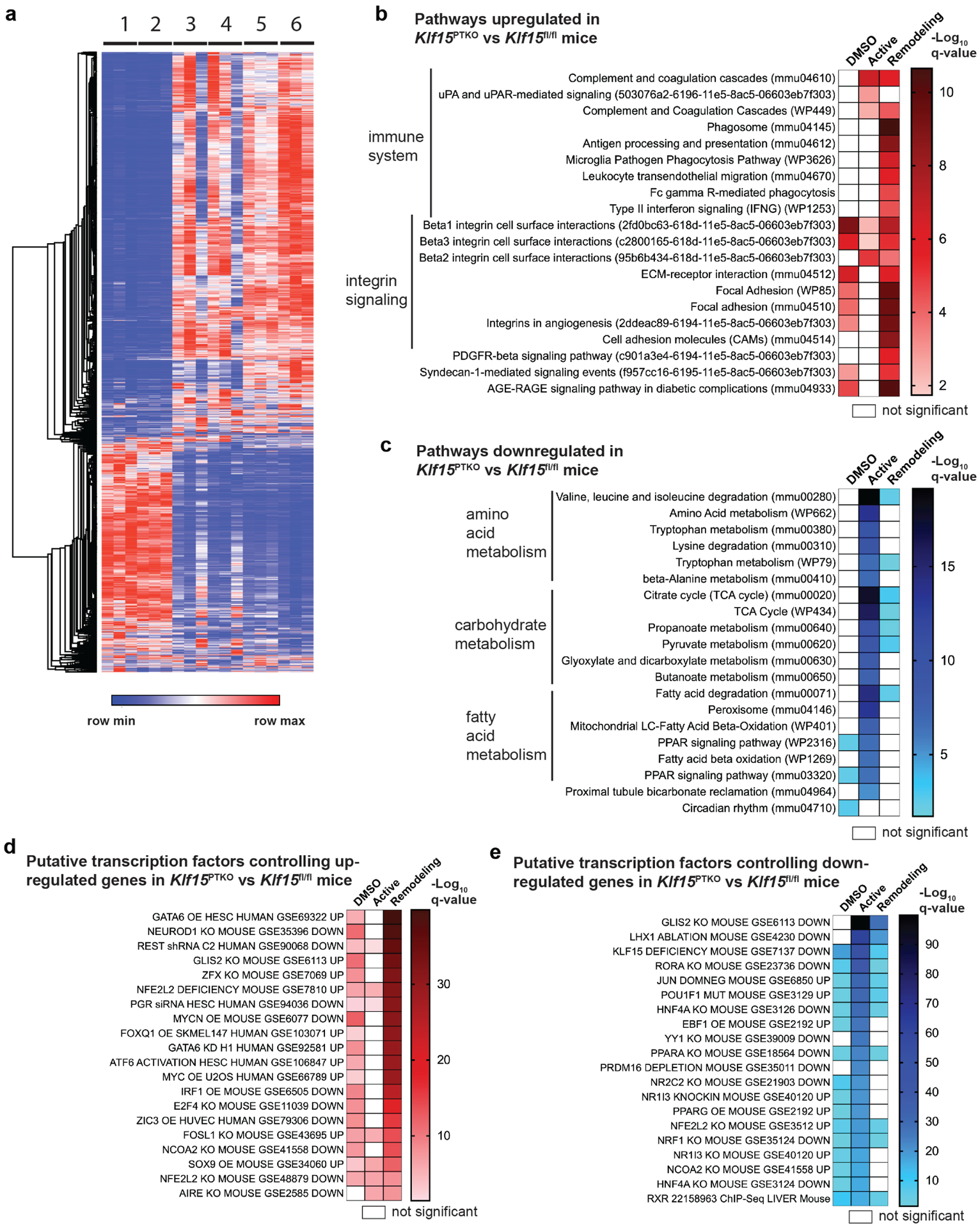
(a) Heat map of combined differentially expressed genes between pairwise genotype- or treatment- comparisons between Klf15fl/fl (lane 1) and Klf15PTKO (lane 2) mice treated with DMSO, Klf15fl/fl (lane 3) and Klf15PTKO (lane 4) mice with AAI in the active phase, and Klf15fl/fl (lane 5) and Klf15PTKO (lane 6) mice with AAI in the remodeling phase. (b,c) A combination of WikiPathway 2019 (mouse), KEGG pathways 2019 (mouse), and NCI-Nature PID enrichment analyses of differentially expressed genes upregulated (b), and downregulated (c), in Klf15PTKO versus Klf15fl/fl mice in the active and remodeling phases. (d,e) Transcription factor enrichment analysis of differentially expressed transcripts upregulated (d), and downregulated (e), in Klf15PTKO versus Klf15fl/fl mice in the active and remodeling phases.
