

Abstract

The electrochemical synthesis of aryl azoles was performed for the first time in a microflow reactor. The reaction relies on the anodic oxidation of the arene partners making these substrates susceptible for C–H functionalization with azoles, thus requiring no homogeneous transition-metal-based catalysts. The synthetic protocol benefits from the implementation of a microflow setup, leading to shorter residence times (10 min), compared to previously reported batch systems. Various azolated compounds (22 examples) are obtained in good to excellent yields.
N-Aryl azoles represent an important recurring motif in various biologically active molecules, relevant for medicinal chemistry,1−4 and crop-protection science.5 Consequently, the C–N cross-coupling between arenes and azoles has been widely studied. Traditionally, its synthesis relied on the use of transition metal catalysts, thus requiring prefunctionalized substrates, meticulous ligand design as well as the addition of oxidants and other additives (Scheme 1A).6−19 Other possibilities20−22 to prepare N-aryl azoles include SNAr-type reactions for electron-deficient arenes23−25 and the use of iodine26,27 or hypervalent iodine species.28,29
Scheme 1. Recent Developments in the Azolation of Arenes.

(A) classical coupling reactions enabled by transition metals; (B) photocatalytic approaches; (C) electrochemical strategies; (D) photoelectrochemical approach; and (E) our work on the development of an electrochemical azolation of electron-rich and -neutral arenes in flow.
In 2015, an important photocatalytic C–H functionalization methodology using an acridinium salt as photocatalyst was reported by Nicewicz.30 The strategy allowed for the regioselective azolation after the initial oxidation of electron-rich arenes. After this work, the development of various photocatalytic azolation strategies of arenes was intensified, using a diverse set of homogeneous and heterogeneous photocatalysts (Scheme 1B).31−36
As an alternative to photocatalytic oxidation strategies, electrochemical methods37 have been developed to enable the amination of C(sp)–H,38 C(sp2)–H,39−43 and benzylic C(sp3)–H bonds.44,45 In fact, synthetic electrochemistry offers several advantages compared to traditional synthetic methods: (i) electrons can be considered as traceless reagents, (ii) no external oxidants or homogeneous metal catalysts are needed, and (iii) the selectivity can be tuned via the applied potential.46−49 Moreover, many technical issues previously related to electrochemical systems are now considered overcome, spurring renewed interest in electrochemical synthetic chemistry.50 Concerning electrochemical azolation reactions (Scheme 1C), several notable strategies have been developed in the past years by the groups of Yoshida,51 Devillers,52 Li,53 Lei,54,55 Wu,56 and Liu.57 Also, electrophotochemical approaches to couple arenes and azoles have been realized by Lambert et al.58 and Hu et al.59 (Scheme 1D).
In these batch-based electrochemical transformations, two recurring drawbacks can be distinguished: (i) the need for extended reaction times and/or higher temperatures, which can lead to reduced reaction selectivity; (ii) the transformation is often tailored to a specific class of substrates.55,57 Considering these insights, we anticipated that flow technology could possibly alleviate those issues (Scheme 1E). Due to the narrow interelectrode gap, the amount of supporting electrolytes can substantially be reduced in electrochemical microreactors.60,61 Furthermore, the high electrode surface-to-volume ratio allows for accelerating the reaction rate significantly, leading to reduced reaction times and less side-product formation.62,63 Here, we describe the first electrochemical dehydrogenative cross-coupling of arenes with azoles in a microflow reactor.64
Our initial investigations commenced by subjecting 1,3,5-trimethylbenzene and pyrazole as model substrates to the electrochemical transformation in a microflow reactor (see Supporting Information). The electrochemical microflow reactor was equipped with a carbon anode and a stainless-steel cathode separated by a Teflon gasket, resulting in an interelectrode gap of 250 μm and a volume of 700 μL (see Supporting Information).65 After optimizing various reaction and process parameters (see Supporting Information for full details), 67% of the desired cross-coupled product 1 could be isolated after exposing a solution of pyrazole and 1,3,5-trimethylbenzene (6 equiv) in hexafluoro-2-propanol (HFIP)/CH2Cl2 (7:3) for only 10 min to the galvanostatic conditions (20 mA, corresponding to 0.71 mA cm–2 and 2.6 F mol–1, Table 1, entry 1). Other solvent systems can be used for the electrochemical azolation of arenes without significant reduction in conversion and yield (Table 1, entries 2 and 3). However, we found that, during the scope exploration, the combination of HFIP/CH2Cl2 (7:3) provided optimal results in terms of conversion, stability, and solubility of the starting materials and the corresponding products. It should be further noted that HFIP is a common solvent in electrochemistry, owing to its acidity, the ability to form hydrogen bonds, and redox stability.66−68 Shortening the residence time was detrimental to the reaction outcome, even at higher current densities (Table 1, entries 4 and 5). This might be due to the formation of larger amounts of byproducts. Further, control experiments revealed that electricity is crucial to induce the observed cross-coupling between 1,3,5-trimethylbenzene and pyrazole (Table 1, entry 6). Finally, the reaction was also carried out in batch using a commercially available Electrasyn, resulting in 65% conversion, with 50% yield for target compound 1 after 1 h (Table 1, entry 7), despite the higher current density.61
Table 1. Optimization of the Reaction Conditions and Control Experiments.

| entry | variations | yieldb (%) |
|---|---|---|
| 1 | nonea | 70 (67)c |
| 2 | LiClO4 (0.1 M),AcOH (10 equiv), CH3CN | 60 |
| 3 | LiClO4 (0.1 M), TFE/CH3OH 4:1 | 71 (67)c |
| 4 | tR = 5 min, 20 mA | 37 |
| 5 | tR = 5 min, 40 mA | 25 |
| 6 | no electricity | |
| 7 | batchd | 50 |
Optimized reaction conditions: pyrazole (0.67 mmol), mesitylene (6.0 equiv), LiClO4 (0.1 M) or Bu4NPF6 (0.05 M), solvent mixture (10 mL), graphite anode/Fe cathode, 20 mA (0.71 mA cm–2, 2.6 F mol–1).
Yield determined by GC-FID with diglyme as an internal standard.
Yield after column chromatography.
Batch reaction conditions: Electrasyn, 7.2 mA cm–2, 2.0 F mol–1, C anode/Fe cathode, pyrazole (0.33 mmol), HFIP/DCM 7:3 (5.0 mL), Bu4NPF6 (0.05 M), 1 h reaction time.
With the optimal conditions in hand, we sought to evaluate the generality of this flow-based electrochemical azolation protocol (Scheme 2). Various electron-neutral arenes could be coupled with pyrazole in good to excellent yields (1–4, 45–90% yields). Product 1 was obtained in 76% yield (219 mg), and also 34% of the mesitylene starting material could be recovered after column chromatography. The reaction is not particularly sensitive to steric hindrance as showcased by compounds 1 and 4. Arenes bearing a free hydroxyl moiety can be readily coupled with pyrazole yielding the targeted cross coupled product in an excellent yield (5, 98% yield). When 1,3,5-methoxybenzene was used as starting material, only homocoupling product was observed by GC–MS, but no product 6 was detected. Anisole and meta-xylene were both efficiently oxidized and coupled with pyrazole, although the corresponding products were isolated as a mixture of inseparable regioisomers (7 and 8, respectively), with a higher selectivity for the ortho-coupled regioisomer in the case of 7 and the 1,4-substituted compound for 8.56,59 The lower yields can be ascribed to the formation of byproducts derived from homocoupling or oxidation of benzylic positions. Various substituted anisole-derivatives bearing formyl, ester, acetyl, and amide functionalities served as competent coupling partners as well (9–12, 50–80% yields). Arenes bearing only electron-withdrawing moieties, e.g., acetophenone (13), did not lead to any product formation, which can be attributed to the high oxidation potential of the arene.
