

Abstract
Filaggrin (FLG), loricrin (LOR), and involucrin are important epidermal barrier proteins. As psoriasis is characterized by overexpression of tumor necrosis factor-α (TNF-α) and impaired skin barrier, we investigated the expression of skin barrier proteins in psoriasis patients and whether their expression was modulated by TNF-α. The expression of FLG and LOR was found to be decreased in lesional and non-lesional skin of psoriasis patients. A correlation was found between the expression of TNF-α and epidermal barrier proteins in psoriasis. TNF-α was found to modulate the expression of FLG and LOR via a c-Jun N-terminal kinase-dependent pathway. Importantly, we report that clinical treatment of psoriasis patients with a TNF-α antagonist results in significant enhancement of epidermal barrier protein expression. Our current study suggests that TNF inhibits barrier protein expression, and TNF-α antagonists may contribute to clinical improvement in patients with psoriasis by improving barrier protein expression.
INTRODUCTION
The epidermis provides an important physical barrier against the environment. Filaggrin (FLG), loricrin (LOR), and involucrin (IVL) are major proteins that have an important role in formation of the epidermal skin barrier (Kalinin et al., 2001;Candi et al., 2005). FLG aggregates keratin filaments and provides a cytoskeleton for the cornified envelope (Candi et al., 2005). LOR is initially expressed in the granular layer and comprises 70% of the total protein mass of the cornified layer (Kalinin et al., 2001; Candi et al., 2005). IVL is an early component in the assembly of cornified envelope and provides a scaffold for cornified envelope (Candi et al., 2005).
Psoriasis is a common chronic inflammatory skin disease and is characterized by an impaired skin barrier (Huffmeier et al., 2007; Proksch et al., 2008). The mechanism for reduced skin barrier function in psoriasis is not known. Tumor necrosis factor-α (TNF-α) is overexpressed in the epidermis of psoriasis skin (Kristensen et al., 1993; Ettehadi et al., 1994) and has a pivotal role in the pathogenesis of psoriasis, as many TNF-α-regulated genes are overexpressed in psoriasis and TNF-α antagonists are highly effective therapeutic agents in most patients (Nickoloff et al., 1991; Richardson and Gelfand, 2008). TNF-α was functionally linked to the IL-23/Th17 pathway in psoriasis by its ability to activate myeloid dendritic cells (Zaba et al., 2007, 2009). This study was conducted to determine whether overexpression of TNF-α in psoriasis may contribute to a deficiency of epidermal barrier proteins.
RESULTS
Deficiency of FLG and LOR in psoriasis skin
We initially determined gene expression of FLG and LOR in skin biopsies from normal subjects and in patients with psoriasis. Using real-time RT-PCR, we found that the gene expression of FLG is significantly decreased in lesional (7.99±2.00 ng FLG per ng of glyceraldehyde-3-phosphate dehydrogenase (GAPDH); P<0.001) and non-lesional (18.04±3.90 ng; P<0.01) skin from psoriasis patients, as compared with skin from normal subjects (45.06±7.78 ng; Figure 1a). In addition, FLG gene expression was significantly decreased in lesional psoriasis skin compared with non-lesional psoriasis skin (P<0.05). LOR gene expression was also significantly decreased in lesional (4.60±1.72 ng LOR per ng of GAPDH; P<0.001) and non-lesional (8.41±2.21 ng; P<0.01) skin from psoriasis patients compared with skin from normal subjects (27.70±6.08 ng; Figure 1b). However, the gene expression of IVL is not decreased in skin from psoriasis patients (data not shown). This was confirmed at the protein level using immunostaining (Figure 1c and d). The composite data for FLG and LOR staining in all samples are shown in Figure 1e and f. The staining intensity of FLG and LOR is significantly decreased in lesional (FLG: P<0.001; LOR: P<0.01) and non-lesional (FLG: P<0.05) psoriasis skin, as compared with skin from normal subjects. In addition, the staining intensity of LOR in lesional psoriasis skin was significantly decreased compared with non-lesional psoriasis skin (P<0.05).
Figure 1. Expression of filaggrin (FLG) and loricrin (LOR) in the skin from normal subjects and patients with psoriasis.
