

Abstract
Background and Objectives
The goal of this work was to investigate the natural history and outcomes after treatment for spontaneous amyloid-related imaging abnormalities (ARIA)-like in cerebral amyloid angiopathy–related inflammation (CAA-ri).
Methods
This was a multicenter, hospital-based, longitudinal, prospective observational study of inpatients meeting CAA-ri diagnostic criteria recruited through the Inflammatory Cerebral Amyloid Angiopathy and Alzheimer's Disease βiomarkers International Network from January 2013 to March 2017. A protocol for systematic data collection at first-ever presentation and at subsequent in-person visits, including T1-weighted, gradient recalled echo–T2*, fluid-suppressed T2-weighted (fluid-attenuated inversion recovery), and T1 postgadolinium contrast-enhanced images acquired on 1.5T MRI, was used at the 3-, 6-, 12-, and 24-month follow-up. Centralized reads of MRIs were performed by investigators blinded to clinical, therapeutic, and time-point information. Main outcomes were survival, clinical and radiologic recovery, intracerebral hemorrhage (ICH), and recurrence of CAA-ri.
Results
The study enrolled 113 participants (10.6% definite, 71.7% probable, and 17.7% possible CAA-ri). Their mean age was 72.9 years; 43.4% were female; 37.1% were APOEε4 carriers; 36.3% had a history of Alzheimer disease; and 33.6% had a history of ICH. A history of ICH and the occurrence of new ICH at follow-up were more common in patients with cortical superficial siderosis at baseline (52.6% vs 14.3%, p < 0.0001 and 19.3% vs 3.6%, p < 0.009, respectively). After the first-ever presentation of CAA-ri, 70.3% (95% confidence interval [CI] 61.6%–78.5%) and 84.1% (95% CI 76.2%–90.6%) clinically recovered within 3 and 12 months, followed by radiologic recovery in 45.1% (95% CI 36.4%–54.8%) and 77.4% (95% CI 67.7%–85.9%), respectively. After clinicoradiologic resolution of the first-ever episode, 38.3% (95% CI 22.9%–59.2%) had at least 1 recurrence within the following 24 months. Recurrence was more likely if IV high-dose corticosteroid pulse therapy was suddenly stopped compared to slow oral tapering off (hazard ratio 4.68, 95% CI 1.57–13.93; p = 0.006).
Discussion
These results from the largest longitudinal cohort registry of patients with CAA-ri support the transient and potentially relapsing inflammatory nature of the clinical-radiologic acute manifestations of the disease and the effectiveness of slow oral tapering off after IV corticosteroid pulse therapy in preventing recurrences. Our results highlight the importance of differential diagnosis for spontaneous ARIA-like events in β-amyloid–driven diseases, including treatment-related ARIA in patients with Alzheimer disease exposed to immunotherapy drugs.
Cerebral amyloid angiopathy (CAA)–related inflammation (CAA-ri) is a rare autoimmune encephalopathy associated with increased CSF concentrations of autoantibodies targeting β-amyloid (Aβ) protein deposited in the walls of cortical and leptomeningeal brain arteries, arterioles, and capillaries.1 The disease is characterized by the acute/subacute presentation of neurologic symptoms that vary from very mild cognitive disturbances and headaches to rapidly progressive cognitive decline, seizures, altered mental state, and focal neurologic deficits. These neurologic symptoms are associated with acute white matter hyperintensities (WMHs) suggestive of vasogenic edema (VE) on MRI, associated with MRI hallmarks of CAA such as cerebral microbleeds (CMBs) and cortical superficial siderosis (cSS).1,2
A diagnosis of definite CAA-ri requires histopathologic confirmation. However, the recent clinical-radiologic criteria2 and the ultrasensitive test for anti-Aβ autoantibody detection in the CSF1 have significantly increased the current diagnostic capacity and reduced the need of an invasive brain biopsy.
There is evidence suggesting CAA-ri as a potentially reversible condition responsive to immunosuppressive therapies, but this is limited to sparse case reports or small retrospective case series deriving from single centers.1,3-9 Although CAA-ri was thought to be restricted only to patients with CAA, the disease is now increasingly recognized and diagnosed in a spectrum of different neurologic and neuroradiologic manifestations,10-15 including the striking radiographic similarities with the amyloid-related imaging abnormalities (ARIA) observed in some participants within immunization clinical trials for Alzheimer disease (AD).1,16-23 Therapies using anti-Aβ monoclonal antibodies are complicated by (1) ARIA edema (ARIA-E), consisting of focal areas of WMH suggestive of parenchymal edema or effusion, and (2) ARIA hemorrhage, consisting of CMBs and cSS.24
Given the very few samples and data available from clinical trials, spontaneously occurring ARIA in CAA-ri (ARIA-like) has been proposed as a valid model for the study of therapy-induced ARIA of such trials.1,18-21,23 This has enforced the establishment of a collaborative research framework, the Inflammatory Cerebral Amyloid Angiopathy and Alzheimer's Disease βiomarkers (iCAβ) International Network initiative,25 aimed at increasing the diagnostic, prognostic, and therapeutic capabilities and reducing variability in the management of these underrecognized diseases.