KLF15 and PPARα likely co-operate to regulate FAO
To determine the potential mechanism by which the loss of KLF15 exacerbates the loss of FAO and PT injury, we performed TRANSFAC position weight matrix analysis14, 43 to identify which of these differentially downregulated genes possesses transcriptional binding sites for KLF15 (Supplemental Data file). Analysis of predicted KLF15 binding sites (BS) showed that multiple genes in the FAO pathway were both significantly downregulated in AAI-treated Klf15PTKO versus Klf15fl/fl mice, and have at least one KLF15 binding site within 10kb of their transcription start site, including Cpt1a (7 BS), Acaa2 (4 BS), Acat1 (1 BS) and Ascl1 (1 BS) (Figure 5a). Furthermore, these genes are also predicted to be PPAR target genes with high confidence,31 or have been shown to have PPARα binding sites in their promoters by ChIP-Seq in mice (GSE113157)32 (Figure 5a). In addition, PPARα is known to be downregulated both in mouse kidney injury models (folic acid nephropathy and overexpression of Notch1 intracellular domain) and human CKD.5 Cpt1a encodes the predominant isoform of mitochondrial carnitine palmitoyltransferase 1 (CPT1), which catalyzes the transfer of long chain fatty acyl-CoA molecules such as palmitoyl-CoA from CoA to carnitine, allowing it to cross the mitochondrial membrane, an essential and rate-limiting step in FAO. Interestingly, Cpt1c, which has 4 predicted KLF15 BS and encodes an isoform of CPT1 that is not usually expressed in the kidney, was upregulated in the setting of injury, but was upregulated significantly less in Klf15PTKO versus Klf15fl/fl mice in the active phase. Acaa2 encodes acyl-CoA acyltransferase 2, that uses CoA to catalyze the removal of 2 carbons from fatty-acyl CoA to generate acetyl-CoA at the end of each round of oxidation, and is therefore used multiple times in each fatty acid molecule oxidation (Figure 5b). Expression levels of Ppara, Cpt1a, and Acaa2 were confirmed to be downregulated both in Klf15PTKO mice compared to Klf15fl/fl mice with DMSO, and after AAI treatment in both genotypes compared to DMSO (Figure 5c). Several genes encoding typical PT markers were expressed at similar levels in AAI-treated mice compared to DMSO-treated mice, and in contrast to Ppara, Cpt1a, and Acaa2, PT markers were not differentially expressed at baseline in Klf15PTKO mice (Figure S6a). Furthermore, as previously reported, some genes encoding glycolytic enzymes were upregulated after AAI treatment (Figure S6b). Thus, reduced expression of Ppara, Cpt1a, and Acaa2 were likely not solely a result of loss of PT cells, but also due to specific downregulation within PT cells. PT specificity of this downregulation in response to AAI treatment was confirmed for Klf15, Ppara, and Cpt1a using RNA-seq data from microdissected S2/S3 PT segments (GSE150897) (Figure S6c). We also determined whether Klf15, Ppara, Cpt1a, and Acaa2 were downregulated in two other fibrotic kidney injury models: human immunodeficiency virus (HIV)-associated nephropathy (HIVAN; Tg26 mice); and mice subjected to unilateral ureteric obstruction (UUO). In both Tg26 and UUO mice, Klf15, Ppara, Cpt1a and Acaa2 expression levels were consistently and strongly downregulated compared to controls (Figure S6d). Since Ppara, Cpt1a, and Acaa2 were downregulated in cortex from Klf15PTKO mice without injury (DMSO-treated), we confirmed this in primary PT cells derived from Klf15fl/fl mice, utilizing adenovirus expressing Cre recombinase to excise Klf15, or adenovirus expressing GFP as a control. Loss of Klf15 led to significantly reduced expression of Ppara, Cpt1a, and Acaa2 (Figure 5d).
Figure 5. Klf15 expression correlates with FAO enzyme expression, and KLF15 binds to CPT1A and ACAA2 promoters.

(a) Heatmap of FAO genes showing average expression for Klf15fl/fl and Klf15PTKO with DMSO (lanes 1, 2), Klf15fl/fl and Klf15PTKO with AAI active phase (lanes 3, 4), and Klf15fl/fl and Klf15PTKO with AAI remodeling phase (lanes 5, 6). Number of predicted KLF15 binding sites (BS), number of ChIP-Seq identified PPARα BS, and prediction score (www.ppargene.org)31 for PPAR-regulated transcription for each gene are shown. (b) Schematic of the mitochondrial fatty acid β-oxidation pathway, representing RNA-Seq Log10 fold changes for Klf15fl/fl with AAI active phase versus Klf15fl/fl with DMSO as arrow colors, and number of KLF15 binding sites as box colors for each enzyme. Two arrows emerging from one enzyme box either denotes a reversible reaction (if the arrows emerge in opposite directions) or a reaction where more than one product is depicted (if the arrows emerge in the same direction). (c) Kidney cortex expression of Ppara, Cpt1a, and Acaa2 in Klf15fl/fl and Klf15PTKO mice treated with DMSO or AAI. *p<0.05, **p<0.01, ***p<0.001; two-way ANOVA with Tukey’s multiple comparisons test. (d) gene expression analysis of primary PT cells from Klf15fl/fl mice treated with adenovirus expressing Cre recombinase (Klf15 KO) or GFP (control). ***p<0.001; t tests with Benjamini Hochberg correction for multiple testing. (e,f) Chromatin immunoprecipitation (ChIP) assay for predicted KLF15 binding sites in the CPT1A (e) and ACAA2 (f) promoters using control IgG or anti-V5 antibodies in cells transiently transfected with V5-empty vector (V5-EV) or V5-tagged KLF15 (V5-KLF15) and treated with DMSO or 20μM AAI. *p<0.05, **p<0.01, ***p<0.001; two-way ANOVA with Tukey’s multiple comparisons test. (g,h) Mapping of open chromatin from ATAC-seq in p2 mouse nephrons and KLF15 and PPARα binding sites with respect to the mouse Cpt1a (g) and Acaa2 (h) loci.
To experimentally validate the predicted binding sites of KLF15 within the CPT1A and ACAA2 promoter regions, we determined direct interactions using chromatin immunoprecipitation (ChIP) assays. HEK293 cells were transiently transfected with V5-tagged KLF15 (V5-KLF15) or V5-empty vector (V5-EV), and ChIP performed after treatment with DMSO or AAI. In V5-EV cells, there was no statistically significant enrichment of the predicted CPT1A and ACAA2 binding sites with anti-V5 antibody compared to control IgG (Figure 5e, f). Induction of KLF15 led to significant enrichment of the predicted binding sites in the CPT1A promoter after IP with anti-V5 antibody compared to control IgG in cells treated both with DMSO and AAI, and compared to V5-EV transfected cells (Figure 5e). Binding of KLF15 to the ACAA2 promoter was less strong, with significant enrichment with anti-V5 compared to IgG pulldown, but with significant enrichment only for the more distal promoter in V5-KLF15 cells compared to V5-EV cells (Figure 5f). In these HEK293 cells overexpressing KLF15, binding of KLF15 to the CPT1A and ACAA2 promoters was maintained even in AAI-treated cells, suggesting that the affinity of binding of KLF15 to these promoters is not altered by injury, but instead that downregulation of KLF15 causes loss of transcriptional activation of CPT1A and ACAA2. KLF15 and PPARα proteins have been shown to physically interact in cardiac myocytes, and mapping of the ChIP-Seq identified PPARα BS in the CPT1A promoter, and predicted PPAR BS in the ACAA2 promoter showed that in both cases, a PPAR(α) binding site is located in close proximity (~100–200bp) to one of the ChIP-validated KLF15 BS in each of the two promoters (Figure 5g, h). Furthermore, these binding sites are located in regions of open chromatin in mouse nephron, thus likely facilitating co-operative binding of both KLF15 and PPARα together to their respective binding site in the CPT1A and ACAA2 promoters.