Scheme 2. Substrate Scope for the Electrochemical Dehydrogenative C–N Coupling between Arenes and Azoles in a Microflow Reactor.
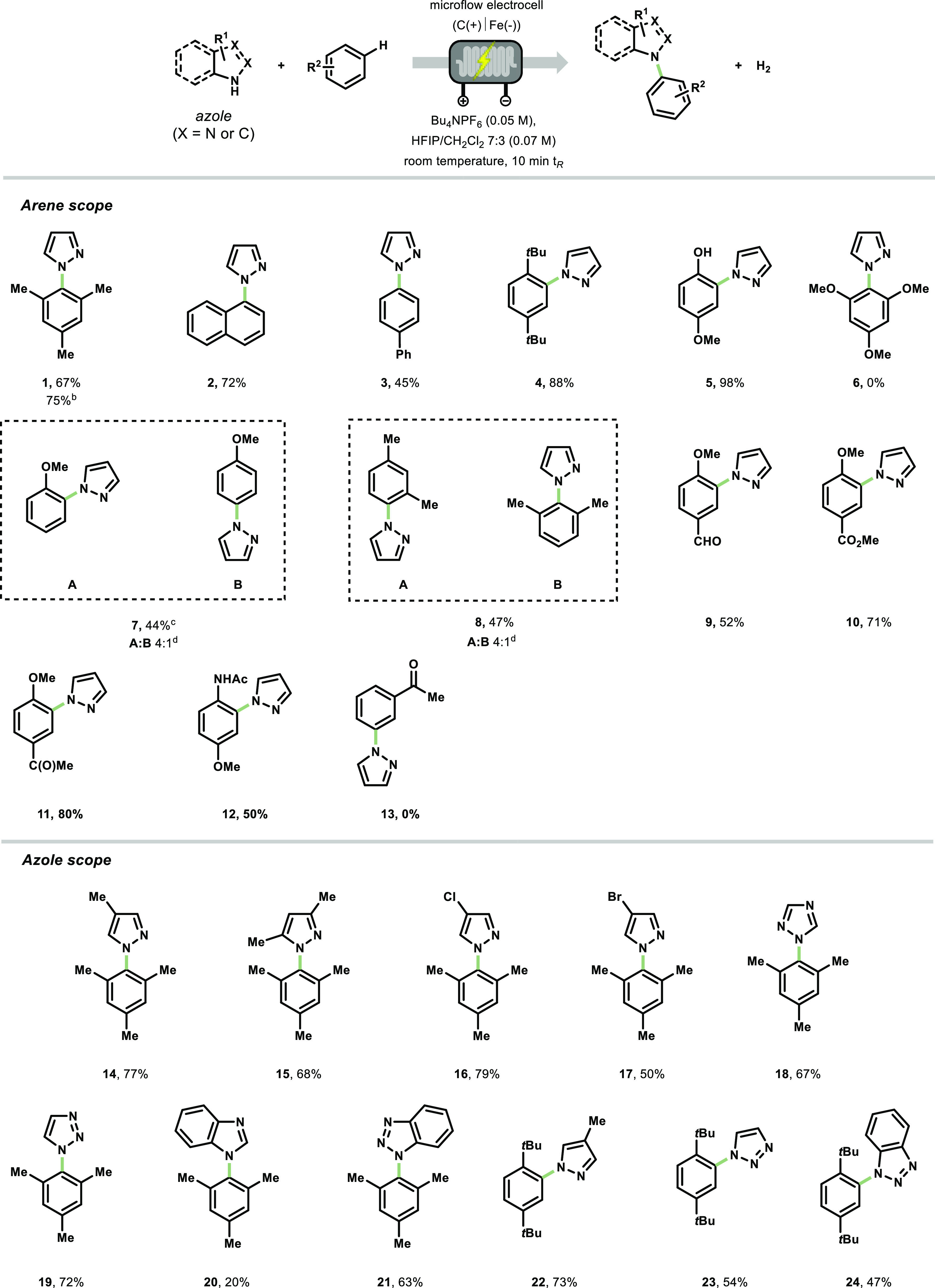
Reaction conditions: The feed contains an azole (0.67 mmol), an arene (6.0 equiv), Bu4NPF6 (0.05 M) in 10 mL of HFIP/CH2Cl2 7:3. The residence time was set at 10 min, and the system was kept at room temperature with I = 20–30 mA (0.71–1.1 mA cm–2). Reported yields are those obtained after column chromatography.
On a 1.54 mmol scale (219 mg of product collected).
The product was prepared in TFE/MeOH 4:1 (0.067 M) using LiClO4 (0.1 M) as a supporting electrolyte.
Regioisomeric ratio measured by 1H NMR of the pure product.
Similarly, we investigated the scope of the azole coupling partner (Scheme 2, azole scope). Alkyl-bearing pyrazoles provided the targeted cross-coupled products in good yields (14–15, 68–77% yields), showing again no particular sensitivity to steric hindrance (e.g., 15). Pyrazoles decorated with halogens were also suitable nucleophiles (16–17, 65–78% yields), providing functional handles in the target molecule to enable further diversification using classical cross-coupling strategies.69 The electrochemical coupling reaction with triazoles afforded products 18 and 19 in comparable yields to those obtained with pyrazoles (67–72% yields). When benzimidazole was employed, product 20 was isolated in 20% yield, whereas the C–N coupled product 21 derived from mesitylene and benzotriazole was obtained in 65% yield. Finally, various azole nucleophiles were combined with the sterically hindered 1,4-di-tert-butylbenzene affording the targeted products in synthetically useful yields (22–24, 47–73% yields).
On the basis of our observations and literature data,56,59 we propose the following mechanism to be operative in the electrochemical flow reactor (Scheme 3): the arene is oxidized at the graphite anode (+2.07 vs saturated calomel electrode (SCE) for mesitylene),70 affording the aryl radical cation I. This species can be subsequently attacked by the nucleophilic azole, leading to the formation of the new C–N bond. Through a deprotonation step, II will generate the radical species III, which will finally afford the azolated product 1, after losing a second proton and an electron. Concerning the cathodic reaction, protons are reduced to form molecular hydrogen as a synthetically useful byproduct.71 This mechanism is also supported by experimental observations, such as the formation of homocoupled products of the arene observed by GC–MS. However, at this stage, we cannot completely rule out the oxidation of both the arene and the azole, which would lead to the product through a radical–radical coupling as suggested by others.57 However, due to the high oxidation potential of pyrazole (+2.21 vs saturated calomel electrode (SCE)),70 we believe this latter scenario is rather unlikely under our reaction conditions for at least the majority of the presented substrates.
Scheme 3. Plausible Mechanism for the Electrochemical Dehydrogenative Azolation of Arenes.

