

ABSTRACT
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the coronavirus disease 19 (COVID-19) pandemic. Despite unprecedented research and developmental efforts, SARS-CoV-2-specific antivirals are still unavailable for the treatment of COVID-19. In most instances, SARS-CoV-2 infection initiates with the binding of Spike glycoprotein to the host cell ACE2 receptor. Utilizing the crystal structure of the ACE2/Spike receptor-binding domain (S-RBD) complex (PDB file 6M0J) in a computer-aided drug design approach, we identified and validated five potential inhibitors of S-RBD and ACE-2 interaction. Two of the five compounds, MU-UNMC-1 and MU-UNMC-2, blocked the entry of pseudovirus particles expressing SARS-CoV-2 Spike glycoprotein. In live SARS-CoV-2 infection assays, both compounds showed antiviral activity with IC50 values in the micromolar range (MU-UNMC-1: IC50 = 0.67 μM and MU-UNMC-2: IC50 = 1.72 μM) in human bronchial epithelial cells. Furthermore, MU-UNMC-1 and MU-UNMC-2 effectively blocked the replication of rapidly transmitting variants of concern: South African variant B.1.351 (IC50 = 9.27 and 3.00 μM) and Scotland variant B.1.222 (IC50 = 2.64 and 1.39 μM), respectively. Following these assays, we conducted “induced-fit (flexible) docking” to understand the binding mode of MU-UNMC-1/MU-UNMC-2 at the S-RBD/ACE2 interface. Our data showed that mutation N501Y (present in B.1.351 variant) alters the binding mode of MU-UNMC-2 such that it is partially exposed to the solvent and has reduced polar contacts. Finally, MU-UNMC-2 displayed high synergy with remdesivir, the only approved drug for treating hospitalized COVID-19 patients.
IMPORTANCE The ongoing coronavirus infectious disease 2019 (COVID-19) pandemic is caused by a novel coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). More than 207 million people have been infected globally, and 4.3 million have died due to this viral outbreak. While a few vaccines have been deployed, a SARS-CoV-2-specific antiviral for the treatment of COVID-19 is yet to be approved. As the interaction of SARS-CoV-2 Spike protein with ACE2 is critical for cellular entry, using a combination of a computer-aided drug design (CADD) approach and cell-based in vitro assays, we report the identification of five potential SARS-CoV-2 entry inhibitors. Out of the five, two compounds (MU-UNMC-1 and MU-UNMC-2) have antiviral activity against ancestral SARS-CoV-2 and emerging variants from South Africa and Scotland. Furthermore, MU-UNMC-2 acts synergistically with remdesivir (RDV), suggesting that RDV and MU-UNMC-2 can be developed as a combination therapy to treat COVID-19 patients.
KEYWORDS: SARS-CoV-2, COVID-19, Spike glycoprotein, ACE2, pseudovirus, remdesivir, drug design
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological agent of coronavirus disease 19 (COVID-19) emerged in early December 2019 in Wuhan City, China (1). Since its emergence, SARS-CoV-2 has claimed more than 4.3 million lives (https://coronavirus.jhu.edu/map.html) and caused unprecedented economic loss globally (2). Fourteen open reading frames (ORFs) of ∼30 kb positive (+) single-stranded SARS-CoV-2 RNA genome encode up to 16 nonstructural proteins, four structural proteins, and nine accessory proteins (3). The Spike glycoprotein (S-protein), membrane protein (M), and envelope protein (E) make the spherical envelope of SARS-CoV-2 (4). The S-protein consists of S1 and S2 subunits (5). The S1 subunit contains an N-terminal domain (NTD) and a receptor-binding domain (RBD). In contrast, the S2 subunit contains fusion peptide (FP), heptad repeat 1 (HR1), central helix (CH), heptad repeat 2 (HR2), connector domain (CD), transmembrane domain (TM), and a cytoplasmic tail (CT) (6). The S-RBD within the S1 subunit binds to the host cell receptor angiotensin-converting enzyme 2 (ACE2) and facilitates viral entry into the host cell (7), while the S2 subunit mediates membrane fusion (8). Although the binding of S-RBD and ACE2 is a well-documented determinant of cellular entry, depending on cell types, the virions may enter the cell through clathrin-mediated endosomal pathways or clathrin-independent nonendosomal pathways (9).
Developing a vaccine against emerging and reemerging viruses is one of the most rational approaches for the containment of their rapid transmission (10). Currently, more than 64 vaccine candidates against SARS-CoV-2 are under development (11). The technology employed for developing these vaccines includes but is not limited to mRNA vaccines, replication-defective viral vector vaccines, inactivated pathogen vaccines, protein subunit vaccines, and virus-like particles, among others (11). USA-FDA has issued emergency use authorization (EUA) to two mRNA-based vaccines: one developed by Pfizer-BioNTech and the other by Moderna. In addition, a nonreplicating viral vector-based vaccine developed by Johnson & Johnson received FDA EUA for use in the United States. These vaccines appear quite successful in SARS-CoV-2 containment. However, the newly emerging variants of concern (VOC) and variants of interest (VOI) of SARS-CoV-2, such as Delta and Delta Plus variants (12) containing multiple mutations within the S-RBD may compromise the efficacy of these vaccines (13, 14). In addition, the effectiveness and durability of these vaccines remain unknown for vulnerable populations, including children, pregnant women, immunocompromised individuals, and people with comorbidities for COVID-19 (15).
Attempts to repurpose several therapeutic candidates to control and/or contain the rapid virus transmission and COVID-19 associated mortality and morbidity have resulted in mild to modest efficacy (16, 17). To date, remdesivir (RDV; GS-5734) is the only antiviral drug that has been approved for the treatment of hospitalized COVID-19 patients (18) in the United States. A randomized, double-blind, placebo-controlled clinical trial conducted by the National Institute of Health (NIH) found a shorter hospitalization period for patients with COVID-19 when treated with RDV compared to the placebo controls (NCT04280705) (19). In contrast, another multicenter randomized clinical trial conducted by the World Health Organization (WHO) reported no superior effect of the RDV on hospitalized COVID-19 patients (NCT04315948) (20). According to the current U.S. Center for Disease Control and Prevention (CDC) guidelines, no specific therapy is recommended for nonhospitalized and hospitalized mild-to-moderate COVID-19 patients who do not require oxygen support. For the hospitalized patients requiring supplemental oxygen (with or without ventilation), RDV alone or RDV with dexamethasone has been recommended depending upon patient condition and availability of the two drugs (www.covid19treatmentguidelines.nih.gov). These facts underscore the necessity of developing potent antivirals that may serve as prophylactic and/or therapeutic agents against COVID-19. In addition, as vaccine hesitancy and availability are a reality, there is likely to be a population that may not receive any vaccination, further necessitating the development of potent small-molecule antivirals.
Developing an effective prophylactic against COVID-19 should preferentially target the viral S-RBD because the first step of the SARS-CoV-2 viral life cycle involves binding S-RBD to the host cell ACE2 receptor. Two high-resolution crystal structure complexes of S-RBD and ACE2 have been reported (21, 22). The crystal structure of S-RBD containing Wuhan-Hu-1 sequence in complex with human ACE2 (PDB entry 6M0J) (21) provides the atomic details of the interaction interface (21). The other crystal structure of the S-RBD/ACE2 complex (PDB entry shows the atomic details of the interaction between ACE2 and S-RBD of P.1 (or Gamma) variant (22). An analysis of PDB entry 6M0J showed that the S-RBD/ACE2 interface contains a pocket of appropriate size that can be targeted for in silico drug discovery endeavors. In our computer-aided drug design (CADD) approach, we docked ∼8 million compounds within this pocket to identify small molecules that have the potential to block S-RBD/ACE2 interaction. We selected five compounds based on the docking score and visual inspection of interactions of these compounds with S-RBD and ACE2 for testing their in vitro potential to block SARS-CoV-2 replication in cell-based assays. Two compounds (MU-UNMC-1 and MU-UNMC-2) blocked viral replication at sub-micromolar IC50. The efficacy of these compounds was then characterized in detail in a human bronchial epithelial cell line (UNCN1T). We also investigated the synergistic effect of the compounds with RDV. Finally, we also found that MU-UNMC-1 and MU-UNMC-2 inhibit replication of emerging variants of SARS-CoV-2 with comparable effectiveness.
RESULTS
Screening library of compounds using a computer-aided drug design (CADD) approach.
The flow chart used for screening the library of compounds using a CADD approach is depicted in Fig. 1A. The residues at the interface of S-RBD and ACE2 targeted for docking the compounds are described in Table 1 and Fig. 1B and C. The residues at the interface of S-RBD and ACE2 are shown in Fig. 1B, whereas Fig. 1C shows a zoomed-in view of the compound binding pocket selected for the docking of the compounds. Out of the ∼8 million compounds screened, for 10 best-fit compounds, their IUPAC name, molecular structure, docking score, and pharmacochemical properties are described in Table 2. Five compounds were selected based on the docking score to test their antiviral potential in the cell-based assays. The docked poses associated with the best docking score of the five compounds at the S-RBD/ACE2 interface are shown in Fig. 1D–I.
FIG 1.

In-silico screening of SARS-CoV-2 entry inhibitors. (A) Flow chart of screening Spike inhibitors using computer-aided drug designing approach. Panels B and C show the residues at the interface of S-RBD and ACE2 and zoomed-in view of the pocket selected for compound docking, respectively. An R programing package Bio3D determined the interface residues, and the figures were generated using PyMol software. The dotted line shows the discontinuous backbone of the protein (S-RBD, cyan, and ACE2, green). (D–I) Molecular docking of five compounds selected based on their glide score for in vitro screening of their antiviral activity against SARS-CoV-2. Panels G and H show the two orientations of MU-UNMC-7. The yellow dotted lines represent the polar interactions formed by the compounds with the nearest protein residues. The ACE2 residues represented as sticks are green carbons, whereas S-RBD residues are colored as cyan sticks. The protein backbone is represented as ribbons (green = ACE2 and cyan = S-RBD). The MU-UNMC compounds are shown in ball-and-stick representations with magenta carbons. All other atoms are colored by atom type (red = oxygen, blue = nitrogen, orange = sulfur, turquoise = fluorine, and light green = chlorine).
