

Abstract
Caging RNA by polyacylation (cloaking) has been developed recently as a simple and rapid method to control the function of RNAs. Previous approaches for chemical reversal of acylation (uncloaking) made use of azide reduction followed by amine cyclization, requiring ~2–4 h for the completion of cyclization. In new studies aimed at improving reversal rates and yields, we have designed novel acylating reagents that utilize quinone methide (QM) elimination for reversal. The QM de-acylation reactions were tested with two bioorthogonally cleavable motifs, azide and vinyl ether, and their acylation and reversal efficiencies were assessed with NMR and mass spectrometry on model small-molecule substrates as well as on RNAs. Successful reversal both with phosphines and strained alkenes was documented. Among the compounds tested, the azido-QM compound A-3 displayed excellent de-acylation efficiency, with t1/2 for de-acylation of less than an hour using a phosphine trigger. To test its function in RNA caging, A-3 was successfully applied to control EGFP mRNA translation in vitro and in HeLa cells. We expect that this molecular caging strategy can serve as a valuable tool for biological investigation and control of RNAs both in vitro and in cells.
Graphical Abstract
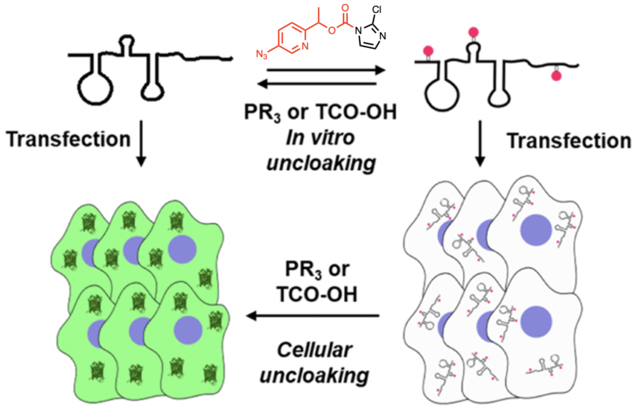
Reversible quinone methide acylating reagents are designed for the rapid control of RNAs (e.g. mRNA) in vitro and in cells.
Introduction
RNA takes part in a multitude of cellular functions, ranging from regulation and expression of proteins (involving mRNA, rRNA, tRNA, and miRNA among others), to gene regulation (e.g. riboswitches and piRNA), and RNA processing and modification (e.g. ribozymes and snoRNA).1–4 To help decipher the complexities of RNA functions in biological settings, caging has proved to be a useful strategy, engendering a pulse of activity that can be studied for its downstream effects. The most popular approach to caging of biomacromolecules has been photocaging due to its high spatiotemporal resolution.5,6 For short RNAs, photocaging groups can be appended to an RNA of interest (ROI) during de novo solid-phase synthesis.5,7–10 However, this technology is inapplicable for longer RNAs which are prepared via transcription, which includes most cellular RNA species including mRNAs and lncRNAs. Moreover, the purely synthetic approach of incorporating caging groups requires access to oligonucleotide solid-phase synthesizers and expertise in modified nucleoside synthesis, limiting widespread use even with shorter RNAs.
To circumvent these limitations, RNA can be photocaged site-specifically through enzymes11–13 or stochastically with diazo compounds.14–16 However, enzymatic approaches are incompatible with most native RNA and require the engineering of multiple substrate motifs into the RNA for inhibition, which can perturb the native function. One exception is a recent report demonstrating chemoenzymatic labeling of mRNA without additional tag sequences,17–22 but this approach requires the presence of a 5 or 9 nt consensus sequence within the RNA of interest, limiting where labeling can occur. An early chemical labeling approach, using diazo compounds stochastically reacting with phosphate groups, is simpler in application, but results in hydrolytically labile internal phosphotriester bonds that lead to RNA degradation.23 Finally, photocaging in general is limited by potential RNA side reactions and to cytotoxicity as a result of the UV irradiation that is often used for reversal.5,24 Chemical uncaging strategies offer the potential to overcome this, but remain underexplored in application to RNA.
As an alternative approach to addressing some of these limitations, RNA 2’-OH polyacylation (“cloaking”) has been reported as a convenient strategy to modify and control RNA (Fig. 1A).25–32 This method typically makes use of acylimidazole reagents that have sufficient solubility and lifetime to yield high levels of ester or carbonate substitutions on an RNA of interest. This method can cage RNA of a wide range of lengths and is compatible with native (unmodified) RNA; in addition, the reaction requires only a single step, and isolation of the caged RNA requires simple ethanol precipitation. Cloaking has been shown to modulate a range of RNA functions including hybridization, folding, catalysis, and binding to proteins.30 The acyl groups tested to date are removed (“uncloaked”) via phosphine-induced intramolecular cyclization or via UV light exposure, restoring the original unmodified RNA. Cellular applications of this technique have been limited by the possible cytotoxicity of the reversal conditions, preventing the use of high phosphine concentrations that would be ideal for rapid, high-yield uncaging, and necessitating optimization to balance reversal and cell damage. This obstacle underscores the need for faster and more biologically benign caging and uncaging chemistry for RNA. Rapid uncaging is desirable because it creates better time resolution in initiating a signal, and is more compatible with the often-short cellular lifetimes of RNAs.
Figure 1.
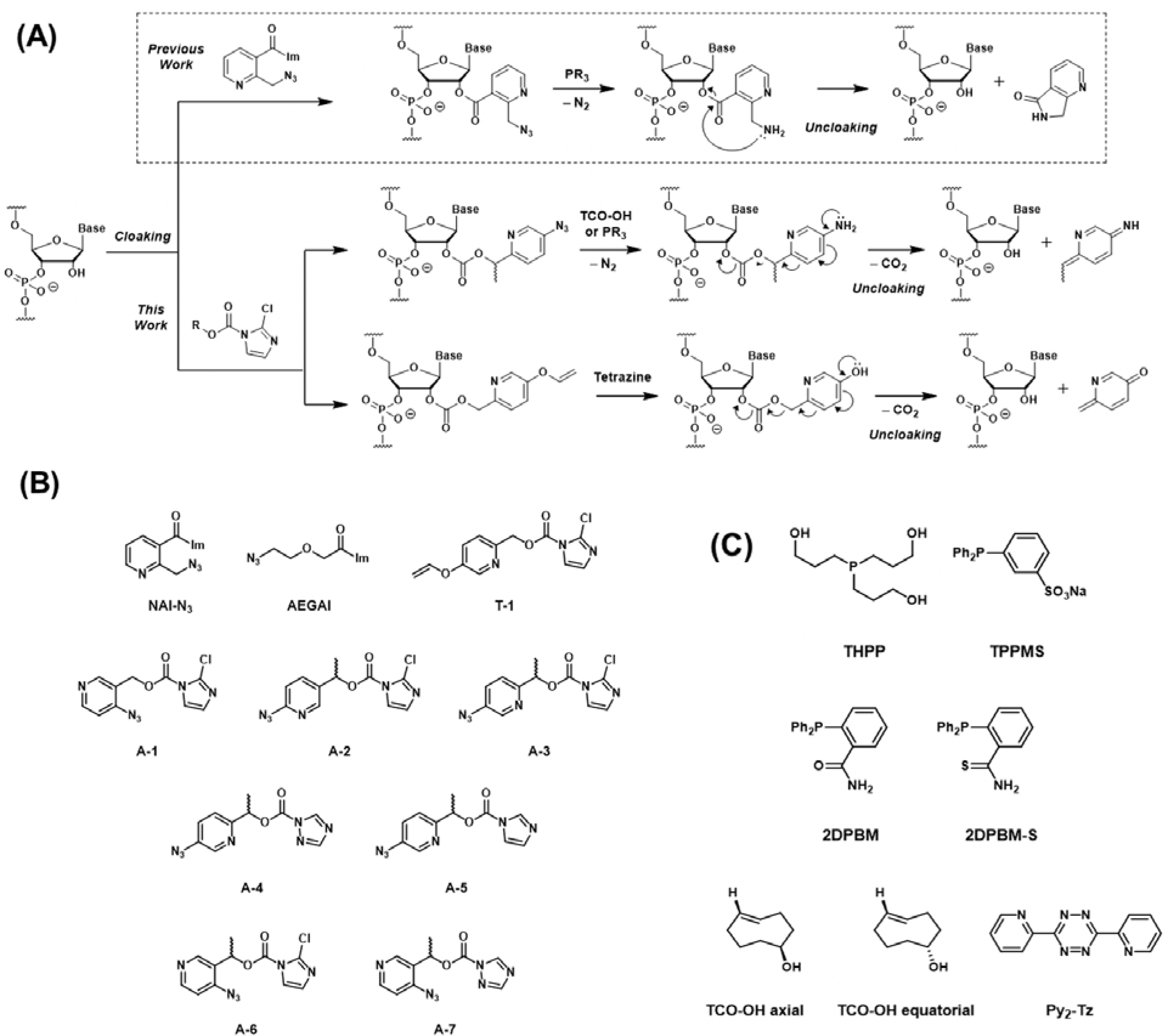
Novel RNA acylating reagent designs based on quinone methide elimination. (A) Scheme of polyacylation (cloaking) and subsequent reversal (uncloaking). PR3 indicates phosphines. (B) Acylating reagent structures studied. (C) Structures of chemical reversal triggers.
