

Abstract
It has been demonstrated that homocysteine (Hcy) can cause inflammatory diseases. Long noncoding RNAs (lncRNA) and microRNAs (miRNAs) are involved in this biological process, but the mechanism underlying Hcy-induced inflammation remains poorly understood. Here, we found that lncRNA TGFB3-AS1 was highly expressed in macrophages treated with Hcy and the peripheral blood monocytes from cystathionine beta-synthase heterozygous knockout (CBS+/−) mice with a high-methionine diet using lncRNA microarray. In vivo and in vitro experiments further confirmed that TGFB3-AS1 accelerated Hcy-induced inflammation of macrophages through the Rap1a/wnt signaling pathway. Meanwhile, TGFB3-AS1 interacted with Rap1a and reduced degradation of Rap1a through inhibiting its ubiquitination in macrophages treated with Hcy. Rap1a mediated inflammation induced by Hcy and serves as a direct target of miR-144. Moreover, TGFB3-AS1 regulated miR-144 by binding to pri-miR-144 and inhibiting its maturation, which further regulated Rap1a expression. More importantly, we found that high expression of TGFB3-AS1 was positively correlated with the levels of Hcy and proinflammatory cytokines in serum of healthy individuals and patients with HHcy. Our study revealed a novel mechanism by which TGFB3-AS1 promoted inflammation of macrophages through inhibiting miR-144 maturation to stay miR-144 regulated inhibition of functional Rap1a expression.
Keywords: homocysteine, inflammation, lncRNA, miRNA, Rap1a
Graphical abstract
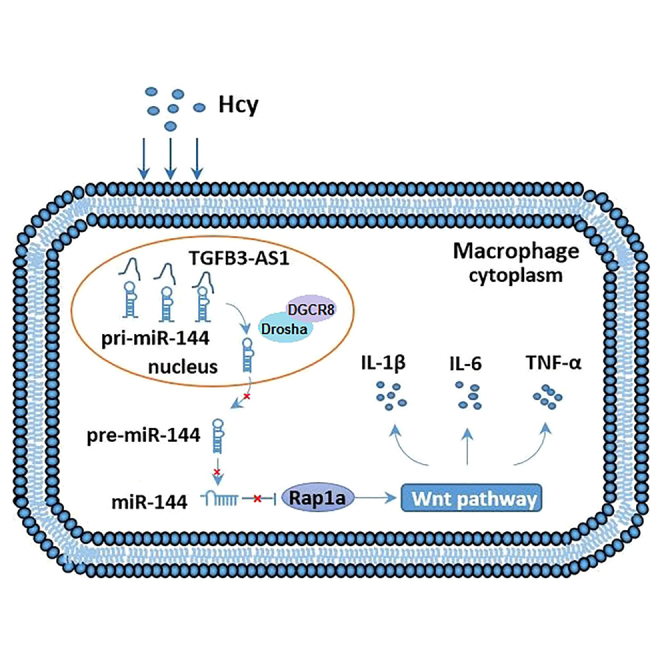
TGFB3-AS1 overexpressed promoted inflammation induced by Hcy. Meanwhile, TGFB3-AS1 suppressed the maturation of miR-144 to promote the expression of its target gene Rap1a, which activated the wnt/β-catenin signaling pathway and participates in Hcy-induced inflammation. These findings propose that inhibition of TGFB3-AS1 might be a potential therapeutic strategy for inflammatory.
Introduction
Accumulating evidence has confirmed that pathological serum level of homocysteine (Hcy) is closely related to the increased frequency of cardiovascular disease.1 Hyperhomocysteinemia (HHcy) is likely related to the enhanced production of proinflammatory cytokines.2 Recent notions indicate that macrophages derived from monocytes are key cells in the inflammatory process.3 However, the detailed mechanism of how Hcy induces proinflammatory effects on macrophages is not well elucidated.
Noncoding RNAs are a cluster of RNAs that do not encode proteins but could regulate gene expression at the posttranscriptional level.4,5 Long noncoding RNAs (lncRNAs) are noncoding transcripts that are more than 200 nucleotides in length. Recent evidence has revealed that lncRNAs play important roles in regulating the functions of multiple biological processes, particularly in inflammatory cells such as macrophages.6, 7, 8 Several lncRNAs, such as lincRNA-Cox2 and FIREE, have been reported to be acutely enhanced inflammatory gene expression via transcriptional and posttranscriptional mechanisms.9,10 In contrast, lnc-IL7R and lincRNA-EPS suppress the inflammatory function of macrophages.11,12 Thus, lncRNAs are important regulators of macrophage activation. MicroRNAs (miRNAs) are another class of ncRNAs that have emerged as major regulators of gene expression by suppressing translation or undermining the mRNAs at the posttranscriptional level.13,14 Recently, the regulating function of lncRNAs on miRNAs has attracted wide interest in multiple biological and pathological processes, such as myocardial infarction, atherosclerosis, and cardiac hypertrophy.15, 16, 17 The biosynthesis of miRNAs initially involves transcription of a primary miRNA (pri-miRNA).18 The pri-miRNA is processed by Drosha/DGCR8 to form a pre-miRNA, which then enters the cytoplasm to form the mature miRNA with the help of Dicer. The mature miRNA associates with members of the Argonaute proteins to form the miRNA-induced silencing complex, which binds to complementary sequences located primarily within the 3′ untranslated region (UTR) of target gene mRNAs.19 Binding of the miRNA-induced silencing complex to a target mRNA leads to degradation of the transcript, and/or inhibition of protein translation.20 In addition, miRNAs can target multiple different transcripts and regulate biological processes, which provides insights into the functions of these noncoding transcripts and their potential as therapeutic targets.21 However, the roles of lncRNAs in accelerating the maturity of miRNAs and regulating HHcy-induced inflammation of macrophages are still unknown.
In this study, we sought to explore the effect and underlying mechanisms of how Hcy induces macrophage inflammation. Our findings revealed a novel mechanism of how lncRNA TGFB3-AS1 suppressed the maturation of miR-144 and further upregulated the expression of Rap1a, which contributed to the activation of the wnt/β-catenin pathway in Hcy-induced inflammation of microphages. Given the causal relationship between macrophage inflammation and atherosclerosis, we propose that TGFB3-AS1 may serve as a potential prognostic biomarker and therapeutic target for Hcy-induced atherosclerosis.
Results
Hcy primarily accelerates inflammation of monocyte-derived macrophages in peripheral blood
It has been reported that monocyte-derived macrophages play a vital role in inflammation.22 To assess the effect of Hcy on inflammation of monocyte-derived macrophages, CBS+/+ and CBS+/− mice were fed with 2.0% methionine for 12 weeks to induce hyperhomocysteinemia. As shown in Figure 1A, the serum levels of Hcy were significantly increased in CBS+/− mice compared with CBS+/+ mice. Simultaneously, the levels of classical proinflammatory cytokines, interleukin-6 (IL-6), IL-1β, tumor necrosis factor alpha (TNF-α), and C-reactive protein (CRP), were remarkably increased in serum of CBS+/− mice (Figure 1B), suggesting that the elevated Hcy in CBS+/− mice fed with a high-methionine diet is associated with induction of monocyte-derived macrophage inflammation.
Figure 1.

Hcy enhances inflammation of monocyte-derived macrophages
(A and B) The serum levels of Hcy, proinflammatory cytokines TNF-α, IL-1β, IL-6, and CRP in CBS+/+ and CBS+/− mice fed with a regular or methionine diet for 12 weeks. (C) Relative spleen weight to body weight (g/g) in CBS+/+ and CBS+/− mice. (D and E) Monocytes population in peripheral blood, spleen, and bone marrow (BM) from CBS+/+ and CBS+/− mice were analyzed by flow cytometry. SSC, side scattered light; FSC, forward-scatter light. (F and G) The percentages of CD11b+ Ly-6C+ inflammatory monocytes in peripheral blood, spleen, and BM isolated from CBS+/+ and CBS+/− mice were analyzed by flow cytometry. (H) The concentration of IL-1β, IL-6, and TNF-α in the supernatants of monocyte-derived macrophages were assayed by ELISA after treatment with different doses of Hcy (0, 50, 100, 200, and 500 μmol/L) for different times (12, 24, and 48 h). ∗p < 0.05, ∗∗p < 0.01, compared with CBS+/+ or control group.
To further identify the distribution of monocytes in Hcy-induced inflammation, we analyzed the monocytes in peripheral blood, spleen, and bone marrow (BM), as BM produces monocytes, and spleen serves as a reservoir for circulating monocytes. The relative spleen weight had a trend to increase in CBS+/− mice compared with CBS+/+ mice fed with a high-methionine diet, but did not reach statistical significance (Figure 1C). Meanwhile, flow cytometry analysis indicated that Hcy significantly increased the population of monocytes in peripheral blood of CBS+/− mice fed with a high-methionine diet compared with those of their controls, but not in other tissues (Figures 1D and 1E). Monocytes can be functionally characterized as inflammatory type (Ly-6Chigh) or resident type (Ly-6Clow) according to Ly-6C expression, we then characterized the CD11b+ monocytes (CD11b is a monocyte marker) as monocyte subsets by flow cytometry analysis using anti-CD11b and anti-Ly-6C antibodies. As shown in Figures 1F and 1G, the CD11b+ Ly-6C+ inflammatory monocyte was the most abundant subset in the peripheral blood of CBS+/− mice fed with a high-methionine diet, indicating that Hcy primarily elevated inflammatory monocyte subsets in peripheral blood. Next, monocyte-derived macrophages were treated with different doses of Hcy to verify the proinflammatory effects of Hcy in vitro. It was observed that Hcy promoted the secretion of proinflammatory factors (IL-1β, IL-6, and TNF-α) in the supernatants of monocyte-derived macrophages, especially under the condition of 100 μmol/L Hcy treatment (Figure 1H). Collectively, these results suggested Hcy exacerbated inflammation in peripheral blood monocyte-derived macrophages.