This study aimed to investigate the natural history of CAA-ri and to provide new evidence into the outcomes associated with corticosteroid treatment in a large, multicenter, longitudinal, prospective observational cohort registry of well-defined patients with first-ever diagnosis of CAA-ri, systematically recruited via the iCAβ International Network initiative.25
Methods
Standard Protocol Approvals and Patient Consents
Patients with CAA-ri diagnosed by clinical presentation, radiologic criteria, or pathologic findings2,26 were prospectively referred to the University of Milano–Bicocca through the iCAβ International Network25 from January 2013 to March 2017. The global initiative involves 35 neurology clinic hospitals worldwide. This study was approved under the University of Milano–Bicocca Ethical Committee protocols for the biomarkARIA and modelCAA research studies. Inpatients provided written informed consent or were included under a waiver of consent for the use of clinical and radiologic data.
iCAβ International Network Cohort Registry of CAA-ri
This multicenter, hospital-based, prospective, longitudinal, observational cohort registry includes a predefined protocol for systematic data collection developed by investigators from the University of Milano–Bicocca coordinating center (F.P., J.C.D.) in 2013 and the Italian Society of Neurology–dementia (SINdem) CAA Study group. According to the protocol, a predefined individual case report form (CRF) for detailed demographic, genetic, pathologic, clinical and medication history, clinical features, MRIs, exposure to immunosuppressive therapy, and clinical and radiologic outcomes at follow-up has been systematically provided at the time of each visit.
Here, we provide a summary of the main methodologic aspects and definitions.
All the CRFs and MRIs have been systematically collected at multiple time points by in-person visits in each center. The baseline CRF was provided at first-ever disease presentation (inpatients). Follow-up CRFs and MRIs were provided at each predefined visit at 3, 6, 12, and 24 months and then annually.
Follow-up time was defined as the time elapsed from the date of CAA-ri diagnosis2 (first visit) to the date of each subsequent visit or MRI rescan.
Given the lack of therapeutic recommendations, the choice of treatment was decided on a case-by-case basis by patients and their physicians. If multiple visits or MRI scans were performed for any clinical or therapeutic need, in addition to those suggested by the protocol, only the closest to the predefined time point was considered for the analyses.
According to radiologic criteria,2 the following specific radiographic sequences were strictly required: T1-weighted, gradient recalled echo (GRE)–T2*, and fluid-suppressed T2-weighted (fluid-attenuated inversion recovery). Additional MRI sequences were suggested but not required: susceptibility-weighted imaging (optional, additional to GRE-T2*) and T1 postgadolinium sequences. All MRIs were acquired on 1.5T imaging systems according to routine clinical acquisition parameters used at each center.
First-ever presentation of a CAA-ri acute clinical inflammatory episode was defined as new, atypical, or worsened headaches, acute/subacute rapidly progressive cognitive decline, behavioral changes, focal neurologic deficits, or seizures in participants with no history of CAA-ri and after exclusion of neoplastic, infectious, or other causes. The presentation was not directly attributed to an acute intracerebral hemorrhage (ICH).2,26
Radiographic evidence of CAA-related bleeding manifestations was based on acute brain MRI findings defined as the presence of ≥1 of the following cortico-subcortical hemorrhagic lesions: CMBs, cSS, and ICH on T1-weighted and GRE-T2* (Figures 1 and 2).2,26
Figure 1. Spontaneous ARIA-like Imaging Findings at Presentation and After Radiologic Remission at 3-Month Follow-up of a Patient With Probable CAA-ri.

(A and B) Fluid-attenuated inversion recovery (FLAIR) images (axial sequences) at presentation show cortico-subcortical regions of hyperintensity suggestive of vasogenic edema and sulcal effacement on both hemispheres (red arrows). (C) Susceptibility-weighted images show microbleeds (red circles). (D and E) MRI 3 months after corticosteroid pulse therapy (5 IV boluses of 1 g/d methylprednisolone followed by 1 mg/kg oral prednisone daily and slow tapering off over several months) showing the disappearance of the acute inflammatory white matter hyperintensity lesions in the corresponding planes of FLAIR sequences indexed at presentation. ARIA = amyloid-related imaging abnormalities; CAA-ri = cerebral amyloid angiopathy–related inflammation.