Prompted by these issues, we have undertaken a new study to explore new approaches for biorthogonal bond-cleavage chemistry for potential application to RNA uncaging. While bond-breaking chemistry has been developed for linkers cleavable by acids, nucleophiles, oxidants, reductants, metals, or UV, the reversal conditions have often limited this chemistry to in vitro applications.33–35 More recent studies have described cleavable motifs with fast reaction rates and low cytotoxicity, including more sophisticated reversal chemical triggers such as alkynes, tetrazines, trans-cyclooctene (TCO), and transition metals.36,37 Based on these advances, we sought to develop RNA cloaking agents with improved biochemical and biological compatibility. Because for the early phosphine-triggered amine cyclization chemistry the rate limiting step was the intramolecular cyclization and required in some cases hours for complete reversal,25,29 we hypothesized that quinone methide (QM) elimination might offer a more rapid reversal mechanism.
Our study explores the use of the QM structural core combined with chemically reversible motifs responsive to three chemical triggers: azide groups reduced by phosphine or TCO, and vinyl phenols dealkylated with tetrazine. Although phosphines can be cytotoxic at high concentrations, optimized aryl azide and phosphine pairs were demonstrated to have exceptionally high reactivity, offering the possibility of lower cytotoxicity.38 TCO and tetrazines have been used with success in cellular and animal experiments.36,39–41 For the new RNA acylating agents, we carried out NMR and mass spectrometry (MS) experiments to optimize QM structure and found QM structures that were reversible with milder conditions at lower phosphine concentrations and shorter reaction times compared to previous amine cyclization-based cloaking agents. Finally, we applied the optimal azido-QM compound A-3 to control EGFP mRNA translation in vitro and in cells. Due to the simplicity and ease of preparation and handling of the reagents, we expect them to serve as convenient tools to control a wide range of RNAs in biological settings.
Results
Design and synthesis
The new RNA acylating reagents were designed around pyridinone methide scaffolds (Fig. 1B) which appeared promising due to their synthetic accessibility and aqueous solubility. Notably, pyridinone methides have also been shown to provide enhanced elimination rates relative to their benzene analogs.42 In the current application, solubility was an important factor, as hydrophobic reagents tend to acylate RNA poorly and can also cause solubility issues for the polyfunctionalized RNA.28,29 For the electrophilic acylating portion of the molecules, imidazole carbamates were chosen due to recent observations of efficient RNA polyacylation as well as synthetic accessibility from alcohols.26–28,32 Specifically, leaving groups consisting of imidazole, 2-chloroimidazole, and two isomers of triazole were examined. The imidazole carbamates displayed a wide range of reactivity with hydrolysis half-lives spanning from 0.5 hours to days (Fig. S1) in water.
Two bioorthogonally-cleavable functional groups were explored: vinyl ether and azide. Aryl vinyl ether groups can be deprotected by tetrazine to phenols, in principle followed by QM elimination and deacylation (Fig. 1A).36 To synthesize vinyl QM compound T-1, methyl 5-hydroxypicolinate was first alkylated with 1,2-dibromoethane (see Supporting Information (SI) for details). Next, the ester group was reduced to alcohol with sodium borohydride, and the remaining bromine was eliminated to form the vinyl product. Finally, the imidazole carbamate group was installed by activating the alcohol with carbonyl-di-imidazole analogs.
Aryl azides can be reduced to amines by phosphines, or they can form unstable adducts with TCO that undergo rearrangement to form imines. In aqueous conditions, the imines can hydrolyze to form amines, which can be harnessed for QM elimination (Fig. 1A).36 To synthesize azido-QM compounds A-1 through A-7, the azido groups were first installed with sodium azide via SNAr reaction (see SI). This was followed by sodium borohydride reduction to form the alcohol, which was subsequently activated to form imidazole carbamates. Multiple isomers were examined to optimize deacylation. The effect of a methyl group at benzylic position to potentially stabilize the partial positive charge during the elimination step and accelerate reversal was also analyzed (Fig. 1A).
NMR experiments with small-molecule mimic of RNA 2’-OH
To compare the relative rates of deacylation of carbonates via the QM mechanism, small-molecule acyl carbonates SM-A-1, 2, 3, and 6 were synthesized and their deacylations observed by NMR (Fig. S2). This model was chosen as a leaving group based on its similar pKa to that of ribose 2’-OH, as well as to avoid spectral overlap that would occur with a ribonucleotide. Comparison was also made with ester adducts of established cloaking agents, azidoethyl glycolic acid imidazole (AEGAI)29and NAI-N325 (Fig. 1B and S2). The model compounds were treated with phosphines and TCO-OH and monitored over time.
A-1 carbonate compound SM-A-1 displayed rapid reduction with phosphine treatment (pseudo-first-order half-life < 8 min), but surprisingly the rate limiting step was the elimination, requiring 1.5 h for 50% release at room temperature (Fig. S3A). The amino intermediate was additionally confirmed with LC/MS analysis (Fig. S3B). Due to the long-lasting nature of the amino intermediate, a mixture of intramolecular cyclization and QM elimination products was observed. SM-A-1 showed slower reversal with TCO-OH treatment, requiring 3 h for 50% release at 37 °C (Fig. S3C). In the absence of uncloaking triggers, carbonate and ester linkages were stable and showed minimal hydrolysis, confirming their potential viability for biological application (Fig. S2).
