

Key Points
Question
What are the characteristics of amyloid-related imaging abnormalities (ARIA) during aducanumab treatment in individuals with early Alzheimer disease?
Findings
In an integrated safety data set of 2 phase 3 clinical trials (EMERGE and ENGAGE) including 3285 participants, 425 patients (41.3%) in the combined 10 mg/kg aducanumab group (n = 1029) experienced ARIA; ARIA-edema occurred in 362 patients (35.2%), and 94 of these patients (26.0%) experienced associated symptoms (eg, headache, confusion, dizziness, and nausea). ARIA-microhemorrhage and ARIA–superficial siderosis occurred in 197 patients (19.1%) and 151 patients (14.7%), respectively.
Meaning
Amyloid-related imaging abnormalities occurred in approximately 40% of participants in the phase 3 studies of aducanumab, and approximately one-quarter of these patients experienced symptoms.
This secondary analysis of 2 phase 3 randomized clinical trials describes the radiographic and clinical characteristics of amyloid-related imaging abnormalities that occurred in the EMERGE and ENGAGE trials.
Abstract
Importance
The EMERGE and ENGAGE phase 3 randomized clinical trials of aducanumab provide a robust data set to characterize amyloid-related imaging abnormalities (ARIA) that occur with treatment with aducanumab, an amyloid-β (Aβ)–targeting monoclonal antibody, in patients with mild cognitive impairment due to Alzheimer disease or mild Alzheimer disease dementia.
Objective
To describe the radiographic and clinical characteristics of ARIA that occurred in EMERGE and ENGAGE.
Design, Setting, and Participants
Secondary analysis of data from the EMERGE and ENGAGE trials, which were 2 double-blind, placebo-controlled, parallel-group, phase 3 randomized clinical trials that compared low-dose and high-dose aducanumab treatment with placebo among participants at 348 sites across 20 countries. Enrollment occurred from August 2015 to July 2018, and the trials were terminated early (March 21, 2019) based on a futility analysis. The combined studies consisted of a total of 3285 participants with Alzheimer disease who received 1 or more doses of placebo (n = 1087) or aducanumab (n = 2198; 2752 total person-years of exposure) during the placebo-controlled period. Primary data analyses were performed from November 2019 to July 2020, with additional analyses performed through July 2021.
Interventions
Participants were randomly assigned 1:1:1 to high-dose or low-dose intravenous aducanumab or placebo once every 4 weeks. Dose titration was used as a risk-minimization strategy.
Main Outcomes and Measures
Brain magnetic resonance imaging was used to monitor patients for ARIA; associated symptoms were reported as adverse events.
Results
Of 3285 included participants, the mean (SD) age was 70.4 (7.45) years; 1706 participants (52%) were female, 2661 (81%) had mild cognitive impairment due to Alzheimer disease, and 1777 (54%) used symptomatic medications for Alzheimer disease. A total of 764 participants from EMERGE and 709 participants from ENGAGE were categorized as withdrawn before study completion, most often owing to early termination of the study by the sponsor. Unless otherwise specified, all results represent analyses from the 10-mg/kg group. During the placebo-controlled period, 425 of 1029 patients (41.3%) experienced ARIA, with serious cases occurring in 14 patients (1.4%). ARIA-edema (ARIA-E) was the most common adverse event (362 of 1029 [35.2%]), and 263 initial events (72.7%) occurred within the first 8 doses of aducanumab; 94 participants (26.0%) with an event exhibited symptoms. Common associated symptoms among 103 patients with symptomatic ARIA-E or ARIA-H were headache (48 [46.6%]), confusion (15 [14.6%]), dizziness (11 [10.7%]), and nausea (8 [7.8%]). Incidence of ARIA-E was highest in aducanumab-treated participants who were apolipoprotein E ε4 allele carriers. Most events (479 of 488 [98.2%]) among those with ARIA-E resolved radiographically; 404 of 488 (82.8%) resolved within 16 weeks. In the placebo group, 29 of 1076 participants (2.7%) had ARIA-E (apolipoprotein E ε4 carriers: 16 of 742 [2.2%]; noncarriers, 13 of 334 [3.9%]). ARIA-microhemorrhage and ARIA–superficial siderosis occurred in 197 participants (19.1%) and 151 participants (14.7%), respectively.
Conclusions and Relevance
In this integrated safety data set from EMERGE and ENGAGE, the most common adverse event in the 10-mg/kg group was ARIA-E, which occurred in 362 of the 1029 patients (35.2%) in the 10-mg/kg group with at least 1 postbaseline MRI scan, with 94 patients (26.0%) experiencing associated symptoms. The most common associated symptom was headache.
Trial Registrations
ClinicalTrials.gov Identifiers: NCT02484547, NCT02477800
Introduction
Amyloid-related imaging abnormalities (ARIA) comprise a spectrum of imaging findings detected on brain magnetic resonance imaging (MRI) and are associated with the investigational use of monoclonal antibodies targeting amyloid-β (Aβ), including aducanumab, in patients with Alzheimer disease (AD).1,2,3,4,5,6,7,8,9 ARIA can manifest as brain edema or sulcal effusion (ARIA-E) or as hemosiderin deposits resulting from hemorrhage in the brain parenchyma or on the pial surface (ARIA-H).6,10 In past clinical trial settings, ARIA-E resolved radiographically over the course of weeks or months, whereas ARIA-H can remain visible on subsequent imaging.6,10,11 Although the biological mechanisms of ARIA have yet to be elucidated, published hypotheses suggest that this phenomenon might be caused by a combination of increased cerebrovascular permeability owing to increased clearance of Aβ neuritic plaques and associated saturation of perivascular drainage, paired with direct antibody interaction with deposited vascular amyloid and weakening of the vessel wall.6,12,13,14
ARIA was the most common adverse event in the phase 1b study of aducanumab (PRIME).15 Following the PRIME trial, 2 identically designed global phase 3 trials, EMERGE and ENGAGE, assessed the safety and efficacy of aducanumab in patients with early AD.
The EMERGE and ENGAGE trials were terminated early based on futility analysis. After termination, it was determined that key assumptions on which the futility decision was based were not valid. The final data were collected per protocol and subsequently analyzed based on the prespecified analysis plan. Primary outcomes of these studies have been reported.16,17
With the recent US Food and Drug Administration (FDA) approval of aducanumab, the EMERGE and ENGAGE trials provide a large safety data set for characterizing ARIA and informing real-world practice. Herein, we describe the radiographic and clinical characteristics of ARIA in the EMERGE and ENGAGE trials and summarize the clinical management of ARIA in these studies.