To determine whether loss of Klf15 leading to a subsequent loss in Cpt1a and Acaa2 expression would alter cellular metabolism, we undertook live cell metabolic assays in the Klf15fl/fl primary PT cells treated with adenoviral Cre or GFP (control), and measured oxygen consumption rates (OCR) as a readout of mitochondrial respiration. Cells were cultured in low glucose (1mM) media, and the FAO substrate palmitate, or BSA (control) were added immediately before the assay. Initial OCR measurements were followed by measurement after addition of etomoxir, an inhibitor of CPT1, to determine the OCR due to FAO, and after addition of oligomycin (inhibitor of ATP synthase) (Figure 6a). Control cells were able to utilize palmitate effectively, as shown by the significant increase in OCR due to FAO in cells treated with palmitate compared to BSA. However, in cells lacking Klf15, there was no significant difference in OCR between cells treated with BSA versus palmitate, showing that cells lacking Klf15 were unable to utilize palmitate as effectively as control cells (Figure 6a, b). To determine whether overexpression of KLF15 could rescue changes in FAO in injured cells, we measured OCR in HK2 EV and KLF15-OE cells treated with DMSO or AAI (Figure 6c). In cells treated with DMSO, there was a trend towards higher FAO in cells supplemented with palmitate compared to BSA (Figure 6d). Palmitate-supplemented EV cells treated with AAI had a significantly decreased FAO compared to cells treated with DMSO, which was not observed in KLF15-OE cells, suggesting partial protection from loss of FAO in injured cells overexpressing KLF15 (Figure 6d, e).
Figure 6. KLF15 alters FAO in vitro.
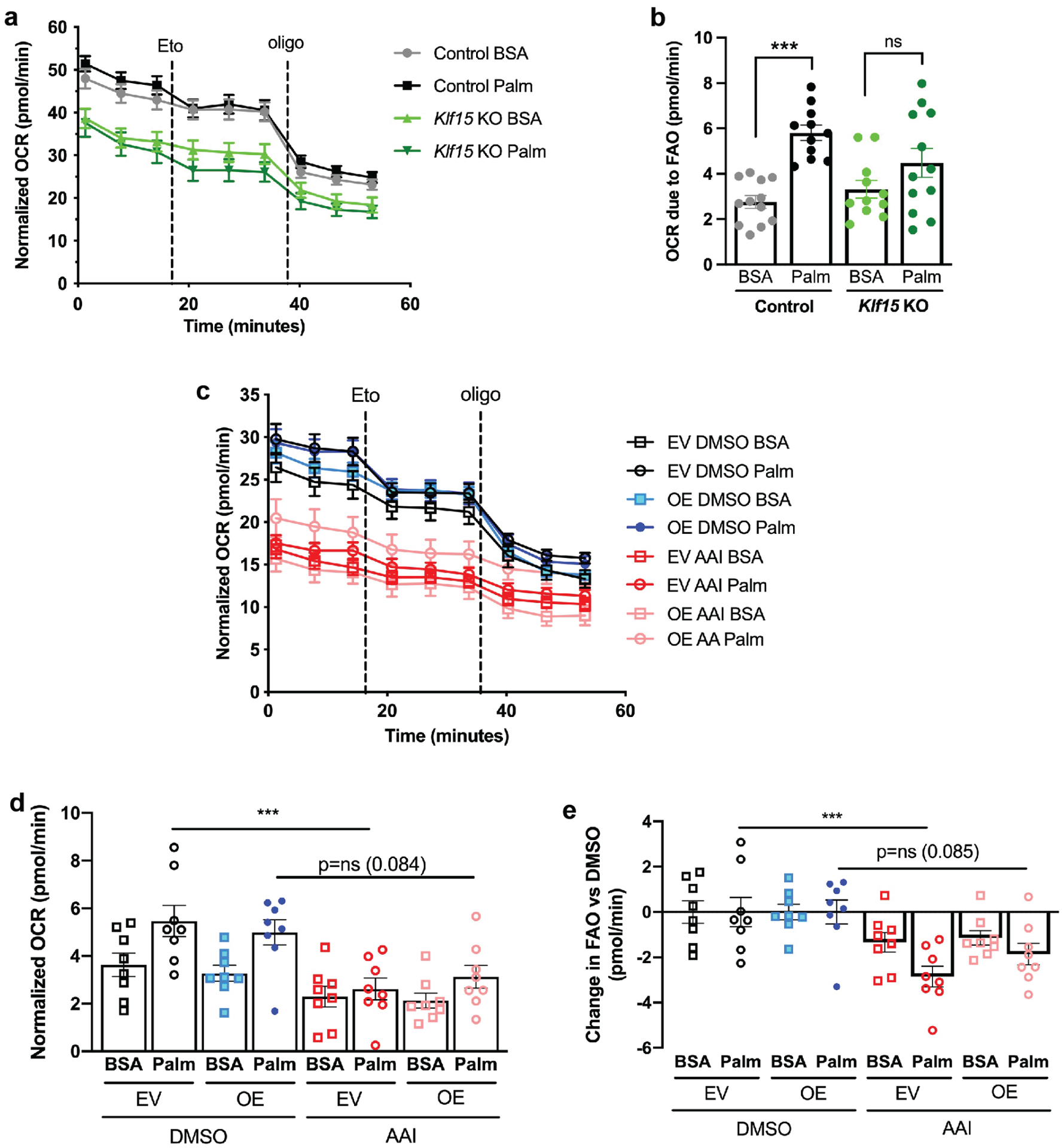
(a) Oxygen consumption rates (OCR) in Klf15fl/fl primary PT cells treated with adenovirus expressing Cre recombinase (Klf15 KO) or GFP (control), and supplemented with FAO substrate palmitate (Palm) or BSA (control). OCR was measured initially, and after addition of etomoxir (Eto) and oligomycin (oligo). (b) Quantification of OCR due to FAO, defined as OCR before etomoxir minus OCR after etomoxir. ***p<0.001; one-way ANOVA with Sidak’s multiple comparisons test. (c) OCR in HK-2 cells expressing empty vector (EV) or KLF15-OE constructs, treated with DMSO (vehicle) or AAI. (d) Quantification of OCR due to FAO. ***p<0.001; one-way ANOVA with Sidak’s multiple comparisons test. (e) Quantification of change in FAO after AAI treatment versus DMSO. ***p<0.001; one-way ANOVA with Sidak’s multiple comparisons test.
