

Abstract
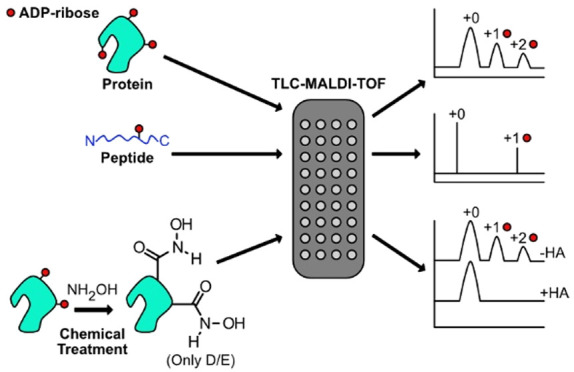
Poly(ADP-ribose) polymerases, PARPs, transfer ADP-ribose onto target proteins from nicotinamide adenine dinucleotide (NAD+). Current mass spectrometric analytical methods require proteolysis of target proteins, limiting the study of dynamic ADP-ribosylation on contiguous proteins. Herein, we present a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) method that facilitates multisite analysis of ADP-ribosylation. We observe divergent ADP-ribosylation dynamics for the catalytic domains of PARPs 14 and 15, with PARP15 modifying more sites on itself (+3–4 ADP-ribose) than the closely related PARP14 protein (+1–2 ADP-ribose)—despite similar numbers of potential modification sites. We identify, for the first time, a minimal peptide fragment (18 amino-acids) that is preferentially modified by PARP14. Finally, we demonstrate through mutagenesis and chemical treatment with hydroxylamine that PARPs 14/15 prefer acidic residues. Our results highlight the utility of MALDI-TOF in the analysis of PARP target modifications and in elucidating the biochemical mechanism governing PARP target selection.
ADP-ribosylation is a widespread and ubiquitous post-translational modification (PTM) across all kingdoms of life.1 Although it was one of the first PTMs described,2 the biochemical selectivity and cellular consequences of ADP-ribosylation is still being uncovered. In humans, 17 related enzymes, known as poly-ADP-ribose polymerases (PARPs) are responsible for the chemical reaction that transfers ADP-ribose (ADPr) from nicotinamide adenine dinucleotide (NAD+) to target protein substrates (Figure 1a).3 The family can be further subdivided on the basis of the PARP’s ability to catalyze poly-ADP-ribosylation (PARPs 1–5), to catalyze mono-ADP-ribosylation (PARPs 6–8, 10–12, 14–16), or its lack of catalytic activity (PARPs 9, 13).4 ADP-ribose transfer occurs on a chemically diverse set of residues: arginine (R), lysine (K), histidine (H), cysteine (C), serine (S), tyrosine (Y), aspartate (D), and glutamate (E).5−10 The biological role for modification at chemically distinct sites is poorly understood. PARPs have emerged as important regulators in a host of cellular pathways;11 they were initially described in the DNA damage repair pathways.12 Further, malfunctioning PARP activity has been linked to a broad array of disease states.13
Figure 1.
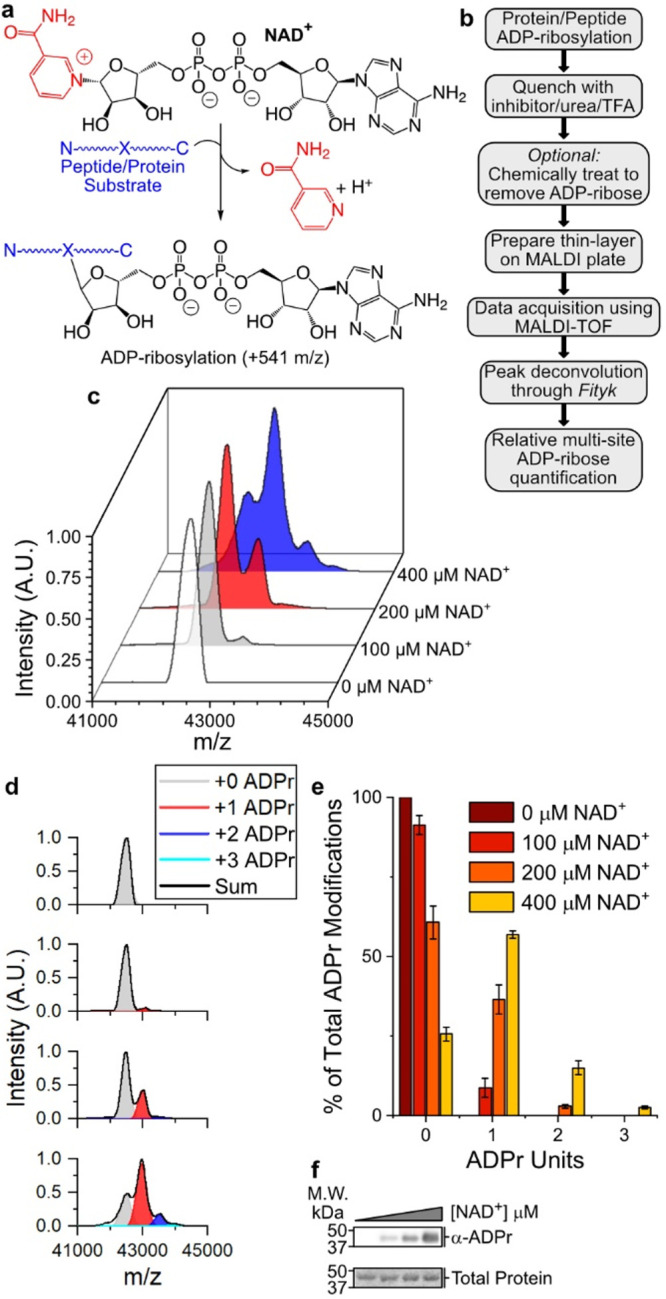
P14 auto-ADP-ribosylates at a single site under physiological conditions. (a) ADP-ribosylation reaction catalyzed by PARP enzymes. (b) TLC-MALDI workflow. (c) P14 was incubated in the presence of increasing concentrations of NAD+ and was subjected to TLC-MALDI to visualize the resulting increase in m/z due to ADPr (+541). (d) MS spectra were integrated to determine the relative levels of auto-ADP-ribosylation. (e) Quantification of the results in (b). The bar graphs depict the fraction of the total P14 protein that has been modified at 0, 1, or 2 distinct sites (mean ± S.E.M., n = 3). (f) In vitro P14 ADP-ribosylation assay.
