

Abstract
Drug conjugates are chemotherapeutic or cytotoxic agents covalently linked to targeting ligands such as an antibody or a peptide via a linker. While antibody-drug conjugates (ADCs) are now clinically established for cancer therapy, peptide-drug conjugates (PDCs) are gaining recognition as a new modality for targeted drug delivery with improved efficacy and reduced side effects for cancer treatment. The linker in a drug conjugate plays a key role in the circulation time of the conjugate and release of the drug for full activity at the target site. Herein, we highlight the main linker chemistries utilized in the design of PDCs, and discuss representative examples of PDCs with different linker chemistries with the related outcome in cell and animal studies.
Keywords: Peptide-Drug Conjugates, Antibody-Drug Conjugates, Linker Chemistry, Therapeutic Efficacy, Targeted Drug Delivery, Cancer Therapy
Graphical Abstract

1. INTRODUCTION
Conventional chemotherapy is still a major way to treat many cancers.1 Chemotherapeutics (CTs), such as doxorubicin, paclitaxel, gemcitabine, and camptothecin, are highly potent agents that utilize different mechanisms to display cytotoxicity. The toxicity, however, is non-specific, leading to reduced efficacy. Different strategies are used to increase the therapeutic efficacy of CT agents and reduce their toxic side effects during cancer treatment. One popular strategy involves the use of targeting ligands for the delivery of CT agents (or the cytotoxic payload/drug) to the cancer site, sparing nonmalignant cells and tissues.2 Active targeting using a ligand allows a higher concentration of the drug to reach the tumor by targeting a specific cell-surface receptor or biomarker at the tumor site. The targeting ligands, such as antibodies or peptides, can be covalently conjugated to the drug or to the surface of a drug carrier system like liposomes, micelles, etc. for site-specific delivery. The covalent conjugation of an antibody or a peptide to a drug via a linker gives an antibody-drug conjugate (ADC) or a peptide-drug conjugate (PDC). Both ADC and PDC are promising therapeutic modalities that are rapidly emerging for cancer treatment.3–7 As of now, at least 8 ADCs have been FDA approved to treat various cancers, highlighting the potential of upcoming ADCs and PDCs that are currently in clinical or pre-clinical stages.3, 8–10
There are a few key differences between an ADC and a PDC. First, the size of an antibody is much larger than the size of a peptide. An antibody (IgG) is typically >1000 amino acids (~ 150 kDa), whereas a peptide used for cancer cell targeting ranges from 5–25 amino acids in length.11 The large size of the antibody and the resulting ADC leads to differences, such as uptake by the cells, plasma half-life, immunogenicity, manufacturing costs, and stability. Second, the conjugation of an antibody to a drug via a linker leads to heterogeneous mixtures where the site of conjugation and the number of conjugated drugs per antibody molecule vary. ADCs typically contain final products where the drug to antibody ratio ranges between 3–8.12 This can cause batch-to-batch variation, and the exact characterization of species present in dosage can be tedious leading to complex pharmacokinetics (PK) after systemic administration. PDCs being small in size, ranging in molecular weight 2–5 kDa, can be easily synthesized as single homogeneous entities that are well characterized for precise large-scale production and can provide improved PK. An additional benefit of small-sized PDCs is their ability to cross the BBB as several cancer types tend to metastasize to the brain. Another difference is the high specificity of an antibody toward the target protein/receptor compared to a smaller peptide.
The goal of both the ADC and PDC is to increase the efficacy of the CT agent and overcome the challenges of short circulation half-life and off-target side effects of the CT agent. ADCs have been extensively reviewed elsewhere, therefore, will be briefly touched upon here.3, 5, 9, 10, 13 The three main components of a conjugate are the targeting ligand, cytotoxic drug, and the linker. The targeting ligand or the peptide in a PDC must be highly specific, which leads to receptor-mediated endocytosis of the conjugate by the target cells only. The use of a highly toxic drug, such as maytansine, camptothecin derivatives, auristatin, or doxorubicin with IC50 values in the sub-nanomolar range, is recommended in the conjugates. Finally, the linker chemistry is selected to allow sufficient circulation time for the conjugate to reach its target cell. Overall, the PDC should be stable enough so that the peptide, linker, and the drug do not get cleaved or metabolized before reaching the target cell, and sufficient concentration of the PDC reaches the target cell to increase the efficacy of the drug for tumor killing.
A perfect linker would release the drug only after intracellular uptake of the conjugate by the cancer cells for selective killing. However, most linkers start getting cleaved the moment they are in the system (upon systemic administration of the PDC), starting with the plasma (blood) and followed by the extracellular milieu of cancer cells. Whatever remains (intact PDC) is then taken up by the cancer cells for the drug’s intracellular delivery. During the synthesis of a drug conjugate, the linker makes two chemical bonds, one between the peptide and the linker and the second between the drug and the linker (Fig. 1). When the conjugate is administered systemically, one of these bonds is generally more susceptible to cleavage, or there could be a third bond (functional group) present in the linker that may lead to peptide drug separation. Ideally, the bond between the drug and the linker should be cleaved to release the unmodified drug at the target site. Also, the bond between the peptide and linker should not influence the affinity of the peptide for its receptor. The common functional groups that are found in the linker region can be broadly divided into 4 groups, enzyme cleavable (ester, amide, and carbamate), acid cleavable (hydrazone and carbonate), reducible disulfide, and non-cleavable (thioether, oxime, and triazole) (Fig. 1). This classification is based on how these functional groups behave after cellular uptake or under in vivo conditions. The linker chemistry plays a major role in the success of a PDC for improving efficacy. This review aims to provide an overview of the linker chemistries used in the design and development of PDCs and the related outcomes in cell and animal studies. The PDCs discussed here are categorized by the bond between the drug and the linker. The lability of this bond is most important for the release of the unmodified drug.
Figure 1.

Schematic of a PDC. The chemical structures of different linker chemistries are shown with the plausible method of release of the drug from PDC after cellular uptake or under in vivo conditions.
2. ENZYME CLEAVABLE LINKER CHEMISTRIES
Enzyme cleavable ester or amide bonds have gained popularity as linkers because they can be designed for selective cleavage in the tumor microenvironment or in the lysosomes. The intracellular compartments like endosomes and lysosomes of cancer cells are rich in esterases and amidases, and upregulation of such enzymes is exploited for the targeted release of the CT inside the cancer cell. Simple ester or amide bonds, or more specific amino acid sequences recognized by enzymes, like cathepsin B and matrix metalloproteinases, have been utilized as linkers. Carbamate linker that gets cleaved by a family of enzymes has also been utilized for PDC development.14–16 However, it is important to note that these linker chemistries are not infallible due to cleavage that occurs prior to reaching the target site. For instance, some cleavage and release of the drug can occur in plasma after systemic administration. Proper manipulation of the linker ensures the stability of much of the conjugate till it reaches the cancer site. The following representative PDCs that utilize ester, amide, or carbamate bond linkages to deliver the drug to the target cancer site highlight the potential of these linker chemistries.
2.1. Ester and Amide.
A study conducted by Karampelas et al. utilized an ester bond to attach gemcitabine with a glutaryl linker, which is further attached to the peptide ([D-Lys6]-GnRH) via an amide bond (1, Fig. 2).17 The overall hypothesis was that the conjugate would be a suitable substrate for the targeted delivery of gemcitabine to tumor cells that overexpress Gonadotropin-Releasing Hormone-Receptor (GnRH-R) on the cell surface. This is because the peptide has a high affinity for the cell surface GnRH-R. Various conjugates varying in length of the linker (4 or 5 carbons utilizing succinyl or glutaryl moiety) and the conjugation site on the drug were synthesized and compared. The gemcitabine structure includes a primary 5’-OH and secondary 3’-OH, which enabled the conjugation of the targeting peptide at either functional group. Evaluation of the antiproliferative effect on two prostate cancer cell lines (DU145 and PC3) showed that the GSG conjugate (Fig. 2) had the greatest potency and sustained gemcitabine levels, and was chosen for subsequent analyses. GSG was characterized by the 4-carbon linker attached to the primary 5’-OH of gemcitabine. When tested in a GnRH-R positive prostate cancer xenograft animal model, mice treated with GSG showed a significant inhibitory effect on tumor growth when compared to control (saline) and equimolar doses of gemcitabine or the peptide alone. A much higher dose of gemcitabine (454.5 μmol/kg) was required to achieve similar tumor reduction efficacy when compared to the GSG dose of choice (18.8 μmol/kg). GSG was also associated with a “slow-down” of gemcitabine inactivation or formation of the inactive metabolite of gemcitabine, resulting in sustained levels of gemcitabine. However, the presence of high levels of gemcitabine following GSG administration in mice suggests that the conjugate was rapidly cleaved at the ester site releasing the free drug from the peptide-linker moiety. Overall, the GSG conjugate allowed higher and sustained levels of gemcitabine in blood for a longer time compared to the direct administration of gemcitabine in mice. The study concluded that the exact mechanism of action for efficacy was not fully determined; however, the conjugate with the ester/amide linkage proved beneficial in inhibiting the tumor growth in mice.
Figure 2.
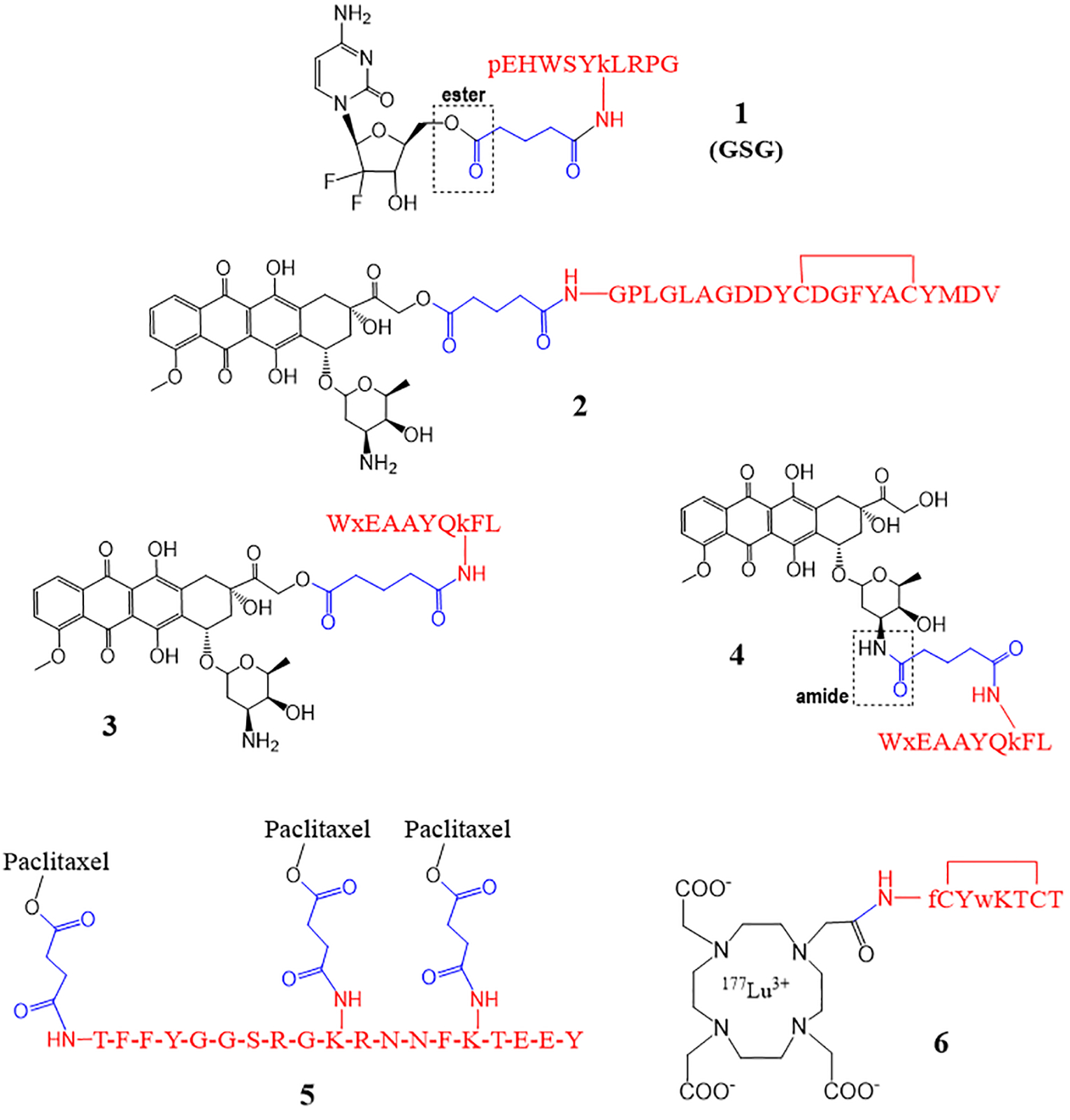
PDCs with an ester and/or an amide as linkers. The peptide is shown in red, the linker in blue, and the drug in black. D-amino acids are shown in lower case.
