

ABSTRACT
Crimean-Congo hemorrhagic fever (CCHF) is a severe disease of humans caused by CCHF virus (CCHFV), a biosafety level (BSL)-4 pathogen. Ticks of the genus Hyalomma are the viral reservoir, and they represent the main vector transmitting the virus to its hosts during blood feeding. We have previously shown that CCHFV can persistently infect Hyalomma-derived tick cell lines. However, the mechanism allowing the establishment of persistent viral infections in ticks is still unknown. Hazara virus (HAZV) can be used as a BSL-2 model virus instead of CCHFV to study virus/vector interactions. To investigate the mechanism behind the establishment of a persistent infection, we developed an in vitro model with Hyalomma-derived tick cell lines and HAZV. As expected, HAZV, like CCHFV, persistently infects tick cells without any sign of cytopathic effect, and the infected cells can be cultured for more than 3 years. Most interestingly, we demonstrated the presence of short viral-derived DNA forms (vDNAs) after HAZV infection. Furthermore, we demonstrated that the antiretroviral drug azidothymine triphosphate could inhibit the production of vDNAs, suggesting that vDNAs are produced by an endogenous retrotranscriptase activity in tick cells. Moreover, we collected evidence that vDNAs are continuously synthesized, thereby downregulating viral replication to promote cell survival. Finally, vDNAs were also detected in CCHFV-infected tick cells. In conclusion, vDNA synthesis might represent a strategy to control the replication of RNA viruses in ticks allowing their persistent infection.
IMPORTANCE Crimean-Congo hemorrhagic fever (CCHF) is an emerging tick-borne viral disease caused by CCHF virus (CCHFV). Ticks of the genus Hyalomma can be persistently infected with CCHFV representing the viral reservoir, and the main vector for viral transmission. Here we showed that tick cells infected with Hazara virus, a nonpathogenic model virus closely related to CCHFV, contained short viral-derived DNA forms (vDNAs) produced by endogenous retrotranscriptase activity. vDNAs are transitory molecules requiring viral RNA replication for their continuous synthesis. Interestingly, vDNA synthesis seemed to be correlated with downregulation of viral replication and promotion of tick cell viability. We also detected vDNAs in CCHFV-infected tick cells suggesting that they could represent a key element in the cell response to nairovirus infection and might represent a more general mechanism of innate immunity against RNA viral infection.
KEYWORDS: Crimean-Congo hemorrhagic fever virus, Hazara virus, tick cell line, viral-derived DNA forms, Orthonairovirus, tick-borne disease, bunyavirus, nairovirus, reverse transcriptase, ticks
INTRODUCTION
Crimean-Congo hemorrhagic fever (CCHF) is an emerging tick-borne viral disease widely distributed across Africa, Southern Europe, the Middle East and Asia (1). It is considered to be one of the major emerging disease threats spreading to, and also within, the European region following increasing circulation of its main vectors, ticks of the genus Hyalomma (2). Furthermore, CCHF is included in the WHO List of Blueprint priority diseases (3). CCHF is caused by Crimean-Congo hemorrhagic fever virus (CCHFV), which belongs to the genus Orthonairovirus of the recently-established family Nairoviridae (4). CCHFV is characterized by an enzootic cycle in asymptomatic mammals and ticks, while human infection represents an accidental event. Although 85% of human cases are subclinical, in symptomatic patients infection begins with fever and other nonspecific clinical signs and can progress to a serious hemorrhagic syndrome with a case fatality rate up to 30% (5, 6).
Although CCHFV has been detected in many tick species, Hyalomma ticks represent the main vectors of the virus and the natural reservoir. In fact, Hyalomma ticks feeding on a viremic host become persistently infected with CCHFV, the virus survives through the subsequent stages of the tick life cycle, and transovarial transmission occurs in this genus (7). To date, very limited information is available about the replication and persistence of CCHFV in ticks due to the requirement for the virus to be handled in high-containment laboratories, compounded by the difficulty in manipulation of infected ticks in a biosafety level (BSL)-4 facility (7, 8). However, we have recently developed a CCHFV infection model based on embryo-derived Hyalomma anatolicum cell lines, providing the opportunity to study virus–vector interaction in an easier-to-handle in vitro system (9, 10).
Hazara virus (HAZV), originally isolated from a pool of adult Ixodes redikorzevi ticks removed from a vole in Pakistan (11), is a member of the family Nairoviridae and is closely related to CCHFV. Although genome sequence analyses clustered CCHFV and HAZV in different species, HAZV was classified in the same serogroup as CCHFV, based on antibody cross-reactivity between antigens of the two viruses (12, 13). Studies reported that CCHFV and HAZV have similar biological characteristics in terms of replication, interaction with cellular partners, and modulation of apoptosis (14–21). Although HAZV is able to simulate a disease similar to that induced by CCHFV in an interferon-deficient mouse model, it has never been associated with human disease and is considered a nonpathogenic virus that can be manipulated under BSL-2 conditions (22).
In the present study, we investigated virus/host interactions and possible mechanisms allowing the establishment of a persistent infection in ticks by applying HAZV, as a safe surrogate for CCHFV, to our tick cell line model. Interestingly, we showed that viral infection is associated with the synthesis of small viral-derived DNA forms (vDNAs), produced by a cellular reverse transcriptase activity, that are required to suppress viral replication and thereby maintain tick cell viability. Finally, we confirmed that vDNAs were also detectable in CCHFV-infected tick cells supporting the hypothesis that they could represent a key element in the cellular response to nairovirus infection. vDNAs might be involved in a general mechanism of innate immunity to counteract RNA virus infections in ticks.
RESULTS
HAZV persistently infects Hyalomma-derived tick cell lines.
To investigate whether HAZV is able to productively infect Hyalomma-derived tick cell lines, the H. anatolicum cell lines HAE/CTVM8 and HAE/CTVM9 (23) were infected with HAZV at a multiplicity of infection (MOI) of 0.1 or 1.0. Viral replication was monitored by progeny titration and by evaluating the intracellular viral RNA yield using a quantitative real-time RT-PCR (qRT-PCR) approach. Titration of viral progeny in supernatants collected on days 1, 2, 3, 4 and 7 postinfection (p.i.) showed that HAZV can productively infect H. anatolicum cells (Fig. 1A). The kinetics of viral progeny release were slightly different in the two cell lines; production of infectious viral particles was faster in HAE/CTVM9 than in HAE/CTVM8 cells. However, at 7 days p.i., prior to subculturing the infected cells, similar HAZV titers were observed in both cell lines (Fig. 1A). No viral particles were detected in supernatants of mock-infected cells (data not shown). The qRT-PCR results confirmed that replication of HAZV was slower in HAE/CTVM8 than in HAE/CTVM9 cells (Fig. 1B). Starting from day 7 p.i., infected tick cell cultures were split every 7–10 days. HAZV was detected in subcultures by viral titration for 70 days suggesting the establishment of a persistent infection (Fig. 1A).