LncRNA TGFB3-AS1 promotes Hcy-induced inflammation of monocyte-derived macrophages
To identify the critical lncRNAs involved in Hcy-induced inflammation of monocyte-derived macrophages, lncRNA microarrays were used to analyze the expression profiles of lncRNAs in monocyte-derived macrophages treated with Hcy. Figure 2A indicates that 33 upregulated and 49 downregulated lncRNAs were differentially expressed in macrophages after Hcy treatment, respectively. The functional roles of the 33 upregulated and 49 downregulated lncRNAs were predicted using KEGG enrichment analysis (Figure 2B). The top 9 significantly upregulated lncRNAs in monocyte-derived macrophages are listed in the volcano plot (Figure 2C), and the expressions were further verified by qRT-PCR assay (Figure 2D). Notably, the expression of (lnc-TGFB3-AS1) was upregulated both in Hcy-treated macrophages and peripheral blood monocytes from CBS+/− mice fed with a high-methionine diet compared with their corresponding controls (Figures 2D, 2E, and S1A). In addition, we also tested the expression of TGFB3-AS1 in heart, liver, lung, kidney, blood, and muscle tissues of CBS+/+ and CBS+/− mice, which indicated that TGFB3-AS1 was particularly enriched in peripheral blood monocytes of CBS+/− mice (Figure 2F). RNA-fluorescent in situ hybridization (FISH) demonstrated that highly expressed TGFB3-AS1 was predominantly localized in the nucleus of macrophages treated with Hcy (Figure 2G). Moreover, qRT-PCR using macrophage nucleic and cytoplasmic RNA as the templates further determined the position of TGFB3-AS1, which showed that TGFB3-AS1 was accumulated in the nucleus of macrophages treated with Hcy (Figure 2H), implying that TGFB3-AS1 mainly exerts its biological function in the nucleus. To further illustrate the function of TGFB3-AS1 in the regulation of Hcy-induced inflammation of macrophages, macrophages were transduced with lenti-TGFB3-AS1 or TGFB3-AS1 siRNAs to overexpress or knockdown of TGFB3-AS1 (Figure S1B). As shown in Figure 2I, overexpression of TGFB3-AS1 promoted the secretion of proinflammatory cytokines IL-1β, IL-6, and TNF-α in macrophages treated with Hcy. In contrast, knockdown of TGFB3-AS1 decreased the secretion of these cytokines in macrophages. Therefore, TGFB3-AS1 is implicated in macrophage inflammation induced by Hcy.
Figure 2.

Hcy enhances monocyte-derived macrophage inflammation via upregulation of TGFB3-AS1
(A) Heatmap of the top 33 up- and 49 downregulated lncRNAs in macrophages treated with Hcy. (B) KEGG enrichment pathway analysis of the lncRNAs with expression significantly changed in Hcy-treated macrophages are shown in the heatmap. (C) Volcano plot of the lncRNAs with significant change in expression (FDR < 0.05, |log2 FC| ≥ 1 cutoff and RPKM ≥ 0.5); the top 9 upregulated lncRNAs were marked out. (D and E) Analysis of the expression of the top 9 upregulated lncRNAs from microarrays data in macrophages under 100 μmol/L Hcy treatment and peripheral blood monocytes from CBS+/+ and CBS+/− mice fed with a 12-week regular or methionine diet. (F) The relative expression of TGFB3-AS1 in tissues of heart, liver, lung, kidney, blood, and muscle of CBS+/+ and CBS+/− mice. (G) RNA-FISH analysis of the subcellular distribution of TGFB3-AS1 in macrophages treated with Hcy. Scale bars, 20 μm. (H) Percentage of cytoplasmic and nucleic RNA levels of GAPDH, U6, and TGFB3-AS1 detected by qRT-PCR in Hcy-treated macrophages. (I) The levels of IL-1β, IL-6, and TNF-α in macrophages transduced with lentivirus expressing TGFB3-AS1 (lenti-TGFB3-AS1) or siRNA targeting TGFB3-AS1 (TGFB3-AS1-siRNA1) were assayed by ELISA, respectively. ∗p < 0.05, ∗∗p < 0.01, compared with control, CBS+/+ + Meth or 100 μmol/L Hcy + lenti-GFP group; #p < 0.05, compared with 100 μmol/L Hcy + TGFB3-AS1-sc group.
TGFB3-AS1 stabilizes Rap1a via inhibiting its ubiquitin-proteasome-mediated degradation
It has been reported that lncRNAs can regulate gene expression through directly interacting with their target proteins.23 We performed an RNA pull-down assay followed by mass spectrometry using in vitro transcribed biotin-labeled TGFB3-AS1 and an antisense control (antisense TGFB3-AS1) to identify the potential proteins interacting with TGFB3-AS1 in macrophages (Figure 3A). The proteins pulled down by TGFB3-AS1 or the antisense TGFB3-AS1 control were separated by silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The result showed that TGFB3-AS1 could co-purify a number of proteins, especially proteins with molecular weight from 15 to 45 kDa, compared with antisense TGFB3-AS1 control (Figure 3B). Based on the analysis of mass spectrometry, we selected the top 3 proteins with highest score and matches (Rap1a, Vim, and Hist1h4) for further validation by RNA pull-down assay (Figure 3C). As shown in Figure 3D, Rap1a could be co-purified by TGFB3-AS1, but not Vim or Hist1h4a. Consistently, RNA-binding protein immunoprecipitation (RIP) assay also showed enrichment of TGFB3-AS1 in the complexes precipitated with antibody against Rap1a as compared with IgG control (Figure 3E). These results indicated that Rap1a is a novel binding partner of TGFB3-AS1. To further disclose the relation between TGFB3-AS1 and Rap1a, we detected changes in Rap1a expression after TGFB3-AS1 expression was enforced or silenced in macrophages by transduction with lenti-TGFB3-AS1 or TGFB3-AS1-siRNA, respectively. The results showed that Rap1a was upregulated in macrophages with TGFB3-AS1 overexpression, while reduced in macrophages with TGFB3-AS1 silence (Figures 3F and 3G), suggesting that TGFB3-AS1 promoted the expression of Rap1a.
Figure 3.

TGFB3-AS1 interacts with Rap1a and protects it from ubiquitin-proteasome-mediated degradation in macrophages
(A) Schematic of RNA pull-down coupled with mass spectrometry analysis for identifying proteins interacting with TGFB3-AS1 in macrophages. (B) Separation of proteins pulled down by TGFB3-AS1 or its antisense RNA control (antisense TGFB3-AS1) using silver-stained SDS-PAGE. The black frame indicates the specific bands of proteins pulled down by TGFB3-AS1 compared with the antisense TGFB3-AS1 control. (C) The top 18 significantly enriched proteins in the RNA pull-down mass spectrometry analysis are listed in the table, with their score, matches, and emPAI displayed. (D) Whole-cell lysate of macrophages was immunoprecipitated with biotinylated TGFB3-AS1 or its antisense control (antisense TGFB3-AS1) followed by immunoblotting with the antibodies against Rap1a, Vim, and Hist1h4. (E) qRT-PCR examined enrichment of TGFB3-AS1 in Rap1a-immunoprecipitated RNA complexes using Rap1a antibodies, isotype identical IgG served as a negative control. (F and G) The mRNA and protein expression of Rap1a in macrophages transduced with either lenti-TGFB3-AS1 or TGFB3-AS1 siRNAs (TGFB3-AS1-siRNA1 and TGFB3-AS1-siRNA2) were detected by qRT-PCR and western blot, respectively. (H) Left panel: western blot was used to detect the expression of Rap1a in macrophages treated with lenti-TGFB3-AS1 and 20 μg/mL cycloheximide (CHX) for various times (0, 15, 30, 60, 120, and 240 min). Right panel: the quantification of Rap1a protein levels in the upper panel. T1/2 denotes an estimated half-life. (I) Western blot was used to detect the expression of Rap1a in macrophages treated with TGFB3-AS1-siRNA or MG132 (5 μmol/L, 24 h). (J) The ubiquitination level of Rap1a was measured by immunoprecipitation with anti-Rap1a antibody and immunoblotting with anti-Ub antibody in macrophages transduced with TGFB3-AS1-siRNA or TGFB3-AS1-sc in the presence of MG132 with or without Hcy. ∗p < 0.05, compared with TGFB3-AS1-sc, lenti-GFP, or 100 μmol/L Hcy. ∗∗p < 0.01, compared with IgG. #p < 0.05, ##p < 0.01, compared with the group of lenti-GFP + 100 μmol/L Hcy or TGFB3-AS1-sc + 100 μmol/L Hcy.