Methods
The design of the phase 3 studies of aducanumab has been previously reported.16 Briefly, the EMERGE trial (n = 1643) and ENGAGE trial (n = 1653) were identically designed double-blind, placebo-controlled, global phase 3 randomized clinical trials. Trial protocols for these trials can be found in Supplement 1. This report includes secondary analyses of the trials. The primary analysis will be reported elsewhere. Most of the secondary analyses for this report were prespecified for the integrated summary of safety but were not included in the trial protocol. Some ad hoc analyses to address regulatory queries are of scientific interest and are therefore included as part of this report.
Trial participants were individuals aged 50 to 85 years who met clinical criteria for mild cognitive impairment due to AD18 or mild dementia due to AD,19 with amyloid pathology confirmed by visual assessment of amyloid positron emission tomography. See the eMethods in Supplement 2 for the inclusion and exclusion criteria. Notably, patients taking nonaspirin antiplatelet agents or anticoagulation treatments were excluded. The original trials were conducted in accordance with the Declaration of Helsinki and the International Conference for Harmonisation and Good Clinical Practice guidelines and was approved by ethics committees or institutional review boards at each site. Study participants provided written informed consent. Safety monitoring included reports of adverse events, assessment of vital signs, physical and neurological examinations, electrocardiography, hematologic and serum chemical testing, urinalysis, and brain MRI scans. An independent data monitoring committee routinely reviewed the safety data.
Participants were randomized in a 1:1:1 ratio to receive low or high doses of aducanumab or placebo, administered by monthly intravenous doses over 76 weeks for a total of 20 doses, and randomization was stratified by apolipoprotein E (ApoE) ε4 status (carrier or noncarrier). This placebo-controlled period was followed by a long-term extension, and data from the long-term extension period are not included in this article. The low dose was either 3 mg/kg (titrated from 1 to 3 mg/kg over 8 weeks) in ApoE ε4 carriers or 6 mg/kg (titrated from 1 to 3 to 6 mg/kg over 24 weeks) in ApoE ε4 noncarriers. Initially, the target dose in the high-dose groups also differed based on participants’ ApoE ε4 carrier status (6 mg/kg for carriers and 10 mg/kg for noncarriers); starting with protocol version 4, the high-dose target was 10 mg/kg (titrated from 1 to 3 to 6 to 10 mg/kg over 24 weeks) regardless of ApoE ε4 status. The EMERGE and ENGAGE trials were terminated early based on futility analysis. Among 3285 patients who had received 1 or more doses of study treatment (placebo or aducanumab), 1812 (55.2%) had completed the placebo-controlled period of the studies (EMERGE, n = 874; ENGAGE, n = 938), and 1473 (44.8%) had not completed the period (EMERGE, n = 764; ENGAGE, n = 709), with the predominant reason being early study termination by the sponsor (EMERGE, 572 patients [35%]; ENGAGE, 491 [30%]). A CONSORT diagram for these studies is available in Figure 1.
Figure 1. CONSORT Diagram for the EMERGE and ENGAGE Trials.
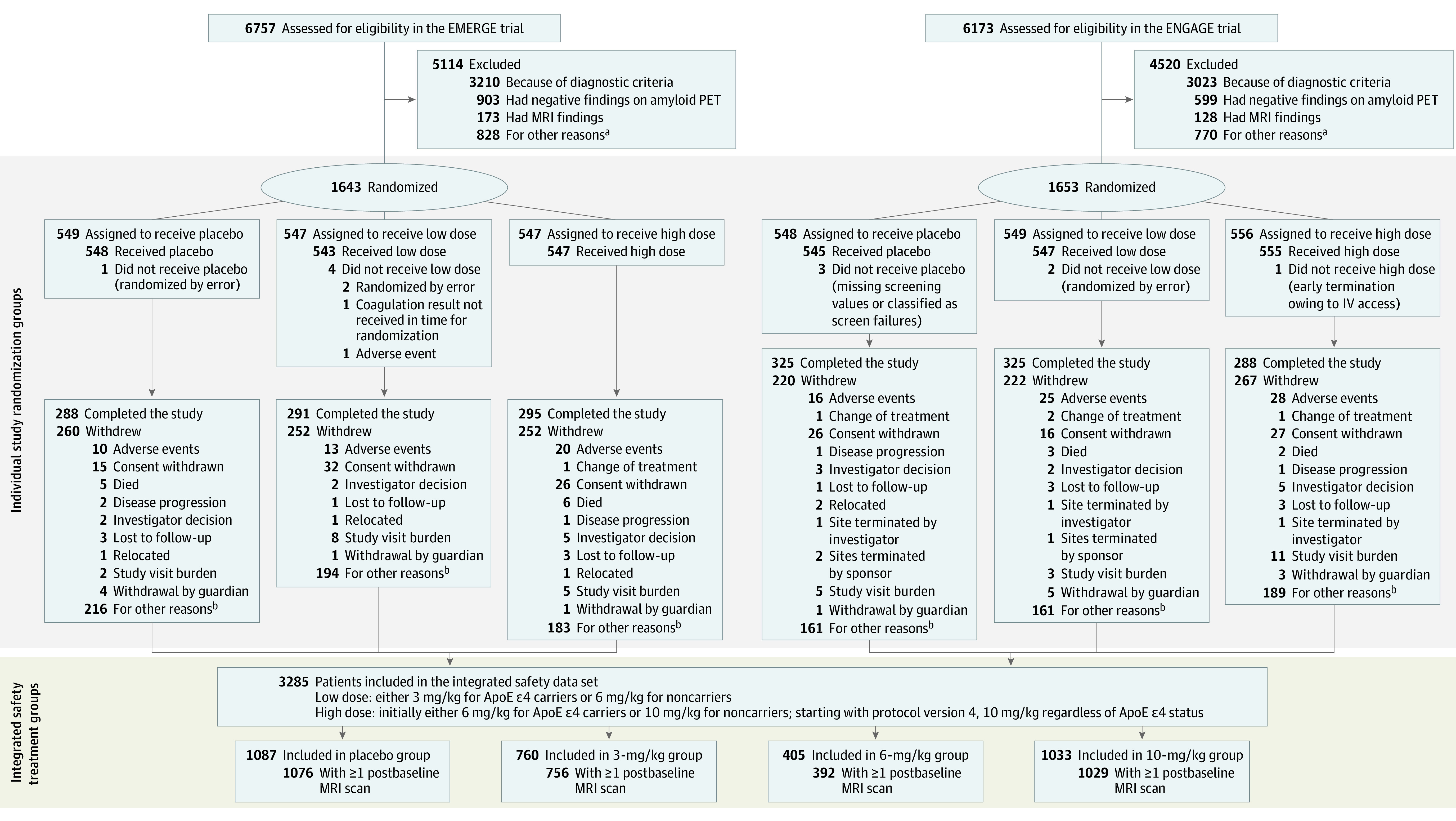
ApoE indicates apolipoprotein E; MRI, magnetic resonance imaging; PET, positron emission tomography.
