

Abstract
Several essential components of the electron transport chain, the major producer of ATP in mammalian cells, are encoded in the mitochondrial genome. These 13 proteins are translated within mitochondria by ‘mitoribosomes’. Defective mitochondrial translation underlies multiple inborn errors of metabolism and has been implicated in pathologies such as aging, metabolic syndrome and cancer. Here, we provide a detailed ribosome profiling protocol optimized to interrogate mitochondrial translation in mammalian cells (MitoRiboSeq), wherein mitoribosome footprints are generated with micrococcal nuclease and mitoribosomes are separated from cytosolic ribosomes and other RNAs by ultracentrifugation in a single straightforward step. We highlight critical steps during library preparation and provide a step-by-step guide to data analysis accompanied by open-source bioinformatic code. Our method outputs mitoribosome footprints at single-codon resolution. Codons with high footprint densities are sites of mitoribosome stalling. We recently applied this approach to demonstrate that defects in mitochondrial serine catabolism or in mitochondrial tRNA methylation cause stalling of mitoribosomes at specific codons. Our method can be applied to study basic mitochondrial biology or to characterize abnormalities in mitochondrial translation in patients with mitochondrial disorders.
Introduction
Mitochondria are responsible for the majority of bioenergy production in most mammalian cells1. In humans, mitochondria are composed of roughly 1,000 different proteins, most of which are encoded in the nuclear genome. These nuclear-encoded mitochondrial proteins are synthesized in the cytosol and then imported into mitochondria2,3. However, there are 13 critical transmembrane proteins that are encoded in the mitochondrial genome. These include core components of complexes I, III, IV and V of the electron transport chain4. Synthesis of these proteins requires dedicated machinery for mitochondrial transcription and translation, including mitoribosomes2,3. Thus, mitochondrial translation is required for effective respiration and oxidative ATP production5.
Defects in mitochondrial translation lead to impaired electron transport chain activity6,7. Prominent examples include the devastating inborn mitochondrial diseases mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonic epilepsy with ragged red fibers (MERRF). These can be caused by mutations in mitochondrial tRNAs and are characterized by severe phenotypes such as cerebellar ataxia, seizures and stroke-like episodes8–10. The importance of mitochondrial translation extends beyond these overt cases. Impaired respiration is a hallmark of both cancer cells and aging11–13, and drug-induced toxicity can result from unexpected interference with mitochondrial function14,15. To date, however, studies of defective mitochondrial translation have been limited by inadequate methodology.
The field of mRNA translation has greatly benefited from the development of ribosome profiling16. Ribosome profiling is a method whereby ribosome-protected mRNA fragments—or ‘footprints’—are generated by ribonuclease treatment and then sequenced in high throughput. The resulting sequencing library contains a snapshot of the ribosome distribution across the transcriptome at single-codon resolution. Numerous scientific advances have been made with ribosome profiling, including the discovery of novel translated regions of the transcriptome17,18, the characterization of translation localized to specific organelles19,20 and the identification of translational bottlenecks caused by insufficient tRNA charging21. Ribosome profiling has been adapted to study translation in almost all kingdoms of life (bacteria22, yeast16, mammals23, plants24). Methods for generating sequencing libraries from small ribosome-protected RNA fragments are broadly generalizable, but specific methods for lysis, footprinting and computational analysis must be optimized for each species and sample type (adherent cells, suspension cells, tissue samples).
Specific methodology is required for mitochondrial ribosomes, which have different features compared with their cytosolic counterparts. First, mitoribosomes are far less abundant: HeLa cells are estimated to contain 9.5 million cytosolic ribosomes and less than half a million mitoribosomes25,26. Second, mitoribosomes are biochemically different from cytosolic ribosomes, enabling separation of the two species but also necessitating reoptimization of various steps, including ribonuclease treatment27. Finally, mitochondrial transcripts are polycistronic28–30, and thus the analysis of mitoribosome profiling data must be adapted to take into account the unique features of the mitochondrial transcriptome. Our method addresses these differences and enables robust, high-resolution characterization of mitochondrial translation in mammalian cells.
Here, we provide a detailed protocol for mammalian mitoribosome profiling at single-codon resolution. We also provide a step-by-step bioinformatics tool that can transform the raw data generated by our MitoRiboSeq protocol into ribosome counts at each nucleotide across the mitochondrial genome. Previously, we used a variant of this pipeline to show that defective serine catabolism impairs mitochondrial tRNA methylation, resulting in defective ‘wobble’ base-pairing of tRNAs to specific codons and consequent mitoribosome stalling31. We also observed this phenotype in MELAS/MERRF patient-derived cells31, providing mechanistic insight into this mitochondrial disease and highlighting the clinical relevance of this method.
Comparison with other methods
Our protocol for mitochondrial ribosome profiling was adapted from standard ribosome profiling protocols designed by Ingolia et al. for yeast and mammalian cells32,33 and by Li et al. for bacteria34 (Fig. 1). A handful of other mitoribosome profiling methods have been developed with various enrichment methods for mitochondrial ribosomes (Table 1). Rooijers et al. applied mitochondrial ribosome profiling to confirm stalling at tryptophan codons in patients with a mutated mitochondrial tryptophan tRNA; they used RNase I at concentrations that we found digested all monosomes (leaving no footprints)35. Following the method of Rooijers et al., Gao et al. found mutant mt-tRNAVal can result in less optimal structure of human mitoribosome and thus changes mitochondrial translation33. Couvillion et al. used mitochondrial ribosome profiling to investigate the coordination of translation of cytosolic and mitochondrial ribosomes during the metabolic transition from fermentation to respiration in yeast36,37; they FLAG-tagged mitoribosomes and immunopurified them (74S yeast mitoribosomes must be purified differently than 55S mammalian mitoribosomes). Pearce et al. used mitoribosome profiling to show that the poly(A)-specific exonuclease PDE12 is critical to maintain charted mt-tRNA pools and prevent mitoribosome stalling38. Here, we compare the methods used in these papers with our protocol, highlighting key differences.
Fig. 1 |. Overview of the workflow for mitochondrial ribosome profiling.
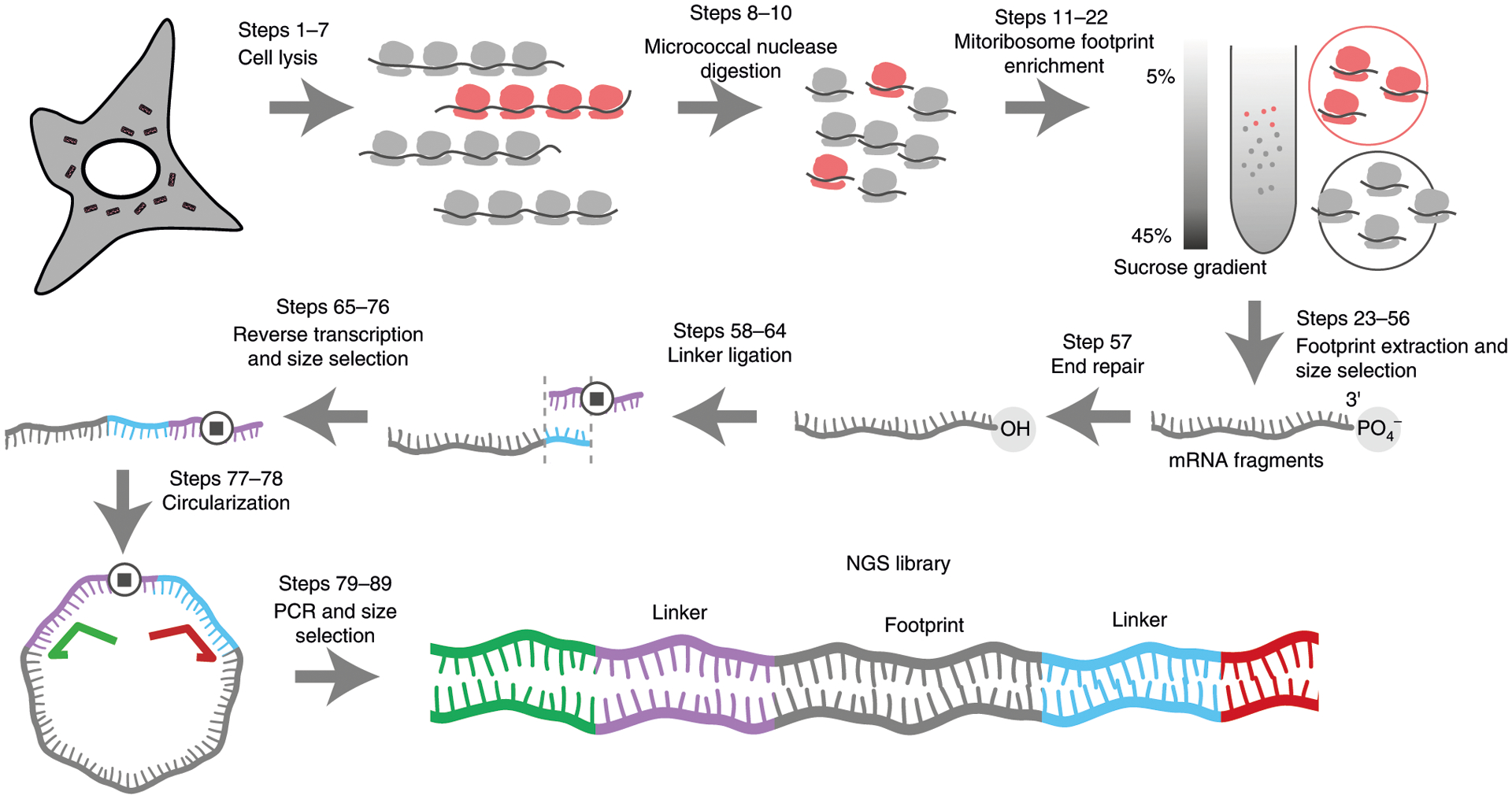
Steps 1–3: mitoribosome enrichment. Cell lysates contain both cytosolic (represented in gray) and mitochondrial (represented in red) ribosomes. After nuclease digestion, the cell lysate is layered onto a 5–45% sucrose gradient and ultracentrifuged. Cytosolic and mitochondrial ribosomes have different sedimentation coefficients and can therefore be separated by ultracentrifugation and collected separately. Steps 4–9: library preparation after the mRNA fragments are extracted. The 3′-end is first repaired to remove the 3′-end phosphate group followed by a 3′-linker ligation (linker in blue). A reverse transcription primer (purple) with a complementary sequence to the linker is used to generate cDNA. The stop symbol represents a spacer to avoid circular amplification during the PCR step. The ssDNA is circularized and amplified to generate NGS library using a universal forward primer (green) and a reverse primer (red) with Illumina barcodes.
Table 1 |.
Comparison of different mitochondrial ribosome profiling methods
| Reference | Species | Antibiotic pretreatmen | Ribosome isolation method | Ribonuclease | Fragment size selected (nt) | rRNA extraction | A-site alignment method | Most common footprint size (nt) |
|---|---|---|---|---|---|---|---|---|
| Standard ribosome profiling: McClincy and Ingolia33 | Yeast | None | Sucrose cushion | RNase I | 15–45, canonical footprint is ~28 | Ribo-Zero Gold (Illumina) | 5’-end offset from start codon for each read length | 28 |
| This paper | Human | None | Sucrose gradient, real-time collection with UV signal | MNase | 15–45 | None | 3’-end offset from start codon | 30 |
| Rooijers et al.35 and Gao et al.67 | Human | Yes | Sucrose gradient, mitoribosome identification with western blot | RNase I | 25–36 | None | 5’-end offset by 16 nt | 33 |
| Couvillion et al.37 and Couvillion & Churchman36 | Yeast | None | Immunoprecipitation using FLAG-tagged mitoribosome subunit | RNase I | 36–42 | Ribo-Zero Gold (Illumina) | 3’-end offset from start codon | 39 |
| Pearce et al.38 | Human | Yes | Sucrose gradient, mitoribosome identification with western blot | RNase I | 25–35 | Duplex strand nuclease treatment at amplicon stage | 5’-offset using lysine stalling in ΔFLP cells | 33 |
In any ribosome profiling method, cell lysis is a key step. Ideally, the positions of all ribosomes along transcripts should be preserved starting immediately after lysis. In an effort to achieve this, previous methods have recommended addition of translation inhibitors even before lysis. However, pretreatment with translation inhibitors has been shown to intervene with the gene expression machinery, affecting some mRNAs differently than others and leading to artifacts39. Therefore, we do not pretreat cells with ribosome elongation inhibitor. Instead, we rapidly wash the cellular monolayer once with ice-cold PBS containing chloramphenicol (which stalls mitochondrial ribosomes40), then add ice-cold lysis buffer also containing chloramphenicol. Similarly, other recent mitoribosome profiling methods avoid pretreatment with antibiotics33,36.