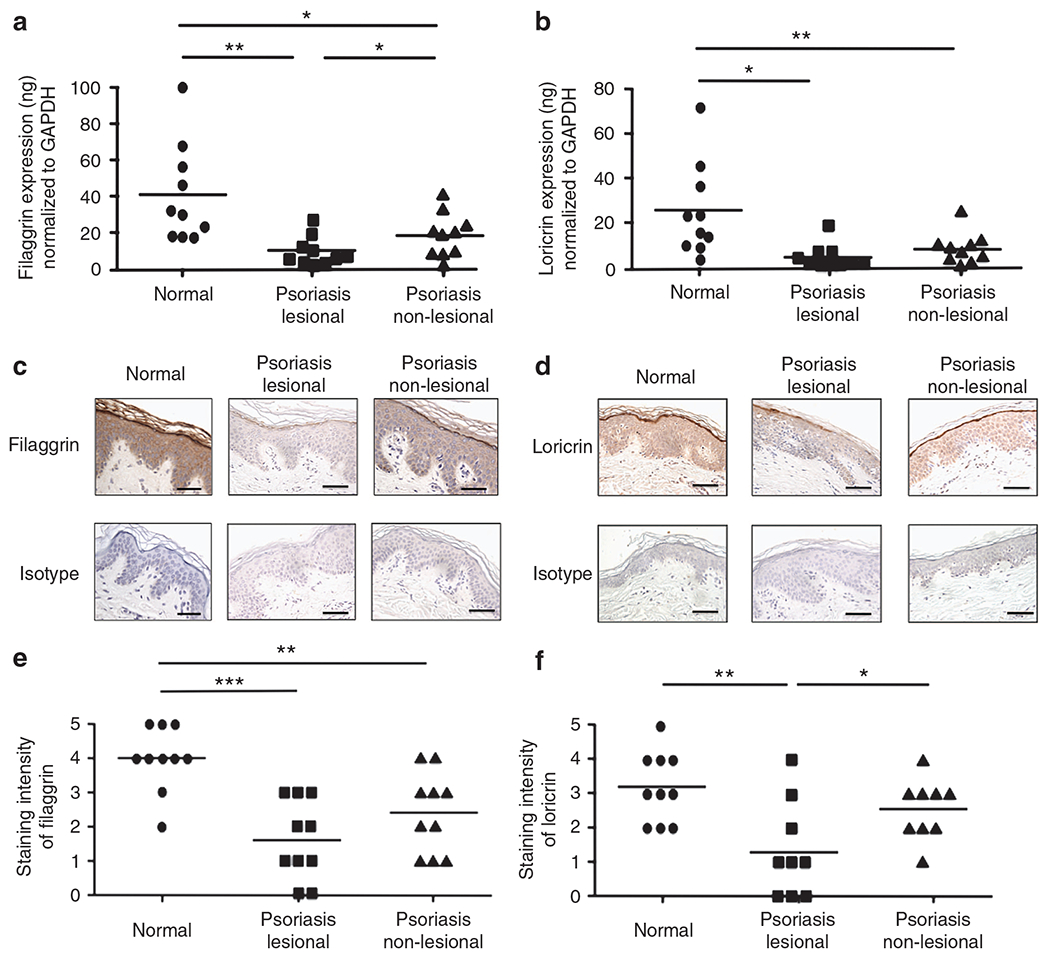
RNA was isolated from the skin of normal subjects and patients with psoriasis. The gene expression FLG (a) and LOR (b) was evaluated using real-time RT-PCR. (c, d) Representative paraffin-embedded skin biopsies from normal subjects (n = 10) and patients with psoriasis (n = 9) stained for FLG (c) and LOR (d) are shown. Images were collected at × 400 magnification. (e, f) The intensities of the staining for FLG (e) and LOR (f) were graded visually on a scale from 0 (no staining) to 5 (the most intense staining). The scale bar represents 50 μm. *P<0.05; **P<0.01; ***P<0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
TNF-α inhibits expression of FLG and LOR
Psoriasis is characterized by overexpression of TNF-α (Kristensen et al., 1993; Ettehadi et al., 1994) and impaired skin barrier (Huffmeier et al., 2007; Proksch et al., 2008). Therefore, we examined whether TNF-α modulates the expression of FLG, LOR, and IVL. We differentiated primary human keratinocytes (KCs) with 1.3 mmol l−1 CaCl2 for 5 days, and then the KCs were incubated with various concentrations of TNF-α for 24 hours. The gene expression of FLG was significantly inhibited by TNF-α with concentration as low as 5 ng ml−1 (3.06±0.48 ng FLG per ng of GAPDH; P<0.05) compared with media alone (5.36±0.07 ng; Figure 2a). Similarly, LOR gene expression was significantly inhibited by TNF-α with concentrations as low as 2 ng ml−1 compared with media alone (data not shown). However, IVL gene expression was not modulated by TNF-α (data not shown), suggesting that TNF-α does not have a global effect on epidermal proteins.
Figure 2. Tumor necrosis factor-α (TNF-α) inhibits expression of filaggrin (FLG).
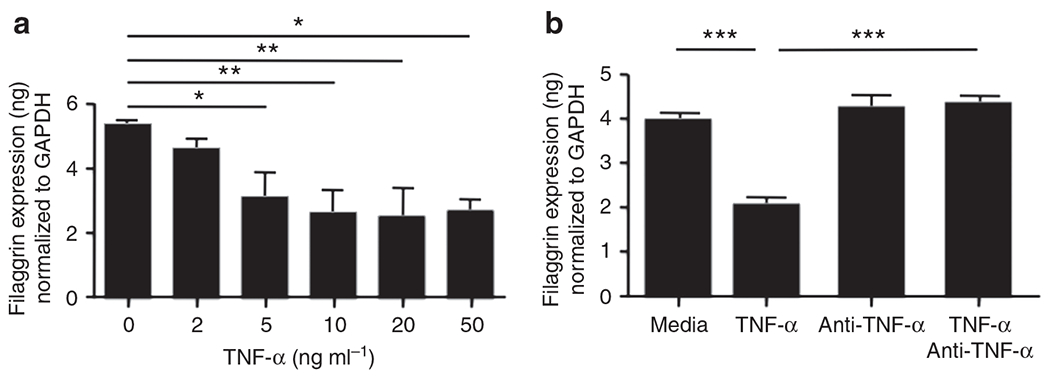
Primary human keratinocytes (KCs) were stimulated in the presence of various concentrations of TNF-α for 24 hours. The gene expression of FLG was examined by real-time RT-PCR (a). In addition, primary human KCs were pre-incubated with 0.1 μg ml−1 of TNF-α-neutralizing antibody before TNF-α (20 ng ml−1) stimulation. The gene expression of FLG was then examined by real-time RT-PCR (b). *P<0.05; **P<0.01; ***P<0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To further understand the modulation of TNF-α on FLG and LOR, human primary KCs were pre-incubated with a TNF-α-neutralizing antibody (Ab) before TNF-α stimulation. We demonstrated that FLG gene expression was not significantly decreased in the KCs treated with a TNF-α-neutralizing Ab (4.39±0.15 ng FLG per ng of GAPDH; P<0.001), as compared with the untreated KCs (2.09±0.16 ng; Figure 2b). Similarly, LOR gene expression was not significantly decreased in the KCs pre-treated with a TNF-α-neutralizing Ab, as compared with the untreated KCs (data not shown).