TABLE 1.
Residues at the interface of S-RBD and ACE2 as deduced from PyMol using PDB file 6M0J
| S-RBD residues | ACE2 residues |
|---|---|
| K417, V445, G446, Y449, Y453, L455, F456, Y473, A475, G476, F486, Y489, Q493, Q498, T500, N501, G502, V503, and Y505 | S19, Q24, T27, F28, D30, K31, H34, E35, E37, D38, Y41, Q42, L45, L79, M82, Y83, K353, G354, D355 and R357 |
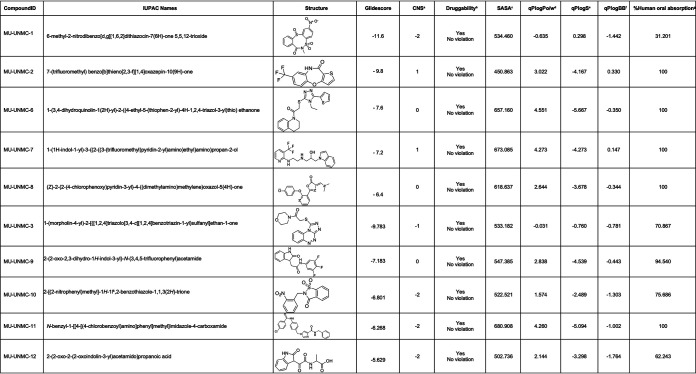
TABLE 2.
List of 10 best hit drugs like compounds obtained from in silico screening of libraries using Computer Aided Drug Designing Approach, Glide Score, and i-silico ADMET screening of the selected compounds
|
aPredicted central nervous system activity from −2 (inactive) to +2 (active). bPrediction of drug likeliness (acceptable range: 0-4 violation). c Total Solvent Accessible Surface Area: SASA (acceptable range: 300–1000). dPredicted octanol/water partition coefficient (acceptable range: −2–6.5). ePredicted aqueous solubility, S in mol/dm3 (acceptable range: −6.5–0.5). fPredicted brain/blood partition coefficient (acceptable range: -3.0–1.2). gPredicted percentage human oral absorption (<25% is poor and >80% is high).
The computed pharmacological properties of the top 10 compounds are shown in Table 2. These ADMET (absorption, distribution, metabolism, excretion, and toxicity) are key elements that determine the safety, uptake, elimination, metabolic behavior, and effectiveness of drugs. The values listed in Table 2 show that all compounds are druggable provided they are active. Since only two compounds (MU-UNMC-1 and MU-UNMC-2) showed inhibitory activities, further investigations focused on these compounds.
The best pose of MU-UNMC-1 and MU-UNMC-2 had a docking score of -11.6 and -9.8, respectively. The docking scores of MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 were -7.6, -7.2, and -6.4, respectively. The best pose (based on the docking score) of the MU-UNMC-1 was docked in a pocket formed by S-RBD residues R403, E406, Q409, K417, Y505, and ACE2 residues N33, H34, E37, R393, F390, P389, Q388, A387 (Fig. 1D). Compound MU-UNMC-2 was also docked in the same pocket. However, the interactions of MU-UNMC-2 with protein residues were different than those of MU-UNMC-2. MU-UNMC-2 docked in a pocket formed by Y505, R403, Y453 (from S-RBD), and N33, H34, E37, P389, F390, Q388, and A387 (Fig. 1E). As can be seen from the docking poses of the two compounds, MU-UNMC-1 has a more significant number of polar interactions (shown as dotted yellow lines) with the proteins than does MU-UNMC-2, which has only four polar interactions with the pocket residues. This may be a reason for a better docking score of MU-UNMC-2 compared with MU-UNMC-1. Compounds MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 were also docked in the same pocket as MU-UNMC-1 (Fig. 1F to I). However, a significant difference in the docking orientation of the compounds MU-UNMC-7 (Fig. 1G and 1H) was noted compared with MU-UNMC-1 or MU-UNMC-2. Thus, one of the two rings of MU-UNMC-7 was exposed to the solvent (Fig. 1H).
The infinite fit docking (IFD) showed that conformation of some sidechains within the binding pocket as the binding mode of the compounds was altered (Fig. 2). Thus, the conformation of S-RBD residues Y505 and R403 was significantly altered upon the flexible docking of MU-UNMC-1 (Fig. 2A). The sidechain conformation change of ACE2 residues N33, H34 and R393 was also altered upon IFD of MU-UNMC-1 within the binding pocket. In addition, the mode of MU-UNMC-1 was also significantly changed (Fig. 2B). The sidechain confirmation of several residues with the binding pocket remained unaltered. These residues include E406 and D405 of the S-RBD, and D30, E37, A386, E387, Q388, and P389 of ACE2. A more significant number of sidechain conformation change was altered upon IFD of MU-UNMC-2 (Fig. 2C). These residues include R403, Y453, Y495, and Y505 (S-RBD) and N33, H34, E37, D38, and K353 (ACE2). Sidechain conformation of S-RBD residues D405 and E406 and ACE2 residue R393 did not change upon IFD of MU-UNMC-2. As seen with MU-UNMC-1, the mode of MU-UNMC-2 was also altered (Fig. 2D).
FIG 2.

Change in sidechain conformation after conducting induced-fit docking of compounds MU-UNMC-1 and MU-UNMC-2. Only sidechains that have significant change upon IFD are shown. (A) Change in conformational change upon docking of MU-UNMC-1. Yellow carbons represent the conformation of S-RBD sidechains before IFD, whereas cyan carbon correspond to the sidechain conformation of S-RBD after IFD. (B) Mode of MU-UNMC-1 binding before (teal carbons) and after IFD (magenta carbons). (C) Change in conformational change upon docking of MU-UNMC-2. Yellow carbons represent the conformation of S-RBD sidechains before IFD, whereas cyan carbon correspond to sidechain conformation of S-RBD after IFD. (D) Mode of MU-UNMC-2 binding before (teal carbons) and after IFD (magenta carbons). Blue = nitrogen, oxygen = red, orange = sulfur, cyan = fluorine.
Screening the impact of the compounds on the binding of ACE2: Spike RBD of SARS-CoV-2.
To screen the impact of the five compounds selected as entry inhibitors using the CADD approach on the binding of ACE2 and SARS-CoV-2 Spike RBD, a Spike/ACE2 Inhibitor Screening Assay was used as described in material and methods. As illustrated in Fig. 3, we observed that all the compounds could reverse the binding between ACE2 and Spike RBD in a dose-dependent manner. While MU-UNMC-1 has an IC50 of <0.25 μM, the other compounds MU-UNMC-2, MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 have an IC50 of 0.45, 1.91, >5.0, and >5.0 μM, respectively. In summary, in this in vitro assay, all five compounds could block the binding of ACE2 and Spike RBD to different extents.
FIG 3.

Screening of potential drug-like compounds selected by a computer-aided drug design approach to block the binding of ACE2: SARS-CoV-2 Spike receptor-binding domain (RBD). MU-UNMC-1, MU-UNMC-2, MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 were tested at different concentrations in triplicate starting from 0.25 to 5 μM to evaluate their ability to inhibit binding of SARS-CoV-2 Spike RBD to immobilized human ACE2 using an ELISA as described in materials and methods. The IC50 values were computed using four-parameter variable slope sigmoidal dose-response models using Graph Pad Prism 8.0 software.
Determination of cellular toxicity of the compounds.
Cytotoxicity of all five compounds was determined in HEK293T-hACE2 cells. In addition, for MU-UNMC-1 and MU-UNMC-2, cytotoxicity was computed in Vero-STAT1 knockout, UNCN1T, and Calu-3 cells using a standard colorimetric MTT assay that measures cell proliferation and viability. The percent viability of the cells is plotted against the increasing concentration of the compounds, and the 50 percent cytotoxic concentration (CC50) of each compound was computed using four-parameter variable slope sigmoidal dose-response models. In HEK293T-hACE2 cells, all five compounds have CC50 values above 100 μM (Fig. 4A to E). In Vero-STAT1 knockout cells, MU-UNMC-1 has a CC50 of 6.21 μM (Fig. 4F), and MU-UNMC-2 has a CC50 of 7.13 μM (Fig. 4G) respectively. On the other hand, in UNCN1T and Calu-3 cells MU-UNMC-1 and MU-UNMC-2 have CC50 values above 100 μM (Fig. 4H-4K).
FIG 4.

Measurement of cytotoxicity of five drug-like compounds using MTT assay. Measurement of cytotoxicity of five drug-like compounds. (A–E) Viability of HEK293T-hACE2 cells in the presence of an indicated concentration of the compounds. (F, G) Measurement of cytotoxicity of MU-UNMC-1 and MU-UNMC-2 in Vero-STAT1 KO cells in the presence of an indicated concentration of the compounds. (H, I) Measurement of cytotoxicity of MU-UNMC-1 and MU-UNMC-2 in UNCN1T cells in the presence of an indicated concentration of the compounds. (J, K) Measurement of cytotoxicity of MU-UNMC-1 and MU-UNMC-2 in Calu-3 cells in the presence of an indicated concentration of the compounds.
Screening entry inhibition potential of compounds using lentiviral pseudovirus expressing SARS-CoV-2 Spike protein.