Acyl products SM-A-2, −3, and −6 showed different responses. 6-Azidopyridine in SM-A-2 displayed poor reactivity to phosphine and no reaction with TCO-OH (Fig. S4). In contrast, the 5-azido isomer SM-A-3 deacylated rapidly with THPP, showing full reversal in under 20 min. Detailed analysis was difficult due to rapid evolution of nitrogen and carbon dioxide gas and peak broadening (Fig. 2A), but the rapid reversal can be attributed to the benzylic methylene group stabilizing the partial positive charge that is generated during the elimination step, which was previously rate limiting for SM-A-1. Deacylation was again slower with TCO-OH and resulted in full release by 6 h at 37 °C (Fig. 2B). The 4-azido isomer SM-A-6 showed the most rapid reversal with THPP, resulting in full release in 10 minutes (Fig 2C). Treatment with TCO-OH resulted in more sluggish reversal, reaching 66% by 8 h (Fig 2D). When SM-A-3 and 6 were treated with a bulkier and less reactive phosphine, TPPMS, there was a change in relative reactivity (Figure 2.10A, B). SM-A-3 fully reversed in 2.5 hours while SM-A-6 required 8 hours, likely due to the azido group in SM-A-6 being more sterically hindered. Reactions with TCO-OH (axial) showed similar reversal as TCO-OH (equatorial) (Figure 2.10C, D).
Figure 2.
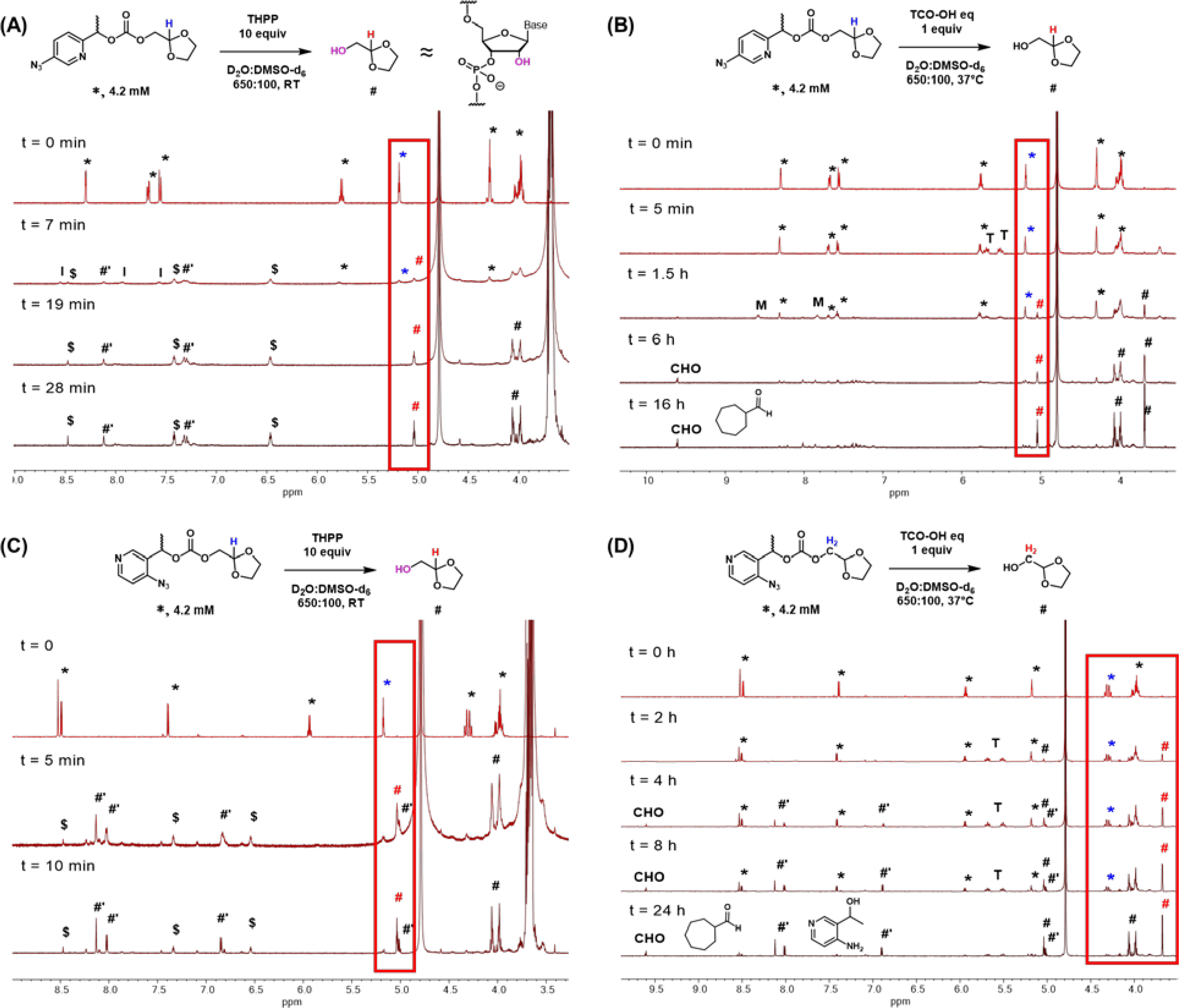
NMR analysis of deacylation of A-3 and 9 carbonate model small molecule adducts. (A) THPP induced deacylation of SM-A-3. The alcohol denoted # is similarly inductively electronic withdrawing as the 2’-OH on RNA and serves as a simpler approximation. (B) TCO-OHeq induced deacylation of SM-A-3. (C) THPP induced deacylation of SM-A-6. (B) TCO-OHeq induced deacylation of SM-A-6. Reversal is monitored with the color-coded methine or methylene peaks (blue: carbonate, red: alcohol) in the indicated red box. *: SM-A-6 or 9, #: released alcohol, #’: released pyridinone methide, $: impurities from THPP stock, I: unidentified short-lived intermediate, T: TCO-OH equatorial, M: cycloadduct or imine intermediate, CHO: cycloheptane carboxaldehyde.
Compared to SM-A-3 and 6, products of NAI-N3 and AEGAI showed relatively slow release rates with THPP treatment, respectively requiring approximately 0.5 and 1 h at room temperature for 50% reversal (Fig. S6). Surprisingly, even by 5 h, the two established compounds failed to fully deacylate despite the presence of excess phosphine. The improved release rate and yield with SM-A-3 and 6 suggested promise of the corresponding acylating agents for cellular application.
Short RNA (Cy5-tRF-3005) cloaking and uncloaking experiments
Experiments were next directed beyond the simple alcohol deacylation model and to RNA acylation. The RNA cloaking and uncloaking efficiency of the new reagents were measured with an 18 nt Cy5 labeled transfer RNA fragment, Cy5-tRF-3005, which is expected to have little or no folded structure.43 6 μM RNA was treated with 0.1 M of cloaking agent in a 3:7 water:DMSO, subsequently isolated by ethanol precipitation, and analyzed with MALDI-MS. The MS data showed that vinyl ether compound T-1 acylated the RNA moderately after 4 h, with up to +2 acyl groups (Fig. S7A). Because T-1 showed no observable deacylation even after overnight treatment with tetrazine, the vinyl ether compounds were determined to undergo reversal too slowly for practical use and were not pursued further, as was azido-QM compound A-2 (Fig. S8A and B). Azido-QM compounds A-1, 3, 6 and 7 displayed a higher degree of RNA acylation with an approximate average of 1 acyl group out of 18 2’-OH groups in the test RNA after 4 h (Fig. 3A and S7). A-4 with its 1,2,4-triazole carbamate, which was designed for improved aqueous solubility, acylated RNA poorly (Fig. S7C). The more hydrolytically stable A-5 also acylated poorly (Fig. S7D).