The effects of TNF α, as compared with Th2 cytokines, on barrier proteins differ
Th2 cytokines have previously been reported to downregulate expression of FLG, LOR, and IVL (Howell et al., 2007; Kim et al., 2008). To determine whether the mechanism for TNF-α differs from effects of Th2 cytokines on barrier proteins, we examined the effects of Th2 cytokines and TNF-α on gene expression of FLG, LOR, and IVL. Primary KCs were treated with Th2 cytokines or TNF-α for 24hours and then mRNA was extracted and evaluated for gene expressions of FLG, LOR, and IVL. The gene expression of FLG, LOR, and IVL was significantly inhibited in the KCs treated with Th2 cytokines (FLG: 0.57±0.04 ng; LOR: 0.72±0.09 ng; IVL: 6.95±1.52 ng) compared with the untreated KCs (FLG: 2.61±0.09 ng; LOR: 3.22±0.31 ng; IVL: 16.95±1.23 ng; P<0.01 for all comparisons; Figure 3a–c). In contrast, the gene expression of FLG and LOR was significantly inhibited in the KCs treated with TNF-α (FLG: 1.22±0.06 ng, P<0.001; LOR: 0.95±0.08 ng, P<0.05) compared with the untreated KCs (FLG: 2.61±0.09 ng; LOR: 3.22±0.31 ng; Figure 3a and b). However, TNF-α did not affect IVL expression (Figure 3c), suggesting that TNF-α modulates barrier proteins in a manner different than Th2 cytokines.
Figure 3. The effects of tumor necrosis factor-α (TNF-α) and Th2 cytokines on barrier proteins.
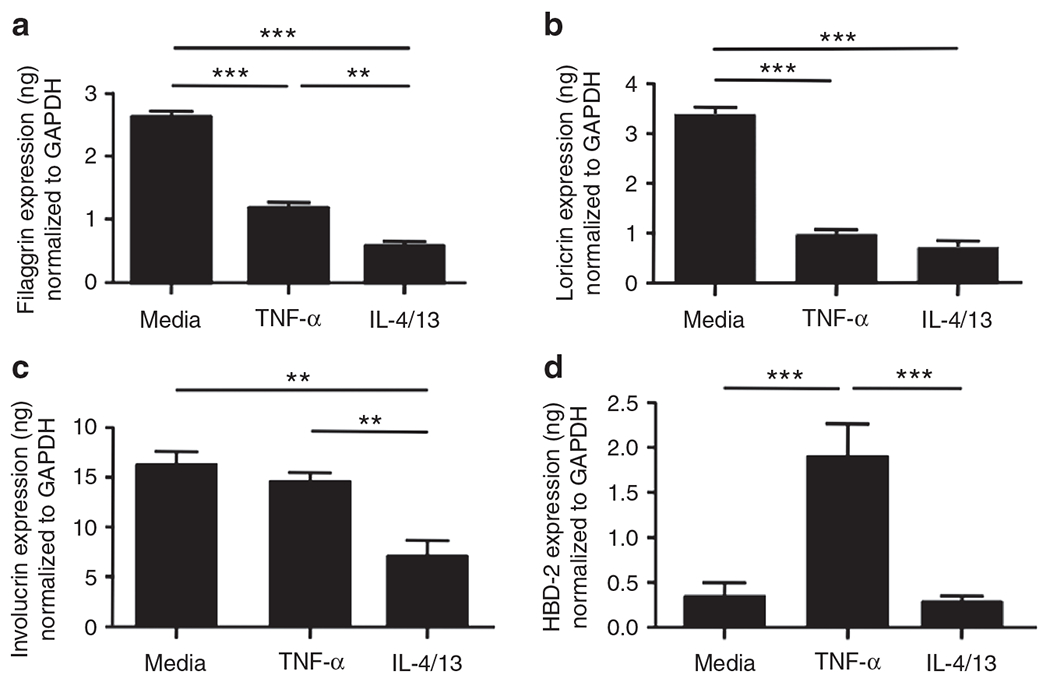
Primary human keratinocytes were stimulated with TNF-α (20 ng ml−1) or a combination of IL-4 (50 ng ml−1) and IL-13 (50 ng ml−1) for 24 hours. The gene expression of filaggrin (FLG) (a), loricrin (LOR) (b), involucrin (IVL) (c), and human-β defensin 2 (HBD-2) (d) were examined by real-time RT-PCR. **P<0.01; ***P<0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
In addition, we determined the effects of TNF-α and Th2 cytokines on human β-defensin 2 (HBD-2), a potent antimicrobial peptide in the epidermis. We found that HBD-2 expression was significantly increased in the KCs treated with TNF-α (1.89±0.21 ng HBD-2 per ng of GAPDH; P<0.001), compared with the untreated KCs (0.36±0.07 ng), whereas Th2 cytokines did not induce HBD-2 (Figure 3d). This suggests that TNF-α potentially induces antimicrobial peptides as well as inhibits barrier proteins in KCs.
c-Jun N-terminal kinase inhibitor blocks TNF-α-mediated inhibition of FLG and LOR
As NF-κB is a downstream target of TNF-α (Hsu et al., 1995; Clarke et al., 2010), we treated primary KCs with NF-κB inhibitor (10 nm) before TNF-α stimulation to examine whether NF-κB inhibitor blocks the effect of TNF-α on barrier proteins. NF-κB inhibitor did not block the TNF-α effects on gene expression of FLG (Figure 4a) and LOR (data not shown), suggesting that TNF-α does not downregulate FLG and LOR by activating NF-κB.