FIG 1.

Hazara virus (HAZV) productively infects Hyalomma anatolicum tick cells without cytopathic effects. (A, B) HAE/CVTVM8 and HAE/CTVM9 cells were infected at MOI 0.1 or MOI 1.0. At the indicated time points, (A) infectious viral particles in the supernatant were titrated in Vero cells, error bars = SD; (B) the relative increase of viral RNA in the infected cells was evaluated by qRT-PCR. Data are the results of a representative experiment; (C) tick cell lines and human SW13 cells were infected with HAZV at MOI 0.1 and 1.0 and cell metabolism was evaluated using the MTT assay at the indicated time points. Data (mean ± SD, N = 3 independent experiments) are percentages of optical density of infected cells with respect to that of uninfected cells set as 100%.
To rule out the possibility that the different pattern of viral replication observed at the early time points (days 2–4 p.i.) was independent of an effect of the virus on cell viability, HAZV-infected HAE/CTVM8 and HAE/CTVM9 cells were monitored by phase-contrast microscopy and an MTT assay was used to evaluate cellular metabolic activity as a measure of viability. HAZV-infected human SW13 cells were used as positive control cells killed by the virus. In fact, HAZV efficiently replicates in SW13 cells (14, 17) producing a robust cytopathic effect as described for CCHFV (18). As expected, the metabolic activity of SW13 cells rapidly decreased over time after viral infection for both MOIs tested (Fig. 1C). In contrast, microscopic analysis of tick cell cultures (data not shown) and MTT assay performed at 2, 4, and 7 days p.i. did not detect any significant effect on the tick cells even at day 7 (Fig. 1C) suggesting that the HAZV infection did not affect tick cell viability.
Taking into account that HAE/CTVM8 cells showed similar kinetics of replication for HAZV and CCHFV (9) and grew more reliably in our hands than HAE/CTVM9 cells, we focused our attention on this cell line for the subsequent experimental steps.
To further characterize the generation of the persistently-infected HAE/CTVM8 cells, we firstly evaluated the percentage of HAZV–infected cells during the initial stages of infection by challenging HAE/CTVM8 cells with HAZV at MOI 0.1. Next, we evaluated infected cells over time by immunostaining of viral nucleoprotein (N). As shown in Fig. 2A, the proportion of N-expressing cells increased from ∼43% at day 6 p.i. to ∼62% at day 15 p.i., while at later time points, when cells are persistently infected, almost all cells stained positive (Fig. 2A, panel showing persistently infected cells). In addition, we confirmed that HAZV did not kill infected cells, as demonstrated by similar growth kinetics of virus-infected and mock-infected control cells (Fig. 2B). These observations suggest that the virus was slowly spreading throughout the culture from an initially low number of infected cells. The persistence of HAZV was further confirmed in infected HAE/CTVM8 cells by RT-PCR at 15, 30, 60, 312, and 715 days p.i. (data not shown) and by the titration of infectious viral progeny (Fig. 2C) indicating continued active viral replication. Persistent infection of HAE/CTVM8 cells was achieved on three independent occasions and one of the persistently-infected cultures was maintained for more than 3 years.
FIG 2.

Hazara virus (HAZV) infects Hyalomma anatolicum tick cells and does not affect cell viability. HAE/CTVM8 cells were infected with HAZV at MOI 0.1 and (A) infected and mock-infected cells were monitored by N protein immunostaining at 6 (6 d) and 15 (15 d) days postinfection (p.i.) and at > 1 year of culture (persistently infected) while (B) the number of live cells in infected and mock-infected control cultures were determined every day up to day 9 p.i. by trypan blue exclusion (three independent experiments) and (C) long term viral progeny release by HAZV-infected HAE/CTVM8 cells was determined by titration in Vero cells. (A) and (C) are representative of the establishment of one persistently-infected cell lines.
Viral-derived DNA forms are detectable in de novo infected and persistently-infected tick cells.
It has been recently reported that in some insect cells RNA virus infection is associated with the synthesis of vDNAs that are involved in the establishment of persistent infection (24, 25). To determine whether HAZV infection of tick cells was associated with the formation of vDNAs, HAE/CTVM8 and HAE/CTVM9 cell were infected with HAZV at MOI 0.1 and 1.0 and the formation of vDNAs was evaluated by PCR. To this end, nine pairs of primers, designed to amplify overlapping sequences of 152–237 bp covering the S segment of the HAZV genome, were adopted (Fig. 3A). Specific PCR amplicons were already obtained from total DNA extracted from infected tick cells at day 1 p.i., with a maximum number of fragments detected at 7 days p.i. (Fig. 3B and C). No vDNAs were detected in mock-infected tick cells or, more significantly, in HAZV-infected mammalian Vero cells (data not shown). Furthermore, while RNase treatment of infected cell DNA extracts did not affect amplification of vDNAs, when DNase was added no PCR products were detected (data not shown). Finally, when combining the primers of contiguous amplicons, we did not detect fragments larger than ∼240 bp, suggesting that each vDNA represent only a small portion of the viral genome segment, and that neither long fragments nor the entire genome segment were synthesized (data not shown). Overall, these results suggest that vDNAs are small DNA fragments likely derived from the HAZV genome.
FIG 3.

Detection of viral-derived DNA (vDNA) in Hyalomma anatolicum tick cells infected with Hazara virus (HAZV) at MOI 0.1 and 1. (A) Schematic representation of nine pairs of primers designed on the genomic S segment of HAZV, used for the detection of vDNAs; (B) Example of vDNAs detected using the nine primer pairs at 1 (upper panel), 7 (middle panel) and 15 (lower panel) days p.i. in HAZV-infected HAE/CTVM8 and HAE/CTVM9 cells; (C) Frequency of vDNA production in the same tick cells. Data (mean ± SD, N = 3 independent experiments) are percentages of vDNAs with respect to the maximum number of detectable amplicons (n = 9), set as 100%, for each sample.
Interestingly, vDNAs were constantly detectable in persistently-infected tick cell cultures, as the presence of vDNAs was periodically confirmed by PCR (up to 10 times per year; data not shown) suggesting that they either represent stable molecules or are continuously synthesized during cell culture.
vDNA synthesis depends on viral RNA replication and is mediated by a cellular reverse transcriptase activity.