Figure 3.
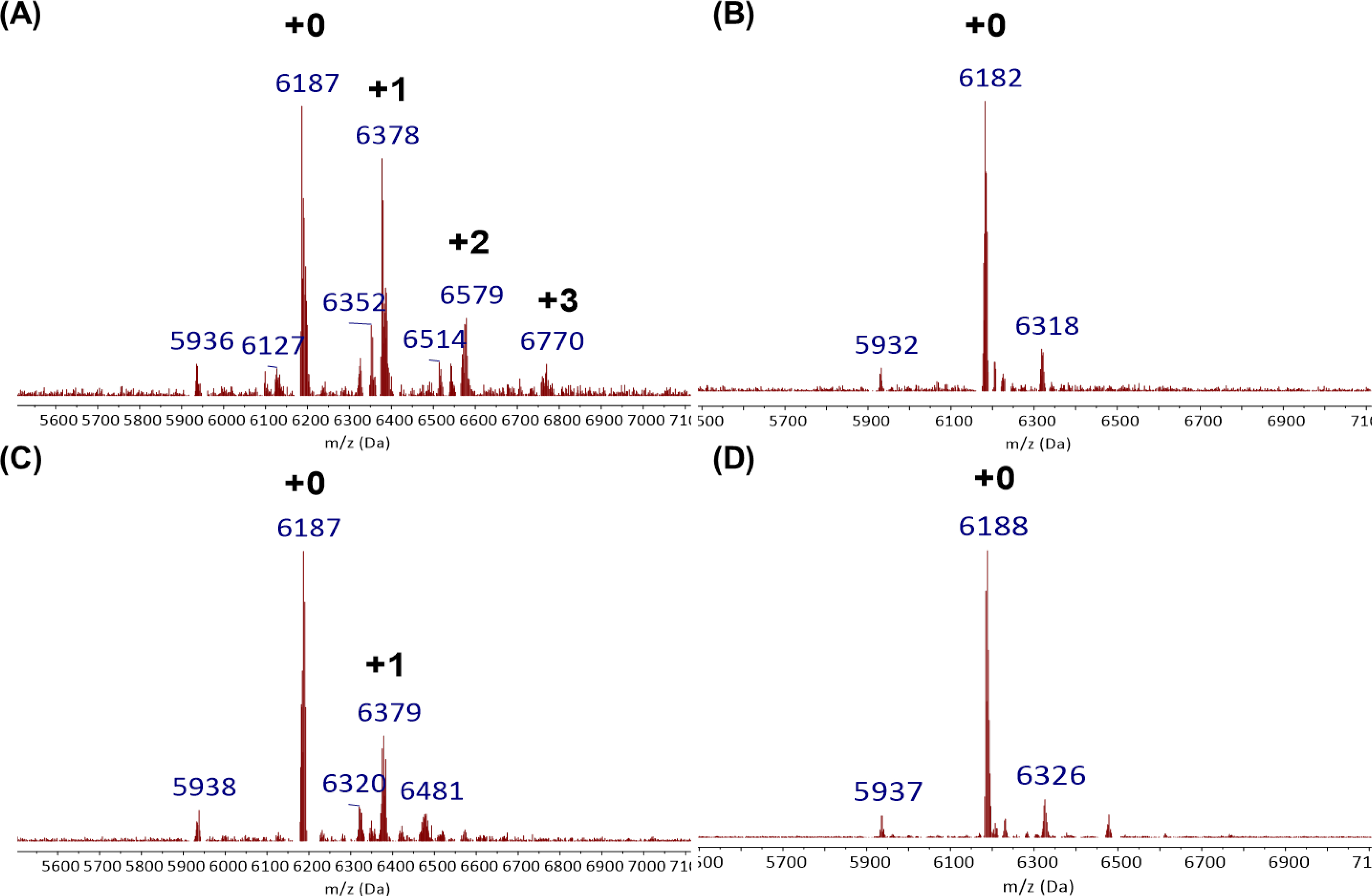
Polyacylation and deacylation efficiency of A-3 with model RNA oligonucleotide, Cy5-tRF-3005. (A) MALDI-TOF spectrum of A-3 polyacylated RNA. 6 μM RNA was treated with 0.1 M of cloaking agent in a 3:7 water:DMSO for 4 h at room temperature. (B) MALDI-TOF spectrum of polacylated RNA reversed with TPPMS. 1 μM RNA was treated with 0.5 mM TPPMS in 50 mM Tris-HCl buffer pH 7.5 (10% DMSO) for 1 h at 37 °C. (C) & (D) MALDI-TOF spectra of polyacylated RNA reversed with TCO-OHeq. 1 μM RNA was treated with 5 mM TCO-OHeq in 50 mM Tris-HCl buffer pH 7.5 (10% DMSO) for 1 h (C) or 4 h (D) at 37 °C. Black numbers indicate the number of adducts.
Having established that the novel azido-QM reagents could successfully polyacylate a model RNA, we next studied deacylation. Consistent with NMR results of the model adduct, A-1 treated RNA was uncaged successfully with the addition of THPP, although some acyl groups persisted after 4 h (Fig. S8B). In contrast, RNA polyacylated with A-3 deacylated rapidly in one hour with 0.5 mM TPPMS (Fig. 3B). This uncloaking condition proceeded at a lower concentration and shorter time than previously used with NAI-N3 and AEGAI, which required 5–20 mM phosphine and 2–4 hours.25,29,30 TCO-OHeq treatment of the same polyacylated RNA showed promising reversal but was less efficient than phosphine and required a relatively high concentration of 5 mM and 4 h for completion (Fig. 3C, D). Interestingly, A-7 acylated RNA failed to reverse with both phosphine or TCO-OH, which we attribute to the azido group being sterically blocked by the RNA near the 2’ acyl group (Fig. S8D, E, F). This contrasts with the azido group in A-3, which lies remote from the RNA 2’ acyl group and should be more accessible. As a control, when cloaked Cy5-tRF-3005 RNA was incubated without the releasing triggers, no deacylation was observed (Fig. S8C). This additionally confirmed the acyl carbonate stability and demonstrated chemical temporal control. Taken as a whole, the data showed that A-3 displayed excellent reversibility on an RNA oligonucleotide and yielded improvements in uncaging speed relative to previous acylating agents. Although the released quinone methide has the potential to react with the nucleobases of RNA due to its electrophilicity, we did not observe a +120 Da adduct, which would correspond to an aza-pyridone methide adduct, in the mass spectrum of uncloaked RNA. We expect that the extremely low concentrations of quinone methide (micromolar for the oligonucleotide) at which it is released reduces the likelihood of bimolecular collisions with RNA.