The choice of ribonuclease is critical for yielding high-resolution ribosome footprints. Ideally, the ribonuclease should degrade unprotected mRNA without digesting ribosomes (which are largely composed of rRNA). The correct choice depends on the kind of sample and the amount of input RNA. RNase I, the classical ribonuclease choice for ribosome profiling, is not ideal for bacterial ribosome profiling because bacterial ribosomes can inhibit RNase I (RNase I is an Escherichia coli enzyme)41. Ribosome profiling implementations in bacteria have conventionally used micrococcal nuclease S7 (MNase) instead. RNase I hydrolyzes RNA nonspecifically, while MNase preferentially cleaves next to adenine and thymine bases, introducing some sequence bias in footprint generation. Analytical methods can be used to account for such biases. Further investigation is needed to perform a robust comparison of the two nucleases in a controlled setting. We recommend MNase, but ultimately, to ensure high data quality, the ribonuclease digestion step should be optimized in each laboratory where this protocol is implemented. We provide guidelines for how to do this in ‘Experimental design’ under ‘Footprinting’.
A final key step is the isolation of mitoribosomes. Since the majority of cellular ribosomes are cytosolic, the extraction of mitoribosomes must be highly specific. One enrichment strategy is to use ultracentrifugation to separate 55S mitochondrial and 80S cytosolic ribosomes on a sucrose gradient followed by fractionation and subsequent purification of mitochondrial ribosomes. Previous methods identify fractions containing mitochondrial ribosomes by western blot, adding a laborious step to the protocol (Rooijers et al., Pearce et al.). Yeast mitoribosomes, which are 74S, sediment too closely to cytosolic ribosomes and must be genetically engineered to express a FLAG tag such that they can be immunopurified (Couvillion et al.). A MitoRibo-Tag mouse has also been developed recently, enabling mitoribosome isolation from mouse tissues using this approach42. The method we describe here uses real-time isolation of mitochondrial ribosomes with a standard fractionator attached to a UV absorbance monitor, without the need for genetic engineering, thereby streamlining the protocol. MNase treatment, which is less harsh compared with RNase I treatment, reliably preserves the 40S and 60S ribosomal subunits during digestion; these subunits are used as landmarks to locate the 55S mitochondrial ribosomes.
Due to the small genome size of mitochondrial DNA, we found that despite substantial rRNA contamination, sufficient read depth and coverage can be achieved. Nevertheless, rRNA depletion can still be applied to minimize sequencing cost. Recently, the Illumina Ribo-Zero Gold for human/mouse/rat (which depletes cytosolic but not mitochondrial rRNA contaminants) was discontinued, but alternatives have emerged. A recent study compared several commercial kits and found that customized rRNA depletion using biotinylated probes engineered to target the major contaminants can work as well as the discontinued Illumina kit or better. Importantly, the authors also suggest that approaches involving RNase H can compromise library integrity. Alternatively, some protocols use duplex-specific nuclease treatment to degrade abundant cDNA derived from rRNA38. Another recent paper provided algorithms to design biotinylated probes compatible with rRNA removal in several bacterial species43. These approaches are particularly amenable to customized rRNA depletion from ribosome profiling libraries, because the number of unique rRNA contaminant species in ribosome profiling libraries is relatively low, and therefore the number of depletion oligos required to remove rRNA contaminants is manageable (~10)34.
Applications, advantages and limitations
The methodology presented here can be leveraged for many interesting future applications. For example, with our methods, simultaneous measurement of cytosolic and mitochondrial ribosome footprints becomes possible. Such measurements would be particularly interesting in the context of translational adaptation. For instance, if cells are starved of oxygen (hypoxic) or other nutrients (glucose, amino acids), they must adapt both cytosolic and mitochondrial translation to meet the demands of their new environment. Couvillion et al. provide one prominent example of coupling between cytosolic and mitochondrial translation: using ribosome profiling data from both cytosolic and mitochondrial ribosomes, they showed that when yeast are switched from glucose (aerobic glycolysis) to glycerol (respiration), both cytosolic and mitochondrial translation rapidly adapt37. To our knowledge, no similar studies have been performed in mammalian systems.
Another future direction using the methodology presented here is in vivo analysis of mitochondrial translation. Standard ribosome profiling has been performed on tissue samples from mice and even humans. These analyses revealed that various tumors (clear cell renal cell carcinoma, breast cancer) have a limited supply of proline21 and that initiating tumor cells depend on noncanonical translation initiation at upstream open reading frames44. These studies provide useful insights into in vivo translation. Similar in vivo analyses of mitochondrial translation are lacking.
One major limitation of all ribosome profiling methods is the inability to infer translation rates directly from footprint density. Li et al. have used ribosome profiling data in E. coli to report translation rates transcriptome wide22, but we and others have observed that translation elongation rates appear to vary dramatically from transcript to transcript, particularly in eukaryotic cells45. Thus, translation rate is a complex function of footprint density and ribosome elongation rate and cannot be inferred reliably from footprint density alone. The biological mechanisms underlying differences in elongation rate are poorly understood and an exciting area for future exploration.
Experimental design
RNase-free environment
General caution is required throughout this protocol to avoid RNase contamination. We recommend frequently wiping down all surfaces (bench, pipettes, and equipment) with RNase-neutralizing solution (e.g., RNaseZap) and using dedicated RNase-free consumables (e.g., tip boxes). All reagents should be stored in an RNase-free environment (store MNase or RNase I separately), and all solutions should be prepared with great care. Diethyl pyrocarbonate (DEPC)-treated water can also be used in place of standard nuclease-free water.
Starting material
We recommend first attempting this method with one ~80% confluent 15 cm plate of mammalian cells per sample. However, yield can vary depending on the user and the batch, and adjustments should be made accordingly to ensure that sample input is sufficient. Experienced practitioners might be able to start with fewer cells (~80% confluent 10 cm plate). Note that the mitochondrial transcriptome is far less complex than the cytosolic transcriptome, and in general, fewer reads are needed to obtain deep coverage as long as clean separation of mitochondrial ribosomes from cytosolic ribosomes is achieved. One can determine whether the amount of starting material was insufficient if more than 12 cycles of PCR are needed at the amplification step. For MitoRiboSeq experiments using tissue samples, starting material should be carefully optimized in the same manner, although even small tissue samples have more material than confluent 15 cm plates.
Footprinting
In this protocol, we focus only on footprinting mitochondrial ribosomes, but cytosolic ribosomes can be extracted from the same sucrose gradients, and their footprints can be prepared for sequencing following the same protocol. Importantly, chloramphenicol is required to stall mitochondrial ribosomes, while cycloheximide is required to stall cytosolic ribosomes. We include both in our buffers, which enables simultaneous extraction of both species, if desired.
To maximize sequencing depth, we digest as much lysate as possible. After setting aside 100 μL of lysate to be used for other analyses, including standard RNA sequencing, we typically digest the rest (~1 mL).
Ribosome profiling results are highly sensitive to both underdigestion and overdigestion. We recommend that this step be optimized in-house by each laboratory that uses this protocol. Ideally, digestion should result in complete footprinting without degradation of ribosomal RNA, which causes ribosome dissociation and loss of RNA footprints. New users should try a range of ribonuclease doses—to start, we recommend 0 μL, 1 μL, 2 μL, 5 μL, 10 μL and 20 μL MNase—on the same pooled lysate, then analyze the resulting digested products on sucrose gradients (Steps 11–23). The presence of polysomes suggests that lysates are underdigested. Overdigested lysates will be missing a large fraction of monosomes. Some monosome degradation might be unavoidable.
For further optimization, sequencing libraries should be prepared using the protocol described here and analyzed with a shallow sequencing run on a MiSeq. One million reads should be sufficient to assess the quality of the digestion. The ribonuclease dose that generates ~28 bp footprints with the lowest rRNA background should be used. We routinely use 10 μL MNase to digest 1 mL cell lysate.
CaCl2 must also be added to enable digestion, as MNase is calcium dependent. The reaction can therefore be quenched by adding ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), which chelates calcium. Nevertheless, we recommend moving on rapidly to ultracentrifugation after digestion, as residual enzyme activity is inevitible even if EGTA is added.
If RNase I is preferred over MNase, optimization can be done in the same manner. We have found that RNase I is much more potent than MNase, and overdigestion can be hard to avoid, even if small amounts of enzyme are added. If this is the case, RNase I digestion can be attempted at 4 °C. Digestion with RNase I does not require calcium. Thus, to quench an RNase I digestion, SUPERase•In should be used instead of EGTA.
Importantly, ribonucleases should be prepared in aliquots and stored at −80 °C upon receipt to ensure reproducibility, as freeze–thaw cycles can dramatically affect enzyme activity.
Sucrose gradient
To isolate mitochondrial ribosomes, digested lysates are loaded onto sucrose gradients. Sucrose gradients can be prepared the previous day and stored at 4 °C; if prepared on the same day, gradients should be allowed to settle at 4 °C for at least 1 h before samples are loaded onto them. We use the combined BioComp Gradient Master and Gradient Fractionator paired with a Model EM-1 Econo UV monitor to both prepare our sucrose gradients and analyze samples after ultracentrifugation. We use the Optima XE with the SW 41 Ti swinging-bucket rotor, both from Beckman Coulter, for ultracentrifugation.
All equipment used to prepare sucrose gradients must first be decontaminated with RNaseZap and thoroughly rinsed with water. This method separates 55S mitochondrial ribosomes from 80S cytosolic ribosomes. Both can be obtained in a single run (Fig. 2).
Fig. 2 |. Typical absorbance trace of sucrose gradient fractionation of lysates treated with micrococcal nuclease.