Figure 2. Spontaneous ARIA-like Imaging Findings of a Patient With Probable CAA-ri at Presentation and After 3 Months of Follow-up With No Radiologic Remission.

(A–C) Fluid-attenuated inversion recovery (FLAIR) axial sequences at presentation show cortico-subcortical hyperintense areas on both frontal lobes with edema and mass effect on the anterior horn of the left lateral ventricle. (E–G) FLAIR sequences 3 months after corticosteroid pulse therapy (5 IV boluses of 1 g/d methylprednisolone followed by 1 mg/kg oral prednisone daily and slow tapering off over several months) treatment showing substantial reduction of the signal abnormalities on the right frontal lobe and persistent hyperintense signal abnormality on the left frontal lobe without mass effect and with atrophy of the corresponding cortico-subcortical region. (D and H) Susceptibility-weighted imaging sequences at presentation show microbleeds (red circle) and diffuse cortical superficial siderosis (red arrows). ARIA = amyloid-related imaging abnormalities; CAA-ri = cerebral amyloid angiopathy–related inflammation.
MRI inflammatory manifestations were identified on the basis of patchy or confluent cortico-subcortical WMH lesions suggestive of VE on fluid-attenuated inversion recovery imaging, with or without mass effect and with or without leptomeningeal involvement (Figures 1 and 2).2,26
Cohort Study Design and Participants
All the participants consecutively referred to the iCAβ International Network initiative25 from January 2013 to March 2017 were considered for this study. All data were centrally checked and verified for consistency at the University of Milano–Bicocca by 2 expert neurologists (J.C.D., M.Z.). Only patients with (1) available complete CRF, (2) available adequate MRIs, and (3) at least 1 follow-up visit (including complete CRFs and MRIs) were included in the analysis.
We followed the Strengthening the Reporting of Observational Studies in Epidemiology reporting guideline.
Visual reading of MRIs was centrally assessed by a trained neuroradiologist (R.P.) without knowledge of clinical, therapeutic, and follow-up stage data. CMB number (manual count) and cSS (presence/absence) were assessed on GRE-T2*. When available, T1 postgadolinium sequences were considered for leptomeningeal involvement (positive/negative).
Validated clinical and radiologic diagnostic criteria for CAA-ri2 were used to centrally assign the diagnosis of definitive (biopsy proven), probable (monofocal/multifocal, asymmetric, and bilateral WMH lesions suggestive of parenchymal VE or sulcal effusion, with or without leptomeningeal enhancement), or possible (unilateral WMH lesions) CAA-ri. The interrater reliability of the central evaluation center (κ = 0.83) was assessed in a subset of 23 randomly selected MRI scans, independently evaluated by a second trained neuroradiologist (G.B.), and compared with the findings of the original reader (R.P.).
Clinical recovery was defined as the stable recovery to neurologic conditions preexisting to the acute inflammatory event or the only persistence of neurologic deficits directly attributable to the brain vascular injury event (i.e., ICH) in the absence of new or worsened MRI findings. Clinical recovery was attributed dichotomously (yes/no) by the same clinical neurologist who evaluated the patient at presentation through in-person visits in each center.
Radiologic recovery was defined as the complete disappearance (full resolution) of the acute inflammatory WMH lesions suggestive of VE2 and sulcal effusion indexed at presentation. This was judged as the absence of WMH signs consistent with CAA-ri2 in the central neuroradiology reading report. Patients with partial radiologic improvement were considered as not resolved at a given time point.
Recurrent episode of CAA-ri (relapse) was defined as the presence of new-onset symptoms associated with MRI signs consistent with a new manifestation of CAA-ri2 and only in participants with ascertained clinical and radiologic recovery of the first presentation of the disease. The radiologic progression without new symptoms or in the absence of a former clinical and radiologic recovery of the first event is not included in this definition because we considered that patients who were not clinically and radiologically resolved would not have the chance to relapse.
Biopsy specimens were processed and stained with standard protocols at each center. APOE genotyping was performed as previously described.1,27
Baseline Features at First-Ever Diagnosis of CAA-ri
Baseline features at presentation were classified as diagnostic certainty (definite, possible, probable CAA-ri)2; onset of symptoms (headache, cognitive or behavioral changes, focal neurologic deficits, and seizures); disease history (mild cognitive impairment or AD dementia, past ICH); APOE genotype; number of CMBs (0, 1 single CMB, 2–4, 5–10, >10); cSS (presence/absence); gadolinium-contrast enhancement (positive/negative); and immunosuppressive therapy (drug dose and type, length of treatment).