Given the emerging role for PARPs in both development and pathophysiology, significant effort has been expended to define the potential pool of ADP-ribosylation targets through high-throughput tandem mass spectrometry (MS/MS).14 MS/MS workflows have been developed to capture whole-cell in vivo ADPr targets,15−17 family member specific targets,18,19 and they are now routinely used to identify the precise site of ADPr modification on protein substrates.5,20−22 Despite these technical advances, the inability to sequence multiple sites on a contiguous protein target (with the obvious exception of a single cleaved peptide containing two distinct acceptor residues) has left an appreciable deficit in our understanding of the biochemical mechanism that governs target selection and the total extent of modification performed on a given substrate. Traditional biochemical assays (e.g., Western blotting) similarly cannot distinguish between a substrate that has been modified by a single ADPr and one that has been modified at multiple sites. García-Saura and co-workers recently described an acid-urea gel system that aids visualization of multisite ADP-ribose modifications; their findings highlight the value of additional technologies to study multisite ADP-ribosylation—especially those that allow for quantification of multisite labeling.23
Matrix-assisted laser-desorption time-of-flight mass spectrometry (MALDI-TOF) presents an intriguing complement to the methods mentioned above as it can be used on an intact substrate. The ionization energy employed with MALDI-TOF has allowed for direct detection of protein–sugar conjugates, which are chemically analogous to ADP-ribosylation.24 Despite this advantage, MALDI-TOF analysis of ADP-ribosylated substrates has been limited primarily to peptides25 and DNA.26 One reason for the underutilization of MALDI-TOF in ADPr-based analysis is the difficulty in identifying matrix conditions capable of ionizing a complex protein–ADPr mixture with sufficient resolution to define and quantify distinct ADP-ribosylated states. Further, the presence of buffer components, detergents, and salts required for biochemical assays often interfere with the downstream MALDI-TOF instrumentation. Recently, ultrathin layer chromatography technologies (TLC-MALDI) have been developed, which significantly improve both the sensitivity and resolution of MALDI-TOF performed with large (≥100 kDa) or complex samples.27−29 The formation of a thin-layer of matrix prior to sample deposition on the MALDI objective aids in crystal formation in the presence of contaminants and results in spectra with higher intensities and improved resolution. Both these features are vital considerations for study of proteins modified by ADP-ribose.
Motivated by a desire to elucidate the biochemical mechanism that governs ADPr target selection and resulting modification, we set out to adapt previous TLC-MALDI analytical methods to study the multisite dynamics of ADP-ribosylation. We hypothesized that this technique would allow us to address three related, ongoing research questions: (1) Do PARP enzymes possess an intrinsic mechanism that governs site-selection by limiting the number of ADPr units attached to a given substrate? (2) Are there proximal interactions between the site of ADPr modification and a specific PARP that dictate site specificity? (3) Can existing chemical analysis techniques be combined with a rapid TLC-MALDI method to reveal preferred acceptor site chemistries within the PARP family? Below, we demonstrate the utility of this technique in addressing these biochemical considerations and we develop this method as a complement to current MS/MS techniques.
Experimental Section
ADP-Ribosylation Assay
P14 or P15 (16 μM) was incubated with NAD+ (Sigma-Aldrich) for 15 min at 30 °C in a 10 μL reaction volume consisting of 50 mM HEPES, pH 7.5, 100 mM NaCl, 12 mM MgCl2, and 0.5 mM TCEP. Reactions were quenched by adding 490 μL of 0.1% TFA prior to loading to a 500 μL capacity 10 000 MWCO concentrator (VWR). P14i1, P14i2, and P14i3 were treated as above except 1 μM of the peptide was incubated with 10 μM enzyme (P14 or P15) in the presence of NAD+ for 30 min at 30 °C. Peptide reactions were quenched by adding 200 μM PJ-34 (Tocris). HA-treated samples were prepared as above, except prior to desalting, the reaction mixture was quenched with 200 μM PJ-34, adjusted to a 20 μL final volume containing 1 M HA, pH 7.0, and incubated for 2 h at 40 °C. Mock treated samples contained 1 M NaCl instead of 1 M HA. Details regarding sample cleanup are included in the Supporting Information.