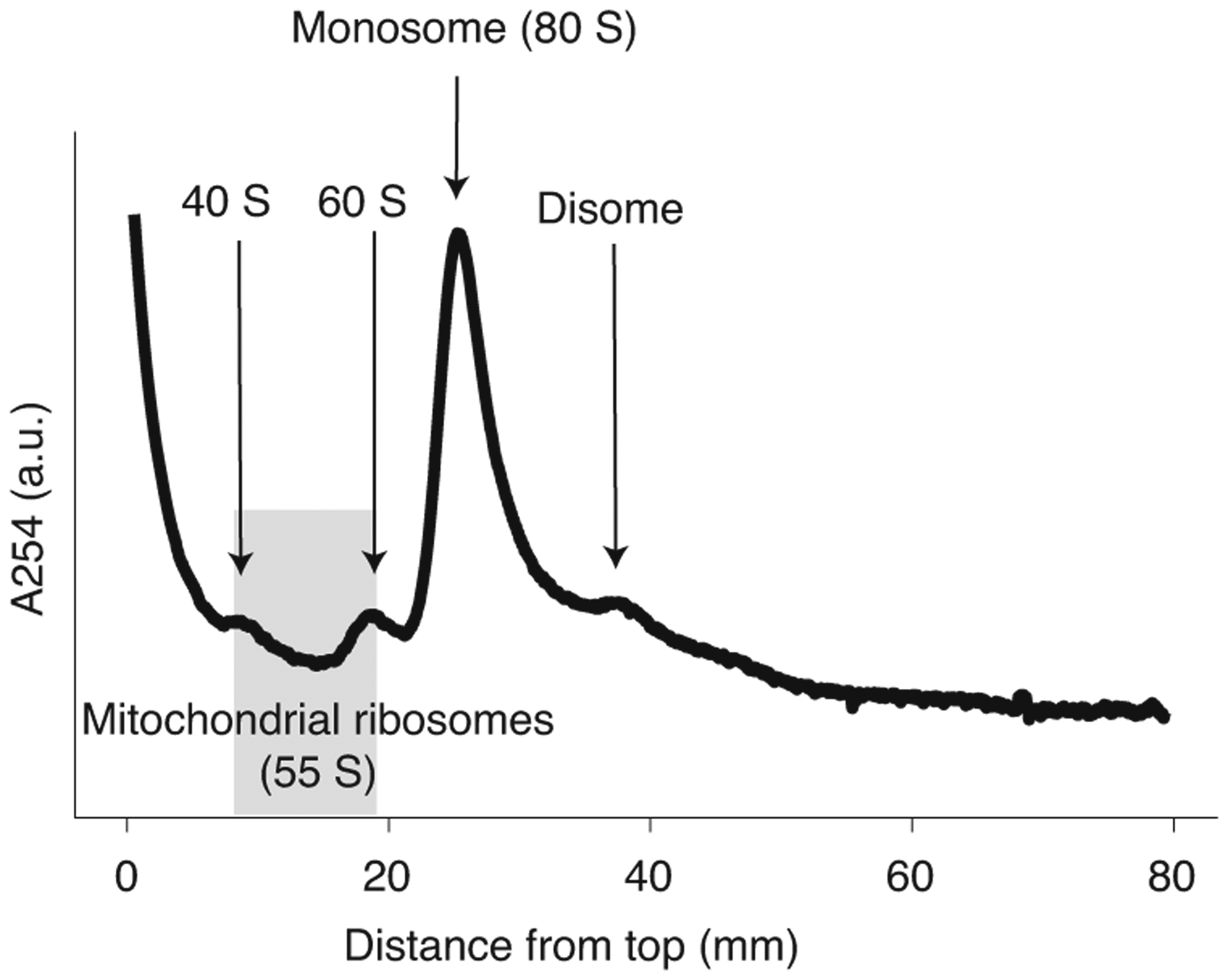
Because they are present at very low abundance relative to cytosolic ribosomes, 55S mitochondrial ribosomes do not appear as a peak but can be reliably isolated by collecting all material eluting between the 40S and 60S subunit peaks (highlighted in gray). a.u., arbitrary units.
The first time this protocol is attempted, we recommend confirming extraction of mitochondrial ribosomes by western blot (see Extended Data fig. 5 of Morscher et al. (ref. 31) for a detailed example).
Footprint size selection
The remaining steps required to sequence mitoribosome footprints are not unique to mitochondrial ribosomes. Rather, they are interchangeable with the corresponding steps of any ribosome profiling protocol. This section of our protocol is similar to the standard protocol from McGlincy and Ingolia33, with various modifications. In general, the protocol has little margin for error; mistakes can be irreparable. Thus, we recommend proceeding with only half of the purified sample from Step 38, and storing the other half at −80 °C as backup.
Footprint size selection is done with a standard denaturing 15% TBE-urea gel. There is some question about what range of RNA sizes to extract. Notably, in bacteria, stalled ribosomes have been reported to protect fewer nucleotides, resulting in shorter fragments than elongating ribosomes46. We have found that the most common footprint size is ~28 nt, but to avoid biasing libraries away from fragments of unexpected length, we recommend extracting a large size range of RNA fragments (15–45 nt, Fig. 3). Unwanted fragments can be removed during bioinformatic analysis. If a certain range of fragment sizes does not include any desired mRNA footprints, as determined experimentally in preliminary experiments, this size range can be excluded in future experiments.
Fig. 3 |. Typical result of a size selection gel for ribosome footprint samples digested with two different nucleases.
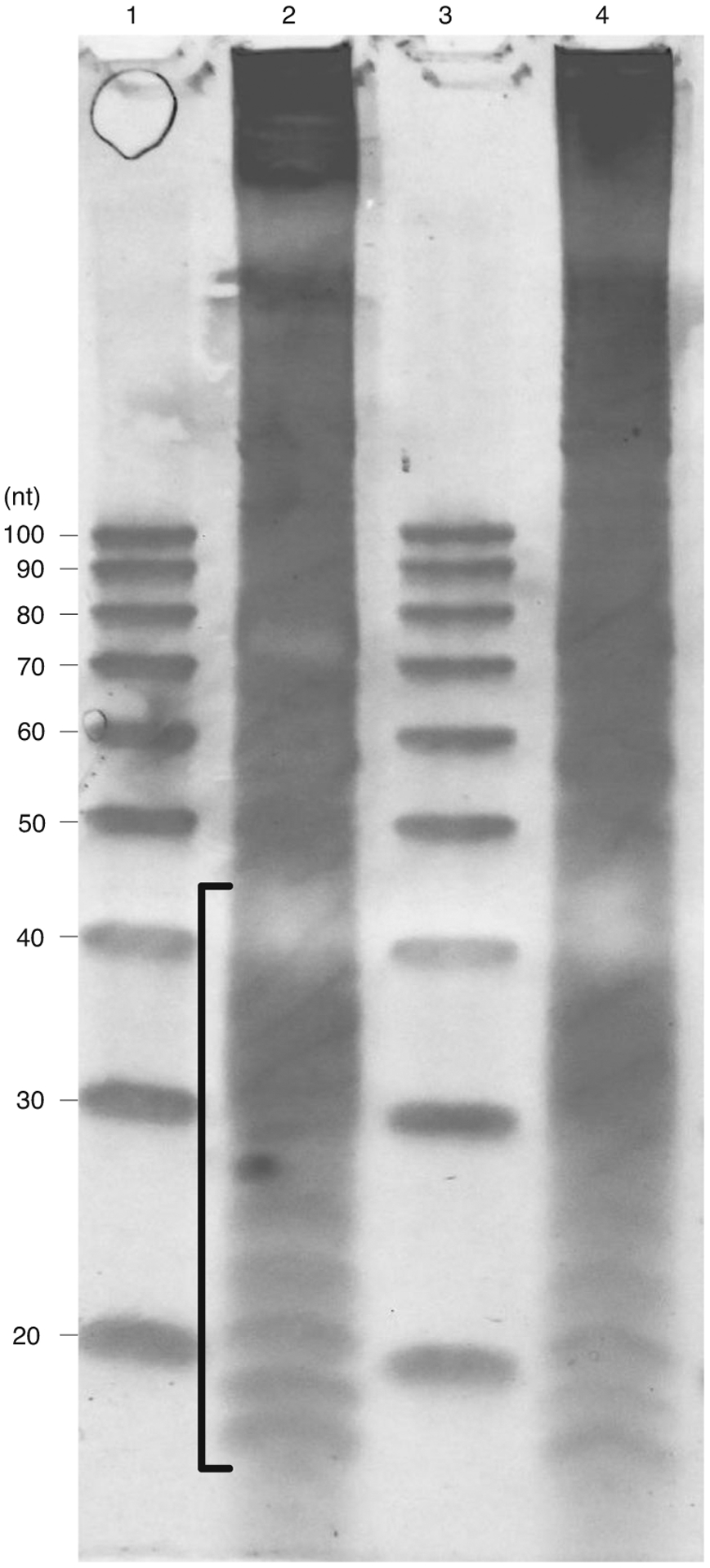
MNase digestion results. Lanes 1 and 3: 20/100 ladder (IDT) with sizes of each band annotated to the left; lanes 2 and 4: two separate ribosome footprint samples. The size of ribosome footprint fragments usually ranges from 15 to 45 nt, as indicated by the bracket for the target area to cut.
End repair and linker ligation
Ribonuclease-mediated hydrolysis yields RNA footprints with 2′,3′-cyclic phosphates that are incompatible with linker ligation. Thus, before linker ligation, an ‘end healing’ step is required, in which T4 polynucleotide kinase (PNK) transfers these 3′ phosphates to itself.
After end healing, the universal miRNA cloning linker and other ligation reaction components (including an engineered variant of T4 RNA ligase 2) are added directly to the first reaction. This linker has special features at both ends. At the 5′ end, the linker is preadenylated, allowing ligation of that end to the 3′ end of another oligonucleotide in the absence of ATP. This adenine is an RNA base; the rest of the bases are DNA. The 3′ base is blocked, preventing linker–linker ligation. Thus, linkers are specifically ligated to the 3′ end of RNA footprints without linker or footprint concatemerization. Others have introduced barcodes into these linkers, enabling pooling of the ligated products for the remainder of the library preparation protocol33.
After the linker ligation reaction, unligated linker is removed by the sequential actions of a 5′ deadenylase and a DNA 5′ to 3′ exonuclease (RecJf).
Assuming an RNA fragment size of 30 nt, the expected product of linker ligation would be 47 nt long; assuming an initial distribution of 15–45 nt, the expected product distribution would be 32–62 nt.
Reverse transcription, circularization and amplification
After linker ligation, RNA-DNA hybrids are reverse-transcribed, circularized and amplified following the standard protocol from McGlincy and Ingolia33. Libraries should be sufficiently amplified after 12 rounds of PCR; further amplification can introduce undesirable biases. If insufficient material is present, the procedure should be repeated with more starting material.
cDNA library quality control and sequencing
Before sequencing libraries are pooled, they should be quantified and analyzed by Bioanalyzer. If yields are high enough and size distributions are correct, libraries can be pooled and sequenced. We have found a single run on the HiSeq 2500 provides sufficient sequencing depth, even when several samples are multiplexed. However, before committing to a high-depth run, we recommend sequencing libraries with a low-depth MiSeq run to confirm appropriate library composition. Deep sequencing can be performed on more recent Illumina sequencers, such as the NovaSeq 6000. Long-read and single-molecule sequencers are not suitable for this application, however.
Single-end standard RNA sequencing
Standard mRNA sequencing might be required to quantify the total abundance of mitochondrial mRNAs. Specifically, mRNA abundances are required if gene-by-gene translation efficiency measurements are desired. On the other hand, total RNA sequencing is not required if one is only interested in the presence or absence of stalled ribosomes at specific codons. If total RNA sequencing is desired, total RNA should first be purified from ~100 μL clarified lysate; this can be achieved with the Direct-zol RNA Miniprep Plus kit, for example. RNA-sequencing libraries can then be prepared using standard methods. We recommend using unique barcoded primers to amplify these libraries so that they can be pooled with ribosome profiling sequencing libraries and sequenced together, if desired.
Bioinformatic analysis
Several bioinformatic tools have emerged for ribosome profiling analysis. Some of these can be accessed easily using the web-based platform Galaxy47 and its derivative RiboGalaxy48. The downstream analysis of mitochondrial ribosome profiling will vary depending on the goal of the study. Here we present our pipeline to generate the codon count table, which is the input for calculating codon occupancy and for plotting cumulative ribosome density along each gene. The codon count table consists of the counts of ribosome footprinting at each sub-codon position, from which we can perform codon occupancy analysis. This codon count table can also be useful for other analyses of interest. We perform this analysis using a Snakemake49 workflow that combines open source software and custom Python and R codes deposited on the GitHub repository (https://github.com/sophiahjli/MitoRiboSeq). Dependencies are installed using the Conda (https://docs.conda.io) package manager and the Bioconda50 package repository. The code for mitoribosome profiling analysis can also be used for other ribosome profiling data as long as proper required input files such as genome sequence files in FASTA format and annotation files in gene transfer format (GTF) or general feature format (GFF) are supplied.
Ribosome A-site determination
The Python package plastid51 is used to analyze fragment read length distribution and perform other conventional analyses such as phasing. A typical mitoribosome profiling experiment using MNase yields a broad length distribution centering around 28 nt. Given the sequence specificity of MNase, it is also known to result in ambiguous read frames and weak periodicity of reads46,52.