Outcomes Evaluation at 3, 6, 12, and 24 Months
Primary outcomes were survival, clinical recovery, and relapse of CAA-ri. Secondary outcome was radiologic recovery. Overall survival was defined as the time elapsed from the date of CAA-ri diagnosis to the date of death or last follow-up. Survival time to clinical recovery was defined as the time elapsed from diagnosis to clinical recovery. Survival time to radiologic recovery was defined as the time elapsed from diagnosis to radiologic recovery. For patients who did not show any recovery, survival time was set equal to the maximum follow-up time available. Survival time to new ICH development was defined as the time elapsed from diagnosis to the radiologic evidence of acute ICH. Relapse-free survival was defined as the time elapsed from the date of ascertained clinical recovery to the date of the first subsequent clinical and radiologic relapse, death, or last follow-up available, whichever occurred first.
Statistical Analysis
Comparisons were obtained with the χ2 test or analysis of variance for categorical and continuous variables, respectively. Overall survival and cumulative incidence/survival probability for clinical recovery, radiologic recovery, and ICH were estimated with the Kaplan-Meier method. Concerning the analysis for clinical recovery, radiologic recovery, and ICH, when the death event was observed in the absence of a previous event, the time to clinical recovery (or radiologic recovery and ICH) was considered censored at the time of death. The probability of clinical recovery related to all the baseline explanatory variables was evaluated through univariate and multivariate logistic regression models. All the available variables were included in the multivariable analysis, without performing any selection criteria model, in an exploratory setting. Relapse-free survival was estimated from a 3-month landmark by the Simon-Makuch method to include the delayed entry of patients with clinical recovery over the entire follow-up. Analyses were performed with STATA 16 (StataCorp, College Station, TX).
Data Availability
Data are available on reasonable request and prior permission from the iCAβ Network.
Results
Of the 150 consecutive enrolled patients, 11 did not meet diagnostic criteria for CAA-ri2 after centralized review (noninflammatory CAA or other diseases), and 26 had no reporting of follow-up data (eTable 1, links.lww.com/WNL/B543), which left 113 participants eligible for the study (Table 1). The mean number of cases per center was 3, ranging from 1 to 16. The geographic distribution of the eligible cases was as follows: Japan 10%, Brazil 4%, Finland 1%, France 4%, Germany 3%, Hungary 1%, Italy 66%, Portugal 1%, Spain 5%, New Zealand 1%, Switzerland 1%, and United States 3%. All patients were from neurology inpatient clinics. Median follow-up time was 12 months. Overall follow-up at each time point was 100% at 3 months, 90.3% at 6 months, 69.9% at 12 months, and 29.2% at 24 months.
Table 1.
Baseline Characteristics at Presentation of the iCAβ International Network Cohort of Patients With First-Ever Diagnosis of CAA-ri

Baseline Cohort Characteristics at First-Ever Diagnosis of CAA-ri.
The characteristics of the baseline cohort are reported in Table 1. Twelve patients had a definite diagnosis of CAA-ri (10.6%), 81 had probable CAA-ri (71.7%), and 20 had possible CAA-ri (17.7%). The mean age at onset was 72.9 years; 28.3% were <70 years old; and 43.4% were female. A history of mild cognitive impairment or AD dementia and radiologic evidence of past ICH were reported in 36.3% and 33.6% of patients, respectively; 37.1% were APOEε4 carriers (22.7% heterozygotes, 14.4% homozygotes). No association between diagnostic category and APOEε4 emerged (p = 0.366). The most frequent symptoms at presentation were cognitive changes (71.7%) and focal neurologic deficits (57.5%). Relatively less frequently reported were seizures (34.5%) and headache (22.1%). It is noteworthy that 37.2% presented with a single symptom. None of them appeared overrepresented compared to patients with multiple symptoms. Among radiologic findings, 60.1% of participants had >10 CMBs. The copresence of CMBs and cSS was observed in 47.8%, whereas 2.7% had only cSS. Of the 93 patients with available T1 postgadolinium MRI sequences, positive contrast enhancement was observed in 63.4%.
Outcomes Evaluation at the 3-, 6-, 12-, and 24-Month Follow-up
Survival
Overall survival and incidence probability of death are reported in Table 2. Thirteen patients died during follow-up: ICH (n = 7), pneumonia (n = 3), cancer (n = 1), status epilepticus (n = 1), and unknown (n = 1). The subanalysis of diagnostic category (possible vs definite and probable CAA-ri)2 did not show any significant effect (p = 0.40) (Figure 3).
Table 2.
Cumulative Incidence Probabilities of Outcomes at the 3-, 6-, 12-, and 24-Month Follow-up in the iCAβ International Network Cohort of 113 Patients With First-Ever Diagnosis of CAA-ri

Figure 3. Kaplan-Meier Estimates of Probability of Survival, Clinical Recovery, Radiologic Recovery, and ICH Development of the iCAβ International Network Cohort Registry25 of 113 Patients With First-Ever Diagnosis of CAA-ri.

