

Abstract
Neurotropic α-herpesvirinae subfamily members, herpes simplex virus type 1 (HSV-1) and bovine herpesvirus 1 (BoHV-1), are important viral pathogens in their respective hosts. Following acute infection on mucosal surfaces, these viruses establish life-long latency in neurons within trigeminal ganglia (TG) and central nervous system. Chronic or acute stress (physiological or psychological) increases the frequency of reactivation from latency, which leads to virus shedding, virus transmission, and recurrent disease. While stress impairs immune responses and inflammatory signaling cascades, we predict stressful stimuli directly stimulate viral gene expression and productive infection during early stages of reactivation from latency. For example, BoHV-1 and HSV-1 productive infection is impaired by glucocorticoid receptor (GR) antagonists but is stimulated by the synthetic corticosteroid dexamethasone. Promoters that drive expression of key viral transcriptional regulatory proteins are cooperatively stimulated by GR and specific Krüppel like transcription factors (KLF) induced during stress induced reactivation from latency. The BoHV-1 immediate early transcription unit 1 promoter and contains two GR response elements (GRE) that are essential for cooperative transactivation by GR and KLF15. Conversely, the HSV-1 infected cell protein 0 (ICP0) and ICP4 promoter as well as the BoHV-1 ICP0 early promoter lack consensus GREs: however, these promoters are cooperatively transactivated by GR and KLF4 or KLF15. Hence, growing evidence suggests GR and stress-induced transcription factors directly stimulate viral gene expression and productive infection during early stages of reactivation from latency. We predict the immune inhibitory effects of stress enhance virus spread at late stages during reactivation from latency.
1. Effects of corticosteroids on gene expression and immune responses
1.1. Regulation of gene expression by GR and corticosteroids
The hypothalamic-pituitary-adrenal (HPA) axis regulate stress responses in mammals. Following stressful stimuli, the HPA axis is activated, resulting in release of glucocorticoids (GCs) from the adrenal gland, reviewed in Smoak and Cidlowski (2004). The predominant GC in humans is cortisol, an essential hormone that also has tissue-specific functions. GCs regulate cell growth, development, metabolism, and in certain situations apoptosis. Stressful stimuli generally increase corticosteroids levels, which enter a cell, and bind to the glucocorticoid receptor (GR) or mineralocorticoid receptor (MR), reviewed in Oakley and Cidlowski (2013) (Fig. 1A). The MR or GR dimer bound to a corticosteroid enters the nucleus and within minutes remodels chromatin and induces transcription. While both receptors are capable of binding GCs, GR has a stronger affinity for the synthetic corticosteroid hormone dexamethasone (DEX), which mimic the effects of stress. These steps occur in the absence of de novo protein synthesis indicating there is a rapid response to increased cortisol levels.
Fig. 1.
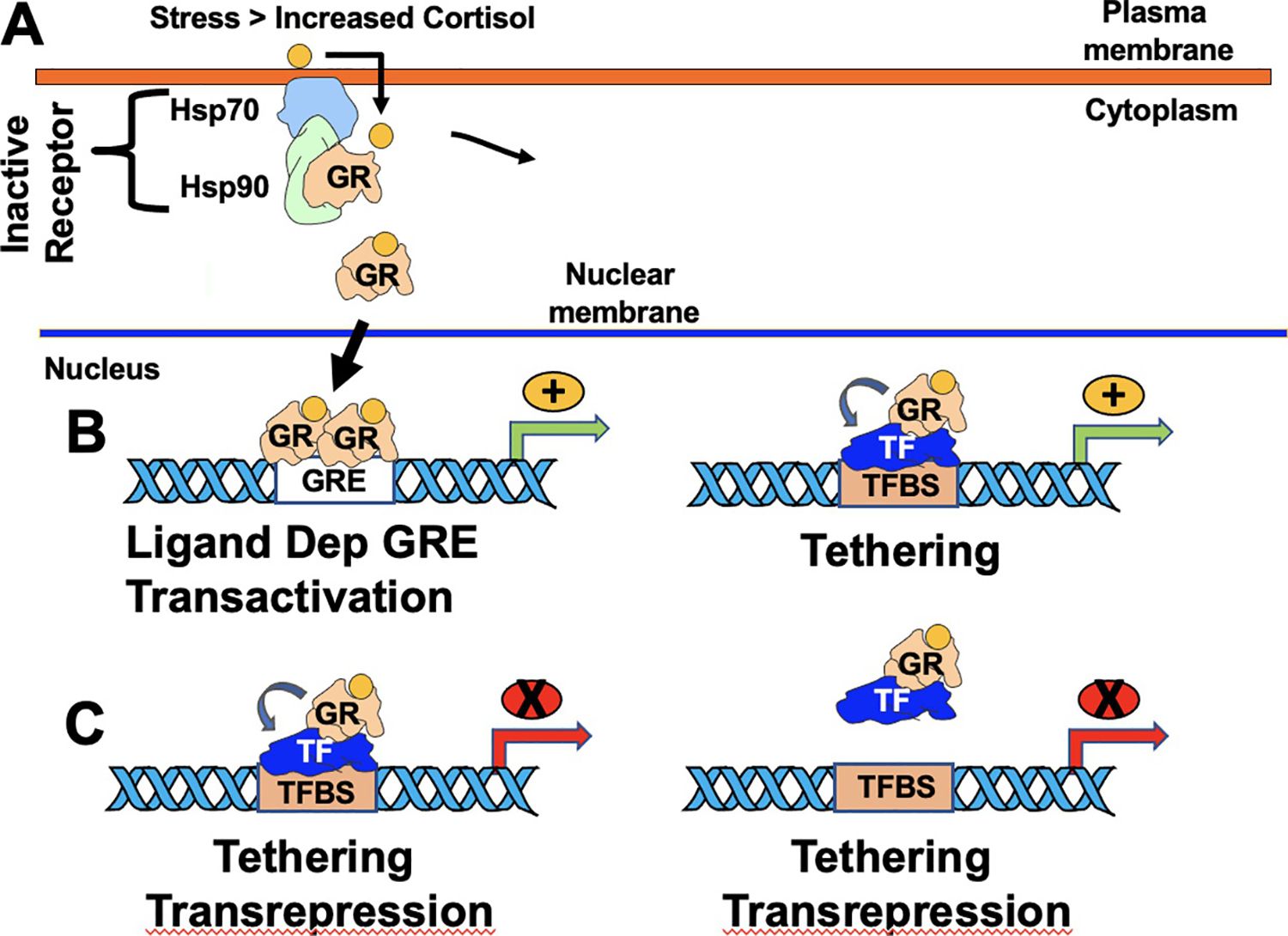
Schematic of how GR regulates gene expression. Panel (A) Inactive GR is sequestered in the cytoplasm by a protein complex containing heat-shock protein 70 (Hsp70) and Hsp90. Increased cortisol, due to increased stress, enters the cell, binds GR, and consequently the GR/hormone complex enters the nucleus. A GR/hormone homodimer subsequently interacts with a GRE. This interaction can lead to ligand dependent GRE transactivation (Panel B; left panel). In addition, GR can interact with other transcription factors (TF) bound to a cognate transcription factor binding site (TFBS), which stimulates transcription. Green arrows denote start site of transcription and orange circles with the plus sign denote transcription is stimulated. Panel (C) denotes two common mechanisms by which GR negatively regulates gene expression by interacting with transcription factors. Arrows denote start site of transcription and red circles with the X denotes transcription is repressed.
GR is a modular transcription factor that contains a DNA binding domain, steroid binding domain, and two transactivation domains (Giguere, Hollenberg, Rosenfeld, & Evans, 1986; Oakley & Cidlowski, 2013). Once translocated to the nucleus, nuclear GR or MR dimers regulate transcription by binding consensus glucocorticoid response elements (GRE) (Giguere et al., 1986; Wang et al., 2004). This process is commonly referred to as ligand-dependent activation (Fig. 1B). GREs contain the consensus sequence gGa/tACANNNTGTc/tCT: capital letters denote conserved nucleotides whereas an N means any nucleotide; a/t or c/t indicates that either of those nucleotides can be found while a lowercase letter denotes a nucleotide not conserved. GR monomers (GGACAN and GG/ANAC/gAT/G) can also bind ½ GREs and activate gene expression (Schiller, Chodankar, Watson, Stallcup, & Yamamoto, 2014; Wang et al., 2004). In addition to ligand-mediated of GR activation, GR can also activate gene expression via a tethering mechanism where GR interacts with transcription factors: however, GR does not directly bind DNA (Fig. 1B). A GR monomer can also stimulate transcription by binding to certain GR half binding sites (Schonevild, Gaemers, & Lamers, 2004; Taniguchi-Yanai et al., 2010). Transrepression by GR can also occur via protein: protein interactions (Fig. 1C; left panel). Finally, GR can interact with other transcription factors and prevent these transcription factors from binding DNA (Fig. 1C; right panel).
In addition to the ligand dependent mechanism of GR-mediated activation of gene expression, several studies revealed GR can stimulate gene expression via an un-liganded mechanism when steroid hormone levels are not dramatically increased (Galliher-Beckley, Williams, & Cidlowski, 2011). GR phosphorylation at serine 134 is crucial for un-liganded GR-induction of gene expression (Galliher-Beckley et al., 2011). For example, serine 134 is hyperphosphorylated is induced by glucose starvation, oxidative stress, UV irradiation, and osmotic shock suggesting cellular stressors in general induce GR phosphorylation at serine 134. Interestingly, serine 134 phosphorylation of GR alters gene expression patterns shown to respond to corticosteroids suggesting levels of cellular stress prior to hormone stimulation allows genes to respond more effectively to glucocorticoids.
1.2. Regulation of immune responses by GR and corticosteroids and its relevance to the latency-reactivation cycle
In addition to stimulating transcription, corticosteroids interfere with immune and inflammatory response (reviewed in Barnes, 1998; Funder, 1997; Schonevild et al., 2004). For example, activated GR interacts with two cellular transcription factors, activating protein 1 (AP-1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), which are crucial for an inflammatory response, reviewed in Oakley and Cidlowski (2013), Rhen and Cidlowski, (2005), Smoak and Cidlowski, (2004). AP-1 and NF-kB activate the beta-interferon enhanceosome following virus infection (Goodbourn, Zinn, & Maniatis, 1985), and beta-interferon has anti-viral activity (Decman, Freeman, Kinchington, & Hendricks, 2005). Interestingly, infiltrating lymphocytes that persist in trigeminal ganglia (TG) of mice or humans latently infected with herpes simplex virus 1 (HSV-1) or calves latently infected with bovine herpesvirus 1 (BoHV-1) (Decman et al., 2005; Knickelbein et al., 2008; Matundan, Mott, & Ghiasi, 2014; Mott, Allen, Zandian, & Ghiasi, 2014) impair reactivation from latency.
2. Introduction to latency-reactivation cycle of α-herpesvirinae subfamily members
2.1. HSV-1 and BoHV-1 are important pathogens
α-herpesvirinae subfamily members, HSV-1 and BoHV-1 for example, are important viral pathogens of their respective hosts. BoHV-1 contributes to bovine respiratory disease complex, the most important disease in cattle ( Jones & Chowdhury, 2007) and can induce abortions in infected cows (Chase et al., 2017). HSV-1 induces stromal keratitis and corneal scarring, ulcerative lesions on the mouth, and encephalitis (Al-Dujaili et al., 2011; Perng & Jones, 2010; Sekizawa & Openshaw, 1984; Tyler et al., 1995). Following acute infection, these viruses establish a life-long latent infection in sensory as well as other neurons, which innervate the site of acute infection, reviewed in Jones (2016), Perng and Jones (2010), Phelan, Barrozo, and Bloom (2017). In contrast to acute infection, viral gene expression is severely restricted during latency and infectious virus is not readily detectable.
2.2. Summary of latency-reactivation cycle
The latency-reactivation cycle is operationally divided into three distinct steps: establishment, maintenance, and reactivation (Jones, 1998, 2003; Perng & Jones, 2010). Following acute infection, latency is established, infectious virus is undetectable, but viral genomes are present in neurons within the peripheral nervous system (Cabrera et al., 1980; Fraser, Lawrence, Wroblewska, Gilden, & Koprowski, 1981; Rock & Fraser, 1983). Maintenance of latency, as with establishment, requires survival of infected neurons, restricted lytic cycle viral gene expression, and little or no virus shedding. A latent infection is periodically interrupted by an external stimulus, which leads to reactivation from latency and shedding of infectious virus at peripheral sites. Stressful stimuli correlate with increasing the incidence of reactivation from latency, which is crucial for virus transmission and with respect to HSV-1 can cause recurrent disease.
