

Deletions of 2p11.2-p12 are exceedingly rare with few reported cases.1,2 Most patients display a mild-to-moderate developmental delay and intellectual disability. Additional manifestations are happy disposition, tendency to obesity, and minor dysmorphic features, such as short stature, prominent forehead, hypertelorism, broad nasal bridge, or large low-set ears. Occasionally congenital malformations are present, such as chest and spine abnormalities, urogenital malformations, or atrial septal defect.1 In this study, we report a patient presenting with early-onset atypical parkinsonism carrying a heterozygous 3.9-Mb deletion on 2p11.2.
The proband, a 35-year-old White woman, was referred to our clinic for further study of parkinsonism. She reported a 2-year history of tremor, generalized stiffness, and progressive gait difficulty. She is the second child of nonconsanguineous parents. She was born preterm by cesarean section because of transverse presentation. Psychomotor development was slightly delayed, but she achieved all milestones without assistance. Medical history was significant for strabismus, amblyopia, and ear surgery for prominent ears. Scoliosis was diagnosed at age 12 years and treated with a brace.
At age 35 years (Video 1), she displayed mild dysmorphic features, such as short stature, high forehead, and mandibular prognathism. Facial expression was markedly reduced with lower face dystonic gesture. Smooth pursuit was impaired, and saccades were slow and inaccurate. Speech was hypophonic and dysarthric. She exhibited high-frequency, low-amplitude tremor, probably with a superimposed distal jerky compound, and severe bradykinesia and rigidity. Dystonic postures emerged with arms outstretched. Inferior limb spasticity, brisk reflexes, and bilateral ankle clonus were observed. Plantar reflexes were equivocal. Gait was unsteady with bilateral flexed-elbow deformity and absent arm swing. When walking, truncal hyperextension and a tendency to invert the left foot appeared. Neuropsychological evaluation was normal. She did not report REM sleep behavior disorder, anosmia, or autonomic symptoms. Dopamine agonists and trihexyphenidyl were not beneficial, but she achieved partial improvement with carbidopa/levodopa 250 mg qid. This dose was stable for the past 5 years without the development of motor fluctuations or dyskinesias. As the disease progressed, foot dystonia worsened, and she received botulinum toxin injections.
Segment 1: Face is markedly hypomimic. Dysmorphic features are evident (short stature, frontal bossing, and mandibular prognathism). She displays postural and intentional tremor, and distal dystonic postures emerge with arms outstretched. Bradykinesia is severe, with left side being more affected. Gait is unstable, with truncal hyperextension, absent arm swing, and flexed-elbow deformity. Segment 2: Smooth pursuit is saccadic. Horizontal and vertical saccades are slow and inaccurate. Speech is hypophonic and dysarthric, but intelligible. Segment 3: Tendon reflexes are easily elicited, with bilateral ankle clonus. Alternating hand movements are slow but precise.Download Supplementary Video 1 (18.6MB, mp4) via http://dx.doi.org/10.1212/00642_Video_1
A thorough laboratory investigation was conducted, including thyroid stimulating hormone, B12 and E vitamins, anti-nuclear antibodies, serum copper, ceruloplasmine, 24-hour urine copper, acanthocytes, plasma amino acids, alpha-fetoprotein, homocysteine, cholestanol, and glucocerebrosidase and sphingomyelinase enzymatic activities, without findings. A nerve conduction study was unremarkable. Brain and spine MRI findings were normal (Figure, A). Presynaptic nigrostriatal imaging with 123-ioflupane confirmed presynaptic denervation (Figure, B). Genes associated with parkinsonism were first screened (PANK2, ATP13A2, PLA2G6, FBXO7, SPG11, PRKRA, ATP1A3, and SLC6A3) using the Ion AmpliSeq Neurological Research Panel (Thermo Fisher Scientific, Waltham, MA) and MAPT by Sanger sequencing. Copy number variations (CNVs) were then investigated by multiplex ligation-dependent probe amplification analysis (MLPA) (SALSA P051/P052 Parkinson, MRC Holland, Amsterdam, the Netherlands), which included PARK2, PINK1, DJ1/PARK7, LRRK2, UCHL1, ATP13A2, LPA, TNFRSF9, CAV2, CAV1, and GCH1. Subsequently, the patient was studied using the panel MovDisord-498,3 which detected the whole REEP1 deletion, later confirmed by MLPA (SALSA P213). The deletion was delineated by SNP-based array analysis (CytoScan HD, Affymetrix Inc., Santa Clara, CA), and a heterozygous 3.9-Mb microdeletion was identified: arr [hg19]2p11.2 (chr2:83,335,425-87,271,924)x1. Parents were studied, and the deletion was confirmed to be de novo.
Figure. Neuroimaging Findings.
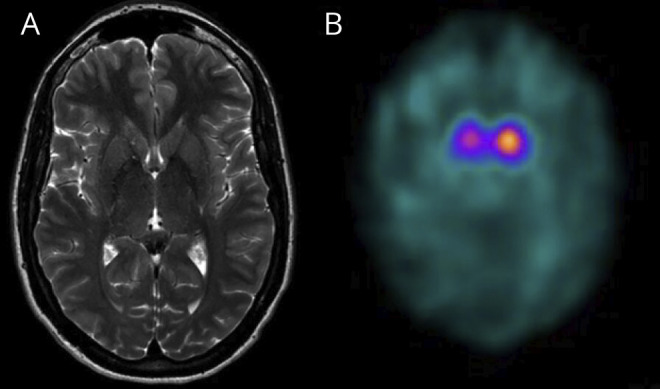
(A) Normal cranial MRI T2 axial image. (B) 123-ioflupane (DatScan) reveals bilateral asymmetrical denervation with right striate showing markedly reduced uptake, contralateral to the most affected side.