(A) Overall survival, (B) clinical recovery, (C) radiologic recovery, and (D) intracerebral hemorrhage (ICH) development. Data are expressed as incidence probability (percentage). Diagnostic category (definite and probable vs possible) diagnosed by clinical presentation, radiologic criteria, or pathologic findings.2 Follow-up time defined as the time elapsed from the date of cerebral amyloid angiopathy–related inflammation (CAA-ri) diagnosis to the date of each in-person visit at 3, 6, 12, and 24 months or last follow-up available. Overall survival defined as the time elapsed from the date of CAA-ri diagnosis to the date of death or last follow-up visit. Any statistically significant effect emerged between the 2 diagnostic category groups (p = 0.40). Clinical recovery defined by stable recovery of the acute neurologic signs or symptoms of CAA-ri2 indexed at presentation. For the patients who did not show any recovery, survival time was set equal to the maximum follow-up time available. Any statistically significant effect emerged between the 2 diagnostic category groups (p = 0.49). Radiologic recovery defined as complete resolution (disappearance) or the almost unperceivable visualization of the acute inflammatory white matter hyperintensity lesions2 indexed at presentation based on the blind-to-clinical-features centralized evaluation. For the patients who did not show any recovery, survival time was set equal to the maximum follow-up time available. Any statistically significant effect emerged between the 2 diagnostic category groups (p = 0.08). ICH development defined as the time elapsed from diagnosis to the radiologic evidence of new ICH at follow-up. For the patients who did not develop any ICH, survival time was set equal to the maximum follow-up time available. Any statistically significant effect emerged between the 2 diagnostic category groups (p = 0.84). Continuous blue line indicates 93 total patients with diagnosis of definitive (n = 12) and probable (n = 81) CAA-ri. Dashed blue lines indicate upper and lower 95% confidence interval (CI) of relapses in definite/probable CAA-ri. Continuous red line indicates 20 patients with diagnosis of possible CAA-ri.
Clinical and Radiological Recovery
The cumulative incidence probability of clinical and radiologic recovery at the 3-, 6-, 12-, and 24-month follow-up is reported in Table 2 and eTable 2 (links.lww.com/WNL/B544).
The majority of patients clinically recovered within 3 months (70.3%, 95% confidence interval [CI] 61.6%–78.5%), with a further increase to 80.2% (95% CI 72.2%–87.1%) and 84.1% (95% CI 76.2%–90.6%) from 3 to 6 months and from 6 to 12 months, respectively. The observed rate of radiologic recovery was slightly slower compared to clinical recovery (45.1% vs 70.3% at 3 months, 59.3% vs 80.2% at 6 months). After 1 year, the probabilities of radiologic and clinical recovery were almost equal (77.4% and 84.1%, respectively). The subanalysis of diagnostic category did not show any significant association with clinical recovery (p = 0.49) or radiologic recovery (p = 0.08) (Figure 3).
Intracerebral Hemorrhage
The observed incidence of acute ICH at follow-up is reported in Table 2 and eTable 2 (links.lww.com/WNL/B544). Overall, new ICHs occurred in 13 patients, 8 of them (7.1%, 95% CI 3.6–13.8) within the first 3 months. The subanalysis of diagnostic category did not show any significant effect (p = 0.84) (Figure 3).
A history of ICH (before CAA-ri) and the occurrence of new ICH at follow-up were more common in patients with cSS (52.6% vs 14.3%, p < 0.0001 and 19.3% vs 3.6%, p < 0.009, respectively). No associations emerged between ICH and CMB number (data not shown).
Relapse and Relapse-Free Survival
The observed probability of relapse was 6.9% (95% CI 2.9%–15.8%) within 3 months, 16.2% (95% CI 9.3%–27.6%) within 12 months, and 38.3% (95% CI 22.9%–59.2%) within 2 years from the ascertained recovery of the first-ever presentation (Table 2). All the relapsed patients had >10 CMBs at baseline. After 3 months from diagnosis (predefined landmark for recovery), 15 of the 90 patients (16.7%) with clinical and radiologic recovery had at least 1 CAA-ri relapse at follow-up. All relapses occurred in patients with definite/probable CAA-ri (Figure 4).
Figure 4. Kaplan-Meier Estimates of Probability of Relapse-Free Survival of the iCAβ International Network Cohort Registry25 of 113 Patients With First-Ever Diagnosis of CAA-ri.2.
