

Abstract
Promising progress has been made in adoptive transfer of allogeneic natural killer (NK) cells to treat relapsed or refractory acute myeloid leukemia (AML). In this regard, chimeric antigen receptor (CAR)-modification of NK cells is considered as a compelling approach to augment the specificity and cytotoxicity of NK cells against AML. Using a non-viral piggyBac transposon technology and human peripheral blood-derived primary NK cells, we generated CAR-NK cells to target NKG2D ligands and demonstrated their in vitro activity in lysing cancer cells expressing the ligands and in vivo efficacy in inhibiting tumor growth in a xenograft KG-1 AML model. We further generated CAR-NK cells co-expressing transgenes for the NKG2D CAR and interleukin-15 (IL-15). The ectopic expression of IL-15 improved the in vitro and in vivo persistence of NKG2D CAR-NK cells, leading to enhanced in vivo tumor control and significant prolongation of mouse survival in the KG-1 AML model. Collectively, our findings demonstrate the ectopic expression of IL-15 as an important means to improve the antileukemic activity of NKG2D CAR-NK cells. Our study further illustrates the feasibility of using the piggyBac non-viral platform as an efficient and cost-effective way for CAR-NK cell manufacturing.
Keywords: PiggyBac transposon, NKG2D CAR-NK cells, K562 APCs, IL-15, acute myeloid leukemia
Graphical Abstract
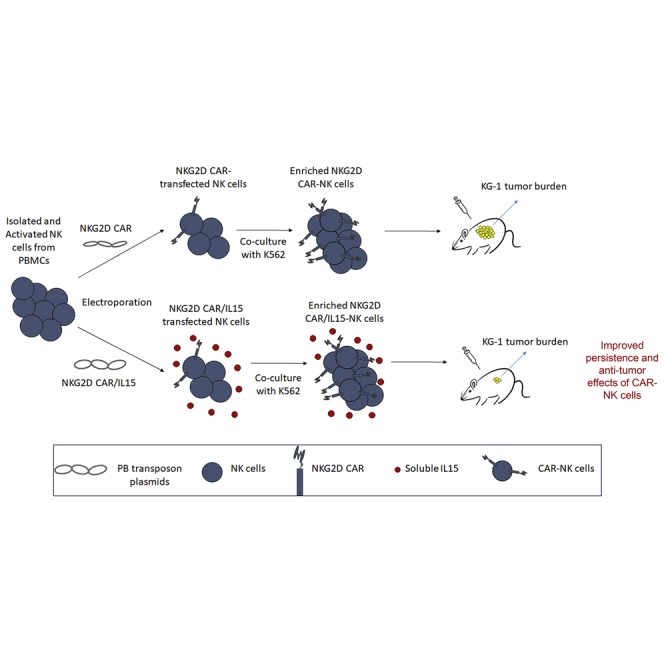
With a non-viral piggyBac transposon system, IL-15 armored NKG2D CAR-NK cells were generated from human PBMCs. These cells displayed superior persistence and anti-AML activity both in vitro and in vivo as compared with NKG2D CAR-NK cells without IL-15.
Introduction
As an aggressive hematopoietic malignancy, acute myeloid leukemia (AML) is characterized by an excess of myeloid blasts in the bone marrow and peripheral blood. AML carries a dismal prognosis, especially in the elderly (aged 65 and older). The only curable option for relapsed or refractory AML (r/r AML) so far is allogeneic hematopoietic cell transplantation (AHSCT).1,2 Since this option is unreachable to most AML patients, adoptive cell therapy of allogeneic peripheral blood natural killer (NK) cells offers an alternative therapeutic option and has shown promise in treating r/r AML.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14
Recently, chimeric antigen receptor (CAR) modification has emerged as an effective approach to augment the specificity and cytotoxicity of NK cells.15, 16, 17 CAR-NK cell therapy combines CAR advantages of targeting specificity to tumor cells and enhanced intracellular activity with the innate antitumor functions and alloreactive potential of NK cells. The cytotoxic function of CAR-NK cells is not CAR-restricted, given the ability of NK cells to lyse cancer cells directly without prior antigen priming. While high levels of interferon-γ and other cytokines can be produced by activated NK cells to shape both innate and adaptive antitumor immune responses, the spectrum of cytokines that NK cells secrete differs from those that can cause cytokine release syndrome, hence are less likely to mediate severe cytokine-related toxicities. With these favorable therapeutic and safety features, CAR-NK cells create a compelling platform to overcome many limitations of the current cell therapies. CD19-specific CAR-NK cells derived from cord blood have been successfully used to treat patients with B-cell acute lymphoblastic leukemia and lymphoma in a phase I/II clinical trial.18
As for AML treatment, identifying a specific target antigen for CAR therapy is challenging given the antigen overlap between AML leukemia stem cells and hematopoietic stem cells.19,20 A special group of CARs has been developed based on a key NK cell activating receptor, the NK group 2D (NKG2D) receptor, for tumor antigen recognition and immune cell activation.21,22 In humans, this receptor recognizes eight stress-induced ligands belonging to two families: two major histocompatibility complex (MHC) class I chain-related proteins MICA and MICB and six HCMV UL16-binding proteins ULBP1-6. The expression of the NKG2D ligands is tightly regulated such that they are not usually present on the cell surface of healthy tissues, but upregulated upon DNA damage, infection, and transformation. Thus, the tumor-associated overexpression feature makes the NKG2D ligands a favorable therapeutic target for anticancer strategies.23 CAR-T cells targeting NKG2D ligands have been tested in earlier stage clinical trials in AML patients.24,25 However, whether NKG2D CAR-NK cells can be used to treat AML, either in preclinical or clinical studies, remains to be an open question.
A key obstacle to CAR-NK cell therapy is genetic modification of primary NK cells to achieve stable CAR expression. For CAR-T cell manufacturing, transduction with a viral vector that integrates the CAR-expressing cassette randomly into the genome for persistent expression has been commonly used. However, the viral procedure is laborious for large-scale virus production, challenging in controlling lot-to-lot variability, and sophisticated in biosafety characterization of clinical-grade viral vectors in compliance with good manufacturing practice, thus being highly expensive. Furthermore, NK cells are difficult to be transduced by approaches commonly used for CAR-T cell transfer, such as lentiviral or retroviral transduction, and low efficiencies of NK cell transduction are often observed.26,27 This is possibly due to the intrinsic antiviral defense mechanisms possessed by NK cells.27,28 Non-viral transposon systems, such as Sleeping Beauty and PiggyBac (PB), are effective for genetic integration and have been used to overcome the limitations associated with the viral manufacturing process for CAR-T cells.29, 30, 31 When looking at the applicability of non-viral transposon in genetic modification of NK cells, we notice that there is only one bioRxiv preprint reporting Tc Buster transposon-mediated CAR modification of peripheral blood NK cells32 and another paper reporting PB transposon-mediated CAR modification of the NK-92 cell line.33
Another hurdle to the success of CAR-NK cell therapy is the short lifespan of NK cells.12,34 Without cytokine support, intravenously injected human NK cells quickly disappeared in the peripheral blood and became barely detectable 1 week later.35 Since interleukin (IL)-15 is the main homeostatic cytokine required for NK cell differentiation, survival, proliferation, activation, and functionality,36 it has been explored clinically to enhance the in vivo persistence and antitumor activity of donor NK cell products.6,37 Concerns have been raised that the prolonged in vivo exposure to injected exogenous IL-15 may stimulate the expansion of CD8-positive T cells to reject donor NK cells or suppress NK cell function.37,38 A strategy that may address the concerns has been developed to include IL-15 in an anti-CD19 CAR construct to ectopically produce IL-15 for autocrine stimulation of CAR-NK cells.18,38,39
Here we present a non-viral PB approach for the stable genetic modification of human peripheral blood-derived NK cells to co-express NKG2D CAR and IL-15. We examined whether these CAR-NK cells could control tumor progression in a mouse AML model.
Results
Generation of NKG2D CAR-NK cells through electroporation of PB transposition plasmids
We have established a PB transposition platform and used it successfully to generate several types of NKG2D CAR-T cells with different activation domains, all of which showed potent ability to eradicate established solid tumors in animal tumor models.40,41 One of the NKG2D CAR transposon plasmids was constructed with the NKG2D ectodomain expressed in tandem with a streptavidin tag (STII tag) for specific detection of NKG2D CAR, 4-1BB as the costimulatory domain, and the CD3 zeta cytoplasmic domain as the activation domain (Figure 1A). The EF1alpha promoter is used to drive the transgene expression in the vector. This transposon plasmid and a plasmid containing the piggyBac transposase gene under the control of the CMV promoter (Figure S1) were used in the current study to generate NKG2D CAR-NK cells.40,41
Figure 1.