aOther reasons for not meeting inclusion and exclusion criteria include inability to comply with study requirements; presence of diabetes that, in the judgment of the investigator, cannot be controlled or adequately managed; inability to understand the purpose and risks of the study and provide signed and dated informed consent and authorization to use protected health information in accordance with national and local subject privacy regulations; other unspecified reasons that, in the opinion of the investigator or sponsor, make the participant unsuitable for enrollment; history of or positive test result at screening for hepatitis C virus antibody or hepatitis B virus (defined as positive for both hepatitis B surface antigen and hepatitis B core antibody); use of allowed chronic medications at doses that have not been stable for at least 4 weeks prior to screening visit 1 and screening up to day 1 or use of AD medications at doses that have not been stable for at least 8 weeks; and unknown/unclear.
bSome categories with fewer than 1% of patients are not displayed, including loss of capacity, pregnancy, and protocol amendment. The predominant reason was early study termination by sponsor.
Brain MRI scans, performed at 1.5-T or 3-T field strengths, were evaluated by central radiologists with expertise in ARIA, and all ARIA events were recorded as adverse events. Patients with more than 4 microhemorrhages or any area of siderosis on gradient echo MRI sequences at baseline were excluded. Each MRI report described the type of ARIA and its radiographic severity (eTable 1 in Supplement 2), including focality and extent of involvement. The potential for functional unblinding due to ARIA is a consideration in studies of anti-Aβ antibodies, and it was considered in the study design. Clinical efficacy raters were different from the raters who assessed and monitored safety, and efficacy raters remained blinded to all other clinical assessments, including ARIA assessments, thus decreasing the potential for unblinding.
The presence of vasogenic edema or effusion was evaluated using T2 fluid-attenuated inversion recovery as the primary diagnostic imaging sequence.6 ARIA-E severity was classified based on the number and size of any edematous regions. A single region less than 5 cm was considered of mild radiographic severity; a single region of 5 to 10 cm or multiple regions all less than 10 cm were considered moderate; and any region more than 10 cm was considered severe.
The presence of any new incident brain microhemorrhages or localized superficial siderosis events was also evaluated using gradient echo sequences. A total of 1 to 4 new incident microhemorrhages were considered mild, 5 to 9 were considered moderate, and 10 or more were considered severe. One new incident area of superficial siderosis was considered mild, 2 was considered moderate, and more than 2 was considered severe.
ARIA risk minimization in the EMERGE and ENGAGE trials included dose titration (based on data from the PRIME study7); routine brain MRI monitoring as well as ad hoc MRI testing as clinically indicated; and dose suspension or permanent discontinuation. Postbaseline routine MRIs were performed at weeks 14, 22, 30, 42, 54, 66, and 78 of the placebo-controlled period (eFigure 1A in Supplement 2). The criteria for dose suspension or discontinuation were based on the type and radiographic severity of ARIA and whether the participant had clinical symptoms (eFigure 1B in Supplement 2). For radiographically mild ARIA, dosing was continued if ARIA was asymptomatic; if symptoms were present, dosing was suspended. Following detection of moderate or severe ARIA-E or detection of moderate ARIA-H, dosing was suspended until resolution of ARIA-E and stabilization of ARIA-H. Severe ARIA-H or a brain hemorrhage more than 1 cm in diameter resulted in permanent discontinuation of dosing. On detection of an ARIA episode, follow-up MRIs were conducted approximately every 4 weeks to document resolution of ARIA-E or stabilization of ARIA-H. Evaluation of any symptoms during ARIA was performed by the investigator.
Statistical Analysis
As the EMERGE and ENGAGE trials were identically designed studies and safety results were consistent between the 2 studies,17 safety data were pooled into an integrated data set. The safety population (n = 3285) consisted of all participants who received at least 1 dose of study treatment (placebo or aducanumab) during the placebo-controlled period. ARIA analyses were based on the MRI safety population (n = 3253), which included all participants in the safety population with at least 1 postbaseline MRI scan.
The treatment groups for the pooled analysis (placebo or 3, 6, or 10 mg/kg of aducanumab) are based on the target dose in the randomized treatment regimen. For participants in the safety population randomized to the high-dose carrier group (n = 746), the target dose was changed from 6 mg/kg to 10 mg/kg in protocol version 4. Most of these participants (674 of 746) received at least 1 dose of 10 mg/kg or provided informed consent to protocol version 4 or higher on or before study day 533 (ie, the target study day for the last dose) and were analyzed in the 10-mg/kg group. The remainder (72 of 746 participants) were analyzed in the 6-mg/kg group.
Adverse events were analyzed based on the principle of treatment emergence and included all events that began or changed after the first dose in the study until the first dose in the long-term extension or last day of study (for participants not entering the extension period). All statistical analyses were performed using SAS software version 9.4 (SAS Institute).
Results
Baseline demographic characteristics were balanced across treatment arms and were representative of the enrolled population with early AD. Of 3285 included participants, the mean (SD) age was 70.4 (7.45) years; 1706 participants (52%) were female, 2661 (81%) had mild cognitive impairment due to Alzheimer disease, and 1777 (54%) used symptomatic medications for Alzheimer disease (eTable 2 in Supplement 2). The placebo-controlled period of the combined EMERGE and ENGAGE trials included 2198 aducanumab-treated participants (2752 total person-years of exposure) and 1087 participants receiving placebo. In the aducanumab 10-mg/kg group, 677 participants (65.5%) were ApoE ε4 carriers compared with 747 (68.7%) in the placebo group. In contrast, because of the study design, the 3-mg/kg group consisted entirely of ApoE ε4 carriers, and the 6-mg/kg group consisted primarily of noncarriers (eTable 2 in Supplement 2).