MNase is known to have sequence bias such that using the 5′ end to determine the A-site offset is unreliable53,54. Studies on E. coli have shown that mapping reads using the 3′ end gives a clear peak at the stop codon46,53. We have found that the same rule can be applied to mitoribosomes with slight modification. Mitochondrial transcription is polycistronic, and mitochondrial translation requires mRNA to be processed into individual genes55,56. Mitochondrial genes differ in their untranslated regions (UTRs) by length and position (5′ or 3′), and the length of poly(A) tail varies as well30,56. Among the 13 genes encoded in mitochondria, ND6 is reported to have a 3′-UTR, and ATP6 a 5′-UTR30. We thus used the Python package plastid for the 3′ end alignment to determine offset by using ND6 at the stop codon and ATP6 at the start codon. We found that there is a peak at 18 nucleotides and 15 nucleotides downstream from the first nucleotide of the start codon and stop codon, respectively. Given the ambiguous read frame determined by the MNase, we use an offset of 14 nucleotides for the A-site to capture the averaged footprints mapped to different read frames. While we found that this offset rarely changes in our experiments, it is advised to run the analysis and determine the offset for every experiment.
Materials
Biological materials
HCT116 cells were purchased from ATCC (CCL-247; https://scicrunch.org/resolver/CVCL_0291), and the HCT116 SHMT2 KO cell line was previously published31 and is readily available from the authors upon request.
Reagents
Cell lysis, ribosome footprinting and sucrose gradient ultracentrifugation
Nuclease-free water (Thermo Fisher Scientific, cat. no. AM9932)
Phosphate-buffered saline suitable for cell culture (Thermo Fisher Scientific, cat. no. 10010023)
Dimethyl sulfoxide (DMSO) suitable for cell culture (Sigma, cat. no. D2650)
1 M Tris-HCl pH 8.0 (Thermo Fisher Scientific, cat. no. AM9856)
1 M Magnesium chloride (MgCl2; Thermo Fisher Scientific, cat. no. AM9530G)
Potassium chloride (KCl; Sigma, cat. no. P9333)
Calcium chloride (CaCl2; Sigma, cat. no. C1016)
DTT (Thermo Fisher Scientific, cat. no. BP172)
Triton X-100 Surfact-Amps Detergent Solution (Thermo Fisher Scientific, cat. no. 28314) ! CAUTION The solution is irritable to eyes and harmful for the environment. Dispose the waste according to the institutional regulation.
NP-40 Surfact-Amps detergent solution (Thermo Fisher Scientific, cat. no. 28324) ! CAUTION The solution is irritable to eyes and harmful for the environment. Dispose the waste according to the institutional regulation.
DNase I (Roche, cat. no. 04716728001)
cOmplete, mini protease inhibitor cocktail (Roche, cat. no. 04693124001)
Chloramphenicol (Sigma, cat. no. C0378) ! CAUTION Chloramphenicol is a suspected carcinogen.
Cycloheximide (Sigma, cat. no. C1988) ! CAUTION Cycloheximide is highly toxic to the body and the environment. Handle it with care, and dispose of the waste according to the institutional regulation.
Micrococcal nuclease (nuclease S7; Roche, 10107921001) ! CAUTION Nuclease activity will affect ribosome footprinting efficiency. Store aliquots at −80 °C, and freeze–thaw only once.
EGTA (Sigma, cat. no. E3889)
Sucrose (Thermo Fisher Scientific, cat. no. AC419760010)
Acid phenol:chloroform (Thermo Fisher Scientific, cat. no. AM9722) ! CAUTION Phenol is very corrosive and will severely burn the skin. Use it in a fume hood with proper safety protection, and dispose the waste according to the institutional regulation.
Chloroform (Sigma, cat. no. 319988) ! CAUTION Chloroform is volatile and toxic. Use it in a fume hood with proper safety protection, and dispose of the waste according to the institutional regulation.
20% (wt/vol) SDS (Thermo Fisher Scientific, cat. no. AM9820)
Library preparation for sequencing
Isopropanol (Sigma, cat. no. 34863) ! CAUTION Isopropanol is highly flammable and volatile.
Ethanol (EtOH; Thermo Fisher Scientific, cat. no. BP2818100) ! CAUTION EtOH is highly volatile and flammable.
GlycoBlue (Invitrogen, cat. no. AM9515)
Glycogen (Invitrogen, cat. no. AM9510)
3 M Sodium acetate pH 5.5 100 mL (Thermo Fisher Scientific, cat. no. AM9740)
0.5 M EDTA pH 8.0 100 mL (Thermo Fisher Scientific, cat. no. AM9260G)
1 M Tris-HCl pH 7.0 (Thermo Fisher Scientific, cat. no. AM9851)
5 M NaCl (Thermo Fisher Scientific, cat. no. AM9759)
20/100 Ladder (IDT, cat. no. 51-05-15-02)
Ultra-low range DNA ladder (Thermo Fisher Scientific, cat. no. 10597012)
Gel loading buffer II (Ambion, cat. no. 8546G)
10× TBE buffer (BioRad, cat. no. 1610770)
15% TBE-urea gel 1.0 mm 12-well (Thermo Fisher Scientific, cat. no. EC68852BOX)
10% TBE-urea gel 1.0 mm 12-well (Thermo Fisher Scientific, cat. no. EC68752BOX)
8% TBE gel 1.0 mm 12-well (Thermo Fisher Scientific, cat. no. EC62152BOX)
SYBR Gold nucleic acid gel stain 500 μL (Thermo Fisher Scientific, cat. no. S11494)
T4 PNK (NEB, cat. no. M0201)
T4 RNA ligase 2, truncated KQ (NEB, cat. no. M0373)
5′ Deadenylase (NEB, cat. no. M0331S)
RecJf (NEB, cat. no. M0264L)
RNA Clean and Concentrator-5 (Zymo Research, cat. no. R1016)
CircLigase (Epicentre, cat. no. CL4111K)
Superscript III reverse transcriptase (Thermo Fisher Scientific, cat. no. 18080044)
dNTP mix (10 mM each) (Thermo Fisher Scientific, cat. no. R0191)
DNA Clean and Concentrator-5 (Zymo Research, cat. no. D4014)
Phusion high-fidelity PCR master mix with HF buffer (NEB, cat. no. M0531L)
Gel loading dye, purple 6× (NEB, cat. no. B7024S)
Direct-zol RNA Miniprep Plus (Zymo Research, cat. no. R2071)
High sensitivity DNA kit (Agilent Technologies, cat. no. 5067–4626)
Oligonucleotides
▲CRITICAL Oligonucleotide sequences are described using standard Integrated DNA Technologies (IDT) nomenclature. Notably, rApp at the 5′ end denotes 5′ adenylation, /5Phos/ denotes 5′ phosphorylation and /iSp18/ denotes an 18-atom hexa-ethyleneglycol spacer. All primers besides the universal miRNA cloning linker can be synthesized by IDT using standard desalting for purification. All primers should be dissolved in water at the appropriate concentration and stored at −20 °C.
Universal miRNA cloning linker, 5′ adenylated, 3′ blocked (NEB S1315S): (5′) rAppCTGTAGGCACCATCAAT-NH2 (3′)
Reverse transcription primer (IDT): /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT/ iSp18/CAAGCAGAAGACGGCATACGAGATATTGATGGTGCCTACAG
Forward library PCR primer (IDT): 5’-CAAGCAGAAGACGGCATACGA-3’
Indexed reverse library PCR primers (IDT): 5’ -AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACNN NNNNACACTCTTTCCCTACAC −3’, where NNNNNN indicates the reverse complement of the index sequence (Table 2) used during Illumina sequencing ▲CRITICAL We use barcodes from the Illumina TruSeq Small RNA Indices A (Table 2). When the plexity is only 2, use RPI6 and RPI12, as recommended by the supplier.
Table 2 |.
Barcodes used for the indexed reverse library PCR primers
| Index | Sequence | Reverse complement |
|---|---|---|
| RPI1 | ATCACG | CGTGAT |
| RPI2 | CGATGT | ACATCG |
| RPI3 | TTAGGC | GCCTAA |
| RPI4 | TGACCA | TGGTCA |
| RPI5 | ACAGTG | CACTGT |
| RPI6 | GCCAAT | ATTGGC |
| RPI7 | CAGATC | GATCTG |
| RPI8 | ACTTGA | TCAAGT |
| RPI9 | GATCAG | CTGATC |
| RPI10 | TAGCTT | AAGCTA |
| RPI11 | GGCTAC | GTAGCC |
| RPI12 | CTTGTA | TACAAG |
Equipment
Centrifuge rotor for 12 mL Seton tubes capable of reaching 35,000 rpm (max 210,000g, min 92,500g; SW 41 Ti, Beckman Coulter, part no. 331362)
Ultracentrifuge, Optima XE-100 (Beckman Coulter, cat. no. A94516)
Gradient station (BioComp, cat. no. 153) ▲CRITICAL Other gradient forming devices such as the gradient former by BioRad can be used. A simple homemade setup can also be used as published by Bellavia et al38. However, the method of freezing layers of sucrose and thawing should be avoided due to the fact that the ion concentrations are not homogeneous throughout the gradient. For gradient fractionation, the gradient fractionator by Brandel, Auto Densi-Flow by LabConco or a homemade setup can be good alternatives.
Model EM-1 Econo UV monitor (BioRad, cat. no. 7318160)
ChemiDoc XRS+ System with Image Lab (Bio-Rad, cat. no. 1708265)
Dark reader (Clare Chemical Research, cat. no. DR46B)
ThermoMixer R (Eppendorf, cat. no. 022670107)
Ultra-hard wear-resistant 440C stainless steel, diameter 12 mm (McMaster-Carr, cat. no. 1598K33)
Cryomill (Retsch, cat. no. 207490001)
10 mL jar stainless steel Cryomill (Retsch, cat. no. 14620331)
SureLock Xcell gel box (Thermo Fisher Scientific, cat. no. EI0001)
PowerEase 90 W (Thermo Fisher Scientific, cat. no. PS0091)
Qubit fluorometer
2100 Bioanalyzer instrument (Agilent, cat. no. G2939BA), 4200 TapeStation system (Agilent, cat. no. G2991BA) or equivalent
MiSeq System (Illumina, cat. no. SY-410-1003) for quality-control sequencing
HiSeq 2500 System (Illumina, obsolesced), NovaSea 6000 System (Illumina, cat. no. 20012850) or equivalent Illumina sequencer
Plastics and other consumables
150 × 20 mm tissue culture treated dish, sterile (Celltreat, cat. no. 229651)
Corning cell lifter (Corning, cat. no. 3008)
Fisherbrand sterile microcentrifuge tubes with screw caps (Thermo Fisher Scientific, cat. no. 02-681-375)
DNA LoBind tubes, 2 mL (Eppendorf, cat. no. 0030108078)
DNA LoBind tubes, 1.5 mL (Eppendorf, cat. no. 0030108051)
Insulin syringe with detachable 26 G needle, 1 mL (BD, cat. no. 329652)
Steriflip disposable vacuum filtration system, 50 mL (Millipore, cat. no. SE1M179M6)
Seton Scientific polyclear centrifuge tube (Thermo Fisher Scientific, cat. no. 7030)
Tips GelWell 0.57 mm 250 μL GT-250-6 (Mettler Toledo, cat. no. 17002379)
Technocut disposable scalpels 10 (HMD Healthcare, cat. no. 6008T-10)
Costar Spin-X centrifuge tube filter (Thermo Fisher Scientific, cat. no. 8160)
Software
For more details of installation, please refer to the GitHub page (https://github.com/sophiahjli/MitoRiboSeq).
Reagent setup
▲CRITICAL Solvents used should be RNase free. Use nuclease-free water, cell culture-grade PBS and cell culture-grade DMSO.
Chloramphenicol stock solution (50 mg/mL) in EtOH
Dissolve 100 mg in 95% (vol/vol) EtOH with total volume of 2 mL, store at −20 °C and protect from light. This stock solution should be used within 30 d.
Cycloheximide stock solution (50 mg/mL) in EtOH
Dissolve 100 mg in 95% (vol/vol) EtOH with total volume of 2 mL, store at −20 °C and protect from light. This stock solution should be used within 30 d.
Wash buffer
PBS with 100 μg/mL chloramphenicol and 100 μg/mL cycloheximide. Add antibiotics to wash buffer immediately before use. Prechill before extraction.