KLF15 and PPARA expression correlate with kidney function in human disease
To determine the correlation between human CKD and PT KLF15 expression, we initially undertook immunofluorescence for KLF15 and AQP1, costaining with the PT brush border-specific lectin Phaseolus vulgaris Erythroagglutinin (PHA-E) in control subjects and six patients with CKD, with or without evidence of exposure to aristolochic acids, as determined by quantification of AL-DNA adducts in the renal cortex (Figures 7a, S7, Supplementary Table 3). Control subjects showed consistent nuclear expression of KLF15 in the PT, whereas all the CKD patients demonstrated markedly decreased or absent nuclear PT KLF15 expression, along with variable loss of PHA-E staining and internalization and aggregation of AQP1, even with only slightly reduced estimated glomerular filtration rate (eGFR) (Supplementary Table 3). To determine quantitative correlations between kidney function, and KLF15, PPARA and CPT1A expression levels in human patients, we interrogated a previously published expression array from the tubulointerstitial compartment of healthy donor nephrectomies, and patients with CKD.35 In healthy control subjects, KLF15 and PPARA expression levels were significantly positively correlated, with a Pearson r correlation coefficient of 0.59 (p<0.01) (Figure 7b). Correlation analysis of eGFR, KLF15, PPARA, and CPT1A expression levels in 59 healthy, HTN, and DKD samples demonstrated that eGFR significantly correlated with both KLF15 and PPARA expression levels (p<0.001), and that CPT1A expression was significantly positively correlated with both PPARA and KLF15 expression (p<0.05) (Figure 7c). We subsequently conducted multivariate linear regression modeling to determine the effects of KLF15, PPARA, and CPT1A expression levels independently on eGFR. The model was highly significant (p<0.0001) with an R-square value of 0.3467, indicating that 34.67% of the variance in eGFR could be predicted from the variables KLF15, PPARA, and CPT1A. While controlling for PPARA and CPT1A expression, there was a significant association between eGFR and KLF15 expression (p=0.0232), suggesting that for every log2 increase in KLF15 expression there is a 46.9ml/min increase in eGFR (Table 2). In addition, while controlling for KLF15 and CPT1A expression, there was a significant association between eGFR and PPARA expression (p=0.0474), suggesting that for every log2 increase in PPARA expression there is a 60.8ml/min increase in eGFR (Table 2). We observed no significant association between eGFR and CPT1A expression (p=0.1619). Extending the analysis to include patients with CKD showed the same correlation between eGFR, and KLF15 and PPARA expression (Figure 7d), with statistically significant decreases in KLF15 and PPARA expression in patients with eGFR of <30ml/min/1.73m2 or 30–60ml/min/1.73m2 versus >90ml/min/1.73m2 (Figure 7e). Expression of several PT marker genes, (CDH20, LRP2, ANPEP, and GGT1) was not decreased in the same patients (Figure S8), suggesting that the decreases in KLF15 and PPARA expression were not solely due to loss of PT. Similarly, we previously reported that KLF15 expression is reduced in the PT in the early stages of DKD, prior to significant tubulointerstitial injury.12 Collectively, these data suggest that the co-operativity between KLF15 and PPARα likely plays a critical role in regulating FAO in the setting of kidney injury.
Figure 7. Kidney function, and KLF15 and PPARA expression levels correlate in human kidneys.

(a) Co-immunofluorescence for KLF15 and AQP1, with co-staining for Phaseolus vulgaris Erythroagglutinin (PHA-E) and DAPI in a control subject and a CKD patient exposed to AA. Scale bars = 100μm. (b) Analysis of correlation between KLF15 and PPARA expression levels from previously reported expression data35 in healthy subjects. (c) Multiple variable Pearson correlation analysis of GFR, and KLF15, PPARA, and CPT1A expression levels using previously reported tubulointerstitial expression data from 59 healthy, HTN and DN samples.35 Pearson r correlation coefficients and p-values are shown for each pairwise comparison. (d) Correlation between eGFR, and KLF15, PPARA, and CPT1A gene expression using expression array data from the tubulointerstitial compartment of 164 human kidney disease patients.35 Each dot in the graph and column in the heatmap represents an individual patient, ranked by eGFR. (e) KLF15 and PPARA gene expression in the 164 patients categorized by eGFR. *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA with Dunnett’s multiple comparisons test.
Table 2.