TLC-MALDI Analysis
Preparation of the steel MALDI target for sample deposition using an ultrathin layer of α-cyano-4-hydroxycinnamic acid (α-CHCA, Sigma-Aldrich) was completed as described.29 Details regarding TLC-MALDI optimization, MS acquisition parameters, and data analysis are included in the Supporting Information.
Results and Discussion
Initially, we incubated the PARP14 catalytic domain (P14) with increasing amounts of NAD+ to isolate individual ADPr modifications (Figure 1b). The resulting spectra clearly show a mass shift of +541 Da as ADPr is attached to P14 (Figure 1c). Notably, there was no statistical difference in the level of PARP14 automodification when samples were quenched with either 6 M urea or 200 μM PJ-34 as compared with 0.1% TFA (Figure S1). Increasing concentrations of NAD+ reveal subsequent mass shifts of +541 Da with each additional ADPr unit that is transferred to the P14 target. To quantify the relative amount of P14 that is modified with 1, 2, or 3 ADPr units, we performed spectral deconvolution for each ADPr modification (Figure 1d). Quantification of the spectra allowed us to compare the percentage of P14 that is ADPr modified within the assayed conditions (Figure 1e). Interestingly, we note that at physiological concentrations of NAD+ (100 μM), P14 is modified at a single site, and this modification represents 8.7% of the protein population. As NAD+ concentrations increase, the fraction of P14 that undergoes modification increases—with 74.4% of P14 modified with at least one ADPr at 400 μM NAD+—but the majority of the protein undergoes only a single modification event. Comparison with a Western blot analysis (Figure 1f) reveals the advantages of the TLC-MALDI method, as it is not possible to distinguish between single and multiple ADPr modifications from the blot. These data demonstrate the utility of our method for quantifying ADPr transfer on contiguous proteins.
Following our observations regarding the activity of P14, we became interested in whether other PARP family members shared P14-like behavior (transferring a single ADPr under physiological concentrations of NAD+) or if they would prove to be more promiscuous. We employed our method to study the behavior of the closely related PARP15 catalytic domain (P15) (Figure 2a,b). Strikingly, P15 is far more promiscuous as compared with P14 in regard to automodification. Whereas we noted minimal labeling of P14 in the presence of 100 μM NAD+, we see robust labeling of P15 in similar conditions (37.9% of P15 is labeled with a single ADPr unit and 2.9% is labeled twice). As we increase the NAD+ concentration, we note that P15 consistently—and significantly—undergoes automodification at more sites and to a greater extent than P14 (Figure 2c). As with P14, this behavior is not observable using traditional Western blot techniques (Figure 2d).
Figure 2.
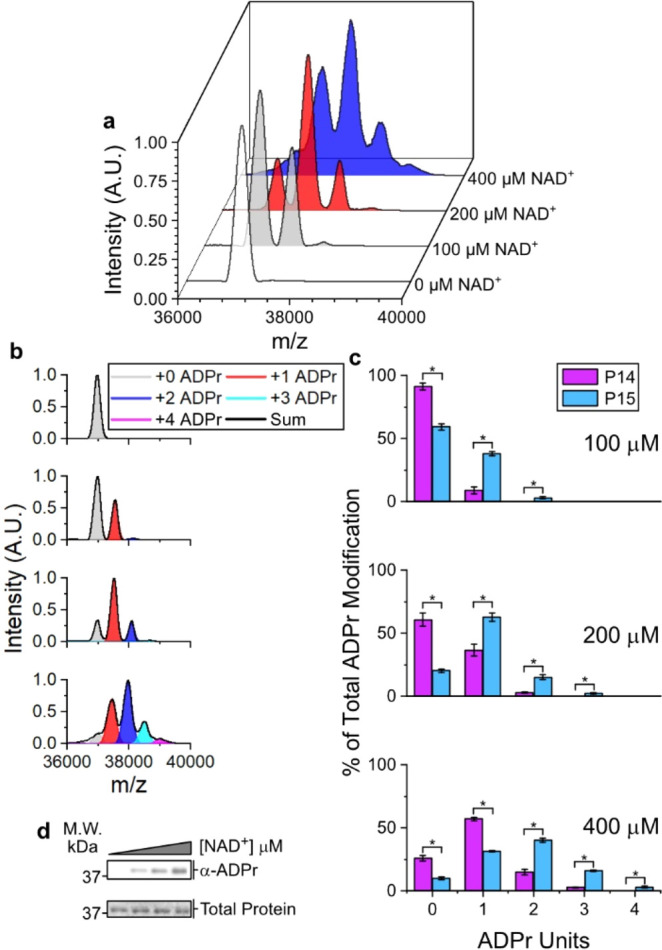
P15 auto-ADP-ribosylates at multiple sites under physiological conditions. (a) P15 was incubated in the presence of increasing concentrations of NAD+ and was subjected to TLC-MALDI to visualize the resulting increase in m/z due to ADP-ribosylation (+541). (b) MS spectra were integrated to determine the relative levels of auto-ADP-ribosylation. (c) Quantification of ADPr levels for both P14 and P15. The bar graphs depict the fraction of the total protein that has been modified at 0, 1, 2, 3, or 4 distinct sites (mean ± S.E.M., n = 3). *represents p-value <0.05, two-tailed Student’s t test. The concentration of NAD+ is given. (d) In vitro P15 ADP-ribosylation assay.