Interestingly, mechanistic model studies suggest that acylation of RNA by A-3 may occur not only at 2’-OH groups but also possibly at exocyclic amines on nucleobases. The reagent shows significant reaction with DNA as judged by gel electrophoresis and mass spectrometry (Fig. S14), suggesting possible amine reactivity on C/A/G (unlike previous reactions with nicotinyl acylimidazoles, which appear to yield exclusively 2’-O-acylation 25). However, comparisons of reactivity between ribonucleotides 3’-UMP and 3’-AMP confirm similar levels of reaction for the two (despite lack of an amine on 3’-UMP), suggesting preferential reaction at 2’-OH groups. Aqueous reactions with nucleobases alone (1-methylU and 9-methyladenine) displayed no measurable reaction (Fig. S15-23, Table S1). Since other imidazolecarbamates seem to prefer primary reaction at 2’-OH groups,27 we hypothesize that the unusually high level of DMSO in the acylation reactions performed here (70% by volume; necessary due to limited aqueous solubility of A-3) may account for this difference. Regardless, reaction at either 2’-OH or exocyclic amines is expected to block pairing, and all group positions on the model RNA are readily reversed by de-acylation conditions.
Control of EGFP mRNA translation in vitro and in cells
Taken together, the data with model small molecules and a short synthetic RNA established that adducts of QM compound A-3 were rapidly and efficiently deacylated, and that the 2-chloroimidazole carbamate derivative can react with RNA in aqueous buffers, leading to multiple acylation adducts. To determine whether these levels of reactivity are sufficient for practical applications with larger biologically active RNAs, we employed mRNA encoding the fluorescent reporter EGFP in tests of modulating translation in vitro and in cells. Note that no previous acylation caging strategy has been reported for mRNA; thus, it was unknown whether acylation would be effective in downregulating translation, and whether uncaging would be efficient and mild enough to restore function to a significant degree. Polyacylation of mRNA with A-3 could have the potential to sterically block one or more of multiple translation factors such as initiation factors, tRNA, or the assembled ribosome, or other factors (such as splicing and posttranscriptional base modification factors) that control RNA biology. Such an ability could have utility in studying and controlling mRNAs for in vitro studies of translation and splicing mechanisms, and if effective in cell culture, might enable a general approach to temporally upregulating gene expression.
Acylation conditions for A-3 and EGFP mRNA were optimized both in water and salt-supplemented MOPS buffer to assess the effects of conditions that might affect mRNA structure on caging and subsequent recovery (Fig. S9). Because RNA acylating agents tend to prefer reaction at unpaired nucleotides,44 the sites and degree of acylation were expected to be different for denatured mRNA in water or folded mRNA in buffer. To maximize reversal efficiency, it was important to identify the minimum amount of acylation that results in strong inhibition of translation. After acylation, mRNA was translated in vitro or in HeLa cells. For cellular experiments, RNA was transfected into HeLa cells via lipofection, and the fluorescence from the translated EGFP was measured. Results showed that cloaking in either buffer or in water suppressed translation; interestingly, acylation in water more potently inhibited EGFP expression compared to that in buffer. Treatment with 10 mM A-3 in water yielded essentially completely blocked translation relative to the DMSO mock-treated mRNA, while 50 mM A-3 in buffer caused approximately 80% inhibition of GFP signal (Fig. S9A and S10A). No significant mRNA degradation from A-3 treatment was detected from gel electrophoretic analysis and thus is not likely a significant contributor to the suppressed signal (Fig. S9B). Thus, the early data suggested that translation could in fact be suppressed strongly by treatment with acylating agent A-3.
A more important test of biomolecular caging effectiveness is recovery. To optimize uncaging, four phosphines, THPP, TPPMS, 2DPBM, and 2DPBM-S, and strained alkenes TCO-OHax/eq were tested with cloaked EGFP mRNA in vitro and in HeLa cells. THPP and TPPMS were previously utilized for CRISPR sgRNA uncloaking with relatively low cytotoxicity,30 while 2DPBM and 2DPBM-S were reported to display fast reduction rates due to neighboring group participation.38 Initial in vitro tests with an earlier nicotinate caging reagent, NAI-N3, revealed cloaked mRNA showed TPPMS to be the most efficient phosphine followed by THPP (Fig. S9E). The relatively poor uncloaking with THPP could potentially be attributed to its sensitivity to oxidation in solutions exposed to air. 2DPBM and 2DPBM-S suffered from low solubility, which may contribute to their poor uncloaking. Based on these findings, we continued our experiments with TPPMS and THPP.
Interestingly, EGFP mRNA cloaked in pure water with A-3 (i.e. in the absence of salts that support folding) failed to recover activity both in vitro and in cellular uncloaking (Fig. S9F, G). On the other hand, EGFP mRNA cloaked in the MOPS buffer upon in vitro uncloaking treatment substantially recovered fluorescence upon transfection into HeLa cells for both phosphines and TCO-OH (Fig. S10A). When cells were transfected with the cloaked mRNA and subsequently uncloaked, phosphine treatment with 2 mM THPP for 4 hours displayed up to 3-fold enhancement of EGFP expression and demonstrated temporal control of mRNA activity (Fig. 4D and S10D). This was a considerable improvement over the previous sgRNA uncloaking condition of 5 mM THPP for 17 hours.30 TCO-OH treatment could not induce significant activation likely due to its lower efficiency as observed in NMR and oligonucleotide experiments.
Figure 4.