To discover whether TGFB3-AS1 upregulated Rap1a expression by enhancing stability of Rap1a protein, macrophages were treated with 20 μg/mL CHX (a protein synthesis inhibitor) for various times before detecting the protein expression of Rap1a. As expected, overexpression of TGFB3-AS1 obviously prolonged the half-life of Rap1a (Figure 3H). Moreover, MG132, a proteasome inhibitor, could recover Rap1a protein expression reduced by TGFB3-AS1 knockdown in macrophages (Figure 3I). Meanwhile, the ubiquitination of Rap1a was significantly increased in TGFB3-AS1-siRNA-transduced macrophages compared with the negative control cells transduced with TGFB3-AS1-sc under MG132 treatment (Figure 3J). Collectively, these results indicated that TGFB3-AS1 interacted with and stabilized Rap1a by inhibiting ubiquitin-proteasome-mediated degradation of Rap1a protein in macrophages.
The Rap1a/wnt pathway is required for TGFB3-AS1-mediated macrophage inflammation
To investigate whether Rap1a is involved in Hcy-induced inflammation, we examined Rap1a expression in peripheral blood monocytes of CBS+/+ and CBS+/− mice fed with a high-methionine diet. The result showed that Rap1a was markedly upregulated in peripheral blood monocytes of CBS+/− mice compared with that of CBS+/+ mice (Figure 4A). Similarly, 100 μmol/L Hcy significantly increased Rap1a expression in macrophages (Figure 4B). To confirm the role of Rap1a in regulation of Hcy-induced inflammation of macrophages, inflammatory factors were measured by ELISA after knockdown of Rap1a. As shown in Figures 4C and 4D, knockdown of Rap1a remarkably reduced the secretion of IL-1β, IL-6, and TNF-α induced by Hcy. To identify the mechanism, we analyzed the changes of associated pathways caused by Hcy. KEGG enrichment analysis showed that the wnt pathway is the most potential signaling pathway in which Rap1a is involved (Figure 4E). In addition, qRT-PCR and western blot analysis showed that the expression of wnt3a, wnt5a, and β-catenin were notably increased in the peripheral blood monocytes of CBS+/− mice fed with a high-methionine diet (Figures 4F, 4G, and 4H), similar results were also observed in macrophages treated with Hcy (Figure 4I). Next, we elucidated whether TGFB3-AS1 induces macrophage inflammation via the Rap1a/wnt signaling pathway. Macrophages were co-transduced with TGFB3-AS1-siRNA and Rap1a-expressing adenoviruses (Ad-Rap1a) with or without Hcy treatment, followed by the determination of expression levels of wnt3a, wnt5a, and β-catenin by western blot. As expected, knockdown of TGFB3-AS1 reduced the expression of wnt3a, wnt5a, and β-catenin, which can be reversed by overexpression of Rap1a (Figure 4I). Conversely, Rap1a knockdown inhibited the elevation of wnt3a, wnt5a, and β-catenin expression induced by TGFB3-AS1 overexpression (Figure 4J). In addition, overexpression of Rap1a significantly rescued the reduction of IL-1β, IL-6, and TNF-α levels induced by TGFB3-AS1 knockdown (Figure 4K); whereas, knockdown of Rap1a had opposite effects (Figure 4L). Taken together, these results demonstrated that TGFB3-AS1 promoted macrophage inflammation induced by Hcy via the Rap1a/wnt pathway.
Figure 4.

Rap1a/wnt pathway is required for TGFB3-AS1-mediated macrophage inflammation
(A) qRT-PCR and western blot were performed to determine the expression of Rap1a in the peripheral monocytes of CBS+/+ and CBS+/− mice fed with a 12-week regular or methionine diet (n = 6). (B) qRT-PCR and western blot were performed to detect the mRNA and protein expression of Rap1a in macrophages treated with different doses of Hcy (0, 50, 100, 200, and 500 μmol/L) for 24 h. (C) Rap1a mRNA and protein were analyzed by qRT-PCR and western blot after macrophages were transfected with si-Rap1a in the presence or absence of Hcy. (D) The contents of IL-1β, IL-6, and TNF-α in the supernatants of macrophages treated with si-Rap1a and Hcy were assayed by ELISA. (E) KEGG enrichment analysis of differentially expressed genes in macrophages treated with Hcy. (F–H) The expression of wnt3a, wnt5a, and β-catenin in the peripheral monocytes of CBS+/+ and CBS+/− mice were examined by qRT-PCR and western blot. (I and J) Western blot analysis of Rap1a, wnt3a, wnt5a, and β-catenin expression in macrophages co-transduced with TGFB3-AS1-siRNA or lenti-TGFB3-AS1 and Ad-Rap1a or si-Rap1a in the presence or absence of Hcy. (K and L) The contents of IL-1β, IL-6, and TNF-α in the supernatants of macrophages were co-transduced with TGFB3-AS-siRNA or lenti-TGFB3-AS1 and Ad-Rap1a or si-Rap1a in the presence of Hcy. ∗p < 0.05, ∗∗p < 0.01, compared with control, CBS+/+ + Meth, Ad-vector or si-NC; #p < 0.05, compared with the group of 100 μmol/L Hcy, Ad-vector + 100 μmol/L Hcy, or si-NC + 100 μmol/L Hcy; ▵p < 0.05, compared with the group of Ad-Rap1a or si-Rap1a; ▿p < 0.05, compared with the group of Ad-vector + TGFB3-AS1-siRNA+ 100 μmol/L Hcy or si-NC + lenti-TGFB3-AS1 + 100 μmol/L Hcy.
miR-144 regulates macrophage inflammation induced by Hcy through directly targeting Rap1a
Since miRNAs are a class of very important posttranscriptional regulators, we sought to investigate miRNAs that regulate inflammation of macrophages induced by Hcy via targeting Rap1a. Based on the TargetScan databases, 19 candidate miRNAs were predicted to target the 3′ UTR of Rap1a (Figure 5A). Analysis of the 19 candidate miRNAs indicated that miR-144 was dramatically downregulated in peripheral blood monocytes of CBS+/− mice fed with a high-methionine diet compared with that of the regular-diet-fed CBS+/− mice (Figure 5B). Consistent with the in vivo results, Hcy dramatically reduced the expression of miR-144 in macrophages (Figure 5C). To prove the reliability of bioinformatics prediction, dual-luciferase-reporter assay was performed and verified that the miR-144 mimic dramatically reduced the luciferase activity in HEK293T cells transfected with wild-type (WT) 3′ UTR of Rap1a, but not in HEK293T cells transfected with mutant (mut) 3′ UTR of Rap1a (Figure 5D), suggesting that Rap1a 3′ UTR has specific sequences bound to miR-144. Subsequently, we knocked down or upregulated miR-144 with miR-144 inhibitor or mimic to illustrate the effect of miR-144 on Rap1a expression (Figure 5E). As depicted in Figure 5F, the miR-144 mimic reduced Rap1a expression, while the miR-144 inhibitor exerted the opposite effect. Furthermore, a rescue experiment was designed to verify the effects of miR-144 on inflammation of macrophages treated with Hcy through Rap1a. As shown in Figure 5G, overexpression of miR-144 significantly decreased the secretion of proinflammatory factors (IL-1β, IL-6, and TNF-α) in macrophages treated with Hcy, which could be alleviated when Rap1a was overexpressed (Figure 5G, left); opposite results were observed when miR-144 was knocked down (Figure 5G, right). These data indicated that miR-144 regulated inflammation of macrophages induced by Hcy via targeting Rap1a.
Figure 5.

miR-144 regulates macrophage inflammation induced by Hcy via targeting Rap1a
(A) Prediction of miRNAs that have the potential to target Rap1a by TargetScan database (http://www.targetscan.org/vert_72/). (B) miR-144 expression was detected by qRT-PCR in peripheral monocytes of CBS+/+ and CBS+/− mice. (C) miR-144 expression was detected by qRT-PCR in macrophages treated with 100 μmol/L Hcy for 24 h. (D) Schematic diagram of miR-144 binding site of Rap1a in various species (left panel). Relative luciferase activities in HEK293T cells co-transfected with plasmids of Rap1a-3′ UTR-WT or Rap1a-3′ UTR-mut and miR-144 mimic (right panel). (E) miR-144 expression was detected by qRT-PCR in macrophages transfected with miR-144 mimics or miR-144 inhibitor. (F) qRT-PCR and western blot analysis of Rap1a expression in macrophages transfected with miR-144 mimic, miR-144 inhibitor, and miR-neg. (G) The contents of IL-1β, IL-6, and TNF-α in macrophages after co-transduction with Ad-Rap1a and miR-144 inhibitor or miR-144 mimic. ∗p < 0.05, ∗∗p < 0.01 compared with CBS+/+ + Meth, control, Rap1a-3′ UTR-WT + miR-neg, miR-neg or 100 μmol/L Hcy group, #p < 0.05 compared with 100 μmol/L Hcy + miR-neg, 100 μmol/L Hcy + miR-144 mimic + Ad-GFP group, or 100 μmol/L Hcy + miR-144 inhibitor + Ad-GFP group.