We hypothesized that this difference in behavior could be due to a greater number of modifiable residues on the P15 protein. On the basis of the reported preference of PARPs for glutamates (E) and aspartates (D),7 and our own chemical analysis of P14 and P15 (see below), we compared the prevalence of D/E residues in both the P14 and P15 protein sequence (Figure S2); P14 has 45 D/Es while P15 has 42. We also compared the number of solvent-exposed residues in P14 and P15 and found 14 in P14 and 19 in P15. Intriguingly, D1611 and D1725 (P14 numbering) are conserved between both PARPs, are solvent exposed, and could be potential shared sites for ADP-ribosylation on the catalytic domain. While future efforts should uncover the exact sites of modification, the difference in promiscuity is not simply due to the number of available acceptor residues, but suggests a more complicated relationship between solvent accessible residues, site preference, and overall transfer mechanism. Nonetheless, these data highlight significant differences in automodification between the closely related P14 and P15 catalytic domains.
Given the paucity of information regarding the mechanism of substrate targeting and potential sequence motifs that direct each PARP to its correct modification site, we sought to expand our method to allow us to directly test the proximal interactions at the peptide level that govern target selection. To this end, we synthesized an 18-mer peptide (P14i1) based on the sequence of a known PARP14-preferred modification site from PARP13.21 P14i1 contains a single glutamate acceptor site that undergoes a single ADPr addition (Figure 3a). As with P14 automodification, the relative level of ADPr increases as NAD+ concentration increases (15.2% labeling at 100 μM versus 46.8% labeling at 1 mM). Removal of P14 results in the loss of labeling of the P14i1 peptide. These data confirm previous reporting of this site as a P14 target. Further, our work suggests that P14 can identify target sites absent any distal interactions with the PARP13 protein.
Figure 3.
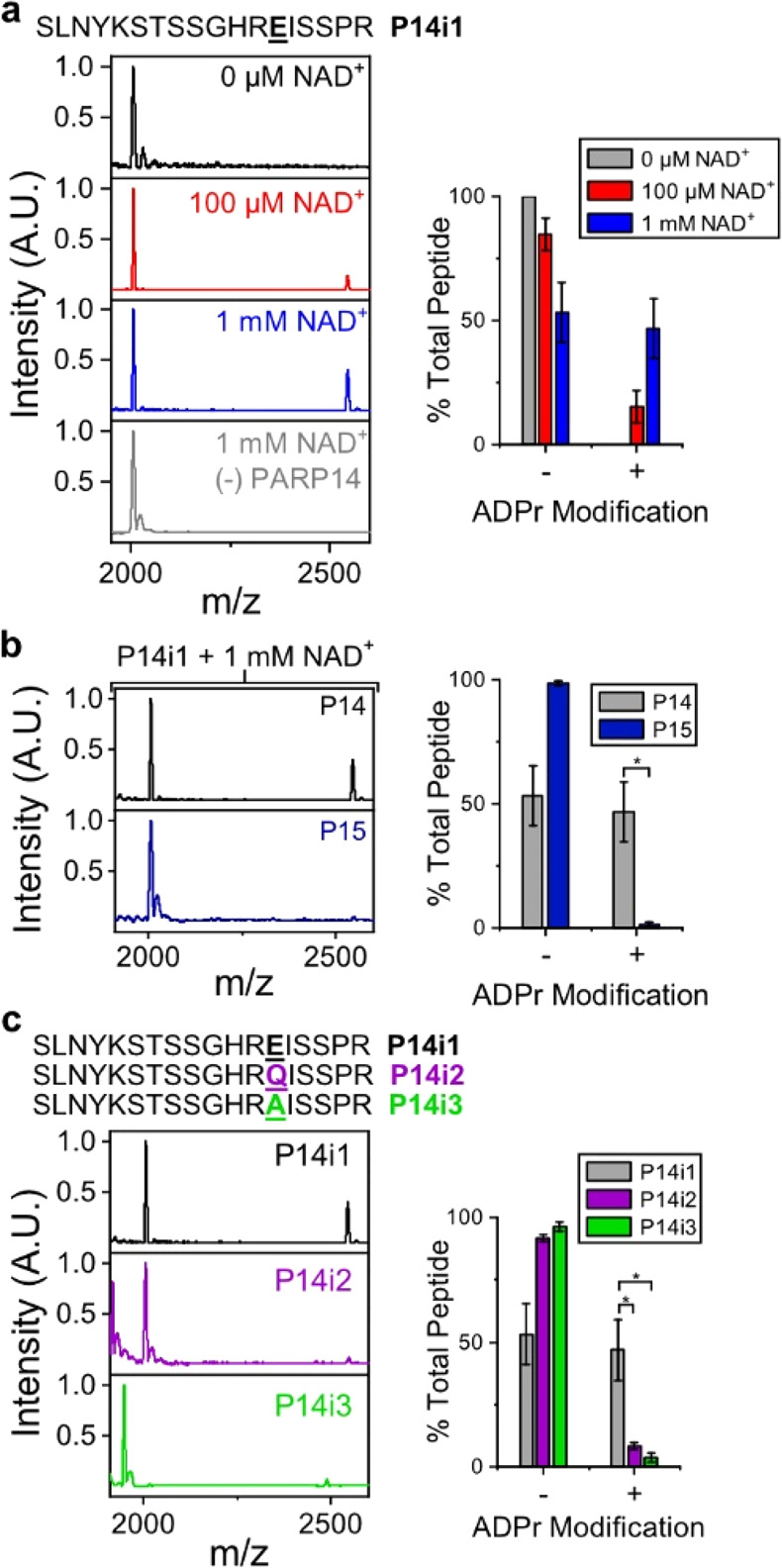
TLC-MALDI performed with minimal peptide fragments reveals proximal PARP selectivity. (a) P14 was incubated with P14i1 in the presence of increasing concentrations of NAD+, and the resulting MS spectra were normalized and plotted against each other to visualize the increase in m/z due to ADPr (+541). The bar graphs depict the fraction of the total peptide that has been modified (mean ± S.E.M., n = 3). The sequence of the P14i1 peptide fragment is provided. (b) P15 is unable to transfer ADPr to the P14i1 peptide. Experiments were performed as in (a) in the presence of 1 mM NAD+. (c) Loss of the acidic modification site prevents ADP-ribosylation. Experiments were performed as in (a) in the presence of 1 mM NAD+. *represents p-value <0.05, two-tailed Student’s t test.