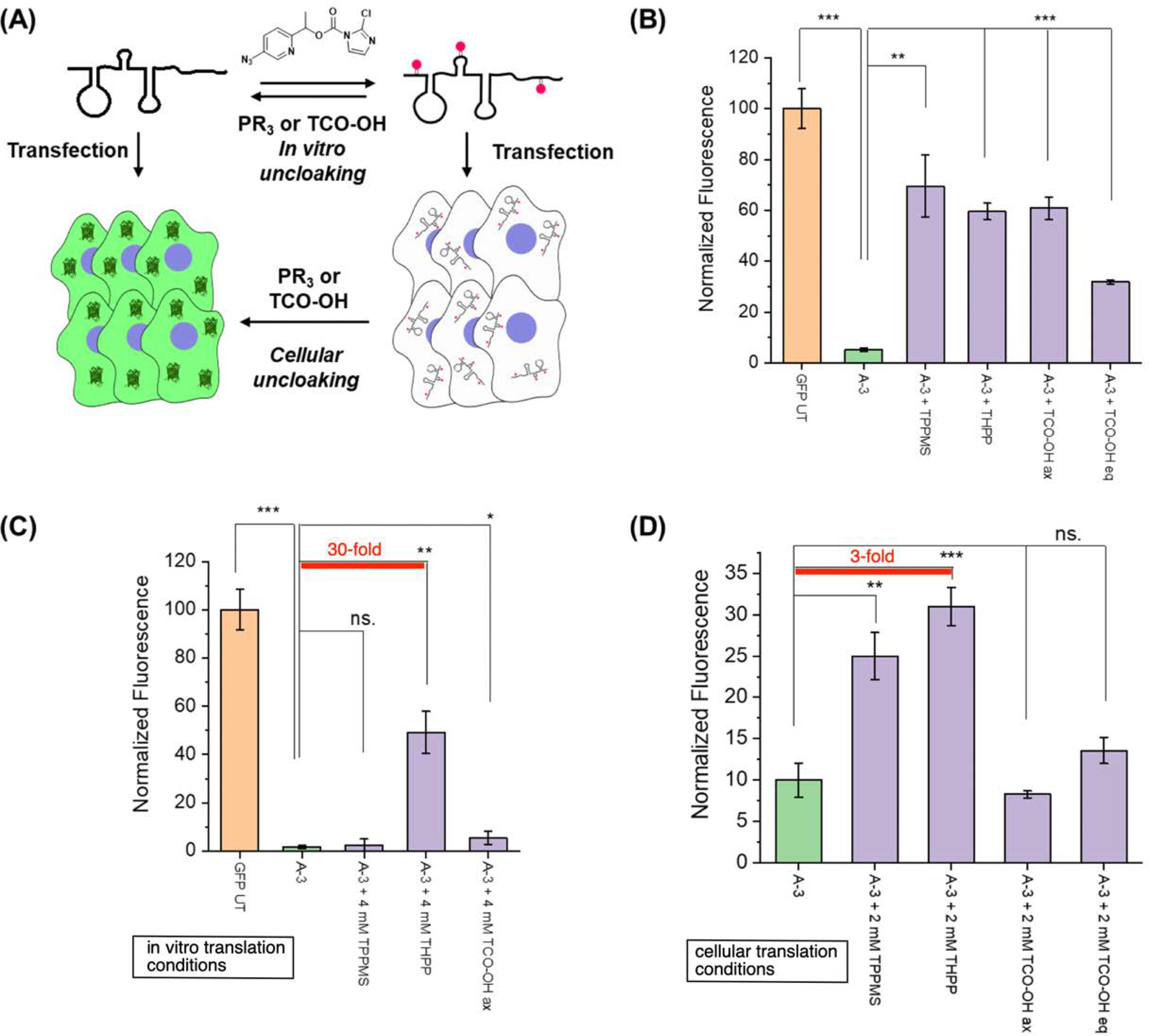
Control of EGFP mRNA translation with QM reagent A-3. (A) Scheme of conditional translation of EGFP mRNA with polyacylation. Red circles represent 2’-OH acyl groups. (B) In vitro translation of uncloaked EGFP mRNA. A-3 cloaked and subsequently uncloaked EGFP mRNA was expressed with WGE for 3 hours at 25 °C. (C) Direct activation of A-3 cloaked EGFP mRNA in WGE. A-3 cloaked EGFP mRNA in WGE was treated with various reversal agents for 3 hours at 25 °C. (D) Cellular control of EGFP mRNA expression. A-3 cloaked mRNA was transfected into HeLa cells for 4 hours and subsequently treated with various reversal agents for 4 hours. Error bars represent standard deviation and p-values: *** p < 0.001, ** p < 0.01, * p < 0.05, ns. – not significant. Orange: untreated RNA, green: cloaked RNA, purple: uncloaked RNA. For panels (B) through (D), the full data sets are displayed in the Fig. S10.
We next examined whether A-3 can be used to control mRNA in an in vitro translation system. When EGFP mRNA was translated with wheat germ extract (WGE), A-3 cloaking strongly inhibited EGFP fluorescence to near background levels and uncloaking prior to translation recovered up to 70% of the original fluorescence (Fig. 4B and S10B). The lower reversal efficiency of TCO-OHeq could be attributed to the lesser reactivity of the equatorial isomer compared to the axial.39 To demonstrate biochemical compatibility, A-3 cloaked mRNA was directly activated in the translation mixture (Fig. 4C and S10C). Interestingly, TPPMS inhibited expression to baseline levels by itself and failed to trigger GFP expression from caged mRNA. In contrast, THPP displayed minimal inhibition of translation and resulted in 30-fold fluorescence enhancement. Although not toxic, TCO-OH treatment resulted in only minor activation similar to the cellular experiments. Taken together, we conclude that the newly developed QM reagent A-3 is capable of controlling RNA function both in vitro and in live cells and shows substantial promise as a convenient chemical tool for the investigation of RNA biology.
Discussion
Our experiments establish that an optimized pyridinone methide framework coupled with a reductively-triggered bond cleavage mechanism offers rapid and high-yield de-acylation both in small molecules and in RNAs. The approach could have general utility in RNA, by enabling caging without the need for engineered domains or recognition sequences. The installation of the caging groups is simple and rapid, and is applied equally readily to short or long RNAs, unlike caging approaches that require chemical RNA synthesis. We show that the combination of a 2-pyridyl benzylic carbonate combined with 5-substitution of an azide group for reductive triggering enables efficient bond cleavage, releasing this blocking group from the original substrate. This cleavage is found to be several-fold more rapid than prior azide/acylation RNA caging strategies,25 which were dependent on slower cyclization steps. Rapid uncaging is potentially important since the goal of caging is often the temporally controlled triggering of a pulse of biological activity. We further show that such a reagent, in activated imidazole carbamate form, retains sufficient water solubility and reactivity to enable multiple acylations on RNAs in vitro. Such acylations have been successfully carried out on a full-length messenger RNA, and the data show that the acylation can strongly suppress translation of the RNA, establishing that bulky substitutions at 2’-OH or exocyclic amines can block this complex process. Upon phosphine treatment, up to 30-fold recovery of translation in vitro and 3-fold in cells has been observed, which is competitive with previously reported methods.13,14,45
The new data show that by employing a QM elimination mechanism, the release rates and conversion yields of reversible RNA acylating compounds were improved. This enhancement ultimately enabled in vitro and cellular application of cloaking methods to a kilobase-length EGFP mRNA; this is one of the first reports of chemical caging of non-engineered mRNA, enabling control of gene expression. These early results suggest the more general potential of the new reagents such as A-3 for caging a broad range of mRNAs. Future experiments will be directed to applying this to other mRNAs and long noncoding RNAs of biological interest. Some limits of this approach are worth noting; one of these is the known cellular toxicity of phosphines.45 While phosphines have been used previously with success in uncaging intracellular proteins and RNAs,30,45 it is prudent to minimize exposure to such species. The greater reactivity of the current caging group A-3 is important as it requires lower concentrations and shorter exposures for uncaging compared with earlier azide cyclization mechanisms.30 For in vitro applications this toxicity is moot, and interestingly, the THPP phosphine shows no deleterious effect on translation in cellular extracts (Fig. 4). We find that the new imidazole carbamate derivatives have lower acylation efficiencies relative to prior acylimidazoles, but reactivity remains sufficient for mRNA inhibition. If a higher degree of acylation is desired, longer times, higher temperatures, or multiple cloaking reactions can be conducted (data not shown).
To our knowledge, the azido QM compounds studied here are the first of the aza-pyridinone methide scaffold for chemically triggered bond breaking. Previously, azaquinone47 or pyridinone methide42 designs were separately adopted for self-immolative linkers. The new structure-activity relationships we observe for reduction and release efficiency may be useful as a guide for future cleavable linkers. One surprising observation was that 6-azidopyridine showed poor reactivity both to phosphine and TCO-OH and that it should be avoided for rapid reversal. The electronically withdrawing effects of pyridine nitrogen seem to reduce reactivity, resulting in 4-azidopyridine being the most the reactive followed by 3-azido isomer. However, 4-azidopyridine displayed much reduced reversibility because of steric hindrance when bulkier reversal triggers were used or larger acyl groups such as RNA were appended. Nevertheless, it shows promise for caging small molecules; for other more sterically demanding substrates, including RNAs, the A-3 scaffold appears to be the most efficient.