In conclusion, we reported the first electrochemical azolation of arenes in flow. The C–H functionalization protocol displays a broad scope, reaches high yields in only 10 min, and does not require the addition of any homogeneous catalysts to forge the targeted carbon–nitrogen bond. Given its operational simplicity and reduced reaction times, we anticipate that this electrochemical azolation strategy will find use in modern medicinal chemistry settings.
Experimental Section
All reagents and solvents were used as received without further purification. Reagents and solvents were bought from Sigma-Aldrich, TCI, and Fluorochem. Technical solvents were bought from VWR International and Biosolve and were used as received. All capillary tubing and microfluidic fittings were purchased from IDEX Health & Science. Disposable syringes were from BD Discardit II or NORMJECT, purchased from VWR Scientific. Syringe pumps were purchased from Chemix Inc. model Fusion 200 Touch. Product isolation was performed manually, using silica (60, F254, Merck). TLC analysis was performed using silica on aluminum foils TLC plates (F254, Supelco Sigma-Aldrich) with visualization under ultraviolet light (254 and 365 nm). 1H (400 MHz) and 13C (100 MHz) spectra were recorded at an ambient temperature using a BrukerAvance 400. 1H NMR spectra are reported in parts per million (ppm) downfield relative to CDCl3 (7.26 ppm), and all 13C NMR spectra are reported in ppm relative to CDCl3 (77.16 ppm), unless stated otherwise. NMR spectra use the following abbreviations to describe the multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, h = hextet, hept = heptet, m = multiplet, dd = double doublet, td = triple doublet. NMR data were processed using the MestReNova 9.0.1 software package. Known products were characterized by comparing to the corresponding 1H NMR and 13C NMR from literature. GC analyses were performed on a GC–MS combination (Shimadzu GC-2010 Plus coupled to a Mass Spectrometer; Shimadzu GCMS-QP 2010 Ultra) with an autosampler unit (AOC-20i, Shimadzu). Melting points were determined with a Buchi B-560 capillary melting point apparatus in open capillaries and are uncorrected. The names of all products were generated using the PerkinElmer ChemBioDraw Ultra v.18.0. software package. For all electrochemical continuous flow reactions, a homemade flow cell was used, together with a Velleman LABPS3005D power supply. The cell consists of a working electrode and a counter electrode, with a PTFE (Polytetrafluoroethylene) gasket (0.25 mm thickness) containing microchannels in between. The material used for the electrodes were the stainless steel electrode (316L) and the Graphite AC-K800 premium grade (purchased by AgieCharmilles). The active reactor volume is 700 μL. This results in an undivided electrochemical cell. In the cell, direct contact between the electrode surface and the reaction mixture is established. The reaction mixture is pumped through the system via a syringe pump and is collected in a glass vial. All the technical data of the electrochemical microreactor are reported elsewhere.65
General Procedure A: Electrochemical C–N Coupling in Flow
The heteroarene (0.665 mmol), tetrabutylammonium hexafluorophosphate (Bu4NPF6, 0.05 M, 194 mg), and the arene, if solid, (3–6 equiv) were dissolved in 10 mL of stock solution (7:3 v/v HFIP/DCM). In the case that the arene was liquid, the heteroarene and the electrolyte were first dissolved in 5 mL of stock solution, and then the arene was added. Finally, the solution was brought up to 10 mL. The mixture was sonicated until homogeneous and taken up in a 10 mL disposable syringe. The solution was pumped through the electrochemical reactor with a fixed flow rate of 0.07 mL/min to give a residence time of 10 min in the active part of the reactor, which was equipped with a graphite anode and steel cathode. After applying the appropriate current, based on the results of a previous voltammogram, the first fraction corresponding to the first 20 min was discarded. The reaction mixture was then collected in a vial, and after an additional 45–70 min, the reaction was stopped. The power supply was turned off, and the reactor was washed with CH3CN (10 at 0.5 mL/min). The crude mixture was evaporated and purified using silica gel column chromatography.
General Procedure B: Electrochemical C–N Coupling in Flow
The heteroarene (0.665 mmol), lithium perchlorate (LiClO4, 0.1 M, 107 mg), and the arene, if solid, (3–6 equiv) were dissolved in 10 mL of stock solution (4:1 v/v TFE/MeOH). In case that the arene was liquid, the heteroarene and the electrolyte were first dissolved in 5 mL of stock solution, the arene was added, and finally, the solution was brought up to 10 mL. The mixture was sonicated until homogeneous and taken in a 10 mL disposable syringe. The solution was pumped through the electrochemical setup with a fixed flow rate of 0.07 mL/min to give a residence time of 10 min in the active part of the reactor, which was equipped with a graphite anode and steel cathode. After applying the appropriate current, based on the results of a previous voltammogram, the first fraction corresponding to the first 20 min was discarded. The reaction mixture was then collected in a vial, and after an appropriate time (45–70 min), the reaction was stopped. The power supply was turned off, and the reactor was washed with CH3CN (10 at 0.5 mL/min, see Supplementary Information for cleaning procedures). The crude mixture was evaporated and purified using silica gel column chromatography.
1-Mesityl-(1H-pyrazole) (1)
Following continuous-flow general procedure B (20 mA), 4.0 mL of the mixture was collected (theoretical yield = 49 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 97:3) to give a white solid (33 mg, 67% yield). The analytical data correspond to the reported ones in literature.581H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 1.8 Hz, 1H), 7.43 (d, J = 2.3 Hz, 1H), 6.94 (s, 2H), 6.43 (t, J = 2.1 Hz, 1H), 2.33 (s, 3H), 1.96 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 140.0, 138.8, 137.0, 136.0, 131.0, 128.9, 105.8, 21.2, 17.3.
1-(Naphthalen-1-yl)-(1H-pyrazole) (2)
Following the general procedure A (20 mA), 3.0 mL of the mixture was collected (theoretical yield = 38.7 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a colorless oil (28 mg, 72% yield). The analytical data correspond to the reported ones in literature.561H NMR (400 MHz, CDCl3): δ 7.94–7.92 (m, 2H), 7.86 (d, J = 1.9 Hz, 1H), 7.82–7.79 (m, 2H), 7.57–7.49 (m, 4H), 6.55 (t, J = 2.1 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 140.8, 137.4, 134.4, 131.8, 129.3, 129.1, 128.2, 127.4, 126.8, 125.2, 123.4, 123.3, 106.7.
1-([1,1′-Biphenyl]-4-yl)-(1H-pyrazole) (3)
Following the general procedure A (15 mA), 3.8 mL of the mixture was collected (theoretical yield = 55.6 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 4:1) to give a yellowish oil (25 mg, 45% yield). The analytical data correspond to the reported ones in literature.591H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 2.5 Hz, 1H), 7.81–7.73 (m, 3H), 7.71–7.65 (m, 2H), 7.65–7.58 (m, 2H), 7.46 (dd, J = 8.4, 6.8 Hz, 2H), 7.42–7.33 (m, 1H), 6.50 (t, J = 2.1 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 141.3, 140.2, 139.5, 139.4, 129.0, 128.2, 127.6, 127.1, 126.8, 119.6, 107.8.