To investigate whether viral RNA replication is required for vDNA synthesis, HAE/CTVM8 cells were infected with UV-inactivated HAZV. Three days later, total DNA was extracted and submitted to PCR analyses and no vDNAs were detected (data not shown). Furthermore, HAE/CTVM8 cells, persistently infected for at least 6 months, were treated with ribavirin (100 mM), a drug known to inhibit viral genome replication. As expected, and shown in Fig. 4A and B, viral progeny production was suppressed and the yield of intracellular viral RNA decreased. In this experimental condition, at 72 h posttreatment, vDNAs disappeared suggesting that viral RNA replication is required for vDNA synthesis (Fig. 4C).
FIG 4.

Effect of ribavirin treatment on Hazara virus (HAZV) replication in persistently-infected Hyalomma anatolicum tick cells. HAE/CTVM8 cells were treated with 100 mM ribavirin and viral replication was evaluated, (A) at different time points by viral titration; (B) at 72 h posttreatment by quantitative RT-PCR. Data are mean ± SD of three independent experiments. ****, P < 0.0001. (C) Furthermore, production of viral-derived DNAs (vDNAs) was suppressed by the ribavirin treatment (Lines 1–9: PCR specific for vDNAs, line 10: negative control, line 11: positive tick DNA extraction control).
In the case of RNA viruses lacking a viral enzyme able to convert genomic RNA to DNA, the formation of vDNAs could be due to the presence of endogenous reverse transcriptases (RTs) encoded by retrotransposons and/or endogenous retroviruses integrated into the genome of tick cells. By performing an RT assay, we detected a clear Mn2+ dependent RT-activity in HAE/CTVM8 cell lysates, with a slightly higher level in HAE/CTVM8 cells persistently infected with HAZV (cells cultured for more than 6 months) compared with uninfected cells (Fig. 5A). Furthermore, to demonstrate that vDNAs were synthesized by cellular RT activity, we treated HAE/CTVM8 cells with the nucleoside RT inhibitor azidothymine triphosphate (AZT). Specifically, 5 mM AZT was selected, as this concentration did not significantly affect cell viability (see next paragraph) and had been already adopted to treat insect cells (24, 25). In addition, three different schedules of AZT administration were adopted: (i) 6 h before virus infection; (ii) at the time of viral infection; and (iii) 6 h before virus infection as well as at the time of infection (Fig. 5B). As shown in Fig. 5C, while a reduction up to 30% in vDNA yield was observed when cells were treated with AZT before viral infection (experimental condition 1), this reduction increased up to 50% when the drug was added during the viral adhesion step (experimental condition 2). A stronger effect (roughly 70% reduction) resulted from the combination of the two treatments (experimental condition 3). Overall, these results suggest that a cellular RT activity is required for the synthesis of vDNA.
FIG 5.

A cellular reverse transcriptase (RT) activity is involved in viral-derived DNA (vDNA) synthesis in Hyalomma anatolicum tick cells infected with Hazara virus (HAZV). (A) Detection of RT activity in uninfected HAE/CTVM8 cells and HAE/CTVM8 cells persistently infected with HAZV (HAE/CTVM8+) using Mg2+ or Mn2+ as enzyme cofactor. Data are mean ± SD of three independent experiments. (B) Schedule of azidothymine triphosphate (AZT) treatment of HAE/CTVM8 cells before (pre) or simultaneously with (post) HAZV infection. (C) Effect of AZT treatment of HAZV-infected HAE/CTVM8 cells on vDNA synthesis at 7 days p.i. Data (mean ± SD, N = 3 independent experiments) are percentages of vDNAs detected in AZT-treated cells with respect to the vDNAs detected in the untreated cells, set as 100%.
vDNAs promote suppression of viral particle production and survival of HAZV-infected tick cells.
To evaluate the effect of vDNAs on HAZV infection in tick cells, we treated persistently infected (>1 year of culture) HAE/CTVM8 cells with 5 mM AZT. Seventy-two hours later, the viral titer obtained from treated cells was roughly four times higher than the one obtained from untreated cells. Furthermore, the number of vDNAs in treated cells was roughly one-third of that seen in the untreated cells (Fig. 6A and B). By contrast, the yield of intracellular viral RNA was not affected by AZT treatment, suggesting that AZT does not interfere with viral genome replication (Fig. 6C). However, immunostaining (Fig. 6D) revealed a clear increase of the amount of HAZV N protein (increase of fluorescence intensity) in AZT-treated cells, which was quantified by Western blotting as 1.5 times greater than that in control cells (Fig. 6E), suggesting a higher level of N protein synthesis.
FIG 6.

Azidothymine triphosphate (AZT) treatment of Hyalomma anatolicum HAE/CTVM8 cells persistently infected with Hazara virus (HAZV) induces an increase in viral replication and decrease in cell metabolism. Cells were treated with 5 mM azidothymine triphosphate (AZT) and incubated for 72 h. Then (A) the viral progeny was quantified by virus titration; (B) the presence of viral-derived DNAs (vDNAs) was evaluated by PCR; and (C) the yield of the intracellular viral RNA was quantified by quantitative RT-PCR. Data are mean ± SD of three independent experiments. ****, P < 0.0001. The nucleoprotein of HAZV was (D) detected by immunostaining and (E) quantified by Western blot to be 1.5× greater in persistently-infected HAE/CTVM8 cells treated with 5 mM AZT compared with untreated cells. PI = persistently-infected cells; C- = uninfected control cells.
Interestingly, an MTT assay showed a significant decrease in the metabolism of AZT-treated cells (Fig. 7A), associated with the increase in viral titer. To demonstrate that the reduction in cell metabolism was associated with viral-mediated cell death, uninfected and persistently-infected HAE/CTVM8 cells were treated with AZT and the number of live cells was counted by trypan blue exclusion assay. Although the AZT caused a slight reduction in the number of uninfected cells over a 72-h period, a significant decrease in live cell numbers was observed only in persistently infected cells (Fig. 7B).
FIG 7.
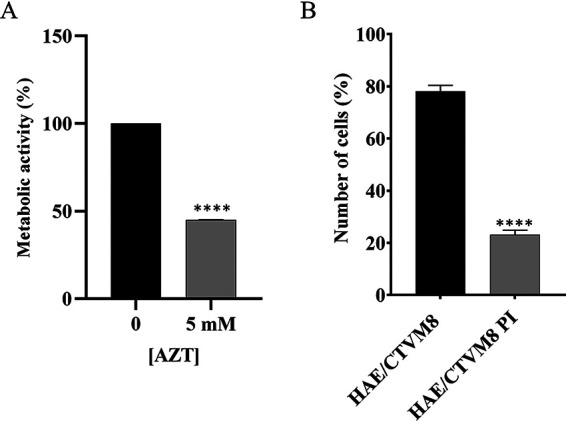
Treatment of Hyalomma anatolicum HAE/CTVM8 cells with azidothymine triphosphate (AZT) induces death in persistently-infected cells. (A) Tick cells persistently infected with Hazara virus were mock-treated or treated with 5 mM AZT, then cell metabolism was measured using the MTT assay at 72 h postinfection (p.i.). (B) Uninfected (HAE/CTVM8) and persistently-infected (HAE/CTVM8 PI) tick cells were treated with 5 mM AZT, then cell viability was determined by trypan blue dye exclusion test at 72 h p.i. Data are mean ± SD of three independent experiments. ****, P < 0.0001.