2.2.1. HSV-1 LAT encodes multiple products that mediate latency
LAT is the only viral transcript abundantly expressed during a latent infection ( Jones, 2013; Perng & Jones, 2010; Phelan et al., 2017). Deleting LAT coding sequences or LAT promoter sequences reduce the efficiency of reactivation in latently infected mice and rabbits. Explant of TG from mice latently infected with HSV-1 is the most reliable method to examine reactivation from latency in mouse models of infection. After TG explant, LAT expression is reduced, chromatin remodeling of the infected cell protein 0 (ICP0) promoter occurs, ICP0 transcription occurs (Amelio, Giordani, Kubat, O’Neil, & Bloom, 2006; Du, Zhou, & Roizman, 2011), and infectious virus is produced (Pesola, Zhu, Knipe, & Coen, 2005). The LAT locus expresses multiple transcripts, six micro-RNAs, and two small non-coding RNAs (Peng, Vitvitskaia, Carpenter, Wechsler, & Jones, 2008; Umbach et al., 2008, 2009). LAT inhibits apoptosis and viral gene expression (Ahmed, Lock, Miller, & Fraser, 2002; Branco & Fraser, 2005; Inman, Lovato, Doster, & Jones, 2001; Inman, Perng, et al., 2001; Inman, Zhang, Geiser, & Jones, 2001; Perng et al., 2000; Thompson & Sawtell, 2001), which protects neurons from cell death and promotes establishment and maintenance of latency (Jones, 1998, 2003; Perng & Jones, 2010). Further support for LAT maintaining latency comes from a study demonstrating that shedding of a LAT null mutant dramatically declines following multiple rounds of heat stress-induced reactivation in a mouse model of latency (Thompson & Sawtell, 2011). Conversely, a LAT positive strain of HSV-1 shed higher levels of virus after multiple rounds of reactivation. The anti-apoptosis functions of LAT requires an active AKT serine/threonine protein kinase (Carpenter et al., 2015; Li, Carpenter, Hsiang, Wechsler, & Jones, 2010). Furthermore, LAT enhances CD8+ T cell exhaustion during latency (Allen et al., 2011) and inhibits granzyme B mediated apoptosis (Jiang et al., 2011). Further evidence that the anti-apoptosis functions of LAT are important during the latency-reactivation cycle comes from studies demonstrating that inserting an anti-apoptosis gene into the LAT locus of a LAT−/− mutant virus restores reactivation to WT like levels (Jin et al., 2008; Jin, Perng, Nesburn, Jones, & Wechsler, 2005; Mott et al., 2003; Perng et al., 2002). LAT may also influence expression of cellular factors that promote establishment and maintenance of latency because LAT is generally considered to express non-coding RNAs.
2.2.2. BoHV-1 LR gene expression promotes maintenance of latency
Abundant expression of the BoHV-1 encoded latency related (LR) gene occurs in latently infected neurons; however, infectious virus is not readily detected (maintenance of latency) (Jones, 1998, 2003; Jones et al., 2006; Kutish, Mainprize, & Rock, 1990; Rock, Beam, & Mayfield, 1987; Rock, Lokensgard, Lewis, & Kutish, 1992). LR-RNA has a unique start site in TG and is anti-sense to and overlaps the bICP0 gene (Bratanich, Hanson, & Jones, 1992; Hossain, Schang, & Jones, 1995). Two LR-specific micro-RNAs are expressed during latency and in transient transfection assays they reduce bICP0 steady state protein levels; but not bICP0 mRNA levels (Jaber, Workman, & Jones, 2010).
The LR gene contains two well-defined open reading frames (ORF1 and ORF2), and two reading frames lacking an initiating ATG (RF-B and RF-C). A LR mutant virus strain with three stop codons at the N-terminus of ORF2 exhibits diminished clinical symptoms, and reduced virus shedding from the eye, TG, or tonsils of infected calves (Inman, Lovato, et al., 2001; Inman, Lovato, Doster, & Jones, 2002; Perez, Inman, Doster, & Jones, 2005). ORF1 and ORF2 are expressed when bovine cells are infected with wild-type the LR-rescued virus, but these proteins have reduced or no expression following infection with the LR mutant virus ( Jiang, Inman, Zhang, Posadas, & Jones, 2004; Meyer et al., 2007, 2007). Wild-type BoHV-1, but not the LR mutant virus, can reactivate from latency (Inman et al., 2002). The anti-apoptosis activity of ORF2 enhances survival of infected neurons (Ciacci-Zanella, Stone, Henderson, & Jones, 1999; Henderson, Perng, Nesburn, Wechsler, & Jones, 2004; Inman, Zhang, et al., 2001; Saira et al., 2008; Shen & Jones, 2008; Zhang & Jones, 2001, 2005; Zhang, Jiang, Zhou, & Jones, 2006). ORF2 also interacts with cellular transcription factors (β-catenin, Notch1, Notch3, and c/EBP-alpha) suggesting these interactions promote the establishment and maintenance of latency by interfering with lytic cycle viral gene expression and productive infection (Meyer, Perez, Geiser, et al., 2007; Meyer, Perez, Jiang, et al., 2007; Workman, Sinani, Pittayakhajonwut, & Jones, 2011; Workman, Zhu, Keel, Smith, & Jones, 2018; Zhu, Workman, & Jones, 2017). ORF2 amino acid sequences that interfere with Notch functions do not overlap with ORF2 sequences necessary for inhibiting apoptosis (Sinani & Jones, 2011). In summary, LR gene products promote establishment and maintenance of latency by promoting neuronal survival and impairing lytic cycle viral gene expression.
3. Stress stimulates reactivation from latency and productive infection
3.1. Stress increases the incidence of HSV-1 reactivation from latency
Physical and psychological stressors suppress immune responses, which can potentially stimulate viral replication and reactivation from latency (Cassidy, Meadows, Catalan, & Barton, 1997; Glaser & Kiecolt-Glaser, 2005; Glaser, Kiecolt-Glaser, Speicher, & Holliday, 1985; Jones, 2014; Padgett et al., 1998; Varnell, Kaufman, Hill, & Thompson, 1995). Reactivation from latency can result in severe recurrent disease and is a major obstacle in developing more effective antiviral therapies. Stress induces reactivation of HSV-1 through physiological means (Glaser et al., 1985; Glaser & Kiecolt-Glaser, 2005; Padgett et al., 1998) or by addition of a synthetic corticosteroid dexamethasone (DEX) (Harrison, Zhu, Thunuguntla, & Jones, 2019). However, many in the field assume the effects of stress on reactivation from latency are due to the anti-inflammatory and immune-inhibitory effects of corticosteroids.
When TG from latently infected mice is explanted in media containing DEX, significantly higher levels of virus shedding are observed relative to TG explanted in media lacking DEX. Addition of the GR-specific antagonist CORT-108297 significantly reduced virus shedding during explant-induced reactivation (Harrison et al., 2019). This study also revealed when latently infected TG are explanted in the presence of DEX for 4 or 8h to stimulate reactivation, higher numbers of TG neurons express the GR protein relative to TG from latently infected mice or TG explanted for 4 or 8h without DEX. Approximately 50% of rat TG sensory neurons express GR (DeLeon et al., 1994) confirming DEX stimulates GR expression in TG neurons and stimulates HSV-1 reactivation from latency. Interestingly, the viral tegument protein (VP16) that stimulates all immediate early (IE) promoters is readily detected prior to viral transcriptional regulatory proteins, ICP0 and ICP4 (Harrison et al., 2019).
Recent studies identified numerous cellular signaling pathways that appear to mediate the latency-reactivation cycle. These include the mTOR pathway, phosphatidylinositol 3-kinase (PI3K)/Akt signaling axis, and Wnt-signaling pathway (Camarena et al., 2010; Harrison, Zhu, Thunuguntla, & Jones, 2020; Kim, Mandarino, Chao, Mohr, & Wilson, 2012; Li et al., 2010). Furthermore, serum and glucocorticoid-regulated protein kinases (SGKs) are serine/threonine protein kinases stimulated by corticosteroids and other cellular stressors (Webster, Goya, Ge, Maiyar, & Firestone, 1993). When SGKs are inhibited, BoHV-1 and HSV-1 virus replication is decreased in a dose-dependent manner (Kook & Jones, 2016); inversely, when PI3K/Akt is inhibited, reactivation from latency is induced (Camarena et al., 2010; Kim et al., 2012). Akt is an important regulator of the Wnt signaling pathway (Zhang et al., 2013; Zhao et al., 2012); Wnt is active during latency (as shown by more TG neurons that express the β-catenin protein). However, during DEX-stimulated explant-induced reactivation reduced numbers of TG neurons express β-catenin (Harrison et al., 2020). The Wnt pathway is directly influenced by stress, as physiological stressors such as UV light enhances expression of soluble Wnt antagonists, which interfere with β-catenin expression (Shou et al., 2002). When TG from HSV-1 latently infected mice are explanted to induce reactivation, addition of the Wnt antagonist iCRT14 significantly reduces virus titers, which confirms the Wnt/β-catenin pathway enhances virus reactivation from latency (Harrison et al., 2019). We do not believe MR plays a significant role during reactivation because a GR-specific antagonist significantly inhibits explant-induced reactivation: furthermore, MR is a weak activator of transcription relative to GR (Funder, 1997).
3.2. GCs rapidly induce BoHV-1 reactivation from latency by stimulating viral and cellular gene expression in TG sensory neurons
Administration of the synthetic corticosteroid DEX to latently infected calves or rabbits initiate BoHV-1 reactivation from latency 100% of the time (Inman et al., 2002; Jones, 1998, 2003; Jones et al., 2000, 2006; Rock et al., 1992). Within 3h after latently infected calves are treated with DEX, lytic cycle viral RNA expression can be detected in TG neurons (Winkler, Doster, & Jones, 2000; Winkler, Doster, Sur, & Jones, 2002). DEX treatment of latently infected calves also induces apoptosis of T cells that persist in TG after infection (Winkler et al., 2002). All proteins expressed from immediate early promoters, bICP0, bICP4, and bICP22, are activated during early stages of reactivation from latency (Frizzo da Silva, Kook, Doster, & Jones, 2013; Guo, Li, & Jones, 2019; Kook, Doster, & Jones, 2015). Interestingly, a viral tegument protein expressed as a late gene (VP16) that activates all immediate early viral promoters is also expressed during early stages of reactivation. In sharp contrast, other late viral proteins (gE and gD) are not readily detected.
Within 3h after DEX treatment, 11 cellular genes are induced more than 10-fold in TG (Workman et al., 2012). Pentraxin 3, a regulator of innate immunity and neurodegeneration, is stimulated more than 30-fold by 3h after DEX treatment. Two transcription factors, promyelocytic leukemia zinc finger (PLZF) and Slug are induced more than 15-fold by 3h after DEX treatment. PLZF or Slug stimulates BoHV-1 productive infection 20-fold or fivefold respectively, and Slug stimulates the late glycoprotein C promoter more than 10-fold. Additional DEX induced transcription factors, SPDEF (Sam-pointed domain containing Ets transcription factor), Krüppel-like transcription factor 15 (KLF15), KLF4, KLF6, and GATA6, stimulate productive infection and certain key viral promoters. The finding that four KLF family members (KLF4, KLF6, KLF15, and PLZF) are stimulated during DEX induced reactivation from latency is intriguing because KLF family members resemble the Sp1 transcription factor family and both family of transcription factors interact with GC rich motifs, reviewed in (Bieker, 2001; Kaczynski, Cook, & Urrutia, 2003). The BoHV-1 genome is GC rich and many viral promoters contain Sp1 consensus binding sites and other GC rich motifs suggesting there are many KLF binding sites in the BoHV-1 genome. Hence, we predict certain KLF transcription factors regulate viral transcription during reactivation from latency.
3.3. Regulation of productive infection by stress and GR
HSV-1 contains numerous GREs throughout the viral genome (unpublished data) and GR activation by DEX stimulate viral gene transcription during productive infection (51). This observation also suggests GR activation directly stimulates viral gene expression and productive infection during reactivation from latency. Additionally, GR-mediated immune suppression provides a permissive environment for HSV-1 productive infection and spread during successful reactivation from latency (Oakley & Cidlowski, 2013; Rhen & Cidlowski, 2005).