Figure 4. c-Jun N-terminal kinase (JNK) inhibitor blocks the tumor necrosis factor-α (TNF-α)-mediated inhibition of filaggrin (FLG).
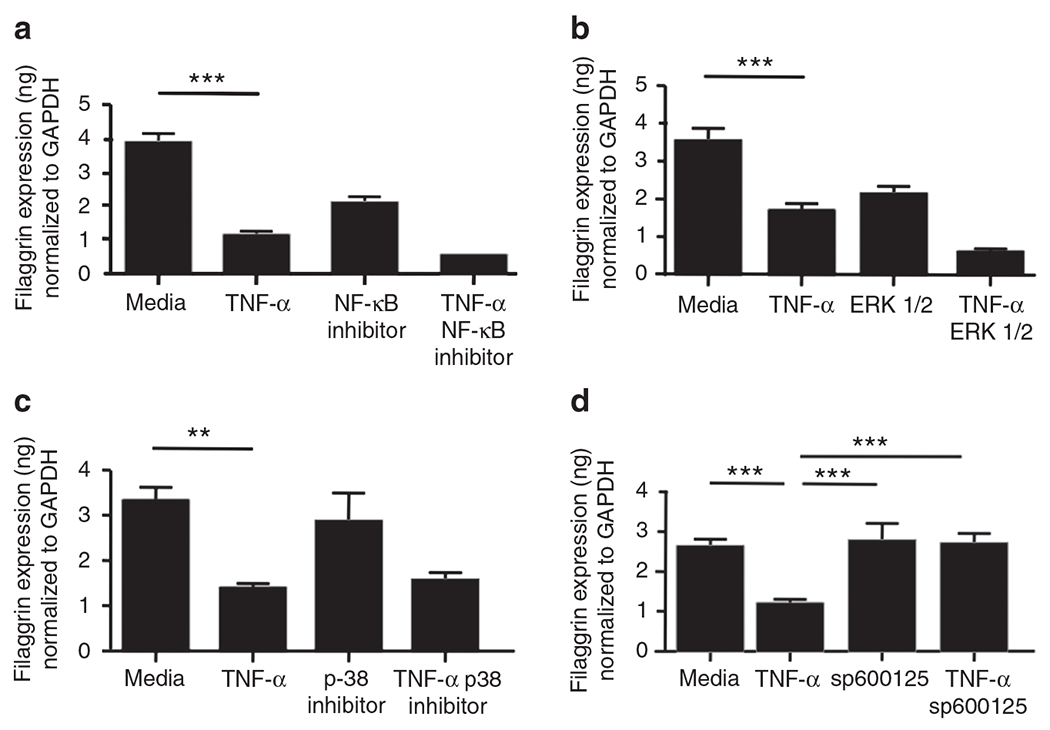
Primary human keratinocytes were pre-incubated with NF-κB inhibitor (10 nm, a), extracellular signal-regulated kinases 1/2 (ERK1/2) inhibitor (5 μm, b), p38 inhibitor (5 μm, c), and JNK inhibitor sp600125 (1 μm, d) before stimulation with TNF-α (20 ng ml−1). The gene expression of FLG was examined by real-time RT-PCR. **P<0.01; ***P<0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
It is known that TNF-α also activates the mitogen-activated protein kinase pathway (Chang and Karin, 2001; Lin, 2003; Zarubin and Han, 2005). Therefore, we pre-treated primary KCs with an extracellular signal-regulated kinases 1/2 inhibitor (5 μm), p38 inhibitor (5 μm), and sp600125 (c-Jun N-terminal kinase inhibitor, 1 μm) before TNF-α (20 ng ml−1) stimulation. Extracellular signal-regulated kinases 1/2 inhibitor and p38 inhibitor did not block the TNF-α effects on gene expression of FLG (Figure 4b and c) and LOR (data not shown). However, FLG gene expression was not significantly decreased in the KCs pre-treated with sp600125 (2.71±0.14 ng FLG per ng of GAPDH; P<0.001), as compared with untreated KCs (1.20±0.06 ng; Figure 4d). Similarly, LOR gene expression was not significantly decreased in the KCs pre-treated with sp600125, as compared with the untreated KCs (data not shown), whereas IVL gene expression was not affected by TNF-α or sp600125 (data not shown).
TNF-α antagonist upregulates FLG and LOR
On the basis of our observations that TNF-α inhibits the in vitro expression of FLG and LOR, we further investigated the clinical significance of this observation by determining whether a TNF-α monoclonal Ab (etanercept) could block the TNF-α-mediated inhibition of these barrier proteins in vivo in humans with psoriasis. For this study, six independent psoriasis patients followed up at the Rockefeller University were given 50mg etanercept, bi-weekly for 12 weeks. We studied skin biopsies, before and after they received etanercept. The gene expression of FLG was significantly increased in lesional skin from after treatment (23.89±5.78 ng FLG per ng of GAPDH; P<0.05) compared with lesional skin from before treatment (4.38±1.77 ng; Figure 5a). Similarly, LOR gene expression was significantly increased in lesional skin from after treatment (27.95±6.92 ng LOR per ng GAPDH; P<0.01) compared with lesional skin from before treatment (4.54±1.43 ng; Figure 5b). Interestingly, the increase in gene expression of FLG and LOR is positively correlated with change in psoriasis area and severity index (data not shown). However, IVL gene expression was not affected (Figure 5c). In addition, we also examined the gene expression of FLG and LOR in lesional and non-lesional psoriasis skin from before treatment. The gene expression of FLG and LOR in lesional psoriasis skin was significantly decreased compared with non-lesional psoriasis skin (FLG: P<0.05; LOR: P<0.01; Figure 5a and b). Interestingly, the gene expression of TNF-α in lesional psoriasis skin was significantly increased compared with non-lesional psoriasis skin (P<0.05; data not shown).