Multivariate linear regression modeling of the effects of KLF15, PPARA, and CPT1A expression on eGFR
| Parameter estimate | Standard Error | p-value | 95% confidence interval for parameter estimate | |
|---|---|---|---|---|
| Intercept | 32.95 | 39.05 | 0.4025 | −45.314 – 111.21 |
| KLF15 | 46.92 | 20.09 | 0.0232 | 6.67 – 87.18 |
| PPARA | 60.82 | 29.99 | 0.0474 | 0.72 – 120.92 |
| CPT1A | −33.19 | 23.41 | 0.1619 | −80.11 – 13.73 |
| R-square | 0.3467 |
DISCUSSION
In this study, we demonstrate the physiological role of PT-specific KLF15 in the setting of DNA damage-induced injury. KLF15 is downregulated during injury and we show that this is a maladaptive response, since Klf15PTKO mice had exacerbated injury in the active phase of AAI toxicity, which also led to worsened fibrosis. In this study, we used a mutagenized Pepck (Pck1) promoter to drive Cre expression. Although analysis of a TdTomato reporter showed expression solely within PT cells, single cell RNA-seq data shows very low expression of endogenous Pck1 in some other cell types, e.g. podocytes.38 Future studies could utilize an alternative PT-specific promoter to drive Cre expression to confirm our findings. We previously demonstrated the role of KLF15 in podocyte injury and in stromal cells during fibrotic injury, but this is the first report showing the role of KLF15 in the PT using a PT-specific injury model. We also demonstrate here that loss of PT Klf15 was detrimental in a second model of injury, IRI. Furthermore, we show by live cell metabolic assays in primary PT cells that the importance of KLF15 is at least partly due to its transcriptional regulation of genes encoding the key FAO enzymes CPT1 and ACAA2, which are critical to maintaining PT cellular metabolism, and this effect is likely through co-operation with PPARα. Furthermore, it has recently been shown that loss of PT FAO is directly linked to PT cell de-differentiation during injury44, and our data shows that KLF15 therefore also likely plays a role in de-differentiation of PT cells through its effects on FAO. KLF15 has previously been shown to be a key determinant of FAO in cardiac muscle,8 but this is the first report demonstrating the importance of KLF15 in kidney cellular, and specifically PT cellular metabolism.
The importance of loss of FAO in PT cells during injury and in contributing to fibrosis is now established, and a number of factors likely play a role in this process, at both the transcriptional and post-translational levels. In the setting of injury, the major transcriptional drivers of FAO, Ppgarc1a (encoding peroxisome proliferator-activated receptor gamma coactivator-1-alpha; PGC1α) and Ppara are frequently downregulated, likely downstream of TGFβ1-induced SMAD3 signaling for PGC1α5 and through suppression by miRNA-21 which is upregulated in injury for PPARα.45 KLF15, which is also downregulated in injury, was previously shown to physically interact with PPARα, and indeed was found to be necessary for the transcriptional activation of FAO genes by PPARα in cardiac myocytes.9 This interaction is likely also to occur in the PT, since PPARα and KLF15 are co-expressed in PT cells. Furthermore, binding sites for KLF15 and PPARα are located within ~200bp of each other in both the CPT1A and ACAA2 gene promoters, which may facilitate co-operative binding of KLF15 and PPARα both to each other and to their target gene promoters. Whilst PPARα and KLF15 positively regulate Cpt1a transcription, hypoxia-inducible factor-1α (HIF-1α) has recently been shown to repress Cpt1a expression in the settings of renal cell carcinoma and ischemic injury.46, 47 Thus, the extent to which Cpt1a and potentially other FAO genes are downregulated during injury is likely to depend on the extent to which PPARα and KLF15 are decreased, and HIF-1α is increased. Indeed, we show here that expression levels of both PPARA and KLF15 are independently associated with eGFR in human samples from healthy and diseased kidneys. Our live-cell metabolic assays showed that overexpression of KLF15 alone did not completely rescue the decrease in FAO after AAI treatment. If indeed both PPARα and KLF15 together are needed for transcription of CPT1A and ACAA2, then activation of PPARα, e.g. using fenofibrate, may be required in addition to KLF15 overexpression, to fully rescue FAO. The therapeutic potential of restoration of FAO in treatment of kidney fibrosis was recently demonstrated in mice with nephron-specific inducible Cpt1a overexpression, that were protected from fibrosis, inflammation, and epithelial cell damage.48
This study demonstrates the importance of KLF15 in a third major kidney cell type, after previous studies in podocytes and stromal cells.12, 14 In each of these, loss of KLF15 is detrimental, but interestingly, this effect appears to have a different mechanism in each cell type. Thus, in podocytes, KLF15 is necessary for expression of podocyte differentiation markers, which are also critical to podocyte function and glomerular filtration barrier maintenance.14 In part, these effects of KLF15 were mediated in combination with the podocyte transcription factor Wilms Tumor 1 (WT1), which is not expressed in PT cells or stromal cells. Loss of KLF15 in Foxd1-positive stromal cells was detrimental through activation of Wnt/β-catenin signaling.12 Although such signaling pathways are also known to be activated in injured PT cells,49 Wnt/β-catenin signaling pathways were not found to be differentially activated in the RNA-Seq data presented here in Klf15PTKO versus Klf15fl/fl mice. Thus, KLF15 likely acts to regulate gene expression in a cell-specific manner, which may rely on different binding partners that are themselves expressed only in specific cell types. Of note, podocytes and probably stromal cells are less reliant on FAO for their main energy source, since they have a lower energy demand than cells that typically rely on FAO, such as PT cells and cardiac myocytes.50 Thus in injury, loss of Cpt1a expression after KLF15 deletion in podocytes and stromal cells would likely have less effect than loss of podocyte differentiation markers, or activation of Wnt/β-catenin signaling, respectively.
We also demonstrate that KLF15 is strongly downregulated in other established murine kidney injury of multiple etiologies, and positively correlates with eGFR in healthy control subjects as well as in individuals with HTN and DKD. Future studies should address whether restoration of KLF15, possibly in conjunction with PPARα activators such as fenofibrate, may protect against kidney injury in vivo. Development of small molecule activators of either KLF15 expression or activity would have the added advantage of potentially increasing KLF15 activity not just in the PT, but also in podocytes and interstitial fibroblasts, in which loss of KLF15 has also been shown to have a detrimental effect. Increasing KLF15 expression in all of these cell types at the same time could show a synergistic effect, particularly in those types of injury that may damage podocytes and proximal tubules, such as HIVAN or DKD.