Adverse events reported in the EMERGE and ENGAGE trial have been described.17 Overall, 425 of 1029 patients (41.3%) in the 10-mg/kg group experienced ARIA (ARIA-E or ARIA-H; Table 1). The incidence of ARIA-E was highest in the aducanumab 10-mg/kg group (362 of 1029 [35.2%]) compared with the 6-mg/kg group (83 of 392 [21.2%]), 3-mg/kg group (223 of 756 [29.5%]), and placebo group (29 of 1076 [2.7%]) (Table 1). The incidence of ARIA-E was higher in aducanumab-treated participants who were ApoE ε4 carriers than in those who were noncarriers (Table 1), and the incidence of ARIA-E was highest in ApoE ε4 carriers (290 of 674 [43.0%]) vs noncarriers (72 of 355 [20.3%]) in the 10-mg/kg group. Among ApoE ε4 carriers in the 10-mg/kg group, the incidence of ARIA-E was 66.0% in homozygous carriers (105 of 159) and 35.9% in heterozygous carriers (185 of 515). In the placebo group, 29 of 1076 participants (2.7%) had ARIA-E (ApoE ε4 carriers, 16 of 742 [2.2%]; noncarriers, 13 of 334 [3.9%]). The incidence of ARIA-E in the 3-mg/kg group was higher than in the 6-mg/kg group, reflecting the higher proportion of ApoE ε4 carriers in the 3-mg/kg group. In the 10-mg/kg group, 109 of 1029 participants (10.6%) had 1 or more ARIA-E events (109 of 362 [30.1%] among those with ARIA-E).
Table 1. Amyloid-Related Imaging Abnormality (ARIA) Incidence.
| Measure | No. (%) | |||
|---|---|---|---|---|
| Placebo (n = 1076) | Aducanumab group | |||
| 3 mg/kg (n = 756) | 6 mg/kg (n = 392) | 10 mg/kg (n = 1029) | ||
| Any ARIA (ARIA-E or ARIA-H) | 111 (10.3) | 274 (36.2) | 104 (26.5) | 425 (41.3) |
| ARIA-E | 29 (2.7) | 223 (29.5) | 83 (21.2) | 362 (35.2) |
| ApoE ε4 carriers, No./total No. (%) | 16/742 (2.2) | 223/756 (29.5) | 25/66 (37.9) | 290/674 (43.0) |
| ApoE ε4 noncarriers, No./total No. (%) | 13/334 (3.9) | NA | 58/326 (17.8) | 72/355 (20.3) |
| Recurrent ARIA-E | 0 | 66 (8.7) | 14 (3.6) | 109 (10.6) |
| ARIA-H | ||||
| Microhemorrhage | 71 (6.6) | 141 (18.7) | 50 (12.8) | 197 (19.1) |
| ApoE ε4 carriers, No./total No. (%) | 57/742 (7.7) | 141/756 (18.7) | 15/66 (22.7) | 153/674 (22.7) |
| ApoE ε4 noncarriers, No./total No. (%) | 14/334 (4.2) | NA | 35/326 (10.7) | 44/355 (12.4) |
| Superficial siderosis | 24 (2.2) | 91 (12.0) | 23 (5.9) | 151 (14.7) |
| ApoE ε4 carriers, No./total No. (%) | 14/742 (1.9) | 91/756 (12.0) | 11/66 (16.7) | 129/674 (19.1) |
| ApoE ε4 noncarriers, No./total No. (%) | 10/334 (3.0) | NA | 12/326 (3.7) | 22/355 (6.2) |
| Hemorrhage >1 cm | 4 (0.4) | 1 (0.1) | 3 (0.8) | 3 (0.3) |
| ApoE ε4 carriers, No./total No. (%) | 1/742 (0.1) | 1/756 (0.1) | 3/66 (4.5) | 1/674 (0.1) |
| ApoE ε4 noncarriers, No./total No. (%) | 3/334 (0.9) | NA | 0/326 (0.0) | 2/355 (0.6) |
| ARIA-H in patients with ARIA-E, No. | 29 | 223 | 83 | 362 |
| Brain microhemorrhage | 4 (13.8) | 103 (46.2) | 32 (38.6) | 146 (40.3) |
| Localized superficial siderosis | 9 (31.0) | 76 (34.1) | 21 (25.3) | 140 (38.7) |
| ARIA-H in patients without ARIA-E, No. | 1047 | 533 | 309 | 667 |
| Brain microhemorrhage | 67 (6.4) | 38 (7.1) | 18 (5.8) | 51 (7.6) |
| Localized superficial siderosis | 15 (1.4) | 15 (2.8) | 2 (0.6) | 11 (1.6) |
| Discontinuations due to ARIA | 6 (0.6) | 47 (6.2) | 21 (5.4) | 64 (6.2) |
| ApoE ε4 carriers, No./total No. (%) | 2/742 (0.3) | 47/756 (6.2) | 14/66 (21.2) | 55/674 (8.2) |
| ApoE ε4 noncarriers, No./total No. (%) | 4/334 (1.2) | NA | 7/326 (2.1) | 9/355 (2.5) |
Abbreviations: ApoE, apolipoprotein E; ARIA-E, amyloid-related imaging abnormality–edema; ARIA-H, amyloid-related imaging abnormality–microhemorrhage or superficial siderosis, NA, not applicable.
Discontinuation of treatment was protocol mandated for serious ARIA-E (regardless of radiographic severity) and radiographically severe or serious ARIA-H (eFigure 1B in Supplement 2). Overall, 64 participants (6.2%) discontinued treatment due to ARIA. Discontinuations due to ARIA were higher for ApoE ε4 carriers than noncarriers (Table 1).
Brain microhemorrhages were the most common type of ARIA-H (placebo group: 71 [6.6%]; 10-mg/kg group: 197 [19.1%]), followed by localized superficial siderosis (placebo group: 24 [2.2%]; 10-mg/kg group: 151 [14.7%]) (Table 1). Larger brain hemorrhages measuring more than 1 cm in diameter were rare and balanced between placebo and aducanumab groups (placebo group: 4 [0.4%]; 10-mg/kg group: 3 [0.3%]).
Analyses of the incidence of ARIA-H stratified by ARIA-E status (Table 1) showed that the incidence of brain microhemorrhages and localized superficial siderosis was increased in participants with ARIA-E compared with participants without ARIA-E. Specifically, in the 362 participants with ARIA-E, the incidence of brain microhemorrhages and localized superficial siderosis was 40.3% (146) and 38.7% (140), respectively, in the 10-mg/kg group. In participants without ARIA-E, the incidence of brain microhemorrhage and localized superficial siderosis was lower and similar between the aducanumab (10-mg/kg group: brain microhemorrhage, 51 of 667 [7.6%]; localized superficial siderosis, 11 of 667 [1.6%]) and placebo groups (brain microhemorrhage, 67 of 1047 [6.4%]; localized superficial siderosis, 15 of 1047 [1.4%]).