You et al. developed a unique approach for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer.18 A peptide fragment from the heavy chain 3 of the full-length antibody trastuzumab was obtained and termed AHNP. The 12-mer AHNP binds the extracellular domain of HER2 with high affinity and displays similar potency as trastuzumab. The peptide mimetic AHNP was then conjugated via an MMP-2 sensitive linker with doxorubicin (Dox) to form MAHNP-Dox conjugate essentially using ester/amide bonds (2, Fig. 2). Conjugate MAHNP-Dox was compared to Dox using HER2 positive breast cancer cell lines BT474 and SKBR3. Cellular toxicity analysis showed that MAHNP-Dox was more potent than free Dox, as indicated by lower IC50 values for both cell lines. The BT474 and SKBR3 cell lines had an IC50 of 746.8 ± 81.5 nM and 110.1 ± 12.7 nM for MAHNP-DOX and 2075.0 ± 368.0 nM and 172.9 ± 19.2 nM for Dox, respectively.
The combination of MAHNP-Dox appeared to be more effective than either agent alone and was potentially synergistic. TUNEL assay was then used to determine the effects of MAHNP-Dox on apoptosis and DNA damage. BT474 and SKBR3 cells had a greater number of apoptotic cells after treatment with MAHNP-Dox compared to free Dox. Data also indicated that Dox is released from the peptide via a specific MMP-2 dependent mechanism but shared similar intracellular distribution to free Dox. The conjugate is mainly cleaved by the MMP-2, which is present in large amounts around tumor cells. In vivo studies were then carried out with the BT474 xenografted mice treated with MAHNP-Dox, free Dox, or saline. MAHP-Dox reduced tumor growth by 74.7 ± 5.1% on day 25 after treatment, whereas free Dox resulted in a reduction of 53.4 ± 6.6%. Tumor weight inhibition was also analyzed. Conjugate MAHNP-Dox treated mice resulted in significant tumor weight inhibition that was 45.4 ± 11.2% higher than free Dox, and the weight loss was less for MAHNP-Dox treated mice. In vivo results paralleled in vitro studies, with more apoptosis at the tumor site and reduced off-target effects associated with MAHNP-Dox administration. Lastly, drug concentration profiles in plasma portrayed that MAHNP-Dox had a longer half-life (17.6 h versus 7.7 h) and slower clearance than free Dox. This study exhibited that the unique hybrid of Dox, ANHP, and an MMP-2 cleavable linker produced a benefit over the use of Dox alone. The conjugate effectively targeted HER2-positive cancer with greater efficacy. The group aims to conduct toxicological and pharmacological studies in the future. Meanwhile, the current data suggest a strong therapeutic potential for the conjugate.
We employed ester and amide bonds to synthesize PDCs (3 and 4, Fig. 2) of Dox that showed promising in vitro results.19 In conjugate 3, the primary alcohol at position 14 of Dox was reacted with a glutaryl linker to form an ester, and the other end of the linker was attached to the lysine side chain of the peptide (18–4) via an amide bond. For conjugate 4, the 3’ amino group of Dox was used to form an amide bond with a glutaryl linker, and the other linkage (amide) was the same as the conjugate 3. The peptide 18–4 (WxEAAYQkFL) used here targets a novel receptor keratin 1 (K1) on the surface of breast cancer cells, and the peptide or PDC enter cells via endocytosis after K1 receptor binding.20 Conjugate 3 had a short half-life in human serum (2 h), most likely due to fast hydrolysis of the ester bond. In contrast, conjugate 4 was relatively stable with a half-life of 48 h when incubated with human serum. The in vitro cell studies showed that conjugate 3 had higher uptake by the breast cancer cells and higher cytotoxicity (IC50 0.9–1.5 μM) compared to conjugate 4 (18.6–19.1 μM). While Dox was uptaken by all cancer and non-cancerous cells, the conjugates showed selective uptake by the cancer cells only. Also, the conjugates showed good uptake by the Dox-resistant cell line (MDA-MB-435R), where Dox showed minimal uptake. Both the conjugates showed low cytotoxicity to non-cancerous cells (IC50 35–51 μM) and were toxic to Dox resistant cells (IC50 5.4 μM). While conjugate 3 (ester/amide) fared better than conjugate 4 (amide/amide) in the cell studies, it is not clear which conjugate will show better efficacy in mice with breast cancer xenografts when used systemically. Upon systemic administration (i.v.), Dox likely will be released quickly from conjugate 3 and, therefore, may display the same efficacy as free Dox (unconjugated Dox); however, this needs to be evaluated with in vivo experiments.
Another study utilized ester/amide bonds to conjugate paclitaxel to a peptide (Angiopep-2) via a succinyl linker.21 Here, three molecules of paclitaxel are covalently linked to a single peptide via the side chain of two lysine residues and an N-terminus amine of the peptide to give a peptide-drug conjugate ANG1005 (5, Fig. 2). Angiopep-2, a 19-mer peptide that targets low-density lipoprotein receptor-related protein 1 (LRP-1) and mediates transcytosis across the BBB, facilitates the uptake of the conjugate into the brain for the treatment of patients with solid tumor with brain metastases. The ester bond in the conjugate is cleaved by the esterases present in lysosomes releasing paclitaxel in the brain. ANG1005 was designed to overcome the main challenge of paclitaxel, which is low BBB permeability due to the multidrug resistance efflux pump P-glycoprotein (P-gp) expression in brain tumor cells. Brain uptake of the conjugate in mouse brains using direct perfusion with [125I]ANG1005 or [3H]paclitaxel showed that total brain uptake of the conjugate was 4.5-fold higher compared to free paclitaxel, and showed that the conjugate could bypass the P-gp system at the BBB when assessed with mdr1a-deficient mice. The in vivo antitumor activity of ANG1005 was investigated in brain tumor (U87 MG glioblastoma) and lung carcinoma tumor (NCI-H460) xenograft models (orthotopic and metastatic) in nude mice, which showed a significant increase in mice survival. In addition, the in vitro cytotoxicity studies with human tumor cell lines carrying glioblastoma, lung, and ovarian carcinoma confirmed that paclitaxel’s activity was maintained in the conjugate ANG1005 where the conjugate was up to 2–3 fold more cytotoxic (2–3 fold lower IC50) towards some cell lines compared to paclitaxel.
ANG1005 has entered several clinical trials (phase I and phase II) in patients with metastatic brain cancers.22–24 Kumthekar et al. reported results of a phase 2 open-label study of ANG1005 in patients with brain metastases from breast cancer (BCBM) with or without Leptomeningeal Carcinomatosis (LC).22 ANG1005 showed significant activity against CNS tumors and improved symptoms with overall increased survival, especially in LC patients with HER2-positive and HER2-negative breast cancers.
A conjugate of a radionuclide with peptide octreotide, called lutetium 177 DOTA-TATE (6, Fig. 2), was FDA approved in 2018 for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs).25 Conjugate 6 belongs to peptide receptor radionuclide therapy (PRRT) and is the first PDC to be FDA approved for cancer treatment.4, 25 In conjugate 6, the radionuclide lutetium 177 (177Lu) is chelated to DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and DOTA is covalently linked to tyrosine-3 octreotate (TATE) via an amide bond.26, 27 Octreotide, a somatostatin analogue, has a high affinity for somatostatin subtype 2 receptor. The conjugate enters the cells via somatostatin receptors that are expressed on the surface of GEP-NET cells, causing damage specifically to the somatostatin receptor-positive cells.
An ester or an amide linker needs to be manipulated to be cleaved within target cancer cells/tissue that have the specific proteases upregulated. There is currently not enough data that underlines what specific manipulations of ester-amide linkers will maximize the tumor reduction efficacy, stability, and patient outcomes. Nonetheless, several PDCs with ester/amide linkers have shown promising in vitro and in vivo results, along with the clinical approval of 177Lu-dotatate to aid future clinical studies.
2.2. Carbamate.
PDCs also utilize carbamate linker that gets cleaved in intracellular endosomes or lysosomes by the enzymatic conditions.14–16 Sun et al. designed several somatostatin receptor (SSTR) targeting conjugates using carbamate linker.28–30 They focused on targeting SSTR subtype 2 with a somatostatin analog peptide. The best conjugate JF-10–81 (7, Fig. 3) showed potent antitumor and anti-angiogenic activity. In vivo studies were performed in rats and mice bearing pancreatic CA20948 tumors, human SCLC NCI-H69 tumors, or human prostate PC-3 tumors. Conjugate 7 inhibited tumor growth in CA20948 and NCI-H69 tumors but not PC-3 tumors. Lack of inhibitory activity in the PC-3 tumors is attributed to an insignificant expression of SSTR2. This finding suggested that the conjugate requires tumor expression of SSTR2 for the drug to be internalized and efficacious. Conjugate 7 also resulted in less off-target toxicity than the cytotoxic agent alone. The stability and chemical characteristics of the carbamate linker were essential for the successful and targeted delivery of the cytotoxic agent. The cleavable linker assisted with drug delivery to the target site and appropriately released free camptothecin without jeopardizing receptor affinity or antitumor activity.
Figure 3.

PDCs with a carbamate linker. D-amino acids are shown in lower case.
Sayyad et al. explored the use of carbamate linker in the design of new derivatives of PDC 1 (Fig. 2).31 The new PDCs 8 and 9 (Fig. 3) utilized the primary or secondary hydroxyls of the drug to attach to the lysine amino group of the peptide via a carbamate linkage. The authors found that the PDCs with the carbamate linker released the drug slowly compared to the PDCs with an ester linkage.31 The direct comparison of the carbamate linker containing conjugates 8 and 9 (Fig. 3) with an ester containing conjugate (GSHG, similar to conjugate 1 in Fig. 2) showed that 8 and 9 were less cytotoxic compared to the GSHG conjugate toward prostate cancer (DU145 and PC3) and breast cancer (MDA-MB-231 and MCF-7) cell lines. This is likely due to the slow release of gemcitabine from 8 and 9 conjugates. Whereas GSHG, which showed rapid release of the free drug when incubated with media (RPMI with 10% FBS and 1% P/S) or human serum, was found to be as cytotoxic as gemcitabine. Further, in vivo intraperitoneal injection of the conjugates in mice showed a higher concentration of the intact conjugate in blood for the mice injected with 8 and 9 compared to the mice injected with GSHG. Mice injected with GSHG showed higher levels of released drug gemcitabine, whereas those administered 8 and 9 had low levels of free drug. The study highlights the in vitro and in vivo stability of a carbamate compared to an ester linker; however, it is unclear which linker chemistry will lead to better efficacy for tumor reduction.
Kumar et al. synthesized peptide-paclitaxel conjugates with carbamate or carbonate linkers and a peptide (MuSSKYQ, Mu = morpholinocarbonyl protecting group) which gets selectively cleaved by prostate-specific antigen (PSA).32 Evaluation of these PDCs revealed that the carbamate containing PDC was more stable compared to the PDC with a carbonate.32 The PDC with a carbamate linker displayed appropriate stability for the specific release of the drug in the presence of PSA, thereby killing PSA-secreting CWR22Rv1 prostate cancer cells. Based on these and other studies, the order of in vivo linker stability from greatest to least is found to be as follows: amide>carbamate>ester>carbonate.33
2.3. Dipeptide or Tripeptide.
The bonds discussed above are cleaved by the enriched proteases in the lysosomes of cancer cells. To further facilitate the release of active drug from the PDC, specific dipeptide or tripeptide sequences have been employed. Liang et al. designed PDC 10 (Fig. 4) with a linker containing dipeptide (Val-Cit or VC) for specific cleavage by a carboxypeptidase, cathepsin B.34 The dipeptide is conjugated to Dox via a para-aminobenzyl carbamate (PABC) spacer that undergoes spontaneous 1,6-elimination after the enzymatic cleavage of the C-terminal amide of citrulline. The dipeptide is conjugated to the cancer cell targeting cyclic RGD peptide via a non-cleavable maleimide thioether. The linker chemistry used in PDC 10 with a dipeptide and thioether is similar to what is found in ADC brentuximab vedotin.9 The dipeptide (VC) is cleaved by enzyme cathepsin B, which is present in excess in lysosomes of tumor cells. This is followed by further removal of the PABC spacer leading to site-specific drug release. The authors found that conjugate 10 displayed superior cellular uptake, cytotoxicity toward integrin αvβ3 overexpressing B16 cells, and in vivo efficacy in tumor carrying c57BL/6 mice compared to another PDC (23, Fig. 9) with a disulfide linker (discussed in section 4) highlighting the potential of a dipeptide linker (VC) containing PDC.
Figure 4.