In conclusion, our data suggest that vDNAs might contribute to controlling HAZV infection in tick cells by suppressing the production of infectious viral progeny and, thereby, promoting the survival of infected cells.
vDNAs are detectable in CCHFV-infected HAE/CTVM8 tick cells.
We have previously demonstrated that CCHFV can persistently infect Hyalomma-derived tick cell lines (9). To demonstrate that CCHFV infection induces vDNA formation in tick cells, HAE/CTVM8 cells were infected with CCHFV at MOI 0.1 and harvested at 3 and 7 days p.i. As for HAZV, we designed a panel of primers covering the entire S segment of CCHFV and the PCR analyses on total DNA extracted from infected cells demonstrated the presence of vDNAs (Fig. 8). This result suggests that vDNA production could be a common tick cellular response following RNA virus infection, as reported for some insects (26).
FIG 8.

Detection of viral DNAs (vDNAs) in Hyalomma anatolicum HAE/CTVM8 cells infected with Crimean-Congo hemorrhagic fever virus (CCHFV). HAE/CTVM8 cells were infected with CCHFV at multiplicity of infection (MOI) 0.1. Three (left panel) and seven (right panel) days after infection, total DNA was extracted and vDNAs were detected by PCR. Lines 1–11: PCR specific for vDNAs, line 12: negative control, line 13: positive tick DNA extraction control.
DISCUSSION
CCHFV is the most important and globally widespread tick-borne hemorrhagic fever virus and its emergence and reemergence highlight the importance of this infectious agent for human health (1). Despite the rapid increase in knowledge of viral biology and the development of diagnostic tools in the last decade (2), there is still a large gap in the characterization of virus/vector interaction.
We and others have shown that CCHFV can persistently infect ticks and tick cell lines without deleterious effects (7, 9, 10, 27, 28); however, the mechanism allowing the persistent infection of CCHFV (and other tick-borne viruses) in ticks has not yet been characterized.
In the present study, we adopted HAZV as a nonpathogenic surrogate for CCHFV, with the aim of studying the mechanisms involved in the establishment of persistent infection of tick vectors. Firstly, we showed that HAZV replicates productively in cell lines derived from tick of a genus (Hyalomma) that was never reported to function as a vector for HAZV (29). In addition, the HAZV infection did not appear to be cytopathic for tick cells and persisted for a long time, as previously reported in the case of CCHFV and other tick-borne viruses (9, 10, 27, 30–32). It is notable that HAZV showed kinetics of replication in H. anatolicum cell lines faster than those previously reported for CCHFV in the same cell lines (9, 27) and produced higher viral titers, suggesting that Hyalomma ticks could support HAZV infection (27).
Previously, Goic and coworkers demonstrated that short vDNAs were detected in vitro and in vivo in insect cells infected by RNA viruses and that they were involved in the control of virus replication allowing persistent infection of cultured cells and insects (24, 25). Remarkably, we detected vDNAs in HAZV-infected tick cells from as early as 24 h p.i. Considering that nairoviruses do not encode a viral RT, the RT-activity required for vDNA synthesis could be provided by endogenous cellular sequences (i.e., retrotransposons and retroviral sequences). No genome sequences are as yet available for any Hyalomma spp. ticks; however a reverse transcriptase-like protein has been described in Amblyomma americanum (UniProtKBQ49P04) while RT activity was detected in three Ixodes scapularis embryo-derived cell lines (33), thus supporting the possibility of RT expression also in Hyalomma derived cell lines. In fact, we detected Mn2+ dependent RT-activity in HAE/CTVM8 cell lysates, and experiments with the RT inhibitor AZT showed that RT is involved in vDNA synthesis in tick cells, as described for insect cells (24, 25).
Moreover, our data showed that vDNAs are always detectable in persistently-infected tick cells and the S segment can be fully used as template for their synthesis. In contrast, Nag and coworkers reported that only two regions of the S segment of La Crosse virus (LACV), another member of the order Bunyavirales, were detectable in infected C6/36 insect cells (34). This apparent discrepancy between HAZV and LACV might be due to biological differences between the two viruses. On the other hand, it should be taken into account that those authors adopted a different PCR strategy, not based on multiple small overlapping amplicons. Furthermore, they analyzed vDNA presence only at one time point after viral infection, a choice that might have negatively impacted the efficiency of detection (34). Interestingly, we observed that vDNA amplification depends on active viral RNA replication, suggesting that vDNAs are not stable but are continuously synthesized during the persistent infection of tick cells.
Reports on insect RNA viruses suggest that vDNAs may be produced via template-switching events, when the RT switches from the retro-elements template to viral RNA forming linear and episomal vDNA-retrotransposon chimeric molecules, while integration events into the genome are rare (24, 25, 35, 36). However, the integration of viral sequences into the arthropod genome, although infrequently detected to date, is considered important in virus/vector co-evolution (10, 37–39). In fact, many bunya- and othomyxo-like sequences have been identified in the I. scapularis genome suggesting that these viruses can produce vDNAs in ticks and occasionally integrate into the genome of germ line cells (40).
The modulation of virus replication mediated by vDNAs in insects is based on an RNAi mechanism; it has been shown that vDNAs are transcribed and used as templates for the synthesis of short RNAs that suppress viral replication (24, 25, 34, 36, 41). Our data are compatible with this mechanism. In fact, we observed that (i) the proportion of infected cells increased over time during the infection while viral titer decreased, and (ii) the inhibition of vDNA synthesis by AZT treatment was associated with an increase in nucleoprotein yield and viral titer without any effect at the level of intracellular viral RNA, thus suggesting posttranscriptional regulation. However, at this stage, a contribution of specific effects on viral genome replication/transcription efficiency cannot be fully ruled out.
The above-described model of vDNA production is compatible with the requirement of active viral RNA replication for vDNA synthesis; continuous virus replication allows the production of RNA templates and the chance of switching events producing new vDNAs. On the other hand, vDNAs may mediate a suppressive effect on virus replication, reducing virus proteins and progeny release, thus inducing an equilibrium between virus replication and generation of new vDNAs.
More interestingly, inhibition of vDNA synthesis induced a reduction in cell viability, as showed by MTT and by the proportion of live cells. According to the literature on insect RNA viruses, increase of viral assembly and budding could be associated with a cytotoxic effect, negatively affecting cell survival (24, 25).