A proportional hazards model to assess potential ARIA risk factors was performed (eFigure 2 in Supplement 2). There was no evidence of an association between the following factors and increased hazard of ARIA-E in the 10-mg/kg group: baseline age, sex, AD stage, and use of antithrombotic medication (including nonaspirin antiplatelet and anticoagulant agents). The same is true of all of these factors for increased hazard of isolated ARIA-H with the exception of baseline age, where there is increased hazard for older participants (hazard ratio [95% CI], 1.06 [1.02-1.09] per additional year). In the 10-mg/kg group, participants with baseline brain microhemorrhages were more likely to be at risk of an ARIA-E event than participants without baseline microhemorrhages (HR 1.7; 95% CI, 1.31-2.27), and ApoE ε4 carriers were more likely to be at risk than noncarriers (HR 2.5; 95% CI, 1.90-3.20); ApoE ε4 carriers and participants with baseline microhemorrhages were not at an increased risk of isolated ARIA-H.
Kaplan-Meier curves of time to first ARIA-E (Figure 2) and ARIA-H (eFigure 3 in Supplement 2) in the 10-mg/kg group illustrate that first ARIA-E and ARIA-H events usually occurred early in treatment. In the 10-mg/kg group, ARIA-E occurred during the first 8 doses of aducanumab among 263 of 362 patients (72.7%), and 298 of all 488 ARIA-E events (61.1%) occurred during first 8 doses.
Figure 2. Kaplan-Meier Analysis of Time to First Amyloid-Related Imaging Abnormality–Edema (ARIA-E) Event in the 10-mg/kg Aducanumab Group.
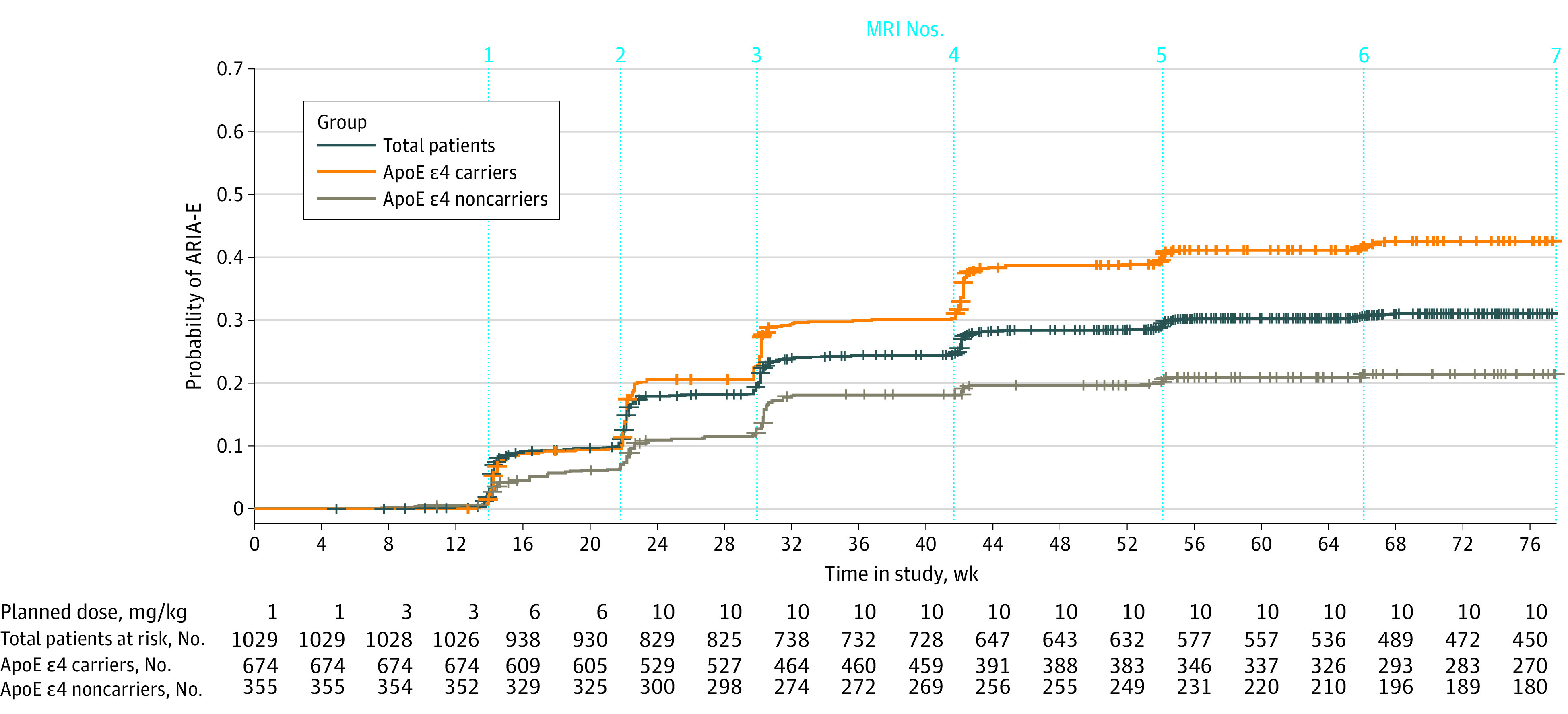
ApoE indicates apolipoprotein E; MRI, magnetic resonance imaging.
Of the 362 participants with ARIA-E in the 10-mg/kg group (Table 2), the maximal radiographic severity was mild in 109 participants (30.1%), moderate in 209 participants (57.7%), and severe in 44 participants (12.2%). Of all ARIA-E events in the 10-mg/kg group, 479 (98.2%) were observed to have resolved (ie, no residual brain edema) during the study; specifically, 336 (68.9%) resolved within 12 weeks and 404 (82.8%) resolved within 16 weeks. Of participants in the 10-mg/kg group with ARIA-H–microhemorrhage and ARIA-H–superficial siderosis, the maximal radiographic severity was severe in 23 of 197 (11.7%) and 33 of 151 (21.9%), respectively (Table 2). Radiographic severity by ApoE ε4 carrier status is detailed in eTable 3 in Supplement 2. Serious ARIA events (including those resulting in hospitalization) were uncommon in the 10-mg/kg group (14 of 1029 total participants [1.4%]; 7 of 674 ApoE ε4 carriers [1.0%]; and 7 of 355 noncarriers [2.0%]; eTable 3 in Supplement 2). Representative MRI images of ARIA-E and ARIA-H cases by radiographic severity are detailed in eFigure 4 in Supplement 2. In the 10-mg/kg group, 178 of 260 ARIA-H–microhemorrhage events (68.5%) and 135 of 200 ARIA-H–superficial siderosis events (92.5%), respectively, stabilized within 8 weeks.