TGFB3-AS1 inhibits the maturation of miR-144 in Hcy-treated macrophages
Next, we explored how TGFB3-AS1 regulates miR-144 expression. It is known that each phase during miRNA biogenesis is subjected to modulation, and that miRNAs are initially transcribed from the genome by RNA polymerase II into primary transcripts (pri-miRNA) and processed to hairpin structures of about ∼70 nucleotide miRNA precursors (pre-miRNAs) by the enzyme Drosha in the nucleus. Considering that TGFB3-AS1 mainly locates in the nucleus of macrophages under Hcy treatment, we wondered whether TGFB3-AS1 impacts the miRNA biogenesis process. Thus, we first determined the distribution of miR-144, pri-miR-144, and pre-miR-144 in macrophages. As shown in Figure 6A, pri-miR-144 and pre-miR-144 were predominantly localized in the nucleus, while miR-144 was predominantly localized in the cytoplasm (Figure 6A). By secondary structure analysis, we found that pri-miR-144 has a complementary sequence with the ultra-conserved region of TGFB3-AS1, which located at the terminal loop region site of the miR-144 primary transcripts (Figure 6B). We then detected changes in the levels of pri-miR-144, pre-miR-144, and miR-144 upon TGFB3-AS1 inhibition and overexpression. As shown in Figure 6C, significantly reduced mature miR-144 and pre-miR-144 and elevated pri-miR-144 were observed in macrophages transduced with lenti-TGFB3-AS1. By contrast, silencing TGFB3-AS1 in the macrophages increased the levels of mature miR-144 and pre-miR-144 and repressed the pri-miR-144 levels.
Figure 6.

TGFB3-AS1 inhibits maturation of miR-144 to upregulate Rap1a expression in macrophages
(A) Localization of mature miR-144, pri-miR-144, and pre-miR-144 in macrophages was detected by qRT-PCR. (B) The stem-loop sequence of pri-miR-144 is partially complemented with TGFB3-AS1. (C) The levels of mature miR-144, pre-miR-144, and pri-miR-144 in macrophages after overexpression of TGFB3-AS1 (lenti-TGFB3-AS1) or knockdown of TGFB3-AS1 (TGFB3-AS1-sc). (D) The expression of Drosha was determined by qRT-PCR and western blot in macrophages after transduction with Drosha-expressing adenoviruses (Ad-Drosha). (E and F) The levels of mature miR-144, pre-miR-144, and pri-miR-144 were analyzed by qRT-PCR in macrophages co-transduced with Ad-Drosha and lenti-TGFB3-AS1 or TGFB3-AS1-siRNA in the presence of 100 μmol/L Hcy treatment. (G) Anchoring of DGCR8 to pri-miR-144 was determined by RIP assay in macrophages after TGFB3-AS1 was overexpressed. (H) The expression of Rap1a in the macrophages co-transduced with lenti-TGFB3-AS1 and miR-144 mimic in the presence of Hcy. (I) The expression of Rap1a in the macrophages after co-transduction with TGFB3-AS1-siRNA and miR-144 inhibitor in the presence of Hcy. ∗p < 0.05, ∗∗p < 0.01 compared with lenti-GFP or control group, #p < 0.05, ##p < 0.01 compared with TGFB-AS1-sc or 100 μmol/L Hcy, ▵p < 0.05, ▵▵p < 0.01 compared with 100 μmol/L Hcy + lenti-TGFB3-AS1 or 100 μmol/L Hcy + TGFB3-AS1-siRNA.
In the nucleus, pri-miRNAs undergo Drosha-dependent cleavage into pre-miRNAs before being exported to the cytoplasm. Given the predicted interaction between TGFB3-AS1 and pri-miR-144 and the observed posttranscriptional downregulation of miR-144 by TGFB3-AS1 overexpression, we hypothesized that TGFB3-AS1 could directly hinder Drosha-mediated processing of pri-miR-144. To address this mechanism, we first analyzed whether TGFB3-AS1 could impact Drosha-mediated miR-144 maturation. As shown in Figures 6D and 6E, overexpression of Drosha decreased the levels of pri-miR-144, while the levels of pre-miR-144 and mature miR-144 increased in Hcy-treated macrophages, and these effects could be suppressed by TGFB3-AS1 overexpression. In addition, knockdown of TGFB3-AS1 could further enhance the effect of Drosha on miR-144 maturation (Figure 6F). As the predicted binding site for TGFB3-AS1 on pri-miR-144 is precisely along the region bound to DGCR8, a critical component of the canonical microprocessor complex for microRNA biogenesis, we next examined whether TGFB3-AS1 impairs the anchoring of DGCR8 to pri-miR-144. With that aim, we performed RIP assay with an antibody against DGCR8. As expected, overexpression of TGFB3-AS1 significantly decreased the levels of pri-miR-144 in the immunoprecipitated RNA with antibody against DGCR8 (Figure 6G). Apart from this, transfection of the miR-144 mimic reversed TGFB3-AS1 overexpression-upregulated Rap1a in macrophages, while the miR-144 inhibitor recovered TGFB3-AS1 knockdown-induced Rap1a suppression (Figures 6H and 6I). Taken together, the aforementioned results indicated that TGFB3-AS1 inhibited the maturation of miR-144 by binding to its primary transcript and impairing the processing of pri-miR-144 by Drosha and DGCR8.
Overexpression of TGFB3-AS1 aggravates the inflammation induced by Hcy in CBS+/− mice
To determine whether TGFB3-AS1 accelerates the inflammation induced by HHcy in vivo, CBS+/− mice fed with a high-methionine diet were given lenti-TGFB3-AS1 injection for 4 weeks to overexpress TGFB3-AS1. As shown in Figure 7A, the TGFB3-AS1 expression was significantly increased in the peripheral blood monocytes from CBS+/− mice after injection with lenti-TGFB3-AS1. ELISA assay revealed that the serum levels of proinflammatory cytokines IL-1β, IL-6, and TNF-α were remarkably increased in CBS+/− mice injected with lenti-TGFB3-AS1 (Figure 7B). In addition, flow cytometry analysis also showed that injection of lenti-TGFB3-AS1 significantly enhanced the percentage of peripheral blood monocytes, especially the CD11b+ Ly-6C+ inflammatory monocyte subset (Figures 7C and 7D). Next, we examined miR-144 expression in peripheral blood monocytes from CBS+/− mice with lenti-TGFB3-AS1 injection or negative control treatment. The results showed that overexpression of TGFB3-AS1 significantly decreased miR-144 expression in peripheral blood monocytes from CBS+/−mice (Figure 7E). Meanwhile, TGFB3-AS1 overexpression increased the expression levels of Rap1a and Wnt signaling pathway-related proteins (wnt3a, wnt5a, and β-catenin) (Figures 7F, 7G, and 7H). Taken together, the above observations suggested that overexpression of TGFB3-AS1 aggravated the inflammatory response induced by Hcy in CBS+/− mice.
Figure 7.

Injection of lenti-TGFB3-AS1 accelerates inflammation induced by Hcy in CBS+/− mice
(A) qRT-PCR analyzed the TGFB3-AS1 expression in peripheral blood monocytes of CBS+/− mice fed with the high-methionine diet after 4-week injection of lenti-TGFB3-AS1, lenti-GFP, or PBS. (B) The contents of proinflammatory cytokines IL-6, IL-1β, and TNF-α in serum of CBS+/− mice injected with lenti-TGFB3-AS1, lenti-GFP, or PBS were measured using an automatic biochemical analyzer. (C) The monocyte populations in BM, peripheral blood, and spleen isolated from CBS+/− mice after injection of lenti-TGFB3-AS1, lenti-GFP, or PBS were analyzed by flow cytometry. CD11b+ and Ly-6C+ monocyte subsets were further characterized using anti-CD11b and anti-Ly-6C antibodies. SSC, side scattered light; FSC, forward-scattered light. (D) The percentages of mononuclear cell (MNC) and CD11b+Ly-6C+ inflammatory monocyte subsets in MNC were analyzed. (E) The expression levels of miR-144 in peripheral monocytes from CBS+/− mice injected with lenti-TGFB3-AS1, lenti-GFP, or PBS were detected by qRT-PCR. (F–H) The expression levels of Rap1a and Wnt signaling pathway-related proteins (wnt3a, wnt5a, β-catenin) in peripheral monocytes from CBS+/− mice injected with lenti-TGFB3-AS1, lenti-GFP, or PBS were analyzed by qRT-PCR and western blot. ∗p < 0.05, ∗∗p < 0.01, compared with lenti-GFP group.