Generation of NKG2D CAR-NK cells with a PB transposon system, antigen-specific expansion stimulated by K562 aAPCs, and phenotypic characterization of the generated CAR-NK cells
(A) The NKG2D CAR construct used in this study and the workflow scheme for CAR-NK cell generation. (B) A representative panel of flow cytometry plots from a single donor to show the antigen-specific enrichment of NKG2D CAR-NK cells stimulated by K562 aAPCs. The CAR-NK cells are gated as CD56+ and Streptavidin II tag (STII)-positive cells. (C) The antigen-specific expansion of NKG2D CAR-NK cells from day 7 to day 28 with K562 aAPCs. Each line represents the data from one donor, n = 5. (D) The expansion of NKG2D CAR-NK cells from day 7 to day 28. Each line represents the data from one donor, n = 5. (E) The populations of CD3-CD56 + NK cells across the expansion procedure. The data shown are mean ± SD from five different donors. (F) The comparison of NK cell surface receptor expression between NKG2D CAR-NK cells and mock-electroporated NK cells. The data shown are mean ± SD from three different donors.
In preparation for future clinical applications of the CAR-NK cell product, we outlined a manufacturing protocol in Figure 1A. We started with the isolation of NK cells from human PBMCs and the NK cell purity following magnetic bead isolation was verified by flow cytometric analysis (Figure S2). The isolated NK cells were activated and expanded for 7 days with gamma-irradiated feeder cells from a K562 cell line engineered to express mbIL-15, mbIL-21 and 4-1BBL established in the lab previously.42 This step increased the amount of NK cells by approximately 5- to 10-fold. After the 7-day expansion, NK cells were electroporated with a Lonza (Basel, Switzerland) 4D-Nucleofector system using a selected program (EN-138, Figure S3) at a transposase:transposon ratio of 1:2 (5 μg piggyBac transposase plasmid:10 μg NKG2D CAR plasmid per 5 × 106 NK cells). Taking the advantage that the gamma-irradiated K562 cells express two of the eight NKG2D ligands (Figure S4) and can be used as artificial antigen-presenting cells (aAPCs), the electroporated NK cells were stimulated with the K562 cells for the enrichment and expansion of NKG2D CAR-NK cells. The K562 cells were replenished every 7 days and eventually eliminated by NK cells. The median CAR-NK transfer efficiency at day 7 post-electroporation was 14% (range 6%–29%; n = 5; Figures 1B and 1C). CAR-positive NK cells were enriched upon the K562 cell stimulation and, after four rounds of weekly stimulation, the percentages of NKG2D CAR-positive cells reached 63% (range 44% to 68%; n = 5) by day 28 post-electroporation (Figures 1B and 1C). CAR-NK cells could be further enriched to approximately 90% if we extended the stimulation further to seven rounds (Figure S5), indicating a stable functionality of the CAR over time. qPCR analysis of isolated genomic DNA (Figure S6) further confirmed CAR transgene integration into genome, with the median vector copy number per diploid genome being two in the CAR-NK cells collected after four rounds of enrichment and expansion (n = 5). The number of NK cells also exponentially increased along the way and the corresponding median expansion was 8,030-fold (range 5,096–10,500, n = 5) by day 28 (Figure 1D), which was consistent with previously reported NK cell expansion range obtained with K562 aAPC methods.12,17 The expansion was NK cell-specific, by day 28 post-electroporation, 95% of cells were CD3-CD56 + cells (Figure 1E) and in particular the expanded NKG2D CAR-NK cell populations were free of CD3-positive cells (Figure S7). The typical NK cell phenotype was not affected by the genetic modification as demonstrated by comparison to that on mock NK cells (Figure 1F) and neither was the expression of checkpoint molecules including TIGIT, PD-1, TIM-3, and LAG-3 (Figure S8). Furthermore, the consistency of TIGIT expression on NK cells from day −7 (immediately after NK cell isolation and before coculture with K562 aAPCs), day 0 (right before and right after electroporation) to day 28 post-electroporation was observed (Figure S8), indicating that our protocol used for CAR-NK cell enrichment and expansion did not induce NK cell exhaustion.43
The PB-generated NKG2D CAR-NK cells exhibit potent in vitro and in vivo antileukemic ability
To validate the antigen-dependent tumor cell lytic activity of NKG2D CAR-NK cells, we used KG-1 and MOLM-14 human AML cell lines, HCT-116 human colon colorectal cell line, SKOV3 human ovarian cancer cell line that expresses NKG2D ligands, and A549 lung cancer cell line that does not express NKG2D ligand as target cells (Figure S4).42 Using the Europium-release method, we performed short-term (3 h) in vitro cytotoxicity assays with effector to target (E:T) cell ratios from 2.5:1 to 20:1. The killing efficiencies of NKG2D CAR-NK cells against all four tested NKG2DL-expressing cancer cell lines significantly increased over those offered by mock NK cells (Figure 2A). In contrast, no significant difference in killing was observed when the NKG2DL-negative A549 cancer cells were included as the target cells (Figure S9). We next investigated the degranulation of NKG2D CAR-modified NK cells upon stimulation by HCT-116 cells. In a typical example, cell surface mobilization of CD107a, a marker of degranulation, increased from 0.2% in the non-stimulated mock NK cells to 7.1% after cancer cell stimulation, whereas on the CAR-NK cells an increase from 0.4% to 16.1% was observed (Figure 2B).
Figure 2.

Assessment of anticancer functionality of NKG2D CAR-NK cells
(A) Cytolytic activity at E:T ratios from 2.5:1 to 20:1. DELFIA EuTDA cytotoxicity assay (3 h EuTDA culturing) was used. Data are presented as mean percent specific killing of target cells ±SD with experiments done in triplicate. The differences between mock NK and NKG2D CAR-NK cells were statistically significant, p < 0.001. (B) Degranulation assay. Mock NK or NKG2D CAR-NK cells were co-cultured with or without target HCT116 cancer cells at 1:1 ratio for 5 h before analysis. Left: Representative samples. CD56 + gated cells were used for CD107a expression analysis. Right: Bar graphs to show the mean percentages of NK cells expressing CD107a. The results shown represent mean ± SD of three independent experiments with three different donors. ∗p < 0.05.
We further validated the in vivo anticancer functionality of NKG2D CAR-NK cells to eradicate established human AML xenografts. For that, luciferase-expressing KG-1 cells (5 × 106 cells per mouse) were intravenously (i.v.) injected into NSG mice through the tail vein 2 weeks before treatment. Between day 14 and day 30 post-tumor inoculation, mice were treated with multiple i.v. injections of NKG2D CAR-NK cells (1 × 107 cells per injection per mouse) for a total of five times, followed by intraperitoneal (i.p.) injections of IL-2 every 3 or 4 days (Figure 3A). Mice treated with the i.v. injections of PBS or mock NK cells plus i.p. injections of IL-2 were used as controls. AML progression was monitored with weekly non-invasive whole-body bioluminescent imaging (BLI) of KG1-Luc cells. The BLI intensities (Figure 3B), indicative of leukemic burden and distribution, demonstrated that KG1 AML progressed aggressively in the control groups treated with PBS and all mice in the group either died or were euthanized due to being moribund by day 42. The injections of mock NK cells slowed down the progression and prolonged mouse survival for 1 more week. The markedly suppressed tumor progression was observed when mice were treated with NKG2D CAR-NK cells. Quantitative analysis of KG1-Luc cell bioluminescence signals demonstrated statistically significant differences (p < 0.0001) in flux values among the three groups at days 28 and 35, with the values in the NKG2D CAR-NK group being significantly lower than those in the two control groups (Figure 3C). By day 42, all mice in the NKG2D CAR-NK group were still alive and their leukemic burdens were even lower than those in the PBS group at day 28, indicating that the treatment delayed the AML progression by about 2 weeks. However, in this well-established human AML xenograft model, in which cancer treatment was delayed till day 14 post-tumor inoculation, the NKG2D CAR-NK cell injections were unable to induce tumor regression and all mice died by day 56. Kaplan-Meier analysis of survival data is shown in Figure 3D. While the median survival time of the PBS group was 41 days and the treatment with mock NK cells increased median survival to 45 days, the treatment with NKG2D CAR-NK cells resulted in median survival of 52 days, prolonging median survival time by 27% as compared with that in the PBS group (P < 0.01). The above results demonstrated that the expression of NKG2D CAR could significantly augment the in vivo antileukemic ability of NK cells.
Figure 3.

NKG2D CAR-NK cells display antitumor effects in vivo
(A) Schematic diagram of a mouse experiment assessing the antitumor effects of NKG2D CAR-modified NK cells in immune-deficient NSG mice. PBS, mock NK cells, or CAR-NK cells were i.v. injected, followed by i.p. injection of IL-2, n = 5 per group. (B) Bioluminescent images prior to treatment (day 10) and following the treatments on day 21 to day 49. (C) Tumor burden over time by BLI after the treatment. Each mouse is represented by one line. ∗∗∗∗p < 0.0001. (D) Kaplan-Meier analysis of survival. Statistical analysis of survival between groups was performed using the log rank test. MS, median survival time. ∗∗p < 0.01.