In addition to controlling RNAs in biological systems, we envision that this caging approach can be applied in vitro for control of RNA-based diagnostics such as CRISPR-Cas13 reporters48 as well as to RNAs in triggerable synthetic gene circuits.49–51 These methods should be compatible with previously established caging methods and could potentially be synergistically combined for additional control and circuit design. Additionally, the appended azide handle substituted at the 4-position of reagent A-3 can be utilized for bioconjugation via the “cloak and click” approach29 and potentially for photocrosslinking applications in mapping RNA interactions as well.52,53
Supplementary Material
Acknowledgement.
We thank the U.S. National Institutes of Health (GM127295) for support.
References
- (1).Müller S; Appel B; Balke D; Hieronymus R; Nübel C Thirty-Five Years of Research into Ribozymes and Nucleic Acid Catalysis: Where Do We Stand Today? F1000Research 2016, 5, 1–11. 10.12688/F1000RESEARCH.8601.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sullenger BA; Nair S From the RNA World to the Clinic. Science 2016, 352 (6292), 1417–1420. 10.1126/science.aad8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Anastasiadou E; Jacob LS; Slack FJ Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2017, 18 (1), 5–18. 10.1038/nrc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cech TR; Steitz JA The Noncoding RNA Revolution - Trashing Old Rules to Forge New Ones. Cell 2014, 157 (1), 77–94. 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- (5).Ruble BK; Yeldell SB; Dmochowski IJ Caged Oligonucleotides for Studying Biological Systems. J. Inorg. Biochem. 2015, 150, 182–188. 10.1016/j.jinorgbio.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ankenbruck N; Courtney T; Naro Y; Deiters A Optochemical Control of Biological Processes in Cells and Animals. Angew. Chemie - Int. Ed. 2018, 57 (11), 2768–2798. 10.1002/anie.201700171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen C; Wang Z; Jing N; Chen W; Tang X Photomodulation of Caged RNA Oligonucleotide Functions in Living Systems. ChemPhotoChem 2021, 5 (1), 12–21. 10.1002/cptc.202000220. [DOI] [Google Scholar]
- (8).Wenter P; Fürtig B; Hainard A; Schwalbe. Kinetics of Photoinduced RNA Refolding by Real-Time NMR Spectroscopy. Angew. Chemie - Int. Ed. 2005, 44 (17), 2600–2603. 10.1002/anie.200462724. [DOI] [PubMed] [Google Scholar]
- (9).Höbartner C; Silverman SK Modulation of RNA Tertiary Folding by Incorporation of Caged Nucleotides. Angew. Chemie - Int. Ed. 2005, 44 (44), 7305–7309. 10.1002/anie.200502928. [DOI] [PubMed] [Google Scholar]
- (10).Seyfried P; Eiden L; Grebenovsky N; Mayer G; Heckel A Photo-Tethers for the (Multi-)Cyclic, Conformational Caging of Long Oligonucleotides. Angew. Chemie - Int. Ed. 2017, 56 (1), 359–363. 10.1002/anie.201610025. [DOI] [PubMed] [Google Scholar]
- (11).Muthmann N; Hartstock K; Rentmeister A Chemo-Enzymatic Treatment of RNA to Facilitate Analyses. Wiley Interdiscip. Rev. RNA 2020, 11 (1), 1–33. 10.1002/wrna.1561. [DOI] [PubMed] [Google Scholar]
- (12).Zhang D; Zhou CY; Busby KN; Alexander SC; Devaraj NK Light-Activated Control of Translation by Enzymatic Covalent MRNA Labeling. Angew. Chemie - Int. Ed. 2018, 57 (11), 2822–2826. 10.1002/anie.201710917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang D; Jin S; Piao X; Devaraj NK Multiplexed Photoactivation of MRNA with Single-Cell Resolution. ACS Chem. Biol. 2020, 15 (7), 1773–1779. 10.1021/acschembio.0c00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ando H; Furuta T; Tsien RY; Okamoto H Photo-Mediated Gene Activation Using Caged RNA/DNA in Zebrafish Embryos. Nat. Genet. 2001, 28 (4), 317–325. 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- (15).Shah S; Rangarajan S; Friedman SH Light-Activated RNA Interference. Angew. Chemie - Int. Ed. 2005, 44 (9), 1328–1332. 10.1002/anie.200461458. [DOI] [PubMed] [Google Scholar]
- (16).Ando H; Kobayashi M; Tsubokawa T; Uyemura K; Furuta T; Okamoto H Lhx2 Mediates the Activity of Six3 in Zebrafish Forebrain Growth. Dev. Biol. 2005, 287 (2), 456–468. 10.1016/j.ydbio.2005.09.023. [DOI] [PubMed] [Google Scholar]
- (17).Ovcharenko A; Weissenboeck FP; Rentmeister A Tag-Free Internal RNA Labeling and Photocaging Based on mRNA Methyltransferases. Angew. Chemie - Int. Ed. 2021, 60 (8), 4098–4103. 10.1002/anie.202013936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Maghami MG; Dey S; Lenz AK; Höbartner C Repurposing Antiviral Drugs for Orthogonal RNA-Catalyzed Labeling of RNA. Angew. Chemie - Int. Ed. 2020, 59 (24), 9335–9339. 10.1002/anie.202001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Maghami MG; Scheitl CPM; Höbartner C Direct in Vitro Selection of Trans-Acting Ribozymes for Posttranscriptional, Site-Specific, and Covalent Fluorescent Labeling of RNA. J. Am. Chem. Soc 2019, 141, 50, 19546–19549. 10.1021/jacs.9b10531. [DOI] [PubMed] [Google Scholar]
- (20).Zhao M; Steffen FD; Börner R; Schaffer MF; Sigel RKO; Freisinger E Site-specific dual-color labeling of long RNAs for single-molecule spectroscopy. Nucleic Acids Res. 2018, 46 (3), e13. 10.1093/nar/gkx1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Knutson SD; Sanford AA; Swenson CS; Korn MM; Manuel BA; Heemstra JM Thermoreversible Control of Nucleic Acid Structure and Function with Glyoxal Caging. J. Am. Chem. Soc. 2020, 142, 41, 17766–17781. 10.1021/jacs.0c08996. [DOI] [PubMed] [Google Scholar]
- (22).Egloff D; Oleinich LA; Zhao M; König SLB; Sigel RKO; Freisinger E Sequence-Specific Post-Synthetic Oligonucleotide Labeling for Single-Molecule Fluorescence Applications. ACS Chem. Biol. 2016, 11, 9, 2558–2567. 10.1021/acschembio.6b00343. [DOI] [PubMed] [Google Scholar]
- (23).Blidner RA; Svoboda KR; Hammer RP; Monroe WT Photoinduced RNA Interference Using DMNPE-Caged 2′-Deoxy-2′-Fluoro Substituted Nucleic Acids in Vitro and in Vivo. Mol. Biosyst. 2008, 4 (5), 431. 10.1039/b801532e. [DOI] [PubMed] [Google Scholar]
- (24).Brash DE; Haseltine WA UV-Induced Mutation Hotspots Occur at DNA Damage Hotspots. Nature 1982, 298 (5870), 1891–1892. 10.1038/298189a0. [DOI] [PubMed] [Google Scholar]