A GR specific antagonist (CORT-108297) reduced virus shedding in productively infected mouse neuroblastoma cells (Neuro-2A) indicating activated GR stimulates productive infection (Ostler, Harrison, Schroeder, Thunuguntla, & Jones, 2019). This result was consistent with a previous study that demonstrated pre-treating human gingival fibroblasts with DEX prior to HSV-1 infection increased GR steady state levels and viral yields (Erlandsson et al., 2002). GR and DEX treatment also stimulate BoHV-1 productive infection in cultured cells (El-mayet, Sawant, Thunuguntla, Zhao, & Jones, 2020; El-Mayet, Sawant, Thungunutla, & Jones, 2017; Kook, Henley, Meyer, Hoffmann, & Jones, 2015a, 2015b).
A GRE was identified within the unique long (UL) origin of replication (oriL): however, it differs from a standard GRE consensus sequence (Hardwicke & Schaffer, 1997). For example, there are 18 undefined “N” center nucleotides instead of three and secondly, the sequence itself is mirrored such that TGTc/tCT is located 5′ to gGa/tACA: (5′-TGTCCTN18 AGGACA-3′). This putative GRE binds GR in vitro and, in the presence of DEX stimulates viral DNA replication in differentiated neuronal cell cultures. Moreover, mutagenesis of the GRE in oriL reduces viral pathogenesis during acute infection and impairs reactivation from latency in a mouse model of infection (Balliet & Schaffer, 2006). The BoHV-1 genome contains two origins of replication: however, there does not appear to be a oriL. Strikingly, both origins of replication in the BoHV-1 genome contain consensus GREs (unpublished studies) suggesting stress also impacts viral replication. In summary, these studies indicate stress directly impacts viral replication and productive infection.
4. Promoters that drive key viral regulatory genes are stimulated by GR and specific stress-induced transcription factors
4.1. Reactivation from latency is stimulated by key viral regulatory proteins
Ectopic expression of infected cell protein 0 (ICP0), ICP4, or virion protein 16 (VP16) induces reactivation from latency using TG cultures derived from mice latently infected with HSV-1 (Halford, Kemp, Isler, Davido, & Schaffer, 2001). ICP0 is a promiscuous transactivator of viral gene expression that also interferes with innate immune responses and disrupts anti-viral promyelocytic leukemia nuclear bodies (Boutell & Everett, 2013; Everett, 2000; Jones, 2009). The ICP4 protein, a 175kD phosphoprotein, is the only viral transcriptional activator that is essential for productive infection because it activates E and L genes (DeLuca, McCarthy, & Schaffer, 1985). ICP4 specifically binds many sites on the viral genome (Kristie & Roizman, 1986) and interacts with the TATA box-binding protein plus RNA pol transcription fact IIB (TFIIB) to stimulate early and late viral gene expression (Smith, Bates, Rivera-Gonzalez, Gu, & DeLuca, 1993). A viral tegument protein, VP16, stimulates IE transcription by interacting with two cellular proteins (Oct1 and host cellular factor 1), and this multi-protein complex binds specific sequences in IE promoters; hence, VP16 transactivates all IE promoters (Misra, Bratanich, Carpenter, & O’Hare, 1994; Misra, Walker, Hayes, & O’Hare, 1995; O’Hare, 1993). VP16, ICP0 and ICP4 proteins are detected in TG neurons of mice latently infected with HSV-1 following explant (Harrison et al., 2019). Expression of these viral proteins are accelerated by DEX treatment.
Given the important roles that ICP0 and ICP4 play in regulating viral replication, these two immediate early genes are predicted to play essential roles during reactivation from latency in humans. Interestingly, VP16, has been reported to induce viral gene expression in HSV-1 quiescently infected embryonic rat neuronal cultures (Camarena et al., 2010; Kim et al., 2012) as well as initiating HSV-1 reactivation in vivo from latently infected mice exposed to hyperthermic stress (Thompson, Preston, & Sawtell, 2009). Since VP16 selectively activates IE gene expression (O’Hare, 1993; O’Hare & Goding, 1988; O’Hare & Hayward, 1985), its expression during early stages of reactivation from latency would enhance expression of lytic cycle viral transcriptional regulatory genes.
4.2. ICP0 promoter is transactivated by stress-induced transcription factors
The ICP0 promoter and gene are in the long internal repeats of HSV-1 (IRL; Fig. 2A and B). There is also a copy of ICP0 in the TRL repeat: both copies of the ICP0 gene overlap and are antisense relative to LAT. The ICP0 promoter spans −800 to +150 nucleotides relative to the transcription initiation site (Fig. 2C) and is considered to be the full-length ICP0 promoter (Kushnir et al., 2009). This promoter is stimulated by heat stress (Kushnir et al., 2009) and strongly transactivated by KLF15 in mouse neuroblastoma cells (Neuro-2A) (Sinani et al., 2013).
Fig. 2.
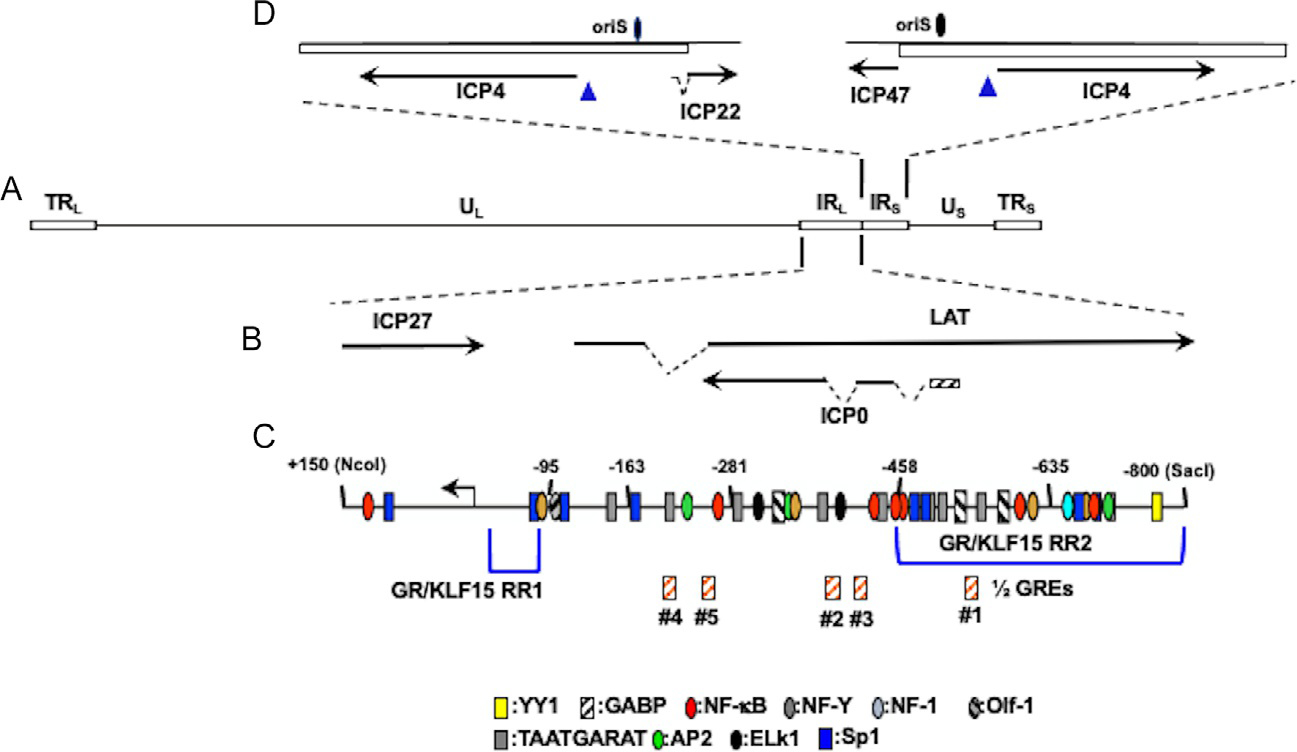
Location of ICP4 gene, ICP0 gene, and adjacent genes within HSV-1 genome. Panel (A) The prototypic HSV-1 genomic structure is shown. Viral repeat regions are shown as open rectangles. TRL is the terminal long repeat. IRL is the internal (or inverted) long repeat. TRS is the terminal short repeat. IRS is the internal (or inverted) short repeat. The unique long (UL) and unique short (US) regions are depicted by a solid line. Panel (B) Location of ICP0 and other genes within IRL. Gene organization within TRL is the same as IRL. Dashed lines denote introns in ICP22, LAT, and ICP0. Panel (C) Schematic of ICP0 promoter (−800 to +150). Numbers assigned correspond to nucleotide positions relative to the transcription initiation site (black arrow). Location of ½ GREs is indicated by orange hatched boxes. GR and KLF15 responsive regions (GR/KLF15 RR1 and GR/KLF15 RR2) previously identified in Neuro-2A and Vero cells are denoted by blue brackets (Ostler et al., 2019; Sinani, Cordes, Workman, Thunuguntia, & Jones, 2013). Location of other potential transcription factor binding sites were previously identified (Kushnir, Davido, & Schaffer, 2009). Panel (D) Location of ICP4 coding regions and flanking genes in IRS (ICP22) or TRS (ICP47). Dashed lines in ICP22 denote the location of an intron. Location of the origin of replication oriS is denoted by black oval.
This initial study was performed using minimal essential media (MEM) that contains 10% fetal bovine serum (FBS). While GATA6, SPDEF, PLZF, SLUG, KLF4, and KLF6 also transactivate the ICP0 promoter, the effects were modest compared to KLF15. A recent study evaluated the ICP0 promoter in MEM containing 2% stripped FBS in the presence or absence of the synthetic corticosteroid DEX. FBS passed through a column that contains “activated” charcoal removes hormones and other lipid-based molecules. However, this treatment does not remove salts, glucose, or most amino acids. In Neuro-2A or Vero (monkey kidney) cells, GR, KLF15, and DEX treatment cooperatively transactivate the ICP0 promoter (Ostler et al., 2019). Cell-type specific differences were seen with the ICP0 promoter in Vero versus Neuro-2A cells. For example, a fragment spanning −635 to +150 (Fig. 2D) is transactivated by GR, KLF15, and DEX at slightly higher levels than the full-length ICP0 promoter in Neuro-2A; but not Vero cells. Consequently, sequences between −458 and −800 appear to be a GR/KLF15 responsive region 2 (RR2) activated by these stress-induced transcription factors. KLF15 also transactivates a −95 to +150 promoter construct approximately 80-fold (Sinani et al., 2013). Transactivation of a −95 to +150 promoter construct is reduced to basal activity when a consensus Sp1 binding site and KLF15 binding site (GGGGGAGGGGG) (Otteson, Lai, Liu, & Zack, 2005) are mutated. Hence, the −95 to −1 promoter proximal sequences are referred to as GR/KLF15 RR1. Collectively, these results indicate that the ICP0 promoter contains at least two separate domains are important for transactivation by GR and KLF15. While there are no consensus glucocorticoid response elements (GREs) in the ICP0 promoter, five putative ½ GREs are present (Fig. 2D). Mutagenesis of ½ GRE #1 reduced transactivation by GR and KLF15: however, a mutant lacking all five ½ GREs was not significantly different than the mutant lacking only ½ GRE #1 (Ostler et al., 2019). ICP0 promoter sequences spanning nucleotides −420 to −70 promote in vivo viral productive infection, ICP0 gene expression in cultured cells (Davido & Leib, 1996), and production of infectious virus during hyperthermic stress induced reactivation from latency (Thompson & Sawtell, 2006) suggesting stress induced transcription factors stimulate ICP0 expression in vivo.
Notably, GR and KLF15 regulate gene expression via a feed-forward loop in response to stress (Mangan & Alon, 2003; Sasse et al., 2013, 2015). A feed-forward loop contains a primary factor (GR) that stimulates expression of a second transcription factor (KLF15). GR is stably associated with KFL15, which correlates with increased transcription of genes not strongly activated by GR or KLF15 alone (Mangan & Alon, 2003; Sasse et al., 2013, 2015). For example, GR and KLF15 cooperatively induce expression of genes that regulate adipogenesis and amino acid-metabolizing enzymes. Neuronal signaling pathways stimulate ICP0 promoter activity because transgenic mice containing the ICP0 promoter upstream of a reporter gene, but not the ICP4 promoter, drives expression of the reporter in specific neuronal populations (Loiacono, Myers, & Mitchell, 2002; Taus & Mitchell, 2001).