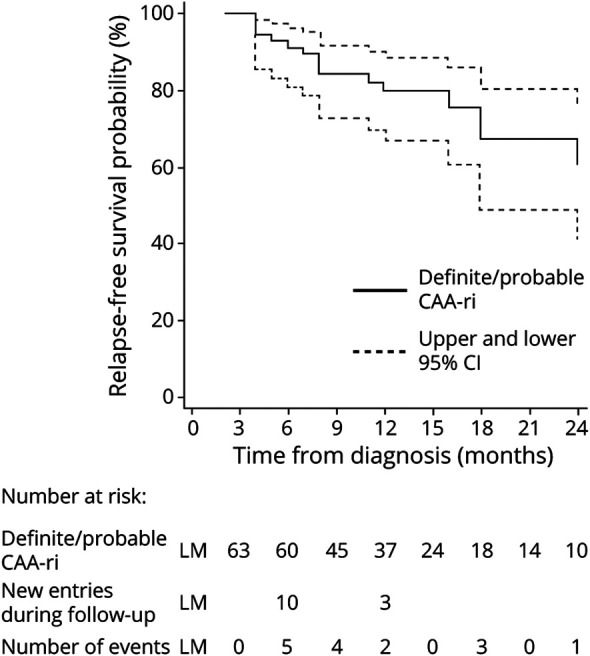
Data are expressed as incidence probability (percentage). Definite and probable cerebral amyloid angiopathy–related inflammation (CAA-ri) (diagnostic category) diagnosed by clinical presentation, radiologic criteria, or pathologic findings.2 Follow-up time defined as the time elapsed from the date of CAA-ri diagnosis to the date of each in-person visit or last follow-up available. Relapse-free survival defined as the time elapsed from the date of ascertained clinical and radiologic recovery to the date of first relapse, death, or last follow-up, whichever occurred first. Relapse-free survival estimated starting from a 3-month landmark (LM) by the Simon-Makuch method to include the delayed entry of patients with clinical recovery over the entire follow-up. Analysis included all the 90 recovered patients (n = 76 with definite/probable CAA-ri; n = 14 with possible CAA-ri with first-ever diagnosis received at baseline). Relapses occurred only in definite/probable CAA-ri (continuous blue line). Dashed blue lines indicate upper and lower 95% confidence interval (CI) of relapses in definite/probable CAA-ri. iCAβ = Inflammatory Cerebral Amyloid Angiopathy and Alzheimer's Disease βiomarkers;
Given the low number of events, additional analyses on prognostic predictors of CAA-ri relapse were limited.
Prognostic Factors
Table 3 reports the univariate and multivariate analyses of factors associated with clinical recovery observed at 3 months by baseline characteristics. Patients presenting with seizures had a significantly reduced probability of recovery (odds ratio 0.17, 95% CI 0.04–0.63; p = 0.01). The sensitivity analysis on diagnostic certainty, included in the univariate and multivariate logistic regression models, did not show any significance.
Table 3.
Logistic Regression Models Relating the Chance of Clinical Recovery (Within 3 Months) to Baseline Characteristics of the iCAβ International Network Cohort of Patients With First-Ever Diagnosis of CAA-ri

Drug Treatment
After the first episode, 87 of 113 patients (77%) received high-dose corticosteroids as 5 IV boluses of 1 g/d methylprednisolone (9 also treated with cyclophosphamide or azathioprine), followed by 1 mg/kg oral prednisone daily and slow tapering off over several months in 73 (83.9%). Although slow oral tapering off showed no statistically significant effects on clinical recovery after first-ever episode of CAA-ri (hazard ratio 0.74, 95% CI 0.39–1.42; p = 0.37), recurrence was more likely if IV corticosteroid pulse therapy was suddenly stopped (hazard ratio 4.68, 95% CI 1.57–13.93; p = 0.006).
Of the remaining 26 patients, 6 of 113 (5.3%) received IV dexamethasone 24 mg for a period of time ranging from 15 to 30 days (2 of 6 followed by tapering off; 1 of 6 also treated with cyclophosphamide); 6 of 113 (5.3%) received oral prednisolone at different dose regimens (25–80 mg) and for different periods of time (7–360 days); 14 of 113 (12.4%) received no treatment for different reasons (2 of 14 asymptomatic/very mild presentations; 2 of 14 refusal of care; n = 1 death; n = 3 contraindications to therapy; n = 1 subjected to chronic corticosteroid therapy for other diseases; n = 5 unspecified).
Statistical analysis between different treatment groups, including those receiving no treatment, was limited given the low number of patients and the high heterogeneity in dose regimens, times, routes for drug administration, or reasons for no therapy.