The ectopic expression of IL-15 improves the anticancer functionality of NKG2D CAR-NK cells
The ectopic expression of IL-15 has been used to improve the in vivo proliferation, persistence, and antitumor activity of CD19-specific CAR-NK cells.18,39 To enhance the in vivo antileukemic activity of the PB-generated NKG2D CAR-NK cells, we moved on to employ the established PB transposition platform to generate CAR-NK cells co-expressing NKG2D CAR and IL-15. The bicistronic vector used for this purpose is shown in Figure 4A. The manufacturing protocol was slightly modified by reducing the initial expansion step from 7 days to 2 days in order to shorten the manufacturing process and improve the transfection efficiency (Figures S10A and S10B). Using this modified protocol, the percentages of CAR-positive cells generated after four rounds of weekly K562 stimulation remained close to 60% (Figure S10C) and the median expansion was 5,758-fold (range 2,536–14,405, n = 5) by day 28 (Figure S10D). It appears that the ectopic expression of IL-15 did not influence the NKG2D CAR-NK cell generation significantly, which is consistent with the finding of the first report on the ectopic expression of IL-15 from CAR-NK cells,39 possibly because of the use of cytokine-expressing K562 feeder cells to promote CAR-NK cell enrichment and expansion. Flow cytometric analysis confirmed that the ectopic expression of IL-15 did not affect the overall phenotypes of CAR-NK cells and NKG2D CAR/IL15-NK cells exhibited a phenotype similar to that of NKG2D CAR-NK cells (Figure S11A) and neither the expression of checkpoint molecules TIGIT, PD-1, TIM-3, and LAG-3 (Figure S11B).
Figure 4.

Effects of ectopically expressed IL-15 on the anticancer functionality of NKG2D CAR-NK cells
(A) The NKG2D CAR/IL15 construct used in this study. (B) ELISA-based detection of IL-15. IL-15 production was examined after CAR-NK cells cultured with or without the HCT-116 target cancer cells for 24 h. The data shown are mean ± SD from three different donors. ∗p < 0.05. (C) IL-15 production after CAR-NK cells cultured with the HCT-116 target cancer cells for 3 h. The data shown are mean ± SD from three different donors. (D) Cytolytic activity at E:T ratios from 0.5:1 to 4:1. DELFIA EuTDA cytotoxicity assay (3 h EuTDA culturing) was used. Data are presented as mean percent specific killing of target cells ±SD with experiments done in triplicate. The differences between NKG2D CAR-NK and NKG2D CAR/IL15-NK cells were statistically significant, p < 0.001. (E) Degranulation assay. NKG2D CAR-NK or NKG2D CAR/IL15-NK cells were co-cultured with or without target HCT116 cancer cells at 1:1 ratio for 5 h before analysis. Left: Representative samples. CD56 + gated cells were used for CD107a expression analysis. Right: Bar graphs to show the mean percentages of NK cells expressing CD107a. The results shown represent mean ± SD of two independent experiments with two different donors. ∗∗p < 0.01.
To measure IL-15 production, NKG2D CAR-NK cells and NKG2D CAR/IL15-NK cells were cultured with or without target cancer cells for 24 h and IL-15 release in the culture supernatant was measured by ELISA. As expected, IL-15 was undetectable in the supernatants collected from the NKG2D CAR-NK cell cultures, either with or without the cancer target cells (Figure 4B). While small amounts of IL-15 were detected in the supernatants collected from the NKG2D CAR/IL15-NK cell culturing alone without the target cells, the IL-15 concentration increased significantly when the NKG2D CAR/IL15-NK cells were co-cultured with HCT-116 target cells, increasing from 7.5 pg/mL in the cultures without HCT-116 to 52.5 pg/mL in the cultures with HCT-116. We further investigated whether a short exposure to target cells would be enough to trigger IL-15 production and indeed observed a low level but consistent production of IL-15 after co-culturing NKG2D CAR/IL15-NK cells with HCT-116 target cells for 3 h (Figure 4C). These results indicate that the IL-15 production by NKG2D CAR/IL15-NK cells could be quick and significantly upregulated upon the interaction with target cancer cells, possibly due to the enhanced proliferation and enrichment of the IL-15 producing CAR-NK cells.
Since short-term IL-15 priming could significantly increase the killing capacity of mouse NK cells and human NK cells, expressing membrane-bound IL-15 displayed a significantly higher cytolytic activity against a set of cancer cell lines in a short-term cytotoxicity assay,44,45 we mixed CAR-NK cells with target cancer cells at low effector to target (E:T) cell ratios from 0.5:1 to 4:1 and performed 3-h in vitro cytotoxicity assays. The killing efficiencies of NKG2D CAR/IL15-NK cells against three tested cancer lines significantly increased over those offered by NKG2D CAR-NK cells (Figure 4D). The degranulation of CAR-NK cells upon stimulation by HCT-116 cells was also investigated, showing that the cell surface mobilization of CD107a increased from 17% on NKG2D CAR-NK cells to 39% on NKG2D CAR/IL15-NK cells (Figure 4E). Taken together, the results in this section illustrate that IL-15 expression by NKG2D CAR/IL15-NK cells or triggered by the interaction of these CAR-NK cells with target cells could quickly increase the cytolytic activity of NK cells.
The ectopic expression of IL-15 supports the persistence of NKG2D CAR-NK cells
To investigate whether the low level of IL-15 released from NKG2D CAR/IL15-NK cells without the interaction with target cells could support the in vitro persistence of the CAR-NK cells, NKG2D CAR-NK cells and NKG2D CAR/IL15-NK cells were cultured with or without a low concentration of IL-2 (10 IU/mL) for 7 days. The left CAR-NK cells were enumerated and cell recovery was calculated as the ratio (%) of the number of cells left to the number of cells initially seeded. We observed significantly higher cell recovery rates in the NKG2D CAR/IL15-NK cell cultures as compared with the NKG2D CAR-NK cell cultures, either with or without IL-2 (Figure 5A). For example, when culturing for 7 days without IL-2, there was only 17% (±8.7%, mean ± SD, n = 4) of the initially seeded NKG2D CAR-NK cells left over, but the leftover NKG2D CAR/IL15-NK cells were 46% (±13.7%, mean ± SD, n = 4) (Figure 5A). We noticed that most of the leftover NKG2D CAR/IL15-NK cells under this condition were NKG2D CAR-positive NK cells, the CAR-positive NK cells increasing from the initial 49% (±5.8%, mean ± SD, n = 4) to 72% (±7.3%, mean ± SD, n = 4) 7 days later (Figure 5B). This result indicates that the ectopic expression of IL-15 preferentially supported the survival or proliferation of genetically modified NK cells.
Figure 5.

Effects of ectopically expressed IL-15 on CAR-NK cell persistence
(A) CAR-NK cell recovery after 7-day culture with or without a low concentration IL-2 (10 IU/mL). Cell recovery was calculated as the ratio (%) of the number of cells left to the number of cells initially seeded. The data shown are mean ± SD from four different donors. ∗p < 0.05. (B) Change in the percentage of CAR-NK cells over the 7-day culture without IL-2 (mean ± SD, n = 4). Left: Representative flow charts from one donor. Right: Bar graphs to show the mean percentages of NK cells expressing NKG2D CAR. The data shown are mean ± SD from four different donors. ∗∗p < 0.01. (C) Persistence of human NK cells in mouse peripheral blood. NKG2D CAR-NK cells or NKG2D CAR/IL15-NK cells were i.v. injected into NSG mice (n = 5 per group) through the tail vein and mouse blood samples were collected 3 and 7 days later for flow cytometric analysis. Left: Representative flow charts from one donor. Right: Dot plot with mean point and error bars. The same results are shown in two different ways: (A) CD45 + CD56 + events within 60-s acquisition; and (B) % Human CD45 + CD56 + cells. ∗p < 0.05. The difference between the two types of CAR-NK cells were statistically significant at day 7 (p < 0.001). (D) Mouse spleens were also collected after euthanization of all mice on day 7 for analysis. Left: Representative flow charts from one donor. Right: Dot plot with mean point and error bars. The same results are shown in two different ways. The difference between the two types of CAR-NK cells was statistically significant at day 7 (p < 0.001).