Buffer base (1×)
20 mM Tris-HCl pH 8.0, 100 mM KCl, 10 mM MgCl2, 100 μg/mL chloramphenicol, 100 μg/mL cycloheximide and 1 mM DTT. We recommend making a 2× buffer base as stock solution. Dilute to 1× and add antibiotics and DTT immediately prior to use.
Lysis buffer
Add 1% (vol/vol) Triton X-100, 0.1% (vol/vol) NP-40, 1× cOmplete protease inhibitor cocktail, 20 U/mL SUPERase•In and 20 U/mL DNase I to the buffer base. Prepare immediately prior to use.
Micrococcal nuclease (375 U/μL)
Dissolve the lyophilized enzyme (15,000 U) in 40 μL 10 mM Tris-HCl pH 8.0. Prepare aliquots into volumes appropriate for single use and store at −80 °C. It should be stable for over 2 months, but we have not tested its activity beyond this duration.
Sucrose solutions (5% and 45% (wt/vol))
Add 5 g or 45 g of sucrose to a concentrated buffer base (2× is recommended) into a 50 mL tube. After dissolving sucrose, top up with water to a final volume of 50 mL to make 1× buffer base. Prepare immediately prior to use.
RNA extraction buffer
300 mM sodium acetate (pH 5.5), 1 mM EDTA and 0.1% (wt/vol) SDS. The solution is stable at room temperature (RT; 22–25 °C).
DNA extraction buffer
10 mM Tris-HCl (pH 8.0), 300 mM NaCl and 1 mM EDTA. This solution is stable at RT.
20/100 Oligo ladder
Dissolve the vial with 100 μL 10 mM Tris-HCl pH 7.0. This solution is stable at RT.
Staining solution
1× TBE with a 10,000× dilution of SYBR Gold. Always use prechilled buffer to avoid diffusion of library from the gel. This solution should be made fresh.
Procedure
Cell lysis ● Timing 1 h
▲CRITICAL Our protocol is intended for adherent mammalian cells. Adjustments are required to accommodate cells growing in suspension. Once cells have reached the desired confluence (~80%), the monolayer is rapidly washed with ice-cold PBS and lysed with a buffer designed to stall ribosomes along mRNAs while keeping these ribosomes intact for long periods of time. We include both chloramphenicol and cycloheximide in our wash buffer and lysis buffer, in case paired analysis of cytosolic ribosome positioning is desired.
-
1
Grow mammalian cells to ~80% confluency in 15 cm culture plates. Ideally, the culture medium should not contain antibiotics (penicillin and streptomycin), which might interfere with translation.
-
2
Once cells have reached the desired confluency, aspirate the medium and wash with 10 mL of ice-cold wash buffer (PBS supplemented with cycloheximide and chloramphenicol).
-
3
Aspirate thoroughly, removing as much wash buffer as possible, and add 1 mL ice-cold lysis buffer (also supplemented with cycloheximide and chloramphenicol). Steps 2 and 3 should be performed as rapidly as possible to minimize translation elongation during lysis.
-
4
Transfer the plate to ice, collect the lysed cells using a cell lifter and transfer the lysate to a 2 mL screw-cap tube. The total lysate volume from one 15 cm dish will exceed 1 mL because of residual wash buffer, but should not exceed 1.6 mL.
-
5
Homogenize the cell lysate by passing it through a 26 G needle three times, taking care not to let the sample warm to RT. Avoid generating bubbles, and return the sample to ice whenever possible.
-
6
Pellet debris at 16,000g for 10 min in a precooled table-top centrifuge (4 °C).
-
7
Transfer clarified lysates to a new set of tubes. Cell lysates can be used immediately for nuclease digestion (recommended) or flash frozen in liquid nitrogen for storage at −80 °C.
■PAUSE POINT Samples can be stored at −80 °C for at least 1 week.
Footprinting with micrococcal nuclease ● Timing 2 h
-
8Prepare the following for nuclease digestion:
Reagent Amount Cell lysate (Step 7) 1 mL MNase [375 U/μL] 10 μL 1 M CaCl2 5 μL -
9
Incubate at RT with gentle shaking for 1 h.
-
10
Quench the reaction by adding 10 μL 0.5 M EGTA.
Mitochondrial ribosome enrichment ● Timing 4 h
-
11
Prepare 50 mL each of 5% and 45% (wt/vol) sucrose solutions.
-
12
Filter each sucrose solution using a 50 mL Steriflip filter. Allow each solution to degas under standard laboratory vacuum for 1 h, tapping the tubes occasionally to release air bubbles.
-
13
Mark the halfway point of each Seton centrifuge tube, using the taller edge of the BioComp marker block as a guide.
-
14
Using a blunt-end metal needle attached to a 50 mL syringe, dispense 5% sucrose up to slightly above the mark line.
-
15
Using the same needle and syringe, add 45% sucrose by carefully moving the needle to the bottom of the tube, then gently dispensing below the 5% sucrose, taking care not to disrupt the separation of the two layers.
-
16
Cap tubes with the rubber caps supplied with the BioComp Gradient Master. Avoid bubbles. Run the GradientMaker program for 5–45% linear sucrose gradients.
▲CRITICAL STEP Do not cool the sucrose solution before mixing, as temperature affects the viscosity and might affect gradient formation.
-
17
After the run, equilibrate the gradients to 4 °C for at least 1 h. We recommend preparing sucrose gradients the previous day, which allows the gradients to equilibrate overnight.
-
18
When digested lysates are ready to be loaded and sitting on ice, precool the ultracentrifuge and ultracentrifuge bucket to 4 °C.
-
19
Gently remove the caps of the sucrose solution tubes. Remove 1 mL of sucrose solution from each tube and add 950 μL of lysate. Balance the tubes such that the mass difference between each pair of tubes to be balanced is at most ~10 mg.
-
20
Spin at 35,000 rpm at 4 °C for 2 h.
-
21
Prepare the Biocomp fractionator by running water and sucrose gradient solution blanks. The 254 nm filter should be used, and the AUFS (Absorbance Units Full Scale) should be set to 2.
-
22
Collect all material eluting between the two visible 40S and 60S cytosolic ribosomal subunit peaks in a cryotube (700 μL to 1.2 mL), and immediately flash freeze the sample in liquid nitrogen.
■PAUSE POINT Samples can be stored at −80 °C for at least a week.
? TROUBLESHOOTING
Mitoribosome footprint extraction ● Timing 4 h
-
23
Thaw samples on ice.
-
24
For each sample, prepare an equal volume of acid-phenol:chloroform in a nuclease-free 2 mL Eppendorf tube. If any sample volume exceeds 700 μL, split the sample into two, extract RNA separately and then combine at the end. Preheat the tubes containing acid-phenol:chloroform to 65 °C.
! CAUTION Working with hot phenol and chloroform is dangerous. Handle only in a chemical hood with appropriate protection.
-
25
Once samples have thawed, add 57 μL 20% (wt/vol) SDS per 1 mL sample, and invert to mix.
-
26
Add each sample to the corresponding preheated tube with acid-phenol:chloroform.
-
27
Incubate at 65 °C for 5 min with vigorous shaking on a thermomixer (1,400 rpm).
-
28
Cool samples on ice for 5 min.
-
29
Spin samples at 16,000g for 2 min at 4 °C. The aqueous layer, which contains the RNA, will be on top. Transfer this layer to a fresh tube, taking care not to distrub the interface. If sample interfaces are disturbed, the 2 min spin can be repeated.
-
30
Add 600 μL chloroform, and vortex for 30 s at RT.
-
31
Spin at 16,000g for 1 min in a microfuge. Transfer top, aqueous layer to a fresh DNA LoBind 1.5 mL Eppendorf tube.
-
32
Precipitate by adding 2 μL GlycoBlue and 0.1 volumes 3 M NaOAc (pH 5.5) (e.g., 78 μL if sample volume is 700 μL), inverting to mix and then adding 750 μL isopropanol. Vortex to mix well.
-
33
Chill samples at −20 °C for 1 h (or at −80 °C overnight).
-
34
Spin at 16,000g for 30 min at 4 °C.
-
35
Add 800 μL ice-cold 80% (vol/vol) EtOH to wash the pellets. Ensure that the 80% EtOH is ice-cold, and work quickly, as small RNAs can solubilize in 80% EtOH.
-
36
Spin at 16,000g for 1 min at 4 °C.
-
37
Carefully remove all residual EtOH.
-
38
Air-dry pellets by leaving the tubes uncapped for ~15 min.
-
39
Resuspend pellets in 20 μL 10 mM Tris-HCl pH 7.0.
■PAUSE POINT The sample can be stored at −80 °C for at least 1 week.
Footprint size selection ● Timing 4 h
-
40
Prerun 15% (wt/vol) TBE-urea gel at 200 V for 15 min in 1× TBE.
-
41
For each sample, mix 10 μL of extracted RNA from Step 38 with 10 μL gel loading buffer II. In addition, prepare a single-strand DNA (ssDNA) ladder by mixing 1 μL 20/100 ladder with 24 μL nuclease-free water and 25 μL gel loading buffer II.
-
42
Denature the samples and the ladder at 80 °C for 90 s, then immediately return to ice.
-
43
Prior to loading any samples, thoroughly rinse the wells of the gel with a 200 μL pipette tip. Load the gel with 10 μL of 20/100 ladder and 20 μL of each sample, leaving an empty well between samples to prevent cross-contamination as the samples migrate. Multiple ladders can be loaded if helpful (it is not necessary for ladders to be separated from samples by an empty well).
-
44
Run the gel at 200 V for 65 min in 1× TBE buffer.
-
45
Stain the gel for ~3 min in prechilled staining solution (1× TBE with SYBR Gold).
-
46
Visualize the gel under a blue-light transilluminator, and excise gel slices between 15 nt and 45 nt, transferring each gel slice to a 2 mL DNA LoBind Eppendorf tube. If possible, we recommend also taking higher-quality images of the gel pre- and postexcision using the SYBR Gold (Safe) program on a BioRad ChemiDoc imager, for documentation purposes.
! CAUTION Strong blue light is harmful to the eyes. Wear appropriate protective goggles.
-
47
Extract the desired band from the gel pieces by first adding 700 μL RNA gel extraction buffer to each tube, then transferring all tubes to dry ice for 30 min.
-
48
Thaw samples overnight at RT with gentle mixing on a nutator. Overnight incubation can be avoided if desired, as most RNA is freed from the gel in the first few hours; if expected yields are high, 1–2 h can suffice.
-
49
Briefly centrifuge gel extractions to collect liquid at the bottom of the tube, and transfer liquid to Costar Spin-X centrifuge tubes. Centrifuge tubes at 12,000g for 3 min at 4 °C. This step removes any remaining gel fragments, which could potentially interfere with the subsequent enzymatic reactions.
-
50
Transfer the eluant to a 1.5 mL DNA LoBind Eppendorf tube.
-
51
Add 2 μL GlycoBlue, invert to mix, then add 750 μL 100% isopropanol. Vortex to mix.
-
52
Chill samples at −20 °C for 1 h (or at −80 °C overnight).
-
53
Centrifuge at 16,000g for 30 min at 4 °C.
-
54
Wash the pellet with 800 μL ice-cold 80% EtOH.
-
55
Air-dry the pellet, as residual EtOH can interfere with the subsequent enzymatic reactions.
-
56
Resuspend samples in 3.5 μL 10 mM Tris-HCl pH 7.0, and transfer to a new set of 1.5 mL DNA LoBind Eppendorf tubes.
■PAUSE POINT The sample can be stored at −80 °C for at least 1 week.
End repair and linker ligation ● Timing 6 h
-
57Combine the following components on ice. Pipette to mix, then briefly spin down contents to collect the reaction mixture at the bottom of the tube. Incubate at 37 °C for 1 h.
Reagent Amount (μL) Sample (Step 56) 3.5 10× PNK buffer without ATP 0.5 SUPERase•In 0.5 T4 PNK 0.5 -
58Directly add the following reagents to each sample. Pipette to mix, then briefly spin down contents to collect the reaction mixture at the bottom of the tube. Incubate at 22 °C for 3 h.