Figure 5. Tumor necrosis factor-α (TNF-α) antagonist increased the gene expression of filaggrin (FLG), loricrin (LOR), and involucrin (IVL).
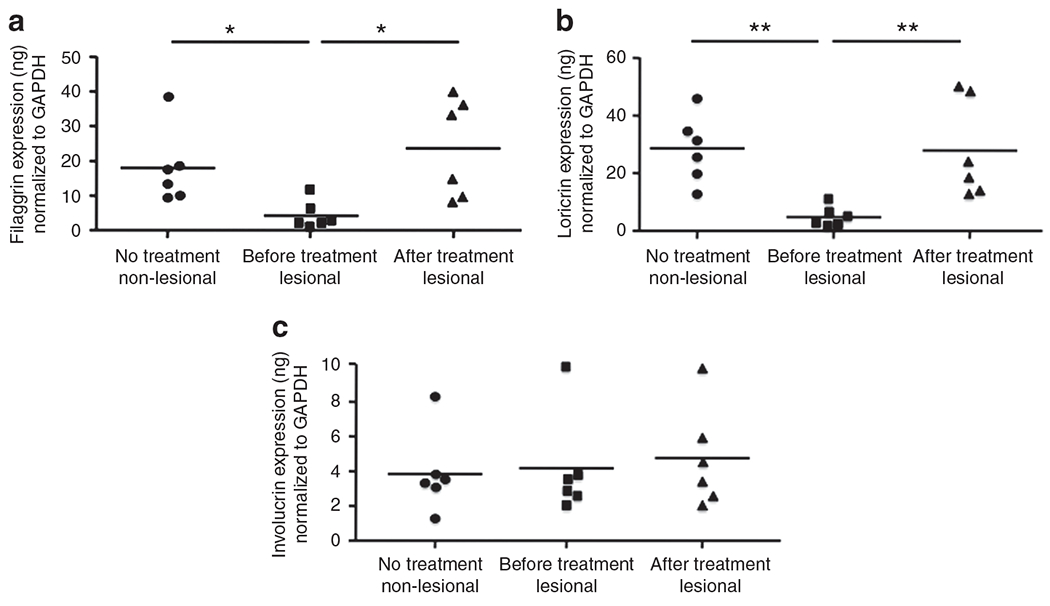
Patients with psoriasis (n = 6) were treated with TNF-α monoclonal antibody for 12 weeks. RNA was isolated from the lesional and non-lesional skin of the patients with psoriasis. Gene expression of FLG (a), LOR (b), and IVL (c) was evaluated using real-time RT-PCR. *P<0.05, **P<0.01. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Immunostaining confirmed increased expression of both FLG and LOR in skin after treatment, as compared with skin from before treatment (Figure 6a and b). The composite data in all samples are shown in Figure 6c and d. The mean fluorescent intensity of FLG and LOR was significantly increased in lesional skin from after treatment compared with lesional skin from before treatment (FLG: P<0.01; LOR: P<0.01). However, the mean fluorescent intensity of IVL was not affected (data not shown). In addition, the mean fluorescent intensity of LOR in lesional psoriasis skin was significantly decreased compared with non-lesional psoriasis skin (P<0.05).
Figure 6. Tumor necrosis factor-α (TNF-α) antagonist increased the protein expression of filaggrin (FLG) and loricrin (LOR).
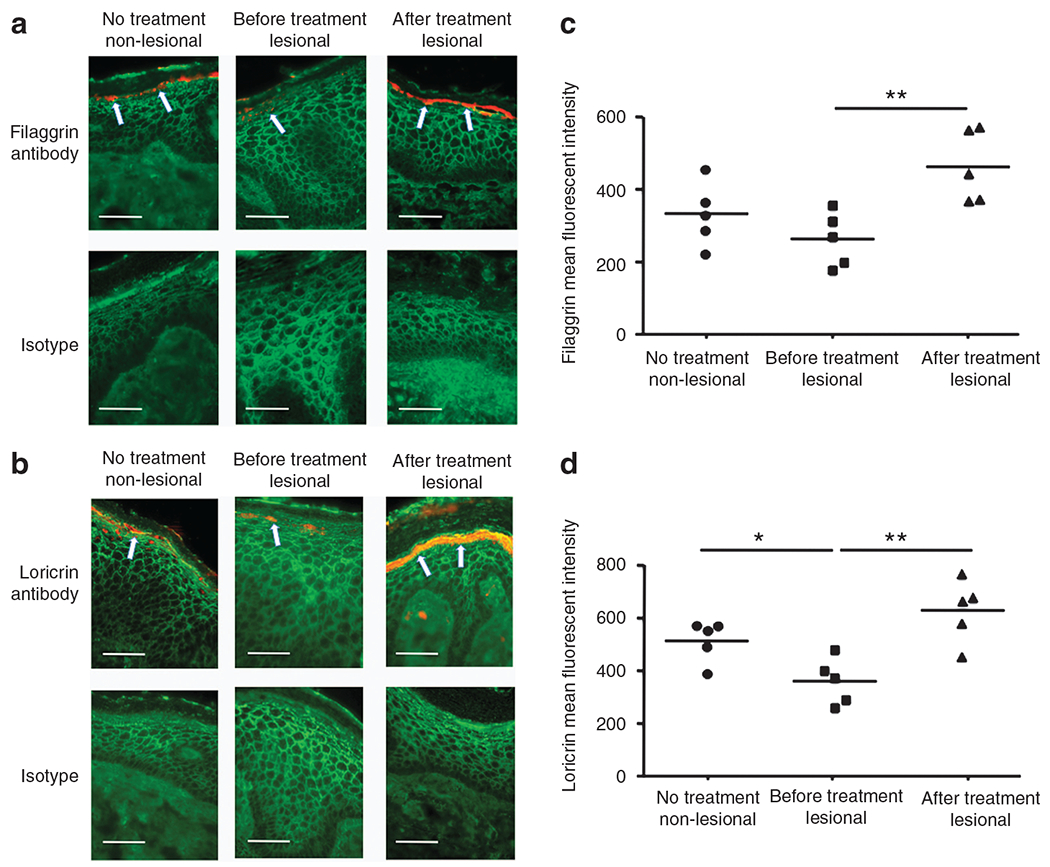
(a, b) Representative skin biopsies from patients with psoriasis stained for FLG (a) and LOR (b) are shown. (c, d) The mean fluorescent intensity is shown for FLG (c) and LOR (d) in the epidermis of each biopsy. The scale bar represents 50 μm. *P<0.05, **P<0.01. Arrows in panel a and b indicate protein expression of filaggrin and loricrin, respectively.