We further examined the difference in in vivo persistence between NKG2D CAR-NK cells and NKG2D CAR/IL15-NK cells. These cells were i.v. injected into normal NSG mice through the tail vein, and mouse blood samples were collected 3 and 7 days later for flow cytometric analysis of human NK cells. On day 3, human NK cells were detectable in both groups of mice but the number of human CD45+CD56+ cells was significantly higher in mice injected with NKG2D CAR/IL15-NK cells than those injected with NKG2D CAR-NK cells (Figure 5C). By day 7, human NK cells could not be detected in the blood samples collected from mice injected with NKG2D CAR-NK cells, but still remained at the same level in the group of mice injected with NKG2D CAR/IL15-NK cells. After euthanization of all mice on day 7, mouse spleens were collected to examine the tissue distribution of human NK cells. Again, human CD45+CD56+ cells were detectable in the mice injected with NKG2D CAR/IL15-NK cells but not in the group of mice injected with NKG2D CAR-NK cells (Figure 5D). The absence of human NK cells in the blood and spleen of immune-deficient mice at day 7 after adoptive transfer without cytokine support was consistent with a previous report by Miller et al. (2014).35 These results validated the applicability of using ectopically expressed IL-15 to support the persistence of NKG2D CAR-NK cells.
The ectopic expression of IL-15 enhances the in vivo antileukemic activity of NKG2D CAR-NK cells
Given the encouraging persistence data above, we moved on to evaluate the in vivo effects of CAR-NK cells co-expressing NKG2D CAR and IL-15 in an AML model similar to that shown in Figure 3. To assess whether the ectopic expression of IL-15 could replace the external support of IL-2, we did not include the IL-2 intraperitoneal injection in this animal experiment. From our previous experience in animal experiments with NK cell therapy (unpublished observations), we anticipated that the lack of the support of exogenous IL-2 would make it much more challenging to observe therapeutic effects of adoptively transferred NK cells. We therefore started CAR-NK cell treatments earlier, on day 3 after i.v. injection of KG1-Luc cells when leukemia engraftment was not clearly manifested. Five groups of mice were tested: one i.v. injection of PBS on day 3, one i.v. injection of NKG2D CAR-NK cells (without IL-15) on day 3, one i.v. injection of NKG2D CAR/IL15-NK cells on day 3, two i.v. injections of NKG2D CAR/IL15-NK cells on days 3 and 10, and three i.v. injections of NKG2D CAR/IL15-NK cells on days 3, 10, and 17 (Figure 6A).
Figure 6.

Enhanced in vivo anti-AML activity of NKG2D CAR/IL15-modified NK cells
(A) Schematic diagram of a mouse experiment assessing the antitumor effects of different CAR-NK cell treatments in immune-deficient NSG mice, n = 4 to 6 per group. (B) Bioluminescent images prior to treatment (day 3) and following the treatments from day 10 to day 45. (C) Total flux values of tumor burden on day 17 were plotted in the bar graph. ∗p < 0.05; ∗∗∗∗p < 0.0001. (D) Time course of BLI signal change from day 3 to day 45. (E) Kaplan-Meier analysis of survival. Statistical analysis of survival between groups was performed using the log rank test. MS, medium survival time.
We observed a trend of KG1 AML progression in the PBS control group similar to what was observed in Figure 3: three out of five mice started to show a moribund condition on day 41 and the rest were euthanized on day 43 (Figure 6B), with a median survival of 41 days, same as the first animal experiment. The treatment with NKG2D CAR-NK cells (without IL-15) delayed the AML progression to some extent as compared with the PBS injection, but still all mice were euthanized by day 45 (Figure 6B). The ectopic expression of IL-15 significantly enhanced the in vivo antileukemic activity of NKG2D CAR-NK cells. A quantitative analysis of tumor burdens imaged on day 17 demonstrated that KG1-Luc cell signals were very strong in the PBS group, relatively lower in the NKG2D CAR-NK cell group, and undebatable in all mice treated with NKG2D CAR/IL15-NK cells, with the statistical significances of the differences between the PBS group versus the NKG2D CAR/IL15-NK cell groups and between the NKG2D CAR-NK group versus the NKG2D CAR/IL15-NK cell groups being <0.0001 and <0.05, respectively (Figure 6C). Figure 6D shows the time course of BLI signal change from day 3 to day 45 for all mice in five groups. While the BLI signals in the PBS group continued increase until animal death/euthanization, the signals in the NKG2D CAR-NK treated mice, after initial increase in the first week, significantly decreased upon treatment. Comparing the effects of one injection of NKG2D CAR-NK cells with and without IL-15 on day 3, we noticed a faster reduction of BLI signals in the NKG2D CAR/IL15 group, possibly because of the IL-15 supported NK cell proliferation. However, tumor regrowth was observed 4 weeks later in all mice in the two groups, indicating that one injection of NKG2D CAR-NK cells was not enough to induce tumor regression. In the group treated with two injections of NKG2D CAR/IL15 cells, two out of four mice showed tumor regression without tumor recurrence over the observation period, whereas all mice in the group treated with three injections of NKG2D CAR/IL15 cells showed substantial tumor regression, as manifested by the finding that only baseline BLI signals were detectable after the treatment.
Both the NKG2D CAR-NK and NKG2D CAR/IL15-NK treatments significantly prolonged the survival of AML-bearing mice, with the increase of median survival from 41 days in the PBS group to 45 days in the NKG2D CAR-NK group (p < 0.01) and 52 days in the NKG2D CAR/IL15-NK cell group with one injection (p < 0.01) (Figure 6E). The difference in median survival time between the NKG2D CAR-NK group and the NKG2D CAR/IL15-NK cell group with one treatment was statistically significant (p < 0.05). When the injection of NKG2D CAR/IL15-NK cells increased to two and three times, no animal death was observed within 60 days. The longest observation period was 100 days, by then all animals treated with three injections of NKG2D CAR/IL15 cells still survived well. Based on these results, we conclude that the ectopic expression of IL-15 could significantly enhance the in vivo antileukemic activity of NKG2D CAR-NK cells and induce tumor regression if given multiple injections.
Discussion
To address the challenge of genetic modification of peripheral blood NK cells for stable expression of a CAR gene, we tested electroporation of a PB transposon system for gene transfer into NK cells in this study. Electroporation of CAR-encoding mRNA has been employed for gene transfer into peripheral blood NK cells and offers high transfection efficiencies, yet causing minimal deleterious effects toward cell viability.27,42,46,47 However, short-term CAR expression could be a limitation of this non-integrating platform to many applications that require longer-term effects of CAR-modified cells. Electroporation with non-viral plasmid vectors has been attempted recently for stable engineering of peripheral blood NK cells and a transfection efficiency of close to 50% was achieved at 5 days after electroporation, representing a major improvement in NK cell transfection.48 The stable transgene integration was not examined in the study, but by day 15 the percentage of the modified NK cells dropped to less than 20%, indicating that the durable persistence of transfected genes is constrained by transfection efficiency.48
The PB transposase can mediate exchange of genetic material between the transposon vector and host genome through recognition of inverted terminal repeat (ITR) sequences present on the vector and the corresponding TTAA sequences present in the genome. Transgenes flanked by the ITRs can hence be integrated stably into TTAA integration sites. PB systems, well characterized as a footprint-free transposon, have been used in a wide range of genetic engineering applications. PB systems offer several advantages, including high cargo capacity of up to 100 Kb DNA fragment for the delivery of multiple transgenes, significantly lower manufacturing costs, and potentially fewer regulatory restrictions in clinical translations.49,50 Considering these appealing features, PB transposons have been recommended as an alternative gene transfer system to viral vectors for CAR-T cell production.31,51,52 Compared with PB-mediated CAR gene transfer in T cells, we noticed a low transfection efficiency for CAR gene transfer into NK cells. With the same PB transposase and NKG2D CAR constructs, we could obtain a CAR transfer efficiency close to 40% in T cells but only 14% of median transfer efficiency in NK cells at day 7 post-electroporation (Figure 1C).40,41 Nevertheless, the observation that the CAR-NK cells were enriched to approximately 60% after four rounds of weekly stimulation with K562 aAPCs indicates that the electroporation of our PB transposon system indeed provides the durable persistence of the transfected CAR gene. Unlike T cells, NK cells cannot be stimulated for expansion by endogenous activating receptor signals alone.53 The enrichment of NKG2D CAR-NK cells by K562 cells observed in this study demonstrated that the activation of a CAR construct could, on the other hand, stimulate the proliferation of the CAR-modified NK cells. Such enrichment was not reported in a previous study in which anti-CD19 CAR-NK cells were stimulated by K562 aAPCs for expansion but the percentage of anti-CD19 CAR-positive NK cells was not changed during the expansion,39 most likely due to no CD19 antigen expression on the K562 aAPCs. The discrepancy in CAR-NK cell enrichment between the two studies reflects the importance to include a target antigen into K562 aAPCs to achieve antigen-stimulated CAR-NK cell expansion.