Table 2. Radiographic Severity of Amyloid-Related Imaging Abnormality (ARIA)a.
| Measure | No. (%) | |||
|---|---|---|---|---|
| Placebo (n = 1076) | Aducanumab group | |||
| 3 mg/kg (n = 756) | 6 mg/kg (n = 392) | 10 mg/kg (n = 1029) | ||
| ARIA-edema, No. | 29 | 223 | 83 | 362 |
| Mild | 21 (72.4) | 66 (29.6) | 33 (39.8) | 109 (30.1) |
| Moderate | 8 (27.6) | 135 (60.5) | 41 (49.4) | 209 (57.7) |
| Severe | 0 | 22 (9.9) | 9 (10.8) | 44 (12.2) |
| ARIA-microhemorrhage, No. | 71 | 141 | 50 | 197 |
| Mild | 65 (91.5) | 103 (73.0) | 38 (76.0) | 147 (74.6) |
| Moderate | 5 (7.0) | 11 (7.8) | 4 (8.0) | 27 (13.7) |
| Severe | 1 (1.4) | 27 (19.1) | 8 (16.0) | 23 (11.7) |
| ARIA–superficial siderosis, No. | 24 | 91 | 23 | 151 |
| Mild | 22 (91.7) | 50 (54.9) | 13 (56.5) | 74 (49.0) |
| Moderate | 0 | 25 (27.5) | 6 (26.1) | 44 (29.1) |
| Severe | 2 (8.3) | 16 (17.6) | 4 (17.4) | 33 (21.9) |
| Serious ARIA | 2 (0.2) | 6 (0.8) | 3 (0.8) | 14 (1.4) |
Severity percentages are based on the number of participants with an event, and participants are counted once at the maximum severity experienced.
Of the 362 participants with ARIA-E in the 10-mg/kg group, 94 (26.0%) were symptomatic (Table 3); most participants who reported ARIA-E did not have symptoms during the episode. The proportion exhibiting symptoms was similar between ApoE ε4 carriers and noncarriers (77 of 290 carriers [26.6%] vs 17 of 72 noncarriers [23.6%]; eTable 4 in Supplement 2). Considering ARIA overall (ARIA-E and ARIA-H), the incidence of symptomatic ARIA is similar to that of ARIA-E (Table 3). For the 99 participants with symptoms during ARIA (either ARIA-E or ARIA-H) in the 10-mg/kg group, the maximal clinical severity of the symptoms, as reported by investigators, was mild for 67 (67.7%), moderate for 28 (28.3%), and severe for 4 (4.0%). Serious ARIA symptoms occurred in 3 of 1029 participants (0.3%) in the 10-mg/kg aducanumab group (Table 3). Among the 103 participants with symptomatic ARIA (symptoms during or not during the ARIA event) in the 10-mg/kg group, the most common symptoms were headache (48 [46.6%]), confusion (15 [14.6%]), dizziness (11 [10.7%]), and nausea (8 [7.8%]). Less frequent symptoms included fatigue, visual impairment, blurred vision, and gait disturbance, each occurring in 5 symptomatic participants (4.9%) in the 10-mg/kg group. The median (IQR) reported duration of symptoms was 5 (1-13) weeks in the 10-mg/kg group. Severe symptoms during ARIA included reports of seizure, although the overall incidence of seizures was balanced between the placebo group (9 of 1087 [0.8%]) and 10-mg/kg aducanumab group (4 of 1033 [0.4%]). Among participants in the 10-mg/kg group, 11 (1.1%) received a concomitant medication for an ARIA event; corticosteroids and acetaminophen were the most commonly administered concomitant medications for ARIA (eTable 5 in Supplement 2). There were no deaths due to ARIA.
Table 3. Symptomatic Amyloid-Related Imaging Abnormality (ARIA) and Maximum Symptom Severitya.
| Measure | No. (%) | |||
|---|---|---|---|---|
| Placebo (n = 1076) | Aducanumab group | |||
| 3 mg/kg (n = 756) | 6 mg/kg (n = 392) | 10 mg/kg (n = 1029) | ||
| Patients with ARIA-E, No. | 29 | 223 | 83 | 362 |
| Asymptomatic | 26 (89.7) | 172 (77.1) | 66 (79.5) | 268 (74.0) |
| Symptomatic | 3 (10.3) | 51 (22.9) | 17 (20.5) | 94 (26.0) |
| Patients with any ARIA, No. | 111 | 274 | 104 | 425 |
| Asymptomatic | 106 (95.5) | 218 (79.6) | 87 (83.7) | 322 (75.8) |
| Symptomatic | 5 (4.5) | 56 (20.4) | 17 (16.3) | 103 (24.2) |
| Patients with symptoms during an ARIA event, No. | 5 | 53 | 17 | 99 |
| Mild | 3 (60.0) | 33 (62.3) | 12 (70.6) | 67 (67.7) |
| Moderate | 1 (20.0) | 16 (30.2) | 3 (17.6) | 28 (28.3) |
| Severe | 1 (20.0) | 4 (7.5) | 2 (11.8) | 4 (4.0) |
| Serious ARIA symptoms | 0 | 3 (0.4) | 0 | 3 (0.3) |
Abbreviation: ARIA-E, amyloid-related imaging abnormalities-edema.
Each participant counted once at maximum symptomatic status and severity.
Based on the study protocol, participants with radiographically mild and asymptomatic ARIA could continue dosing without interruptions (eFigure 1B in Supplement 2). A total of 161 ARIA-E events started as radiographically mild and asymptomatic in the 10-mg/kg group, with participants receiving at least 1 aducanumab dose during the ARIA-E event. Most of these ARIA-E events (93 of 161 [57.8%]) remained radiographically mild and asymptomatic (eTable 6 in Supplement 2). The radiographic severity of ARIA-E changed to moderate or severe in 64 events (39.8%) and 1 event (0.6%), respectively, and 11 of 161 events (6.8%) were subsequently associated with symptoms (eTable 6 in Supplement 2). Reporting of these clinical symptoms did not always coincide with a change in the radiographic severity of ARIA-E.