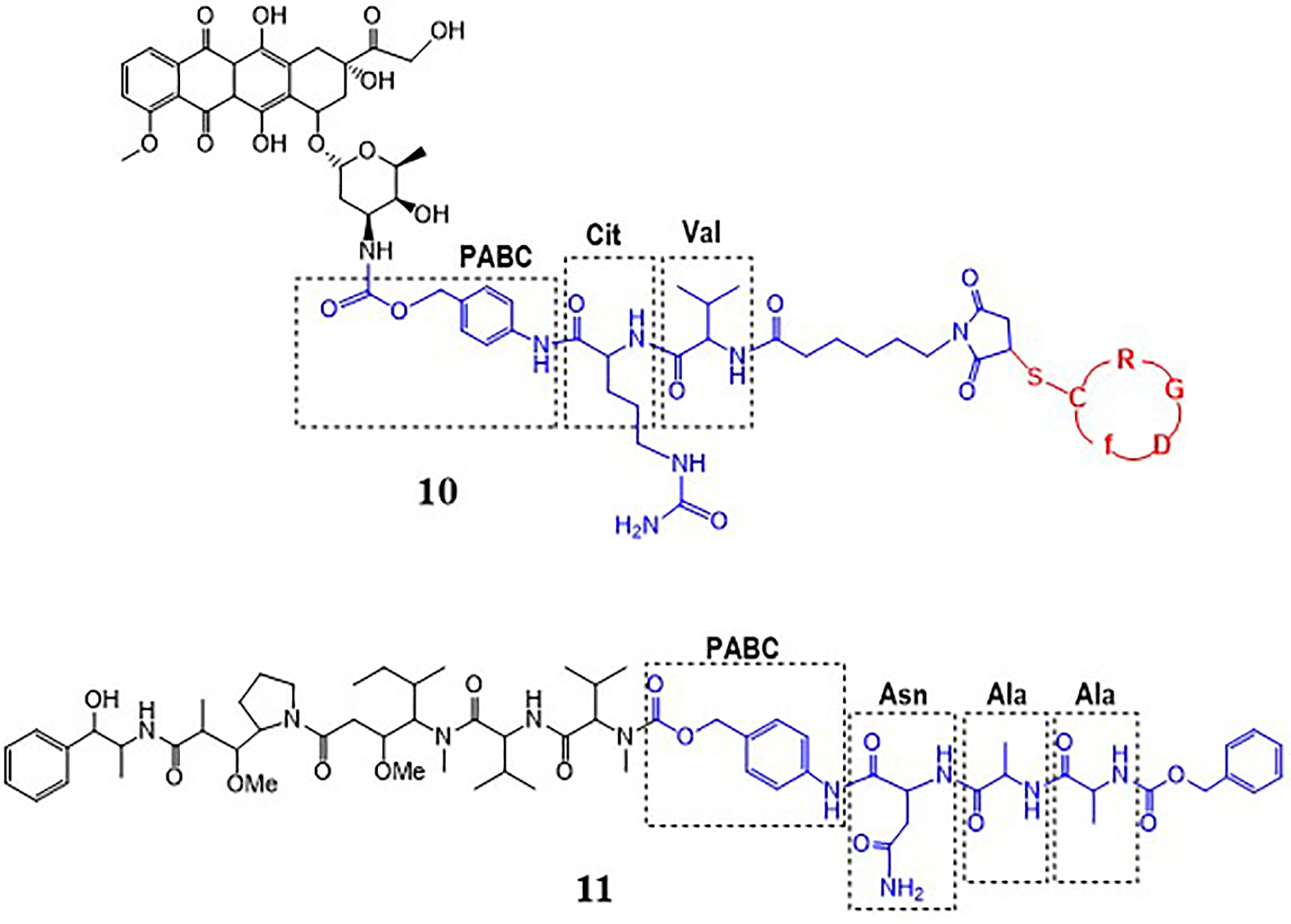
PDCs with a dipeptide or tripeptide linker.
Figure 9.
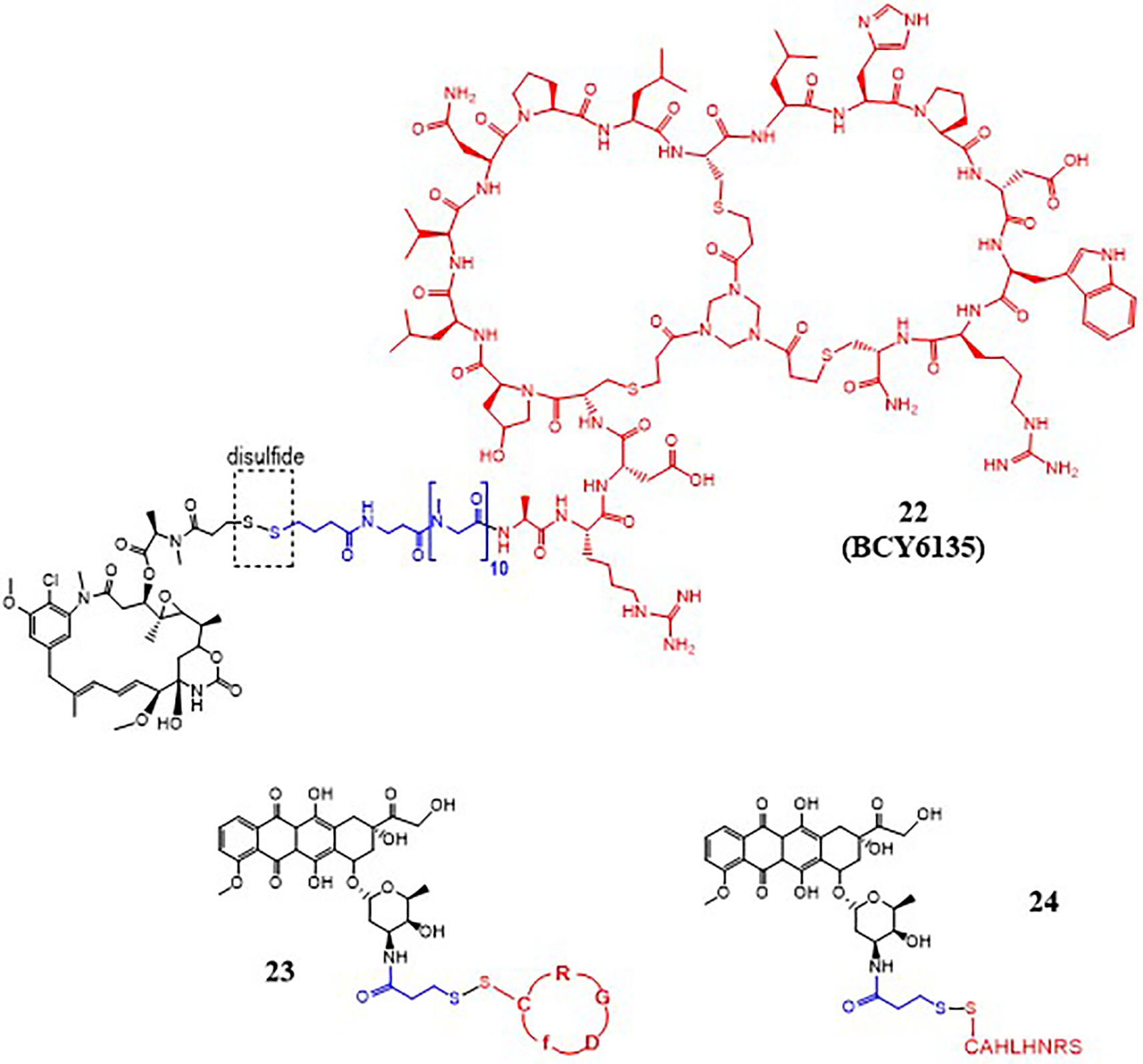
PDCs with a disulfide as a linker.
Another peptide sequence that is used for specific release of the drug at the tumor site is the Ala-Ala-Asn (AAN) tripeptide cleaved by enzyme legumain.35–37 Legumain is an asparaginyl endopeptidase reported to be overexpressed in tumor cells and microenvironment that specifically cleaves at the C-terminal of asparagine. Inhibition of legumain’s proteolytic activity and its catalytic property for prodrug activation is being utilized for developing cancer therapy. To employ the later approach for cancer therapy, Bajjuri et al. prepared several prodrugs with cytotoxic di-desmethylauristatin E (DDAE) or monomethylauristatin E (MMAE) and AAN tripeptide where the drug is linked to the peptide via a carbamate or amide bond.35 There is no targeting peptide in these prodrugs, however, these prodrugs could be conjugated to a targeting peptide to enhance cancer specificity and prepare peptide-drug conjugates. Among different prodrugs that were evaluated, prodrug 11 (Pro-MMAE-2, Fig. 4) was found to be most effective and mice treated with 11 (0.5 mg/kg) inhibited 4T1 breast cancer by 57% in comparison to mice treated with buffer alone.35 Further, the prodrug showed no toxicity in contrast to the high mortality rate in MMAE treated mice.
2.4. Amide and a non-cleavable thioether.
Succinimidyl thioether gained a lot of attention in recent years after the approval of several ADCs that contain this functional group as part of the linker.3 The succinimidyl thioether bond is rapidly formed by Michael addition of a thiol to the maleimide group, making it a popular choice for conjugating a peptide to a drug. The thioether is quoted as “non-cleavable”, even though this bond is reversible and not very stable as the linker breaks over time.38–40 Nonetheless, the drug is attached to the linker via a cleavable bond, like amide, to form the PDCs (12 and 13, Fig. 5), likely allowing the release of the unmodified drug in the lysosomes by the action of amidases. A similar succinimidyl thioether is also found in PDC 10 (Fig. 4) discussed above.
Figure 5.

PDCs with an amide and a succinimidyl thioether as linkers.
We synthesized a thioether containing PDC 12 (Fig. 5) using a bifunctional cross-linker sulfo-SMCC.41 Dox was attached to the linker via an amide bond, whereas the thiolated breast cancer targeting peptide (18–4) was conjugated to the maleimide group of the linker to form the succinimidyl thioether bond. The conjugate showed a half-life of ~18 hours when incubated with human serum. It was found that the instability was mainly due to the retro-Michael reaction that gave back the thiolated peptide and the MCC-Dox (maleimide). The release of the thiolated peptide (peptide-SH) was confirmed by the presence of peptide dimer in the human serum aliquots, as observed by mass spectrometry. We also found that conjugate 12 was not very stable under acidic conditions (half-life 24 h).41 At pH 5, hydrolysis of the acetal group present in doxorubicin led to the loss of intact conjugate. The conjugate was highly cytotoxic toward TNBC cells, namely, MDA-MB-231 (IC50 = 1.3 μM) and MDA-MB-468 (IC50 = 4.7 μM), and showed much less toxicity toward non-cancerous MCF-10A breast cells (IC50 = 38.6 μM). The cytotoxicity of the conjugate was in the same range as free Dox (IC50 range 0.4–1.5 μM), suggesting that the drug was released from the PDC inside the cells and/or the PDC itself was active.
Liang et al. 34 compared three PDCs containing Dox and cyclic RGD peptide (cRGDfC) linked via three different linkers. The linkers used were (i) dipeptide with thioether (RVCDOX or cRGDfC-VC-DOX or 10, Fig. 4), (ii) a succinimidyl thioether (RSDOX or cRGDfC-S-DOX or 13, Fig. 5), and (iii) a disulfide (RSSDOX or cRGDfC-SS-DOX or 23, Fig. 9). The three conjugates self-assemble into nanoparticles in water with RSDOX forming the largest particles. The average diameters of RSDOX, RVCDOX, and RSSDOX were 384 nm, 207 nm, and 154 nm, respectively. The cellular uptake via receptor-mediated endocytosis was higher for the RVCDOX than the RSDOX and RSSDOX, which showed a similar presence by flow cytometry analysis. The colocalization with the lysosomes (using confocal microscopy) was highest for the RVCDOX followed by RSDOX and the least for RSSDOX. The cytotoxicity of the conjugates on integrin αvβ3 overexpressing B16 cells also followed the same order, RVCDOX > RSDOX > RSSDOX. RVCDOX likely released the free drug faster than the other two conjugates in the lysosomes due to the presence of dipeptide and PABC. RSDOX and RSSDOX could also release free Dox after the amide bond cleavage by amidases in lysosomes. The authors concluded that both RVCDOX and RSDOX delivered a higher concentration of DOX to cytoplasm which led to better efficacy for tumor inhibition in c57BL/6 mice tumor models compared to disulfide containing RSSDOX. The antitumor efficacy of treatment groups RVCDOX and RSDOX was 1.7– 2-fold of the treatment group RSSDOX. Based on the reduction in tumor volumes and average tumor weight, the authors found that the order of efficacy was RVCDOX and RSDOX > DOX.HCl > RSSDOX.