Replication of defective lentiviral particles expressing coronavirus Spike glycoprotein effectively mimics coronavirus host cell entry (33). Therefore, we produced lentiviral particles that incorporated surface-expressed SARS-CoV-2 Spike protein and determined the relative efficiency of viral entry without compound using human ACE2 expressing HEK-293T cells. HEK-293T-hACE2 cells were treated with increasing concentrations of the compounds (0.25 to 5 μM), transduced with pseudotyped lentiviral particles, and pseudotype viral entry was subsequently calculated relative to vehicle controls (DMSO) after 48 h. MU-UNMC-1 displayed a low level of viral entry inhibition with a slight increase at higher concentrations (Fig. 5A). Simultaneously, MU-UNMC-2 treatment resulted in significant and robust viral entry inhibition across a broad concentration range (Fig. 5B). In contrast, MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 failed to prevent the pseudovirus entry (Fig. 5C–E). Finally, the IC50 value was calculated for MU-UNMC-2, resulting in an IC50 value of 0.84 μM based upon a four-parameter variable slope sigmoidal dose-response model (Fig. 5F).
FIG 5.

Screening of entry inhibition potential of five drug-like compounds using pseudovirus assay. Screening entry inhibition potential of drug-like compounds in HEK-293T cells expressing human ACE2. (A–E) HEK-293T-hACE2 cells were pretreated with the indicated concentration of compounds and then inoculated with pseudotyped lentiviral particles expressing Spike glycoprotein of SARS-CoV-2. At 48 h postransduction, pseudotype entry was analyzed after normalization against untreated cells by determining luciferase activity in cell lysates. (F) The percentage inhibition of entry of pseudotyped lentiviral particles expressing Spike glycoprotein of SARS-CoV-2 was measured for MU-UNMC-2 at indicated concentration of the compounds (cells that received DMSO were considered vehicle controls). The IC50 value was computed using four-parameter variable slope sigmoidal dose-response models using Graph Pad Prism 8.0 software.
Antiviral efficacy of MU-UNMC-1 and MU-UNMC-2 against live SARS-CoV-2 isolates.
Next, we evaluated the antiviral efficacy of MU-UNMC-1 and MU-UNMC-2 against live SARS-CoV-2 in UNCN1T and Vero-STAT1 knockout cells. UNCN1T is a human bronchial epithelial cell line and serves as a disease-relevant context for the target tissue of SARS-CoV-2, lung epithelium. The Vero-STAT1 knockout cells are highly susceptible to viral infection due to the absence of STAT1, a transcription factor involved in the cellular antiviral interferon-mediated response. Therefore, these cells serve as a positive control for robust viral infection. The SARS-CoV-2 replication kinetics at 24 and 48 hpi were determined in the presence of increasing concentrations of our two compounds using the two cellular models. Based on SARS-CoV-2 viral loads in the culture supernatant of UNCN1T cells, MU-UNMC-1 and MU-UNMC-2 showed antiviral activity with an IC50 value of 0.67 and 1.72 μM at 24 hpi (Fig. 6A). In contrast, the compounds had an IC50 value of 1.16 and 0.89 μM, respectively, at 48 hpi (Fig. 6B). In Vero-STAT1 knockout cells, 24 hpi MU-UNMC-1 and MU-UNMC-2 have an IC50 value of 5.35 and 1.63 μM (Fig. 6C); while at 48 hpi, the compounds have an IC50 value of 2.94 and 0.54 μM, respectively (Fig. 6D). We also tested the antiviral efficacy of MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8 against live SARS-CoV-2 infected UNCN1T cells. We did not see any reduction in viral replication kinetics compared to DMSO vehicle control (data not shown). As new mutant variants of SARS-CoV-2 continue to emerge globally, we evaluated the antiviral efficacy of MU-UNMC-1 and MU-UNMC-2 against variants of SARS-CoV-2. As a proof of concept and based on their availability from BEI resources, we selected two mutant variants emerging from South Africa (linage: B.1.351) and Scotland (linage: B.1.222), respectively. Based on SARS-CoV-2 viral loads in the culture supernatant of Calu-3 cells, MU-UNMC-1 and MU-UNMC-2 showed comparable antiviral activity against the newly emerging variant strains compared to wild-type virus. MU-UNMC-1 has an IC50 value of 9.27 μM and 2.64 μM for South African (linage: B.1.351) variant (Fig. 7A) and Scotland (linage: B.1.222) variant (Fig. 7B) at 24 hpi, whereas MU-UNMC-2 has an IC50 value of 3.00 μM for South African (linage: B.1.351) variant (Fig. 7C) and 1.39 μM against Scotland (linage: B.1.222) variant at 24 hpi (Fig. 7D) respectively.
FIG 6.

SARS-CoV-2 dose-response curve in MU-UNMC-1 and MU-UNMC-2 treated and SARS-CoV-2 infected UNCN1T and Vero-STAT1 knockout cells. (A, B) MU-UNMC-1 (in blue) and MU-UNMC-2 (in green) dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 and 48 hpi in UNCN1T cells with indicated drug concentrations. (C, D) MU-UNMC-1 (in blue) and MU-UNMC-2 (in green) dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 and 48 hpi in Vero-STAT1 knockout cells with indicated compound concentrations.
FIG 7.

SARS-CoV-2 dose-response curve in MU-UNMC-1 and MU-UNMC-2 treated and SARS-CoV-2 variant of concern infected Calu-3 cells. (A) MU-UNMC-1 dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 hpi in Calu-3 cells infected with South Africa variant (linage: B.1.351) with indicated drug concentrations. (B) MU-UNMC-1 dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 hpi in Calu-3 cells infected with Scotland variant (linage: B.1.222) with indicated drug concentrations. (C) MU-UNMC-2 dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 hpi in Calu-3 cells infected with South Africa variant (linage: B.1.351) with indicated drug concentrations. (D) MU-UNMC-2 dose-response curve by percentage inhibition of SARS-CoV-2 replication 24 hpi in Calu-3 cells infected with Scotland variant (linage: B.1.222) with indicated drug concentrations.
Optimal binding mode of MU-UNMC-1 and MU-UNMC-2 to Wuhan-Hu-1 and B.1.351.
Because of the inherent rigid-body nature of our initial docking, it was not clear if the docked pose of MU-UNMC-1 and MU-UNMC-2 in a pocket at the S-RBD and ACE2 interface were the real binding modes. Therefore, we used IFD to obtain the optimal binding modes of MU-UNMC-1 (43 poses) and MU-UNMC-2 (39 poses) in Wuhan-Hu-1 S-RBD/ACE2 complex, and in B.1.351 S-RBD/ACE2 complex. In the IFD, the sidechains of the protein are optimized to obtain the best binding pose of a given compound. The best-docked poses obtained in the IFD of MU-UNMC-1 and MU-UNMC-2 in Wuhan-Hu-1 S-RBD/ACE2 complex are shown in Fig. 8A, B, E, to F, and in S-RBD/ACE2 complex of B.1.351 are shown in Fig. 8C, D, G, to H. As an example, the comparison of sidechain conformations near the MU-UNMC-2 binding site in Wuhan-Hu-1 S-RBD/ACE2 complex and B.1.135 S-RBD/ACE2 complex after IFD of MU-UNMC-2 in two viruses is shown in Fig. 9. It is clear from these figures that MU-UNMC-2 binds to the Wuhan-Hu-1 S-RBD/ACE2 complex in a different conformation than in B.1.351 S-RBD/ACE2 complex. While MU-UNMC-2 binds in the same pocket, the trifluoromethyl moiety of the compound was more exposed to the solvent in B.1.351 S-RBD/ACE2 complex than in the Wuhan-Hu-1 S-RBD/ACE2 complex (Fig. 8E, G). Increased exposure of MU-UNMC-2 to the solvent in B.1.351 appears is due to mutation N501Y, which reduced the size of the binding pocket. This exposure would most likely reduce the binding affinity of MU-UNMC-2 to the B.1.351 variant, which is reflected in the reduced efficacy of MU-UNMC-2 for B.1.351. MU-UNMC-2 was flipped ∼180° in B.1.351 compared with Wuhan-Hu-1 S-RDB/ACE2 complex (Fig. 8E–H). This flipping of MU-UNMC-2 decreased polar interactions (dotted lines) in B.1.351 S-RBD/ACE2 complex compared to the Wuhan-Hu-1 S-RDB/ACE2 complex (4 versus 5). A cumulative effect of increased exposure of MU-UNMC-2 and reduced polar interactions in the B.1.351 variant may be a reason for its reduced efficacy against B.1.351. Additionally, we evaluated the hydrophobic and Van der Waals interactions between the compouds and S-RBD/ACE2 complexes (Fig. 8I, J) using LigPlot+ (34). The hydrophobic and Van der Waals interactions between MU-UNMC-1 and S-RBD/ACE2 complex and MU-UNMC-2 and S-RBD/ACE2 complex were comparable.
FIG 8.

Optimal binding mode of MU-UNMC-1 and MU-UNMC-2 with S-RBD/ACE2 complexes of Wuhan-Hu-1 and B.1.351. The best docking pose of MU-UNMC-1 and MU-UNMC-2 was obtained from the IFD. Panels A and C show surface representation of Wuhan-Hu-1 and B.1.351 S-RBD (cyan)/ACE2 (green) complexes, respectively for MU-UNMC-1. (B, D) The details of interactions of MU-UNMC-1 with S-RBD/ACE2 complexes of Wuhan-Hu-1 and B.1.351 are shown in panels B and D. Panels E and G show surface representation Wuhan-Hu-1 and B.1.351 S-RBD (cyan)/ACE2 (green) complexes, respectively for MU-UNMC-2. (F, H) The details of interactions of MU-UNMC-2 with S-RBD/ACE2 complexes of Wuhan-Hu-1 and B.1.351 are shown in panels F and H. The yellow dotted lines represent the polar interactions between MU-UNMC-2 and proteins. All colors and the representations are the same as in Fig. 1, except the teal carbons of MU-UNMC-1/2 in the S-RBD/ACE2 complex of the B.1.351 variant. The two panels represent the different modes of binding of MU-UNMC-1 and MU-UNMC-2 in the two variants. (Panels I and J show the hydrophobic and Van der Waals interactions of MU-UNMC-1 and MU-UNMC-2, respectively, with the Wuhan S-RBD/ACE2 complex. The S-RBD residues are distinguished from ACE2 by marking RBD after the residue number. R393 of ACE2 and Y505 of S-RBD also form the H-bond with MU-UNMC-2, and green dotted lines represent them. Similarly, Y453 of S-RBD forms an H-bond with the N1 atom MU-UNMC-2 and is shown as a green dotted line. The circle size is proportional to the interacting atoms from each residue and the atoms that interact are represented by ‘rays’ emerging from the atoms.