Discussion
These results from the largest international, prospective, longitudinal cohort registry of patients with CAA-ri highlight the acute, transient, but potentially relapsing inflammatory nature of the disease and the effectiveness of corticosteroids in preventing subsequent inflammatory relapses. Our findings suggest that the spontaneous ARIA-like presentations occurring in CAA-ri could be part of the widely recognized inflammatory spectrum in Aβ-driven pathologies of aging. This opens the possibility that CAA-ri is not a rare manifestation restricted to a subset of patients with CAA as originally thought. Furthermore, our results points to the importance of differential diagnosis for CAA-ri in the general AD population, in particular in patients exposed to immunotherapy, in whom it may be challenging to separate the natural incidence of spontaneous ARIA-like presentations from drug-related ARIA.
We found that at presentation 60% of our patients had >10 CMBs, 50% had cSS, and 34% had a history of ICH. The high prevalence of these well-established surrogate markers of CAA severity28 further highlights the contributing role of underlying CAA pathology in the onset of CAA-ri.
Of note, sporadic CAA is a frequent pathologic finding in the elderly, and at least some degree of CAA is evident in almost all pathology studies on AD (depending on the method used), in which it is correlated with age and disease severity.29-32
We found that 36% of our cohort had a previous diagnosis of mild cognitive impairment or AD dementia before CAA-ri presentation and 37% were APOEε4 carriers, a prevalence never reported before in CAA-ri.
APOEε4 is the major single genetic risk factor for sporadic AD and CAA,33 and there is evidence for its contributing role in Aβ deposition and clearance, including a regulatory effect on tau phosphorylation,34,35 neurovascular unit maintenance and repair,36,37 neuroinflammation,38 and impaired lymphatic drainage of the brain.39
Several reasons may explain the lower prevalence of APOEε4 in our cohort compared to the 75% recently reported in a retrospective, monocentric, series of 33 patients with CAA-ri.9 First, the latter were diagnosed retrospectively from a clinical cohort of patients with CAA recruited from 1998 in a highly specialized center for CAA, where milder cases may be less likely to be referred. Second, it is important to note that the majority of them were found in patients with pathology confirmation. This raises the possibility that the findings might not generalize because biopsies are likely reserved for challenging diagnostic cases such as younger patients with atypical or more aggressive presentations. Third, the mean age at presentation of our cohort was 73 years, which is 4 years older than reported in previous studies4,9 and similar to that in population studies of AD. While the high prevalence of APOEε4 in sporadic AD is well known (19%–39%, depending on ethnicity), there is evidence that this prevalence decreases with increasing age, particularly from the seventh decade of life. On the basis of these considerations, our data are likely to reflect the increased awareness of CAA-ri in the general neurology clinic setting, and not only by the highly specialized CAA community.
Although CAA-ri has formerly been reported as a severe presentation characterized by multiple clinical symptoms, it is noteworthy that 37% of our cohort presented with a single symptom. This raises the question of the actual underrecognition of ARIA-like manifestations in the general CAA and AD population, which carry the risk of CAA-ri. This also highlights the importance of differential diagnosis for CAA and CAA-ri in patients with dementia in memory clinics, particularly for very mild or asymptomatic manifestations of CAA-ri, which may often be missed if not properly investigated.40
The immune-mediated mechanisms of spontaneous ARIA-like and therapy-induced ARIA reported in clinical trials are likely to be generated by a complex combination of genetic, vascular, and immunologic risk factors, closely related to the balance of Aβ intramural periarterial drainage/vascular deposition and to the dose- and time-related downstream effects generated by anti-Aβ (auto)antibodies.1,15,23,24,39,41 When ARIA occur, (1) the severity of preexisting underlying CAA pathology, (2) the displacement of Aβ from parenchymal plaques to the vessel walls, and (3) the rapid disaggregation of Aβ at high doses of the drug, particularly in patients carrying APOEε4, may play a key role in determining or sustaining the inflammatory and immune response.19,23,42-44
The contributing role of underlying CAA pathology finds further support in the acute and focal nature of WMHs in both CAA-ri and ARIA-E,23,41 which present mainly in posterior occipital regions of the brain where there is a likely higher CAA burden.45,46
Of note, although ARIA-E presents in brain areas with extensive removal of Aβ on amyloid-PET, with a mechanism presumably involving microglial activation,41 similar consistently large amyloid reductions have been observed also in patients without ARIA-E in open-label extension studies.47,48 This raises the possibility that ARIA-E may not merely represent a treatment-related complication but may be proof of an exacerbated drug engagement and efficacy in at-risk patients. It is likely that future pretreatments or combination therapies with drugs tailored against CAA will lower the incidence of ARIA.19 Further studies are needed to provide more generalized evidence for understand the role of CAA on ARIA onset, progression, and response to therapy.