Leukemia cells of most AML patients express at least one NKG2D ligand at the surface, albeit the expression levels could be low.54, 55, 56, 57, 58, 59 NKG2D CAR-T cells have been evaluated in seven patients with AML and five patients with multiple myeloma in a first-in-human, phase I dose-escalation study and showed good safety without identifying a maximum tolerated dose at the tested doses ranging from 1 × 106 to 3 × 107 total CAR-T cells, although a robust efficacy signal was not observed.24 Described in a case report by Sallman et al.,25 NKG2D CAR-T cells, given at the dose level of 3 × 108 cells (flat dose) per injection every 2 weeks for three administrations, induced remission in a patient with r/r AML who had resolution of symptoms with improved hematopoiesis that continued to improve until the patient was treated with an AHSCT. A recent paper shows that even low-level expression of NKG2D ligands in primary AML samples results in robust NKG2D-CAR activity, demonstrating that these ligands are viable targets for CAR-T cell therapy in AML.54 Although it has not been tested in AML patients, NKG2D CAR-NK cells produced by mRNA electroporation have been evaluated in a single-center pilot study in patients with late-stage, metastatic colorectal cancer and, especially, the intratumoral injection of the RNA CAR-NK cells in one of the patients led to a complete metabolic response in the injected tumor lesion.47 While local injection of CAR-NK cells is not feasible in AML therapy, this case showed a promising potential for CAR-NK cell therapy targeting NKG2D ligands in cancer treatment.
Even though NKG2D CAR-modified immune cells have been tested in patients, their antitumoral effects in AML models are not reported as per our knowledge. We established a robust AML xenograft model by tail vein injection of a well-established AML cell line KG-1 for the evaluation of therapeutic effects of NKG2D CAR-NK cells. Phosphotyrosine profiling has identified the cell line as a model for the study of fibroblast growth factor receptor-1 (FGFR1) fusions in AML.60 As compared with the use of primary patient samples, the advantages of using a well-established AML cell line include the availability of a large amount of human AML cells to generate a reproducible animal model and a faster engraftment in immunodeficient mice.61 The therapeutic effects of NKG2D CAR-NK cells in controlling in vivo progression of KG-1 tumors were confirmed in two mouse experiments using CAR-NK cells expressing NKG2D CAR, and CAR-NK cells expressing both NKG2D CAR and IL-15, respectively. The first mouse experiment was performed with IL-2 supplementation in order to sustain in vivo survival and expansion of infused human NK cells, especially to activate NK cells in the immunosuppressive tumor microenvironment.62 The main purpose of the second animal experiment was to investigate whether the ectopic expression of IL-15 could improve the in vivo antileukemic activity of NKG2D CAR-NK cells and could be used to replace the systemic injection of exogenous IL-2, which might stimulate the generation and homeostasis of immunosuppressive Tregs that can antagonize NK cells by reducing their expansion capacity and effector functions.63,64 In contrast to IL-2, IL-15 lacks the capability to stimulate Treg cells. Of note, the expression of IL-15 observed in this study was predominantly driven by the interaction between the NKG2D CAR-NK cells and target cells (Figure 5A). The IL-15 produced in this way may provide an autocrine stimulation without significantly increasing circulating IL-15 concentrations and, when used for AML therapy, might favorably support the local expansion of CAR-NK cells in the bone marrow where AML blasts are produced. The targeted delivery of IL-15 to NK cells may also be important clinically for an improved safety profile.37
The current study was inspired by the use of a viral construct co-expressing anti-CD19 CAR and IL-15 to generate CAR-NK cells reported by Liu et al.,18,39 in which the secreted form of IL-15 was expressed ectopically from the same CAR-modified NK cells. The infusion of the CAR-NK cells with the ectopically expressed IL-15 has been successfully used as a single agent-based therapy to treat CD19-positive lymphoid tumors.18 Heczey et al. recently reported the use of IL-15 armored, anti-GD2 CAR-NKT cells to treat three patients with relapsed or refractory neuroblastoma and observed stable disease in two patients and partial response in one patient who showed an objective response with regression of bone metastatic lesions.65 Several means other than the ectopic expression of the secreted form of IL-15 have been developed to support the in vivo expansion and persistence of NK cells without the use of exogenous cytokines.12 Imamura et al.44 reported the expression of tethered IL-15 in a membrane-bound form to sustain NK cell survival and expansion in vitro and in vivo, although no CAR-NK cells were tested. They demonstrated that IL-15 secretion was negligible and the membrane-bound IL-15 expressed on NK cells preferentially stimulates in cis rather than in the trans mode, that is, preferentially engaging IL-15 receptors on the same cells and resulting in autocrine stimulation. They further demonstrated that this on-board IL-15 approach conferred a substantial survival and growth advantage over the expression of non-membrane-bound IL-15 from NK cells. In a recently reported clinical trial using iPSC-derived CD19 CAR-NK cells, an IL-15/IL-15 receptor fusion protein was constitutively expressed on the CAR-NK cells to promote cytokine-autonomous persistence.66 Furthermore, a recent paper has demonstrated that the activation of inducible MyD88/CD40 could be coupled with ectopic IL-15 to enhance antitumor efficacy of CAR-NK cells.67 These IL-15-based approaches could be considered to further improve the performance and safety profile of the NKG2D CAR-NK cells developed in this study.
Transposon-engineered CAR-T cells have been tested in clinical trials and shown promising antitumor results.29 However, similar to integrating viral transduction, non-viral transposon technology faces safety issues, such as potential insertional mutagenesis and oncogenesis. Recently, in a first-in-human clinical trial (registered at www.anzctr.org.au as ACTRN12617001579381), an unforeseen development of lymphoma derived from T cells modified with a PB CAR against CD19 was reported in two of the nine patients who experienced complete remissions.68 The detailed analysis of integration sites of the PB CAR revealed no insertion into typical oncogenes and the overall integration pattern was not different from those previously reported for piggyBac or viral vectors, suggesting that the lymphoma development was not related to insertional mutagenesis/dysregulation.69 While the mechanism underlying the lymphoma development remains under investigation, in one of the two patients who developed lymphoma and was investigated for the number of vector copy number (VCN) integrants, the authors observed a VCN of 24 in malignant cells, which, as the authors suggested, could be related to their production methodology that included a single high-voltage pulse, high concentrations of transposon materials used, and the subsequent expansion of the CAR-T cells on feeder cells.69 The authors further confirmed that marked global changes in transcription mainly correlated with a high transgene copy number per cell rather than insertion sites.69 A significantly higher VCN suggests that CAR overexpression could have contributed to a survival advantage of individual PB CAR-T cell clones, especially given the extensive presence of the CD19 antigen in vivo.70 Overall, the unforeseen development of lymphoma alerts us that future clinical studies using CAR-modified immune cells should aim for low transgene copy numbers (probably <5) per transduced cell to reduce the potential risk for cell transformation.69,70
NK cells act in a non-MHC restricted manner and do not cause graft-versus-host disease when used in an allogeneic setting, enabling them as attractive candidates to generate “off-the-shelf” products from healthy donors to ensure timely availability of cell therapeutics and improve their clinical applicability.15 Off-the-shelf NK cell therapeutics can be generated from multiple cell sources ranging from peripheral blood cells, umbilical cord blood cells, bone marrow cells, and NK cell lines to embryonic and induced pluripotent stem cells. However, similar to other allogeneic donor cells such as off-the-shelf CAR-T cell products,71 allogeneic CAR-NK cells are expected to elicit host-derived immune responses that result in immune-mediated elimination of the transplanted cells. On the other hand, NK cells can function as natural regulators to modulate T cell immunity.72, 73, 74, 75 Under the transplantation condition, alloreactive NK cells in donor transplants can deplete host dendritic cells, therefore modulating their ability to prime T cell responses.72 In addition to the indirect dendritic cell-killing mechanism, NK cells can directly lyse activated T cells, which upregulate NKG2D ligands during activation, thus becoming targets of NK cell-mediated killing.75 It would be interesting to investigate in future studies whether the regulation of T cell responses by allogeneic NK cells, especially by NKG2D CAR-NK cells developed in this study, could be beneficial to off-the-shelf NK cell therapy.
In conclusion, we have successfully developed a non-viral PB transposon-based platform technology for CAR engineering of NK cells, which meets the crucial requirement of simplifying the manufacturing process for cost-effective production of CAR effector cells, an essential feature favors the widespread use of CAR-NK cells. CAR-NK cells expressing both NKG2D CAR and IL-15 were generated and shown to be superior to CAR-NK cells expressing NKG2D CAR only in controlling tumor progression in an AML xenograft model. Together, our findings suggest the infusion of non-viral NKG2D CAR/IL15-NK cells as a promising cell therapy option for AML treatment. A clinical trial has been planned to evaluate the safety and efficacy of the CAR-NK cells.