1-(2,5-Di-tert-butylphenyl)-(1H-pyrazole) (4)
Following the general procedure A (25 mA), 4.0 mL of the mixture was collected (theoretical yield = 68.1 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a white solid (60 mg, 88% yield). Mp: 47.5–48 °C. 1H NMR (400 MHz, CDCl3): δ 7.58 (d, J = 1.8 Hz, 1H), 7.45 (d, J = 2.3 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.33 (dd, J = 8.5, 2.3 Hz, 1H), 6.98 (d, J = 2.3 Hz, 1H), 6.31 (t, J = 2.1 Hz, 1H), 1.21 (s, 9H), 1.06 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3): δ 149.5, 144.0, 139.2, 139.1, 132.5, 127.7, 127.2, 126.4, 105.8, 35.4, 34.3, 31.4, 31.2. HRMS (ESI) m/z [M + H]+ calcd for C17H25N2, 257.2018; found, 257.2005.
4-Methoxy-2-(1H-pyrazol-1-yl)phenol (5)
Following the general procedure A (30 mA), 3.6 mL of the mixture was collected (theoretical yield = 45.5 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane/ethyl acetate 9:1 to 1:1) to give a white solid (45 mg, 98% yield). The analytical data correspond to the reported ones in literature.571H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 2.6 Hz, 1H), 7.72 (d, J = 1.9 Hz, 1H), 7.03 (d, J = 8.9 Hz, 1H), 6.92 (d, J = 2.9 Hz, 1H), 6.76 (dd, J = 8.9, 2.8 Hz, 1H), 6.49 (t, J = 2.3 Hz, 1H), 3.80 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 152.7, 143.3, 139.2, 126.9, 124.9, 119.4, 112.8, 107.0, 104.5, 56.1.
1-(2-Methoxyphenyl)-(1H-pyrazole (7A) and 1-(4-Methoxyphenyl)-(1H-pyrazole) (7B)
Following the general procedure B (20 mA), 2.4 mL of the mixture was collected (theoretical yield = 27.8 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 97:3) to give a yellowish oil (12 mg, 44%, A/B 4:1). The analytical data correspond to the reported ones in literature.72 Compound 7A1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 2.4 Hz, 1H), 7.72 (m, 2H), 7.30 (td, J = 7.9, 1.7 Hz, 1H), 7.06–7.03 (m, 2H), 6.43 (t, J = 2.1 Hz, 1H), 3.88 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, mixture of A and B): δ 158.3, 151.4, 140.7, 140.2, 131.7, 129.9, 128.1, 126.9, 125.4, 121.3, 121.0, 114.6, 112.4, 107.3, 106.3, 56.1, 55.7. Compound 7B1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 1.8 Hz, 1H), 7.61–7.54 (m, 2H), 6.98–6.93 (m, 2H), 6.44 (m, 1H), 3.84 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, mixture of A and B) 158.3, 151.4, 140.7, 140.2, 131.7, 129.9, 128.1, 126.9, 125.4, 121.3, 121.0, 114.6, 112.4, 107.3, 106.3, 56.1, 55.7.
1-(2,4-Dimethylphenyl)-(1H-pyrazole (8A) and 1-(2,6-Dimethylphenyl)-(1H-pyrazole) (8B)
Following the general procedure A (15 mA), 4.5 mL of the mixture was collected (theoretical yield = 51.5 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a yellowish oil (24 mg, 47%, A/B 4:1). The analytical data correspond to the reported ones in literature.58 Compound 8A1H NMR (400 MHz, CDCl3): δ 7.73 (s, 1H), 7.59–7.58 (m, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.14 (s, 1H), 7.09 (d, J = 8.1 Hz, 1H), 6.44 (s, 1H), 2.39 (s, 3H), 2.21 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, mixture of A and B): δ 140.1, 138.5, 137.7, 136.4, 133.6, 132.0, 130.8, 129.1, 128.3, 127.2, 126.1, 106.1, 21.2, 18.0, 17.4. Compound 8B1H NMR (399 MHz, CDCl3): δ 7.76 (s, 1H), 7.48 (s, 1H), 7.28 (s, 2H), 7.16 (s, 1H), 6.47 (s, 1H), 2.02 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3, mixture of A and B): δ 140.1, 138.5, 137.7, 136.4, 133.6, 132.0, 130.8, 129.1, 128.3, 127.2, 126.1, 106.1, 21.2, 18.0, 17.4.
4-Methoxy-3-(1H-pyrazol-1-yl)benzaldehyde (9)
Following the general procedure A (20 mA), 4.0 mL of the mixture was collected (theoretical yield = 53.7 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane/ethyl acetate 9:1 to 4:1) to give a yellow oil (28 mg, 52% yield). 1H NMR (400 MHz, CDCl3): δ 9.95 (s, 1H), 8.29 (d, J = 2.1 Hz, 1H), 8.07 (d, J = 2.5 Hz, 1H), 7.87 (dd, J = 8.5, 2.1 Hz, 1H), 7.74 (d, J = 1.9 Hz, 1H), 7.18 (d, J = 8.6 Hz, 1H), 6.46 (t, J = 2.1 Hz, 1H), 4.00 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 190.4, 155.9, 140.7, 131.6, 130.3, 130.2, 129.3, 127.4, 112.5, 106.9, 56.6. HRMS (ESI) m/z [M + H]+ calcd for C11H11N2O2, 203.0821; found, 203.0816.
Methyl 4-Methoxy-3-(1H-pyrazol-1-yl)-benzoate (10)
Following the general procedure A (20 mA), 4.3 mL of the mixture was collected (theoretical yield = 66 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a white solid (47 mg, 71% yield). Mp: 95–96 °C. 1H NMR (399 MHz, CDCl3): δ 8.40 (s, 1H), 8.03–8.01 (m, 2H), 7.73 (s, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.44 (s, 1H), 3.95 (s, 3H), 3.90 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 166.3, 154.9, 140.5, 131.6, 130.0, 129.5, 126.9, 123.4, 111.8, 106.6, 56.3, 52.2. HRMS (ESI) m/z [M + H]+ calcd for C12H13N2O3, 233.0926; found, 233.0924.
1-(4-Methoxy-3-(1H-pyrazol-1-yl)phenyl)ethan-1-one (11)
Following the general procedure A (30 mA), 4.2 mL of the mixture was collected (theoretical yield = 60.3 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a white solid (48 mg, 80% yield). Mp 71.5–72.3 °C. 1H NMR (400 MHz, CDCl3): δ 8.31 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 2.5 Hz, 1H), 7.94 (dd, J = 8.7, 2.3 Hz, 1H), 7.72 (d, J = 1.8 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.44 (t, J = 2.2 Hz, 1H), 3.95 (s, 3H), 2.58 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 196.4, 154.9, 140.5, 131.6, 130.7, 129.5, 128.5, 126.0, 111.9, 106.7, 56.3, 26.6. HRMS (ESI) m/z [M + H]+ calcd for C12H13N2O2, 217.0977; found, 217.0965.
N-(4-Methoxy-2-(1H-pyrazol-1-yl)phenyl)acetamide (12)
Following the general procedure A (30 mA), 4.6 mL of the mixture was collected (theoretical yield = 70 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane/ethyl acetate 1:1) to give a yellow oil (34 mg, 50% yield). The analytical data correspond to the reported ones in literature.571H NMR (400 MHz, CDCl3): δ 9.91 (s, 1H), 8.28 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 2.2 Hz, 2H), 6.89 (dd, J = 9.0, 2.8 Hz, 1H), 6.84 (d, J = 2.8 Hz, 1H), 6.49 (t, J = 2.2 Hz, 1H), 3.82 (s, 3H), 2.10 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 168.4, 156.1, 141.2, 130.3, 130.2, 124.9, 124.7, 112.7, 109.0, 107.3, 55.8, 24.9.