DISCUSSION
In our current study, we demonstrated that there is a deficiency of FLG and LOR in lesional and non-lesional psoriasis skin. IVL expression, however, was not decreased in psoriasis skin. We demonstrated these findings using both real-time RT-PCR and immunostaining. This finding is similar to our previous data showing a relative decrease of FLG and LOR in psoriasis skin in comparison with normal skin (Guttman-Yassky et al., 2009). Therefore, our data suggest that the well-established skin barrier defect (Huffmeier et al., 2007;Proksch et al., 2008), known to occur in psoriasis, is the result of a deficiency in epidermal barrier proteins. In contrast, it is well known that psoriasis is rarely associated with microbial infection and overexpress antimicrobial peptides. Therefore, this skin disease has a different profile of epidermal barrier proteins than found in atopic dermatitis (AD; Ong et al., 2002).
KCs are the primary cells of the epidermis and express skin barrier proteins including FLG, LOR, and IVL (Watt, 1989; Candi et al., 2005). For this reason, we used primary human KCs to examine why FLG and LOR are decreased in the skin of psoriasis patients. Psoriasis skin is characterized by overexpression of TNF-α (Kristensen et al., 1993; Ettehadi et al., 1994). Therefore, we investigated whether skin barrier proteins are modulated by TNF-α. We found that the gene expression of FLG and LOR was significantly inhibited in the KCs treated with TNF-α following a dose-dependent pattern. The importance of TNF-α in downregulating these barrier proteins was supported by the observation that a TNF-α-neutralizing Ab blocked the in vitro TNF-α-mediated inhibition of FLG and LOR.
It has been reported that <5% of patients with psoriasis have FLG mutations and 80% of psoriasis patients have FLG deficiency in their skin (Huffmeier et al., 2007). Our current data suggest that the deficiency of FLG and LOR in most patients with psoriasis is acquired as the TNF-α modulation. In addition, our laboratory has previously demonstrated that deficiencies of barrier proteins in AD skin are acquired rather than constitutive (Howell et al., 2007; Kim et al., 2008).
On the basis of our current data and previous data (Howell et al., 2007; Kim et al., 2008), both TNF-α and Th2 cytokines modulate barrier proteins. To determine whether the mechanisms for cytokine modulation of epidermal proteins are different, we stimulated KCs with TNF-α or Th2 cytokines and examined the effects of these cytokines on barrier proteins. Interestingly, TNF-α only reduced FLG and LOR. In contrast, Th2 cytokines inhibited FLG, LOR, and IVL. Therefore, we could suggest that TNF-α modulates barrier proteins in a manner different than Th2 cytokines. Indeed previous reports have demonstrated that Th2 cytokines inhibit barrier proteins via the signal transducer and activator of transcription 6 in vitro in KCs and in vivo in mice that overexpress signal transducer and activator of transcription 6 (Kim et al., 2008; Sehra et al., 2010).
Both AD and psoriasis are common chronic inflammatory skin diseases and are characterized by impaired barrier proteins. Importantly, 30% of AD patients suffer from recurrent skin infections, whereas only 6.7% of psoriasis patients suffer from skin infection (Christophers and Henseler, 1987). To explain this discrepancy, we examined the expression of HBD-2, a potent antimicrobial peptide, in the KCs treated with TNF-α or Th2 cytokines, which are overexpressed in AD skin. Importantly, HBD-2 was increased by TNF-α; however, Th2 cytokines did not increase HBD-2. Therefore, we postulate that the reason psoriasis patients have a barrier defect, but are less susceptible to microbial infections, than AD patients is that TNF-α only inhibits barrier proteins while inducing antimicrobial peptides. Indeed our laboratory has previously demonstrated that TNF-α induces HBD-2 through signal transducer and activator of transcription 1 and NF-κB (Albanesi et al, 2007). Furthermore, our data recently showed that TNF-α synergizes with IL-17 in induction of antimicrobial peptides in human KCs (Chiricozzi et al, 2010). In contrast, in AD, Th2 cytokines inhibit both barrier proteins and antimicrobial peptides needed to fight infection.