4.3. The ICP4 promoter is transactivated by stress-induced transcription factors
A copy of the ICP4 promoter and gene is located within the short internal repeat (IRS) and short terminal repeat (TRS; Fig. 2D). PLZF, SLUG and KLF4 modestly transactivate an ICP4 promoter construct in Neuro-2A cells containing 10% FBS (Sinani et al., 2013). An ICP4 enhancer fragment, nucleotides −330 to −110 (pα4R; Fig. 3A and B), cloned upstream of a minimal promoter in a luciferase reporter vector was examined in cells incubated with stripped FBS to test whether stress-mediated transactivation of ICP4 sequences occurs in a heterologous promoter. The pα4R construct exhibited strong enhancer activity in Neuro-2A and Vero cells and was cooperatively transactivated by GR and specific stress-induced transcription factors (KLF4, KLF15, PLZF, and SLUG) (Ostler et al., 2021). Interestingly, several key differences were observed in Neuro-2A versus Vero cells. The most striking difference was GR+KLF15 cooperatively transactivated the pα4R construct in Vero but not in Neuro-2A cells. GR and KLF4 also cooperatively transactivated the pa4R construct in Vero cells in the absence of DEX treatment, however, DEX increased transactivation in transfected Neuro-2A cells. Thirdly, PLZF mediated transactivation is ligand dependent in Neuro-2A but not Vero cells. Finally, GR, SLUG and DEX treatment significantly stimulated pα4R promoter activity in in Neuro-2A cells, but not in Vero cells.
Fig. 3.
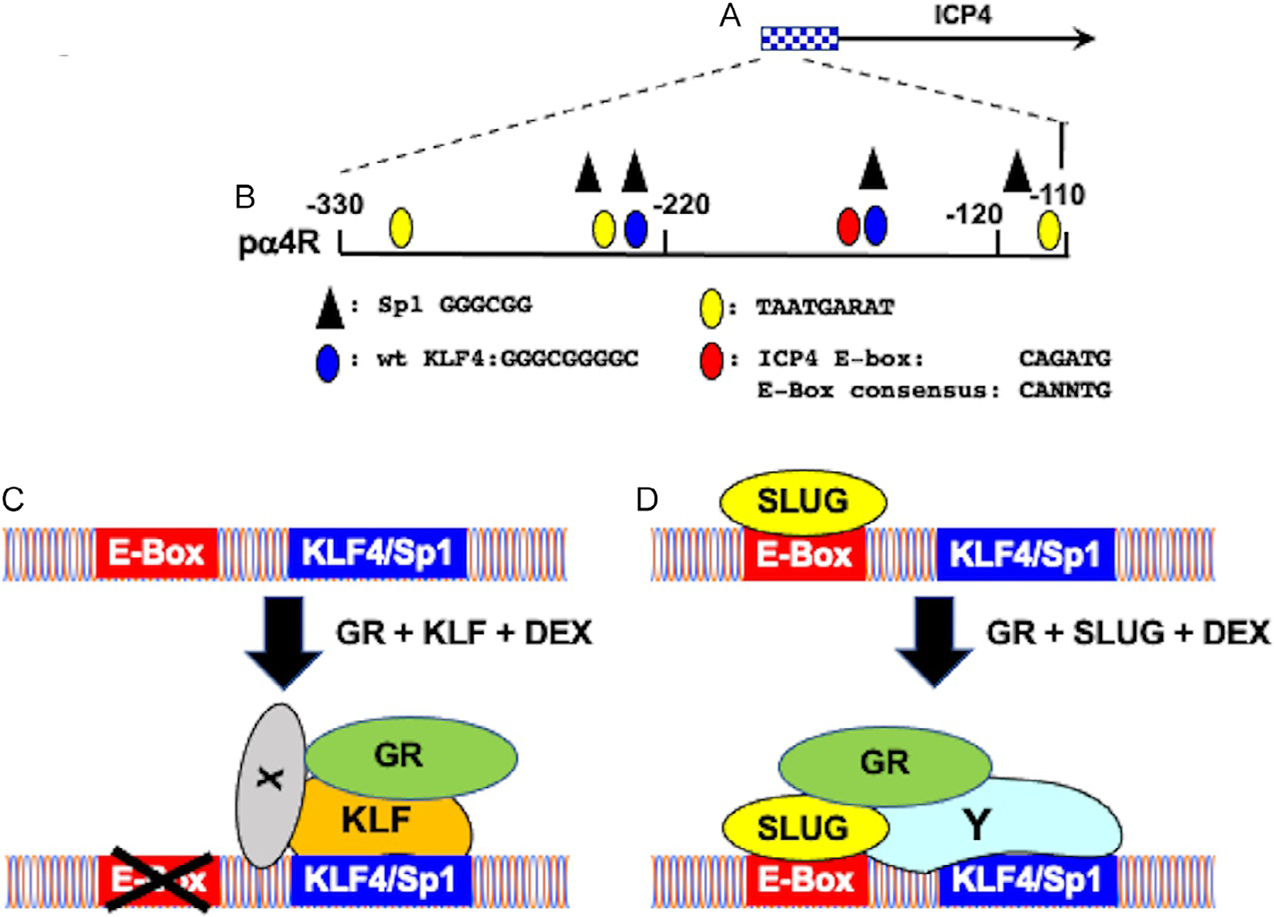
Schematic of ICP4 promoter/enhancer and mechanism by which GR and stress-induced transcription factors stimulate ICP4 enhancer sequences. Panel (A) Schematic of ICP4 promoter/enhancer sequences and ICP4 coding sequences. Panel (B) Schematic of ICP4 enhancer fragment (pα4R) inserted upstream of the minimal promoter within the firefly luciferase reporter plasmid, pGL4.24[luc2/minP] (Ostler, Thunguntla, Hendrickson, & Jones, 2021). Nucleotide position is given relative to the ICP4 transcription initiation site. Location of transcription factor binding sites are shown below the schematic. Panel (C) Potential model summarizing how GR, KLF family members (KLF4, KLF15, and PLZF) and DEX cooperatively transactivate the ICP4 enhancer. X denotes unknown factors believed to facilitate tethering GR to KLF family members. The X that crosses out the E-Box denotes these sequences are not important for transactivation by GR and KLF family members in the context of pa4R construct. Panel (D) Potential model summarizing how GR, SLUG, and DEX cooperatively transactivate the ICP4 enhancer. Y denotes Sp1 or KLF family members that are important for GR and SLUG mediated transactivation in the context of the pa4R construct.
Three consensus KLF4 binding sites (Soufi et al., 2015) are located in the ICP4 enhancer fragment: two are frequently bound by KLF4 (Fig. 3B; denoted by a blue oval). The KLF4 binding site overlaps a consensus Sp1 binding site: thus, mutating these KLF4 binding sites also destroys the Sp1 binding site. Mutating both KLF4 binding sites reduced GR, DEX and KLF4, PLZF, or SLUG cooperative transactivation to basal levels. Interestingly, the 3′ KLF4 binding site adjacent to the E-Box is more important than the 5′ KLF4 binding site. Collectively, these studies revealed consensus KLF4/Sp1 binding sites were essential for GR and stress-induced mediated transactivation of the ICP4 enhancer activity.
Based on these observations, we developed a model to explain how GR and stress-induced transcription factors strongly transactivate the ICP4 enhancer in the absence of a consensus GRE (Fig. 3C and D). The E-Box that is 8 bases from the 3′ consensus KLF4 binding site is crucial for GR, DEX, and Slug mediated transactivation of the pα4R construct: strikingly, this mutation does not influence cooperative transactivation mediated by GR, DEX, and KLF4 or PLZF indicating specific binding to the E-Box by an E-Box enhancer binding protein is not important (Fig. 3C). This model further predicts GR is tethered to the 3′ KLF4/Sp1 binding site via a KLF family member (KLF4, KLF15, or PLZF) (Bieker, 2001; Black, Black, & Azizkhan-Clifford, 2001). Mutating consensus KLF4 binding sites impairs GR, DEX, and SLUG mediated transactivation suggesting SLUG interacts with Sp1 or a KLF family member to cooperatively transactivate pα4R promoter activity (Fig. 3D; denoted by Y). Although SLUG occupancy of E-boxes can lead to transcriptional activation (Wels, Koefinger, Joshi, Bergler, & Schaider, 2011), SLUG is generally considered to be a transcriptional repressor (Hemavathy, Ashraf, & Ip, 2000; Hemavathy, Guru, Harris, Chen, & Ip, 2000). Hence, SLUG may occupy ICP4 enhancer sequences and repress ICP4 expression during latency. Following a stressful stimulus, GR and other coactivators can convert SLUG into a transcriptional activator and participate in activating lytic cycle viral gene expression. Notably, a consensus E-Box binding site is less than 50 nucleotides upstream of the HSV-1 VP16 TAATA box (Sawtell & Thompson, 2016). Since the VP16 promoter is proposed to induce reactivation from latency, this E-Box may also be important for stimulating VP16 expression following stressful stimuli.
Chromatin immunoprecipitation (ChIP) assays demonstrated GR occupancy of ICP4 enhancer sequences is significantly reduced when KLF4/Sp1 binding sites or the E-Box is mutated relative to the wild-type pα4R construct in transfected Neuro-2A cells (Ostler et al., 2021). ChIP assays also revealed SLUG occupancy with the E-Box mutant was similar to negative controls: however, as expected SLUG readily binds wild-type ICP4 enhancer sequences. These studies reveal there is a correlation between occupancy of ICP4 enhancer sequences by GR and the respective stress-induced transcription factors with KLF4/Sp1 or E-Box motifs and transcriptional activation.
4.4. Activation of BoHV-1 promoters by GR and stress-induced transcription factors
For successful BoHV-1 reactivation from latency to occur, the quiescent viral genome must be remodeled into actively transcribing genome to produce infectious virus. BoHV-1 proteins encoded by all three immediate early (IE) genes (bICP0, bICP4, and bICP22) as well as the viral tegument protein that specifically activates IE promoters (VP16) are detected in TG neurons within hours after DEX treatment (Frizzo da Silva et al., 2013; Guo et al., 2019; Kook, Doster, & Jones, 2015). There are over 100 consensus GREs in the BoHV-1 genome (Kook, Henley, et al., 2015a, 2015b), which partially explains why stress, as mimicked by DEX treatment, consistently induces reactivation from latency (Sheffy & Davies, 1972; Workman, Perez, Doster, & Jones, 2009a, 2009b). Consequently, activation of GR by corticosteroids is expected to directly stimulate viral gene expression and productive infection, a prerequisite for reactivation from latency.
The organization of the BoHV-1 bICP0 and bICP4 coding regions is unique relative to other a-herpesvirinae subfamily members. For instance, the IEtu1 promoter drives IE expression of IE/2.9 and IE/4.2 mRNAs, which are translated into the bICP0 or bICP4 proteins respectively (Wirth et al., 1992; Wirth, Gunkel, Engels, & Schwyzer, 1989; Wirth, Vogt, & Schwyzer, 1991) (Fig. 4A). IEtu2 drives expression of the bICP22 protein, which suppresses certain viral promoters, including the IEtu1 promoter (Koppel et al., 1997). The bICP0 E promoter drives expression of E/2.6, an early transcript translated into the bICP0 protein (Fraefel et al., 1994; Geiser, Zhang, & Jones, 2005; Saira et al., 2008; Wirth et al., 1989, 1991, 1992), presumably to maintain high levels of bICP0 protein expression throughout productive infection. In spite of these differences, the BoHV-1 regulatory proteins are considered functional analogs of HSV-1 encoded ICP0, ICP4, and ICP22 (Jones, 2003).
Fig. 4.