1-Mesityl-4-methyl-(1H-pyrazole) (14)
Following the general procedure A (20 mA), 3.5 mL of the mixture was collected (theoretical yield = 46.5 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a colorless oil (36 mg, 77% yield). 1H NMR (399 MHz, CDCl3): δ 7.50 (s, 1H), 7.15 (s, 1H), 6.86 (s, 2H), 2.25 (s, 3H), 2.11 (s, 3H), 1.91 (s, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 140.5, 138.6, 137.2, 136.0, 129.6, 128.8, 116.2, 21.2, 17.3, 9.0 HRMS (ESI) m/z [M + H]+ calcd for C13H17N2, 201.1392; found, 201.1383.
1-Mesityl-3,5-dimethyl-(1H-pyrazole) (15)
Following the general procedure A (10 mA), 3.0 mL of the mixture was collected (theoretical yield = 42.7 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 95:5) to give a colorless oil (29 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 6.92 (s, 2H), 5.95 (s, 1H), 2.31 (s, 3H), 2.28 (s, 3H), 1.96 (s, 3H), 1.92 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 148.6, 140.1, 138.6, 136.5, 135.4, 128.8, 104.5, 21.2, 17.3, 13.8, 11.0. HRMS (ESI) m/z [M + H]+ calcd for C14H19N2, 215.1548; found, 215.1538
4-Chloro-1-mesityl-(1H-pyrazole) (16)
Following the general procedure A (20 mA), 3.8 mL of the mixture was collected (theoretical yield = 55.7 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a dark yellow oil (44 mg, 79% yield). The analytical data correspond to the reported ones in literature.581H NMR (400 MHz, CDCl3): δ 7.65 (s, 1H), 7.43 (s, 1H), 6.94 (s, 2H), 2.33 (s, 3H), 1.99 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 139.4, 138.6, 136.6, 135.8, 129.0, 128.9, 110.5, 21.2, 17.3.
1-Mesityl-4-bromo-(1H-pyrazole) (17)
Following the general procedure A (10 mA), 4.2 mL of the mixture was collected (theoretical yield = 74 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 98:2) to give a yellow oil (37 mg, 50% yield).581H NMR (399 MHz, CDCl3): δ 7.68 (s, 1H), 7.46 (s, 1H), 6.94 (s, 2H), 2.33 (s, 3H), 1.98 (s, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 140.8, 139.4, 136.6, 135.8, 131.1, 129.0, 93.7, 21.2, 17.4.
1-Mesityl-(1H-1,2,4-triazole) (18)
Following the general procedure A (20 mA), 4.6 mL of the mixture was collected (theoretical yield = 56.3 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane/ethyl acetate 4:1 to 1:1) to give a white solid (38 mg, 67% yield).731H NMR (400 MHz, CDCl3): δ 8.21 (s, 1H), 8.19 (s, 1H), 6.98 (s, 2H), 2.35 (s, 3H), 1.97 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 151.9, 144.4, 140.2, 135.6, 133.0, 129.3, 21.2, 17.4.
1-Mesityl-(1H-1,2,3-triazole) (19)
Following the general procedure A (20 mA), 5.0 mL of the mixture was collected (theoretical yield = 62.2 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane/ethyl acetate 9:1 to 4:1) to give a white solid (45 mg, 72% yield). Mp: 188–189 °C. 1H NMR (400 MHz, CDCl3): δ 7.87 (s, 1H), 7.62 (s, 1H), 6.98 (s, 2H), 2.34 (s, 3H), 1.93 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 140.1, 135.2, 133.7, 133.5, 129.2, 125.5, 21.2, 17.3. HRMS (ESI) m/z [M + H]+ calcd for C11H14N3, 188.1188; found, 188.1187.
1-Mesityl-(1H-benzo[d]imidazole) (20)
Following the general procedure A (20 mA), 3.8 mL of the mixture was collected (theoretical yield = 59.6 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a yellow oil (12 mg, 20% yield). The analytical data correspond to the reported ones in literature.741H NMR (400 MHz, CDCl3): δ 7.89 (d, J = 8.0 Hz, 1H), 7.87 (s, 1H), 7.35–7.24 (m, 2H), 7.05 (s, 2H), 7.03 (d, J = 7.7 Hz, 1H), 2.39 (s, 3H), 1.92 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 143.4, 143.2, 139.4, 136.4, 134.3, 131.2, 129.5, 123.6, 122.5, 120.5, 110.3, 21.3, 17.6.
1-Mesityl-(1H-benzo[d][1,2,3]triazole) (21)
Following the general procedure A (25 mA), 3.4 mL of the mixture was collected (theoretical yield = 53.6 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a yellowish oil (34 mg, 63% yield). The analytical data correspond to the reported ones in literature.741H NMR (400 MHz, CDCl3): δ 8.16 (d, J = 8.2, 1H), 7.44 (dddd, J = 24.5, 8.0, 6.9, 1.1 Hz, 2H), 7.20 (d, J = 8.2, 1H), 7.06 (s, 2H), 2.40 (s, 3H), 1.87 (s, 6H). 13C{1H} NMR (101 MHz, CDCl3): δ 145.5, 140.3, 136.3, 134.0, 131.7, 129.4, 128.1, 124.1, 120.2, 109.9, 21.3, 17.5.
1-(2,5-Di-tert-butylphenyl)-4-methyl-(1H-pyrazole) (22)
Following the general procedure A (20 mA), 4.7 mL of the mixture was collected (theoretical yield = 84.5 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 98:2) to give a white solid (62 mg, 73% yield). Mp: 94–94.5 °C. 1H NMR (400 MHz, CDCl3): δ 7.50–7.44 (m, 2H), 7.40 (dd, J = 8.5, 2.3 Hz, 1H), 7.31 (s, 1H), 7.06 (d, J = 2.3 Hz, 1H), 2.17 (s, 3H), 1.29 (s, 9H), 1.17 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3): δ 149.4, 144.1, 139.7, 139.4, 131.4, 127.6, 127.3, 126.3, 116.3, 35.5, 34.3, 31.5, 31.3, 9.0. HRMS (ESI) m/z [M + H]+ calcd for C18H27N2, 271.2174; found, 271.2165.
1-(2,5-Di-tert-butylphenyl)-(1H-1,2,3-triazole) (23)
Following the general procedure A (20 mA), 3.0 mL of the mixture was collected (theoretical yield = 51.3 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a white solid (28 mg, 54% yield). Mp: 115–115.5 °C. 1H NMR (400 MHz, CDCl3): δ 7.84 (s, 1H), 7.74 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.49 (dd, J = 8.5, 2.2 Hz, 1H), 7.01 (d, J = 2.2 Hz, 1H), 1.30 (s, 9H), 1.12 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3): δ 150.0, 143.8, 135.4, 133.3, 128.1, 127.6, 127.4, 126.6, 35.6, 34.4, 31.5, 31.2. HRMS (ESI) m/z [M + H]+ calcd for C16H24N3, 258.1970; found, 258.1975.