In 2014, three pharmaceutical companies reported on the instability of the ring-closed succinimidyl thioether linker, and showed that the opening of the thiosuccinimide ring could lead to a stable thioether conjugate (Fig. 6).38–40 Lyon et al. from Seattle Genetics proposed the hydrolysis of the ring with a proximal amino group (primary amine), which acts as an intramolecular base catalyst, to yield ring-opened stable thioether 39. Fontaine et al. from ProLynx reported that succinimidyl thioethers with electron-withdrawing N-substituents accelerated the ring-opening reaction leading to the stable ring-opened products with t1/2 of >2 years.38 In addition, Tumey et al. from Pfizer reported a simple method for the ring-opening hydrolysis of the succinimidyl thioether using slightly basic conditions like buffer with pH 7.4–9.2 and leaving the reaction at 37–45 °C for 14–48 hours 40. The conjugates with succinimidyl thioether linker appear to perform well where stability is required for a shorter duration like 1–2 days; however, if prolonged circulation of the PDC is desired, a ring-opened stable thioether linker may be more desirable. While the stability of the thioether bond is an issue, the resulting thioether conjugates (ADCs) have progressed well into the clinics as the conjugates display a favorable pharmacokinetic profile.3, 9
Figure 6.
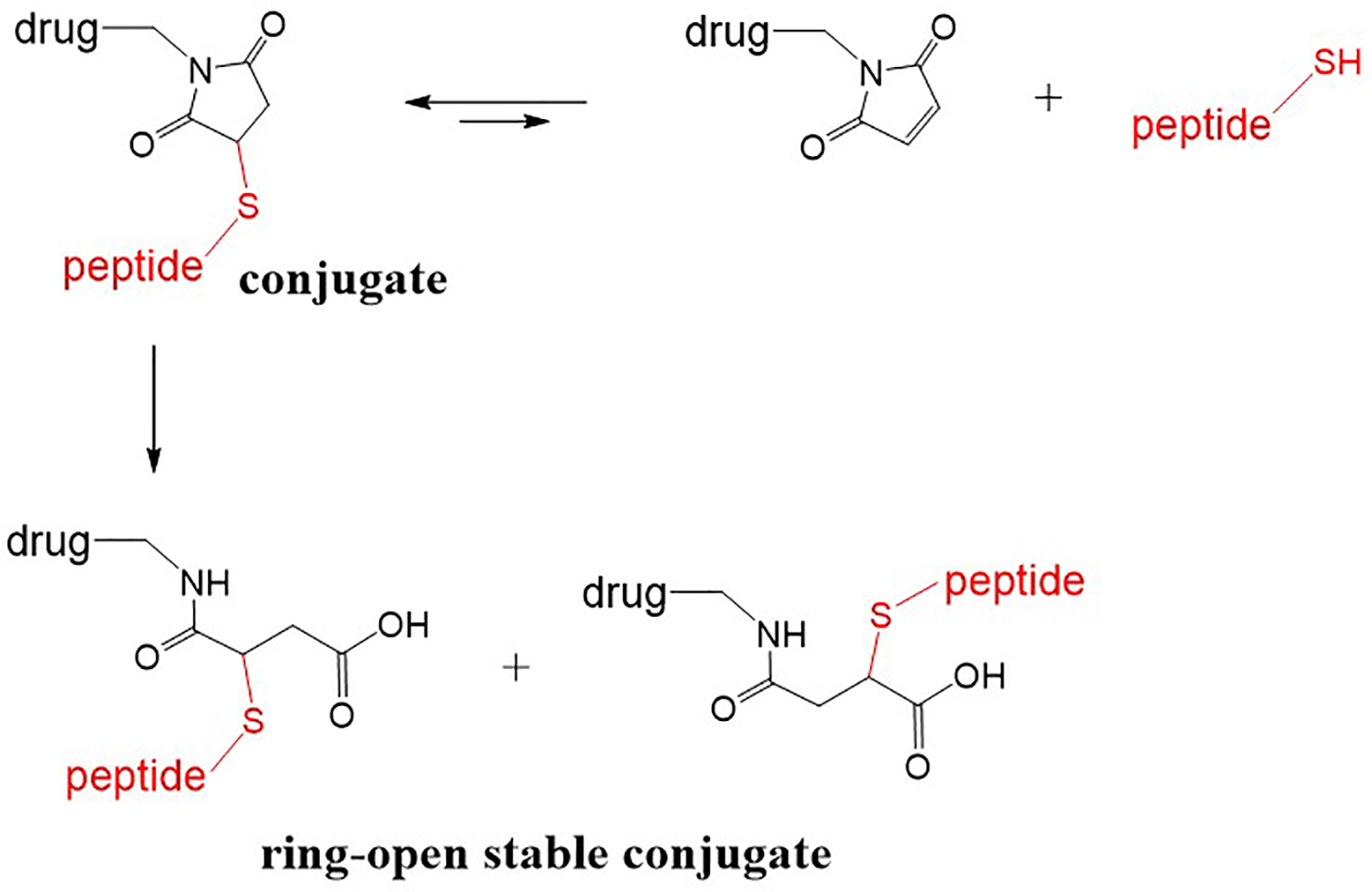
Peptide-drug conjugate with the ring closed succinimidyl thioether and ring-open stable thioether.
2.5. Ester and a non-cleavable triazole.
An azide-alkyne cycloaddition to form a triazole has been applied to link peptide with a drug in PDCs due to its ease and efficiency. Although triazole formation using click chemistry is chemically favorable, after systemic administration this bond is not subjected to enzyme degradation. Therefore, the release of the drug from the peptide is likely due to the presence of other enzyme cleavable groups such as esters, amides, or other labile groups in the linker region.
The group of Pellecchia utilized click chemistry to synthesize a number of erythropoietin-producing hepatocellular receptor A2 (EphA2)-targeting PDCs (14–16, Fig. 7).42, 43 EphA2 is a protein from the receptor tyrosine kinase family that serves as a known target for cancer treatment due to its overexpression in tumors and low expression in healthy tissues.44, 45 Initially, the team synthesized two peptides, YSA and 123B9, and found that peptide 123B9 is more stable in plasma. The 123B9 peptide was shown to target the EphA2 receptor and simultaneously functioned as an agonist. Agonist activity towards the receptor resulted in the inhibition of several pathways and tumor suppression effect. The dimeric 123B9 was found to activate the receptor at several-fold lower concentration (down to 1 μM) compared to the monomeric 123B9. Next, the authors’ utilized triazole linker via click chemistry for synthesizing PDCs with the dimeric (123B9)2. As a result, the PDC (123B9)2−L2−paclitaxel (14, Fig. 7) was developed. Efficacy studies were conducted to determine whether the optimized EphA2-agonistic agent was able to retain binding and selectivity for EphA2 and activate intracellular receptors. Conjugate 14 was directly compared to Abraxane, which is an injectable albumin-bound formulation of paclitaxel used for the treatment of various cancers. The synthesized PDC (14) was superior to Abraxane in its ability to prevent lung metastases, as indicated by a 75% reduction of the gross lung-metastasis count, compared with that of Abraxane in an intracardiac breast-cancer metastasis model. Abraxane did not show success in the reduction of metastases in organs other than the lungs, whereas the PDC did. Additionally, conjugate 14 reduced the metastasis number and size through various mechanisms, such as targeting of primary tumor cells, diminishing CTCs, and causing an evident reduction in tumor vasculature, in comparison to Abraxane.
Figure 7.

PDCs with an ester and a triazole as linkers.
Conjugates 15 and 16 with an EphA2 targeting peptide (123B9 or YNH, respectively) linked to cytotoxic gemcitabine via ester and triazole bonds showed promising results in the treatment of pancreatic cancer.42 Athymic mice carrying MIA PaCa-2-luc cells subcutaneous xenografts, when treated with either PBS, gemcitabine, or conjugate 15, showed the greatest tumor growth inhibition in the conjugate treated mice. Similarly, an efficacy study with gemcitabine and conjugate 16 in mice showed that conjugate 16 led to significant more suppression in tumor growth compared to gemcitabine alone. The conjugates displayed no adverse side effects, the body weight was maintained in mice, and the median survival time of conjugate 16 treated mice was significantly longer than gemcitabine or untreated mice groups.
3. ACID CLEAVABLE LINKER CHEMISTRIES
Acid-sensitive linkers have been employed for the development of several PDCs.41, 46–48 While linker chemistries like hydrazone have been utilized often, other acid-sensitive groups like acetal, ketal, and carbonate have been less studied.49, 50 The acid-labile bonds get cleaved primarily in the acidic tumor microenvironment (pH 6.5–6.9) or in the acidic cellular compartments, endosomes (pH 5.5–6.2) and lysosomes (pH 4.5–5.0), of cancer cells releasing unmodified drug, while maintaining stability in blood circulation (pH 7.4).9 Although the acid-sensitive linkers display good stability in buffer solutions (pH 7.4), the plasma stability has been variable for such linkers making it difficult to predict the clinical outcome of the conjugate. The following PDCs help understand some of the pros and cons of acid-sensitive linkers.
3.1. Hydrazone.
Hydrazone is a popular acid-sensitive linker utilized in the design of several drug conjugates.41, 46–48 The hydrolysis/stability of the hydrazone to give ketone and hydrazide can vary based on the original ketone from which the hydrazone is derived. The examples discussed here contain doxorubicin’s aliphatic C-13 ketone-derived hydrazone linkers. The cytotoxic agent doxorubicin is then released in the tumor microenvironment or after uptake inside the cellular compartments, like endosomes or lysosomes, of the target cancer cells. This allows specific targeting, thereby reducing the damage to healthy cells.
To introduce an acid-sensitive hydrazone, most studies utilize the same linker as found in aldoxorubicin. Aldoxorubicin (17, Fig. 8) is a derivative of Dox where the 13-carbonyl group of Dox is substituted with a hydrazone or more specifically, an EMCH (N-ε-maleimidocaproic acid hydrazide) linker. When administered intravenously, aldoxorubicin reacts with endogenous albumin to form a protein-drug conjugate.51, 52 The conjugate is formed by a covalent (thioether) bond between the maleimide of aldoxorubicin and exposed cysteine-34 (C34) thiol of the protein albumin (Mwt ~ 66 kDa). The albumin-drug conjugate formed by aldoxorubicin allows for site-selective delivery of Dox to the tumor.53 This is because albumin, a major circulating protein in the blood (mM concentration), pertains to be a nutrition source for the growth of cancer cells, and therefore tends to accumulate in tumors. When aldoxorubicin is administered systemically for cancer treatment, the albumin-aldoxorubicin conjugate formed in the blood allows the release of Dox by hydrolysis of the hydrazone linker, either intracellularly in the acidic environment of lysosomes/endosomes after uptake by tumor cells or extracellularly in the slightly acidic environment of tumor tissues. The clinical results indicated the ability to administer greater doses (3–4 times more) of Dox with minimal cardiotoxicity using aldoxorubicin. A disadvantage of aldoxorubicin is the possible reactions with cysteine and lysine of other proteins that can lead to off-site toxicities. Aldoxorubicin is currently in advanced phase clinical trials for the treatment of soft tissue sarcoma.54, 55 Other investigations with aldoxorubicin include combination treatment with immunotherapy and early-stage clinical trials for other cancers, including brain cancer.56, 57
Figure 8.
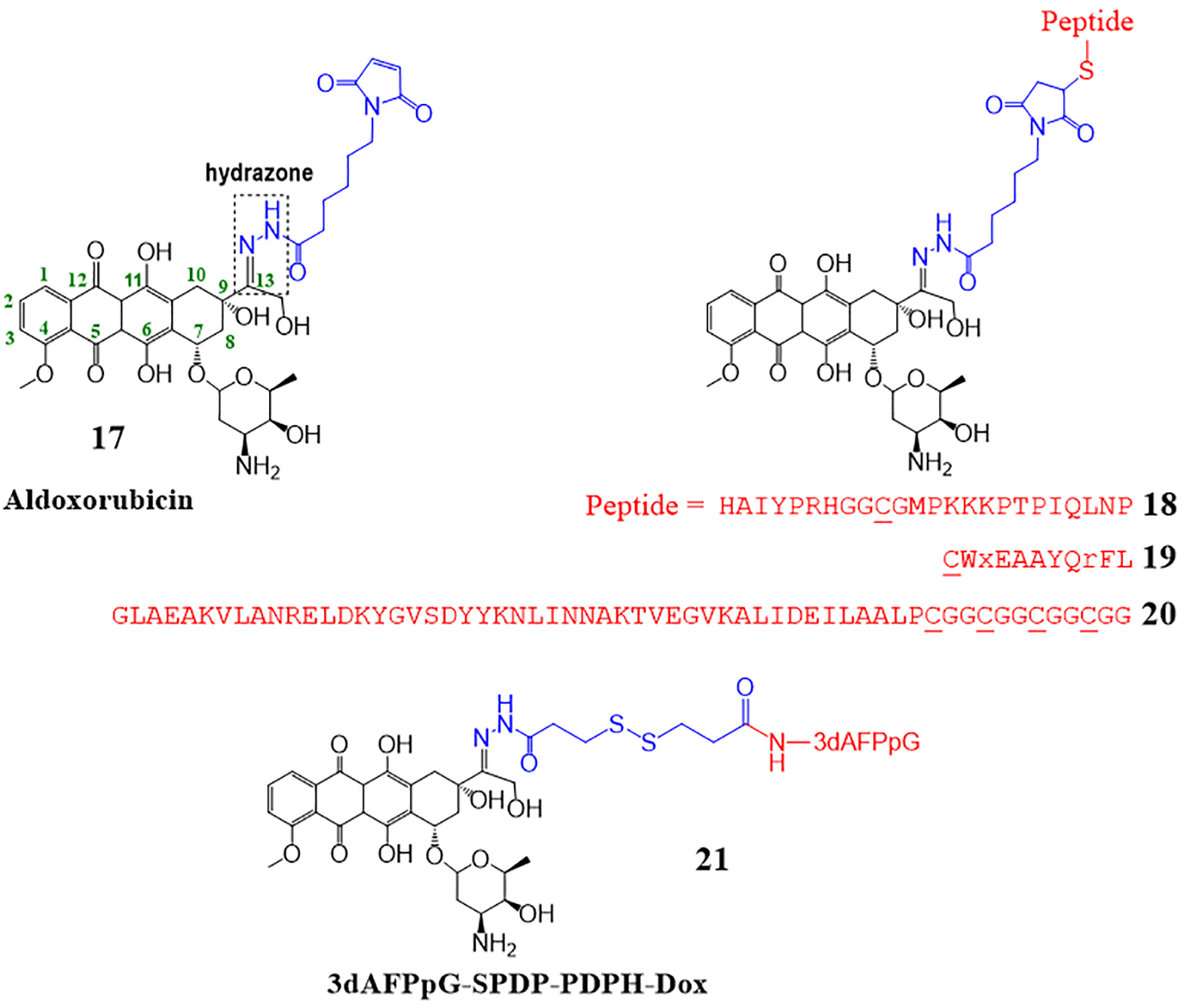
Drug conjugates with a hydrazone bond as a linker.