Reagent Amount (μL) 50% wt/vol PEG 8000 3.5 10× T4 RNA ligase 2 buffer 0.5 20 μM Universal miRNA cloning linker 0.5 T4 RNA ligase 2, truncated K227Q 0.5 -
59
Add 0.5 μL of 5′ deadenylase (NEB) and 0.5 μL of RecJf exonuclease (NEB), and incubate at 30 °C for 45 min.
-
60
Add 89 μL nuclease-free water to each sample, bringing the total volume to 100 μL.
-
61
Purify samples using the RNA Clean and Concentrator-5 kit, according to the manufacturer’s instructions.
-
62
Elute the sample by adding 10 μL 10 mM Tris-HCl pH 7.0 to the column, incubating for 1 min at RT and centrifuging for 1 min at 16,000g. Note if any part of the sample is precipitated on the plastic surface of the column (not on the membrane). Take care to resuspend the entire sample by resuspending the material on the side of the column with the elution buffer. The sample should be blue, as the GlycoBlue from the previous precipitation has not yet been removed.
-
63
Transfer the eluant to a new 1.5 mL DNA LoBind Eppendorf tube.
-
64
Move on to the next step with 5 μL of each sample. Store the rest at −80 °C.
■PAUSE POINT The sample can be stored at −80 °C for at least 1 week.
Reverse transcription and size selection ● Timing 1 h for reverse transcription + 4 h for size selection
-
65
Transfer samples to PCR tubes, and add 2 μL 1.25 μM reverse transcription primer to each. Pipette to mix.
-
66
Denature at 65 °C for 5 min, and rapidly return to ice.
-
67Add the following components to each sample. Pipette to mix.
Reagent Amount (μL) Final concentration 5× First-strand buffer 4 1× dNTP mix 1 0.5 mM each 0.1 M DTT 1 5 mM SUPERase•In (20 U/μL) 1 1 U/ μL Superscript RT III (200 U/μL) 1 10 U/ μL -
68
Incubate at 55 °C for 30 min in a thermocycler.
-
69
Add 2.2 μL 1 M NaOH, and incubate at 70 °C for 20 min to hydrolyze the RNA template.
-
70
Prerun a 10% (wt/vol) TBE-urea gel at 200 V for 15 min in 1× TBE.
-
71
Add 23 μL gel loading buffer II to each sample. Also prepare a 20/100 ssDNA ladder. Denature the samples and ladder at 80 °C for 90 s.
-
72
Thoroughly rinse the wells, then add the samples and ladder onto a denaturing 10% TBE-urea gel. Because of the large sample volume, add each sample into two adjacent wells.
-
73
Run at 200 V for 80 min in 1× TBE buffer.
-
74
Excise the reverse-transcribed products, which, assuming an initial distribution of 15–45 nt, should be between 89–119 nt (lane 2, Fig. 4).
? TROUBLESHOOTING
-
75
Extract the cDNA as described in Steps 47–56, but use DNA extraction buffer instead of RNA extraction buffer.
-
76
Resuspend cDNA in 15 μL 10 mM Tris-HCl pH 8.0.
■PAUSE POINT The sample can be stored at −80 °C for at least 1 month.
Fig. 4 |. A typical result of a cDNA gel for ribosome footprint samples.
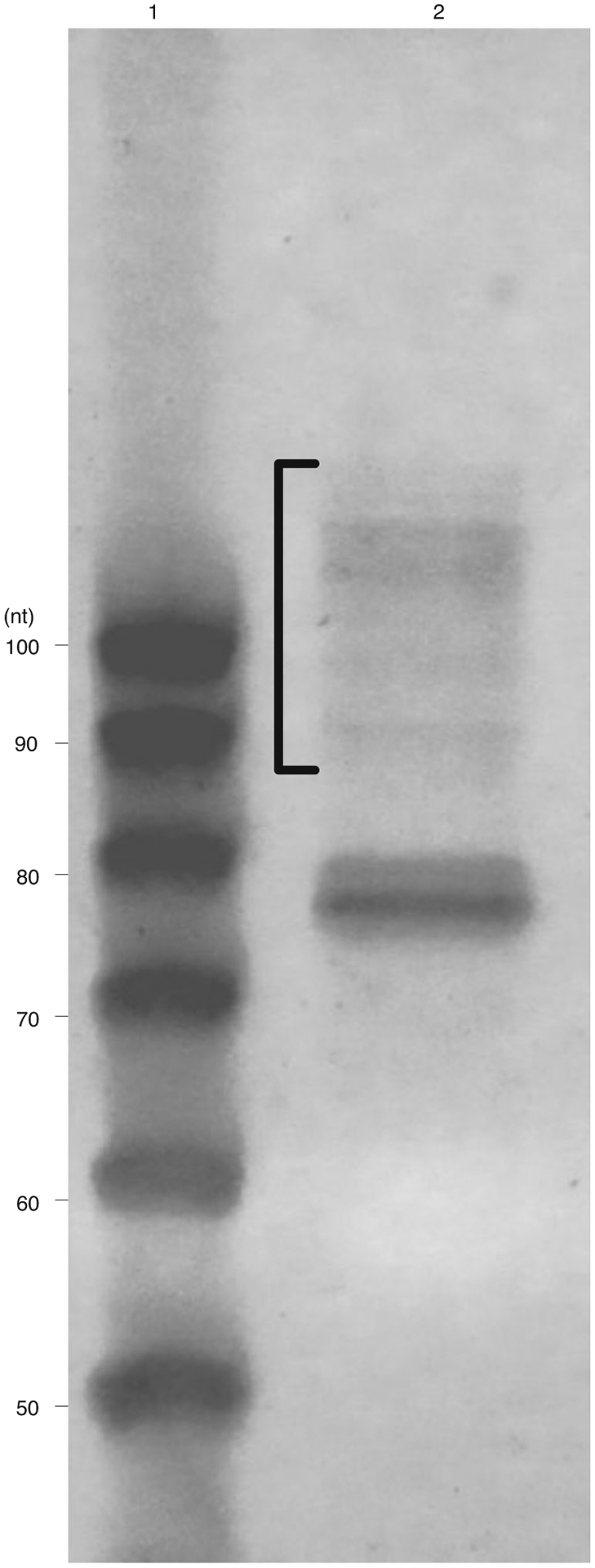
The region highlighted by the bracket is the target reverse transcribed cDNA product size. Lane 1: 20/100 ladder (IDT); lane 2: Reverse-transcribed ribosome footprint sample; the oligonucleotide at 74 nt is the reverse transcription primer.
Circularization ● Timing 2 h
-
77Mix the following in a PCR tube:
Reagent Amount (μL) Final concentration Sample 15 - 10× CircLigase buffer 2 1× 1 mM ATP 1 50 μM 50 mM MnCl2 1 2.5 mM CircLigase (100 U/μL) 1 5 U/μL Total 20 -
78
Incubate at 60 °C for 2 h, then inactivate the enzyme at 80 °C for 10 min.
■PAUSE POINT The sample can be stored at −20 °C for weeks, or at −80 °C for longer. Note that cDNA is much more stable than RNA.
PCR and size selection ● Timing 5 h
▲CRITICAL Circularized libraries must be amplified to produce sufficient material for sequencing, but PCR-based amplification is known to introduce sequence biases. To mitigate this bias, libraries should be amplified only as much as required. Thus, we recommend performing several PCR reactions for each sample, varying the number of amplification cycles between 8 and 14, then running the products of these reactions on a gel and extracting only the amplified library with the desired yield without unspecific amplification. A clean band like the one shown in lane 3 of Fig. 5 is desired.
Fig. 5 |. A typical result of a size selection gel for PCR products.
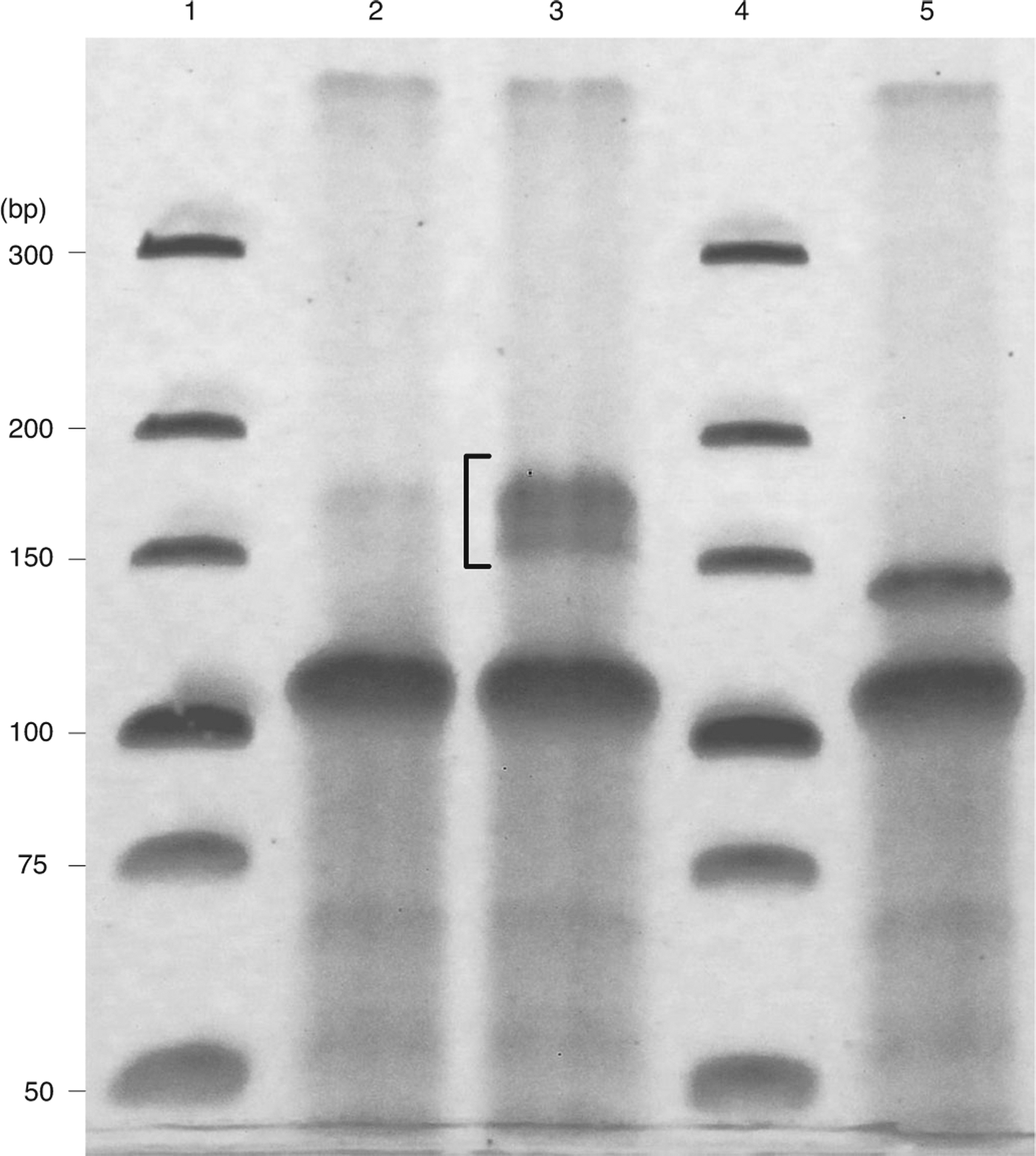
Lanes 1 and 4: Invitrogen ultra-low range DNA ladder; lanes 2 and 3: PCR amplified products using 5× diluted template (2) and undliluted template (3) amplified for nine cycles. The bracket marks the desired product size. Lane 5: PCR amplified linker-only products.
-
79Prepare a PCR master mix as follows:
Reagent Amount (μL) Final concentration 2× Phusion high-fidelity PCR master mix with HF buffer 40 1× 10 μM Forward library PCR primer 4 0.5 μM 10 μM Indexed reverse library PCR primer 4 0.5 μM Nuclease-free H2O 27 - Total 75 -
80
Add 5 μL template cDNA from Step 77 to the master mix. Use a different indexed reverse primer for each sample.