- (25).Kadina A; Kietrys AM; Kool ET RNA Cloaking by Reversible Acylation. Angew. Chemie - Int. Ed. 2018, 57 (12), 3059–3063. 10.1002/anie.201708696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Velema WA; Kietrys AM; Kool ET; Kadina A; Kietrys AM; Kool ET RNA Control by Photoreversible Acylation. J. Am. Chem. Soc 2018, 140, 3491–3495. 10.1002/anie.201708696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Habibian M; Velema WA; Kietrys AM; Onishi Y; Kool ET Polyacetate and Polycarbonate RNA: Acylating Reagents and Properties. 2019, 21 (14), 5413–5416. 10.1021/acs.orglett.9b01526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Velema WA; Kool ET Water-Soluble Leaving Group Enables Hydrophobic Functionalization of RNA. Org. Lett. 2018, 20 (20), 6587–6590. 10.1021/acs.orglett.8b02938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Park HS; Kietrys AM; Kool ET Simple Alkanoyl Acylating Agents for Reversible RNA Functionalization and Control. Chem. Commun. 2019, 55, 5135–5138. 10.1039/C9CC01598A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Habibian M; Mckinlay C; Blake TR; Kietrys AM; Waymouth RM; Wender PA; Kool ET Reversible RNA Acylation for Control of CRISPR–Cas9 Gene Editing. Chem. Sci. 2020, 11, 1011–1016. 10.1039/C9SC03639C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang S; Wu L; Huang H; Xiong W; Liu J; Wei L; Yin P; Tian T; Zhou X Conditional Control of RNA-Guided Nucleic Acid Cleavage and Gene Editing. Nat. Commun. 2020, 11, 91. 10.1038/s41467-019-13765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Velema WA; Kool ET The Chemistry and Applications of RNA 2′-OH Acylation. Nature Reviews Chemistry. Springer; US: 2020, pp 22–37. 10.1038/s41570-019-0147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Leriche G; Chisholm L; Wagner A Cleavable Linkers in Chemical Biology. Bioorganic Med. Chem. 2012, 20 (2), 571–582. 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- (34).Rudolf GC; Heydenreuter W; Sieber SA Chemical Proteomics: Ligation and Cleavage of Protein Modifications. Curr. Opin. Chem. Biol. 2013, 17 (1), 110–117. 10.1016/j.cbpa.2012.11.007. [DOI] [PubMed] [Google Scholar]
- (35).Bargh JD; Isidro-Llobet A; Parker JS; Spring DR Cleavable Linkers in Antibody-Drug Conjugates. Chem. Soc. Rev. 2019, 48 (16), 4361–4374. 10.1039/c8cs00676h. [DOI] [PubMed] [Google Scholar]
- (36).Tu J; Xu M; Franzini RM Dissociative Bioorthogonal Reactions. ChemBioChem 2019, 20 (13), 1615–1627. 10.1002/cbic.201800810. [DOI] [PubMed] [Google Scholar]
- (37).Li J; Chen PR Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol. 2016, 12 (3), 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- (38).Lukasak B; Morihiro K; Deiters A Aryl Azides as Phosphine-Activated Switches for Small Molecule Function. Sci. Rep. 2019, 9, 1470. 10.1038/s41598-018-37023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Matikonda SS; Orsi DL; Staudacher V; Jenkins IA; Fiedler F; Chen J; Gamble AB Bioorthogonal Prodrug Activation Driven by a Strain-Promoted 1,3-Dipolar Cycloaddition. Chem. Sci. 2015, 6 (2), 1212–1218. 10.1039/c4sc02574a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jiménez-Moreno E; Guo Z; Oliveira BL; Albuquerque IS; Kitowski A; Guerreiro A; Boutureira O; Rodrigues T; Jiménez-Osés G; Bernardes GJL Vinyl Ether/Tetrazine Pair for the Traceless Release of Alcohols in Cells. Angew. Chemie - Int. Ed. 2017, 56 (1), 243–247. 10.1002/anie.201609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rossin R; Versteegen RM; Wu J; Khasanov A; Wessels HJ; Steenbergen EJ; Ten Hoeve W; Janssen HM; Van Onzen AHAM; Hudson PJ; Robillard MS Chemically Triggered Drug Release from an Antibody-Drug Conjugate Leads to Potent Antitumour Activity in Mice. Nat. Commun. 2018, 9 (1), 1–11. 10.1038/s41467-018-03880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Perry-feigenbaum R; Baran S; Shabat D The Pyridinone-Methide Elimination. Org. Biomol. Chem. 2009, 7, 4825–4828. 10.1039/b915265b. [DOI] [PubMed] [Google Scholar]
- (43).Lee YS; Shibata Y; Malhotra A; Dutta A A Novel Class of Small RNAs: TRNA-Derived RNA Fragments (TRFs). Genes Dev. 2009, 23 (22), 2639–2649. 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lee B; Flynn RA; Kadina A; Guo JK; Kool ET; Chang HY Comparison of SHAPE Reagents for Mapping RNA Structures inside Living Cells. Rna 2017, 23 (2), 169–174. 10.1261/rna.058784.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ogasawara S Control of Cellular Function by Reversible Photoregulation of Translation. ChemBioChem 2014, 15 (18), 2652–2655. 10.1002/cbic.201402495. [DOI] [PubMed] [Google Scholar]
- (46).Luo J; Liu Q; Morihiro K; Deiters A Small-Molecule Control of Protein Function through Staudinger Reduction. Nat. Chem. 2016, 8 (11), 1027–1034. 10.1038/nchem.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Erez R; Shabat D The Azaquinone-Methide Elimination: Comparison Study of 1,6- and 1,4-Eliminations under Physiological Conditions. Org. Biomol. Chem. 2008, 6, 2669–2672. 10.1039/b808198k. [DOI] [PubMed] [Google Scholar]
- (48).Myhrvold C; Freije CA; Gootenberg JS; Abudayyeh OO; Metsky HC; Durbin AF; Kellner MJ; Tan AL; Paul LM; Parham LA; Garcia KF; Barnes KG; Chak B; Mondini A; Nogueira ML; Isern S; Michael SF; Lorenzana I; Yozwiak NL; MacInnis BL; Bosch I; Gehrke L; Zhang F; Sabeti PC Field-Deployable Viral Diagnostics Using CRISPR-Cas13. Science 2018, 360 (6387), 444–448. 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gardner L; Deiters A Light-Controlled Synthetic Gene Circuits. Curr. Opin. Chem. Biol. 2012, 16 (3–4), 292–299. 10.1016/j.cbpa.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Isaacs FJ; Dwyer DJ; Collins JJ RNA Synthetic Biology. Nat. Biotechnol. 2006, 24 (5), 545–554. 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- (51).Ausländer S; Fussenegger M Engineering Gene Circuits for Mammalian Cell-Based Applications. Cold Spring Harb. Perspect. Biol. 2016, 8 (7), 1–18. 10.1101/cshperspect.a023895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Buchmueller KL; Hill BT; Platz MS; Weeks KM RNA-Tethered Phenyl Azide Photocrosslinking via a Short-Lived Indiscriminant Electrophile. J. Am. Chem. Soc. 2003, 125 (36), 10850–10861. 10.1021/ja035743+. [DOI] [PubMed] [Google Scholar]
- (53).Smith E; Collins I Photoaffinity Labeling in Target- and Binding-Site Identification. Future Med. Chem. 2015, 7 (2), 159–183. 10.4155/fmc.14.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.