To determine whether a peptide substrate can act as a selective target for different PARP family members, we tested the activity of P15 with P14i1 (Figure 3b). In contrast to the robust labeling we see in the presence of P14 and 1 mM NAD+, we observed almost no ADPr transfer during P15 treatment (1.5% of total P14i1 is ADPr modified). This decrease in ADP-ribosylation is not due to a difference in overall enzyme activity in the presence of P14i1 as both P14 and P15 are fully active in the presence of the peptide (Figure S3a, b). Therefore, the proximal interactions present in P14i1 retain selectivity when comparing the two closely related PARP catalytic domains.
A significant challenge with MS/MS-based ADPr modification site assignment is the difficulty in correctly localizing ADPr. This difficulty is compounded further by conflicting reports of PARP site preference (R, K, H, C, S, Y, D, and E have all been proposed as acceptor sites),5−10 which presents a computational problem when multiple potential acceptor sites occur in close proximity. Using the P14i1 peptide—which has multiple potential acceptor sites in close proximity—we confirm that ADPr transfer is occurring on the sole glutamate site.
After mutating the glutamate to either glutamine (Q, P14i2) or alanine (A, P14i3), residues that cannot accept ADPr, we evaluated ADP-ribosylation in the presence of P14 and 1 mM NAD+ (Figure 3c). In contrast to the labeling we see with P14i1, both peptides lost significant ADP-ribosylation following removal of the E acceptor site (8.3% labeling of P14i2 and 3.7% of P14i3). In the absence of the primary acceptor site, there is some off-target labeling, but it is clearly not preferred to the original acceptor site. Taken together, these data strongly suggest that the previously identified PARP13 site is a glutamate and highlight the utility of TLC-MALDI for rapidly confirming MS/MS identified modification sites.
To confirm that D/E residues were the likely targets of P14 and P15 modification, we employed a chemical approach involving the removal of the ADPr moiety from esters in the presence of hydroxylamine (HA), which has been widely used to confirm ADP-ribosylation on D/E residues (Figure 4a).30,31 We hypothesized that if the ADPr modifications were confined to D/E sites, we could use our method to confirm chemical removal of ADPr from D/E residues based on the loss of one or more ADPr spectral peaks. In the case of both automodified P14 (Figure 4b) and P15 (Figure 4c), treatment of the protein with HA resulted in a near-complete removal of ADPr. Our results were confirmed using Western blot (Figure S4a, b), which displayed a similar pattern of ADPr removal. Further, treatment of the known E acceptor site on P14i1 using HA resulted in the complete loss of the ADPr peak (Figure 4d). Therefore, we surmise that the sites of automodification on both P14 and P15 are predominately D/E residues as their HA-dependent removal mirrors the removal of ADPr from the PARP13 glutamate acceptor site on P14i1. These data demonstrate the feasibility of combining the described TLC-MALDI method with chemical treatment of ADPr-modified substrates to rapidly screen the identity of modified residues (e.g., D/E versus S/C).
Figure 4.