Patients carrying 2p11.2-p12 deletions exhibit neurodevelopmental delay, minor dysmorphic features, and congenital malformations.1,2 Our patient showed early-onset atypical parkinsonism with dystonia and lower limb spasticity. She displayed subtle facial dysmorphia, short stature, and scoliosis, but no congenital malformations nor cognitive impairment.
The size of the 2p11.2-2p12 deletion was smaller than that previously reported, ranging from 7.5 to 11.4 Mb.1 Among the 47 genes included in the deleted region, the most remarkable were POLR1A, involved in the Cincinnati type of acrofacial dysostosis (AFDCIN; MIM 616462) and REEP1 in spastic paraplegia type 31 (SPG31; MIM 610250), both autosomal dominant conditions (eTable, links.lww.com/NXG/A496). Our proband's phenotype seemed to be closer to SPG31-associated phenotypes. Most patients manifest a pure SPG, although SPG31 complex clinical pictures can include peripheral neuropathy, cerebellar ataxia, tremor, and cognitive impairment.4,5 Moreover, REEP1 variations may be responsible for distal hereditary motor neuropathy and congenital spinal muscular atrophy with respiratory distress type 1.6 No cases with movement disorders are known, likely because of delayed expression of additional clinical features.4 Nevertheless, recent evidence suggests that REEP1 variations may impair the function of DRP1, a mitochondrial protein involved in Parkinson disease pathogenesis.7 Identification of 2p11.2-p12 cases and complex SPG31 is needed to unravel genotype-phenotype correlations and further delineate the REEP1 biological role.
In conclusion, this case expands the 2p11.2-p12 deletion-associated phenotypes, including parkinsonism, and underscores that CNVs must be considered when analyzing next-generation sequencing data.
Ethical Compliance Statement
Written informed consent was obtained from the proband, and this study was approved by the ethic committee of the Hospital U. i P. La Fe (2019/0052).
Acknowledgment
The authors thank the patient and her parents for their support of this work.
Appendix. Authors
Contributor Information
Raquel Baviera-Muñoz, Email: raquelbaviera@gmail.com.
Dolores Martínez-Rubio, Email: mdmartinez@cipf.es.
Isabel Sastre-Bataller, Email: isa.sastre99@gmail.com.
Marina Campins-Romeu, Email: marinacampinsromeu@gmail.com.
Mireya Losada-López, Email: mireya_losada@hotmail.com.
Julia Pérez-García, Email: perez_margari@gva.es.
Edurne Novella-Maestre, Email: novella_edu@gva.es.
Irene Martínez-Torres, Email: irenemto@hotmail.com.
Study Funding
This work was supported by the Instituto de Salud Carlos III (ISCIII)—Subdirección General de Evaluación y Fomento de la Investigación [PI18/00,147] and by the Generalitat Valenciana [PROMETEO/2018/135], within the framework of the National R + D + I Plan cofunded with ERDF funds. Part of the equipment employed in this work has been funded by Generalitat Valenciana and cofinanced with ERDF funds (OP ERDF of Comunitat Valenciana 2014–2020).
Disclosure
The authors report no disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Silipigni R, Cattaneo E, Baccarin M, Fumagalli M, Bedeschi MF. Rare interstitial deletion of chromosome 2p11.2p12. Report of a new patient with developmental delay and unusual clinical features. Eur J Med Genet. 2016;59(1):39-42. [DOI] [PubMed] [Google Scholar]
- 2.Stevens SJ, Blom EW, Siegelaer IT, Smeets EE. A recurrent deletion syndrome at chromosome bands 2p11.2-2p12 flanked by segmental duplications at the breakpoints and including REEP1. Eur J Hum Genet. 2015;23(4):543-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correa-Vela M, Lupo V, Montpeyo M, et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann Clin Transl Neurol. 2020;7(8):1436-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beetz C, Schule R, Deconinck T, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131(pt 4):1078-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goizet C, Depienne C, Benard G, et al. REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat. 2011;32(10):1118-1127. [DOI] [PubMed] [Google Scholar]
- 6.Maroofian R, Behnam M, Kaiyrzhanov R, Salpietro V, Salehi M, Houlden H. Further supporting evidence for REEP1 phenotypic and allelic heterogeneity. Neurol Genet. 2019;5(6):e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Liu L, Jiang X, et al. The essential role of DRP1 and it sregulation by S-nitrosylation of Parkin in dopaminergic neurodegeneration: implications for Parkinson's disease. Antioxid Redox Signal. 2016;25(11): 609-622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Segment 1: Face is markedly hypomimic. Dysmorphic features are evident (short stature, frontal bossing, and mandibular prognathism). She displays postural and intentional tremor, and distal dystonic postures emerge with arms outstretched. Bradykinesia is severe, with left side being more affected. Gait is unstable, with truncal hyperextension, absent arm swing, and flexed-elbow deformity. Segment 2: Smooth pursuit is saccadic. Horizontal and vertical saccades are slow and inaccurate. Speech is hypophonic and dysarthric, but intelligible. Segment 3: Tendon reflexes are easily elicited, with bilateral ankle clonus. Alternating hand movements are slow but precise.Download Supplementary Video 1 (18.6MB, mp4) via http://dx.doi.org/10.1212/00642_Video_1