TGFB3-AS1 is a potential biomarker for evaluating inflammation in patients with HHcy
To explore the potential clinical significance of TGFB3-AS1, we examined whether the identified TFGB3-AS1/miR-144/Rap1a axis was clinically relevant to patients with HHcy. The basic clinical characteristic of the enrolled 50 people in each group (HHcy patients and healthy individuals) in this study are presented in Table 1. There was no significant difference in age, gender, the percentage of hyperlipidemia, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides (TG), and total cholesterol (CHO) between the healthy individuals and HHcy patients; while the body mass index (BMI) in HHcy patients was significantly higher than that in healthy individuals (Figure 8A; Table 1). Meanwhile, the levels of proinflammatory cytokines (IL-6, IL-1β, TNF-α, and CRP) were remarkably elevated in serum of patients with HHcy in comparison with healthy individuals (Figure 8B). Next, analysis of the expression of TGFB3-AS1, miR-144, and Rap1a revealed that TGFB3-AS1 and Rap1a were significantly increased in the peripheral blood monocytes from HHcy patients compared with those from healthy individuals, while miR-144 was significantly downregulated in the peripheral blood monocytes from HHcy patients (Figure 8C). Moreover, statistical analyses found that expression of TGFB3-AS1 was positively correlated with the levels of Hcy and proinflammatory cytokines in serum of healthy individuals and patients with HHcy (r = 0.4636, p < 0.01; r = 0.2276, p < 0.05; r = 0.2634, p < 0.01; r = 0.2348, p < 0.05) (Figures 8D and 8E). When analyzing correlations of TGFB3-AS1, miR-144, and Rap1a expression in all samples from healthy individuals and patients with HHcy, we observed a negative correlation between the expression of TGFB3-AS1 and miR-144 (r = −0.1986, p < 0.05), and a positive correlation between TGFB3-AS1 and Rap1a expression (r = 0.5449, p < 0.01) (Figure 8F). Accordingly, we also found that miR-144 expression was negatively correlated with the Rap1a expression (r = −0.208, p < 0.05) (Figure 8G). Of note, no correlation was observed between TGFB3-AS1 expression and serum lipids levels, including LDL-C, HDL-C, TG, and CHO (Figure 8H). Collectively, these results indicated that elevated TGFB3-AS1 expression in peripheral blood monocytes may be a potential and reliable biomarker for evaluating the degree of inflammation in patients with HHcy.
Table 1.
Comparison of clinical parameters between HHcy patients and the healthy group
| Characteristic | HHcy patients (n = 50) | Healthy group (n = 50) | p Value |
|---|---|---|---|
| Age, years (mean ± SD) | 66.39 ± 2.32 | 63.19 ± 1.73 | 0.2737 |
| Gender (male/female) | 29/21 | 20/30 | 0.072 |
| BMI (kg/m2, mean ± SD) | 9.85 ± 0.51 | 8.39 ± 0.83 | 0.0009 |
| Hyperlipidemia (n, %) | 21 (42%) | 13 (26%) | 0.091 |
| LDL-C (mmol/L) | 2.62 ± 0.13 | 2.57 ± 0.13 | 0.7798 |
| HDL-C (mmol/L) | 1.09 ± 0.05 | 1.05 ± 0.04 | 0.5804 |
| TG (mmol/L) | 1.70 ± 0.17 | 1.46 ± 0.14 | 0.2652 |
| CHO (mmol/L) | 4.78 ± 0.16 | 4.67 ± 0.17 | 0.6511 |
HHcy, hyperhomocysteinemia; SD, standard deviation; BMI, body mass index; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triglyceride; CHO, cholesterol.
Figure 8.

Clinical significance of TFGB3-AS1 in patients with HHcy
(A and B) The serum levels of Hcy, proinflammatory cytokines TNF-α, IL-1β, IL-6, and CRP in healthy individuals and patients with HHcy. n = 50 per group. (C) The expression levels of TGFB3-AS1, miR-144, and Rap1a in peripheral blood monocytes of healthy individuals and patients with HHcy were determined by qRT-PCR. (D and E) Correlations between TFGB3-AS1 expression and serum levels of Hcy or proinflammatory cytokines (IL-6, IL-1β, TNF-α, and CRP) in all enrolled subjects were evaluated by Pearson correlation analysis. (F) The correlation between TFGB3-AS1 expression and expression of miR-144 or Rap1a in peripheral monocytes of all enrolled subjects. (G) Correlation analysis of miR-144 and Rap1a expression in peripheral monocytes of all enrolled subjects. (H) Correlations between TFGB3-AS1 expression and the serum lipid levels (LDL-C, HDL-C, TG, and CHO) in all enrolled subjects. Pearson correlation coefficients (r) and p values are presented in the graphs. ∗p < 0.05, ∗∗p < 0.01, compared with healthy group.
Discussion
Abundant clinical and epidemiological research has revealed that HHcy is an independent risk factor for cardiovascular disease.24 The studies from our laboratory and others have demonstrated that Hcy can accelerate vascular inflammation by regulating monocyte-derived inflammatory gene expression. Recent studies indicated that Hcy induces the differentiation of an inflammatory Ly6Chigh monocyte subset and increases the accumulation of inflammatory monocytes/macrophages in the formation of atherosclerotic lesions, which finally results in vessel wall inflammation.25 Thus, our results support the notion that Hcy plays a vital role in the pathogenesis of inflammation by promoting the aggregation of the CD11b+ Ly-6C+ inflammatory monocyte subset. However, previous studies are mainly focused on the phenomenon assessment, lacking integrative analysis and mechanistic assessment. Therefore, it is necessary to disclose the precise molecular mechanisms and the biological function of target genes underlying Hcy-induced proinflammatory effects on macrophages.
LncRNAs are one type of widely studied biomolecules, which have been emerging as critical regulators in various tissues and biological processes.26 Thus, they can be used as molecular targets for diagnosis or therapy of disease. However, little is known about their role in Hcy-induced inflammation of macrophages. Recently, Li et al. reported the changed expression profiles of lncRNAs in Hcy-induced vascular endothelial injury.27 Here, macrophages with Hcy treatment were used to identify dysregulated lncRNAs, which is helpful for uncovering the mechanism of HHcy-induced macrophage inflammation. Notably, Hcy promoted the expression of TGFB3-AS1 in macrophages both in vitro and in vivo. Several lncRNAs have been shown to be involved in inflammation- or inflammatory response-related biological processes. Yan et al. demonstrated that lncRNA HIX003209 was dysregulated in macrophages, and promoted the proliferation and inflammatory cytokine secretion of macrophages through the IκBα/NF-κB pathway in rheumatoid arthritis.28 Others reported that lncRNA PTPRE-AS1 modulated M2 macrophage activation and inflammatory diseases by epigenetically promoting PTPRE.29 As a sulfur-containing amino acid, a connection between lncRNAs and inflammatory can be established through Hcy, because it can induce oxidative stress and mediate inflammation in many cell types.30,31 In addition, evidence also indicated that alteration of lncRNA-H19 promoter methylation through TNF-α/NF-κB pathway was associated with Hcy-induced oxidative stress.32, 33, 34 A previous study demonstrated that LOC646329-variant D exerted its suppression effect on colorectal cancer progression via regulating Wnt and TGFB signaling pathways, which characterized LOC646329 as a novel potential therapy target.35 In this study, we demonstrated that TGFB3-AS1 was increased in peripheral blood monocytes of patients with HHcy, and had a positive correlation with proinflammatory cytokines in serum of patients with HHcy. These findings supported TGFB3-AS1 is a powerful therapeutic and diagnostic target in inflammatory diseases induced by Hcy.
As a member of the RAS oncogene family, aberrant expression of Rap1a is vital for tumor cell proliferation, apoptosis, migration, invasion, and metastasis in several cancer types.36 In addition, it was reported that lncRNA NR-026690 can regulate inflammation by targeting Rap1a in COPD patients.37 In this study, we demonstrated that Rap1a interacted with TGFB3-AS1 in macrophages treated with Hcy. Furthermore, we confirmed the molecular mechanisms that interaction of Rap1a with TGFB3-AS1 could decrease the ubiquitination levels of Rap1a by preventing its proteasome-mediated degradation. Rap1a was reported to mediate the hepatitis inflammation induced by HBV through the PI3K/p38/NF-κB signaling pathway.38 In a heart failure model, pathological concentrations of Hcy were found to stimulate NF-κB activation, and thereby induce proinflammatory cytokines in THP-1-derived macrophages.39 Here, we found that knockdown of TGFB3-AS1 reduced IL-1β, IL-6, and TNF-α levels in macrophages under Hcy treatment via the wnt/β-catenin pathway. These results confirmed that TGFB3-AS1 promoted inflammatory responses of macrophages induced by Hcy through the Rap1a/wnt signaling pathway, which is consistent with previous studies that reported that Rap1a is required for activation of the nuclear transport of β-catenin.
In addition, we demonstrated that aberrant miR-144 expression involved in the inflammatory response of macrophages treated with Hcy and peripheral blood monocytes from CBS+/− mice fed a high-methionine diet. As a sulfur-containing endogenous amino acid, Hcy produced in the methionine cycle of protein metabolism, participated in maintaining the redox balance, monocyte chemoattractant protein-1 secretion, and the production of multiple inflammatory cytokines in monocytes/macrophages.40,41 It was reported that miR-144-relieved activation of the inflammatory ROCK1/MLC pathway in vascular ECs as a potential therapeutic strategy to counter inflammatory lung injury.42 Generally, miRNAs are vital small endogenous ncRNAs that inhibit gene expression by binding to the 3′ UTR of genes transcripts. We found that Rap1a could be negatively regulated by miR-144. In this study, overexpression of miR-144 observably reduced the secretion of proinflammatory factors. Furthermore, miR-144 expression was significantly suppressed in the peripheral blood monocytes of patients with HHcy, suggesting that miR-144 might be a promising diagnostic and therapeutic target for Hcy-induced inflammation.