Materials and methods
Generation and expansion of CAR-NK cells
Fresh human PBMCs were isolated from the healthy donors' leukocyte reduction system cones (National University of Singapore, NUS-IRB H-17-046) with Ficoll-Paque Plus (GE Healthcare, Little Chalfont, UK) through density gradient centrifugation. To generate CAR-NK cells, human NK cells were magnetically sorted from human PBMCs through negative selection according to the manufacturer's instructions (Miltenyi, Bergisch Gladbach, Germany). The isolated NK cells then were co-cultured with 100 Gy gamma-irradiated K562 aAPCs with membrane-bound IL-15, IL-21, and 41BBL at a 1:1 effector to target ratio for 7 days or 2 days for initial expansion and activation.42 The culture medium used was AIM-V (Invitrogen, Carlsbad, CA) supplemented with 5% AB serum (Valley Biomedical, Winchester, VA), with 50 IU/mL IL-2 (PeproTech, Rocky Hill, NJ) supplied on the first day. Every 2 to 3 days, half volume of the media was changed with IL-2 replenished with full volume. Subsequently, NK cells were harvested (at D0), washed by Opti-MEM (Invitrogen) three times and re-suspended in P3 Primary Cell Nucleofector Solution (Lonza) at the concentration of 5 × 106 cells per 90 μL solution; 5 μg of piggyBac transposase plasmid and 10 μg of NKG2D CAR or NKG2D CAR-IL15 plasmid were added in the cell suspension and transferred together to the Nucleocuvette Vessel (Lonza).40 NKG2D CAR/IL15 is a bicistronic vector generated by inserting IRES-IL15 into the original NKG2D CAR plasmid. The electroporation was conducted with a 4D-Nucleofector system (Lonza) by following the manufacturer's protocol. The expansion started immediately after the electroporation by co-culturing the electroporated NK cells with K562 feeder cells at a 1:1 ratio with 50 IU/mL IL-2 (PeproTech) in AIM-V (Invitrogen) supplemented with 5% AB serum (Valley Biomedical) for four rounds, with 7 days per round. The culture medium and supplements were replenished every 2 to 3 days.
Cancer cell culture
AML cell lines KG-1 and MOLM-14 were cultured in IMDM (Invitrogen) supplemented with 20% FBS (GE Healthcare) and RPMI (Invitrogen) supplemented with 10% FBS (GE Healthcare), respectively. Human colorectal cancer cell line HCT-116 and ovarian cancer line SKOV-3 were cultured in McCoy's 5A Medium (Invitrogen) supplemented with 10% FBS (GE Healthcare). All cells were maintained at 5% C02 in a humidified 37°C incubator.
Antibody staining and flow cytometry analysis
The antibodies and related isotype controls used in this study are listed in Table S1. The antibody staining procedure followed the manufacturer's protocols and the cells were re-suspended in 500 μL autoMACS Running Buffer (Miltenyi). Flow cytometry analysis was performed with BD Accuri C6 Flow Cytometer (BD Biosciences, Franklin Lakes, NJ). Cells in the lymphocyte gate were used for analysis. In total, 10,000 events were collected for each sample and the generated data were analyzed by CFlow Sampler software (BD Biosciences).
Quantitative PCR
The genomic DNA was first isolated from NK cells by the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and qPCR analysis was performed with ABI TaqMan technology (Thermo Fisher Scientific, Waltham, MA). To determine the total CAR copy number in each reaction (100 ng genomic DNA/reaction), the forward/reverse primers and probe that specifically recognize the CAR sequences were designed and applied together with the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific). To determine the total number of cells, TaqMan Copy Number Reference Assay, human, TERT was used by following the manufacturer's protocol. The plasmid generated by IDT (Coralville, IA) contains both the CAR and TERT sequence fragments that were used to plot a standard curve for the quantification. The reactions were run in a Bio-Rad CFX96, C1000 Touch Thermal Cycler (Bio-Rad Laboratories, Hercules, CA). The average CAR copy number was calculated by using the following formula:
Cytotoxicity assay
A 3-h Europium-release assay by the DELFIA EuTDA Cytotoxicity Reagents kit (PerkinElmer, Waltham, MA) was used for assessing the immune cell-mediated cytotoxicity. Target cells (1 × 106 cells) were first re-suspended in 1 mL cell culture medium and labeled with 2 μL of bis(acetoxymethyl)2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (BATDA) in a 37°C, 5% CO2 incubator for 20 min with two times of PBS (GE Healthcare) washing subsequently. The target cells were then suspended in AIM-V (Invitrogen) supplemented with 5% AB serum (Valley Biomedical) at the concentration of 5 × 104 cells/mL. The CAR-NK cells, as the effector cells, were mixed with the target cells in triplicate at different Effector:Target (E:T) ratios ranging from 20:1 to 2.5:1 (or 4:1 to 0.5:1) and seeded into 96-well U-bottom plates (Thermo Fisher Scientific) with 200 μL of culture medium (5 × 103 target cells each well). The spontaneous release of labeled cancer cells was prepared by seeding the target cells alone in triplicate. The maximal release was prepared by adding 10 μL DEILFA lysis buffer into the 190 μL culture medium with the target cells in triplicate. After 3-h incubation in a 37°C, 5% CO2 incubator, the culture plate was centrifuged and 20 μL of the supernatant was transferred into a flat bottom reading plate (PerkinElmer) with 200 μL DELFIA Europium solution (PerkinElmer) added. The plate was subsequently shaken for 15 min by an orbital shaker at the speed of 250 rpm before reading values with the Victor3 plate reader (PerkinElmer). The killing efficacy was calculated by using the following formula:
CD107a degranulation assay
NK cells were first washed with 1x PBS (Hyclone) once and re-suspended into 1 mL AIM-V (Invitrogen) supplemented with 5% AB serum (Valley Biomedical). APC-conjugated CD107a antibody (BD Biosciences) and Golgi stop containing monensin (BD Biosciences) were added in and mixed up well with the cells. Target cells were washed and re-suspended into AIM-V (Invitrogen) supplemented with 5% AB serum (Valley Biomedical), and subsequently seeded into a U-bottom 96-well plate (Thermo Fisher Scientific) at the concentration of 2 × 105 cells/100 μL/well. NK cells were then co-cultured at a 1:1 E: T ratio with target cells in a 37°C, 5% CO2 incubator for 5 h. After incubation, the cells were used for anti-CD56 PE antibody staining (Miltenyi). The cells were then suspended in 150 μL autoMACS Running Buffer (Miltenyi) for the detection of the CD107a surface expression on NK cells by BD Accuri C6 Flow Cytometer (BD Biosciences).
In vitro IL-15 production by NK cells and NK cell recovery assay
CAR-NK cells or CAR/IL15-NK cells (4 × 106/mL) were cultured with or without HCT-116 cells (E:T = 5:1) in 24-well plates (Thermo Fisher Scientific) for either 3 h or 24 h. The supernatant was collected for measuring of IL-15 production with the Human IL-15 Quantikine ELISA Kit (R&D System, Minneapolis, MN) following the manufacturer's protocol.
To test NK cell recovery in vitro, CAR-NK cells or CAR/IL15-NK cells (1 × 106/mL) were seeded into the 24-well plates (Thermo Fisher Scientific) with AIM-V (Invitrogen) supplemented with 5% AB serum (Valley Biomedical) and cultured with low concentration (10 IU/mL) of IL-2 or without IL-2 for 7 days. The IL-2 was replenished every 2 to 3 days. The viable cell number was determined by trypan blue exclusion assay (Lonza) and the CAR percentage was analyzed by flow cytometry (BD Biosciences).
Animal experiments
Animal experiments were performed according to protocols reviewed and approved by Institutional Animal Care and Use Committee (IACUC), the Biological Resource Center (BRC), the Agency for Science, Technology and Research (A∗STAR), Singapore (Permit number BRC IACUC#181324). Male non-obese diabetic/severe combined immunodeficiency/IL-2Rγcnull (NSG) mice at the age of 8 weeks (The Jackson Laboratory, Bar Harbor, ME) were maintained and used in all studies. To examine therapeutic effects of CAR-NK cells, a mouse xenograft model of human AML was established by i.v. injection of 5 × 106 KG1-Luc cells through the tail vein. On either day 10 or day 3 post-tumor inoculation, mice were randomly divided into different groups for the subsequent treatments with PBS, mock NK cells, NKG2D CAR-NK cells or NKG2D CAR/IL15-NK cells. NK cells, 1 × 107 per mouse, were i.v. injected into NSG mice through the tail vein. Tumor progression was monitored by BLI and luminescent images were acquired and analyzed using the Xenogen living imaging software v2.5. Behavior and survival of the mice were monitored closely. Humane endpoints were used and mice were euthanized by cervical dislocation under sodium pentobarbital anesthesia. The survival curves were established based on the dates when mice were found dead or euthanized. To examine the in vivo CAR-NK cell persistence, NKG2D CAR-NK cells or NKG2D CAR/IL15-NK cells (1 × 107 per mouse) were i.v. injected into NSG mice through the tail vein. Blood samples were collected on day 3 and day 7 for analysis. The mice were euthanized on day 7 and kidneys were collected to examine the tissue distribution of human NK cells.
Statistical analysis
Data are presented as mean ± SD. Statistics were computed using GraphPad Prism 7.0 (GraphPad, La Jolla, CA). Statistical differences were marked by ∗, ∗∗, and ∗∗∗ for p values of <0.05, <0.01, and <0.001, respectively.