Because TNF-α activates NF-κB (Hsu et al., 1995;Clarke et al., 2010) and mitogen-activated protein kinase (Chang and Karin, 2001; Lin, 2003), we further investigated the effect of a NF-κB inhibitor or mitogen-activated protein kinase inhibitors on TNF-α modulation of barrier proteins. Of note, only the c-Jun N-terminal kinase inhibitor blocked the TNF-α-mediated inhibition of FLG and LOR in the KCs. This observation strongly suggests that TNF-α modulates the expression of FLG and LOR through the c-Jun N-terminal kinase-dependent pathway.
Perhaps, the most important observation made in our current study was to examine the role of TNF antagonism in vivo on skin barrier protein expression in the skin of patients with psoriasis. Treatment of patients with psoriasis for 12 weeks with anti-TNF-α significantly increased the expression of FLG and LOR in their psoriasis lesions. Therefore, our data strongly suggest that TNF-α antagonists contribute to clinical improvement in patients with psoriasis by improving barrier protein expression. We also demonstrate directly in a clinically relevant setting that TNF-α has a key role in driving barrier dysfunction in psoriasis. We conclude that barrier protein deficiency in patients with psoriasis can be acquired via TNF-α-mediated downregulation.
MATERIALS AND METHODS
Subjects
Subjects included 10 healthy persons with no history of skin disease and 10 psoriasis patients with moderate-to-severe psoriasis. In addition, we studied skin biopsies from six adult psoriasis patients with moderate-to-severe psoriasis (mean psoriasis area and severity index: 25.9±4.4) who received 12 weeks of the anti-TNF-α etanercept therapy in vivo under a Rockefeller University Institutional Review Board-approved protocol. Skin biopsies (6 mm) were obtained from lesional and non-lesional skin of these patients before etanercept therapy was initiated and after 12 weeks of treatment. The patients were treated with 50 mg of etanercept, bi-weekly, as previously described (Zaba et al., 2007). Overall, a 69% decrease in psoriasis area and severity index in all study participants was observed with etanercept therapy, as previously described (Zaba et al., 2007, 2009). None of the patients had previously received systemic corticosteroids or cyclosporine, and none had received topical corticosteroid or calcineurin inhibitors for at least 1 week before enrollment. These studies were conducted according to the Declaration of the Helsinki Guidelines and were approved by the institutional review board at the National Jewish Health in Denver. All subjects gave written informed consent before participation in these studies.
From all other normal subjects and patients with lesional and non-lesional psoriasis, 2 mm punch skin biopsies were obtained. The skin biopsies were immediately submerged in 1 ml Tri Reagent (Molecular Research Center, Cincinnati, OH) and frozen at −80 °C for future RNA isolation and immunostaining, or in 1 ml of 10% buffered formalin for immunohistochemistry.
RNA preparation and real-time RT-PCR
Total RNA was isolated from skin biopsies by chloroform/phenol extraction and isopropanol precipitation according to manufacturer’s guidelines (Sigma Chemical, St Louis, MO). RNeasy Mini Kits (Qiagen, Valencia, CA) were used according to the manufacturer’s protocol to isolate RNA from cell cultures and to further purify RNA from skin biopsies. RNA was reverse transcribed into cDNA and analyzed by real-time RT-PCR by using an ABI Prism 7000 sequence detector (Applied Biosystems, Foster City, CA) described earlier (Nomura et al., 2003). Primers and probes for human GAPDH, FLG, LOR, IVL, and HBD-2 were purchased from Applied Biosystems. To allow for comparisons between samples and group, quantities of all targets in test samples were normalized to the corresponding GAPDH levels in the skin biopsies and cultured KCs, and expressed as target gene normalized to GAPDH.
Immunohistochemical staining
Paraffin-embedded tissues were cut at 5 μm and placed on frosted microscope slides. Using toluene and a series of ethanol washes slides were deparaffinized and rehydrated. Slides were incubated with a monoclonal mouse anti-human Ab directed against FLG (1:500 dilution; Abcam, Cambridge, MA) or a polyclonal rabbit anti-human Ab direct against LOR (1:500 dilution; Abcam) at 4 °C overnight. The cell and tissue staining kits (R&D systems, Minneapolis, MN) were used according to the manufacturer’s protocol. Ab specificity was determined by replacing the primary Ab with an isotype-matched control (purified non-immune mouse IgG or rabbit IgG; Southern Biotechnology, Birmingham, AL). All slides were coded before the samples were evaluated so that the identity of the study subjects was not revealed. Images were collected at × 400 magnification and the intensity of the immunostaining was scored on a scale from 0 to 5, with 0 indicating no staining and 5 the most intense staining.
Immunofluorescent staining
Frozen tissues from six patients were cut at 5 μm and fixed in 4% paraformaldehyde for 10 minutes at room temperature. Skin sections were then blocked as described above. Slides were then stained with a monoclonal mouse anti-human Ab directed against FLG (1:500 dilution; Abcam), a polyclonal rabbit anti-human Ab direct against LOR (1:500 dilution; Abcam), and a monoclonal mouse anti-human Ab directed against IVL (1:500 dilution; Abcam) at 4 °C overnight. Slides were then washed with phosphate-buffered saline/Tween (0.1%), followed by incubation with a Cy3-conjugated donkey anti-mouse IgG (Jackson Laboratories, West Grove, PA) or a Cy3-conjugated donkey anti-rabbit IgG (Jackson Laboratories). The slides were visualized with confocal microscopy (Leica, Wetzlar, Germany). Slides were coded to ensure patient anonymity. Images were collected at × 40, and levels of mean fluorescence intensity were measured with Slidebook 5.0 (Intelligent Imaging Innovations, Denver, CO). Mean fluorescence intensity was determined for each exposure group and was reported as mean mean fluorescence intensity ± SE.