In conclusion, downregulation of PT-specific Klf15 during kidney injury is a maladaptive response, likely through loss of transcriptional activation of essential FAO genes in conjunction with PPARα. This effect is consistent across nephrotoxic, HIV-associated, and UUO nephropathies, and in human HTN and DKD patients.
Supplementary Material
Supplementary Figure 1: Quantification of AAI-induced kidney injury parameters and KLF15 expression. (a) Quantification for number of AL-positive nuclei per high power field (HPF). (b-d) Quantification of area staining for LL (b), COL1α1 (c), and αSMA (d), expressed as fold change (fc) versus DMSO treated mice. (e) Quantification of Klf15 mRNA expression in kidney cortex expressed as fc versus DMSO treated mice. (f) Quantification of PT KLF15 expression, defined as KLF15-positive nuclei within LL-positive tubules, expressed as fc versus DMSO treated mice. *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA with Sidak’s correction. (g) Representative images of KLF15 immunostaining extracted from the merged images in Figure 1E. Scale bars = 100μm. (h) Transcription factors with ChIP-seq determined binding sites in the Klf15 promoter region. (i) Mapping of open chromatin from ATAC-seq in p2 mouse nephrons and transcription factor binding sites with respect to the Klf15 locus, with RNA-seq data from microdissected S2/S3 PT segments after 1 injection of DMSO or AAI. *p<0.05, **p<0.01.
Supplementary Figure 2: Validation of KLF15 knockdown in proximal tubules in Klf15PTKO mice. (a) Immunofluorescent staining for tdTomato expression (red), with counterstaining for Lotus lectin (green) and DAPI (blue). Scale bar = 100μm. (b) Immunofluorescent staining for KLF15 (red), with counterstaining for Lotus lectin (green) and DAPI (blue). Representative images provided from each group (n=6 per group). Scale bars = 100μm. (c) Quantification of PT KLF15 protein expression, defined as KLF15-positive nuclei within LL-positive tubules. *p<0.05; unpaired t test. (d) Quantification of Klf15 mRNA expression in kidney cortex. *p<0.05; unpaired t test.
Supplementary Figure 3: Inflammatory, fibrotic and PT markers in the remodeling phase of AAI-treated mice. (a-e) Quantification of immunostaining for: Gr1+ cells per high power field (HPF) (a), CD68+ cells per HPF (b), proliferating myofibroblasts (Ki67 and α-SMA positive cells) (c), α-SMA deposition (d), and COL1α1 deposition (e). (f) Quantification of Col1a1 mRNA expression in kidney cortex. *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA with Sidak’s correction.
Supplementary Figure 4: Loss of PT-specific Klf15 exacerbates IRI. Klf15PTKO and control mice were subjected to uninephrectomy (UNx) of the right kidney with simultaneous IRI of the left kidney. (a,b) Serum creatinine (a) and urea nitrogen (b) concentrations. $p=0.050, *p<0.05, **p<0.01, ***p<0.001; multiple t tests with Benjamini Hochberg correction for multiple testing. (c) Histological analysis using Hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) staining. Scale bars = 250μm/50μm for insets. (d) Examination of PT by cytokeratin-20 immunofluorescence with Lotus lectin staining. Scale bars = 500μm.
Supplementary Figure 5: Expression of PT transporters after DMSO or AAI (active phase). (a) S1 transporters. (b) S2/S3 transporters. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; one-way ANOVA with Sidak’s correction.
Supplementary Figure 6: Expression of PT markers and metabolic genes in kidney cortex and isolated PT from control and injured mice. (a) Expression of 9 genes encoding mouse PT markers (Slc5a2, Nox4, Slc34a1, Slc4a4, Cubn, Lrp2, Aqp1, Anpep, and Ggt1) in Klf15fl/fl and Klf15PTKO mice treated with DMSO and AAI. (b) Heatmap of glycolytic genes showing average expression for Klf15fl/fl and Klf15PTKO with DMSO (lanes 1, 2), Klf15fl/fl and Klf15PTKO with AAI active phase (lanes 3, 4), and Klf15fl/fl and Klf15PTKO with AAI remodeling phase (lanes 5, 6). Genes in bold have KLF15 binding sites in their promoters. (c) mRNA expression of Klf15, Ppara, Cpt1a, and Acaa2 in S2/S3 PT segments microdissected from mice after 1 injection of DMSO or AAI. *p<0.05, **p<0.01; t test with Benjamini Hochberg correction for multiple testing. (d) Kidney cortex expression of Klf15, Ppara, Cpt1a, and Acaa2 in control mice and models of human immunodeficiency virus (HIV)-associated nephropathy (Tg26 mice), and mice subject to unilateral ureteric obstruction. ***p<0.001; t test with Benjamini Hochberg correction for multiple testing.
Supplementary Figure 7: Histological and immunofluorescent analysis of human AAI-nephropathy patients. (a) PAS staining of kidney cortex samples from patients with (red labels) and without (black labels) evidence of exposure to AA, with images captured at 10x (top) and 40x (bottom) magnification. Scale bars = 250μm (top), 50μm (bottom). (b) Co-immunofluorescence for KLF15 and AQP1, with co-staining for Phaseolus vulgaris Erythroagglutinin (PHA-E) and DAPI in a control subject and CKD patients. The corresponding images for Patient #42 are shown in Figure 8a. Scale bars = 100μm.
Supplementary Figure 8: Expression of genes encoding human PT markers. Expression of genes encoding human PT markers in tubulointerstitial expression array data from patients with CKD.35
Supplementary Table 1. Primer sequences for qRT-PCR
Supplementary Table 2. Primer sequences for qPCR for ChIP
Supplementary Table 3. CKD patient data
Supplemental Data File. Differentially expressed genes
Translational statement:
AKI causes loss of fatty acid oxidation, the main cellular energy source for the proximal tubule (PT). Here, we report that loss of the transcription factor KLF15 specifically in the PT is detrimental in AKI and worsens the resulting fibrosis. We show that this correlates with loss of fatty acid oxidation, and that KLF15 and PPARα likely interact to transcriptionally regulate key enzymes required for FAO. Our results reveal that KLF15 is a potential therapeutic target in AKI.