In other examples of drug conjugates with hydrazone linkers (18–20, Fig. 8), the maleimide of aldoxorubicin is reacted with a targeting agent (e.g. peptide) to form a peptide-drug conjugate before systemic administration.41, 46–48 These PDCs also contain the succinimidyl thioether linkage, however, the hydrazone bond is more labile than the thioether so it takes priority in releasing the drug from the conjugate for targeted activity. For instance, conjugate 18 consists of an aldoxorubicin molecule linked to a hybrid 24-mer peptide via both the hydrazone and the thioether linkages.47 The N-terminal (HAIYPRH) of the peptide facilitates targeting tumor cells as it specifically binds to transferrin receptor (TfR) overexpressed in tumor cells leading to TfR-mediated endocytosis. While the C-terminal portion is an ERK peptide inhibitor. The N- and C-terminal peptides have a middle GGCG sequence, where the thiol of cysteine allows conjugation to the aldoxorubicin (Fig. 8). In vitro studies supported the release of Dox from the rest of the conjugate in cells (likely in acidic endosomes or lysosomes) as it was found that the amount of Dox was significantly higher inside the whole cells for the conjugate 18 compared to the free Dox. In this experiment, MCF-7/ADR cells were incubated with conjugate 18 or free Dox and the amount of Dox in whole cells was determined using HPLC. Efficacy studies in MCF-7/ADR tumor bearing mice showed that a group of mice treated with the conjugate 18 inhibited tumor growth (72.2 ± 4.6% inhibition) more significantly than mice treated with free Dox (45.7 ± 2.8% inhibition). The biodistribution of Dox was higher in the tumor of conjugate treated mice compared to the free Dox treated mice, further indicating the targeting and release of Dox from the conjugate in the tumor environment.
We reported a PDC, peptide 18–4 – aldoxorubicin conjugate (19, Fig. 8) for targeting Dox to triple-negative breast cancer (TNBC) cells.41 Conjugate 19, which consists of a breast cancer cell targeting peptide 18–4 and doxorubicin, was synthesized by reacting aldoxorubicin with an N-terminally thiolated peptide. Due to the presence of an acid-sensitive hydrazone linker, the conjugate was not stable in media and human serum. In DMEM/F12 media with HEPES, 19 was completely hydrolyzed in <3 hours. The half-life in human serum was 6 hours, and it was confirmed using mass spectrometry that the hydrazone linker was the susceptible linkage during these conditions. The conjugate, however, showed specific toxicity toward the TNBC cells with an IC50 of 1.2–2.2 μM, almost the same as the free doxorubicin (IC50 0.4–1.5 μM). The PDC was about 9-times less cytotoxic to non-cancerous breast tissue-derived MCF-10A cells (IC50 15.1 μM), whereas Dox was still highly toxic (IC50 0.24 μM). The in vitro cell studies suggest that the hydrazone linker played a major role in the activity of the conjugate, and the thioether linker added to the stability of the conjugate.
The albumin binding approach is gaining recognition for the targeted delivery of chemotherapeutics. To develop an ex vivo approach for Dox delivery using albumin, Yousefpour et al. synthesized a PDC (20 or ABD-Dox, Fig. 8) using aldoxorubicin and an albumin-binding peptide.48 An albumin-binding protein domain (ABD) consisting of 47 amino acid residues is designed. ABD strongly interacts with albumin by non-covalent binding in contrast to the aldoxorubicin that binds to albumin by a covalent thioether bond. The non-covalent interaction of the ABD peptide would facilitate easier dissociation of the ABD (or the ABD conjugate) for release into the tumor. ABD was synthesized with additional GGC repeats (four or eight) in the C-terminal. The reaction of ABD-(CGG)8 with aldoxorubicin gave a mixture of conjugates with 3–4 Dox molecules per peptide, which were highly hydrophobic and insoluble under aqueous conditions. However, ABD-(CGG)4 reacted with aldoxorubicin to give conjugates with 1–2 Dox molecules that were soluble in PBS at pH 7.4 and used for subsequent in vitro and in vivo experiments. The in vivo PK (via monitoring plasma Dox concentration) in BALB/c mice showed a plasma elimination half-life of 29.4 ± 0.8 h for ABD-Dox, whereas for free Dox it was only in the order of minutes. The biodistribution studies showed that the level of Dox in the tumor at 2, 24, and 72 h was significantly higher for the ABD-Dox treated mice compared to the free Dox treated mice. At 72 h, Dox in the tumor was found to be ≈120-fold greater for the ABD-Dox treated mice compared to the free Dox group. Overall, the results showed that ABD-Dox has better therapeutic efficacy compared to free Dox in both colon and pancreatic mice models, and treatment with ABD-Dox led to the complete disappearance of tumors in both cases. The albumin-binding approach of this study can be used for other anticancer drugs that have limited clinical applications due to poor pharmacokinetic properties such as short circulation half-life, and the use of hydrazone and thioether to attach the drug to the peptide proved to be beneficial.
Mollaev et al. reported a protein-Dox conjugate with an acid-sensitive hydrazone and a disulfide bond in the linker.46 The protein used in the conjugate was the receptor-binding domain of human alpha-fetoprotein. The protein was linked to Dox via three functional groups. First, the C-13 ketone of Dox is modified into hydrazone, which is followed by a disulfide bond, and finally, the other end of the linker ends with an amide bond linking the protein (21 or PDPH-DOX, Fig. 8). A similar linker with a hydrazone and a disulfide can be found in approved ADCs gemtuxumab ozogamicin and inotuzumab ozogamicin.9 The authors found that the conjugate shared almost equivalent cytotoxicity as unmodified or free Dox and overcame resistance to anthracycline resistant MCF7Adr cells. Conjugate was not toxic to the lymphocytes, whereas Dox was highly cytotoxic. The conjugate also fared well in the in vivo efficacy studies showing a substantial decrease in tumor volumes and higher percent survival for the conjugate treated mice versus Dox or PBS treated mice. It was concluded that the hydrolysis of hydrazone along with the thiol reductase activity in lysosomes expedited hydrazone and disulfide bond hydrolysis, and Dox release from the conjugate. The pharmacological profile of the conjugate suggested its potential in cases when the rapid release of anticancer agents is necessary in tumor tissue.
4. REDUCIBLE DISULFIDE LINKER CHEMISTRY
The rationale behind using a disulfide linker is that the excessive reductive environment in tumor cells leads to the release of the free drug at the cancer site. In general, the intracellular glutathione concentration is several-fold (~1000) higher than the extracellular concentration. The glutathione concentration in cancer cells may be further augmented due to oxidative stress and hypoxia in the tumor microenvironment facilitating the rapid release of the thiolated drug inside cancer cells.58
Disulfide linker is utilized by BicycleTx Limited (Cambridge, UK) in the design of PDCs, such as 22 (Fig. 9), for targeting EphA2 receptor that is highly expressed in several solid tumors.59 The targeting peptides called Bicycles are developed by phage display screening while the bicycle peptide is attached to the bacteriophage, and this later allows conjugation of the Bicycle to toxic payloads without affecting EphA2 receptor affinity. A Bicycle consists of a peptide with three thioether bonds between the peptide and a central scaffold, such as 1,3,5-tris(bromomethyl)benzene (TBMB) and 1,3,5-triacryoyl-1,3,5-triazinane (TATA). The thioether bonds provide the targeting peptide a constraint structure leading to high selectivity and nanomolar affinity for the EphA2 receptor. A Bicycle is linked to a cytotoxic mertansine (DM1) via a disulfide bond to obtain PDCs that release the drug inside the cancer cells. The conjugates with the Bicycle peptide showed potent antitumor activity with one undesirable property i.e. high liver uptake. Chemical optimization of the lead Bicycle peptide guided the design of a new Bicycle (BCY6099) with TATA scaffold and polar or charged unnatural amino acids, providing increased hydrophilic character. This led to the design of conjugate 22 with the new Bicycle peptide BCY6099. Evaluation of conjugate 22 in an HT-1080 xenograft mouse model displayed comparable efficacy to earlier conjugates. Complete regression of the tumor was observed in 2 weeks at a dose of 3 mg/kg, which was well tolerated. Overall, conjugate 22 displayed faster renal elimination and low biodistribution to the liver leading to reduced liver toxicity and improved therapeutic efficacy. Clinical evaluation of similar conjugates with BCY6099 Bicycle peptide is currently underway.59
Liang et al. designed several PDCs to compare different linkers with respect to the antitumor efficacy.34 PDC 23 (Fig. 9) was designed with a disulfide linker between Dox and cyclic RGD peptide and was found to release Dox slowly overtime with 39.2% drug released at 48 h. The drug release, however, was dramatically increased in the presence of DTT (50 mM) to almost completion (~100%) in 48 hours. PDC 23 was compared to PDC 13 (Fig. 5) containing a thioether linker. The drug release profile (22% in 48 h) of PDC 13 showed no significant change in the presence of DTT and the conjugate was found to be more stable than 23. Furthermore, the cellular uptake, cytotoxicity, and in vivo efficacy were better for the PDC 13 with the thioether linker compared to the disulfide carrying PDC 23.
Li et al. 60 designed a reduction-responsive peptide-drug conjugate (BP9a-SS-DOX) where a modified analog of peptide BP9a with a sequence of CAHLHNRS was linked to Dox through disulfide linker to form BP9a-SS-DOX or PDC 24 (Fig. 9). The uptake studies showed that BP9a peptide analog has a high affinity for transferrin receptors (TfR) that are overexpressed in HepG2 liver cancer cells; therefore, it can be used as a promising carrier for selective drug delivery. The disulfide bond between BP9a and Dox is cleaved by reduced glutathione (GSH) present in tumor cells to release Dox-SH. The conjugate showed good selectivity but low potency compared to free Dox. The use of disulfide linker in PDCs may continue, however, with the availability of other linkers that provide higher selectivity and in vivo efficacy the use of disulfide must be carefully implemented. For instance, disulfide in combination with hydrazone has been successfully used in two of the FDA approved ADCs.9
5. NON-CLEAVABLE LINKER CHEMISTRIES
Non-cleavable linkers have been used for the synthesis of PDCs. Non-cleavable chemistries such as succinimidyl thioether, oxime, and triazole have been employed for conjugating a chemotherapeutic drug to a targeting peptide. However, as mentioned earlier, there can be several functional groups present in the linker, such as group conjugating the drug to the linker, linker to the peptide and sometimes an additional functional group in the middle of the linker which may be cleavable. After systemic administration of a PDC, the most labile group is cleaved first followed by other groups. This results in a mixture with different concentrations of the unmodified drug, drug with a portion of the linker, or drug with linker and amino acid(s) from the peptide. The advantage of the non-cleavable linkers is that these likely do not cleave in the plasma unlike other linker chemistries, and prematurely release some drug in plasma before reaching the target cancer site.