Although more research is required to further characterize the biology of vDNA production and function, our in vitro data supported the view that vDNAs are linked to the establishment of persistent viral infections, reducing the production of viral progeny and protecting tick cells from deleterious effects. In vivo experiments could investigate the relevance of vDNA inhibition for tick survival, as demonstrated in the case of mosquitoes infected with arboviruses (24, 25). Indeed, the development of tools to interfere with vDNA synthesis might represent a strategy to reduce the fitness of infected ticks and lower the risk of transmission of the virus in endemic areas.
In conclusion, we show for the first time that vDNAs are detectable in tick cells infected with HAZV and CCHFV. As described for insect-borne RNA viruses, vDNAs seem to be associated with the establishment of persistent infection in ticks, which are classified in a different subphylum of the Arthropoda from insects. Also, in this context vDNAs appear to control viral replication and promote cell survival, thus allowing persistence of the virus in the environment. Overall these findings suggest that vDNA synthesis might represent a common strategy to control viral infections in arthropods.
MATERIALS AND METHODS
Cells and viruses.
All culture media and supplements were obtained from Gibco unless otherwise indicated. SW13 cells (human adrenal cortex adeno-carcinoma cells, ATCC CCL-105™), were cultured in Leibovitz’s L-15 medium (L-15) supplemented with 10% heat-inactivated fetal bovine serum (FBSi) and 100 U/ml penicillin and 100 μg/ml streptomycin (p/s). Vero cells (African green monkey kidney cells, ATCC CCL-81) were grown in Dulbecco’s modified Eagle’s medium containing 10% FBSi and p/s. Vero cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in air while SW13 cells were maintained at 37°C in ambient air.
The H. anatolicum embryo-derived cell lines HAE/CTVM8 and HAE/CTVM9 were grown in, respectively, L15/H-Lac medium (equal volumes of L-15 supplemented with 10% tryptose phosphate broth [TPB] and Hanks’ balanced salt solution with 0.5% lactalbumin hydrolysate [Sigma]) and L-15/MEM medium (equal volumes of L-15 and Minimal Essential Medium with Hank’s salts supplemented with 10% TPB), both supplemented with 2 mM l-glutamine, 20% FBSi and p/s, and incubated in sealed flat-sided culture tubes (Nunc, Thermo-Fisher Scientific) in ambient air at 32°C (23).
Uninfected and HAZV-infected SW13 and tick cell metabolic activity was tested with an assay based on the reduction of a tetrazolium salt (MTT Cell Proliferation Assay ATCC 30-1010K™) in a 96-well plate format according to the manufacturer’s instructions. Tick cells were grown in sealed 96-well plates for the MTT assay.
The HAZV JC280 and the CCHFV IbAr10200 strains produced in SW13 cells were used in the experiments (21).
Viral stock preparation.
SW13 cells were seeded in T75 flasks and then infected with HAZV or CCHFV (MOI of ∼ 0.1). At 48–72 h p.i., supernatants were collected, centrifuged at 896 × g for 10 min, then 10-fold serially diluted in L-15 with 2% FBSi and titrated on Vero cells in 96-well plates. After 24 h of incubation, cells were fixed with methanol-acetone and stained for immunofluorescence assay using a rabbit polyclonal anti-CCHFV nucleoprotein antibody (21), that also recognized HAZV-N, and Alexa FluorTM 488 goat anti-rabbit IgG (Invitrogen), according to the manufacturer’s instructions. The fluorescent foci in each well were counted and viral titer was expressed as focus-forming units per ml (FFU)/ml.
Infection of tick cell lines.
Tick cells (2 × 106) were seeded in flat-sided tubes and cultured for 48 h in 2.5 ml of complete medium. Then, 1.5 ml medium was removed and retained, and cells were incubated for 1 h with HAZV, at the appropriate MOI, in a final volume of ∼1 ml of complete medium. Cells were carefully washed once with phosphate-buffered saline (PBS) and cultured in 2.5 ml of conditioned medium (retained old medium and fresh medium in a ratio of 1:2). In studies of kinetics of viral progeny release, 200 μl of supernatant medium were collected at the appropriate time points for viral titration on Vero cells as above, and an equal volume of fresh medium was replaced in the culture tubes. To evaluate the viral RNA yield, cells were harvested, centrifuged at 7,168 × g for 10 min and washed once with PBS before lysis.
Infections of tick cells with UV-inactivated HAZV were performed using a viral stock inactivated as previously described (42). Briefly, an aliquot of 1 ml virus stock in a well of a 6-well plate was irradiated with UV (UV Mineral light lamp, model UVG-54, 254 nm, UVP, Upland, CA) at a distance of 17 mm for 1 min.
Immunostaining.
Tick cells were collected and centrifuged at 206 × g for 7 min and washed once with PBS, and finally resuspended in PBS. Cells (0.6 × 106 in 200 μl) were applied to cleaned microscope slides using a Shandon III cytocentrifuge (3 min at 1000 rpm). After centrifugation, cells were fixed in 70% ethanol at 4°C for 30 min and an immunofluorescence assay was performed. Nonspecific sites were blocked using 2.5% bovine serum albumin (BSA, Sigma) in PBS and incubation for 1 h at room temperature. Subsequently, slides were incubated for 1.5 h at 37°C with the above-mentioned anti-N antibody diluted 1:200 in PBS with 2.5% BSA and 0.1% Tween 20. After this incubation, slides were washed with PBS and incubated for 1 h at 37°C in the dark with Alexa FluorTM 488 goat anti-rabbit IgG (Invitrogen) diluted 1:1000 in PBS with 2.5% BSA and 0.1% Tween 20, and nuclei were stained with a 1:1000 dilution of 5 mM DRAQ5 solution (Thermo-Fisher Scientific).
After this last incubation, infected and mock-infected control cells were examined under a Nikon A1RSi Laser Scanning inverted confocal microscope equipped with NIS-Elements Advanced Research software (Nikon Instruments Inc., Tokyo, Japan) and blue (488 nm) and green (561 nm) lasers.
Western blot analysis.