ACKNOWLEDGMENTS
The authors would like to acknowledge the technical support provided by the Research Histology Core Laboratory, Department of Pathology, Stony Brook Medicine; and Stony Brook University Department of Medicine Biostatistical Core support.
Funding
This work was supported by funds from the UAB-UCSD O’Brien Center (award number P30DK079337 from the NIDDK) and an American Society of Nephrology KidneyCure Joseph V. Bonventre Research Scholar Grant to S.E.P, NIH/NIDDK grant (DK112984, DK121846) to S.K.M., VA Merit award (I01BX003698, IS1BX004815) to S.K.M., and NIH grants (U54HL127624 and U24CA224260) to A.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors have no financial conflicts of interest to disclose.
DATA AVAILABILITY STATEMENT
The RNA-seq data supporting the findings of this study are openly available in the Gene Expression Omnibus database (GSE150656).
REFERENCES
- 1.Liu BC, Tang TT, Lv LL, et al. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–579. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543, 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou D, Fu H, Zhang L, et al. Tubule-Derived Wnts Are Required for Fibroblast Activation and Kidney Fibrosis. J Am Soc Nephrol. 2017;28:2322–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou D, Li Y, Zhou L, et al. Sonic hedgehog is a novel tubule-derived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol. 2014;25:2187–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bialkowska AB, Yang VW, Mallipattu SK. Krüppel-like factors in mammalian stem cells and development. Development. 2017;144:737–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollak NM, Hoffman M, Goldberg IJ, et al. Kruppel-like factors: Crippling and un-crippling metabolic pathways. JACC Basic Transl Sci. 2018;3:132–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prosdocimo DA, Anand P, Liao X, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem. 2014;289:5914–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prosdocimo DA, John JE, Zhang L, et al. KLF15 and PPARα Cooperate to Regulate Cardiomyocyte Lipid Gene Expression and Oxidation. PPAR Res. 2015;2015:201625–201625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao X, Huang L, Grosjean F, et al. Low-protein diet supplemented with ketoacids reduces the severity of renal disease in 5/6 nephrectomized rats: a role for KLF15. Kidney Int. 2011;79:987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao X, Wu G, Gu X, et al. Kruppel-Like Factor 15 Modulates Renal Interstitial Fibrosis by ERK/MAPK and JNK/MAPK Pathways Regulation. Kidney and Blood Pressure Research. 2013;37:631–640. [DOI] [PubMed] [Google Scholar]
- 12.Gu X, Mallipattu SK, Guo Y, et al. The loss of Kruppel-like factor 15 in Foxd1(+) stromal cells exacerbates kidney fibrosis. Kidney Int. 2017;92:1178–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallipattu SK, Liu R, Zheng F, et al. Kruppel-Like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem. 2012;287:19122–19135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Y, Pace J, Li Z, et al. Podocyte-Specific Induction of Krüppel-Like Factor 15 Restores Differentiation Markers and Attenuates Kidney Injury in Proteinuric Kidney Disease. J Am Soc Nephrol. 2018;29:2529–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallipattu SK, Guo Y, Revelo MP, et al. Krüppel–Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J Am Soc Nephrol. 2017;28:166–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu Y, Zhang L, Liao X, et al. Kruppel-like factor 15 is critical for vascular inflammation. J Clin Invest. 2013;123:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piret SE, Guo Y, Attallah AA, et al. Kruppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang L, Scarpellini A, Funck M, et al. Development of a chronic kidney disease model in C57BL/6 mice with relevance to human pathology. Nephron extra. 2013;3:12–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skrypnyk NI, Harris RC, de Caestecker MP. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Vis Exp. 2013;78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang SY, Weber EJ, Sidorenko VS, et al. Human liver-kidney model elucidates the mechanisms of aristolochic acid nephrotoxicity. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torre D, Lachmann A, Ma’ayan A. BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst. 2018;7:556–561 e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark NR, Hu KS, Feldmann AS, et al. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics. 2014;15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lachmann A, Xu H, Krishnan J, et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilliard S, Song R, Liu H, et al. Defining the dynamic chromatin landscape of mouse nephron progenitors. Biol Open. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Huang J, Chen T, et al. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008;18:1177–1189. [DOI] [PubMed] [Google Scholar]
- 30.Siersbaek R, Nielsen R, John S, et al. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011;30:1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang L, Zhang M, Li Y, et al. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016;2016:6042162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang N, Damdimopoulos A, Goni S, et al. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARalpha. Nat Commun. 2019;10:1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wieser M, Stadler G, Jennings P, et al. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am J Physiol Renal Physiol. 2008;295:F1365–1375. [DOI] [PubMed] [Google Scholar]
- 34.Chen CH, Dickman KG, Moriya M, et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc Natl Acad Sci U S A. 2012;109:8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ju W, Nair V, Smith S, et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med. 2015;7:316ra193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickman KG, Sweet DH, Bonala R, et al. Physiological and molecular characterization of aristolochic acid transport by the kidney. J Pharmacol Exp Ther. 2011;338:588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirita Y, Wu H, Uchimura K, et al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A. 2020;117:15874–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Kirita Y, Donnelly EL, et al. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J Am Soc Nephrol. 2019;30:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seidman JG, Seidman C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest. 2002;109:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–1024. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Kumar S, Dolzhenko E, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marable SS, Chung E, Adam M, et al. Hnf4a deletion in the mouse kidney phenocopies Fanconi renotubular syndrome. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matys V, Fricke E, Geffers R, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dhillon P, Park J, Hurtado Del Pozo C, et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. 2021;33:379–394 e378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chau BN, Xin C, Hartner J, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4:121ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du W, Zhang L, Brett-Morris A, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8:1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ke Q, Yuan Q, Qin N, et al. UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 2020;11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miguel V, Tituana J, Herrero JI, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo C, Zhou S, Zhou Z, et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J Am Soc Nephrol. 2018;29:1238–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinkkoetter PT, Bork T, Salou S, et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell reports. 2019;27:1551–1566 e1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Quantification of AAI-induced kidney injury parameters and KLF15 expression. (a) Quantification for number of AL-positive nuclei per high power field (HPF). (b-d) Quantification of area staining for LL (b), COL1α1 (c), and αSMA (d), expressed as fold change (fc) versus DMSO treated mice. (e) Quantification of Klf15 mRNA expression in kidney cortex expressed as fc versus DMSO treated mice. (f) Quantification of PT KLF15 expression, defined as KLF15-positive nuclei within LL-positive tubules, expressed as fc versus DMSO treated mice. *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA with Sidak’s correction. (g) Representative images of KLF15 immunostaining extracted from the merged images in Figure 1E. Scale bars = 100μm. (h) Transcription factors with ChIP-seq determined binding sites in the Klf15 promoter region. (i) Mapping of open chromatin from ATAC-seq in p2 mouse nephrons and transcription factor binding sites with respect to the Klf15 locus, with RNA-seq data from microdissected S2/S3 PT segments after 1 injection of DMSO or AAI. *p<0.05, **p<0.01.