5.1. Oxime.
In a series of papers, Gabor Mezo and coauthors showed the potential of an oxime as a linker for the design of PDCs.61–64 The group made several PDCs by introducing an oxime bond at the C-13 carbonyl of doxorubicin or daunorubicin (Fig. 10) and evaluated in vitro and in vivo properties for targeted therapy of breast, colorectal, and pancreatic cancers. The authors found that the oxime bond was more stable compared to ester linkage for conjugating a drug to the targeting peptide allowing longer circulation half-life. The drug is released after uptake by the cancer cells by the lysosomal enzymes. As oxime is a stable and non-cleavable linker, the PDC is likely cleaved at the amide bond or at the peptide portion. Incubation of different conjugates, with a variety of linkers including oxime, with rat liver lysosomal homogenate and comparison of the released fragments showed that the oxime-linked conjugates do not release the free drug, whereas conjugates with the drug linked via ester or dipeptide bond release the free drug in addition to other fragments.65
Figure 10.

PDCs with an oxime as a linker.
Fourteen oxime linked daunorubicin (Dau)-GnRH-III conjugates were prepared in which a variety of unnatural and D-amino acids were used in the peptide sequence.63 The peptides used were GnRH peptide derivatives to target the overexpressed GnRH-receptors found in many cancers. All fourteen Dau-GnRH-III conjugates showed activity against MCF-7 breast cancer cells (IC50 0.14–6.64 μM) as well as against HT-29 colon cancer cells (IC50 3.31–18.00 μM). The lead conjugates 25 and 26 (Fig. 10) showed stability in 90% human serum for at least 24 hours. Ligand competition assay of 25 and 26 to GnRH-receptors showed both compounds have a high binding affinity for the GnRH-receptors. The degradation of conjugates 25 and 26 was analyzed in rat liver lysosome homogenate, in which 26 showed higher stability compared to 25. In general, oxime was found to be highly stable to chemical or enzymatic degradation.64 The conjugates got cleaved at the peptide portion giving the drug with small peptide fragments attached as the smallest active metabolites. The cellular uptake of 26 was reported higher than 25 when treated with MCF-7 and HT-29 for 6 hours via flow cytometry. Further, incubation of the MCF-7 breast cancer cells with 26 and observation of the cellular uptake of the PDC using confocal microscope images taken at different time intervals (5 sec- 60 min) showed that in the first minute Dau signal was present mainly in the cytosol and small vesicles. At 5 mins, however, the Dau fluorescence was observed everywhere in the cell, including the nuclei, suggesting the drug reached its site of action within 5 minutes.
Randelovic et al. continued this work and showed in vivo activity of conjugates 25 and 26 on breast and colorectal cancer bearing mice using orthotopic models 4T1 and MDA-MB-231 breast cancer, and HT-29 colorectal cancer in BALB/s and SCID mice.62 The results showed a significant tumor volume reduction as well as metastases inhibition effect with less toxic side effects such as weight and behavior of the animals compared to the free drug. The results of this study prove that GnRH-III-Dau conjugates 25 and 26 are promising candidates for targeted tumor therapy in breast and colon cancers.
Next, oxime was used to construct PDCs for targeting Wnt/β-catenin pathway found to be hyperactivated in pancreatic cancers. The targeting peptide SKAAKN was conjugated to daunomycin via oxime linkage to prepare several PDCs.61 PDC 27 (Fig. 10), with a cathepsin B cleavable peptide (GFLG) inserted at the N-terminal of targeting peptide sequence and two drug molecules conjugated per peptide, was most efficient in uptake by PANC-1 cells and displayed high cytotoxicity. Incubation of 27 with lysosome homogenate led to the identification of several active Dau-containing metabolites with one or more amino acids while there was no detection of free Dau. In vivo evaluation in PANC-1 tumor bearing SCID male mice showed that tumor volume inhibition was significant for the conjugate 27 treated mice (17.5% for the dose of 2 mg/kg group and 39.9% for the dose of 10 mg/kg group) compared to the control group (treated with sterile water) on day 70. At the end of the study, on day 74, the tumor weights for the conjugate treated group was significantly lower (27.3% for the dose of 2 mg/kg group and 30.4% for the dose of 10 mg/kg group) than the control group. Mice treated with free Dau were terminated on day 28 due to significant weight loss (21%). Conjugate 27 was able to inhibit the tumor growth significantly even at the low dose of 2 mg/kg with minimal side effects, further highlighting the potential of conjugate 27, with enzyme cleavable linker and higher drug to peptide ratio of 2, for pancreatic cancer treatment. The study indicated the influence of factors like cellular uptake and effective release of the active metabolite (active drug) on improved efficacy of PDCs.
Feni et al. utilized oxime to attach cytotoxic daunorubicin to the linker and additional chemistries like triazole and amide were utilized to put together an integrin targeting PDC (28, Fig. 10). Briefly, an alkyne-azide click reaction was used to develop PDC 28 with an integrin-targeting cyclic diketopiperazine (DKP)-RGD peptide and a cell-penetrating peptide (CPP) to promote effective and targeted cellular uptake.66 Integrins are found to be overexpressed on the surface of some tumor cells and thereby are an appropriate target for cancer drug delivery. Cyclic RGD peptide was conjugated to the CPP using azide PEG4-linker via click chemistry. The CPP termed sC18 included an additional four amino acids Gly-Phe-Leu-Gly (GFLG) attached to the side chain of lysine which were then conjugated to the drug daunorubicin via an oxime bond. As both oxime and triazole are non-cleavable bonds, the tetrapeptide GFLG was inserted for facilitating cleavage by cathepsin B in the lysosomes for efficient drug release after cellular uptake. In addition, the linker also incorporates several amides which can be cleaved by lysosomal amidases. The affinity of 28 for tumor-associated integrin receptors, αvβ3 and αvβ5, was determined using an in vitro binding assay. The conjugate expressed good affinity, and further cytotoxicity studies with 28 revealed that although the conjugate was toxic (EC50 range 2.7–11.0 μM) to all cell lines (U87, HT-29, and MCF-7), it was much less toxic than the free Dau. The conjugate showed no selectivity toward cells with higher expression of integrin αvβ3 (U87).
5.2. Triazole.
As discussed above, several PDCs utilize the alkyne-azide click reaction for the facile linking of a drug to the peptide. The non-cleavable triazole formed by the alkyne-azide reaction provides stability to the PDC structure, and other bonds in the linker region likely facilitate the release of the drug from the PDC after cellular uptake.
An interesting example of a conjugate with a triazole linker is a PDC developed for cancer treatment using photodynamic therapy (PDT). PDT incorporates a photosensitizer, molecular oxygen, and appropriate light to produce reactive oxygen to destroy tumor cells. Zheng et al. explored two variables in the conjugate design for targeted PDT, linker length and number of peptides.67 The click reaction was used to synthesize silicon phthalocyanines axially substituted with cyclic peptide, cRGD (29 a-c, Fig. 11). Different ethylene glycol units (n=2–4; named a-c, respectively) were utilized as a linker between silicon phthalocyanines and the peptides to obtain conjugates with varied lengths and properties. The silicon phthalocyanine that did not contain a peptide was used as a control. Intermediates were reacted to develop the final non-symmetric and asymmetric product conjugates with either one or two peptide(s). Human colon carcinoma HT-29 cells and human breast adenocarcinoma MCF-7 cells were used for in vitro studies to evaluate the photodynamic activity of the products.
Figure 11.

PDC with triazole linkers.
Different cytotoxicities towards the two cell lines occurred with red light (λ > 610 nm). The compounds modified with oligomeric ethylene glycols (no peptide) had the highest potency, and enhanced photocytotoxicity correlated with an increase of ethylene glycol chains (n = 3>2>1). Contrastingly, conjugates with cRGD moieties had reduced photocytotoxicity, likely due to hindrance and varying cellular uptake mechanisms, and these conjugates were selective. Compounds 29a-29c (Fig. 11) had significantly different potency, which is likely due to the varying distances between the phthalocyanine and the peptide. Conjugate 29c was most cytotoxic towards both the cell lines; however, conjugate 29b was more selective with higher toxicity toward HT-29 compared to MCF-7. Further studies suggested that the uptake process of 29b involved receptor-mediated endocytosis. In vivo studies were conducted to analyze the biodistribution of 29b in KM mice with an H22 tumor, which has high integrin expression. Fluorescence images of mice injected with 29b showed selective accumulation at the tumor site, and 29b was seen to persist at the tumor site up to 24 h. At 24 h, mice were sacrificed, and the fluorescence from isolated organs showed that the tumor region had higher fluorescence (~ 2-fold) than the liver and kidney. The two cRGD moieties seemed to attribute to the high specificity of 29b towards H22 tumor and led to a reduction of tumor growth by approximately 75%. The study shows the potential of designing a PDC with a cancer cell targeting peptide to improve the outcome of PDT for cancer treatment. For PDT, it is not required that the drug be released free from the conjugate after cellular uptake, so in this case, the use of non-cleavable linkers is appropriate.
6. FUTURE OUTLOOK AND CONCLUDING REMARKS
With the current progress in PDC development and an enormous knowledge base from ADCs approved for clinical use,3–7 it is just a matter of time that PDCs will prove to be a novel modality for cancer treatment. As addressed above, several factors influence the efficacy of drug conjugates, including the target receptor specificity, cellular uptake, half-life of the conjugate, release of an active drug or drug metabolite, and concentration of the drug at the target site. A higher concentration of drug at the cancer site has been achieved frequently with ADCs due to the inherent conjugation of several drug molecules at different sites on an antibody, and ADCs typically end up with a high drug-to-antibody ratio (DAR) of 3–8. This, however, leads to heterogeneous mixtures with differences in solubility, aggregation, and clearance. The use of high DAR is recommended for better efficacy, and this strategy is also utilized for PDCs as that can easily achieve homogeneity. PDCs such as 5 (Fig. 2) and 27 (Fig. 10) with a drug-to-peptide ratio of 3 and 2, respectively, were found to be significantly more efficacious delivering a higher concentration of the drug at the target site than PDCs with a drug-to-peptide ratio of 1.21, 61
The release of the active drug or the drug metabolite from the conjugate depends on the linker. All linker chemistries discussed above for the PDCs have been successful in the realm of ADCs.3, 9 Based on the recent ADC approvals, however, it appears that enzyme cleavable linkers are more effective (Table 1). Non-cleavable succinimidyl thioether proved to be useful as it is extensively used to conjugate the antibody to the linker via the thiol group present in the cysteine residues of the antibody, such as ADCs brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin, and trastuzumab deruxtecan,68–70 and in acid/base cleavable ADC sacituzumab govitecan (Fig. 12). Trastuzumab emtansine is the only ADC where succinimidyl thioether is the main non-cleavable linker, however this ADC also has additional amide bonds in the linker region for possible cleavage by amidases. It has been postulated that trastuzumab emtansine likely undergoes proteolytic cleavage at the lysine residue from the antibody portion to give lysine-linker-drug as a major active metabolite after lysosomal degradation 71. Further, Phillips et al. found that the trastuzumab emtansine displayed superior in vivo efficacy and PK properties compared to similar ADCs with disulfide bond linker when evaluated in HER2-positive breast cancer mice models.71 Trastuzumab emtansine displayed favorable pharmacokinetic profile making all the way to the clinics. Several PDCs, such as 12 or 13 (Fig. 5), with the same linker chemistry as trastuzumab emtansine have thus been synthesized and are under development.34, 41
Table 1.
Linker Chemistries of the ADCs Approved for Clinical Use
| ADC | Linker Chemistry | Year approved |
|---|---|---|
|
| ||
| Enzyme cleavable | ||
| Brentuximab vedotin (Adcetris) | carbamate, dipeptide, thioether | 2011 |
| Polatuzumab vedotin (Polivy) | carbamate, dipeptide, thioether | 2019 |
| Enfortumab vedotin (Padcev) | carbamate, dipeptide, thioether | 2019 |
| Trastuzumab deruxtecan (Enhertu) | amide, tripeptide, thioether | 2019 |
| Trastuzumab emtansine (Kadcyla) | thioether, amide | 2013 |
|
| ||
| Enzyme or acid cleavable/ Reducible disulfide | ||
| Gemtuzumab ozogamicin (Mylotarg) | disulfide, hydrazone, amide | 2000* |
| Inotuzumab ozogamicin (Besponza) | disulfide, hydrazone, amide | 2017 |
|
| ||
| Acid cleavable | ||
| Sacituzumab govitecan (Trodelvy) | carbonate, triazole, thioether | 2020 |
It was taken off market in 2010 and reapproved in 2017
Figure 12.