FIG 9.
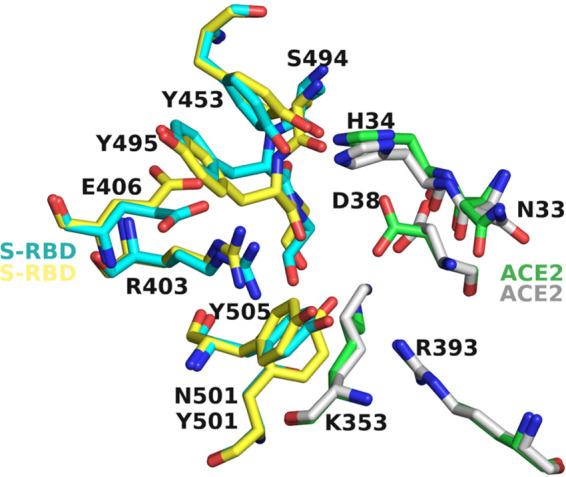
Comparison of the sidechain conformations near the binding site of MU-UNMC-2 in Wuhan-Hu-1 and B.1.351. Cyan and green carbons represent the S-RBD and ACE2 residues of Wuhan-Hu-1, whereas yellow and gray carbons represent the sidechain conformations of S-RBD and ACE2 in the B.1.351 variant. For clarity, the MU-UNMC-2 compound has been removed.
Antiviral efficacy of MU-UNMC-1 and MU-UNMC-2 after different times of addition to SARS-CoV-2 infected cells.
To determine at what stage of the viral life cycle the compounds MU-UNMC-1 and MU-UNMC-2 impart their antiviral effect, we performed a time of addition assay. The experimental schema of the assay is described in Fig. 10A. We observed that when both the compounds were added -2 hpi, there was more than an 80% reduction in SARS-CoV-2 infectivity compared to vehicle controls when the compounds were added at the time of infection, there was around 40% reduction in SARS-CoV-2 infectivity, and when the compounds were added +4 hpi, there was no significant difference in SARS-CoV-2 infection compared with vehicle controls (Fig. 10B). In summary, this result confirms that MU-UNMC-1 and MU-UNMC-2 are entry inhibitors that predominantly block the entry of the virus to host cells by interacting with the ACE2 and SARS-CoV-2 Spike RBD binding interface.
FIG 10.

Impact of time addition of the compounds on replication of SARS-CoV-2 in Vero-STAT1 knockout cells. (A) An experimental outline describing the time of adding MU-UNMC-1 and MU-UNMC-2 to the cells, SARS-CoV-2 infection, and measurement of viral replication kinetics at the termination of the experiment. (B) Percentage of SARS-CoV-2 replication in the presence of vehicle control (DMSO), MU-UNMC-1 (5 μM) and MU-UNMC-2 (5 μM) at -2 hpi, +0 hpi and +4 hpi in Vero-STAT1 knockout cells, respectively.
Chemical properties of MU-UNMC-2.
We used the SwissADME web portal to compute the biophysical properties of all the compounds tested in this study. Here we discuss the properties of MU-UNMC-2, which showed the synergistic effect with RDV. The Log Po/w value of this compound was 2.27 suggesting a high permeability of the compound. The predicted solubility of the compound was moderate. However, the pharmacokinetics data showed that the compound MU-UNMC-2 has high gastrointestinal absorption. It was expected not to inhibit CYP2C9, CYP2D6, and CYP3A4 suggesting low toxicity of the compound. The compound followed all Lipnski’s Rules of Five, and it was predicted to exhibit high drug likeliness with no PAINS (Pan-assay interference compounds).
Combinational antiviral efficacy of RDV/MU-UNMC-1 and RDV/MU-UNMC-2.
After confirming the effectiveness of MU-UNMC-1 and MU-UNMC-2 in blocking the cellular entry of SARS-CoV-2, we evaluated the combinational antiviral effect of MU-UNMC-1/MU-UNMC-2 with RDV. As described in Material and Methods, the dose-response curve of RDV was determined at different fixed-dose combinations of MU-UNMC-1, and MU-UNMC-2 using SARS-CoV-2 infected UNCN1T cells (Fig. 11A and 12A). Similarly, the dose-response curves of MU-UNMC-1 and MU-UNMC-2 were determined at different fixed-dose combinations of RDV using SARS-CoV-2 infected UNCN1T cells (Fig. 11B and 12B). The dose-response percent inhibition matrix of single and combination treatment of RDV/MU-UNMC-1 and RDV/MU-UNMC-2 is described in Fig. 11C and 12C, respectively. The 3-D interaction landscape of the combinational treatment was computed based on Loewe additive model using SynergyFinder v.2 in SARS-CoV-2 infected UNCN1T cells 24 hpi. Synergy maps highlight synergistic and antagonistic dose regions in red and green, respectively. A negative Loewe synergy score indicates an antagonistic drug combination, a score between 0 to 10 indicates the additive effect of drug combinations, and a score above 10 indicates a synergistic drug combination. RDV/MU-UNMC-1 combination has a Loewe synergy score of -30.69, indicating an antagonistic effect (Fig. 11D). On the other hand, RDV/MU-UNMC-2 has a Loewe synergy score of 26.64, indicating a synergistic effect (Fig. 12D). The synergistic/antagonistic effect of drug combinations are reconfirmed using Chou and Talalay combination index (CI) theorem (30). As per Chou and Talalay’s combination index theorem, CI < 1, = 1, and >1 show synergism, additive effect, and antagonism, respectively. Using CompuSyn, we computed CI values of RDV/MU-UNMC-1 as >1 and CI value of RDV/MU-UNMC-2 as CI < 1 confirming their antagonistic and synergistic effect respectively. Using CompuSyn, in UNCN1T cells, we obtained a dose reduction index (DRI) of 28.30 for MU-UNMC-2 and 2.33 for RDV, respectively. (DRI is the estimation of fold reduction in dose of a drug when used in combination with other drugs [having a synergistic effect] to achieve a similar level of effect that are comparable with those achieved with individual drugs).
FIG 11.

Combinational effect of remdesivir and MU-UNMC-1 treatment against SARS-CoV-2 infected UNCN1T cells at 24 h postinfection. (A) Dose response curve of remdesivir in SARS-CoV-2 infected UNCN1T cells at 24 hpi in the presence of different fixed concentrations of MU-UNMC-1; (B) Dose-response curve of MU-UNMC-1 in SARS-CoV-2 infected UNCN1T cells at 24 hpi in the presence of a different fixed concentration of remdesivir; (C) Dose-response percent inhibition matrix of single and combined treatment of remdesivir and MU-UNMC-1 in SARS-CoV-2 infected UNCN1T cells at 24 hpi. (D) 3-D interaction landscape between remdesivir and MU-UNMC-2 calculated based on Loewe additive model using SynergyFinder v.2 in SARS-CoV-2 infected UNCN1T cells at 24 hpi (Loewe synergy score -30.69; with most synergistic area score of -21.34).
FIG 12.

Combinational effect of remdesivir and MU-UNMC-2 treatment against SARS-CoV-2 infected UNCN1T cells at 24 h postinfection. (A) Dose-response curve of remdesivir in SARS-CoV-2 infected UNCN1T cells at 24 hpi in the presence of a different fixed concentration of MU-UNMC-2; (B) dose-response curve of MU-UNMC-2 in SARS-CoV-2 infected UNCN1T cells at 24 hpi in the presence of a different fixed concentration of remdesivir; (C) dose-response percent inhibition matrix of single and combined treatment of remdesivir and MU-UNMC-2 in SARS-CoV-2 infected UNCN1T cells at 24 hpi. (D) 3-D interaction landscape between remdesivir and MU-UNMC-2 calculated based on Loewe additive model using SynergyFinder v.2 in SARS-CoV-2 infected UNCN1T cells at 24 hpi (Loewe synergy score 26.63; with most synergistic area score of 37.25).
DISCUSSION
Using the in silico workflow, we screened ∼8 million compounds for their potential to bind at the interface of the SARS-CoV-2 S-RBD and human ACE2. Five compounds were selected based on the docking score and visual inspection to test their efficacy in SARS-CoV-2 replication cell-based assays. Initially, using an ELISA-based biophysical assay, we confirmed that the compounds could block the binding of S-RBD/ACE2 with varying concentrations. One compound, MU-UNMC-2, exhibited potent entry inhibition in lentiviral-based pseudovirus assays. In contrast, another compound, MU-UNMC-1, showed only a modest inhibition activity against virus entry even though MU-UNMC-1 showed high binding and a high docking score. The modest inhibition of MU-UNMC-1 in pseudotyped appears to be related to the low stability of its mono-sulfonyl group. The other three compounds (MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8) did not block virus entry or replication. The antiviral activity of the two compounds active in pseudovirus assays was also tested to block SARS-CoV-2 replication in a physiologically relevant human bronchial epithelial cell line (UNCN1T). In the 2-D in vitro culture system, both MU-UNMC-1 and MU-UNMC-2 showed antiviral activity with IC50 values in the sub-micromolar range. In addition, our drug addition assay confirmed that MU-UNMC-1 and MU-UNMC-2 act at the entry of SARS-CoV-2 infection. These results demonstrated that both MU-UNMC-1 and MU-UNMC-2 are potential SARS-CoV-2 entry inhibitors.