Supplementary Figure 2: Validation of KLF15 knockdown in proximal tubules in Klf15PTKO mice. (a) Immunofluorescent staining for tdTomato expression (red), with counterstaining for Lotus lectin (green) and DAPI (blue). Scale bar = 100μm. (b) Immunofluorescent staining for KLF15 (red), with counterstaining for Lotus lectin (green) and DAPI (blue). Representative images provided from each group (n=6 per group). Scale bars = 100μm. (c) Quantification of PT KLF15 protein expression, defined as KLF15-positive nuclei within LL-positive tubules. *p<0.05; unpaired t test. (d) Quantification of Klf15 mRNA expression in kidney cortex. *p<0.05; unpaired t test.
Supplementary Figure 3: Inflammatory, fibrotic and PT markers in the remodeling phase of AAI-treated mice. (a-e) Quantification of immunostaining for: Gr1+ cells per high power field (HPF) (a), CD68+ cells per HPF (b), proliferating myofibroblasts (Ki67 and α-SMA positive cells) (c), α-SMA deposition (d), and COL1α1 deposition (e). (f) Quantification of Col1a1 mRNA expression in kidney cortex. *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA with Sidak’s correction.
Supplementary Figure 4: Loss of PT-specific Klf15 exacerbates IRI. Klf15PTKO and control mice were subjected to uninephrectomy (UNx) of the right kidney with simultaneous IRI of the left kidney. (a,b) Serum creatinine (a) and urea nitrogen (b) concentrations. $p=0.050, *p<0.05, **p<0.01, ***p<0.001; multiple t tests with Benjamini Hochberg correction for multiple testing. (c) Histological analysis using Hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) staining. Scale bars = 250μm/50μm for insets. (d) Examination of PT by cytokeratin-20 immunofluorescence with Lotus lectin staining. Scale bars = 500μm.
Supplementary Figure 5: Expression of PT transporters after DMSO or AAI (active phase). (a) S1 transporters. (b) S2/S3 transporters. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; one-way ANOVA with Sidak’s correction.
Supplementary Figure 6: Expression of PT markers and metabolic genes in kidney cortex and isolated PT from control and injured mice. (a) Expression of 9 genes encoding mouse PT markers (Slc5a2, Nox4, Slc34a1, Slc4a4, Cubn, Lrp2, Aqp1, Anpep, and Ggt1) in Klf15fl/fl and Klf15PTKO mice treated with DMSO and AAI. (b) Heatmap of glycolytic genes showing average expression for Klf15fl/fl and Klf15PTKO with DMSO (lanes 1, 2), Klf15fl/fl and Klf15PTKO with AAI active phase (lanes 3, 4), and Klf15fl/fl and Klf15PTKO with AAI remodeling phase (lanes 5, 6). Genes in bold have KLF15 binding sites in their promoters. (c) mRNA expression of Klf15, Ppara, Cpt1a, and Acaa2 in S2/S3 PT segments microdissected from mice after 1 injection of DMSO or AAI. *p<0.05, **p<0.01; t test with Benjamini Hochberg correction for multiple testing. (d) Kidney cortex expression of Klf15, Ppara, Cpt1a, and Acaa2 in control mice and models of human immunodeficiency virus (HIV)-associated nephropathy (Tg26 mice), and mice subject to unilateral ureteric obstruction. ***p<0.001; t test with Benjamini Hochberg correction for multiple testing.
Supplementary Figure 7: Histological and immunofluorescent analysis of human AAI-nephropathy patients. (a) PAS staining of kidney cortex samples from patients with (red labels) and without (black labels) evidence of exposure to AA, with images captured at 10x (top) and 40x (bottom) magnification. Scale bars = 250μm (top), 50μm (bottom). (b) Co-immunofluorescence for KLF15 and AQP1, with co-staining for Phaseolus vulgaris Erythroagglutinin (PHA-E) and DAPI in a control subject and CKD patients. The corresponding images for Patient #42 are shown in Figure 8a. Scale bars = 100μm.
Supplementary Figure 8: Expression of genes encoding human PT markers. Expression of genes encoding human PT markers in tubulointerstitial expression array data from patients with CKD.35
Supplementary Table 1. Primer sequences for qRT-PCR
Supplementary Table 2. Primer sequences for qPCR for ChIP
Supplementary Table 3. CKD patient data
Supplemental Data File. Differentially expressed genes
Data Availability Statement
The RNA-seq data supporting the findings of this study are openly available in the Gene Expression Omnibus database (GSE150656).