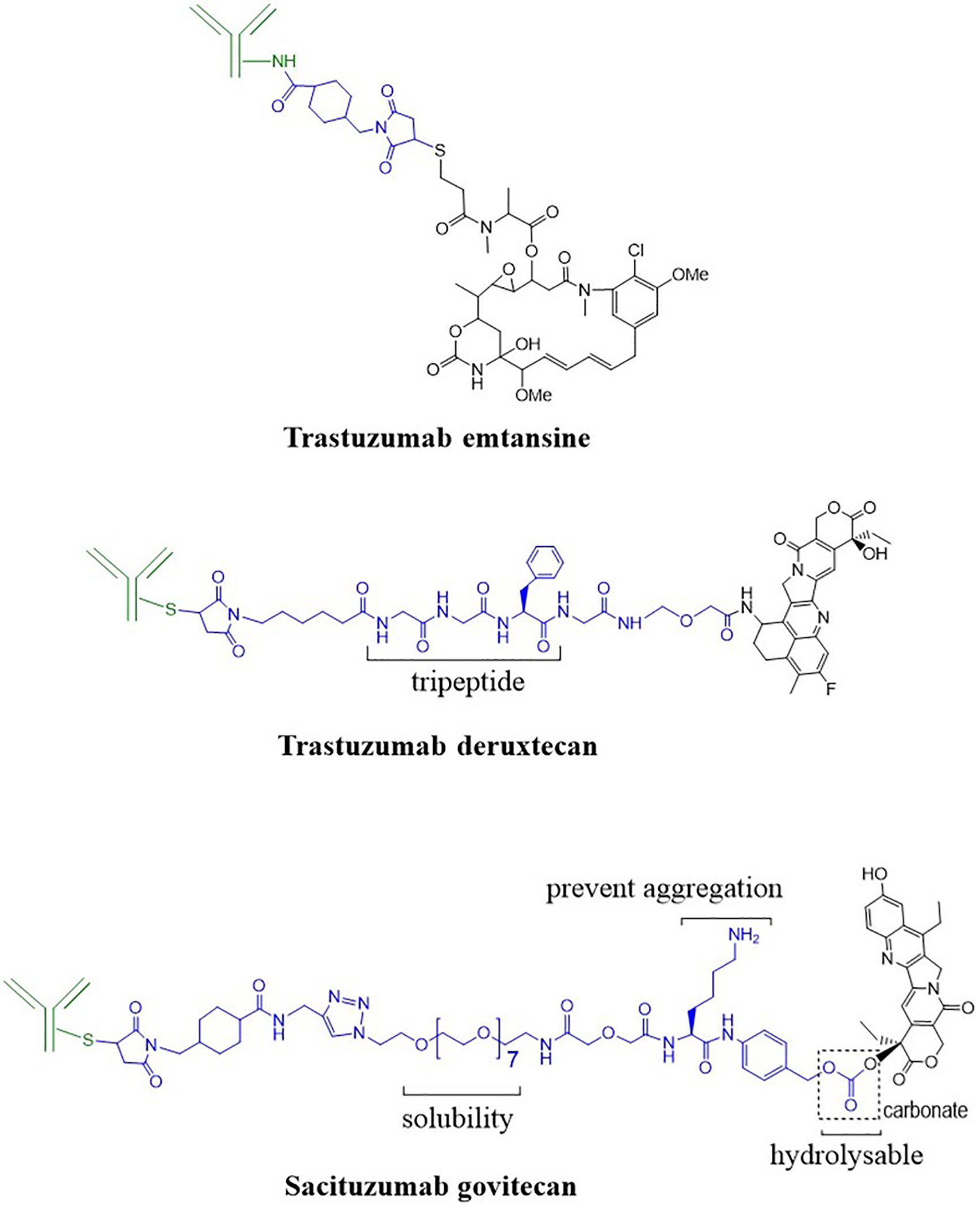
Structures of some approved ADCs with different linker chemistries. The antibody is shown in green, the linker in blue, and the drug in black.
Sacituzumab govitecan (Table 1 and Fig. 12) is the most recent ADC that came into clinics.8, 72 It consists of a hydrolysable carbonate linker that breaks under the acidic (low pH) conditions of lysosomes and tumor microenvironment.72, 73 Lysine is inserted in the linker region to prevent aggregation, and hydrophilic PEG facilitates the solubility of ADC. The less toxic SN-38 drug (e.g., compared to Dxd in trastuzumab deruxtecan) allowed the conjugation of up to eight SN-38 molecules per antibody for this ADC. A carbonate linker is less utilized in the design of PDCs. With the success of sacituzumab govitecan, we hope to see more use of this linker in the development of future PDCs.
In this review, chemical advances made in the linker technology for the development of PDCs are highlighted. Clues from the linker chemistries utilized for ADCs are helpful in the design and successful development of PDCs. Due to the simplicity of peptides compared to antibodies and the ease of possible reactions during PDC synthesis, it is envisioned that PDCs with other chemistries (not seen in ADCs) that give rise to different linkers and structures will be explored. The key role played by the linker chemistry in PDCs for enhancing efficacy, increasing circulation time, and lowering toxicity makes it worth exploring. Strategic implementation of linker functional groups will allow more PDCs to enter clinical trials and, eventually, clinics for cancer treatment and beyond.
ACKNOWLEDGEMENTS
The National Cancer Institute of the National Institutes of Health (Award Number R15CA208656) supported this research.
ABBREVIATIONS
- ADC
antibody-drug conjugate
- BBB
blood brain barrier
- CT
chemotherapeutic
- Dau
daunorubicin
- DDAE
di-desmethylauristatin E
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- Dox
doxorubicin
- EMCH
N-ε-maleimidocaproic acid hydrazide
- EphA2
erythropoietin-producing hepatocellular receptor A2
- GEP-NETs
gastroenteropancreatic neuroendocrine tumors
- GnRH-R
gonadotropin releasing hormone-receptor
- HER2
human epidermal growth factor receptor 2
- i.v.
intravenous
- K1
keratin 1
- LC
leptomeningeal carcinomatosis
- 177Lu
lutetium 177
- MMAE
monomethylauristatin E
- PABC
para-aminobenzyl carbamate
- PDC
peptide-drug conjugate
- PDT
photodynamic therapy
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- PSA
prostate-specific antigen
- TATE
tyrosine-3 octreotate
Biographies
Mona Alas is a Pharm.D. student at the Chapman University School of Pharmacy (CUSP) anticipated to graduate in 2021. Before joining CUSP, she was accepted into the pre-pharmacy Freshman Early Assurance Program (FEAP) at Chapman University. She began her research during her pre-pharmacy days and continues to show her commitment to research by enrolling in a Capstone project while at CUSP. Her research interests have grown to include medicinal chemistry and the development of novel oncology therapeutics.
Azam Saghaeidehkordi is a final year Ph.D. student at Chapman University School of Pharmacy. She also received her master’s in pharmaceutical sciences from Chapman University (2018). Both her master and Ph.D., under the supervision of Professor Kamaljit Kaur, focus on the development of peptide-drug conjugates for breast cancer treatment. Azam received her BS in pharmaceutical sciences from the University of California, Irvine, in 2015 and worked for a pharmaceutical company in diagnostic clinical trials for a year (2015–2016) before joining Chapman. Her interests remain in the areas of biomedical/clinical sciences and cancer treatment.
Kamaljit Kaur is an Associate Professor of Targeted Drug Delivery and Biomedical Diagnostics at the Chapman University School of Pharmacy. She started her academic career in 2004 as an Assistant Professor of Medicinal Chemistry at the Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta (Canada). She moved to Chapman University (USA) in 2014 to accept a part-time position as a Distinguished Chancellor Fellow and in 2015 as a full-time founding member of the CUSP faculty. Her education is from India and the US with her B.Sc. (Chemistry Honors) from University of Delhi, M.Sc. (Organic Chemistry) from Indian Institute of Technology, Kanpur, and Ph.D. (Bioorganic Chemistry) from Case Western Reserve University, Cleveland, Ohio. Her lab combines organic and analytical chemistry methods with biology to engineer peptides for biomedical applications.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Gotwals P; Cameron S; Cipolletta D; Cremasco V; Crystal A; Hewes B; Mueller B; Quaratino S; Sabatos-Peyton C; Petruzzelli L; Engelman JA; Dranoff G, Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer 2017, 17 (5), 286–301. [DOI] [PubMed] [Google Scholar]
- 2.Mitra AK; Agrahari V; Mandal A; Cholkar K; Natarajan C; Shah S; Joseph M; Trinh HM; Vaishya R; Yang X; Hao Y; Khurana V; Pal D, Novel delivery approaches for cancer therapeutics. J Control Release 2015, 219, 248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chau CH; Steeg PS; Figg WD, Antibody-drug conjugates for cancer. Lancet 2019, 394 (10200), 793–804. [DOI] [PubMed] [Google Scholar]
- 4.He R; Finan B; Mayer JP; DiMarchi RD, Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules 2019, 24 (10), 1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas A; Teicher BA; Hassan R, Antibody-drug conjugates for cancer therapy. Lancet Oncol 2016, 17 (6), e254–e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vrettos EI; Mezo G; Tzakos AG, On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J Org Chem 2018, 14, 930–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y; Cheetham AG; Angacian G; Su H; Xie L; Cui H, Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev 2017, 110–111, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardia A; Mayer IA; Vahdat LT; Tolaney SM; Isakoff SJ; Diamond JR; O’Shaughnessy J; Moroose RL; Santin AD; Abramson VG; Shah NC; Rugo HS; Goldenberg DM; Sweidan AM; Iannone R; Washkowitz S; Sharkey RM; Wegener WA; Kalinsky K, Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med 2019, 380 (8), 741–751. [DOI] [PubMed] [Google Scholar]
- 9.Bargh JD; Isidro-Llobet A; Parker JS; Spring DR, Cleavable linkers in antibody-drug conjugates. Chem Soc Rev 2019, 48 (16), 4361–4374. [DOI] [PubMed] [Google Scholar]
- 10.Trail PA; Dubowchik GM; Lowinger TB, Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol Ther 2018, 181, 126–142. [DOI] [PubMed] [Google Scholar]
- 11.Li Z; Krippendorff BF; Shah DK, Influence of molecular size on the clearance of antibody fragments. Pharm Res 2017, 34 (10), 2131–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagh A; Song H; Zeng M; Tao L; Das TK, Challenges and new frontiers in analytical characterization of antibody-drug conjugates. MAbs 2018, 10 (2), 222–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopp A; Thurber GM, Severing ties: quantifying the payload release from antibody drug conjugates. Cell Chem Biol 2019, 26 (12), 1631–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang TL; Szekacs A; Uematsu T; Kuwano E; Parkinson A; Hammock BD, Hydrolysis of carbonates, thiocarbonates, carbamates, and carboxylic esters of alpha-naphthol, beta-naphthol, and p-nitrophenol by human, rat, and mouse liver carboxylesterases. Pharm Res 1993, 10 (5), 639–648. [DOI] [PubMed] [Google Scholar]
- 15.Laizure SC; Herring V; Hu Z; Witbrodt K; Parker RB, The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 2013, 33 (2), 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vacondio F; Silva C; Mor M; Testa B, Qualitative structure-metabolism relationships in the hydrolysis of carbamates. Drug Metab Rev 2010, 42 (4), 551–589. [DOI] [PubMed] [Google Scholar]
- 17.Karampelas T; Argyros O; Sayyad N; Spyridaki K; Pappas C; Morgan K; Kolios G; Millar RP; Liapakis G; Tzakos AG; Fokas D; Tamvakopoulos C, GnRH-Gemcitabine conjugates for the treatment of androgen-independent prostate cancer: pharmacokinetic enhancements combined with targeted drug delivery. Bioconjug Chem 2014, 25 (4), 813–823. [DOI] [PubMed] [Google Scholar]
- 18.You Y; Xu Z; Chen Y, Doxorubicin conjugated with a trastuzumab epitope and an MMP-2 sensitive peptide linker for the treatment of HER2-positive breast cancer. Drug Deliv 2018, 25 (1), 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soudy R; Chen C; Kaur K, Novel peptide-doxorubucin conjugates for targeting breast cancer cells including the multidrug resistant cells. J Med Chem 2013, 56 (19), 7564–7573. [DOI] [PubMed] [Google Scholar]
- 20.Soudy R; Etayash H; Bahadorani K; Lavasanifar A; Kaur K, Breast cancer targeting peptide binds keratin 1: a new molecular marker for targeted drug delivery to breast cancer. Mol Pharm 2017, 14 (3), 593–604. [DOI] [PubMed] [Google Scholar]
- 21.Regina A; Demeule M; Che C; Lavallee I; Poirier J; Gabathuler R; Beliveau R; Castaigne JP, Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol 2008, 155 (2), 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumthekar P; Tang SC; Brenner AJ; Kesari S; Piccioni DE; Anders C; Carrillo J; Chalasani P; Kabos P; Puhalla S; Tkaczuk K; Garcia AA; Ahluwalia MS; Wefel JS; Lakhani N; Ibrahim N, ANG1005, a brain-penetrating peptide-drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin Cancer Res 2020, 26 (12), 2789–2799. [DOI] [PubMed] [Google Scholar]
- 23.Kurzrock R; Gabrail N; Chandhasin C; Moulder S; Smith C; Brenner A; Sankhala K; Mita A; Elian K; Bouchard D; Sarantopoulos J, Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol Cancer Ther 2012, 11 (2), 308–316. [DOI] [PubMed] [Google Scholar]
- 24.O’Sullivan CC; Lindenberg M; Bryla C; Patronas N; Peer CJ; Amiri-Kordestani L; Davarpanah N; Gonzalez EM; Burotto M; Choyke P; Steinberg SM; Liewehr DJ; Figg WD; Fojo T; Balasubramaniam S; Bates SE, ANG1005 for breast cancer brain metastases: correlation between (18)F-FLT-PET after first cycle and MRI in response assessment. Breast Cancer Res Treat 2016, 160 (1), 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bushnell DL; Bodeker KL, Overview and current status of peptide receptor radionuclide therapy. Surg Oncol Clin N Am 2020, 29 (2), 317–326. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee S; Pillai MR; Knapp FF, Lutetium-177 therapeutic radiopharmaceuticals: linking chemistry, radiochemistry, and practical applications. Chem Rev 2015, 115 (8), 2934–2974. [DOI] [PubMed] [Google Scholar]