As with all RNA viruses, SARS-CoV-2 has been mutating. While most mutations are destructive for viral fitness and survival, a minor fraction may evolve with the altered transmission, pathogenicity, and immune escape capability. Several SARS-CoV-2 variants that appear to have high transmission, pathogenicity, and immune escape capability emerged in different parts of the world (35). WHO classified some of them as variants of concern that includes Delta (B.1.617.2) from India, Gamma (P1) from Brazil, Beta (B.1.351) from South Africa, and Alpha (B.1.1.7) from the U.K (13, 36, 37). We tested the efficacy of MU-UNMC-2 against South African variants (B.1.351 or Beta) and the Scotland variant (B.1.222). To assess if the two lead compounds can be docked with a significant docking score, we generated a molecular model of S-RBD/ACE2 using the S-RBD/ACE2 complex crystal structure of P.1 (Gamma) variant (PDB entry 7NXC) (22) and docked MU-UNMC-1 and MU-UNMC-2 into this model. The model of S-RBD/ACE2 complex generated using ‘Prime’ (Schrödinger Suite) had ∼0.5 Å root-mean-square deviation (RMSD) when compared with the crystal structure of Wuhan-Hu-1 S-RBD/ACE2 complex (PDB entry 6M0J). This is not surprising as the RMSD between P.1 variant S-RBD/ACE2 complex and Wuhan-Hu-1 S-RBD/ACE2 complex is also less than ∼0.5 Å. A low RMSD between two crystal structure complexes of S-RBD/ACE2 strongly suggests that mutation does not significantly cause overall structural changes. Hence, the impact of the mutant appears to change in the local geometry of the Spike protein.
In the CADD, rigid body docking is conducted for virtual screening of compounds (i.e., without optimizing the sidechain conformation). In the IFD, the sidechain and ligand conformations are optimized to get an optimal binding pose. However, the rigid-body docking does not provide the optimal configuration of the binding pocket and the ligand. The sidechain conformational change upon ligand binding can also be assessed by the molecular dynamics (MD) simulations, a proven computational technique to determine protein structure changes under various conditions (temperature, pH, ion strength, etc.). However, MD simulations are not widely used in CADD, primarily for two reasons: (i) The MD simulations are not computationally cost-effective in CADD, and (ii) it is cumbersome to obtain specific sidechain conformation. Hence, the CADD tools such as IFD are employed. As shown in Fig. 9E and G, the conformation of the sidechains of Y453 and Y505 of S-RBD in the WT (Wuhan-Hu-1) and H34 of ACE2 change to interact with MU-UNMC-2, whereas the conformation of R403 (S-RBD) and K353 (ACE2) in B.1.351 is adjusted to remove the steric clashes. For example, the steric clash between the ring O atom of MU-UNMC-2 and the Nζ atom of K351 in ACE2 (1.9 Å) is removed by reorienting the K351 sidechain. This kind of specific interaction details can be obtained only after computationally expensive MD simulations.
Our docking showed that MU-UNMC-2 binds in a pocket formed by Y505, R403, Y453 (from S-RBD), and N33, H34, E37, P389, F390, Q388, and A387 (ACE2). None of these residues in B.1.351 directly interact with MU-UNMC-2, confirming that the signature mutations of B.1.351 (N501Y) present at the S-RBD/ACE2 interface may not impact the efficacy of this compound. A slight reduction in the effectiveness of MU-UNMC-2 against these two variants may be due to the altered geometry of the binding pocket as Y501 is bulkier than N501, as noted in our IFD docking (Fig. 9). Our in silico analysis showed that none of the emerging SARS-CoV-2 variants, including the UK (Alpha), the Brazil/Japan (Gamma), or California variant (Eta/Iota) have mutations within the MU-UNMC-2 binding pocket (data not shown) (13).
The crystal structure S-RBD/ACE2 complex solution triggered the drug-discovery efforts to discover entry inhibitors, including peptide analogs, monoclonal antibodies, and protein chimeras (38–46). Despite these new technologies and macromolecules as therapeutic agents, small molecules remain the preferred choice for drug development due to their better pharmacokinetic profile, stability, and dosage logistics (47, 48). Also, small molecules have the advantage of logistics to remote locations, and their production cost is economical compared to peptide-based therapeutics. Thus, several studies are under way to develop small molecules and peptide inhibitors to block the human ACE2 (49–52). ACE2 is a principal regulator of the renin-angiotensin system (RAS) that regulates the cardiovascular and renal systems. Still, it is not a preferable therapeutic target, as its inhibition may disrupt the physiological homeostasis of the systems (53).
In addition, the ACE2/S-RBD interface is distal to the protease active site of ACE2. Hence, inhibitors developed against the ACE2 protease activity would most likely not affect S protein binding to ACE2. ACE2 protects lung injury in acute respiratory distress syndrome caused by respiratory viral infections (54). In this respect, our strategy of screening small molecules that block S protein and ACE2 interaction are superior to protein-based therapeutics.
In the arena of antiviral therapeutics, the use of combination/repurposing drugs that target different stages of the viral life cycle and host factors is well documented, resulting in superior virological and physiological response compared to monotherapy (17, 55). This approach increases the overall efficacy of the treatment, reduces the dosage requirement of individual drugs, improves toxicity profile, and lowers the chances of developing drug resistance (56). This approach is time-tested during the development of combinational antiretroviral therapy to treat human immunodeficiency virus-1 (17, 57). During the early days of the COVID-19 pandemic, investigational combination therapy approaches have been tested in severely ill hospitalized patients. These strategies only yielded mild to moderate responses in different clinical settings (58, 59). In a similar pursuit, we examined the synergy between RDV (an RdRp inhibitor) and MU-UNMC-1 or MU-UNMC-2 using human bronchial epithelial cell line and SARS-CoV-2 virus. Our finding showed a 28.3-fold and 2.3-fold reduction in dosages for MU-UNMC-2 and RDV, respectively, thereby supporting the combinatorial use of the two compounds against monotherapies. On the contrary, we observed an antagonistic effect MU-UNMC-1/RDV combinational treatment. The combination therapy targeting two critical stages of the viral life cycle, namely, cell entry (MU-UNMC-2) and replication (RDV), enhances the overall therapeutic efficacy and reduces the chances of resistance associated with one drug. The reduction in RDV dose is also expected to reduce its adverse side effects that substantially limit its use in clinical settings (19, 60).
Our strategy combines in silico drug screening and in vitro validation in the cell-based system, which has a significant advantage over the classical drug-discovery approaches. It substantially reduces the resources and expenses for initial screening of drug-like compounds. It increases the probability of selecting the best lead compounds with desired therapeutic profile with fewer trials. Our strategy also shortens the lead identification to optimization, preclinical test, and translational application to clinical trials. Although we have not optimized the lead compound, we are in the process of doing so, and the results will be presented elsewhere. However, initial theoretical estimation of the ADME profile suggests that the compound MU-UNMC-2 has a favorable physicochemical profile and can be quickly considered in pre-clinical trials. With the emergence of new variants of SARS-CoV-2, particularly the increase in transmission of delta and delta plus (12, 37) that are supposed to have accelerated transmission, new potent antivirals are warranted.
One of the limitations of the present compounds is their relatively narrow selectivity index. However, these are lead compounds, and using their design and synthesis of new molecules with a better selectivity index is under investigation in our laboratories. The SARS-CoV-2 Spike can be activated by early plasma membrane or late endosomal membrane entry as per the availability of proteases (61). The Vero cells utilize the late endosomal pathway using TMPRSS2 and cathepsin L for Spike cleavage and viral entry to host cells used in the present study. While in Calu-3 cells, the early pathway is followed, where furin-based cleavage of Spike mediates the viral entry in host cells (8). Recent studies reported that in alpha and delta variants of SARS-CoV-2 isolates, mutations in the furin cleavage sites make the sequence more basic, facilitating more efficient furin cleavage responsible for their higher transmissibility in vivo (62, 63). For this reason, we tested our compounds in Calu-3 cells against the mutant variants for better understanding their antiviral activity against these viral isolates.
In summary, herein, we report the identification of drug-like compounds (MU-UNMC1 and MU-UNMC-2) that are potential SARS-CoV-2 entry inhibitors. MU-UNMC-2 alone or in combination with RDV showed antiviral activity against the viral isolates from the early days of the pandemic and against emerging mutant variants of SARS-CoV-2 at micromolar concentrations. In addition, these compounds’ ADME toxicity properties were well within the criteria to fall into the drug-likeliness category. Hence, MU-UNMC-2 may potentially be a prophylactic/therapeutic drug against COVID-19 once validated in vivo.
MATERIALS AND METHODS
Identification of potential inhibitors using the computer-aided drug design approach.
To identify potential SARS-CoV-2 entry inhibitors, we docked ∼8 million drug-like compounds from an in-house library of compounds. The library consists of compounds from MayBridge Hitfinder compounds, small molecules from the Zinc database (zinc.docking.org), ChEMBL, Bingo, JChemforExcel, ChemDiff, and BindingMOAD (https://www.click2drug.org/index.php#Databases), and all compounds have been prepared in their docking-ready conformation using ‘LigPrep’ program of Schrödinger Suite (Schrödinger LLC, NY). The S-RBD/ACE2 complex [Protein Databank Entry 6M0J; Lan et al. 2020 (21)] was used for in silico screening. A docking-ready structure was generated by the “Protein Preparation Wizard” of the Schrödinger Suite (Schrödinger LLC, NY), which adds the hydrogen atoms, missing sidechains, and assigns protonation states to histidine, glutamine, and asparagine residues together with the optimization of the hydrogen atoms’ orientation. The resulting structure was energy minimized using OPLS_2005 forcefield for 10,000 iterations to remove steric conflicts. Potential compound binding sites were identified by SiteMap (Schrödinger Suite) and SiteID (SybylX-2.1, Certera, Princeton, NJ). We selected a binding pocket present at the interface of S-RBD/ACE2 for the docking of the library compounds (details of interface residues and pocket are given in ‘Results’ section. The Glide program of Schrödinger Suite (23) with SP (Simple Precision) was used in initial docking in a grid box of 20 × 20 × 20 Å3 size. The top 500 compounds based on the docking score were re-docked using the XP (Extra Precision) (24) option of Glide. These results were then manually visualized for the interactions between compounds and protein (S-RBD and ACE2). Five compounds were finally selected for testing their in vitro inhibitory activity in cell-based assays (Table 2). To gain insight into the binding mode of these compounds and accessing the flexibility of the binding site residues, we also conducted flexible docking using the IFD (25) program of the Schrödinger Suite.