Tick cell cultures were harvested and washed in PBS by centrifugation at 335 × g for 7 min at 4°C and then lysed in 100 μl of 1X radioimmunoprecipitation assay (RIPA) buffer (PBS containing 1% Nonidet P-40, 0.5% deoxycholate and 0.05% SDS) in the presence of protease inhibitors (0.1 mM Nα-p-tosyl-L-lysine chloromethyl ketone, 0.1 mM tosylsulfonyl phenylalanyl chloromethyl ketone, Complete Protease Inhibitor Cocktail Tablets, Roche). Samples were boiled for 5 min at 100°C and directly resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4.5% stacking gel and 10% resolving gel). Proteins were electroblotted onto a Hybond ECL nitrocellulose membrane (GE Healthcare Life Sciences). The N protein was detected by employing the above-mentioned rabbit polyclonal anti-CCHFV nucleoprotein antibody followed by anti-rabbit HRP-conjugated IgG. The loading control was evaluated using a rabbit polyclonal anti-calnexin antibody (In-house, Agrisera). Blots were developed with enhanced chemiluminescence reagents (Amersham Pharmacia) (43). Western blot quantification was performed using the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific).
Drug treatments.
AZT (Sigma) was dissolved in DMSO to a final concentration of 5M while ribavirin (Sigma) was dissolved in ultrapure H2O (MilliQ, Merck) to a final concentration of 100 mM. Aliquots were stored at −20°C. For treatment of cell cultures, half the volume of culture medium was removed and replaced with fresh medium containing the drug. Cells were incubated until the indicated time points. Then, cells were detached by pipetting, centrifuged at 2,205 × g for 10 min and washed once with PBS. Cell pellets were then processed for DNA or RNA extraction.
Nucleic acid isolation and qRT-PCR analysis.
DNA and RNA were extracted using a DNeasy blood and tissue kit and a RNeasy minikit (Qiagen) respectively, according to the manufacturer's instructions. HAZV RNA was amplified using the Superscript III Platinum One-step kit (Invitrogen) following the standard PCR conditions for TaqMan probes using primers and probe targeting the S segment (21). Kinetics of intracellular viral RNA replication were evaluated using the ΔΔCt method for the relative quantification of RNA (set to 1 at day 0) using the putative translation elongation factor EF-1 alpha/Tu endogenous gene of H. anatolicum tick cells to normalize the viral RNA (9, 44).
The yield of intracellular HAZV RNA in the ribavarin and AZT experiments was evaluated using a standard curve generated from six serial dilutions (from 5 × 106 to 50 copies) of a control plasmid containing the region amplified by the primers. The HAZV RNA copy number of the samples was calculated automatically with the software of the ABI 7900HT Sequence Detection Systems (Thermo Fisher Scientific) and then expressed as numbers of viral RNA copies per 0.2 μg of total RNA.
Detection of vDNAs.
PCRs were performed on total DNA extracted from infected HAE/CTVM8 cells with nine pairs of primers mapping within the S segment of the HAZV genome (GenBank: KP406725.1) and the CCHFV genome (GenBank: U88410.1). Primers used in this study were designed using the program “Primer3” available online (http://bioinfo.ut.ee/primer3-0.4.0/). The primer sequences are available on request.
Each PCR mixture contained 5 μl of 10× PCR buffer with 15 mM MgCl2, 1 μl of 0.625 mM dNTPs mix, 2 μl of each primer (10 μM), 0.5 μl of TaqGold, 50 ng of DNA and PCR grade water up to the final reaction volume of 50 μl (all reagents were purchased from Thermo Scientific). Cycling conditions were 1 cycle of 10 min at 95°C; 40 cycles of 15 s at 95°C, 30 s at 60°C and 45 s at 72°C; 5 min at 72°C. Twenty microliters of each PCR product were loaded onto a 2% (wt/vol) agarose gel containing the GelRed Nucleic Acid Gel Stain (Biotium).
In vitro reverse transcriptase assay.
To extract proteins, cells were lysed in CHAPS lysis buffer (10 mM Tris-HCl pH 7.5, 400 mM NaCl, 0.7 mM MnCl2, 1 mM MgCl2, 1 mM EGTA, 0.5% CHAPS, 10% glycerol, freshly supplemented with complete EDTA-free protease inhibitors cocktail [Roche] and 1 mM DTT). After incubation at 4°C for 10 min, cell debris was removed by centrifugation at 16,900 × g for 10 min at 4°C. Supernatants were transferred to clean tubes. Total protein concentration was determined using a Micro BCA protein assay kit (Thermo Scientific) following the manufacturer’s instructions.
Reverse transcriptase assays were carried out for 15 min at 25°C in a total reaction volume of 50 μl containing 4 μg of protein sample, 320 ng of PAGE-purified oligo(dT)18, 500 ng of poly(rA), and 1 μl of 84 Ci/mmol 3H-dTTP in 50 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 5 mM DTT, and 0.1% Triton X-100. After this, the entire 50 μl of each reaction was spotted on an ion paper (Amersham Hybond-N+, GE Healthcare) that retains incorporated nucleotides but not free dNTPs. Papers were washed 3 times (10 min for each wash) with saline-sodium citrate buffer (0.3 M NaCl, 0.03 M sodium citrate pH 7.2) and immersed in 4 ml of liquid scintillation cocktail Ultima Gold (PerkinElmer). Radioactivity was measured using a Scintillator (TRI-carb 2819 TR, Perkin Elmer).
Statistical analyses.
Graphs and statistical comparisons, applying Student's t test, were performed with the GraphPad Prism 8 software (21). Data subjected to statistical analyses have been replicated in at least three independent experiments. Differences were considered to be statistically significant at P < 0.05.
ACKNOWLEDGMENTS
The tick cell lines HAE/CTVM8 and HAE/CTVM9 were provided by the Tick Cell Biobank. This work was supported by University of Padova grants (DOR 2017-2019 and PRID 2017) to C.S., ArboNET (2015–01885), a Swedish Research Council grant (2017–03126) to A.M., and United Kingdom Biotechnology and Biological Sciences Research Council grants BB/N023889/2 and BB/P024270/1 to L.B.-S. M.V.S was supported by a fellowship of the Ph.D. School in Molecular Medicine, University of Padova.
Contributor Information
Ali Mirazimi, Email: ali.mirazimi@ki.se.
Cristiano Salata, Email: cristiano.salata@unipd.it.
Rebecca Ellis Dutch, University of Kentucky College of Medicine.