We next explored the mechanism of how TGFB3-AS1 and miR-144 regulate expression of Rap1a. By overexpression or knockdown of TGFB3-AS1, we found that TGFB3-AS1 negatively controlled the expression of miR-144. In addition, the intronic ultra-conserved regions of lncRNAs are generally related to the regulation of transcription and DNA binding, prompting their potential role as regulators of miRNAs.43 Recent studies have confirmed lncRNA-mediated regulatory networks for miRNA processing, as ceRNA can function as miRNA sponges and decrease the activity of target miRNAs with changing their biogenesis.44 For instance, lower-stem strand invasion by lncRNA uc.283 + A impairs microprocessor recognition and efficient pri-miR-195 cropping.45 The processing requirement of pre-miRNA during miRNA maturation is the direct interaction of pri-miRNA with the Drosha-DGCR8 microprocessor complex46. The pri-miRNA/pre-miRNA hairpin structure consists of mismatches, internal loops, and bulges. The terminal loop of these hairpins demonstrates a variable structure, which may be important for the rate of miRNA processing. In this study, in situ hybridization and nuclear/cytoplasmic fractionation analysis demonstrated that TGFB3-AS1 was mainly located in the nucleus of macrophages with Hcy treatment. The nuclear localization of TGFB3-AS1 implies the possibility of binding mRNA to modulate gene transcription. As expected, we found that TGFB3-AS1 could suppress processing of pri-miR-144 by Drosha, and RIP assay further revealed that TGFB3-AS1 could interfere with DGCR8 anchoring to the miR-144 primary transcript (pri-miR-144). Together with the secondary structure analysis between TGFB3-AS1 and pri-miR-144, a complementary binding sequence was shown between them. Our findings demonstrated that TGFB3-AS1 regulates pri- to- pre-miRNA cleavage of miR-144 by binding to pri-miR-144 and interfering in the function of Drosha and DGCR8, leading to decreased levels of mature miR-144 and thereby increased Rap1a expression in macrophages under Hcy treatment.
In summary, our study reveals a novel mechanism by which TGFB3-AS1 promotes inflammatory response by suppressing the maturation of miR-144, thus upregulating expression of its target gene Rap1a, which activates the wnt/β-catenin signaling pathway and participates in HHcy-induced proinflammatory response in monocyte-derived macrophages (see the Graphical abstract). These findings propose that inhibition of TGFB3-AS1 might be a potential therapeutic strategy for inflammation induced by Hcy.
Materials and methods
This research was performed at the Ningxia Medical University and General Hospital of Ningxia Medical University, and conformed to the principles of the Declaration of Helsinki.
Animal models
Cystathionine beta-synthase heterozygous knockout (CBS+/−, C57BL/6J background) mice and C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, USA), and the breeding colony is established in Ningxia Medical University. CBS+/− mice were crossbred to C57BL/6J mice for at least eight generations to generate heterozygous (CBS+/−) and wild-type (CBS+/+) littermates. Mice genotypes for the targeted CBS allele were determined by PCR of DNA obtained from tail biopsies with a specific set of primers.47,48 CBS+/+ mice and CBS+/− mice were housed individually under a temperature-controlled environment with food and water ad libitum. The 6-week-old male CBS+/+ mice and CBS+/− mice were fed with standard mouse chow (regular diet: 20% protein, 4.5% fat, 55.5% carbohydrate) (Xiao Shu You Tai [Beijing] Biotechnology) or a high-methionine diet (methionine diet: 2.0% methionine, Xiao Shu You Tai [Beijing] Biotechnology) for an additional 12 weeks. The lentivirus (lenti-TGFB-AS1 and lenti-GFP, virus titer: 2 × 107 TU/mL) or PBS injections were started at week 8 after initiation of CBS+/− mice being fed the high-methionine diet. Animal handling protocols were conducted following the National Institutes of Health (NIH) guidelines and approved by the Animal Ethics Committee for General Hospital of Ningxia Medical University (no. 2016-052). Mice were euthanasia with an overdose of 1% pentobarbital sodium (100 mg/kg, i.p.) at the endpoint of the experiment.
Patients and clinical samples
Data from the health examination database in the General Hospital of Ningxia Medical University between January 2016 and December 2019 were extracted. Individuals with missing anthropometric or biochemical measurements were excluded, leaving 100 patients retained in this study. According to the Guidelines for the Prevention of Cardiovascular Diseases in China, these patients were categorized into the control group (Hcy < 15 μmol/L) and the hyperhomocysteinemia group (Hcy ≥ 15 μmol/L).49 Information including gender, age, height, and weight were obtained by clinical data. BMI was calculated by using the formula as weight in kilograms divided by the height in meters squared. Blood samples were collected for measuring IL-1β, IL-6, TNF-α, CRP, Hcy, TG, CHO, LDL-C, and HDL-C. This research was approved by the Medical Ethics Committee of the General Hospital of Ningxia Medical University. Written informed consent was obtained from all participants enrolled in this study.
Cell culture and PMA-induced differentiation of monocytes
The THP-1 cell line (FDCC, ShangHai, China) was grown in RPMI-1640 (Gibco, USA) supplemented with 10% FBS (Gibco, USA). THP-1 monocytes were treated with 100 nmol/L phorbol 12-myristate 13-acetate (PMA) (Promega, USA) for 24 h to induce differentiation into macrophages before stimulation with 50, 100, 200, and 500 μmol/L Hcy (L-homocysteine, Sigma-Aldrich, USA).
Cell transfection
Replication recombinant lentivirus including the intact coding sequence of TGFB3-AS1 (lenti-TGFB3-AS1) was provided by Hanbio Biotechnology (Shanghai, China). Lentiviruses expressing GFP were negative control (lenti-GFP). In addition, three siRNAs targeting human TGFB3-AS1 were designed and inserted into lentivirus vector to generate lentivirus constructs (TGFB3-AS1-siRNA); lentiviruses expressing nontargeting siRNA (TGFB3-AS1-sc) were the negative control. To establish stable cell lines, cells were transduced with lenti-TGFB3-AS1 or TGFB3-AS1-siRNA. After 72 h of transduction, cells were selected with 2.5 μg/mL puromycin.
Cells transfected with adenoviral constructs (Ad-GFP, Ad-Rap1a, si-NC, and si-Rap1a) were purchased from Hanbio Biotechnology. Cells were incubated in RPMI-1640 and 10% FBS and transduced with the adenovirus for 6 h.
miR-144 mimics, miR-144 inhibitors, and controls were obtained from RiboBio (China). To increase or reduce expression of MiR-144, the cells were cultured with serum-free medium and transfected with miRNA mimics and inhibitors using Lipofectamine 3000 (Invitrogen, USA) for 8 h. The cells were incubated for 24 h for the following experiments.
Determination of Hcy levels in blood samples
Blood samples from each mouse were collected and centrifuged at 3,000 rpm immediately. The serum Hcy levels were determined using an Automatic Biochemistry analyzer AU5821 (Beckman Coulter, Japan).50
Flow cytometry analysis
Cells in peripheral blood, spleen, and BM were isolated from mice. After combination staining with fluorochrome-conjugated antibodies against CD11b (0.25 μg/100 μL, BD Biosciences, USA) and Ly-6C (0.25 μg/100 μL, BD Biosciences), cells were examined by FACS (BD Biosciences) and analyzed using FlowJo 7.6 software (TreeStar, Ashland, OR). Live cells were first appraised after eliminating red blood cells. The inflammatory monocyte as further defined as CD11b+Ly-6C+ MNC.
ELISA analysis of IL-1β, IL-6, and TNF-α
On the basis of the manufacturer's instructions, human inflammatory factors ELISA kits (R&D Systems, USA) were used to analyze the levels of IL-1β, IL-6, and TNF-α in samples.
LncRNA microarray assay
Total RNA was isolated from macrophages using TRIzol reagent (Invitrogen, USA), and cDNA libraries were established. All lncRNA microarray experiments, including labeling with fluorescent dye, microarray scan, and data analyses were provided by Biomarker Technologies (China). Differential expression analysis of lncRNAs in macrophages (n = 3) treated with or without Hcy were performed using the R package “limma” (version 3.42.0). Those lncRNAs with |log2 (FC)| > 1 and false discovery rate adjusted p value < 0.05 were considered as differentially expressed lncRNAs. Finally, differential expression data of 82 lncRNAs were obtained for KEGG pathway analyses using the cluster ProFiler package in the R platform.51
Quantitative real-time PCR
Total RNAs from cultured cells from mice were isolated using the RNA Isolation Kit (QIAGEN, Germany). Reverse transcription and real-time PCR (SYBER Green Dye, Takara, Japan) for mRNA, miRNA, and lncRNA were performed following the manufacturer's instructions. The following conditions were used in PCR: 95°C for 5 min (initial activation step), followed by 45 cycles, 5 s at 95°C (denaturing temperature), 15 s at 58°C (annealing temperature), and 15 s at 72°C (extension temperature and fluorescence data collection). Specific primers for TGFB3-AS1, Rap1a, miR-144, wnt3a, Wnt5a, β-catenin U6, and GAPDH used in the study are listed in Table S1.