Molecular modeling of S-RBD of B.1.351 variant in complex with ACE2.
In the absence of a high-resolution crystal structure of the S-RBD of B.1.351 complex with ACE2, we used the S-RBD/ACE2 complex crystal structure of the P.1 variant [(22); PDB entry 7NXC]. Mutation K417T in the P.1 variant was altered to K471N to generate the structure of the B.1.351 S-RBD variant using the Prime (26) modeling program of Schrodinger Suite. This structure was then used in ‘Induced Fit Docking’ of MU-UNMC-1 and MU-UNMC-2.
Chemical properties of the compounds.
The pharmacochemical properties of the top 10 compounds were determined by QikProp program of the Schrodinger Suite (Schrödinger, LLC, NY). The input data were provided in the form of the three-dimensional structure of the compounds.
Reagents and cell lines.
RDV (GS-5734) was obtained from Selleck Chemicals LLC (Houston, TX). The SARS-CoV-2 entry inhibitors were obtained from MolPort (Riga, Latvia). Calu-3 (ATCC HTB-55), Vero E6 (CRL-1586), and Vero-STAT1 knockout cells (CCL-81-VHG) were obtained from ATCC. Vero E6 and Vero-STAT1 knockout cells were cultured in DMEM containing 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 units/ml penicillin, 100 units/ml streptomycin, and 10 mM HEPES (pH 7.4). Calu-3 cells were cultured in Eagle's Minimum Essential Medium (ATCC 30–2003) containing 10% FBS. UNCN1T cells (a human bronchial epithelial cell line; Kerafast catalog number ENC011) were cultured in BEGM media (Bronchial Epithelial Cell Growth Medium; Lonza catalog number CC-3170) in FNC (Athena Enzyme Systems catalog number 0407) coated 96-well plates. All other reagents (molecular biology grad fine chemicals) used in the study were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise mentioned.
ACE2: SARS-CoV-2 Spike RBD inhibitor screening assay.
All five compounds (MU-UNMC-1, MU-UNMC-2, MU-UNMC-6, MU-UNMC-7, and MU-UNMC-8) were tested in triplicate in the concentration range from 0.25 to 5.00 μM using the ACE2: SARS-CoV-2 Spike Inhibitor Screening assay kit (BPS Bioscience catalog number 79936) per the manufacturer’s instruction. In brief, the ACE2 protein was thawed on ice, diluted to 1 μg/ml using 1X PBS, applied at 50 μl/well to the 96-well nickel-coated plate provided in the kit, and incubated for 1 h at room temperature with gentle shaking. The plate was then washed three times with 1X immune buffer 1, 100 μl of 1X blocking buffer 2 was added/well and incubated at room temperature for 10 min with slow shaking. Next, 10 μl of the compounds was added/well in triplicate and incubated for 1 h at room temperature with slow shaking. Ten μl of 5% DMSO was used as vehicle control. After that, 5 nM SARS-CoV-2 Spike (RBD)-Fc (20 μl) was added/well and incubated at room temperature for 10 min with slow shaking. The plate was rewashed three times with 1X immune buffer 1, 100 μl of 1X blocking buffer 2 was added/well and incubated at room temperature for 10 min with slow shaking. Next, 1:1000 diluted anti-mouse-Fc-HRP was added/well of the plate and incubated for 1 h at room temperature with slow shaking. Finally, the plate was rewashed as described above and HRP substrate was added to produce chemiluminescence, which was measured using a SpectraMax i3x multi-mode plate reader (Molecular Devices, San Jose, CA).
MTT cell viability assay.
HEK-293T-hACE2, UNCN1T, Vero-STAT1 knockout, and Calu-3 cells were seeded at the density of 20,000 cells/well in a 96-well plate containing 100 μl complete media specific for each cell type. Cells were incubated for 12 h at 37°C in a humidified 5% CO2 incubator for adherence. After 12-h incubation, the media was replaced with fresh media, and HEK-293T-hACE2 cells were treated with the five compounds at concentrations ranging between 0.001 to 100 μM. The Calu-3, UNCN1T, and Vero STAT1 knockout cells were treated with MU-UNMC-1 and MU-UNMC-2. Untreated cells were considered a negative control, and DMSO treated cells were considered vehicle controls. After the treatment, cells were incubated at 37°C in humidified 5% CO2 incubator. Seventy-two hour posttreatment, 20 μl of MTT substrate (5 mg/ml) was added to each well and incubated for 4 additional hours at 37°C in the dark. Then the culture media was carefully removed, and blue formazan crystals were dissolved in 200 μl of DMSO, and the purple color was read at 595 nm with a reference filter of 620 nm.
Production and titration of lentiviral-based pseudovirus expressing SARS-CoV-2 Spike glycoprotein.
For pseudotyping, lentiviral particles expressing SARS-CoV-2 Spike protein were generated as described by Crawford et al. (27). In brief, 3 × 106 HEK-293T cells were co-transfected with a plasmid containing a lentiviral backbone expressing luciferase and ZsGreen (BEI catalog number NR-52516), a lentiviral helper plasmid expressing HIV Gag-Pol (BEI catalog number NR-52517), a lentiviral helper plasmid expressing HIV Tat (BEI catalog number NR-52518), and a lentiviral helper plasmid expressing HIV Rev (BEI catalog number NR-52519) along with a plasmid expressing the Spike protein of SARS-CoV-2 (7) using jetPRIME transfection reagent (Polyplus-transfection; NY) per the manufacturer’s instructions. The culture supernatant containing pseudovirus particles was harvested at 48 h posttransfection, by centrifugation at 1200 rpm for 10 min and filtration through a 0.45 μM filter to remove cellular debris and then stored at −80°C freezer in aliquots for downstream applications. The viral titers were determined using engineered HEK-293T cells expressing the human ACE2 receptor. For this purpose, 12,500 HEK293T-hACE2 cells were seeded per well in a poly-l-lysine-coated 96-well plate. 24 h after seeding, lentiviral particles were serially diluted with complete DMEM supplemented with Polybrene (5 mg/ml), and 50 μl of each dilution were added in four replicate wells. 48 h post addition, pseudoviral transduction efficiency was determined by measuring firefly luciferase activity in cell lysates using a bright-glo luciferase assay system (Promega, Madison, WI; catalog number E2610). The luminescence was measured using a SpectraMax i3x multi-mode plate reader (Molecular Devices, San Jose, CA) and relative luminescence units (RLUs) were plotted against virus dilutions.
SARS-CoV-2 entry inhibitor screening assay.
For screening SARS-CoV-2 entry inhibitors, 24 h before starting the assay, 20,000 HEK-293T-hACE2 cells were seeded per well in a poly-l-lysine-coated 96-well plate. On the day of the assay setup, different concentrations of the five compounds were mixed with the lentiviral particles expressing SARS-CoV-2 Spike protein (1.00 × 105 RLU/well) and incubated at 37°C for 30 min followed by the addition of 50 μl lentiviral particle-compound complex in the cells supplemented with Polybrene (5 mg/ml). After 48 h, the activity of firefly luciferase was measured as described above to calculate the ability of the compounds to block the entry of transduced pseudoviral particles in HEK-293T-hACE2 cells.
Production and titration of SARS-CoV-2 stocks.
SARS-CoV-2 isolates USA-WI1/2020 (BEI catalog number NR-52384), hCoV-19/South Africa/KRISP-EC-K005321/2020 (BEI catalog number NR-54008), and hCoV-19/Scotland/CVR2224/2020 (BEI catalog number NR-53945) were passaged in Vero-STAT-1 knockout cells. The viral titer was determined using the plaque assay (28). In brief, Vero E6 cells were seeded in 6-well plates. After 24 h, cells were washed with sterile 1X PBS. The viral stock was serially diluted and added to the cells in duplicate with fresh media, and the plates were incubated at 37°C for 1 h with occasional shaking every 15 min. Then, 2 ml of 0.5% agarose in minimal essential media (MEM) containing 5% FBS and antibiotics was added per well. Plates were incubated at 37°C for 72 h. Then, the cells were fixed with 4% paraformaldehyde overnight, followed by removing the overlay and staining with 0.2% crystal violet to visualize PFU. All assays were performed in a BSL-3 laboratory setting. The viral stocks used for all antiviral assays were generated in passage 1–2 of the initial stock obtained from BEI.
Assessment of antiviral activity of selected compounds.
The antiviral potential of the compounds were screened as reported earlier (29). In brief, UNCN1T or Vero-STAT1 knockout cells were seeded in 96-well plates 24 h before infection at 20,000 cells/well, or 48 h prior to infection for Calu-3 cells at the same seeding density as before. Different compounds ranging between 0.001 μM and 10 μM were added to the cells 2 h prior to the infection. The cells were infected with 0.1 MOI of SARS-CoV-2 using Opti-MEM I reduced serum medium (Thermo Fisher catalog number 31985062) and incubated for 1 h at 37°C with 5% CO2. For vehicle control, cells were treated with the same concentration of DMSO. Mock-infected cells received only Opti-MEM I reduced serum medium. At the end of the incubation of the virus, the inoculum was removed, cells were washed with 1X PBS three times, and fresh media was added supplemented with the same concentration of compounds. Culture supernatant was collected at 24 hpi (hour postinfection) and 48 hpi. The SARS-CoV-2 viral load was quantified in the culture supernatant using RT-QPCR with primer probes targeting the E gene of SARS-CoV-2 using PrimeDirect Probe RT-qPCR Mix (TaKaRa Bio USA, Inc) and Applied Biosystems QuantStudio3 real-time PCR system (Applied Biosystems, Waltham, MA, USA) per manufacturer’s instructions. Primers and probes used for SARS-CoV-2 RNA quantification were as follows: E_Sarbeco_F1: 5′–ACAGGTACGTTAATAGTTAATAGCGT–3′ (400 nM), E_Sarbeco_R2: 5′–ATATTGCAGCAGTACGCACACA–3′ (400 nM) and E_Sarbeco_P1: 5′– FAM–ACACTAGCCATCCTTACTGCGCTTCG-BHQ1–3′ (200 nM) as recommended by the WHO. The SARS-CoV-2 genome equivalent copies were calculated using quantitative PCR (qPCR) control RNA from heat-inactivated SARS-CoV-2, isolate USA-WA1/2020 (BEI catalog number NR-52347). The percentage inhibition of SARS-CoV-2 replication in MU-UNMC-1 and MU-UNMC-2 treated cells was calculated with respect to viral loads in vehicle control wells that received DMSO (considered 0% inhibition) and negative control wells (uninfected cells). IC50 values were calculated using four-parameter variable slope sigmoidal dose-response models using Graph Pad Prism 8.0 software.