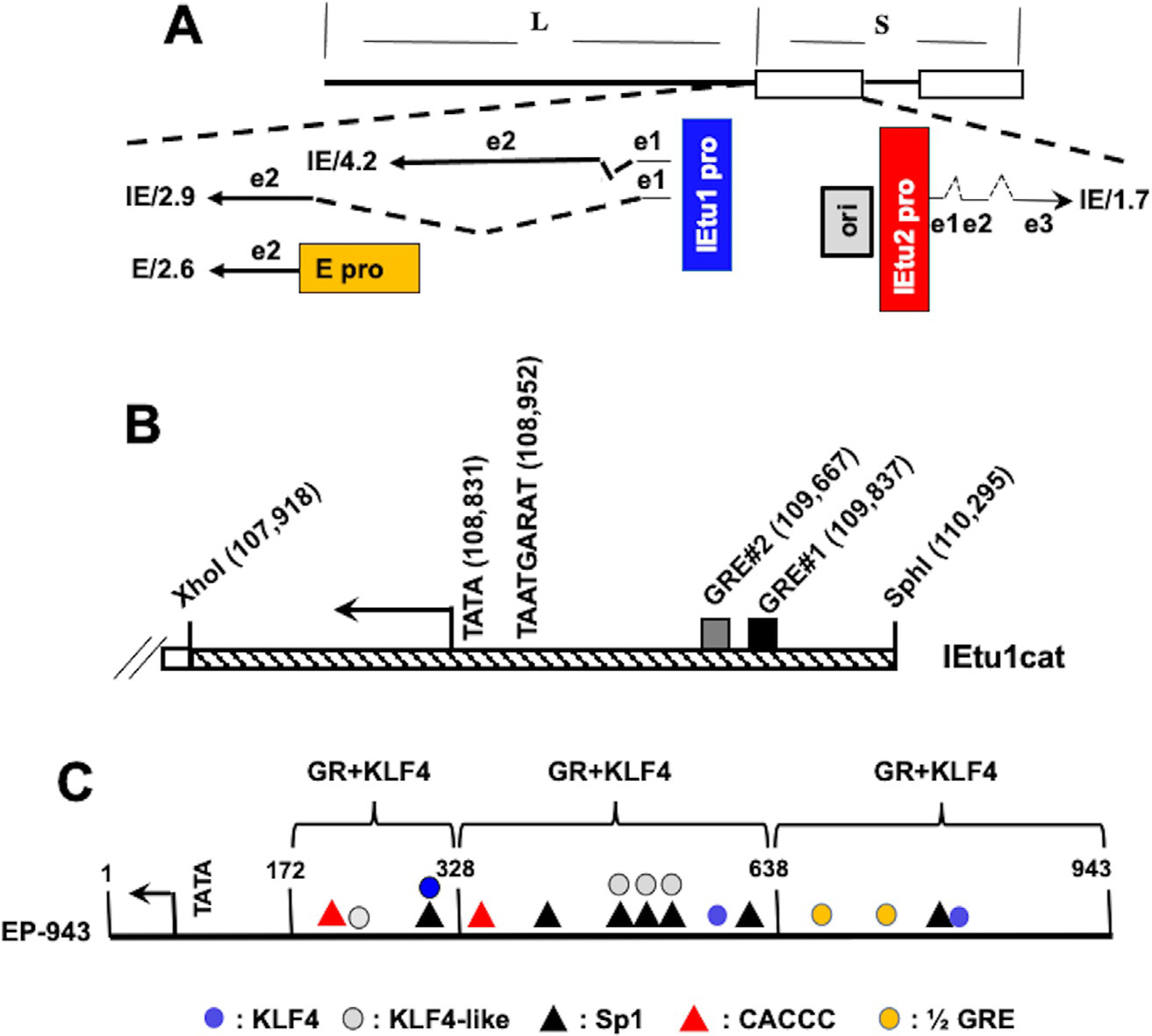
Schematic of BoHV-1 genome and key regulatory genes located in the repeats. Panel (A) Location of IE transcripts and IE promoters that drive expression of key viral regulatory proteins that stimulate productive infection (Koppel, Vogt, & Schwyzer, 1997; Wirth et al., 1989, 1991, 1992). IE/4.2 mRNA encodes the bICP4 protein and IE/2.9 mRNA encodes the bICP0 protein. One IE promoter activates expression of IE/4.2 and IE/2.9 and is designated IEtu1 (blue rectangle). E/2.6s the early mRNA that encodes bICP0 and an early promoter activates expression of this transcript (E pro; orange rectangle). Exon 2 (e2) of bICP0 contains all of the protein coding sequences. The origin of replication (ORI; gray box) separates IEtu1 from IEtu2. The IEtu2 promoter (IEtu2 pro; red box) regulates expression of the IE1.7 mRNA that is translated into the bICP22 protein. Solid lines in the transcript position map represent exons (e1, e2, or e3) and dashed lines denote introns. Panel (B) Schematic of IEtu1 promoter. Start site of transcription (arrow), TATA box, binding site for VP16/Oct1 denoted TAATGARAT (Misra et al., 1994), and location of GRE#1 and GRE#2 (black and gray rectangles respectively) are shown. Numbers are genomic coordinates of the first nucleotide of each respective motif or restriction enzyme site. Panel (C) Schematic of bICP0 E promoter. Position and definition of consensus transcription factor binding sites in bICP0 E promoter are shown; description of these binding sites is below schematic. Location of TATA box and three major start sites of transcription (denoted by arrow) are 22–24 nucleotides downstream of the TATA box (Wirth et al., 1992). The number 1 denotes the last nucleotide prior to the initiating methionine of the bICP0 protein. Brackets denote enhancer domains of bICP0 E promoter transactivated by GR and KLF4 (El-mayet et al., 2020, 2020).
The IEtu1 promoter is an unusual viral promoter because the TATA box and single TAATGARAT motif are separated more than 700 bases from GRE#2 and GRE#1 (Fig. 4B) (Kook, Doster, & Jones, 2015; Kook, Henley, et al., 2015a, 2015b). The IEtu1 promoter drives immediate early expression of bICP0 and bICP4 and is stimulated by GR and DEX unless the two consensus GREs are mutated (El-Mayet et al., 2017; Kook, Doster, & Jones, 2015; Kook, Henley, et al., 2015a, 2015b). GR, DEX, and KLF15 cooperatively stimulate productive infection, and transactivate IEtu1 promoter activity in a ligand dependent manner (El-Mayet et al., 2017). Since GR and KLF15 form a feed-forward loop, it was not surprising to find that GR and KLF15 formed a stable complex in transfected cells (El-Mayet et al., 2017). Host cellular factor 1 (HCF-1), which binds VP16 and Oct1 and then interacts with the immediate early specific enhancer core via the TAATGARAT motif (Kristie, 2007; Vogel & Kristie, 2000) cooperates with GR and DEX to stimulate IEtu1 promoter activity (Sawant, Kook, Vogel, Kristie, & Jones, 2018). Collectively, these findings suggest reactivation from latency is facilitated by GR, KLF15, and perhaps HCF-1 in certain neurons.
Previous studies provided evidence that bICP0 RNA is detected more frequently than bICP4 RNA in TG of calves during DEX induced reactivation from latency (Workman et al., 2009a, 2009b). Furthermore, the bICP0 protein is detected prior to bICP4 in TG neurons during DEX induced reactivation (Guo et al., 2019) suggesting the bICP0 E promoter is activated by stress induced cellular transcription factors. Unlike the IEtu1 promoter, the bICP0 E promoter contains no obvious GREs: however, there are two ½ GREs (Fig. 4C). Strikingly, GR and KLF4 cooperatively transactivate the bICP0 E promoter and stimulate productive infection (El-mayet et al., 2020). GR+KLF15 also transactivates the bICP0 E promoter; but not as efficiently as GR+KLF4. Surprisingly, DEX is not required for GR+KLF4 or GR+KLF15 mediated transactivation of the bICP0 E promoter suggesting transactivation occurred via an unliganded mechanism. ChIP studies revealed KLF4, KLF15, and GR occupied bICP0 E promoter sequences: however, only GR and KLF15 occupied ICP0 E promoter sequences late during productive infection (El-mayet et al., 2020). The bICP0 E promoter has 7 KLF4 as well as KLF4-like binding sites (Soufi et al., 2015), 6 Sp1 binding sites, and many GC and CA-rich motifs in the bICP0 E promoter that may be important for KLF4 and/or KLF15 mediated transactivation (Bieker, 2001; Ghaleb & Yang, 2017) (Fig. 4C). Studies designed to identify sequences in the bICP0 E promoter that impair GR and KLF4 mediated transactivation are in pogress.
Based on our results derived from the IEtu1 and bICP0 E promoters, it seems clear stress-induced transcription occurs by distinct mechanisms. For example, the IEtu1 promoter contains two well-defined GREs that are crucial for GR and KLF15 mediated transactivation via a liganded mechanism. In stark contrast, the bICP0 E promoter does not contain consensus GREs and the two putative 1/2 GREs are not important for GR and KLF4 mediated transactivation (El-mayet et al., 2020). Furthermore GR+KLF4, not KLF15, strongly stimulates promoter activity and GR+KLF4 mediated transactivation occurs by an un-liganded mechanism. Based on these published studies, we propose a model that fits the current results (see schematic in Fig. 5). It is well established that GR contains a well-defined DNA binding domain, steroid binding domain, and at least two transactivation domains (Giguere et al., 1986; Oakley & Cidlowski, 2013). We suggest GR interacts with numerous transcriptional coactivators and specific transcription factors, including certain KLF family members (Knoedler & Denver, 2014) (Fig. 5A) to transactivate the IEtu1 promoter. We further suggest HCF-1 directly or indirectly interacts with an unknown transcriptional coactivator (denoted as X) that promotes interactions with the GR/KLF15 complex.
Fig. 5.
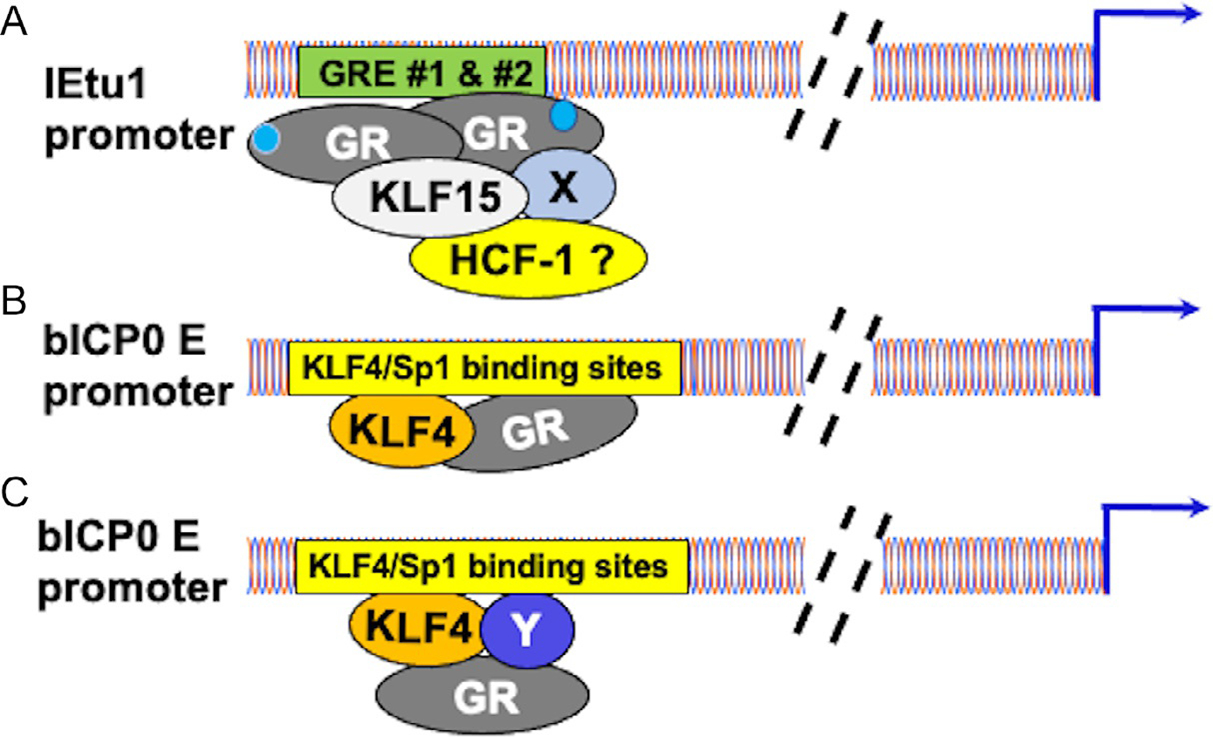
Model illustrating differences between GR mediated transactivation of IEtu1 versus GR+KLF4 mediated transactivation of bICP0 E promoter. Blue arrows denote start sites of transcription. Dashed lines denote sequences between GREs (Panel A) or a KLF4/Sp1 binding sites in the bICP0 promoter (Panels B and C) relative to the start site of transcription. With respect to Panel (A) X denotes an unknown transcriptional cofactor or cofactors associated with the GR transcription complex that transactivates IEtu1 promoter activity. HCF-1? may interact with this transcription complex: this prediction is supported by a previous study (Sawant et al., 2018). Light blue circles associated with GR denotes a GR/hormone complex, which indicates transactivation is mediated by a ligand dependent mechanism (El-Mayet et al., 2017; Kook, Doster, & Jones, 2015; Kook, Henley, et al., 2015a, 2015b). Panel (B) A KLF4 complex associates with certain KLF4/Sp1 binding sites in the bICP0 E promoter independent of a GRE or ½ GRE. This interaction occurs in an unliganded mechanism because DEX significantly reduces transactivation by GR and KLF4 (El-mayet et al., 2020). Panel (C) Y denotes an unknown transcriptional coactivator that associates with GR and KLF4 transcription complex. If this model is correct, GR does not directly interact with DNA.