REFERENCES
- 1.Spengler JR, Bente DA, Bray M, Burt F, Hewson R, Korukluoglu G, Mirazimi A, Weber F, Papa A. 2018. Second International Conference on Crimean-Congo Hemorrhagic Fever. Antiviral Res 150:137–147. 10.1016/j.antiviral.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papa A, Mirazimi A, Köksal I, Estrada-Pena A, Feldmann H. 2015. Recent advances in research on Crimean-Congo hemorrhagic fever. J Clin Virol 64:137–143. 10.1016/j.jcv.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehand MS, Al-Shorbaji F, Millett P, Murgue B. 2018. The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antiviral Res 159:63–67. 10.1016/j.antiviral.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maes P, Alkhovsky SV, Bào Y, Beer M, Birkhead M, Briese T, Buchmeier MJ, Calisher CH, Charrel RN, Choi IR, Clegg CS, de la Torre JC, Delwart E, DeRisi JL, Di Bello PL, Di Serio F, Digiaro M, Dolja VV, Drosten C, Druciarek TZ, Du J, Ebihara H, Elbeaino T, Gergerich RC, Gillis AN, Gonzalez JPJ, Haenni AL, Hepojoki J, Hetzel U, Hô' T, Hóng N, Jain RK, Jansen van Vuren P, Jin Q, Jonson MG, Junglen S, Keller KE, Kemp A, Kipar A, Kondov NO, Koonin EV, Kormelink R, Korzyukov Y, Krupovic M, Lambert AJ, Laney AG, LeBreton M, Lukashevich IS, Marklewitz M, Markotter W, et al. 2018. Taxonomy of the family Arenaviridae and the order Bunyavirales: update 2018. Arch Virol 163:2295–2310. 10.1007/s00705-018-3843-5. [DOI] [PubMed] [Google Scholar]
- 5.Akıncı E, Bodur H, Leblebicioglu H. 2013. Pathogenesis of Crimean-Congo hemorrhagic fever. Vector Borne Zoonotic Dis 13:429–437. 10.1089/vbz.2012.1061. [DOI] [PubMed] [Google Scholar]
- 6.Bente DA, Forrester NL, Watts DM, McAuley AJ, Whitehouse CA, Bray M. 2013. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antiviral Res 100:159–189. 10.1016/j.antiviral.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Xia H, Beck AS, Gargili A, Forrester N, Barrett ADT, Bente DA. 2016. Transstadial transmission and long-term association of Crimean-Congo Hemorrhagic fever virus in ticks shapes genome plasticity. Sci Rep 6:35819. 10.1038/srep35819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thangamani S, Bente D. 2014. Establishing protocols for tick containment at Biosafety Level 4. Pathog Dis 71:282–285. 10.1111/2049-632X.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salata C, Monteil V, Karlberg H, Celestino M, Devignot S, Leijon M, Bell-Sakyi L, Bergeron É, Weber F, Mirazimi A. 2018. The DEVD motif of Crimean-Congo hemorrhagic fever virus nucleoprotein is essential for viral replication in tick cells. Emerg Microbes Infect 7:190. 10.1038/s41426-018-0192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salata C, Moutailler S, Attoui H, Zweygarth E, Decker L, Bell-Sakyi L. 2021. How relevant are in vitro culture models for study of tick-pathogen interactions? Pathog Glob Health: 1–19. 10.1080/20477724.2021.1944539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Begum F, Wisseman CL, Casals J. 1970. Tick-borne viruses of West Pakistan. II. Hazara virus, a new agent isolated from Ixodes redikorzevi ticks from the Kaghan Valley, W. Pakistan. Am J Epidemiol 92:192–194. 10.1093/oxfordjournals.aje.a121197. [DOI] [PubMed] [Google Scholar]
- 12.Kuhn JH, Wiley MR, Rodriguez SE, Bào Y, Prieto K, Travassos da Rosa APA, Guzman H, Savji N, Ladner JT, Tesh RB, Wada J, Jahrling PB, Bente DA, Palacios G. 2016. Genomic characterization of the genus nairovirus (Family Bunyaviridae). Viruses 8:164. 10.3390/v8060164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalkan-Yazıcı M, Karaaslan E, Çetin NS, Hasanoğlu S, Güney F, Zeybek Ü, Doymaz MZ. 2021. Cross-reactive anti-nucleocapsid protein immunity against Crimean-Congo hemorrhagic fever virus and Hazara virus in multiple species. J Virol 95:e02156-20. 10.1128/JVI.02156-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuller J, Surtees RA, Shaw AB, Álvarez-Rodríguez B, Slack GS, Bell-Sakyi L, Mankouri J, Edwards TA, Hewson R, Barr JN. 2019. Hazara Nairovirus elicits differential induction of apoptosis and nucleocapsid protein cleavage in mammalian and tick cells. J Gen Virol 100:392–402. 10.1099/jgv.0.001211. [DOI] [PubMed] [Google Scholar]
- 15.Fuller J, Surtees RA, Slack GS, Mankouri J, Hewson R, Barr JN. 2019. Rescue of infectious recombinant Hazara Nairovirus from cDNA reveals the nucleocapsid protein DQVD caspase cleavage motif performs an essential role other than cleavage. J Virol 93:e00616-19. 10.1128/JVI.00616-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto Y, Nouchi T, Ohta K, Nishio M. 2019. Regulation of Hazara virus growth through apoptosis inhibition by viral nucleoprotein. Arch Virol 164:1597–1607. 10.1007/s00705-019-04236-7. [DOI] [PubMed] [Google Scholar]
- 17.Surtees R, Dowall SD, Shaw A, Armstrong S, Hewson R, Carroll MW, Mankouri J, Edwards TA, Hiscox JA, Barr JN. 2016. Heat shock protein 70 family members interact with Crimean-Congo Hemorrhagic fever virus and Hazara virus nucleocapsid proteins and perform a functional role in the Nairovirus replication cycle. J Virol 90:9305–9316. 10.1128/JVI.00661-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlberg H, Tan Y-J, Mirazimi A. 2011. Induction of caspase activation and cleavage of the viral nucleocapsid protein in different cell types during Crimean-Congo hemorrhagic fever virus infection. J Biol Chem 286:3227–3234. 10.1074/jbc.M110.149369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlberg H, Tan Y-J, Mirazimi A. 2015. Crimean-Congo haemorrhagic fever replication interplays with regulation mechanisms of apoptosis. J Gen Virol 96:538–546. 10.1099/jgv.0.000011. [DOI] [PubMed] [Google Scholar]
- 20.Carter SD, Surtees R, Walter CT, Ariza A, Bergeron E, Nichol ST, Hiscox JA, Edwards TA, Barr JN. 2012. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J Virol 86:10914–10923. 10.1128/JVI.01555-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monteil V, Salata C, Appelberg S, Mirazimi A. 2020. Hazara virus and Crimean-Congo hemorrhagic fever virus show a different pattern of entry in fully-polarized Caco-2 cell line. PLoS Negl Trop Dis 14:e0008863. 10.1371/journal.pntd.0008863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dowall SD, Findlay-Wilson S, Rayner E, Pearson G, Pickersgill J, Rule A, Merredew N, Smith H, Chamberlain J, Hewson R. 2012. Hazara virus infection is lethal for adult type I interferon receptor-knockout mice and may act as a surrogate for infection with the human-pathogenic Crimean-Congo hemorrhagic fever virus. J Gen Virol 93:560–564. 10.1099/vir.0.038455-0. [DOI] [PubMed] [Google Scholar]