Discussion
Radiographic findings of ARIA-E and ARIA-H have been reported as adverse effects of treatment in multiple clinical studies of anti-Aβ antibodies in patients with AD.1,2,3,4,5,15 A radiographic finding of ARIA-E was the most common adverse event in aducanumab-treated participants in the EMERGE and ENGAGE phase 3 randomized clinical trials, and ARIA-E was only sporadically reported in the placebo groups. Consistent with prior aducanumab studies and with data from other anti-Aβ immunotherapies,3,5,7,11,15,20 the incidence of ARIA-E was highest in the high-dose (10 mg/kg) group vs lower doses (3 mg/kg and 6 mg/kg). ARIA-E incidence was also higher in ApoE ε4 carriers than ApoE ε4 noncarriers. When ARIA incidence was assessed by ApoE ε4 carrier status, the incidence was also dose dependent (Table 1). Among other demographic and baseline characteristics, only pretreatment brain microhemorrhages and ApoE ε4 carrier status were found to be associated with an increase in the incidence of ARIA-E. These same factors did not confer an increased risk of ARIA-H. Such an association between pretreatment microhemorrhages and ARIA-E was also observed in phase 3 bapineuzumab studies.11,20
Most ARIA-E events occurred early in treatment, during dose titration or shortly after attaining the target dose. These results are consistent with clinical studies of other anti-Aβ antibodies in which ARIA also primarily occurred within the first months of treatment, while the risk of ARIA-E decreased during the subsequent treatment course.5,21
Approximately 30% of ARIA-E events in the 10-mg/kg group were limited to a single brain region and measured less than 5 cm (mild ARIA-E), whereas approximately 58% of the events in the group were observed in one or multiple brain locations but each area of edema measured less than 10 cm (moderate ARIA-E). ARIA-E measuring more than 10 cm was reported in approximately 12% of patients in the 10-mg/kg group. ARIA-E was transient, with episodes resolved typically within 12 to 16 weeks after initial detection, with no areas of residual edema.
Brain microhemorrhages and localized superficial siderosis (ARIA-H in clinical studies) have also been observed in patients with AD, commonly in the setting of small-vessel angiopathy.22 The integrated data set from the EMERGE and ENGAGE trials presented here furthers our knowledge of the association between brain microhemorrhages or superficial siderosis and ARIA-E. Specifically, brain microhemorrhages or superficial siderosis tended to occur more frequently in patients who also had ARIA-E, whereas in the absence of ARIA-E, the incidence of brain microhemorrhages and siderosis equated to the previously observed background rate of placebo groups. Therefore, although patients with AD might be at risk of microhemorrhages or superficial siderosis, such imaging findings can be observed more frequently in aducanumab-treated patients with ARIA-E. Lastly, brain hemorrhages more than 1 cm were rare in the 10-mg/kg aducanumab group (0.3%) and similar to placebo (0.4%), and participants with brain hemorrhages discontinued treatment as required by the study protocols. Important to the interpretation of ARIA-H incidence in this study is the fact that patients with preexisting microhemorrhages (more than 4) or areas of superficial siderosis were excluded from the phase 3 trials, which may have contributed to the overall observed incidence of ARIA-H.
In the participants from the 10-mg/kg aducanumab group who experienced ARIA-E, 26.0% reported symptoms. When present, common clinical symptoms included headache, confusion, dizziness, and nausea. Reports of seizures were overall balanced between the aducanumab and placebo groups, and there were no fatal events due to ARIA. The frequency and type of symptoms in the EMERGE and ENGAGE trials are consistent with prior clinical trial experiences in which symptoms tended to be nonspecific and headache was the most frequently reported symptom in the setting of ARIA.3,5,7,12,15 A 2020 report23 detailed the experience of the clinical course and management of 1 male ApoE ε4 carrier who received a total of 4 doses of aducanumab (2 monthly doses at 1 mg/kg and 2 monthly doses at 3 mg/kg). He subsequently experienced symptoms, headache, and encephalopathy in the presence of ARIA-E and ARIA-H as well as severe malignant hypertension and epileptiform activity on electroencephalography. He was treated with corticosteroids and experienced a decrease in radiographic severity of ARIA-E and resolution of symptoms over the course of 6 months.23 More real-world data on the treatment and management of ARIA, outside of the clinical trial protocols, are required to assess which medications may be effective in treating symptomatic patients with ARIA.
In the EMERGE and ENGAGE trials, risk management for ARIA included routine MRI monitoring. Dose management included titration that, compared with fixed aducanumab dosing, was shown to reduce the incidence of ARIA-E in the phase 1b trial of aducanumab7 and was implemented in all phase 3 trial participants. Aducanumab dosing was suspended in participants with radiographically moderate or severe ARIA-E, until resolution of the episode. Participants without clinical symptoms who had radiographically mild ARIA-E could continue treatment without interruptions. In some cases, ARIA-E in these participants progressed radiographically (40.4%) or symptomatically (6.8%). To our knowledge, the EMERGE and ENGAGE trials are the first phase 3 randomized clinical trials in which continued dosing during asymptomatic and radiographically mild ARIA-E was implemented, and the results indicate that continued dosing for asymptomatic and radiographically limited ARIA-E is feasible since, during uninterrupted dosing, most ARIA-E events did not increase in size or involve multiple brain locations and patients remained typically asymptomatic.
Limitations
This study has limitations. Potential limitations of this work include the early termination of the trials (35% of participants in the EMERGE trial and 30% in the ENGAGE trial withdrew owing to early study termination), which led to fewer patients completing a full course of dosing and could potentially impact the incidence of adverse events. Additionally, the global phase 3 study populations lacked racial and ethnic diversity; therefore, very limited data about the frequency and severity of ARIA in diverse populations are available. In the EMERGE and ENGAGE trials, treatment was initiated in patients with mild cognitive impairment due to AD or mild AD dementia. Data are not available for initiation of treatment at other stages of disease (ie, preclinical) or later (ie, moderate to severe dementia) stages of AD, and initiation of aducanumab is not indicated in those populations.24
Conclusions
The EMERGE and ENGAGE results constitute one of the largest ARIA data sets and shed light on the clinical and radiographic characteristics of ARIA during treatment with aducanumab. In the EMERGE and ENGAGE trials, screening for ARIA was executed through brain MRI monitoring, and in some cases, per protocol, dosing adjustments were made. With the recent FDA approval of aducanumab for the treatment of AD, ARIA will be monitored and managed regularly outside of clinical trials for the first time. Although ARIA can occur at any time, clinical suspicion for ARIA should be highest early in treatment, and routine surveillance MRIs should be supplemented with ad hoc MRIs in patients with new-onset symptoms potentially associated with ARIA. Guidance on the timing of surveillance MRIs for detection of asymptomatic ARIA is described in the Aduhelm US prescribing information (obtain MRI prior to the 7th and 12th infusions, in addition to as clinically indicated). As seen in Figure 2, most first ARIA-E events occur prior to the scheduled 12th dose (ie, week 44). A 2021 publication references monitoring for asymptomatic ARIA using the EMERGE/ENGAGE MRI schedule.24 In patients with identified ARIA, prescribers should carefully assess the radiographic and clinical findings when deciding whether to continue dosing. Safety data collected while treating patients with aducanumab in real-world settings as well as clinical settings through the phase 3b EMBARK redosing trial25 and forthcoming FDA-mandated confirmatory clinical studies will continue to inform best practices and characterization of ARIA in the clinic.