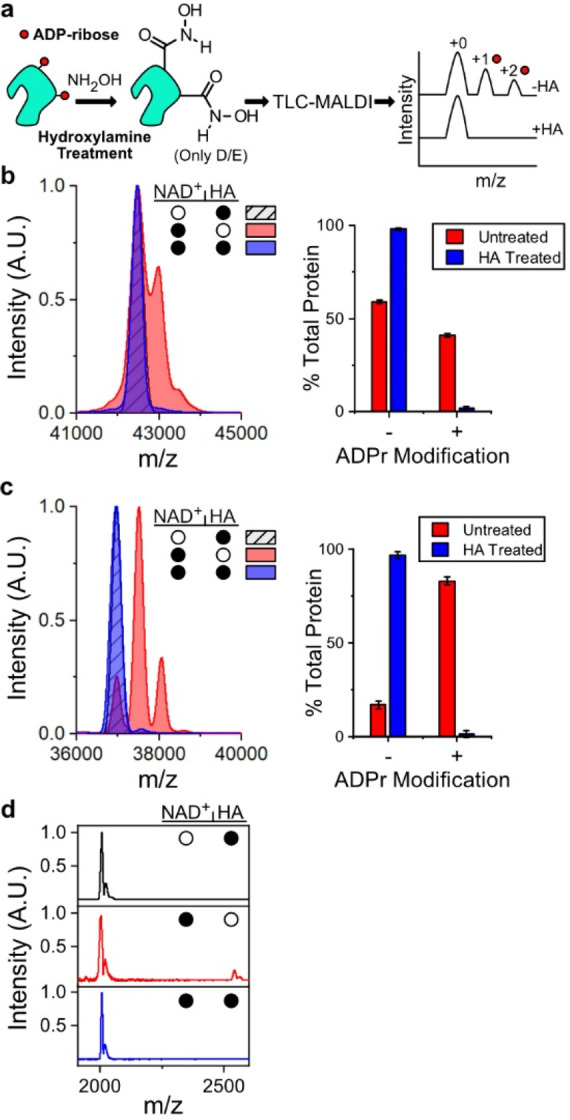
TLC-MALDI performed following HA treatment confirms acidic residues as the targets of P14 and P15 ADP-ribosylation. (a) HA treatment workflow. (b) P14 was incubated with 400 μM NAD+ prior to treatment with HA and was subjected to TLC-MALDI to visualize the resulting increase in m/z due to ADP-ribosylation (+541). The bar graphs depict the fraction of the total protein that has been modified (mean ± S.E.M., n = 3). (c) The same experiment from (a) was performed with P15. (d) P14 was incubated with P14i1 and treated as in (a). The filled circles represent treatment with NAD+, HA, or both.
Herein we demonstrated the adaptability of TLC-MALDI toward proteins and peptides that have undergone ADP-ribosylation. By examining the dynamics of this modification using the closely related catalytic domains of PARP14 and PARP15, we discovered divergent ADPr profiles. P14 vastly prefers a single automodification, while P15 is far more promiscuous. It will be important to determine in the future if this preference is hard-wired into the specific protein (i.e., modification profiles are simply dictated by acceptor residue availability) or if it reflects a governing mechanism that controls PARP activity (i.e., automodification on P14 is inhibitory). Optimizing our method for systems that contain multiple potential substrates will help us address this question and serves as an exciting future adaptation of our technique. The utilization of our method will allow us to start uncovering the fundamental principles that govern PARP activity in the cell.
In addition, TLC-MALDI provides a robust platform for interrogating peptide modification in the presence of various PARPs. We demonstrate a family-member-specific preference for the P14i1 peptide, and researchers can use this technique to screen for the various determinants that govern proximal target discovery within the PARP family—through an alanine scan of P14i1, for example. This will bolster ongoing efforts to discover peptide-based antagonists/agonists for the PARP family and could even find usage in developing site-specific anti-ADPr antibodies.
Finally, our method is compatible with post-ADPr chemical treatments. Using this technique, we were able to confirm a single E residue as the acceptor site on P14i1, and we demonstrated that both P14 and P15 exclusively modify D/E residues in vitro. Interestingly, Nielsen and colleagues recently reported the preference of P14 for Y residues in vivo.6 D/E and Y are biochemically dissimilar potential acceptor sites. The recently elucidated mechanism for histone parylation factor 1 (HPF1)-dependent acceptor site switching with PARP1 provides a potential reconciliation of these differences. HPF1 binds to PARP1 and alters the selection of D/E sites toward S modification.10 Recent structural work by Sun and co-workers demonstrated that HPF1 remodels the PARP1 active site by creating a composite active site shared by PARP1 and HPF1.32 While the switch from D/E to S is not as dramatic as D/E to Y, our findings are compatible with an adapter protein that alters P14 site preference in the cell under certain conditions—similar to HPF1. We envision expanding this technique to include enzymatic treatments of ADP-ribose targets (e.g., incubating treated samples with a glycohydrolase) to evaluate the dynamics of ADP-ribose removal. Taken together, we have demonstrated the utility of a TLC-MALDI approach toward analyzing ADP-ribosylation, and we propose using this technique as a complement in ongoing proteomic-based efforts.
Acknowledgments
We thank K. Wheeler, P. Abbyad, and M. Cohen for many helpful discussions regarding the manuscript and experimental design. This work was funded by the National Institutes of Health (NIH 1R15GM139150) to I.C.-O. and A.A.F.; L.Y.C., S.R.W., M.Y., B.Y., Y.T., D.R., Z.J., S.T., M.Z., and D.L. were supported by internal funding from Santa Clara University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00542.