Time of compound addition assay.
Vero-STAT1 knockout cells were seeded in 24-well plates and incubated overnight. The next day, MU-UNMC-1 (5 μM) and MU-UNMC-2 (5 μM) were added to the cells -2 h prior to infection, during infection (0 h), and +4 hpi. Then cells were infected with 0.1 MOI SARS-CoV-2. The culture supernatants were collected at 24 hpi, and percent inhibition of viral replication was calculated under different exposure conditions using RT-qPCR.
Measuring the combinational antiviral potential of MU-UNMC-1/MU-UNMC-2 and RDV.
To determine the possible synergistic antiviral effect of MU-UNMC-1/RDV and MU-UNMC-2/RDV against SARS-CoV-2 replication, we tested combined doses of MU-UNMC-1/RDV and MU-UNMC-2/RDV in SARS-CoV-2-infected UNCN1T cells. For these assays, the cells were seeded in 96-well plates (20,000 cells/well) 24 h before infection and then treated with respective combinations of compounds 2 h before infection. The cells were infected with 0.1 MOI of SARS-CoV-2 isolate USA-WI1/2020 as described above. The percentage inhibition of viral replication for each dose-combination was determined by RT-qPCR as described above. The percent inhibition of viral replication for a 1:1 fixed-dose combination of the compounds was used to generate dose-response plots. The CI was calculated using multiple drug effect equations developed by Chou and Talalay using the CompuSyn algorithm (https://www.combosyn.com). CI values of <1 indicate synergy, CI values >1 indicate antagonism, and values equal to 1 indicate additive effects (30, 31). Dose-response percent inhibition matrix of single and combined treatment of RDV/MU-UNMC-1 and RDV/MU-UNMC-2 in SARS-CoV-2 infected UNCN1T Vero-STAT1 knockout cells 24 hpi and 3-D interaction landscape were calculated based on Loewe additive model using SynergyFinder v.2 (32).
Statistical analysis.
The CC50 and IC50 values were computed using four-parameter variable slope sigmoidal dose-response models using GraphPad Prism (version 8.0). The CI was calculated using Chou and Talalay's multiple drug effect equation using the CompuSyn algorithm (https://www.combosyn.com). The 3-D interaction landscape between RDV and MU-UNMC-2 was calculated based on Loewe’s additive model incorporated within SynergyFinder v.2.
ACKNOWLEDGMENTS
We acknowledge the University of Nebraska Medical Center (UNMC BSL-3) core facility for allowing us to perform all in vitro experiments involving SARS-CoV-2. The UNMC BSL-3 core facility is administered through the Office of the Vice-Chancellor for Research and supported by the Nebraska Research Initiative (NRI). The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: (i) SARS-related coronavirus 2 isolate USA-WI1/2020 (BEI catalog number NR-52384), hCoV-19/South Africa/KRISP-EC-K005321/2020 (BEI catalog number NR-54008), and hCoV-19/Scotland/CVR2224/2020 (BEI catalog number NR-53945); (ii) quantitative PCR (qPCR) control RNA from heat-inactivated SARS-related coronavirus 2 isolate USA-WA1/2020 (BEI catalog number NR-52347); (iii) HEK-293T-hACE2; and (iv) a plasmid containing a lentiviral backbone expressing luciferase and ZsGreen (BEI catalog number NR-52516), a lentiviral helper plasmid expressing HIV Gag-Pol (BEI catalog number NR-52517), a lentiviral helper plasmid expressing HIV Tat (BEI catalog number NR-52518), and a lentiviral helper plasmid expressing HIV Rev (BEI catalog number NR-52519). K. Singh acknowledges the computation facilities of the Molecular Interactions Core of the University of Missouri, Columbia, MO.
Each author approved the manuscript before submission for publication.
This work was partially supported by National Institute of Allergy and Infectious Diseases grants R01 AI129745, R01 AI113883, and DA052845 and by the Frances E. Lageschulte and Evelyn B. Weese New Frontiers in Medical Research Fund to S.N.B. S.N.B. acknowledges independent research and development (IRAD) funding from the National Strategic Research Institute (NSRI) at the University of Nebraska. K. Singh was partially funded by the Bond Life Sciences Center (Early Concept grant) and by the National Institute of Allergy and Infectious Diseases (R37 AI076119).
A provisional patent describing the drugs used in this study has been filed. C.L.L. is the co-founder and CSO of Shift Pharmaceuticals. K. Singh is a consultant at Sanctum Therapeutics Corporation, Sunnyvale, CA.
Contributor Information
Kamal Singh, Email: singhka@missouri.edu.
Siddappa N. Byrareddy, Email: sid.byrareddy@unmc.edu.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Wang C, Horby PW, Hayden FG, Gao GF. 2020. A novel coronavirus outbreak of global health concern. Lancet 395:470–473. 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma A, Tiwari S, Deb MK, Marty JL. 2020. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int J Antimicrob Agents 56:106054. 10.1016/j.ijantimicag.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. 2020. The proximal origin of SARS-CoV-2. Nat Med 26:450–452. 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, Nakane T, Zivanov J, Neufeldt CJ, Cerikan B, Lu JM, Peukes J, Xiong X, Krausslich HG, Scheres SHW, Bartenschlager R, Briggs JAG. 2020. Structures and distributions of SARS-CoV-2 Spike proteins on intact virions. Nature 588:498–502. 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raghuvamsi PV, Tulsian NK, Samsudin F, Qian X, Purushotorman K, Yue G, Kozma MM, Hwa WY, Lescar J, Bond PJ, MacAry PA, Anand GS. 2021. SARS-CoV-2 S protein:ACE2 interaction reveals novel allosteric targets. Elife 10 10.7554/eLife.63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell 181:281–292 e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280 e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang T, Jaimes JA, Bidon MK, Straus MR, Daniel S, Whittaker GR. 2021. Proteolytic activation of SARS-CoV-2 Spike at the S1/S2 boundary: Potential role of proteases beyond furin. ACS Infect Dis 7:264–272. 10.1021/acsinfecdis.0c00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang N, Shen HM. 2020. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int J Biol Sci 16:1724–1731. 10.7150/ijbs.45498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardi N, Weissman D. 2020. Development of vaccines and antivirals for combating viral pandemics. Nat Biomed Eng 4:1128–1133. 10.1038/s41551-020-00658-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyriakidis NC, Lopez-Cortes A, Gonzalez EV, Grimaldos AB, Prado EO. 2021. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines 6:28. 10.1038/s41541-021-00292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan SR, Spratt AN, Cohen AR, Naqvi SH, Chand HS, Quinn TP, Lorson CL, Byrareddy SN, Singh K. 2021. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J Autoimmun 124:102715. 10.1016/j.jaut.2021.102715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spratt AN, Kannan SR, Woods LT, Weisman GA, Quinn TP, Lorson CL, Sonnerborg A, Byrareddy SN, Singh K. 2021. Evolution, correlation, structural impact and dynamics of emerging SARS-CoV-2 variants. Comput Struct Biotechnol J 19:3799–3809. 10.1016/j.csbj.2021.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hossain MK, Hassanzadeganroudsari M, Apostolopoulos V. 2021. The emergence of new strains of SARS-CoV-2. What does it mean for COVID-19 vaccines? Expert Rev Vaccines 20:635–638. 10.1080/14760584.2021.1915140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connors M, Graham BS, Lane HC, Fauci AS. 2021. SARS-CoV-2 vaccines: Much accomplished, much to learn. Ann Intern Med 174:687–690. 10.7326/M21-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cusinato J, Cau Y, Calvani AM, Mori M. 2021. Repurposing drugs for the management of COVID-19. Expert Opin Ther Pat 31:295–307. 10.1080/13543776.2021.1861248. [DOI] [PubMed] [Google Scholar]
- 17.Trivedi J, Mohan M, Byrareddy SN. 2020. Drug repurposing approaches to combating viral infections. J Clin Med 9 10.3390/jcm9113777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin HXJ, Cho S, Meyyur Aravamudan V, Sanda HY, Palraj R, Molton JS, Venkatachalam I. 2021. Remdesivir in coronavirus disease 2019 (COVID-19) treatment: A review of evidence. Infection 49:401–410. 10.1007/s15010-020-01557-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez de Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh MD, Ruiz-Palacios GM, Benfield T, Fatkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, Lane HC, Members A-SG. 2020. Remdesivir for the treatment of Covid-19: Final report. N Engl J Med 383:1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan H, Peto R, Henao-Restrepo A-M, Preziosi M-P, Sathiyamoorthy V, Abdool Karim Q, Alejandria MM, Hernández García C, Kieny M-P, Malekzadeh R, Murthy S, Reddy KS, Roses Periago M, Abi Hanna P, Ader F, Al-Bader AM, Alhasawi A, Allum E, Alotaibi A, Alvarez-Moreno CA, Appadoo S, Asiri A, Aukrust P, Barratt-Due A, Bellani S, Branca M, Cappel-Porter HBC, Cerrato N, Chow TS, Como N, Eustace J, García PJ, Godbole S, Gotuzzo E, Griskevicius L, Hamra R, Hassan M, Hassany M, Hutton D, Irmansyah I, Jancoriene L, Kirwan J, Kumar S, Lennon P, Lopardo G, Lydon P, Magrini N, Maguire T, Manevska S, Manuel O, Consortium WHOST, et al. 2021. Repurposed antiviral Ddrugs for Covid-19: Interim WHO solidarity trial results. N Engl J Med 384:497–511. 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. 2020. Structure of the SARS-CoV-2 Spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 22.Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM, Zhao Y, Duyvesteyn HME, Tuekprakhon A, Nutalai R, Wang B, Lopez-Camacho C, Slon-Campos J, Walter TS, Skelly D, Costa Clemens SA, Naveca FG, Nascimento V, Nascimento F, Fernandes da Costa C, Resende PC, Pauvolid-Correa A, Siqueira MM, Dold C, Levin R, Dong T, Pollard AJ, Knight JC, Crook D, Lambe T, Clutterbuck E, Bibi S, Flaxman A, Bittaye M, Belij-Rammerstorfer S, Gilbert SC, Carroll MW, Klenerman P, Barnes E, Dunachie SJ, Paterson NG, Williams MA, Hall DR, Hulswit RJG, Bowden TA, Fry EE, Mongkolsapaya J, Ren J, Stuart DI, Screaton GR. 2021. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 184:2939–2954 e9. 10.1016/j.cell.2021.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. 2004. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 24.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. 2006. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 25.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. 2006. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49:534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. 2004. A hierarchical approach to all-atom protein loop prediction. Proteins 55:351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 27.Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, Chu HY, Tortorici MA, Veesler D, Murphy M, Pettie D, King NP, Balazs AB, Bloom JD. 2020. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 Spike protein for neutralization assays. Viruses 12 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendoza EJ, Manguiat K, Wood H, Drebot M. 2020. Two detailed plaque assay protocols for the quantification of infectious SARS-CoV-2. Curr Protoc Microbiol 57:ecpmc105. 10.1002/cpmc.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acharya A, Pandey K, Thurman M, Challagundala KB, Vann KR, Kutateladze TG, Morales GA, Durden DL, Byrareddy SN. 2021. Blockade of SARS-CoV-2 infection in vitro by highly potent PI3K-alpha/mTOR/BRD4 inhibitor. bioRxiv 10.1101/2021.03.02.433604. [DOI]
- 30.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 31.Chou TC. 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446. 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 32.Ianevski A, Giri AK, Aittokallio T. 2020. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 48:W488–W493. 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleine-Weber H, Pohlmann S, Hoffmann M. 2019. Spike proteins of novel MERS-coronavirus isolates from North- and West-African dromedary camels mediate robust viral entry into human target cells. Virology 535:261–265. 10.1016/j.virol.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laskowski RA, Swindells MB. 2011. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51:2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 35.Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, Ludden C, Reeve R, Rambaut A, Consortium C-GU, Peacock SJ, Robertson DL, COVID-19 Genomics UK (COG-UK) Consortium. 2021. SARS-CoV-2 variants, Spike mutations and immune escape. Nat Rev Microbiol 19:409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raman R, Patel KJ, Ranjan K. 2021. COVID-19: Unmasking emerging SARS-CoV-2 variants, vaccines and therapeutic strategies. Biomolecules 11. 10.3390/biom11070993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kannan SR, Spratt AN, Cohen AR, Naqvi SH, Chand HS, Quinn TP, Lorson CL, Byrareddy SN, Singh K. 2021. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J Autoimmun 124:102715. 10.1016/j.jaut.2021.102715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schutz D, Ruiz-Blanco YB, Munch J, Kirchhoff F, Sanchez-Garcia E, Muller JA. 2020. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv Drug Deliv Rev 167:47–65. 10.1016/j.addr.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karoyan P, Vieillard V, Gomez-Morales L, Odile E, Guihot A, Luyt CE, Denis A, Grondin P, Lequin O. 2021. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun Biol 4:197. 10.1038/s42003-021-01736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan DC, Morris C, Mahindra A, Blair CM, Tejeda G, Herbert I, Turnbull ML, Lieber G, Willett BJ, Logan N, Smith B, Tobin AB, Bhella D, Baillie G, Jamieson AG. 2021. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 Spike protein do not prevent virus internalization. Pept Sci (Hoboken) :e24217. 10.1002/pep2.24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.VanPatten S, He M, Altiti A, K FC, Ghanem MH, Al-Abed Y. 2020. Evidence supporting the use of peptides and peptidomimetics as potential SARS-CoV-2 (COVID-19) therapeutics. Future Med Chem 12:1647–1656. 10.4155/fmc-2020-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus A, van Kuppeveld FJM, Haagmans BL, Grosveld F, Bosch BJ. 2020. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun 11:2251. 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Li R, Pan Z, Qian C, Yang Y, You R, Zhao J, Liu P, Gao L, Li Z, Huang Q, Xu L, Tang J, Tian Q, Yao W, Hu L, Yan X, Zhou X, Wu Y, Deng K, Zhang Z, Qian Z, Chen Y, Ye L. 2020. Human monoclonal antibodies block the binding of SARS-CoV-2 Spike protein to angiotensin converting enzyme 2 receptor. Cell Mol Immunol 17:647–649. 10.1038/s41423-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang S, Zhang X, Du L. 2021. Therapeutic antibodies and fusion inhibitors targeting the Spike protein of SARS-CoV-2. Expert Opin Ther Targets 25:415–421. 10.1080/14728222.2020.1820482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X. 2020. Inhibitors of SARS-CoV-2 entry: Current and future opportunities. J Med Chem 63:12256–12274. 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barh D, Tiwari S, Silva Andrade B, Giovanetti M, Almeida Costa E, Kumavath R, Ghosh P, Goes-Neto A, Carlos Junior Alcantara L, Azevedo V. 2020. Potential chimeric peptides to block the SARS-CoV-2 Spike receptor-binding domain. F1000Res 9:576. 10.12688/f1000research.24074.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurevich EV, Gurevich VV. 2014. Therapeutic potential of small molecules and engineered proteins. Handb Exp Pharmacol 219:1–12. 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ngo HX, Garneau-Tsodikova S. 2018. What are the drugs of the future? Medchemcomm 9:757–758. 10.1039/c8md90019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huentelman MJ, Zubcevic J, Hernandez Prada JA, Xiao X, Dimitrov DS, Raizada MK, Ostrov DA. 2004. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension 44:903–906. 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- 50.Beeraka NM, Sadhu SP, Madhunapantula SV, Rao Pragada R, Svistunov AA, Nikolenko VN, Mikhaleva LM, Aliev G. 2020. Strategies for targeting SARS CoV-2: Small molecule inhibitors-—the current status. Front Immunol 11:552925. 10.3389/fimmu.2020.552925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, Parry T, Kent R, Enright J, Wu QL, Conley G, DeOliveira D, Morganelli L, Ducar M, Wescott CR, Ladner RC. 2003. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem 278:15532–15540. 10.1074/jbc.M212934200. [DOI] [PubMed] [Google Scholar]
- 52.Han Y, Kral P. 2020. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 14:5143–5147. 10.1021/acsnano.0c02857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. 2000. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87:E1–9. 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 54.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. 2005. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116. 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ianevski A, Yao R, Biza S, Zusinaite E, Mannik A, Kivi G, Planken A, Kurg K, Tombak E-M, Ustav M, Shtaida N, Kulesskiy E, Jo E, Yang J, Lysvand H, Løseth K, Oksenych V, Aas PA, Tenson T, Vitkauskienė A, Windisch MP, Fenstad MH, Nordbø SA, Ustav M, Bjørås M, Kainov DE. 2020. Identification and tracking of antiviral drug combinations. Viruses 12 10.3390/v12101178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 57.Tseng A, Seet J, Phillips EJ. 2015. The evolution of three decades of antiretroviral therapy: challenges, triumphs and the promise of the future. Br J Clin Pharmacol 79:182–194. 10.1111/bcp.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rayner CR, Dron L, Park JJH, Decloedt EH, Cotton MF, Niranjan V, Smith PF, Dodds MG, Brown F, Reis G, Wesche D, Mills EJ. 2020. Accelerating clinical evaluation of repurposed combination therapies for COVID-19. Am J Trop Med Hyg 103:1364–1366. 10.4269/ajtmh.20-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, Ng YY, Lo J, Chan J, Tam AR, Shum HP, Chan V, Wu AK, Sin KM, Leung WS, Law WL, Lung DC, Sin S, Yeung P, Yip CC, Zhang RR, Fung AY, Yan EY, Leung KH, Ip JD, Chu AW, Chan WM, Ng AC, Lee R, Fung K, Yeung A, Wu TC, Chan JW, Yan WW, Chan WM, Chan JF, Lie AK, Tsang OT, Cheng VC, Que TL, Lau CS, Chan KH, To KK, Yuen KY. 2020. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet 395:1695–1704. 10.1016/S0140-6736(20)31042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan Q, Zhang B, Ma J, Zhang S. 2020. Safety profile of the antiviral drug remdesivir: An update. Biomed Pharmacother 130:110532. 10.1016/j.biopha.2020.110532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang T, Bidon M, Jaimes JA, Whittaker GR, Daniel S. 2020. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res 178:104792. 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teyssou E, Delagreverie H, Visseaux B, Lambert-Niclot S, Brichler S, Ferre V, Marot S, Jary A, Todesco E, Schnuriger A, Ghidaoui E, Abdi B, Akhavan S, Houhou-Fidouh N, Charpentier C, Morand-Joubert L, Boutolleau D, Descamps D, Calvez V, Marcelin AG, Soulie C. 2021. The Delta SARS-CoV-2 variant has a higher viral load than the Beta and the historical variants in nasopharyngeal samples from newly diagnosed COVID-19 patients. J Infect 10.1016/j.jinf.2021.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Callaway E. 2021. Delta coronavirus variant: scientists brace for impact. Nature 595:17–18. 10.1038/d41586-021-01696-3. [DOI] [PubMed] [Google Scholar]