With respect to the bICP0 E promoter, we suggest two scenarios by which GR+KLF4 transactivates the bICP0 E promoter: The first model predicts KLF4, in part due to interactions with GR binds KLF4/Sp1 binding sites. If this prediction is correct, GR is expected to interact with this site or adjacent sequences. The second model suggests KLF4 and other transcriptional coactivators (denoted by Y) interact with certain KLF4/Sp1 binding sites in the bICP0 E promoter and GR is tethered to this complex but does not directly interact with bICP0 E promoter sequences (Fig. 5C). Studies designed to resolve these scenarios are in progress.
5. Summary and conclusions
While we demonstrated that the synthetic corticosteroid DEX initiates BoHV-1 reactivation in calves and rabbits ( Jones, 2014) and enhances HSV-1 reactivation using TG explants prepared from latently infected mice (Harrison et al., 2019), certain external reactivation stimuli may not increase corticosteroid levels. For example, fever and UV light increase the incidence of reactivation from latency in humans but these stimuli are not typically associated with increased corticosteroids (Cassidy et al., 1997; Glaser et al., 1985; Jones, 1998, 2003; Padgett et al., 1998; Perng & Jones, 2010; Rooney et al., 1992). However, UV light can induce ligand independent GR phosphorylation and transcriptional activation (Davies et al., 2018; Galliher-Beckley et al., 2011). Support for these findings comes from a report demonstrating UV light induces expression of certain enzymes regulated by GR activation (Skobowiat, Sayre, Dowdy, & Slominski, 2013). Interestingly, UV light and stress activate a serine/threonine protein kinase, JNK (c-Jun N-terminal kinase) (Davies et al., 2018), which is crucial for remodeling HSV-1 chromatin in an in vitro neuronal model of reactivation from latency (Cliffe et al., 2015). Finally, inhibitors of cortisol production (Noisakran, Halford, Veress, & Carr, 1998) significantly reduce heat-shock induced HSV-1 reactivation in an in vivo mouse model of latency (Sawtell & Thompson, 1992). Hence, unrelated reactivation stimuli may share common factors, including GR, that mediate early stages of reactivation from latency.
Mammals, including humans and cattle, experience low levels of stress every day; however, reactivation from latency does not occur every day. Hence, we predict viral and/or cellular factors prevent low levels of stress from stimulating viral gene expression and reactivation from latency. Support for this prediction comes from a recent study demonstrating Akt1 and Akt2 (both serin/threonine protein kinases) reduce GR mediated transactivation of the IEtu1 promoter when cultures are treated with DEX (Zhao, Zhu, Wijesekera, & Jones, 2020). Furthermore, GR and KLF15-dependent transactivation of the HSV-1 ICP0 promoter is inhibited by Akt1 when cultures are treated with DEX. While Akt3 had no effect on either promoter, Akt3, but not Akt1 or Akt2, enhances neurite formation of Neuro-2A cells, which is a hallmark of neuronal differentiation.
The three Akt family members are predicted to regulate certain aspects of the BoHV-1 and HSV-1 latency-reactivation cycle. For instance, Akt3 protein levels are significantly higher in TG neurons of calves latently infected with BoHV-1 relative to TG from uninfected calves. Within 30min after DEX is administered to initiate reactivation, Akt3 RNA expression is reduced more than 50-fold and the number of Akt3 positive TG neurons are significantly reduced (Workman et al., 2018).
Several studies also provided evidence that Akt signaling is important for the HSV-1 latency-reactivation cycle. For example, inhibiting the phosphatidylinositol 3-kinase (PI3K)/Akt signaling axis induces HSV-1 reactivation from latency in primary rat sympathetic neurons (Camarena et al., 2010; Carpenter et al., 2015; Kim et al., 2012; Li et al., 2010) and Lund Human Mesencephalic (LUHMES) neuronal cells (Edwards & Bloom, 2019). Secondly, interfering with Akt kinase functions blocks the ability of HSV-1 LAT to interfere with apoptosis and trigger neurite formation (Carpenter et al., 2015; Li et al., 2010). Mice latently infected with wild-type HSV-1 contain significantly more TG neurons that express β-catenin when compared to a LAT null mutant (Harrison et al., 2020). Akt family members and the Wnt signaling pathway form a positive regulatory loop (Fukumoto et al., 2001; Kim, Lee, & Choi, 2007; Langhammer et al., 2013; Tenbaum et al., 2007; Zhang et al., 2013; Zhao et al., 2012) indicating these signaling pathways promote establishment and maintenance of a life-long latent infections. In conclusion, the latency-reactivation cycle of neurotropic herpesviruses must co-opt key cellular signaling pathways to maintain and reactivate from latency in their respective natural hosts.
Acknowledgments
This research was supported by grants to C. Jones from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number R01NS111167, two USDA NIFA competitive grants (2016-09370, 2018-06668, and 2021-67015-34463), and funds from the Sitlington Endowment. Dr. J. Ostler is supported by a postdoctoral fellowship from the USDA NIFA 2019-07214. The Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence Grant #P20GM103648) also supported part of these studies.
References
- Ahmed M, Lock M, Miller CG, & Fraser NW (2002). Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. Journal of Virology, 76(2), 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dujaili LJ, Clerkin PP, Clement C, McFerrin HE, Bhattacharjee PS, Varnell ED, et al. (2011). Ocular herpes simplex virus: How are latency, reactivation, recurrent disease, and therapy interrelated. Future Microbiology, 6, 877–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Hamrah P, Gate D, Mott KR, Mantopoulos D, Zheng L, et al. (2011). The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. Journal of Virology, 85(9), 4184–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Giordani NV, Kubat NJ, O’Neil JE, & Bloom DC (2006). Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. Journal of Virology, 80, 2063–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balliet JW, & Schaffer PA (2006). Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. Journal of Virology, 80, 440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (1998). Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clinical Science, 94, 557–572. [DOI] [PubMed] [Google Scholar]
- Bieker JJ (2001). Kruppel-like factors: Three fingers in many pies. The Journal of Biological Chemistry, 276, 34355–34358. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD, & Azizkhan-Clifford J (2001). Sp1 and Kruppel-like transcription factor family of transcription factors in cell growth and cancer. Journal of Cellular Physiology, 188, 143–160. [DOI] [PubMed] [Google Scholar]
- Boutell C, & Everett RD (2013). Regulation of alphaherpesvirus infections by the ICP0 family of proteins. The Journal of General Virology, 94, 465–481. [DOI] [PubMed] [Google Scholar]
- Branco FJ, & Fraser NW (2005). Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. Journal of Virology, 79, 9019–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratanich AC, Hanson ND, & Jones C (1992). The latency-related gene of bovine herpesvirus 1 inhibits the activity of immediate-early transcription unit 1. Virology, 191(2), 988–991. [DOI] [PubMed] [Google Scholar]
- Cabrera C, Wohlenberg C, Openshaw H, Rey-Mendez M, Puga A, & Notkins AL (1980). Herpes simplex virus DNA sequences in the CNS of latently infected mice. Nature, 288, 228–290. [DOI] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JK, Roehm P, Perez R, Gardner J, et al. (2010). Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host & Microbe, 8, 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter D, Hsiang C, Jiang X, Osorio N, BenMohamed L, Jones C, et al. (2015). The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) protects cells against cold shock induced apoptosis by maintaining phosphorylation of protein kinase B (AKT). Journal of Neurovirology, 21, 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy L, Meadows J, Catalan J, & Barton S (1997). Are stress and coping style associated with frequent recurrence of genital hereps? Genitourinary Medicine, 73, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase C, Fulton RW, O’Toole D, Gillette B, Daly RF, Perry G, et al. (2017). Bovine herpesvirus 1 modified live vaccines for cattle reproduction: Balancing protection with undesired effects. Veterinary Microbiology, 206, 69–77. [DOI] [PubMed] [Google Scholar]
- Ciacci-Zanella J, Stone M, Henderson G, & Jones C (1999). The latency-related gene of bovine herpesvirus 1 inhibits programmed cell death. Journal of Virology, 73(12), 9734–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe A, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, et al. (2015). Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host & Microbe, 18, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davido DJ, & Leib DA (1996). Role of cis-acting sequences of the ICP0 promoter of herpes simplex virus type 1 in viral pathogenesis, latency, and reactivation. The Journal of General Virology, 77, 1853–1863. [DOI] [PubMed] [Google Scholar]
- Davies L, Karthikeyan N, Lynch JT, Sial E-A, Gkourtsa A, Demonacos C, et al. (2018). Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Molecular Endocrinology, 22, 1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Freeman ML, Kinchington PR, & Hendricks RL (2005). Immune control of HSV-1 latency. Viral Immunology, 18(3), 466–473. [DOI] [PubMed] [Google Scholar]
- DeLeon M, Covenas R, Chadi G, Narvaez JA, Fuxe K, & Cintra A (1994). Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal gnaglia. Brain Research, 14, 338–342. [DOI] [PubMed] [Google Scholar]
- DeLuca NA, McCarthy AM, & Schaffer PA (1985). Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. Journal of Virology, 56(2), 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, & Roizman B (2011). HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proceedings of the National Academy of Sciences of the United States of America, 108, 18820–18824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T, & Bloom DC (2019). Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. Journal of Virology, 93, e02210–e02218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mayet FS, Sawant L, Thungunutla P, & Jones C (2017). Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus1 transcription and productive infection. Journal of Virology, 91(91), e00904–e00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-mayet F, Sawant L, Thunuguntla P, Zhao J, & Jones C (2020). Two pioneer transcription factors, Krüppel-like transcription factor 4 and glucocorticoid receptor, cooperatively transactivate the bovine herpesvirus 1 ICP0 early promoter and stimulate productive infection. Journal of Virology, 94(4), e01670–01619. 10.1128/jvi.01670-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsson AC, Bladh L-C, Stierna P, Yucel-Lindberg T, Hammersten O, Modeer T, et al. (2002). Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kB levels. Journal of Endocrinology, 175, 165–176. 10.1677/joe.0.1750165. [DOI] [PubMed] [Google Scholar]
- Everett RD (2000). ICP0, a regulator of herpes simplex virus during lytic and latent infection. BioEssays, 22(8), 761–770. [DOI] [PubMed] [Google Scholar]
- Fraefel C, Zeng J, Choffat Y, Engels M, Schwyzer M, & Ackermann M (1994). Identification and zinc dependence of the bovine herpesvirus 1 transactivator protein BICP0. Journal of Virology, 68(5), 3154–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser N, Lawrence WC, Wroblewska Z, Gilden DH, & Koprowski H (1981). Herpes simplex virus type 1 DNA in human brain tissue. Proceedings of the National Academy of Sciences of the United States of America, 78, 6461–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzo da Silva L, Kook I, Doster A, & Jones C (2013). Bovine herpesvirus 1 regulatory proteins, bICP0 and VP16, are readily detected in trigeminal ganglionic neurons expressing the glucocorticoid receptor during the early stages of reactivation from latency. Journal of Virology, 87, 11214–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S, Hsieh C-M, Maemura K, Layne MD, Yet S-F, Lee K-H, et al. (2001). Akt participation in the Wnt signaling pathway through dishevelled. The Journal of Biological Chemistry, 276, 17479–17483. [DOI] [PubMed] [Google Scholar]
- Funder JW (1997). Glucocorticoids and mineralocorticoid receptors: Biology and clinical relevance. Annual Review of Medicine, 48, 231–240. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley A, Williams JG, & Cidlowski JA (2011). Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Molecular and Cellular Biology, 31, 4663–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser V, Zhang Y, & Jones C (2005). Analysis of a bovine herpesvirus 1 recombinant virus that does not express the bICP0 protein. The Journal of General Virology, 86(Pt. 7), 1987–1996. 10.1099/vir.0.80921-0. [DOI] [PubMed] [Google Scholar]
- Ghaleb A, & Yang VW (2017). Krüppel-like factor 4 (KLF4): What we currently know. Gene, 61, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V, Hollenberg SM, Rosenfeld MG, & Evans RM (1986). Functional domains of the human glucocorticoid receptor. Cell, 46, 645–652. [DOI] [PubMed] [Google Scholar]
- Glaser R, & Kiecolt-Glaser JK (2005). Stress-induced immune dysfunction: Implications for health. Nature Reviews Immunology, 5, 243–251. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK, Speicher CE, & Holliday JE (1985). Stress, loneliness, and changes in herpesvirus latency. Journal of Behavioral Medicine, 8, 249–260. [DOI] [PubMed] [Google Scholar]
- Goodbourn S, Zinn K, & Maniatis T (1985). Human beta-interferon gene expression is regulated by an inducible enhancer element. Cell, 41, 509–520. [DOI] [PubMed] [Google Scholar]