Acknowledgments
This work was supported by Singapore Ministry of Health's National Medical Research Council (NMRC/OFLCG/003/2018; MOH-000465-01) and Agency for Science, Technology and Research, Singapore (IAF-PP:H19/01/a0/022).
Author contributions
S.W. conceived and designed the experiments. Z.D., Y.Y.N., and S.Z. performed the experiments. Z.D., Y.Y.N., and S.Z. analyzed the data. Z.D. and S.W. wrote the manuscript.
Declaration of interests
The authors S.W. and Y.Y.N. have filed patent applications related to CAR technology and could potentially receive licensing royalties in future.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.10.014.
Supplemental information
References
- 1.Bittencourt M.C.B., Ciurea S.O. Recent advances in allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia. Biol. Blood Marrow Transpl. 2020;26:e215–e221. doi: 10.1016/j.bbmt.2020.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Arteaga A., Gyurkocza B. Recent advances in allogeneic hematopoietic cell transplantation for acute myeloid leukemia. Curr. Opin. Hematol. 2020;27:115–121. doi: 10.1097/MOH.0000000000000572. [DOI] [PubMed] [Google Scholar]
- 3.Bachanova V., Cooley S., Defor T.E., Verneris M.R., Zhang B., McKenna D.H., Curtsinger J., Panoskaltsis-Mortari A., Lewis D., Hippen K., et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855–3863. doi: 10.1182/blood-2013-10-532531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baragano Raneros A., Lopez-Larrea C., Suarez-Alvarez B. Acute myeloid leukemia and NK cells: two warriors confront each other. Oncoimmunology. 2019;8:e1539617. doi: 10.1080/2162402X.2018.1539617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjorklund A.T., Carlsten M., Sohlberg E., Liu L.L., Clancy T., Karimi M., Cooley S., Miller J.S., Klimkowska M., Schaffer M., et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 2018;24:1834–1844. doi: 10.1158/1078-0432.CCR-17-3196. [DOI] [PubMed] [Google Scholar]
- 6.Cooley S., He F., Bachanova V., Vercellotti G.M., DeFor T.E., Curtsinger J.M., Robertson P., Grzywacz B., Conlon K.C., Waldmann T.A., et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019;3:1970–1980. doi: 10.1182/bloodadvances.2018028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curti A., Ruggeri L., D'Addio A., Bontadini A., Dan E., Motta M.R., Trabanelli S., Giudice V., Urbani E., Martinelli G., et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118:3273–3279. doi: 10.1182/blood-2011-01-329508. [DOI] [PubMed] [Google Scholar]
- 8.Miller J.S., Soignier Y., Panoskaltsis-Mortari A., McNearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J., et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 9.Romee R., Rosario M., Berrien-Elliott M.M., Wagner J.A., Jewell B.A., Schappe T., Leong J.W., Abdel-Latif S., Schneider S.E., Willey S., et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl Med. 2016;8:357ra123. doi: 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubnitz J.E., Inaba H., Ribeiro R.C., Pounds S., Rooney B., Bell T., Pui C.H., Leung W. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010;28:955–959. doi: 10.1200/JCO.2009.24.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaffer B.C., Le Luduec J.B., Forlenza C., Jakubowski A.A., Perales M.A., Young J.W., Hsu K.C. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transpl. 2016;22:705–709. doi: 10.1016/j.bbmt.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimasaki N., Jain A., Campana D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- 13.Wang C.J., Huang X.J., Gong L.Z., Jia J.S., Liu X.H., Wang Y., Yan C.H., Chang Y.J., Zhao X.S., Shi H.X., et al. Observation on the efficacy of consolidation chemotherapy combined with allogeneic natural killer cell infusion in the treatment of low and moderate risk acute myeloid leukemia. Zhonghua Xue Ye Xue Za Zhi. 2019;40:812–817. doi: 10.3760/cma.j.issn.0253-2727.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J., Niu T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020;13:167. doi: 10.1186/s13045-020-00996-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basar R., Daher M., Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Blood Adv. 2020;4:5868–5876. doi: 10.1182/bloodadvances.2020002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daher M., Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11:45–58. doi: 10.1158/2159-8290.CD-20-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yilmaz A., Cui H., Caligiuri M.A., Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J. Hematol. Oncol. 2020;13:168. doi: 10.1186/s13045-020-00998-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abadir E., Gasiorowski R.E., Silveira P.A., Larsen S., Clark G.J. Is hematopoietic stem cell transplantation required to unleash the full potential of immunotherapy in acute myeloid leukemia? J. Clin. Med. 2020;9:554. doi: 10.3390/jcm9020554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehninger A., Kramer M., Rollig C., Thiede C., Bornhauser M., von Bonin M., Wermke M., Feldmann A., Bachmann M., Ehninger G., et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obajdin J., Davies D.M., Maher J. Engineering of chimeric natural killer cell receptors to develop precision adoptive immunotherapies for cancer. Clin. Exp. Immunol. 2020;202:11–27. doi: 10.1111/cei.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sentman C.L., Meehan K.R. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–159. doi: 10.1097/PPO.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frazao A., Rethacker L., Messaoudene M., Avril M.F., Toubert A., Dulphy N., Caignard A. NKG2D/NKG2-ligand pathway offers new opportunities in cancer treatment. Front Immunol. 2019;10:661. doi: 10.3389/fimmu.2019.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumeister S.H., Murad J., Werner L., Daley H., Trebeden-Negre H., Gicobi J.K., Schmucker A., Reder J., Sentman C.L., Gilham D.E., et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol. Res. 2019;7:100–112. doi: 10.1158/2326-6066.CIR-18-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sallman D.A., Brayer J., Sagatys E.M., Lonez C., Breman E., Agaugue S., Verma B., Gilham D.E., Lehmann F.F., Davila M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica. 2018;103:e424–e426. doi: 10.3324/haematol.2017.186742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantesso S., Geerts D., Spanholtz J., Kucerova L. Genetic engineering of natural killer cells for enhanced antitumor function. Front Immunol. 2020;11:607131. doi: 10.3389/fimmu.2020.607131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt P., Raftery M.J., Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front Immunol. 2020;11:611163. doi: 10.3389/fimmu.2020.611163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutlu T., Nystrom S., Gilljam M., Stellan B., Applequist S.E., Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum. Gene Ther. 2012;23:1090–1100. doi: 10.1089/hum.2012.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magnani C.F., Tettamanti S., Alberti G., Pisani I., Biondi A., Serafini M., Gaipa G. Transposon-based CAR T cells in acute leukemias: where are we going? Cells. 2020;9:1337. doi: 10.3390/cells9061337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh H., Moyes J.S., Huls M.H., Cooper L.J. Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015;22:95–100. doi: 10.1038/cgt.2014.69. [DOI] [PubMed] [Google Scholar]
- 31.Wilson M.H. Consider changing the horse for your CAR-T? Mol. Ther. 2018;26:1873–1874. doi: 10.1016/j.ymthe.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pomeroy E.J., Lahr W.S., Chang J.W., Krueger J., Wick B.J., Slipek N.J., Skeate J.G., Webber B.R., Moriarity B.S. Non-Viral Engineering of CAR-NK and CAR-T cells using the Tc Buster Transposon System™. bioRxiv. 2021 doi: 10.1101/2021.08.02.454772. [DOI] [Google Scholar]
- 33.Wang J., Lupo K.B., Chambers A.M., Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J. Immunother. Cancer. 2018;6:136. doi: 10.1186/s40425-018-0441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Wallace D.L., de Lara C.M., Ghattas H., Asquith B., Worth A., Griffin G.E., Taylor G.P., Tough D.F., Beverley P.C., et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121:258–265. doi: 10.1111/j.1365-2567.2007.02573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller J.S., Rooney C.M., Curtsinger J., McElmurry R., McCullar V., Verneris M.R., Lapteva N., McKenna D., Wagner J.E., Blazar B.R., et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol. Blood Marrow Transpl. 2014;20:1252–1257. doi: 10.1016/j.bbmt.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfefferle A., Jacobs B., Haroun-Izquierdo A., Kveberg L., Sohlberg E., Malmberg K.J. Deciphering natural killer cell homeostasis. Front Immunol. 2020;11:812. doi: 10.3389/fimmu.2020.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiore P.F., Di Matteo S., Tumino N., Mariotti F.R., Pietra G., Ottonello S., Negrini S., Bottazzi B., Moretta L., Mortier E., et al. Interleukin-15 and cancer: some solved and many unsolved questions. J. Immunother. Cancer. 2020;8:e001428. doi: 10.1136/jitc-2020-001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurney M., O'Dwyer M. Realizing innate potential: CAR-NK cell therapies for acute myeloid leukemia. Cancers (Basel) 2021;13:1568. doi: 10.3390/cancers13071568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A., et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng Y.Y., Tay J.C.K., Li Z., Wang J., Zhu J., Wang S. T cells expressing NKG2D CAR with a DAP12 signaling domain stimulate lower cytokine production while effective in tumor eradication. Mol. Ther. 2021;29:75–85. doi: 10.1016/j.ymthe.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tay J.C.K., Wang J., Du Z., Ng Y.Y., Li Z., Ren Y., Zhang C., Zhu J., Xu X.H., Wang S. Manufacturing NKG2D CAR-T cells with piggyBac transposon vectors and K562 artificial antigen-presenting cells. Mol. Ther. Methods Clin. Dev. 2021;21:107–120. doi: 10.1016/j.omtm.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng Y.Y., Tay J.C.K., Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol. Ther. Oncolytics. 2020;16:75–85. doi: 10.1016/j.omto.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Judge S.J., Murphy W.J., Canter R.J. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol. 2020;10:49. doi: 10.3389/fcimb.2020.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imamura M., Shook D., Kamiya T., Shimasaki N., Chai S.M., Coustan-Smith E., Imai C., Campana D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124:1081–1088. doi: 10.1182/blood-2014-02-556837. [DOI] [PubMed] [Google Scholar]