-
81
Distribute the master mix into four separate reactions on separate PCR strips.
-
82Run the following program on a thermocycler, repeating Steps 2–4 13 times (14 cycles total).
No Step Temperature Duration 1 Initial denaturation 98 °C 1 min 2 Denature 98 °C 20 s 3 Annealing 60 °C 20 s 4 Extension 72 °C 10 s 5 Final extension 72 °C 3 min -
83
At the end of cycle 8, remove the first PCR strip. Remove the second after cycle 10 and the third after cycle 12.
-
84
Add 4 μL of 6× DNA loading dye (nondenaturing) to each PCR reaction. Also prepare a ladder by combining: 0.5 μL 10 bp ultra-low range DNA ladder, 16 μL 10 mM Tris-HCl pH 8.0 and 3.5 μL 6× DNA loading dye.
-
85
Load all of each sample and 3.5 μL ladder on an 8% (wt/vol) TBE gel.
-
86
Run the gel at 200 V for 40 min in 1× TBE buffer.
-
87
Excise the amplified library from the first reaction where a clear band is present. A 30 nt footprint should produce a 172 nt cDNA, and 15–45 nt library should produce amplified cDNAs 157–187 nt long (Fig. 5). Self-ligated universal miRNA cloning linker and self-circularized reverse transcription primer produce products lower than 150 nt (Fig. 5, lane 5). Take care to exclude these bands when extracting libraries.
? TROUBLESHOOTING
-
88
Extract DNA as described in Step 75 using glycogen in place of GlycoBlue, as GlycoBlue can interfere with library quantification.
-
89
Resuspend in 11 μL 10 mM Tris-HCl pH 8.0.
■PAUSE POINT The sample can be stored at −80 °C for at least 1 month.
cDNA library quality control and sequencing
-
90
Quantify cDNA library concentrations using a Qubit fluorometer. Each sample should be roughly 1 ng (5 ng is required for deep sequencing after pooling). If yield is not sufficent, libraries should be reamplified with more amplification.
-
91
Analyze library size distributions on an Agilent 2100 Bioanalyzer. Libraries should be between 157 and 187 bp (Fig. 6).
-
92
Pool libraries such that each sample is present at the same concentration.
-
93
(Optional) Before running a high-depth sequencing run, sequence libraries on an Illumina MiSeq to confirm appropriate library composition.
-
94
Sequence on an Illumina HiSeq 2500 or NovaSeq 6000 in rapid mode for 75 cycles (single-end reads).
Fig. 6 |. A typical result of a MitoRiboSeq library quality control assessment using the high sensitivity DNA assay on an Agilent 2100 Bioanalyzer.
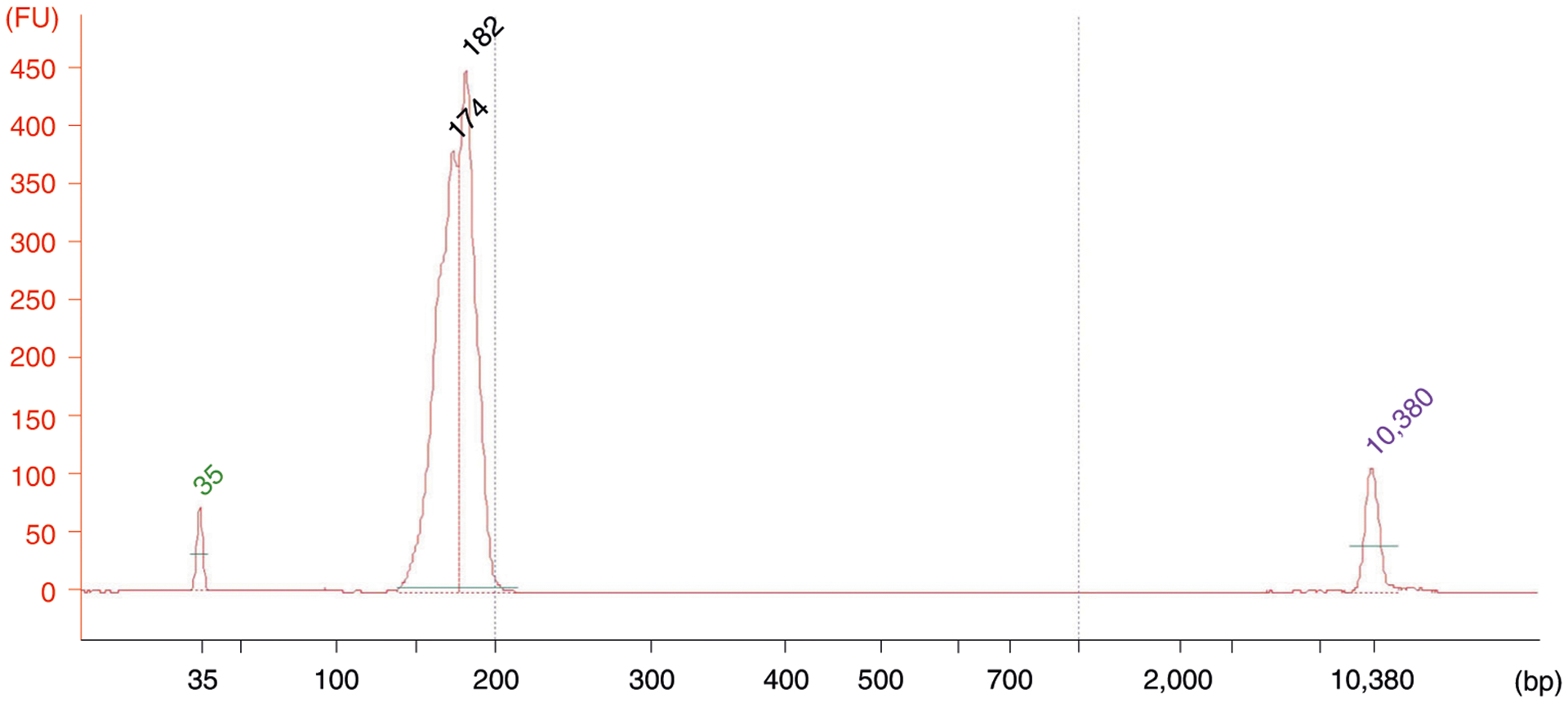
Libraries should be between 157 and 187 bp. The peaks at 35 bp and 10380 bp are standards.
Processing and aligning the sequencing data
-
95
The analysis starts with a FASTQ file generated from the sequencer. The mito_readphasing_metagene Snakemake pipline provided in the MitoRiboSeq package will process the samples pooled according to Steps 92–94. In a pooled run, first split each sample by the index barcodes used for each sample during PCR. Next, trim off the common 3′ adapter sequence CTGTAGGCACCAT CAATATCTCGTATGCCGTCTTCTGCTTG using Cutadapt57.
-
96
Align the processed sequences to the whole human genome assembly Ensembl GRCh38.p1358, which contains the mitochondrial genome, using Burrows-Wheeler Aligner (BWA)59. Corresponding QC files are summarized and output into a single report by MultiQC60. We have seen that many MitoRiboSeq reads do not uniquely map to the mitochondrial genome; they also map to sites in the nuclear genome. We do not exclude these nonuniquely mapped reads, reasoning that the mitoribosome enrichment step likely removed most cytosolic mRNAs associated with cytosolic ribosomes.
? TROUBLESHOOTING
Quality check and determination of 3′-offset to infer mitoribosome A-site
-
97
Determine the ribosome A-site by aligning the footprints’ 3′-end to the stop codon of ND6 and the start codon of ATP6.
Generation of codon count tables
-
98
After the quality check and offset determination, generate a codon count table using the aligned BAM file (from Step 92), a genome reference sequence in FASTA format and a genome annotation in GFF. To remove extraneous reads, we only use mapped reads between 15 and 45 bp long in the codon count calculations. The codon count table outputs the mitochondrial A-site counts from each codon of each gene into independent rows. The same pipeline also outputs the read coverage across the genome in bedgraph formatted files for visualization using a genome browser such as the Integrative Genomics Viewer61.
Analysis of codon occupancy and cumulative ribosome counts along the transcript
-
99
Calculate the occupancy by comparing the counts of codons from all genes with the expected counts. Codon occupancy analysis reveals any biased ribosome stalling at a particular codon, which might be associated with translational regulation or defects. We define codon occupancy as the ratio of measured ribosome counts to the expected, which is dependent only on the codon frequency. The ratio should center around one if there is no ribosome stalling. The mito_codontable Snakemake pipline provided in the MitoRiboSeq package takes input from the codon count table in txt format and outputs a text file containing occupancy values and a plot in PDF format. In the same pipeline, we also output the cumulative ribosome count plot, which reveals the ribosome distribution along the transcript (see ‘Anticipated results’). Codon occupancy and the cumulative ribosome count plots are generated using R62 and the R packages: tidyverse63, pheatmap64, ggrepel65 and cowplot66.
Troubleshooting
Troubleshooting advice can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 22 | No mitoribosome peak appears in UV absorption | Mitoribosomes exist at much lower abundance than cytosolic ribosomes. Sometimes the mitoribosome peak in UV absorption is not apparent | Collect the fractions between the 40S and 60S cytosolic ribosome subunit peaks and extract RNA from all of them, or check for mitochondrial ribosomes by western blot |
| 74 | No reverse transcription product observed in gel | Depending on sample input and technique during library preparation, the amount of sample can be very low at this stage. Nevertheless, even if a band is not visible, there might be enough cDNA to be amplified by PCR | Proceed by cutting the desired size range |
| 87 | No targeted band observed in PCR gel | Sample input at the start of the procedure might be too low, or the sample was lost during a previous step | If this continues to occur, we recommend increasing sample input and analyzing sample loss at each stage to identify the problem |
| 93 | Issues in mapping and computation | - | Refer to the GitHub page for some common problems in the bioinformatics analysis |
Timing
Steps 1–7, cell lysis: 1 h
Steps 8–10, micrococcal nuclease digestion: 2 h
Steps 11–22, mitochondrial ribosome enrichment: 4 h
Steps 23–39, mitoribosome footprint extraction: 4 h
Steps 40–56, footprint size selection: 4 h
Steps 57–64, end repair and linker ligation: 6 h
Steps 65–76, reverse transcription and size selection: 5 h
Steps 77–78, circularization: 2 h
Steps 79–89, PCR and size selection: 5 h
Steps 90–94, cDNA library quality control and sequencing: 1–8 d
Steps 95–99, data analysis: 1–3 d
Anticipated results
To demonstrate the protocol outlined above, we performed MitoRiboSeq on wild-type (WT) and serine hydroxymethyl transferase 2 (SHMT2)-knockout (KO) HCT116 human colon cancer cells. SHMT2 is a mitochondrial enzyme (encoded in the nuclear genome) that transfers one carbon from serine to a folate carrier. Morscher et al.31 used a variant of this protocol to demonstrate that such serine-derived one-carbon units are required to methylate mitochondrial tRNAs and thus sustain mitochondrial translation. Here, we reproduce the key experiments in that study with our most up-to-date version of the method. There are many ways to report the results of ribosome profiling (Fig. 7). One can analyze the data by codon, for example. Some codons occur more frequently than others in the mitochondrial genome; this is true, of course, for both WT and SHMT2 KO cells. After normalizing for codon abundance, the SHMT2-dependent differences emerge. In SHMT2 KO cells, mitoribosomes stall at codons that ‘wobble’ base-pair to tRNAs that are methylated at the wobble position, such as UUG and AAG (Fig. 7a,b). Importantly, such results might not be quantitatively robust, and in general, ribosome profiling is a method that mostly has enabled qualitative discoveries. However, the specific codons and even the specific stall sites (Fig. 7c,d) are reproducible from experiment to experiment. Curiously, stalling occurs at the codon expected and sometimes at one or two codons downstream; it remains to be seen whether this observed stalling represents true stalling within cells, or whether mitoribosomes are shifting during extraction or digestion. Overall, stalling at codons encoding amino acids is comparable to the natural pausing at stop codons, which is seen also in healthy mitochondria. Importantly, we find that MitoRiboSeq is reproducible between biological replicates, particularly for reads per gene (aggregated over all codons) and reads per codon (aggregated over all genes), but we typically do not achieve coverage sufficient to yield reproducible results at each specific mitochondrial codon. To achieve that, further methodological enhancements will be required.