- Guo J, Li Q, & Jones C (2019). The bovine herpesvirus 1 regulatory proteins, bICP4 and bICP22, are expressed during the escape from latency. Journal of Neuovirology, 25, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Kemp CD, Isler JA, Davido DJ, & Schaffer PA (2001). ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. Journal of Virology, 75(13), 6143–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwicke MA, & Schaffer PA (1997). Differential effects of nerve growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. Journal of Virology, 71(5), 3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison K, Zhu L, Thunuguntla P, & Jones C (2019). Antagonizing the glucocorticoid receptor impairs explant-induced reactivation in mice latently infected with herpes simplex virus 1. Journal of Virology, 93(13), e00418–e00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison K, Zhu L, Thunuguntla P, & Jones C (2020). Herpes simplex virus 1 regulates beta-catenin expression in TG neurons during the latency-reactivation cycle. PLoS One, 15, e0230870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemavathy K, Ashraf SI, & Ip IT (2000). Snail/slug family of repressors: Slowly going into the fast lane of development and cancer. Gene, 257, 1–12. [DOI] [PubMed] [Google Scholar]
- Hemavathy K, Guru SC, Harris J, Chen JD, & Ip YT (2000). Human slug is a repressor that localizes to sites of active transcription. Molecular and Cellular Biology, 26, 5087–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G, Perng G-C, Nesburn A, Wechsler S, & Jones C (2004). The latency related gene of bovine herpesvirus 1 can suppress caspase 3 and caspase 9 during productive infection. Journal of Neurovirology, 10, 64–70. [DOI] [PubMed] [Google Scholar]
- Hossain A, Schang LM, & Jones C (1995). Identification of gene products encoded by the latency-related gene of bovine herpesvirus 1. Journal of Virology, 69(9), 5345–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Lovato L, Doster A, & Jones C (2001). A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. Journal of Virology, 75, 8507–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Lovato L, Doster A, & Jones C (2002). A mutation in the latency related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. Journal of Virology, 76, 6771–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Perng G-C, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, et al. (2001). Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. Journal of Virology, 75(8), 3636–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Zhang Y, Geiser V, & Jones C (2001). The zinc ring finger in the bICP0 protein encoded by bovine herpes virus-1 mediates toxicity and activates productive infection. The Journal of General Virology, 82, 483–492. [DOI] [PubMed] [Google Scholar]
- Jaber T, Workman A, & Jones C (2010). Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). Journal of Virology, 84(13), 6297–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Chentoufi A, Hsiang C, Carpenter D, Osorio N, BenMohamed L, et al. (2011). The herpes simplex virus type 1 latency associated transcript (LAT) can protect cells from Granzyme B induced apoptosis and CD8 T-cell killing. Journal of Virology, 85, 2325–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Inman M, Zhang Y, Posadas NA, & Jones C (2004). A mutation in the latency related gene of bovine herpesvirus 1 (BHV-1) inhibits protein expression of a protein from open reading frame 2 (ORF-2) and an adjacent reading frame during productive infection. Journal of Virology, 78, 3184–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, et al. (2008). Cellular FLIP can substitute for the herpes simplex virus type 1 LAT gene to support a wild type virus reactivation phenotype in mice. Journal of Neurovirology, 14, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Perng G-C, Nesburn AB, Jones C, & Wechsler SL (2005). The baculovirus inhibitor of apoptosis gene (cpIAP) can restore reactivation of latency to a herpes simplex virus type 1 that does not express the latency associated transcript (LAT). Journal of Virology, 79, 12286–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C (1998). Alphaherpesvirus latency: Its role in disease and survival of the virus in nature. Advances in Virus Research, 51, 81–133. [DOI] [PubMed] [Google Scholar]
- Jones C (2003). Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clinical Microbiology Reviews, 16(1), 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C (2009). Regulation of innate immune responses by bovine herpesvirus 1 and infected cell protein 0. Viruses, 1, 255–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C (2013). Bovine herpes virus 1 (BHV-1) and herpes simplex virus type 1 (HSV-1) promote survival of latently infected sensory neurons, in part by inhibiting apoptosis. Journal of Cell Death, 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C (2014). Reactivation from latency by alpha-herpesvirinae submfamily members: A stressful situation. Current Topics in Virology, 12, 99–118. [Google Scholar]
- Jones C (2016). Latency of bovine herpesvirus 1 (BoHV-1) in sensory neurons. In Omgradi J (Ed.), Vol. 1. Herpesviridae (1st ed., p. 24). 10.5772/63750. [DOI] [Google Scholar]
- Jones C, & Chowdhury S (2007). A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex, and development of improved vaccines. Animal Health Research Reviews, 8, 187–205. [DOI] [PubMed] [Google Scholar]
- Jones C, Geiser V, Henderson G, Jiang Y, Meyer F, Perez S, et al. (2006). Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Veterinary Microbiology, 113, 199–210. [DOI] [PubMed] [Google Scholar]
- Jones C, Newby TJ, Holt T, Doster A, Stone M, Ciacci-Zanella J, et al. (2000). Analysis of latency in cattle after inoculation with a temperature sensitive mutant of bovine herpesvirus 1 (RLB106). Vaccine, 18(27), 3185–3195. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, & Urrutia R (2003). Sp1- and Kruppel-like transcription factors. Genome Biology, 4, 206.201–206.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-E, Lee W-J, & Choi K-Y (2007). The PI3 kinase-Akt pathway mediates Wnt3a-induced proliferation. Cellular Signaling, 19, 511–518. [DOI] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, & Wilson AC (2012). Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of HSV-1 in neurons. PLoS Pathogens, 8, e1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, & Hendricks RL (2008). Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science, 322, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoedler JR, & Denver RJ (2014). Kruppel-like factors are effectors of nuclear receptor signaling. General and Comparative Endocrinology, 203, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook I, Doster A, & Jones C (2015). Bovine herpesvirus 1 regulatory proteins are detected in trigeminal ganglionic neurons during the early stages of stress-induced escape from latency. Journal of Neurovirology, 21, 585–591. [DOI] [PubMed] [Google Scholar]
- Kook I, Henley C, Meyer F, Hoffmann FG, & Jones C (2015a). Bovine herpesvirus 1 productive infection and immediate early transcription unit 1 promoter are stimulated by the synthetic corticosteroid dexamethasone. Virology, 484, 377–385. 10.1016/j.virol.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Kook I, Henley C, Meyer F, Hoffmann F, & Jones C (2015b). Bovine herpesvirus 1 productive infection and the immediate early transcription unit 1 are stimulated by the synthetic corticosteroid dexamethasone. Virology, 484, 377–385. [DOI] [PubMed] [Google Scholar]
- Kook I, & Jones C (2016). The serum and glucocorticoid-regulated protein kinases (SGK) stimulate bovine herpesvirus 1 and herpes simplex virus 1 productive infection. Virus Research, 222, 106–112. [DOI] [PubMed] [Google Scholar]
- Koppel R, Vogt B, & Schwyzer M (1997). Immediate-early protein BICP22 of bovine herpesvirus 1 trans-represses viral promoters of different kinetic classes and is itself regulated by BICP0 at transcriptional and posttranscriptional levels. Archives of Virology, 142(12), 2447–2464. [DOI] [PubMed] [Google Scholar]
- Kristie TM (2007). Early pre-initiation of alphaherpesvirus viral gene expression. In Arvin GC-FA, Mocarski E, Moore PS, Whitley R, & Yamanishi K (Eds.), Vol. 1. Human herpesviruses: Biology, therapy, and immunoprophylaxis (pp. 112–127). Cambridge, NY: Cambridge University Press. [PubMed] [Google Scholar]
- Kristie TM, & Roizman B (1986). Alpha 4, the major regulatory protein of herpes simplex virus type 1, is stably and specifically associated with promoter-regulatory domains of alpha genes and of selected other viral genes. Proceedings of the National Academy of Sciences of the United States of America, 83, 3218–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir AS, Davido DJ, & Schaffer PA (2009). Role of nuclear factor Y in stress-induced activation of the herpes simplex virus type 1 ICP0 promoter. Journal of Virology, 84, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutish G, Mainprize T, & Rock D (1990). Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. Journal of Virology, 64(12), 5730–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhammer T-S, Roolf C, Krohn S, Kretzchmar C, Huebner R, Rolfs A, et al. (2013). PI3/K/Akt signalling interacts with Wnt/Beta-catenin signaling but does not induce an accumulation of beta-catenin in the nucleus of acute lymphoblastic leukemia cell lines. Blood, 122, 4886. [Google Scholar]
- Li S, Carpenter D, Hsiang C, Wechsler SL, & Jones C (2010). The herpes simplex virus type 1 latency-associated transcript (LAT) locus inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells following serum starvation by maintaining active AKT (protein kinase B). The Journal of General Virology, 91, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiacono CM, Myers R, & Mitchell WJ (2002). Neurons differentially activate the herpes simplex virus type 1 immediate-early gene ICP0 and ICP27 promoters in transgenic mice. Journal of Virology, 76(5), 2449–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, & Alon U (2003). Structure and function of the feed-forward loop network motif. Proceedings of the National Academy of Sciences of the United States of America, 100,11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matundan H, Mott KR, & Ghiasi H (2014). Role of CD8+ T cells and lymphoid dendritic cells in protection from ocular herpes simplex virus 1 challenge in immunized mice. Journal of Virology, 88, 8016–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, Perez S, Geiser V, Sintek M, Inman M, & Jones C (2007). A protein encoded by the bovine herpes virus 1 (BHV-1) latency related gene interacts with specific cellular regulatory proteins, including the CCAAT enhancer binding protein alpha (C/EBP-a). Journal of Virology, 81, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, Perez S, Jiang Y, Zhou Y, Henderson G, & Jones C (2007). Identification of a novel protein encoded by the latency-related gene of bovine herpesvirus 1. Journal of Neurovirology, 13, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra V, Bratanich AC, Carpenter D, & O’Hare P (1994). Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha gene trans-inducing factor. Journal of Virology, 68(8), 4898–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra V, Walker S, Hayes S, & O’Hare P (1995). The bovine herpesvirus alpha gene trans-inducing factor activates transcription by mechanisms different from those of its herpes simplex virus type 1 counterpart VP16. Journal of Virology, 69(9), 5209–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott KR, Allen SJ, Zandian M, & Ghiasi H (2014). Coregulatory interactions among CD8alpha dendritic cells, the latency-associated transcript, and programmed death 1 contribute to higher levels of herpes simplex virus 1 latency. Journal of Virology, 88, 6599–6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott K, Osorio N, Jin L, Brick D, Naito J, Cooper J, et al. (2003). The bovine herpesvirus 1 LR ORF2 is crucial for this gene’s ability to restore the high reactivation phenotype to a herpes simplex virus-1 LAT null mutant. The Journal of General Virology, 84, 2975–2985. [DOI] [PubMed] [Google Scholar]
- Noisakran S, Halford WP, Veress L, & Carr DJJ (1998). Role of the hypothalmic pituitary adrenal axis and IL-6 in stress-induced reactivation of latent herpes simplex virus type 1. Journal of Immunology, 160, 5441–5447. [PubMed] [Google Scholar]
- Oakley RH, & Cidlowski JA (2013). The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. The Journal of Allergy and Clinical Immunology, 132, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare P (1993). The virion transactivator of herpes simplex virus. Seminars in Virology, 4, 145–155. [Google Scholar]
- O’Hare P, & Goding CR (1988). Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell, 52(3), 435–445. [DOI] [PubMed] [Google Scholar]
- O’Hare P, & Hayward GS (1985). Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. Journal of Virology, 56(3), 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostler J, Harrison KS, Schroeder K, Thunuguntla P, & Jones C (2019). The glucocorticoid receptor (GR) stimulates Herpes Simplex Virus 1 productive infection, in part because the infected cell protein 0 (ICP0) promoter is cooperatively transactivated by the GR and Krüppel-like transcription factor 15. Journal of Virology, 93, e02063–02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostler JB, Thunguntla P, Hendrickson BY, & Jones C (2021). Transactivation of HSV-1 infected cell protein 4 (ICP4) enhancer by glucocorticoid receptor and stress-induced transcription factors requires overlapping Krüppel like transcription factor 4/Sp1 binding sites. Journal of Virology, 95, e01776–20. 10.1128/JVI.01776-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otteson DC, Lai H, Liu Y, & Zack DJ (2005). Zinc-finger domains of the transcriptional repressor KLF15 binds multiple sites in rhodopsin and IRBP promoters including the CRS-1 and G-rich elements. BMC Molecular Biology, 6, 1–16. 10.1186/1471-2199-1186-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett DA, Sherida JF, Dorne J, Berntson GG, Candelora J, & Glaser R (1998). Social stress and the reactivation of latent herpes simplex virus type 1. Proceedings of the National Academy of Sciences of the United States of America, 95, 7231–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, & Jones C (2008). Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1 (HSV-1) encoded latency-associated transcript (LAT). Journal of Neurovirology, 14, 41–52. [DOI] [PubMed] [Google Scholar]