1-(2,5-Di-tert-butylphenyl)-(1H-benzo[d][1,2,3]triazole) (24)
Following the general procedure A (20 mA), 4.3 mL of the mixture was collected (theoretical yield = 87.9 mg). The reaction mixture was purified by flash chromatography on silica (cyclohexane to cyclohexane/ethyl acetate 9:1) to give a white solid (42 mg, 47% yield). Mp: = 122–123 °C. 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.3 Hz, 1H), 7.57 (d, J = 8.6 Hz, 1H), 7.47 (dd, J = 8.5, 2.3 Hz, 1H), 7.40 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 7.33 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 7.23–7.15 (m, 1H), 6.96 (d, J = 2.3 Hz, 1H), 1.21 (s, 9H), 1.00 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3): δ 150.3, 145.4, 145.3, 136.0, 133.9, 128.5, 128.0, 127.6, 127.1, 124.1, 119.9, 110.7, 35.5, 34.4, 31.6, 31.2. HRMS (ESI) m/z [M + H]+ calcd for C20H26N3, 308.2127; found, 308.2114.
Acknowledgments
L.B. would like to acknowledge the European Union for a Marie Sklodowska Curie IF Grant (ELECTROSULF, grant no. 840724).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01409.
Description of reaction setups, radical trapping experiments, and spectral data of all products (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Khanna I. K.; Yu Y.; Huff R. M.; Weier R. M.; Xu X.; Koszyk F. J.; Collins P. W.; Cogburn J. N.; Isakson P. C.; Koboldt C. M.; Masferrer J. L.; Perkins W. E.; Seibert K.; Veenhuizen A. W.; Yuan J.; Yang D.-C.; Zhang Y. Y. Selective Cyclooxygenase-2 Inhibitors: Heteroaryl Modified 1,2-Diarylimidazoles Are Potent, Orally Active Antiinflammatory Agents. J. Med. Chem. 2000, 43 (16), 3168–3185. 10.1021/jm0000719. [DOI] [PubMed] [Google Scholar]
- Hili R.; Yudin A. K. Making Carbon-Nitrogen Bonds in Biological and Chemical Synthesis. Nat. Chem. Biol. 2006, 2 (6), 284–287. 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Peng X.-M.; Damu G. L. V.; Geng R.-X.; Zhou C.-H. Comprehensive Review in Current Developments of Imidazole-Based Medicinal Chemistry. Med. Res. Rev. 2014, 34 (2), 340–437. 10.1002/med.21290. [DOI] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Vicentini C. B.; Romagnoli C.; Andreotti E.; Mares D. Synthetic Pyrazole Derivatives as Growth Inhibitors of Some Phytopathogenic Fungi. J. Agric. Food Chem. 2007, 55 (25), 10331–10338. 10.1021/jf072077d. [DOI] [PubMed] [Google Scholar]
- Wolfe J. P.; Wagaw S.; Marcoux J.-F.; Buchwald S. L. Rational Development of Practical Catalysts for Aromatic Carbon-Nitrogen Bond Formation. Acc. Chem. Res. 1998, 31 (12), 805–818. 10.1021/ar9600650. [DOI] [Google Scholar]
- Hartwig J. F. Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides. Acc. Chem. Res. 2008, 41 (11), 1534–1544. 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambiagio C.; Marsden S. P.; Blacker A. J.; McGowan P. C. Copper Catalysed Ullmann Type Chemistry: From Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43 (10), 3525–3550. 10.1039/C3CS60289C. [DOI] [PubMed] [Google Scholar]
- He J.; Shigenari T.; Yu J.-Q. Palladium(0)/PAr3-Catalyzed Intermolecular Amination of C(sp3)-H Bonds: Synthesis of β-Amino Acids. Angew. Chem., Int. Ed. 2015, 54 (22), 6545–6549. 10.1002/anie.201502075. [DOI] [PubMed] [Google Scholar]
- Sadhu P.; Punniyamurthy T. Copper(II)-Mediated Regioselective N-Arylation of Pyrroles, Indoles, Pyrazoles and Carbazole via Dehydrogenative Coupling. Chem. Commun. 2016, 52 (13), 2803–2806. 10.1039/C5CC08206D. [DOI] [PubMed] [Google Scholar]
- Pawar G. G.; Wu H.; De S.; Ma D. Copper(I) Oxide/ N, N’ -Bis[(2-Furyl)Methyl]Oxalamide-Catalyzed Coupling of (Hetero)Aryl Halides and Nitrogen Heterocycles at Low Catalytic Loading. Adv. Synth. Catal. 2017, 359 (10), 1631–1636. 10.1002/adsc.201700026. [DOI] [Google Scholar]
- Surry D. S.; Buchwald S. L. Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew. Chem., Int. Ed. 2008, 47 (34), 6338–6361. 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet F.; Dodd R. H.; Dauban P. Catalytic C-H Amination: Recent Progress and Future Directions. Chem. Commun. 2009, (34), 5061–5074. 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]
- Armstrong A.; Collins J. C. Direct Azole Amination: C-H Functionalization as a New Approach to Biologically Important Heterocycles. Angew. Chem., Int. Ed. 2010, 49 (13), 2282–2285. 10.1002/anie.200906750. [DOI] [PubMed] [Google Scholar]
- Pan J.; Su M.; Buchwald S. L. Palladium(0)-Catalyzed Intermolecular Amination of Unactivated C(sp3)-H Bonds. Angew. Chem., Int. Ed. 2011, 50 (37), 8647–8651. 10.1002/anie.201102880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda S.; Su M.; Buchwald S. L. Highly N2-Selective Palladium-Catalyzed Arylation of 1,2,3-Triazoles. Angew. Chem., Int. Ed. 2011, 50 (38), 8944–8947. 10.1002/anie.201103882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda S.; Su M.; Buchwald S. L. Completely N 1 -Selective Palladium-Catalyzed Arylation of Unsymmetric Imidazoles: Application to the Synthesis of Nilotinib. J. Am. Chem. Soc. 2012, 134 (1), 700–706. 10.1021/ja2102373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda S.; Buchwald S. L. Catalyst-Controlled Chemoselective Arylation of 2-Aminobenzimidazoles. Angew. Chem., Int. Ed. 2012, 51 (41), 10364–10367. 10.1002/anie.201204710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirunavukkarasu V. S.; Kozhushkov S. I.; Ackermann L. C-H Nitrogenation and Oxygenation by Ruthenium Catalysis. Chem. Commun. 2014, 50 (1), 29–39. 10.1039/C3CC47028H. [DOI] [PubMed] [Google Scholar]
- Louillat M.-L.; Patureau F. W. Oxidative C-H Amination Reactions. Chem. Soc. Rev. 2014, 43 (3), 901–910. 10.1039/C3CS60318K. [DOI] [PubMed] [Google Scholar]
- Louillat-Habermeyer M.-L.; Jin R.; Patureau F. W. O2-Mediated Dehydrogenative Amination of Phenols. Angew. Chem., Int. Ed. 2015, 54 (13), 4102–4104. 10.1002/anie.201500089. [DOI] [PubMed] [Google Scholar]
- Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Recent Advances in Radical C-H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117 (13), 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- Bambal R.; Hanzlik R. P. Synthesis of N. Epsilon.-(p-Bromophenyl)-L-Lysine and N. Tau.-(p-Bromophenyl)-L-Histidine as Models for Adducts of Bromobenzene 3,4-Oxide to Protein. Observation of an Unusual Pd-Catalyzed N. Tau. to N. Pi.-Aryl Substituent Migration. J. Org. Chem. 1994, 59 (4), 729–732. 10.1021/jo00083a011. [DOI] [Google Scholar]
- Ji P.; Atherton J. H.; Page M. I. The Kinetics and Mechanisms of Aromatic Nucleophilic Substitution Reactions in Liquid Ammonia. J. Org. Chem. 2011, 76 (9), 3286–3295. 10.1021/jo200170z. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Hong X.; Cui H.-Z.; Huang S.; Yi Y.; Hou X.-F. The Construction of C-N, C-O, and C(sp2)-C(sp3) Bonds from Fluorine-Substituted 2-Aryl Benzazoles for Direct Synthesis of N-, O-, C-Functionalized 2-Aryl Benzazole Derivatives. J. Org. Chem. 2018, 83 (12), 6363–6372. 10.1021/acs.joc.8b00587. [DOI] [PubMed] [Google Scholar]
- Wu W.-B.; Huang J.-M. Highly Regioselective C-N Bond Formation through C-H Azolation of Indoles Promoted by Iodine in Aqueous Media. Org. Lett. 2012, 14 (23), 5832–5835. 10.1021/ol302609m. [DOI] [PubMed] [Google Scholar]
- Tanimoto K.; Okai H.; Oka M.; Ohkado R.; Iida H. Aerobic Oxidative C-H Azolation of Indoles and One-Pot Synthesis of Azolyl Thioindoles by Flavin-Iodine-Coupled Organocatalysis. Org. Lett. 2021, 23 (6), 2084–2088. 10.1021/acs.orglett.1c00241. [DOI] [PubMed] [Google Scholar]
- Gonda Z.; Novák Z. Transition-Metal-Free N -Arylation of Pyrazoles with Diaryliodonium Salts. Chem. - Eur. J. 2015, 21 (47), 16801–16806. 10.1002/chem.201502995. [DOI] [PubMed] [Google Scholar]
- Teskey C. J.; Sohel S. M. A.; Bunting D. L.; Modha S. G.; Greaney M. F. Domino N -/ C -Arylation via In Situ Generation of a Directing Group: Atom-Efficient Arylation Using Diaryliodonium Salts. Angew. Chem., Int. Ed. 2017, 56 (19), 5263–5266. 10.1002/anie.201701523. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Margrey K. A.; Tay N. E.; Nicewicz D. A. Site-Selective Arene C-H Amination via Photoredox Catalysis. Science (Washington, DC, U. S.) 2015, 349 (6254), 1326–1330. 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- Niu L.; Yi H.; Wang S.; Liu T.; Liu J.; Lei A. Photo-Induced Oxidant-Free Oxidative C-H/N-H Cross-Coupling between Arenes and Azoles. Nat. Commun. 2017, 8 (1), 14226. 10.1038/ncomms14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Natarajan P.; König B. Teaching Old Compounds New Tricks: DDQ-Photocatalyzed C-H Amination of Arenes with Carbamates, Urea, and N-Heterocycles. Chem. - Eur. J. 2017, 23 (72), 18161–18165. 10.1002/chem.201705442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margrey K. A.; McManus J. B.; Bonazzi S.; Zecri F.; Nicewicz D. A. Predictive Model for Site-Selective Aryl and Heteroaryl C-H Functionalization via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139 (32), 11288–11299. 10.1021/jacs.7b06715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J.; Le C. Chip; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1 (7), 52. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Samanta S.; Ravi C.; Rao S. N.; Joshi A.; Adimurthy S. Visible-Light-Promoted Selective C-H Amination of Heteroarenes with Heteroaromatic Amines under Metal-Free Conditions. Org. Biomol. Chem. 2017, 15 (45), 9590–9594. 10.1039/C7OB02504A. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Ghosh I.; König B.; Wang X. Metal-Free Semiconductor Photocatalysis for Sp 2 C-H Functionalization with Molecular Oxygen. ChemCatChem 2019, 11 (2), 703–706. 10.1002/cctc.201801948. [DOI] [Google Scholar]
- Kärkäs M. D. Electrochemical Strategies for C-H Functionalization and C-N Bond Formation. Chem. Soc. Rev. 2018, 47 (15), 5786–5865. 10.1039/C7CS00619E. [DOI] [PubMed] [Google Scholar]
- Hou Z.; Mao Z.; Zhao H.; Melcamu Y. Y.; Lu X.; Song J.; Xu H. Electrochemical C-H/N-H Functionalization for the Synthesis of Highly Functionalized (Aza)Indoles. Angew. Chem., Int. Ed. 2016, 55 (32), 9168–9172. 10.1002/anie.201602616. [DOI] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Electrochemical C-H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J. Am. Chem. Soc. 2013, 135 (13), 5000–5003. 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Heterocyclization Approach for Electrooxidative Coupling of Functional Primary Alkylamines with Aromatics. J. Am. Chem. Soc. 2015, 137 (31), 9816–9819. 10.1021/jacs.5b06526. [DOI] [PubMed] [Google Scholar]
- Hayashi R.; Shimizu A.; Song Y.; Ashikari Y.; Nokami T.; Yoshida J. Metal-Free Benzylic C-H Amination via Electrochemically Generated Benzylaminosulfonium Ions. Chem. - Eur. J. 2017, 23 (1), 61–64. 10.1002/chem.201604484. [DOI] [PubMed] [Google Scholar]
- Wesenberg L. J.; Diehl E.; Zähringer T. J. B.; Dörr C.; Schollmeyer D.; Shimizu A.; Yoshida J.; Hellmich U. A.; Waldvogel S. R. Metal-Free Twofold Electrochemical C-H Amination of Activated Arenes: Application to Medicinally Relevant Precursor Synthesis. Chem. - Eur. J. 2020, 26 (72), 17574–17580. 10.1002/chem.202003852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ošeka M.; Laudadio G.; van Leest N. P.; Dyga M.; Bartolomeu A. de A.; Gooßen L. J.; de Bruin B.; de Oliveira K. T.; Noël T. Electrochemical Aziridination of Internal Alkenes with Primary Amines. Chem. 2021, 7 (1), 255–266. 10.1016/j.chempr.2020.12.002. [DOI] [Google Scholar]
- Lin M.-Y.; Xu K.; Jiang Y.-Y.; Liu Y.-G.; Sun B.-G.; Zeng C.-C. Intermolecular Electrochemical C(sp3)-H/N-H Cross-Coupling of Xanthenes with N-Alkoxyamides: Radical Pathway Mediated by Ferrocene as a Redox Catalyst. Adv. Synth. Catal. 2018, 360 (8), 1665–1672. 10.1002/adsc.201701536. [DOI] [Google Scholar]