Additional experimental details, materials, and methods; comparison of quenching reagents (Figure S1), sequence alignment of P14 and P15 (Figure S2), confirmation of P14 and P15 activity in the presence of P14i1 (Figure S3), and in vitro HA ADPr removal assay (Figure S4) (PDF)
Author Contributions
‡ S.R.W. and L.Y.C. contributed equally. The manuscript was written by I.C.-O. Experiments were designed by I.C.-O., L.Y.C., and S.R.W. Experiments were performed by I.C.-O., L.Y.C., S.R.W., M.Y., B.T.Y., Y.M.T., and D.R. and they analyzed the resulting data. Z.J., S.T., and M.Z. performed data analysis. D.R.L. and A.A.F. synthesized/purified the peptide. All authors have given approval to the final version of the manuscript. .
The authors declare no competing financial interest.
Supplementary Material
References
- Citarelli M.; Teotia S.; Lamb R. S. Evolutionary History of the Poly(ADP-Ribose) Polymerase Gene Family in Eukaryotes. BMC Evol. Biol. 2010, 10 (1), 308. 10.1186/1471-2148-10-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger K. M.; Barbieri J. T. The Family of Bacterial ADP-Ribosylating Exotoxins. Clin. Microbiol. Rev. 1995, 8 (1), 34–47. 10.1128/CMR.8.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Hassa P. O.; Lüscher B.; Schüler H.; Koch-Nolte F. Toward a Unified Nomenclature for Mammalian ADP-Ribosyltransferases. Trends Biochem. Sci. 2010, 35 (4), 208–219. 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Vyas S.; Matic I.; Uchima L.; Rood J.; Zaja R.; Hay R. T.; Ahel I.; Chang P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5, 4426. 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidecker O.; Bonfiglio J. J.; Colby T.; Zhang Q.; Atanassov I.; Zaja R.; Palazzo L.; Stockum A.; Ahel I.; Matic I. Serine Is a New Target Residue for Endogenous ADP-Ribosylation on Histones. Nat. Chem. Biol. 2016, 12 (12), 998–1000. 10.1038/nchembio.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch-Larsen S. C.; Hendriks I. A.; Lodge J. M.; Rykær M.; Furtwängler B.; Shishkova E.; Westphall M. S.; Coon J. J.; Nielsen M. L. Mapping Physiological ADP-Ribosylation Using Activated Ion Electron Transfer Dissociation. Cell Rep. 2020, 32 (12), 108176. 10.1016/j.celrep.2020.108176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. S.; Chang P. Insights into the Biogenesis, Function, and Regulation of ADP-Ribosylation. Nat. Chem. Biol. 2018, 14 (3), 236–243. 10.1038/nchembio.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S. C.; Hendriks I. A.; Lyon D.; Jensen L. J.; Nielsen M. L. Systems-Wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24 (9), 2493–2505.e4. 10.1016/j.celrep.2018.07.083. [DOI] [PubMed] [Google Scholar]
- Leslie Pedrioli D. M.; Leutert M.; Bilan V.; Nowak K.; Gunasekera K.; Ferrari E.; Imhof R.; Malmström L.; Hottiger M. O. Comprehensive ADP-Ribosylome Analysis Identifies Tyrosine as an ADP-Ribose Acceptor Site. EMBO Rep. 2018, 19 (8), e45310. 10.15252/embr.201745310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L.; Leidecker O.; Prokhorova E.; Dauben H.; Matic I.; Ahel I. Serine Is the Major Residue for ADP-Ribosylation upon DNA Damage. eLife 2018, 7, e34334. 10.7554/eLife.34334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R.; Liu Z.; Kraus W. L. PARPs and ADP-Ribosylation: Recent Advances Linking Molecular Functions to Biological Outcomes. Genes Dev. 2017, 31 (2), 101–126. 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal J. M. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair 2018, 71, 177–182. 10.1016/j.dnarep.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J.; Li L.; Fattah F. J.; Dong Y.; Bey E. A.; Patel M.; Gao J.; Boothman D. A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryotic Gene Expression 2014, 24 (1), 15–28. 10.1615/CritRevEukaryotGeneExpr.2013006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels C. M.; Ong S.-E.; Leung A. K. L. The Promise of Proteomics for the Study of ADP-Ribosylation. Mol. Cell 2015, 58 (6), 911–924. 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagné J.-P.; Isabelle M.; Lo K. S.; Bourassa S.; Hendzel M. J.; Dawson V. L.; Dawson T. M.; Poirier G. G. Proteome-Wide Identification of Poly(ADP-Ribose) Binding Proteins and Poly(ADP-Ribose)-Associated Protein Complexes. Nucleic Acids Res. 2008, 36 (22), 6959–6976. 