- Perez S, Inman M, Doster A, & Jones C (2005). Latency-related gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. Journal of Clinical Microbiology, 43, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng G-C, & Jones C (2010). Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdisciplinary Perspectives on Infectious Diseases, 2010, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng G-C, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, et al. (2000). Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript (LAT). Science, 287, 1500–1503. [DOI] [PubMed] [Google Scholar]
- Perng G-C, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, et al. (2002). A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. Journal of Virology, 76, 1224–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesola JM, Zhu J, Knipe DM, & Coen DM (2005). Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. Journal of Virology, 79, 14516–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan D, Barrozo ER, & Bloom DC (2017). HSV1 latent transcription and non-coding RNA: A critical retrospective. Journal of Neuroimmunology, 308, 65–101. [DOI] [PubMed] [Google Scholar]
- Rhen T, & Cidlowski JA (2005). Antiinflammatory action of glucocorticoids—New mechanisms of old drugs. New England Journal of Medicine, 353, 1711–1723. [DOI] [PubMed] [Google Scholar]
- Rock DL, Beam SL, & Mayfield JE (1987). Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits. Journal of Virology, 61(12), 3827–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock D, & Fraser NW (1983). Detection of HSV-1 genome in central nervous system of latently infected mice. Nature, 302, 523–525. [DOI] [PubMed] [Google Scholar]
- Rock D, Lokensgard J, Lewis T, & Kutish G (1992). Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. Journal of Virology, 66(4), 2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JT, Straus SE, Mannix ML, Wohlenberg CR, Banks S, Jagannath S, et al. (1992). UV light-induced reactivation of herpes simplex virus type 2 and prevention by acyclovir. The Journal of Infectious Diseases, 166, 500–506. [DOI] [PubMed] [Google Scholar]
- Saira K, Chowdhury S, Gaudreault N, da Silva L, Henderson G, Doster A, et al. (2008). The zinc RING finger of bovine herpesvirus 1-encoded bICP0 protein is crucial for viral replication and virulence. Journal of Virology, 82(24), 12060–12068. 10.1128/jvi.01348-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse S, Mailoux CM, Barczak AJ, Wang Q, Altonsy MO, Jain MK, et al. (2013). The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Molecular and Cellular Biology, 33, 2104–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse S, Zuo Z, Kadiyala V, Zhang L, Pufall MA, Jain MK, et al. (2015). Response element composition governs correlations between binding site affinity and transcription in glucocorticoid receptor feed-forward loops. The Journal of Biological Chemistry, 290, 19756–19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant L, Kook I, Vogel JL, Kristie TM, & Jones C (2018). The cellular coactivator HCF-1 is required for glucocorticoid receptor-mediated transcription of bovine herpesvirus 1 immediate early genes. Journal of Virology, 92, e00987–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, & Thompson RL (1992). Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. Journal of Virology, 66(4), 2150–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell N, & Thompson RL (2016). De novo herpes simplex virus VP16 expression gates a dynamic programmatic transition and sets the latent/lytic balance during acute infection in trigeminal ganglia. PLoS Pathogens, 12(9), e1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller B, Chodankar R, Watson LC, Stallcup MR, & Yamamoto KR (2014). Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biology, 15, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonevild OJLM, Gaemers IC, & Lamers WH (2004). Mechanisms of glucocorticoid signalling. Biochemica et Biophysica Acta, 1680, 114–128. [DOI] [PubMed] [Google Scholar]
- Sekizawa T, & Openshaw H (1984). Encephalitis resulting from reactivation of latent herpes simplex virus in mice. Journal of Virology, 50, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffy BE, & Davies DH (1972). Reactivation of a bovine herpesvirus after corticosteroid treatment. Proceedings of the Society for Experimental Biology and Medicine, 140(3), 974–976. [DOI] [PubMed] [Google Scholar]
- Shen W, & Jones C (2008). Open reading frame 2 encoded by the latency related gene of bovine herpesvirus 1 has anti-apoptosis activity in transiently transfected neuroblastoma cells. Journal of Virology, 82, 10940–10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou J, Ali-Osman F, Multani AS, Pathak S, Fedi P, & Srivenugopal KS (2002). Human Dkk-1, a gene encoding a Wnt antagonist, responds to DNA damage and its overexpression sensitizes brain tumor cells to apoptosis following alkylation damage of DNA. Oncogene, 21, 878–889. [DOI] [PubMed] [Google Scholar]
- Sinani D, Cordes E, Workman A, Thunuguntia P, & Jones C (2013). Stress induced cellular transcription factors expressed in trigeminal ganglionic neurons stimulate the herpes simplex virus type 1 (HSV-1) infected cell protein 0 (ICP0) promoter. Journal of Virology, 87, 1183–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinani D, & Jones C (2011). Localization of sequences in a protein encoded by the latency related gene of bovine herpesvirus 1 (ORF2) that inhibits apoptosis and interferes with Notch1 mediated trans-activation of the bICP0 promoter. Journal of Virology, 85, 12124–12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobowiat C, Sayre RM, Dowdy JC, & Slominski AT (2013). Ultraviolet radiation regulates cortisol activity in a waveband dependent manner in human skin ex-vivo. The British Journal of Dermatology, 168, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Bates P, Rivera-Gonzalez R, Gu B, & DeLuca NA (1993). ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. Journal of Virology, 67(8), 4676–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak KL, & Cidlowski JA (2004). Mechanisms of glucocorticoid receptor signaling during inflammation. Mechanisms of Aging and Development, 125, 697–706. [DOI] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewcz A, Osman N, Pellegrini M, & Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell, 161, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi-Yanai K, Koike Y, Hasegawa T, Furuta Y, Serizawa M, Ohshima N, et al. (2010). Identification and characterization of glucocorticoid receptor-binding sited in the human genome. Journal of Receptors and Signal Transduction, 30, 88–105. [DOI] [PubMed] [Google Scholar]
- Taus NS, & Mitchell WJ (2001). The transgenic ICP4 promoter is activated in Schwann cells in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. Journal of Virology, 75(21), 10401–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, et al. (2007). B-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nature Medicine, 18, 892–902. [DOI] [PubMed] [Google Scholar]
- Thompson RL, Preston CM, & Sawtell NM (2009). De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathogens, 5(3), e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, & Sawtell NM (2001). Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. Journal of Virology, 75(14), 6660–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, & Sawtell NM (2006). Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. Journal of Virology, 80, 10919–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, & Sawtell NM (2011). The herpes simplex virus type 1 latency associated transcript is required for the maintenance of reactivation competent latent infection. Journal of Neurovirology, 17, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler K, Tedder DG, Yamamoto LJ, Klapper JA, Ashley R, Lichtenstein KA, et al. (1995). Recurrent brainstem encephalitis associated with herpes simplex virus type 1 DNA in cerebrospinal fluid. Neurology, 45, 2246–2250. [DOI] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, & Cullen BR (2008). MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature, 454, 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jural I, Karnowski HW, Coen DM, & Cullen BR (2009). Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. Journal of Virology, 83, 10677–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnell E, Kaufman H, Hill J, & Thompson H (1995). Cold stress-induced recurrences of herpetic keratitis in the squirrel monkeys. Investigative Ophthalmology & Visual Science, 36, 1181–1183. [PubMed] [Google Scholar]
- Vogel JL, & Kristie TM (2000). The novel coactivator C1 (HCF) coordinates multi-protein enhancer formation and mediates transcription activation by GABP. The EMBO Journal, 19, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, & Yamamoto KR (2004). Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proceedings of the National Academy of Sciences of the United States of America, 101, 15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, Goya L, Ge Y, Maiyar AC, & Firestone GL (1993). Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Molecular and Cellular Biology, 13, 2031–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wels C, Koefinger P, Joshi S, Bergler H, & Schaider H (2011). Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the EMT like phenotype in melanoma. The Journal of Investigative Dermatology, 131, 1877–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MT, Doster A, & Jones C (2000). Persistence and reactivation of bovine herpesvirus 1 in the tonsil of latently infected calves. Journal of Virology, 74, 5337–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MT, Doster A, Sur JH, & Jones C (2002). Analysis of bovine trigeminal ganglia following infection with bovine herpesvirus 1. Veterinary Microbiology, 86(1–2), 139–155. [DOI] [PubMed] [Google Scholar]
- Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, & Schwyzer M (1992). Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′ coterminal and encode a putative zinc finger transactivator protein. Journal of Virology, 66(5), 2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth UV, Gunkel K, Engels M, & Schwyzer M (1989). Spatial and temporal distribution of bovine herpesvirus 1 transcripts. Journal of Virology, 63(11), 4882–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth UV, Vogt B, & Schwyzer M (1991). The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. Journal of Virology, 65(1), 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, et al. (2012). Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. Journal of Virology, 86, 2459–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Perez S, Doster A, & Jones C (2009a). Dexamethasone treatment of calves latently infected with bovine herpesvirus 1 leads to activation of the bICP0 early promoter, in part by the cellular transcription factor C/EBP-alpha. Journal of Virology, 83(17), 8800–8809. 10.1128/jvi.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Perez S, Doster A, & Jones C (2009b). Dexamethasone treatment of calves latently infected with bovine herpesvirus 1 (BHV-1) leads to activation of the bICP0 early promoter, in part by the cellular transcription factor C/EBP-alpha. Journal of Virology, 83, 8800–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Sinani D, Pittayakhajonwut D, & Jones C (2011). A protein (ORF2) encoded by the latency related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. Journal of Virology, 85, 2536–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Zhu L, Keel BN, Smith TPL, & Jones C (2018). The Wnt signaling pathway is differentially expressed during the bovine herpesvirus 1 latency-reactivation cycle: Evidence that two proteinkinases associated with neuronal survival, Akt3 and BMPR2, are expressed at higher levels during latency. Journal of Virology, 92(7), e01937–01917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang Y, Zhou J, & Jones C (2006). The bovine herpes virus 1 (BHV-1) immediate early protein (bICP0) interacts with the histone acetyltransferase p300, and these interactions correlate with stimulation of gC promoter activity. The Journal of General Virology, 87, 1843–1851. [DOI] [PubMed] [Google Scholar]
- Zhang Y, & Jones C (2001). The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. Journal of Virology, 75(20), 9571–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, & Jones C (2005). Identification of functional domains within the bICP0 protein encoded by BHV-1. The Journal of General Virology, 86, 879–886. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shemezis JR, McQuinn ER, Wang J, Sverdlov M, & Chenn A (2013). AKT activation by N-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural Development, 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Fu J, Liu X, Wang T, Zhang J, & Zhao Y (2012). Activation of Akt/GSK-3 beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurological Research, 34, 400–407. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhu L, Wijesekera N, & Jones C (2020). Specific Akt family members impair stress mediated transactivation of viral promoters and enhance neuronal differentiation: Important functions for maintaining latency. Journal of Virology, 94, e00901–e00920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Workman A, & Jones C (2017). A potential role for a beta-catenin coactivator (high mobility group AT-hook 1 protein) during the latency-reactivation cycle of bovine herpesvirus 1. Journal of Virology, 91, e02132–e20136. [DOI] [PMC free article] [PubMed] [Google Scholar]