- 27.Jamous M; Haberkorn U; Mier W, Synthesis of peptide radiopharmaceuticals for the therapy and diagnosis of tumor diseases. Molecules 2013, 18 (3), 3379–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L; Fuselier JA; Coy DH, Effects of camptothecin conjugated to a somatostatin analog vector on growth of tumor cell lines in culture and related tumors in rodents. Drug Deliv 2004, 11 (4), 231–238. [DOI] [PubMed] [Google Scholar]
- 29.Sun LC; Coy DH, Somatostatin receptor-targeted anti-cancer therapy. Curr Drug Deliv 2011, 8 (1), 2–10. [DOI] [PubMed] [Google Scholar]
- 30.Sun LC; Mackey LV; Luo J; Fuselier JA; Coy DH, Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin Med Oncol 2008, 2, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sayyad N; Vrettos EI; Karampelas T; Chatzigiannis CM; Spyridaki K; Liapakis G; Tamvakopoulos C; Tzakos AG, Development of bioactive gemcitabine-D-Lys(6)-GnRH prodrugs with linker-controllable drug release rate and enhanced biopharmaceutical profile. Eur J Med Chem 2019, 166, 256–266. [DOI] [PubMed] [Google Scholar]
- 32.Kumar SK; Williams SA; Isaacs JT; Denmeade SR; Khan SR, Modulating paclitaxel bioavailability for targeting prostate cancer. Bioorg Med Chem 2007, 15 (14), 4973–4984. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh AK; Brindisi M, Organic carbamates in drug design and medicinal chemistry. J Med Chem 2015, 58 (7), 2895–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang Y; Li S; Wang X; Zhang Y; Sun Y; Wang Y; Wang X; He B; Dai W; Zhang H; Wang X; Zhang Q, A comparative study of the antitumor efficacy of peptide-doxorubicin conjugates with different linkers. J Control Release 2018, 275, 129–141. [DOI] [PubMed] [Google Scholar]
- 35.Bajjuri KM; Liu Y; Liu C; Sinha SC, The legumain protease-activated auristatin prodrugs suppress tumor growth and metastasis without toxicity. ChemMedChem 2011, 6 (1), 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mai CW; Chung FF; Leong CO, Targeting legumain as a novel therapeutic strategy in cancers. Curr Drug Targets 2017, 18 (11), 1259–1268. [DOI] [PubMed] [Google Scholar]
- 37.Stern L; Perry R; Ofek P; Many A; Shabat D; Satchi-Fainaro R, A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjug Chem 2009, 20 (3), 500–510. [DOI] [PubMed] [Google Scholar]
- 38.Fontaine SD; Reid R; Robinson L; Ashley GW; Santi DV, Long-term stabilization of maleimide-thiol conjugates. Bioconjug Chem 2015, 26 (1), 145–152. [DOI] [PubMed] [Google Scholar]
- 39.Lyon RP; Setter JR; Bovee TD; Doronina SO; Hunter JH; Anderson ME; Balasubramanian CL; Duniho SM; Leiske CI; Li F; Senter PD, Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat Biotechnol 2014, 32 (10), 1059–1062. [DOI] [PubMed] [Google Scholar]
- 40.Tumey LN; Charati M; He T; Sousa E; Ma D; Han X; Clark T; Casavant J; Loganzo F; Barletta F; Lucas J; Graziani EI, Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjug Chem 2014, 25 (10), 1871–1880. [DOI] [PubMed] [Google Scholar]
- 41.Ziaei E; Saghaeidehkordi A; Dill C; Maslennikov I; Chen S; Kaur K, Targeting triple negative breast cancer cells with novel cytotoxic peptide-doxorubicin conjugates. Bioconjug Chem 2019, 30 (12), 3098–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinn BA; Wang S; Barile E; Das SK; Emdad L; Sarkar D; De SK; Morvaridi SK; Stebbins JL; Pandol SJ; Fisher PB; Pellecchia M, Therapy of pancreatic cancer via an EphA2 receptor-targeted delivery of gemcitabine. Oncotarget 2016, 7 (13), 17103–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salem AF; Wang S; Billet S; Chen JF; Udompholkul P; Gambini L; Baggio C; Tseng HR; Posadas EM; Bhowmick NA; Pellecchia M, Reduction of circulating cancer cells and metastases in breast-cancer models by a potent EphA2-agonistic peptide-drug conjugate. J Med Chem 2018, 61 (5), 2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ireton RC; Chen J, EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr Cancer Drug Targets 2005, 5 (3), 149–157. [DOI] [PubMed] [Google Scholar]
- 45.Walker-Daniels J; Hess AR; Hendrix MJ; Kinch MS, Differential regulation of EphA2 in normal and malignant cells. Am J Pathol 2003, 162 (4), 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mollaev M; Gorokhovets N; Nikolskaya E; Faustova M; Zabolotsky A; Zhunina O; Sokol M; Zamulaeva I; Severin E; Yabbarov N, Type of pH sensitive linker reveals different time-dependent intracellular localization, in vitro and in vivo efficiency in alpha-fetoprotein receptor targeted doxorubicin conjugate. Int J Pharm 2019, 559, 138–146. [DOI] [PubMed] [Google Scholar]
- 47.Sheng Y; You Y; Chen Y, Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int J Pharm 2016, 512 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- 48.Yousefpour P; Ahn L; Tewksbury J; Saha S; Costa SA; Bellucci JJ; Li X; Chilkoti A, Conjugate of doxorubicin to albumin-binding peptide outperforms aldoxorubicin. Small 2019, 15 (12), e1804452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillies ER; Goodwin AP; Frechet JM, Acetals as pH-sensitive linkages for drug delivery. Bioconjug Chem 2004, 15 (6), 1254–1263. [DOI] [PubMed] [Google Scholar]
- 50.Tomlinson R; Heller J; Brocchini S; Duncan R, Polyacetal-doxorubicin conjugates designed for pH-dependent degradation. Bioconjug Chem 2003, 14 (6), 1096–1106. [DOI] [PubMed] [Google Scholar]
- 51.Di Stefano G; Lanza M; Kratz F; Merina L; Fiume L, A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci 2004, 23 (4–5), 393–397. [DOI] [PubMed] [Google Scholar]
- 52.Kratz F, DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs 2007, 16 (6), 855–866. [DOI] [PubMed] [Google Scholar]
- 53.Kratz F; Warnecke A; Schmid B; Chung DE; Gitzel M, Prodrugs of anthracyclines in cancer chemotherapy. Curr Med Chem 2006, 13 (5), 477–523. [DOI] [PubMed] [Google Scholar]
- 54.Chawla SP; Papai Z; Mukhametshina G; Sankhala K; Vasylyev L; Fedenko A; Khamly K; Ganjoo K; Nagarkar R; Wieland S; Levitt DJ, First-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma: a phase 2b randomized clinical trial. JAMA Oncol 2015, 1 (9), 1272–1280. [DOI] [PubMed] [Google Scholar]
- 55.Gong J; Yan J; Forscher C; Hendifar A, Aldoxorubicin: a tumor-targeted doxorubicin conjugate for relapsed or refractory soft tissue sarcomas. Drug Des Devel Ther 2018, 12, 777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levitt D Cytotoxic Agents for the Treatment of Cancer. U.S. Patent US20190350952A1 November 21, 2019.
- 57.Mita MM; Natale RB; Wolin EM; Laabs B; Dinh H; Wieland S; Levitt DJ; Mita AC, Pharmacokinetic study of aldoxorubicin in patients with solid tumors. Invest New Drugs 2015, 33 (2), 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forman HJ; Zhang H; Rinna A, Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 2009, 30 (1–2), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mudd GE; Brown A; Chen L; van Rietschoten K; Watcham S; Teufel DP; Pavan S; Lani R; Huxley P; Bennett GS, Identification and optimization of EphA2-selective bicycles for the delivery of cytotoxic payloads. J Med Chem 2020, 63 (8), 4107–4116. [DOI] [PubMed] [Google Scholar]
- 60.Li S; Zhao H; Mao X; Fan Y; Liang X; Wang R; Xiao L; Wang J; Liu Q; Zhao G, Transferrin receptor targeted cellular delivery of doxorubicin via a reduction-responsive peptide-drug conjugate. Pharm Res 2019, 36 (12), 168. [DOI] [PubMed] [Google Scholar]
- 61.Dokus LE; Lajko E; Randelovic I; Mezo D; Schlosser G; Kohidai L; Tovari J; Mezo G, Phage display-based homing peptide-daunomycin conjugates for selective drug targeting to PANC-1 pancreatic cancer. Pharmaceutics 2020, 12 (6), 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Randelovic I; Schuster S; Kapuvari B; Fossati G; Steinkuhler C; Mezo G; Tovari J, Improved in vivo anti-tumor and anti-metastatic effect of GnRH-III-daunorubicin analogs on colorectal and breast carcinoma bearing mice. Int J Mol Sci 2019, 20 (19), 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schuster S; Biri-Kovacs B; Szeder B; Buday L; Gardi J; Szabo Z; Halmos G; Mezo G, Enhanced in vitro antitumor activity of GnRH-III-daunorubicin bioconjugates influenced by sequence modification. Pharmaceutics 2018, 10 (4), 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuster S; Biri-Kovacs B; Szeder B; Farkas V; Buday L; Szabo Z; Halmos G; Mezo G, Synthesis and in vitro biochemical evaluation of oxime bond-linked daunorubicin-GnRH-III conjugates developed for targeted drug delivery. Beilstein J Org Chem 2018, 14, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schlage P; Mezo G; Orban E; Bosze S; Manea M, Anthracycline-GnRH derivative bioconjugates with different linkages: synthesis, in vitro drug release and cytostatic effect. J Control Release 2011, 156 (2), 170–178. [DOI] [PubMed] [Google Scholar]
- 66.Feni L; Parente S; Robert C; Gazzola S; Arosio D; Piarulli U; Neundorf I, Kiss and run: promoting effective and targeted cellular uptake of a drug delivery vehicle composed of an integrin-targeting diketopiperazine peptidomimetic and a cell-penetrating peptide. Bioconjug Chem 2019, 30 (7), 2011–2022. [DOI] [PubMed] [Google Scholar]
- 67.Zheng BY; Yang XQ; Zhao Y; Zheng QF; Ke MR; Lin T; Chen RX; Ho KKK; Kumar N; Huang JD, Synthesis and photodynamic activities of integrin-targeting silicon(IV) phthalocyanine-cRGD conjugates. Eur J Med Chem 2018, 155, 24–33. [DOI] [PubMed] [Google Scholar]
- 68.Deeks ED, Polatuzumab vedotin: first global approval. Drugs 2019, 79 (13), 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenberg JE; O’Donnell PH; Balar AV; McGregor BA; Heath EI; Yu EY; Galsky MD; Hahn NM; Gartner EM; Pinelli JM; Liang SY; Melhem-Bertrandt A; Petrylak DP, Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J Clin Oncol 2019, 37 (29), 2592–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamura K; Tsurutani J; Takahashi S; Iwata H; Krop IE; Redfern C; Sagara Y; Doi T; Park H; Murthy RK; Redman RA; Jikoh T; Lee C; Sugihara M; Shahidi J; Yver A; Modi S, Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol 2019, 20 (6), 816–826. [DOI] [PubMed] [Google Scholar]
- 71.Lewis Phillips GD; Li G; Dugger DL; Crocker LM; Parsons KL; Mai E; Blattler WA; Lambert JM; Chari RV; Lutz RJ; Wong WL; Jacobson FS; Koeppen H; Schwall RH; Kenkare-Mitra SR; Spencer SD; Sliwkowski MX, Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008, 68 (22), 9280–9290. [DOI] [PubMed] [Google Scholar]
- 72.Sahota S; Vahdat LT, Sacituzumab govitecan: an antibody-drug conjugate. Expert Opin Biol Ther 2017, 17 (8), 1027–1031. [DOI] [PubMed] [Google Scholar]
- 73.Goldenberg DM; Cardillo TM; Govindan SV; Rossi EA; Sharkey RM, Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6 (26), 22496–22512. [DOI] [PMC free article] [PubMed] [Google Scholar]