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello R.; Leutert M.; Jungmichel S.; Bilan V.; Larsen S. C.; Young C.; Hottiger M. O.; Nielsen M. L. Proteome-Wide Identification of the Endogenous ADP-Ribosylome of Mammalian Cells and Tissue. Nat. Commun. 2016, 7 (1), 12917. 10.1038/ncomms12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S.; Rosenthal F.; Altmeyer M.; Lukas J.; Hottiger M. O.; Nielsen M. L. Proteome-Wide Identification of Poly(ADP-Ribosyl)Ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 52 (2), 272–285. 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Carter-O’Connell I.; Jin H.; Morgan R. K.; David L. L.; Cohen M. S. Engineering the Substrate Specificity of ADP-Ribosyltransferases for Identifying Direct Protein Targets. J. Am. Chem. Soc. 2014, 136 (14), 5201–5204. 10.1021/ja412897a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B. A.; Zhang Y.; Jiang H.; Hussey K. M.; Shrimp J. H.; Lin H.; Schwede F.; Yu Y.; Kraus W. L. Chemical Genetic Discovery of PARP Targets Reveals a Role for PARP-1 in Transcription Elongation. Science 2016, 353 (6294), 45–50. 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wang J.; Ding M.; Yu Y. Site-Specific Characterization of the Asp- and Glu-ADP-Ribosylated Proteome. Nat. Methods 2013, 10 (10), 981–984. 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- Carter-O’Connell I.; Vermehren-Schmaedick A.; Jin H.; Morgan R. K.; David L. L.; Cohen M. S. Combining Chemical Genetics with Proximity-Dependent Labeling Reveals Cellular Targets of Poly(ADP-Ribose) Polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13 (10), 2841–2848. 10.1021/acschembio.8b00567. [DOI] [PubMed] [Google Scholar]
- Rodriguez K. M.; Buch-Larsen S. C.; Kirby I. T.; Siordia I. R.; Hutin D.; Rasmussen M.; Grant D. M.; David L. L.; Matthews J.; Nielsen M. L.; et al. Chemical Genetics and Proteome-Wide Site Mapping Reveal Cysteine MARylation by PARP-7 on Immune-Relevant Protein Targets. eLife 2021, 10, e60480. 10.7554/eLife.60480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Saura A. G.; Schüler H. PARP10 Multi-Site Auto- and Histone MARylation Visualized by Acid-Urea Gel Electrophoresis. Cells 2021, 10 (3), 654. 10.3390/cells10030654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Carbohydrates. Mass Spectrom. Rev. 1999, 18 (6), 349–450. . [DOI] [PubMed] [Google Scholar]
- Ando Y.; Elkayam E.; McPherson R. L.; Dasovich M.; Cheng S.-J.; Voorneveld J.; Filippov D. V.; Ong S.-E.; Joshua-Tor L.; Leung A. K. L. ELTA: Enzymatic Labeling of Terminal ADP-Ribose. Mol. Cell 2019, 73 (4), 845–856.e5. 10.1016/j.molcel.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhaoui I.; Lebedeva N. A.; Zarkovic G.; Saint-Pierre C.; Kutuzov M. M.; Sukhanova M. V.; Matkarimov B. T.; Gasparutto D.; Saparbaev M. K.; Lavrik O. I.; et al. Poly(ADP-Ribose) Polymerases Covalently Modify Strand Break Termini in DNA Fragments in Vitro. Nucleic Acids Res. 2016, 44 (19), 9279–9295. 10.1093/nar/gkw675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadene M.; Chait B. T. A Robust, Detergent-Friendly Method for Mass Spectrometric Analysis of Integral Membrane Proteins. Anal. Chem. 2000, 72 (22), 5655–5658. 10.1021/ac000811l. [DOI] [PubMed] [Google Scholar]
- Fenyo D.; Wang Q.; DeGrasse J. A.; Padovan J. C.; Cadene M.; Chait B. T. MALDI Sample Preparation: The Ultra Thin Layer Method. J. Visualized Exp. 2007, 3, 192. 10.3791/192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signor L.; Boeri Erba E. Matrix-Assisted Laser Desorption/Ionization Time of Flight (MALDI-TOF) Mass Spectrometric Analysis of Intact Proteins Larger than 100 KDa. J. Visualized Exp. 2013, 79, 50635. 10.3791/50635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia J. A.; Tsai S. C.; Adamik R.; Yost D. A.; Hewlett E. L.; Moss J. Amino Acid-Specific ADP-Ribosylation. Sensitivity to Hydroxylamine of [Cysteine(ADP-Ribose)]Protein and [Arginine(ADP-Ribose)]Protein Linkages. J. Biol. Chem. 1985, 260 (30), 16187–16191. 10.1016/S0021-9258(17)36219-1. [DOI] [PubMed] [Google Scholar]
- Zhen Y.; Zhang Y.; Yu Y. A Cell-Line-Specific Atlas of PARP-Mediated Protein Asp/Glu-ADP-Ribosylation in Breast Cancer. Cell Rep. 2017, 21 (8), 2326–2337. 10.1016/j.celrep.2017.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.-H.; Zhao P.; Zhang N.; Kong L.-L.; Wong C. C. L.; Yun C.-H. HPF1 Remodels the Active Site of PARP1 to Enable the Serine ADP-Ribosylation of Histones. Nat. Commun. 2021, 12 (1), 1028. 10.1038/s41467-021-21302-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.