Fig. 7 |. Typical analysis results of MitoRiboSeq from HCT116 cells in wild type (WT) and SHMT2 KO mutant.
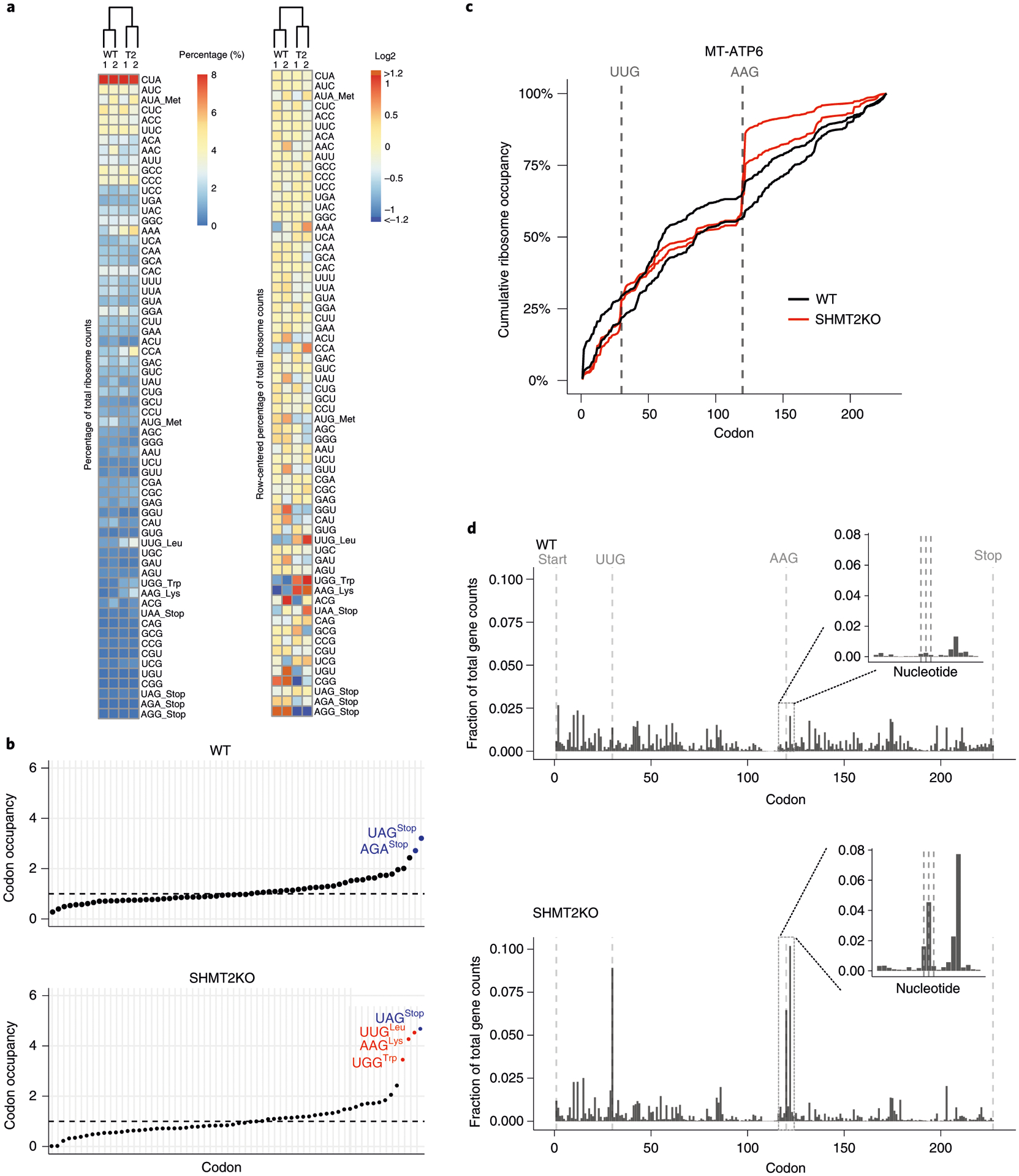
a, Clustering results of codon counts. The columns represent biological replicates and conditions, while the rows are organized by decreasing codon frequency in the mitochondrial genome. The heat map on the right is row-mean centered. b, Codon occupancy of wild type and SHMT2 KO. The colors indicate the codon type. Blue: stop codon; red: stalling sites. c, Normalized cumulative ribosome counts along the transcript of MT-ATP6 from two biological replicates from each condition. The vertical lines highlight the AAG and UUG codons. d, Fraction of codon counts along the transcript of MT-ATP6. The vertical lines highlight the start, stop, AAG and UUG codons. The inlet highlights the ribosome counts at nucleotide level around the AAG.
Acknowledgements
We thank R. J. Morscher for his help with establishing the initial MitoRiboSeq method. We thank the Gitai lab and the Rabinowitz lab for their comments and suggestions. We thank S. Vianello for his support with the artistic illustration of the protocol workflow. This work is supported by funding to J.D.R. from the US National Institutes of Health (NIH) (R01CA163591 and DP1DK113643) and StandUp to Cancer (SU2CAACR-DT-20-16). Z.G. was supported by the NIH (DP1AI124669).
Footnotes
Code availability
All related codes required for making the plots shown in this protocol are provided on the GitHub page at https://github.com/sophiahjli/MitoRiboSeq. These files are sufficient for readers to reproduce the bioinformatics results shown in this manuscript. The user can use the readme file with a step-by-step guide to set up the computation environment and to run the codes.
Competing interests
The authors declare no competing interests.
Data availability
The raw FASTQ data files are deposited at NCBI’s Sequence Read Archive (SRA). The sequencing results used in the bioinformatics analysis are available under BioProject PRJNA590503 and SRA accession numbers SRR10491343, SRR10491342, SRR10491341 and SRR10491340.
References
- 1.Spinelli JB & Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol 20, 745–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernández-Vizarra E, Tiranti V & Zeviani M Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim. Biophys. Acta 1793, 200–211 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Fox TD Mitochondrial protein synthesis, import, and assembly. Genetics 192, 1203–1234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvo SE & Mootha VK The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. G 11, 25–44 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greber BJ & Ban N Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem 85, 1–30 (2015). [DOI] [PubMed] [Google Scholar]
- 6.DiMauro S & Schon EA Mitochondrial respiratory-chain diseases. N. Engl. J. Med 348, 2656–2668 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Boczonadi V & Horvath R Mitochondria: impaired mitochondrial translation in human disease. Int. J. Biochem. Cell Biol 48, 77–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rötig A Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta 1807, 1198–1205 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Keilland E et al. The expanding phenotype of MELAS caused by the m.3291T > C mutation in the MT-TL1 gene. Mol. Genet. Metab. Rep 6, 64–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirino Y, Goto Y, Campos Y, Arenas J & Suzuki T Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl Acad. Sci. USA 102, 7127–7132 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Škrtić M et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace DC Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dykens JA & Will Y The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 12, 777–785 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Kalghatgi S et al. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci. Transl. Med 5, 192ra85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingolia NT, Ghaemmaghami S, Newman JRS & Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji Z, Song R, Regev A & Struhl K Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife 4, e08890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jan CH, Williams CC & Weissman JS Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams CC, Jan CH & Weissman JS Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 346, 748–751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loayza-Puch F et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Li G-W, Burkhardt D, Gross C & Weissman JS Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia NT, Lareau LF & Weissman JS Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Juntawong P, Girke T, Bazin J & Bailey-Serres J Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl Acad. Sci. USA 111, E203–E212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itzhak DN, Tyanova S, Cox J & Borner GH Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5, e16950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf SF & Schlessinger D Nuclear metabolism of ribosomal RNA in growing, methionine-limited, and ethionine-treated HeLa cells. Biochemistry 16, 2783–2791 (1977). [DOI] [PubMed] [Google Scholar]
- 27.Amunts A, Brown A, Toots J, Scheres SHW & Ramakrishnan V The structure of the human mitochondrial ribosome. Science 348, 95–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kummer E et al. Unique features of mammalian mitochondrial translation initiation revealed by cryo-EM. Nature 560, 263–267 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Montoya J, Ojala D & Attardi G Distinctive features of the 5′-terminal sequences of the human mitochondrial mRNAs. Nature 290, 290465a0 (1981). [DOI] [PubMed] [Google Scholar]
- 30.Temperley RJ, Wydro M, Lightowlers RN & Chrzanowska-Lightowlers ZM Human mitochondrial mRNAs—like members of all families, similar but different. Biochim. Biophys. Acta 1797, 1081–1085 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morscher RJ et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingolia NT, Brar GA, Rouskin S, McGeachy AM & Weissman JS The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGlincy NJ & Ingolia NT Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson GE & Li G-W Genome-wide quantitation of protein synthesis rates in bacteria. Methods Enzymol. 612, 225–249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rooijers K, Loayza-Puch F, Nijtmans LG & Agami R Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat. Commun 4, 2886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couvillion MT & Churchman LS Mitochondrial ribosome (mitoribosome) profiling for monitoring mitochondrial translation in vivo. Curr. Protoc. Mol. Biol 119, 4.28.1–4.28.25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couvillion MT, Soto IC, Shipkovenska G & Churchman LS Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearce SF et al. Maturation of selected human mitochondrial tRNAs requires deadenylation. eLife 6, e27596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos DA, Shi L, Tu BP & Weissman JS Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res. 47, 4974–4985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones CN, Miller C, Tenenbaum A, Spremulli LL & Saada A Antibiotic effects on mitochondrial translation and in patients with mitochondrial translational defects. Mitochondrion 9, 429–437 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Datta AK & Burma DP Association of ribonuclease I with ribosomes and their subunits. J Biol. Chem 247, 6795–6801 (1972). [PubMed] [Google Scholar]
- 42.Busch JD et al. MitoRibo-Tag mice provide a tool for in vivo studies of mitoribosome composition. Cell Rep. 29, 1728–1738.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culviner PH, Guegler CK & Laub MT A simple, cost-effective, and robust method for rRNA depletion in RNA-sequencing studies. mBio 11, e00010–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sendoel A et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature 541, 494–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riba A et al. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc. Natl Acad. Sci. USA 116, 15023–15032 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohammad F, Green R & Buskirk AR A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8, e42591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afgan E et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michel AM et al. RiboGalaxy: a browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 13, 316–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Köster J & Rahmann S Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 34, 3600–3600 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Grüning B et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dunn JG & Weissman JS Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang J-Y & Buskirk AR A ribosome profiling study of mRNA cleavage by the endonuclease RelE. Nucleic Acids Res. 45, 327–336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woolstenhulme CJ, Guydosh NR, Green R & Buskirk AR High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oh E et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147, 1295–1308 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taanman J-W The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123 (1999). [DOI] [PubMed] [Google Scholar]
- 56.D’Souza AR & Minczuk M Mitochondrial transcription and translation: overview. Essays Biochem. 62, 309–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 58.Yates AD et al. Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H & Durbin R Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ewels P, Magnusson M, Lundin S & Käller M MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Core Team. R: a language and environment for statistical computing. https://www.r-project.org
- 63.Wickham H et al. Welcome to the tidyverse. J. Open Source Softw 4, 1686 (2019). [Google Scholar]
- 64.Kolde R pheatmap: pretty heatmaps. https://rdrr.io/cran/pheatmap/
- 65.Slowikowski K ggrepel: automatically position non-overlapping text labels with “ggplot2” (2020).
- 66.Wilke CO cowplot: streamlined plot theme and plot annotations for “ggplot2” (2019).
- 67.Gao F et al. Using mitoribosomal profiling to investigate human mitochondrial translation. Wellcome Open Res. 2, 116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw FASTQ data files are deposited at NCBI’s Sequence Read Archive (SRA). The sequencing results used in the bioinformatics analysis are available under BioProject PRJNA590503 and SRA accession numbers SRR10491343, SRR10491342, SRR10491341 and SRR10491340.