- 45.Luu T.T., Schmied L., Nguyen N.A., Wiel C., Meinke S., Mohammad D.K., Bergo M., Alici E., Kadri N., Ganesan S., et al. Short-term IL-15 priming leaves a long-lasting signalling imprint in mouse NK cells independently of a metabolic switch. Life Sci. Alliance. 2021;4:e202000723. doi: 10.26508/lsa.202000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang Y.H., Connolly J., Shimasaki N., Mimura K., Kono K., Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–1786. doi: 10.1158/0008-5472.CAN-12-3558. [DOI] [PubMed] [Google Scholar]
- 47.Xiao L., Cen D., Gan H., Sun Y., Huang N., Xiong H., Jin Q., Su L., Liu X., Wang K., et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther. 2019;27:1114–1125. doi: 10.1016/j.ymthe.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ingegnere T., Mariotti F.R., Pelosi A., Quintarelli C., De Angelis B., Tumino N., Besi F., Cantoni C., Locatelli F., Vacca P., et al. Human CAR NK cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. Front Immunol. 2019;10:957. doi: 10.3389/fimmu.2019.00957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar D., Anand T., Talluri T.R., Kues W.A. Potential of transposon-mediated cellular reprogramming towards cell-based therapies. World J. Stem Cell. 2020;12:527–544. doi: 10.4252/wjsc.v12.i7.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tschorn N., Berg K., Stitz J. Transposon vector-mediated stable gene transfer for the accelerated establishment of recombinant mammalian cell pools allowing for high-yield production of biologics. Biotechnol. Lett. 2020;42:1103–1112. doi: 10.1007/s10529-020-02889-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakazawa Y., Huye L.E., Dotti G., Foster A.E., Vera J.F., Manuri P.R., June C.H., Rooney C.M., Wilson M.H. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J. Immunother. 2009;32:826–836. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Neil R.T., Saha S., Veach R.A., Welch R.C., Woodard L.E., Rooney C.M., Wilson M.H. Transposon-modified antigen-specific T lymphocytes for sustained therapeutic protein delivery in vivo. Nat. Commun. 2018;9:1325. doi: 10.1038/s41467-018-03787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felices M., Lenvik T.R., Kodal B., Lenvik A.J., Hinderlie P., Bendzick L.E., Schirm D.K., Kaminski M.F., McElmurry R.T., Geller M.A., et al. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol. Res. 2020;8:1139–1149. doi: 10.1158/2326-6066.CIR-19-0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Driouk L., Gicobi J.K., Kamihara Y., Rutherford K., Dranoff G., Ritz J., Baumeister S.H.C. Chimeric antigen receptor T cells targeting NKG2D-ligands show robust efficacy against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Front Immunol. 2020;11:580328. doi: 10.3389/fimmu.2020.580328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilpert J., Grosse-Hovest L., Grunebach F., Buechele C., Nuebling T., Raum T., Steinle A., Salih H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J. Immunol. 2012;189:1360–1371. doi: 10.4049/jimmunol.1200796. [DOI] [PubMed] [Google Scholar]
- 56.Paczulla A.M., Rothfelder K., Raffel S., Konantz M., Steinbacher J., Wang H., Tandler C., Mbarga M., Schaefer T., Falcone M., et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572:254–259. doi: 10.1038/s41586-019-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salih H.R., Antropius H., Gieseke F., Lutz S.Z., Kanz L., Rammensee H.G., Steinle A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102:1389–1396. doi: 10.1182/blood-2003-01-0019. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez-Correa B., Morgado S., Gayoso I., Bergua J.M., Casado J.G., Arcos M.J., Bengochea M.L., Duran E., Solana R., Tarazona R. Human NK cells in acute myeloid leukaemia patients: analysis of NK cell-activating receptors and their ligands. Cancer Immunol. Immunother. 2011;60:1195–1205. doi: 10.1007/s00262-011-1050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlegel P., Ditthard K., Lang P., Mezger M., Michaelis S., Handgretinger R., Pfeiffer M. NKG2D signaling leads to NK cell mediated lysis of childhood AML. J. Immunol. Res. 2015;2015:473175. doi: 10.1155/2015/473175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu T.L., Goss V.L., Reeves C., Popova L., Nardone J., Macneill J., Walters D.K., Wang Y., Rush J., Comb M.J., et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108:4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- 61.Saland E., Boutzen H., Castellano R., Pouyet L., Griessinger E., Larrue C., de Toni F., Scotland S., David M., Danet-Desnoyers G., et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015;5:e297. doi: 10.1038/bcj.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ben-Shmuel A., Biber G., Barda-Saad M. Unleashing natural killer cells in the tumor microenvironment-the next generation of immunotherapy? Front Immunol. 2020;11:275. doi: 10.3389/fimmu.2020.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bachmann M.F., Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. 2007;8:1142–1148. doi: 10.1038/sj.embor.7401099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Isidori A., Cerchione C., Daver N., DiNardo C., Garcia-Manero G., Konopleva M., Jabbour E., Ravandi F., Kadia T., Burguera A.F., et al. Immunotherapy in acute myeloid leukemia: where we stand. Front Oncol. 2021;11:656218. doi: 10.3389/fonc.2021.656218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heczey A., Courtney A.N., Montalbano A., Robinson S., Liu K., Li M., Ghatwai N., Dakhova O., Liu B., Raveh-Sadka T., et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat. Med. 2020;26:1686–1690. doi: 10.1038/s41591-020-1074-2. [DOI] [PubMed] [Google Scholar]
- 66.Bachanova V., Cayci Z., Lewis D., Maakaron J.E., Janakiram M., Bartz A., Payne S., Wong C., Cooley S., Valamehr B., et al. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. 2020;136:8. [Google Scholar]
- 67.Wang X., Jasinski D.L., Medina J.L., Spencer D.M., Foster A.E., Bayle J.H. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4:1950–1964. doi: 10.1182/bloodadvances.2020001510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bishop D.C., Clancy L.E., Simms R., Burgess J., Mathew G., Moezzi L., Street J.A., Sutrave G., Atkins E., McGuire H.M., et al. Development of CAR T-cell lymphoma in two of ten patients effectively treated with piggyBac modified CD19 CAR T-cells. Blood. 2021 doi: 10.1182/blood.2021010813. [DOI] [PubMed] [Google Scholar]
- 69.Micklethwaite K.P., Gowrishankar K., Gloss B.S., Li Z., Street J.A., Moezzi L., Mach M.A., Sutrave G., Clancy L.E., Bishop D.C., et al. Investigation of product derived lymphoma following infusion of piggyBac modified CD19 chimeric antigen receptor T-cells. Blood. 2021 doi: 10.1182/blood.2021010858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schambach A., Morgan M., Fehse B. Two cases of T cell lymphoma following Piggybac-mediated CAR T cell therapy. Mol. Ther. 2021;29:2631–2633. doi: 10.1016/j.ymthe.2021.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wagner D.L., Fritsche E., Pulsipher M.A., Ahmed N., Hamieh M., Hegde M., Ruella M., Savoldo B., Shah N.N., Turtle C.J., et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021;18:379–393. doi: 10.1038/s41571-021-00476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crouse J., Xu H.C., Lang P.A., Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. 2015;36:49–58. doi: 10.1016/j.it.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 73.Pallmer K., Oxenius A. Recognition and regulation of T cells by NK cells. Front Immunol. 2016;7:251. doi: 10.3389/fimmu.2016.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schuster I.S., Coudert J.D., Andoniou C.E., Degli-Esposti M.A. "Natural Regulators": NK cells as modulators of T cell immunity. Front Immunol. 2016;7:235. doi: 10.3389/fimmu.2016.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simonetta F., Alvarez M., Negrin R.S. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. 2017;8:465. doi: 10.3389/fimmu.2017.00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.