- 23.Bell-Sakyi L. 1991. Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J Parasitol 77:1006–1008. 10.2307/3282757. [DOI] [PubMed] [Google Scholar]
- 24.Goic B, Vodovar N, Mondotte JA, Monot C, Frangeul L, Blanc H, Gausson V, Vera-Otarola J, Cristofari G, Saleh MC. 2013. RNA-mediated interference and reverse transcription control the persistence of RNA viruses in the insect model Drosophila. Nat Immunol 14:396–403. 10.1038/ni.2542. [DOI] [PubMed] [Google Scholar]
- 25.Goic B, Stapleford KA, Frangeul L, Doucet AJ, Gausson V, Blanc H, Schemmel-Jofre N, Cristofari G, Lambrechts L, Vignuzzi M, Saleh MC. 2016. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat Commun 7:12410–12410. 10.1038/ncomms12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han Y, Wu Q, Ding SW. 2018. Templating antiviral RNAi in insects. Cell Host Microbe 23:290–292. 10.1016/j.chom.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Bell-Sakyi L, Kohl A, Bente DA, Fazakerley JK. 2012. Tick cell lines for study of Crimean-Congo hemorrhagic fever virus and other arboviruses. Vector Borne Zoonotic Dis 12:769–781. 10.1089/vbz.2011.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logan TM, Linthicum KJ, Bailey CL, Watts DM, Dohm DJ, Moulton JR. 1990. Replication of Crimean-Congo hemorrhagic fever virus in four species of Ixodid ticks (Acari) infected experimentally. J Med Entomol 27:537–542. 10.1093/jmedent/27.4.537. [DOI] [PubMed] [Google Scholar]
- 29.Honig JE, Osborne JC, Nichol ST. 2004. The high genetic variation of viruses of the genus Nairovirus reflects the diversity of their predominant tick hosts. Virology 318:10–16. 10.1016/j.virol.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 30.Růžek D, Bell-Sakyi L, Kopecký J, Grubhoffer L. 2008. Growth of tick-borne encephalitis virus (European subtype) in cell lines from vector and non-vector ticks. Virus Res 137:142–146. 10.1016/j.virusres.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Kholodilov IS, Litov AG, Klimentov AS, Belova OA, Polienko AE, Nikitin NA, Shchetinin AM, Ivannikova AY, Bell-Sakyi L, Yakovlev AS, Bugmyrin SV, Bespyatova LA, Gmyl LV, Luchinina SV, Gmyl AP, Gushchin VA, Karganova GG. 2020. Isolation and characterisation of Alongshan virus in Russia. Viruses 12:362. 10.3390/v12040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belova OA, Litov AG, Kholodilov IS, Kozlovskaya LI, Bell-Sakyi L, Romanova LI, Karganova GG. 2017. Properties of the tick-borne encephalitis virus population during persistent infection of ixodid ticks and tick cell lines. Ticks Tick Borne Dis 8:895–906. 10.1016/j.ttbdis.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Bell-Sakyi L, Attoui H. 2016. Article Commentary: Virus discovery using tick cell lines. Evol Bioinform Online 12s2:EBO.S39675. 10.4137/EBO.S39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nag DK, Brecher M, Kramer LD. 2016. DNA forms of arboviral RNA genomes are generated following infection in mosquito cell cultures. Virology 498:164–171. 10.1016/j.virol.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 35.Olson KE, Bonizzoni M. 2017. Nonretroviral integrated RNA viruses in arthropod vectors: An occasional event or something more? Curr Opin Insect Sci 22:45–53. 10.1016/j.cois.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Poirier EZ, Goic B, Tomé-Poderti L, Frangeul L, Boussier J, Gausson V, Blanc H, Vallet T, Loyd H, Levi LI, Lanciano S, Baron C, Merkling SH, Lambrechts L, Mirouze M, Carpenter S, Vignuzzi M, Saleh MC. 2018. Dicer-2-dependent generation of viral DNA from defective genomes of RNA viruses modulates antiviral immunity in insects. Cell Host Microbe 23:353–365. 10.1016/j.chom.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houé V, Gabiane G, Dauga C, Suez M, Madec Y, Mousson L, Marconcini M, Yen PS, de Lamballerie X, Bonizzoni M, Failloux AB. 2019. Evolution and biological significance of flaviviral elements in the genome of the arboviral vector Aedes albopictus. Emerg Microbes Infect 8:1265–1279. 10.1080/22221751.2019.1657785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houé V, Bonizzoni M, Failloux AB. 2019. Endogenous non-retroviral elements in genomes of Aedes mosquitoes and vector competence. Emerg Microbes Infect 8:542–555. 10.1080/22221751.2019.1599302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forth JH, Forth LF, Lycett S, Bell-Sakyi L, Keil GM, Blome S, Calvignac-Spencer S, Wissgott A, Krause J, Höper D, Kampen H, Beer M. 2020. Identification of African swine fever virus-like elements in the soft tick genome provides insights into the virus’ evolution. BMC Biol 18:136. 10.1186/s12915-020-00865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russo AG, Kelly AG, Enosi Tuipulotu D, Tanaka MM, White PA. 2019. Novel insights into endogenous RNA viral elements in Ixodes scapularis and other arbovirus vector genomes. Virus Evol 5:vez010. 10.1093/ve/vez010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nag DK, Kramer LD. 2017. Patchy DNA forms of the Zika virus RNA genome are generated following infection in mosquito cell cultures and in mosquitoes. J Gen Virol 98:2731–2737. 10.1099/jgv.0.000945. [DOI] [PubMed] [Google Scholar]
- 42.Andersson I, Karlberg H, Mousavi-Jazi M, Martínez-Sobrido L, Weber F, Mirazimi A. 2008. Crimean-Congo hemorrhagic fever virus delays activation of the innate immune response. J Med Virol 80:1397–1404. 10.1002/jmv.21222. [DOI] [PubMed] [Google Scholar]
- 43.Calistri A, Munegato D, Toffoletto M, Celestino M, Franchin E, Comin A, Sartori E, Salata C, Parolin C, Palù G. 2015. Functional interaction between the ESCRT-I Component TSG101 and the HSV-1 tegument ubiquitin specific protease. J Cell Physiol 230:1794–1806. 10.1002/jcp.24890. [DOI] [PubMed] [Google Scholar]
- 44.Salata C, Monteil V, Leijon M, Bell-Sakyi L, Mirazimi A. 2020. Identification and validation of internal reference genes for real-time quantitative polymerase chain reaction-based studies in Hyalomma anatolicum ticks. Ticks Tick Borne Dis 11:101417. 10.1016/j.ttbdis.2020.101417. [DOI] [PMC free article] [PubMed] [Google Scholar]