Trial protocols.
eMethods. Inclusion and exclusion criteria.
eFigure 1. ARIA risk minimization: MRI monitoring (A) and dose management (B).
eFigure 2. Forest plot of hazard ratios risk factors in the 10-mg/kg aducanumab group for potential ARIA-E (A), isolated ARIA-H (B), and ARIA-E/ARIA-H (C).
eFigure 3. Kaplan-Meier analysis of time to first ARIA-H event in the 10-mg/kg aducanumab group.
eFigure 4. Representative ARIA-E and ARIA-H MRI images by radiographic severity.
eTable 1. Descriptions of ARIA-E and ARIA-H MRI severity.
eTable 2. Baseline characteristics by dose group for the integrated safety data set.
eTable 3. Radiographic severity of ARIA.
eTable 4. Symptomology during an ARIA episode and serious ARIA events.
eTable 5. Concomitant medications administered for ARIA.
eTable 6. Continued dosing of radiographically mild and asymptomatic ARIA-E in 10-mg/kg group.
References
- 1.Black RS, Sperling RA, Safirstein B, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24(2):198-203. doi: 10.1097/WAD.0b013e3181c53b00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson C, Siemers E, Hake A, et al. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer’s disease. Alzheimers Dement (Amst). 2016;2:75-85. doi: 10.1016/j.dadm.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salloway S, Sperling R, Gilman S, et al. ; Bapineuzumab 201 Clinical Trial Investigators . A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061-2070. doi: 10.1212/WNL.0b013e3181c67808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrero J, Williams L, Stella H, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y). 2016;2(3):169-176. doi: 10.1016/j.trci.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrowitzki S, Lasser RA, Dorflinger E, et al. ; Scarlet Road Investigators . A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9(1):95. doi: 10.1186/s13195-017-0318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sperling RA, Jack CR Jr, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367-385. doi: 10.1016/j.jalz.2011.05.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budd Haeberlein S, O’Gorman J, Chiao P, et al. Clinical development of aducanumab, an anti-aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4(4):255-263. [DOI] [PubMed] [Google Scholar]
- 8.Cummings JL, Cohen S, van Dyck CH, et al. ABBY: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90(21):e1889-e1897. doi: 10.1212/WNL.0000000000005550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doody RS, Thomas RG, Farlow M, et al. ; Alzheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group . Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311-321. doi: 10.1056/NEJMoa1312889 [DOI] [PubMed] [Google Scholar]
- 10.Barakos J, Sperling R, Salloway S, et al. MR imaging features of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol. 2013;34(10):1958-1965. doi: 10.3174/ajnr.A3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arrighi HM, Barakos J, Barkhof F, et al. Amyloid-related imaging abnormalities-haemosiderin (ARIA-H) in patients with Alzheimer’s disease treated with bapineuzumab: a historical, prospective secondary analysis. J Neurol Neurosurg Psychiatry. 2016;87(1):106-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30-42. doi: 10.1038/s41582-019-0281-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241-249. doi: 10.1016/S1474-4422(12)70015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zago W, Schroeter S, Guido T, et al. Vascular alterations in PDAPP mice after anti-Aβ immunotherapy: implications for amyloid-related imaging abnormalities. Alzheimers Dement. 2013;9(5)(suppl):S105-S115. doi: 10.1016/j.jalz.2012.11.010 [DOI] [PubMed] [Google Scholar]
- 15.Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50-56. doi: 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- 16.Budd Haeberlein S, Salloway S, Aisen P, et al. Evaluation of aducanumab efficacy in early Alzheimer’s disease. Paper presented at: 15th International Conference on Alzheimer's & Parkinson's Diseases virtual conference; March 9-14, 2021. [Google Scholar]
- 17.Chalkias S, Mummery C, Salloway S, et al. Evaluation of aducanumab safety in early Alzheimer’s disease. Paper presented at: 15th International Conference on Alzheimer's & Parkinson's Diseases virtual conference; March 9-14, 2021. [Google Scholar]
- 18.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270-279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ketter N, Brashear HR, Bogert J, et al. Central review of amyloid-related imaging abnormalities in two phase III clinical trials of bapineuzumab in mild-to-moderate Alzheimer’s disease patients. J Alzheimers Dis. 2017;57(2):557-573. doi: 10.3233/JAD-160216 [DOI] [PubMed] [Google Scholar]
- 21.Salloway S, Sperling R, Fox NC, et al. ; Bapineuzumab 301 and 302 Clinical Trial Investigators . Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322-333. doi: 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74(17):1346-1350. doi: 10.1212/WNL.0b013e3181dad605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.VandeVrede L, Gibbs DM, Koestler M, et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement (Amst). 2020;12(1):e12101. doi: 10.1002/dad2.12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398-410. doi: 10.14283/jpad.2021.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.A study to evaluate safety and tolerability of aducanumab in participants with Alzheimer's disease who had previously participated in the aducanumab studies 221AD103, 221AD301, 221AD302 and 221AD205. ClinicalTrials.gov identifier: NCT04241068. Updated September 5, 2021. Accessed September 5, 2021. https://clinicaltrials.gov/ct2/show/NCT04241068
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocols.
eMethods. Inclusion and exclusion criteria.
eFigure 1. ARIA risk minimization: MRI monitoring (A) and dose management (B).
eFigure 2. Forest plot of hazard ratios risk factors in the 10-mg/kg aducanumab group for potential ARIA-E (A), isolated ARIA-H (B), and ARIA-E/ARIA-H (C).
eFigure 3. Kaplan-Meier analysis of time to first ARIA-H event in the 10-mg/kg aducanumab group.
eFigure 4. Representative ARIA-E and ARIA-H MRI images by radiographic severity.
eTable 1. Descriptions of ARIA-E and ARIA-H MRI severity.
eTable 2. Baseline characteristics by dose group for the integrated safety data set.
eTable 3. Radiographic severity of ARIA.
eTable 4. Symptomology during an ARIA episode and serious ARIA events.
eTable 5. Concomitant medications administered for ARIA.
eTable 6. Continued dosing of radiographically mild and asymptomatic ARIA-E in 10-mg/kg group.