- Hou Z.; Liu D.; Xiong P.; Lai X.; Song J.; Xu H. Site-Selective Electrochemical Benzylic C-H Amination. Angew. Chem., Int. Ed. 2021, 60 (6), 2943–2947. 10.1002/anie.202013478. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Wright D. L. The Application of Cathodic Reductions and Anodic Oxidations in the Synthesis of Complex Molecules. Chem. Soc. Rev. 2006, 35 (7), 605. 10.1039/b512308a. [DOI] [PubMed] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117 (21), 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M.; Shinde V. S.; Rueping M. A Review of Asymmetric Synthetic Organic Electrochemistry and Electrocatalysis: Concepts, Applications, Recent Developments and Future Directions. Beilstein J. Org. Chem. 2019, 15, 2710–2746. 10.3762/bjoc.15.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston C.; Palkowitz M. D.; Takahira Y.; Vantourout J. C.; Peters B. K.; Kawamata Y.; Baran P. S. A Survival Guide for the “Electro-Curious. Acc. Chem. Res. 2020, 53 (1), 72–83. 10.1021/acs.accounts.9b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem., Int. Ed. 2018, 57 (16), 4149–4155. 10.1002/anie.201707584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Direct C-N Coupling of Imidazoles with Aromatic and Benzylic Compounds via Electrooxidative C-H Functionalization. J. Am. Chem. Soc. 2014, 136 (12), 4496–4499. 10.1021/ja501093m. [DOI] [PubMed] [Google Scholar]
- de Robillard G.; Makni O.; Cattey H.; Andrieu J.; Devillers C. H. Towards Sustainable Synthesis of Pyren-1-Yl Azoliums via Electrochemical Oxidative C-N Coupling. Green Chem. 2015, 17 (9), 4669–4679. 10.1039/C5GC01142F. [DOI] [Google Scholar]
- Yang Y.-Z.; Song R.-J.; Li J.-H. Intermolecular Anodic Oxidative Cross-Dehydrogenative C(sp3)-N Bond-Coupling Reactions of Xanthenes with Azoles. Org. Lett. 2019, 21 (9), 3228–3231. 10.1021/acs.orglett.9b00947. [DOI] [PubMed] [Google Scholar]
- Wan Z.; Wang D.; Yang Z.; Zhang H.; Wang S.; Lei A. Electrochemical Oxidative C(sp3)-H Azolation of Lactams under Mild Conditions. Green Chem. 2020, 22 (12), 3742–3747. 10.1039/D0GC00687D. [DOI] [Google Scholar]
- Yu Y.; Yuan Y.; Liu H.; He M.; Yang M.; Liu P.; Yu B.; Dong X.; Lei A. Electrochemical Oxidative C-H/N-H Cross-Coupling for C-N Bond Formation with Hydrogen Evolution. Chem. Commun. 2019, 55 (12), 1809–1812. 10.1039/C8CC09899A. [DOI] [PubMed] [Google Scholar]
- Wang J.-H.; Lei T.; Nan X.-L.; Wu H.-L.; Li X.-B.; Chen B.; Tung C.-H.; Wu L.-Z. Regioselective Ortho Amination of an Aromatic C-H Bond by Trifluoroacetic Acid via Electrochemistry. Org. Lett. 2019, 21 (14), 5581–5585. 10.1021/acs.orglett.9b01910. [DOI] [PubMed] [Google Scholar]
- Feng P.; Ma G.; Chen X.; Wu X.; Lin L.; Liu P.; Chen T. Electrooxidative and Regioselective C-H Azolation of Phenol and Aniline Derivatives. Angew. Chem., Int. Ed. 2019, 58 (25), 8400–8404. 10.1002/anie.201901762. [DOI] [PubMed] [Google Scholar]
- Huang H.; Strater Z. M.; Rauch M.; Shee J.; Sisto T. J.; Nuckolls C.; Lambert T. H. Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication. Angew. Chem., Int. Ed. 2019, 58 (38), 13318–13322. 10.1002/anie.201906381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Liardet L.; Luo J.; Ren D.; Grätzel M.; Hu X. Photoelectrocatalytic Arene C-H Amination. Nat. Catal. 2019, 2 (4), 366–373. 10.1038/s41929-019-0231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgueiras-Amador A. A.; Wirth T. Perspectives in Flow Electrochemistry. J. Flow Chem. 2017, 7 (3–4), 94–95. 10.1556/1846.2017.00020. [DOI] [Google Scholar]
- Noël T.; Cao Y.; Laudadio G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52 (10), 2858–2869. 10.1021/acs.accounts.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudadio G.; Straathof N. J. W.; Lanting M. D.; Knoops B.; Hessel V.; Noël T. An Environmentally Benign and Selective Electrochemical Oxidation of Sulfides and Thiols in a Continuous-Flow Microreactor. Green Chem. 2017, 19 (17), 4061–4066. 10.1039/C7GC01973D. [DOI] [Google Scholar]
- Laudadio G.; Barmpoutsis E.; Schotten C.; Struik L.; Govaerts S.; Browne D. L.; Noël T. Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 2019, 141 (14), 5664–5668. 10.1021/jacs.9b02266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govaerts S.; Nyuchev A.; Noel T. Pushing the Boundaries of C-H Bond Functionalization Chemistry Using Flow Technology. J. Flow Chem. 2020, 10 (1), 13–71. 10.1007/s41981-020-00077-7. [DOI] [Google Scholar]
- Laudadio G.; de Smet W.; Struik L.; Cao Y.; Noël T. Design and Application of a Modular and Scalable Electrochemical Flow Microreactor. J. Flow Chem. 2018, 8 (3), 157–165. 10.1007/s41981-018-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer I.; Chamberlain A. E. R.; Haughey M. B.; Donohoe T. J. Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem. 2017, 1 (11), 0088. 10.1038/s41570-017-0088. [DOI] [Google Scholar]
- Röckl J. L.; Dörr M.; Waldvogel S. R. Electrosynthesis 2.0 in 1,1,1,3,3,3-Hexafluoroisopropanol/Amine Mixtures. ChemElectroChem 2020, 7 (18), 3686–3694. 10.1002/celc.202000761. [DOI] [Google Scholar]
- Ramos-Villaseñor J. M.; Rodríguez-Cárdenas E.; Barrera Díaz C. E.; Frontana-Uribe B. A. Review—Use of 1,1,1,3,3,3-Hexafluoro-2-Propanol (HFIP) Co-Solvent Mixtures in Organic Electrosynthesis. J. ElectroChem. Soc. 2020, 167 (15), 155509. 10.1149/1945-7111/abb83c. [DOI] [Google Scholar]
- Johansson Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem., Int. Ed. 2012, 51 (21), 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27 (05), 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- Cao Y.; Soares C.; Padoin N.; Noël T. Gas Bubbles Have Controversial Effects on Taylor Flow Electrochemistry. Chem. Eng. J. 2021, 406, 126811. 10.1016/j.cej.2020.126811. [DOI] [Google Scholar]
- Sikari R.; Sinha S.; Chakraborty G.; Das S.; Leest N. P.; Paul N. D. C-N Cross-Coupling Reactions Under Mild Conditions Using Singlet Di-Radical Nickel(II)-Complexes as Catalyst: N-Arylation and Quinazoline Synthesis. Adv. Synth. Catal. 2019, 361 (18), 4342–4353. 10.1002/adsc.201900545. [DOI] [Google Scholar]
- Abramovitch R. A.; Beckert J. M.; Gibson H. H.; Belcher A.; Hundt G.; Sierra T.; Olivella S.; Pennington W. T.; Solé A. The 1,2,4-Triazolyl Cation: Thermolytic and Photolytic Studies. J. Org. Chem. 2001, 66 (4), 1242–1251. 10.1021/jo001382u. [DOI] [PubMed] [Google Scholar]
- Hirano K.; Biju A. T.; Glorius F. Copper-Catalyzed Synthesis of 2-Unsubstituted, N-Substituted Benzimidazoles. J. Org. Chem. 2009, 74 (24), 9570–9572. 10.1021/jo902160y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.