FISH
Subcellular localization of TGFB3-AS1 in macrophages was analyzed using FISH kit (RiboBio, China), which includes an locked nucleic acid oligodeoxynucleotide probe labeled with Alexa Fluor 594. In brief, macrophages were cultured on glass coverslips, washed with PBS, and fixed in 4% paraformaldehyde at room temperature for 30 min. Then, the cells were prehybridized at 37°C for 30 min. Oligodeoxynucleotide probes of TGFB3-AS1, U6, and 18S rRNA were combined with the hybridization buffer in a dark and humid environment at 37°C overnight. The nucleus was stained with DAPI. Images were obtained using a confocal laser-scanning microscope (Zeiss, Germany).
Immunoblotting analysis
Cells were washed with ice-cold PBS and lysed in Nonidet P 40 (NP-40, Solarbio, China) lysis buffer with 10 mg/mL phenylmethanesulfonyl fluoride (Solarbio, China). The concentration of protein was determined using the BCA assay (KEYGEN, China) according to the manufacturer's instructions. Protein lysates were loaded and separated by 8% SDS-PAGE, and transferred to 0.22 μm polyvinylidene fluoride membranes (Millipore, Germany) membranes. After being blocked with 5% non-fat milk for 2 h at room temperature, the membranes were incubated with the appropriate primary antibodies, including antibodies against wnt3a (ab81614, 1:1,000, Abcam, UK), Wnt5a (ab179824, 1:1,000, Abcam), β-catenin (ab32572, 1:1,000, Abcam), Vim (ab92547, 1:1,000, Abcam), Dorsha (ab183732, 1:10,000, Abcam), Hist1h1a (CSB-PA010429OA91nbutHU, 1:1,000, CUSABIO, CN), Rap1a (sc-373968, 1:1,000, Santa Cruz Biotechnology, USA), and β-actin (sc-47778, 1:5000, Santa Cruz Biotechnology) overnight at 4°C. Then, the membranes were later incubated with secondary antibody: horseradish peroxidase (HRP)-labeled rabbit antibodies to IgG (ab97051, 1:5,000, Abcam, USA) or HRP-labeled mouse antibodies to IgG (ab6728, 1:5,000, Abcam, USA) for 2 h at room temperature. The membranes were developed with chemiluminescent substrates from ECL (Millipore, Germany). Densitometry was done on immunoblots using Bio-Rad (Hercules, CA) equipped with Image Lab5.0.
RNA pull-down assay
Cells were harvested, lysed, and sonicated. Biotinylated TGFB3-AS1 was synthesized by RiboBio (China). In brief, 50 pmol biotinylated TGFB3-AS1 was incubated with streptavidin beads (Invitrogen, USA) at 4°C overnight. Then, the cell lysates were incubated with streptavidin agarose beads (Invitrogen) for 1 h. After washing with wash buffer, RNA-associated proteins were stained by silver, and eluted proteins were detected by western blot.
RIP assay
Magnetic beads (Thermo Fisher Scientific, USA) were mixed with the anti-Rap1a antibody, anti-DGCR8 (ab191875, Abcam, UK) or normal rabbit IgG for 30 min and the cell lysates was immunoprecipitated with beads for 6 h at 4°C. Beads were washed five times and RNAs were isolated from protein-antibody-agarose complexes. RT-PCR was performed following the manufacturer's protocol to detect the enrichment of RNA in the immunoprecipitated samples.
Ubiquitination assay
In brief, cells were treated with or without 5 μmol/L MG132 before they were collected. The cells were lysed using radio-immunoprecipitation assay buffer and incubated with Dynabeads Protein G (Thermo Fisher Scientific, USA), and subjected to immunoprecipitation with anti-Rap1a antibody at 4°C for 1 h. The precipitated proteins were then released from the beads by boiling for 10 min. Then, the ubiquitination of Rap1a was detected by western blotting with anti-ubiquitin antibody (Cell Signaling Technology; no. 3936, 1:1,000).
Dual-luciferase assays
The luciferase reporter constructs containing wild-type or mutant 3′ UTR of Rap1a (Rap1a-3′ UTR-WT and Rap1a 3′ UTR-mut, respectively) were provided by GenePharma (China). Rap1a-3′ UTR-WT or Rap1a 3′ UTR-mut, and miR-144 mimic or miR-neg, were co-transfected into HEK293T cells using Lipofectamine 3000 reagent (Invitrogen). Cell lysates were collected after 48 h of transfection, and luciferase activity was analyzed using the Dual-Luciferase Assay Kit (Promega, USA).
Statistical analysis
GraphPad prism version 8.0 (GraphPad, USA) and SPSS 22.0 (SPSS, USA) software were used for statistical analysis. All quantitative data are presented as the mean ± standard deviation (SD). Differences in gene expression levels were analyzed using the Student's t test between two groups. Multiple groups were compared with one-way ANOVA. The differences of categorical variables between the control (healthy) and the HHcy groups were analyzed through the chi-square test. The correlation between the Hcy levels and inflammatory cytokine or lipid levels was assessed by Pearson correlation analysis. p value < 0.05 was considered as statistically significant.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (grant nos. 81870225, 81860044, 81870332, 81760090, and 81760095), a major research project from the Ningxia Science and Technique of Ningxia Hui autonomous region (grant no. 2018BEG02004), Basic scientific research operating expenses from the public welfare research institutes at the central level of the Chinese Academy of Medical Sciences (grant no. 2019PT330002).
Author contributions
Y.J., Hui Zhang, Huiping Zhang, and Y.H. designed the project and wrote the manuscript. Hui Zhang, A.Y., L. Xie, N.D., L. Xu, and Y.W. performed most of the experiments. Y.W., Y.Y., and Y.B. collected and analyzed the clinical data. All of the authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.031.
Contributor Information
Huiping Zhang, Email: zhp19760820@163.com.
Yideng Jiang, Email: jydeng@nxmu.edu.cn.
Supplemental information
References
- 1.Gao F., Zhang J., Ni T., Lin N., Lin H., Luo H., et al. Herpud1 deficiency could reduce amyloid-β40 expression and thereby suppress homocysteine-induced atherosclerosis by blocking the JNK/AP1 pathway. J. Physiol. Biochem. 2020;76:383–391. doi: 10.1007/s13105-020-00741-5. [DOI] [PubMed] [Google Scholar]
- 2.Lazzerini P.E., Capecchi P.L., Selvi E., Lorenzini S., Bisogno S., Galeazzi M., et al. Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmun. Rev. 2007;6:503–509. doi: 10.1016/j.autrev.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Hegedus J., Kahnt A.S., Ebert R., Heijink M., Ioan-Facsinay A. Toll-like receptor signaling induces a temporal switch towards a resolving lipid profile in monocyte-derived macrophages. Biochim. Biophys. Acta. 2020;1865:158740. doi: 10.1016/j.bbalip.2020.158740. [DOI] [PubMed] [Google Scholar]
- 4.Fasolo F., Di Gregoli K., Maegdefessel L., Johnson J.L. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc. Res. 2019;115:1732–1756. doi: 10.1093/cvr/cvz203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulte C., Barwari T., Joshi A., Zeller T., Mayr M. Noncoding RNAs versus protein biomarkers in cardiovascular disease. Trends Mol. Med. 2020;26:583–596. doi: 10.1016/j.molmed.2020.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Xie Q., Li F., Shen K., Luo C., Song G. LOXL1-AS1/miR-515-5p/STAT3 positive feedback loop facilitates cell proliferation and migration in atherosclerosis. J. Cardiovasc. Pharmacol. 2020;76:151–158. doi: 10.1097/FJC.0000000000000853. [DOI] [PubMed] [Google Scholar]
- 7.Ou M., Li X., Zhao S., Cui S., Tu J. Long non-coding RNA CDKN2B-AS1 contributes to atherosclerotic plaque formation by forming RNA-DNA triplex in the CDKN2B promoter. EBioMedicine. 2020;55:102694. doi: 10.1016/j.ebiom.2020.102694. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Uchida S., Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015;116:737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 9.Hu G., Gong A.Y., Wang Y., Ma S., Chen X., Chen J., et al. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J. Immunol. 2016;196:2799–2808. doi: 10.4049/jimmunol.1502146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang P., Cao L., Zhou R., Yang X., Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019;10:1495. doi: 10.1038/s41467-019-09482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao F.,Z.J., Ni T., Lin N., Lin H., Luo H., Guo H., Chi J. Impaired lnc-IL7R modulatory mechanism of Toll-like receptors is associated with an exacerbator phenotype of chronic obstructive pulmonary disease. FASEB J. 2020;34:383–391. doi: 10.1096/fj.202000632R. [DOI] [PubMed] [Google Scholar]