KC cell culture
Primary human KCs were grown in serum-free EpiLife cell culture medium (Cascade Biologics, Portland, OR) containing 0.06 mmol l−1 calcium chloride, 1% human KCs growth supplement V2 (Cascade Biologics), and 1% antibiotics (penicillin/streptomycin) under standard tissue culture conditions. To investigate the effects of the TNF-α on barrier proteins, primary KCs were differentiated with 1.3 mmol l−1 CaCl2 for 5 days and then incubated in various concentrations of TNF-α (R&D systems) for 24 hours. To further examine the modulation of TNF-α on barrier proteins, KCs were stimulated in the presence and absence of TNF-α (20 ng ml−1), anti-TNF-α (0.1 μg ml−1, R&D systems), NF-κB activation inhibitor (10 nM, EMD Chemicals, Darmstadt, Germany), IL-4 and IL-13 (50 ng ml−1, R&D systems), or their combination for 24 hours. In addition, the primary KCs were stimulated with TNF-α in the presence or absence of mitogen-activated protein kinase inhibitors (EMD Chemicals), including extracellular signal-regulated kinases 1/2 inhibitor (5 μm), p38 inhibitor (5 μm), and c-Jun N-terminal kinase inhibitor (1 μm). Total RNA was isolated from KCs by using RNeasy kits according to the manufacturers guidelines (Qiagen) for real-time RT-PCR.
Statistical analysis
Statistical analysis was conducted using Graph Pad Prism, version 4.03 (San Diego, CA). Statistical differences in gene expression or protein staining between multiple groups was determined by using one-way analysis of variance, and significant differences were determined by a Tukey-Kramer test.
ACKNOWLEDGMENTS
This research was supported by NIAMS RO1 AR41256. We thank Maureen Sandoval for her assistance in the preparation of this manuscript.
Abbreviations:
- Ab
antibody
- AD
atopic dermatitis
- FLG
filaggrin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HBD
human-β defensin
- IVL
involucrin
- KC
keratinocyte
- LOR
loricrin
- TNF
tumor necrosis factor
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Albanesi C, Fairchild HR, Madonna S et al. (2007) IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol 179:984–92 [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, Melino G (2005) The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 6:328–40 [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410:37–40 [DOI] [PubMed] [Google Scholar]
- Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M et al. (2010) Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131:677–87 [DOI] [PubMed] [Google Scholar]
- Christophers E, Henseler T (1987) Contrasting disease patterns in psoriasis and atopic dermatitis. Arch Dermatol Res 279(Suppl):S48–51 [DOI] [PubMed] [Google Scholar]
- Clarke DL, Clifford RL, Jindarat S et al. (2010) TNF\{alpha\} and IFN\{gamma\} synergistically enhance transcript. J Biol Chem 285: 29101–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettehadi P, Greaves MW, Wallach D et al. (1994) Elevated tumour necrosis factor-alpha (TNF-alpha) biological activity in psoriatic skin lesions. Clin Exp Immunol 96:146–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman-Yassky E, Suárez-Fariñas M, Chiricozzi A et al. (2009) Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol 124:1235–44 [DOI] [PubMed] [Google Scholar]
- Howell MD, Kim BE, Gao P et al. (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120:150–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81:495–504 [DOI] [PubMed] [Google Scholar]
- Huffmeier U, Traupe H, Oji V et al. (2007) Loss-of-function variants of the filaggrin gene are not major susceptibility factors for psoriasis vulgaris or psoriatic arthritis in German patients. J Invest Dermatol 127:1367–70 [DOI] [PubMed] [Google Scholar]
- Kalinin A, Marekov LN, Steinert PM (2001) Assembly of the epidermal cornified cell envelope. J Cell Sci 114:3069–70 [DOI] [PubMed] [Google Scholar]
- Kim BE, Leung DY, Boguniewicz M et al. (2008) Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol 126:332–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen M, Chu CQ, Eedy DJ et al. (1993) Localization of tumour necrosis factor-alpha (TNF-alpha) and its receptors in normal and psoriatic skin: epidermal cells express the 55-kD but not the 75-kD TNF receptor. Clin Exp Immunol 94:354–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A (2003) Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25:17–24 [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Karabin GD, Barker JN et al. (1991) Cellular localization of interleukin-8 and its inducer, tumor necrosis factor-alpha in psoriasis. Am J Pathol 138:129–40 [PMC free article] [PubMed] [Google Scholar]
- Nomura I, Goleva E, Howell MD et al. (2003) Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol 171:3262–9 [DOI] [PubMed] [Google Scholar]
- Ong PY, Ohtake T, Brandt C et al. (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–60 [DOI] [PubMed] [Google Scholar]
- Proksch E, Brandner JM, Jensen JM (2008) The skin: an indispensable barrier. Exp Dermatol 17:1063–72 [DOI] [PubMed] [Google Scholar]
- Richardson SK, Gelfand JM (2008) Update on the natural history and systemic treatment of psoriasis. Adv Dermatol 24:171–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehra S, Yao Y, Howell MD et al. (2010) IL-4 regulates skin homeostasis and the predisposition towards allergic skin inflammation. J Immunol 184:3186–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM (1989) Terminal differentiation of epidermal keratinocytes. Curr Opin Cell Biol 1:1107–15 [DOI] [PubMed] [Google Scholar]
- Zaba LC, Cardinale I, Gilleaudeau P et al. (2007) Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 204:3183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaba LC, Suarez-Farinas M, Fuentes-Duculan J et al. (2009) Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol 124:1022–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–8 [DOI] [PubMed] [Google Scholar]