- 12.Atianand M., Hu W., Satpathy A., Shen Y., Ricci E., Alvarez-Dominguez J., et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell. 2016;165:1672–1685. doi: 10.1016/j.cell.2016.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai Y., Wang S., Chang S., Ren D., Shali S., Li C. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J. Mol. Cell Cardiol. 2020;142:65–79. doi: 10.1016/j.yjmcc.2020.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Canfrán-Duque A., Rotllan N., Zhang X., Fernández-Fuertes M., Ramírez-Hidalgo C., Araldi E., et al. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017;9:1244–1262. doi: 10.15252/emmm.201607492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan M., Liu Q., Jiang Y., Wang B., Xie Y. Long non-coding RNA LNC_000898 alleviates cardiomyocyte apoptosis and promotes cardiac repair after myocardial infarction via modulating miR-375/PDK1 axis. J. Cardiovasc. Pharmacol. 2020;76:77–85. doi: 10.1097/FJC.0000000000000845. [DOI] [PubMed] [Google Scholar]
- 16.Hua Z., Ma K., Liu S., Yue Y., Cao H., Li Z. LncRNA ZEB1-AS1 facilitates ox-LDL-induced damage of HCtAEC cells and the oxidative stress and inflammatory events of THP-1 cells via miR-942/HMGB1 signaling. Life Sci. 2020;247:117334. doi: 10.1016/j.lfs.2020.117334. [DOI] [PubMed] [Google Scholar]
- 17.Yang X., Tao L., Zhu J., Zhang S. Long noncoding RNA FTX reduces hypertrophy of neonatal mouse cardiac myocytes and regulates the PTEN/PI3K/akt signaling pathway by sponging microRNA-22. Med. Sci. Monit. 2019;25:9609–9617. doi: 10.12659/MSM.919654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y., Jeon K., Lee J.T., Kim S., Kim V.N. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pong S.K., Gullerova M. Noncanonical functions of microRNA pathway enzymes—Drosha, DGCR8, Dicer and Ago proteins. FEBS Lett. 2018;592:2973–2986. doi: 10.1002/1873-3468.13196. [DOI] [PubMed] [Google Scholar]
- 20.Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 21.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 22.Davis F.M., Gallagher K.A. Epigenetic mechanisms in monocytes/macrophages regulate inflammation in cardiometabolic and vascular disease. Arteriosclerosis, Thromb. Vasc. Biol. 2019;39:623–634. doi: 10.1161/ATVBAHA.118.312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francesca A. Long non-coding RNA and Polycomb: an intricate partnership in cancer biology. Front. Bioence. 2018;23:2106–2132. doi: 10.2741/4693. [DOI] [PubMed] [Google Scholar]
- 24.Rong D., Liu J., Jia X., Al-Nafisee D., Jia S., Sun G., et al. Hyperhomocysteinaemia is an independent risk factor for peripheral arterial disease in a Chinese Han population. Atherosclerosis. 2017;263:205–210. doi: 10.1016/j.atherosclerosis.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D., Pu F., Jiang X., Nelson J., Wang H. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ. Res. 2012;111:37–49. doi: 10.1161/CIRCRESAHA.112.269472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarroux J., Morillon A., Pinskaya M. History, discovery, and classification of lncRNAs. Adv. Exp. Med. Biol. 2017;1008:1–46. doi: 10.1007/978-981-10-5203-3_1. [DOI] [PubMed] [Google Scholar]
- 27.Jian L., Ming L., Xie N., Wang H., Wang J. Bioinformatics-based analysis of the involvement of AC005550.3, RP11-415D17.3, and RP1-140K8.5 in homocysteine-induced vascular endothelial injury. Am. J. Transl. Res. 2018;10:2126–2136. [PMC free article] [PubMed] [Google Scholar]
- 28.Yan S., Wang P., Wang J., Yang J., Lu H., Jin C., et al. Long non-coding RNA HIX003209 promotes inflammation by sponging miR-6089 via TLR4/NF-κB signaling pathway in rheumatoid arthritis. Front Immunol. 2019;10:2218. doi: 10.3389/fimmu.2019.02218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han X., Huang S., Xue P., Fu J., Zhou Y. LncRNA PTPRE-AS1 modulates M2 macrophage activation and inflammatory diseases by epigenetic promotion of PTPRE. Sci. Adv. 2019;5:eaax9230. doi: 10.1126/sciadv.aax9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S., Zhang E., Senapati P., Amaram V., Reddy M.A., Stapleton K., et al. A novel angiotensin II-induced long noncoding RNA giver regulates oxidative stress, inflammation, and proliferation in vascular smooth muscle cells. Circ. Res. 2018;123:1298–1312. doi: 10.1161/CIRCRESAHA.118.313207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng M., Zheng Y., Gao M., Ma H., Zhang X., Li Y., et al. Expression and clinical value of lncRNA MALAT1 and lncRNA ANRIL in glaucoma patients. Exp. Ther. Med. 2020;19:1329–1335. doi: 10.3892/etm.2019.8345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L., Xie J., Zhang M., Wang S. Homocysteine harasses the imprinting expression of IGF2 and H19 by demethylation of differentially methylated region between IGF2/H19 genes. Acta Biochim. Biophys. Sinica. 2009;41:464–471. doi: 10.1093/abbs/gmp033. [DOI] [PubMed] [Google Scholar]
- 33.Bing Y., Jennifer W., Nathan D., Yao T., Wuerzberger-Davis S.M., Moon-Hee L., et al. A novel pathway links oxidative stress to loss of insulin growth factor-2 (IGF2) imprinting through NF-κB activation. Plos One. 2014;9:e88052. doi: 10.1371/journal.pone.0088052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J., Yang L., Zhong T., Mueller M., Men Y., Zhang N., et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015;6:10221. doi: 10.1038/ncomms10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javanmard A.R., Dokanehiifard S., Bohlooli M., Soltani B.M. LOC646329 long non-coding RNA sponges miR-29b-1 and regulates TGFβ signaling in colorectal cancer. J. Cancer Res. Clin. Oncol. 2020;146:1205–1215. doi: 10.1007/s00432-020-03145-6. [DOI] [PubMed] [Google Scholar]
- 36.Simanshu D.K., Nissley D.V., McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qi X., Chen H., Fu B., Huang Z., Mou Y., Liu J., et al. LncRNAs NR-026690 and ENST00000447867 are upregulated in CD4(+) T cells in patients with acute exacerbation of COPD. Int. J. Chron. Obstruct Pulmon Dis. 2019;14:699–711. doi: 10.2147/COPD.S191815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu A., Chen H., Xu C., Zhou J., Chen S., Shi Y., et al. miR-203a is involved in HBx-induced inflammation by targeting Rap1a. Exp. Cell Res. 2016;349:191–197. doi: 10.1016/j.yexcr.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Lv J., Chen J., Wang M., Yan F. Klotho alleviates indoxyl sulfate-induced heart failure and kidney damage by promoting M2 macrophage polarization. Aging (Albany NY) 2020;12:9139–9150. doi: 10.18632/aging.103183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Q., Liu L.Z., Yin Y., He J., Li Q., Qian X., et al. Regulatory circuit of PKM2/NF-κB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene. 2015;34:5482–5493. doi: 10.1038/onc.2015.6. [DOI] [PubMed] [Google Scholar]
- 41.Karimian M.S., Pirro M., Majeed M., Sahebkar A. Curcumin as a natural regulator of monocyte chemoattractant protein-1. Cytokine Growth Factor Rev. 2017;33:55–63. doi: 10.1016/j.cytogfr.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Siddiqui M.R., Akhtar S., Shahid M., Tauseef M., McDonough K., Shanley T.P. miR-144-mediated inhibition of ROCK1 protects against LPS-induced lung endothelial hyperpermeability. Am. J. Respir. Cell. Mol. Biol. 2019;61:257–265. doi: 10.1165/rcmb.2018-0235OC. [DOI] [PubMed] [Google Scholar]
- 43.Li L.J., Leng R.X., Fan Y.G., Pan H.F., Ye D.Q. Translation of noncoding RNAs: focus on lncRNAs, pri-miRNAs, and circRNAs. Exp. Cell Res. 2017;361:1–8. doi: 10.1016/j.yexcr.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Paraskevopoulou M.D., Hatzigeorgiou A.G. Analyzing MiRNA-LncRNA interactions. Methods Mol. Biol. (Clifton, NJ) 2016;1402:271–286. doi: 10.1007/978-1-4939-3378-5_21. [DOI] [PubMed] [Google Scholar]
- 45.Liz J., Portela A., Soler M., Gómez A., Ling H., Michlewski G., et al. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol. Cell. 2014;55:138–147. doi: 10.1016/j.molcel.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Han J., Lee Y., Yeom K.H., Kim Y.K., Jin H., Kim V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tyagi N., Qipshidze N., Munjal C., Vacek J.C., Metreveli N., Givvimani S., et al. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 2012;47:128–138. doi: 10.1007/s12031-011-9695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe M., Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M.R., et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu, Dayi The Chinese Society of Cardiology. Jpn. Circ. J. 2012;76:265–267. [Google Scholar]
- 50.Liu Z., Luo H., Zhang L., Huang Y., Liu B., Ma K., et al. Hyperhomocysteinemia exaggerates adventitial inflammation and angiotensin II-induced abdominal aortic aneurysm in mice. Circ. Res. 2012;111:1261–1273. doi: 10.1161/CIRCRESAHA.112.270520. [DOI] [PubMed] [Google Scholar]
- 51.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics-a J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.