Our longitudinal study on CAA-ri, which can be considered a human spontaneous model of ARIA, confirms the transient nature of the disease, with 70% and 80% of clinical recovery within 3 and 6 months, respectively, and 84% of full clinical and radiologic recovery within 1 year. From our data, it is reasonable to advise first MRI monitoring after 3 months from CAA-ri presentation, followed by subsequent control MRIs at 6 and 12 months and then yearly. This approach is supported by the very good prognosis of our cohort and the relatively small probability of early relapses within 3 months, especially with a slow tapering of oral corticosteroids. Nevertheless, the 38% of recurrences within 24 months highlight the importance of maintaining close follow-up monitoring after first presentation of CAA-ri and of not underestimating new neurologic symptoms.49
Regarding the prognostic predictors of CAA-ri, our study shows that neither the MRI hallmarks of CAA nor the presence of APOEε4 is associated with a worse outcome. This suggests that although underlying CAA pathology remains the most accredited risk factor for the onset of CAA-ri, the prognosis is most likely to be driven by the autoimmune and focal inflammatory response.
In our work, CAA-ri emerged as a treatment-reversible disease with a good response to immunomodulatory drugs. Our data suggest that the prompt management of the inflammatory process affects clinical outcomes. Although formal statistical analyses of recovery with respect to treatment were limited given the high heterogeneity within each group, our data suggest the use of high-dose corticosteroid pulse therapy for individuals presenting with CAA-ri. This is in line with recent observations in a small retrospective study on 27 patients treated with (any) corticosteroids vs no treatment.9
Our subanalysis of the 87 patients treated with 5 IV boluses of 1 g/d methylprednisolone, followed by 1 mg/kg oral prednisone and subsequent slow tapering off for several months until radiologic remission, highlights the effectiveness of oral steroid taper for the prevention of subsequent recurrences. Unlike CAA and AD, which have no effective treatments, these results further demonstrate the importance of differential diagnosis for this potentially treatable and reversible condition, including the potential use of corticosteroids for managing symptomatic ARIA-E in patients exposed to immunotherapy with monoclonal antibodies targeting Aβ.
This study has several strengths. This is by far the largest longitudinal study for this rare condition, including patients with definite, probable, and possible CAA-ri diagnosis,2 as well outcome comparisons between diagnostic categories. To date, only single case reports and few small single-center case series are available, mostly based on anecdotal and retrospective observations, making it difficult to interpret the findings.1,3-9 Here, the risks of selection and information bias are minimized by the predefined protocol, including prespecified CRFs systematically recorded at each consecutive in-person visit through geographically distributed recruiting centers,25 the central reading of MRIs, and the central assessment of diagnostic categories.
The main limitation is the lack of validated clinical and radiologic rating scales to quantify disease severity and outcomes in patients with CAA-ri, mirroring the situation in current clinical practice. This could have influenced treatment decisions and interpretations of outcomes. Properly weighted placebo-controlled randomized clinical trials are mandatory to confirm our data and to exclude potential Hawthorn effects of prospective observational studies, as well as potential regional bias in recruitment and management of patients to confirm generalizability to other populations. The heterogeneity of patients and the lack of specific treatment and clinical-radiological guidelines make it impossible to further compare between treatment groups or to study predictors of relapse.
Our patients were enrolled according to current diagnostic criteria for CAA-ri, which include routine CSF analyses to exclude other causes.2,9 The Model CAA Biomarkers Discovery and Validation Study on large, independent, international cohorts with systematic collection of longitudinal CSF and plasma samples and an extended imaging protocol is the focus of current research in the iCAβ International Network.25
This study suggests that spontaneous ARIA-like as observed in CAA-ri may not be a rare inflammatory manifestation of the widely recognized and diffused Aβ-driven pathologies of aging.
Our data highlight that the identification of ARIA-like events in the AD population will be critical in light of primary and secondary prevention studies of AD and the recent approval of aducanumab as part of future treatment regimens for AD in clinical practice. Given the precautions and warnings of potential ARIA side effects associated with such drugs, an increased understanding of the ARIA phenomena and increased knowledge about how to recognize, treat, manage, and separate the incidence of spontaneous ARIA-like events from drug-related ARIA will be critically important for both treating clinicians and the patient's community.50
To optimize benefits and reduce potential risks to patients, reliable, timely accessible and less costly biomarkers for the prediction, early diagnosis, response to therapy, and longitudinal monitoring of ARIA are urgently needed.1,16,19,23
If viable clinical and therapeutic guidelines are to be issued, a wide consensus based on a collaborative effort such as the iCAβ International Network initiative25 is the only way to achieve this, avoiding data fragmentation in small case series and thus conferring a higher potential and impact.
Our data suggest that prompt management of the ARIA-E–like inflammatory process affects clinical outcomes and reasonably support first-line treatment with 5 IV boluses of 1 g/d methylprednisolone, followed by 1 mg/kg of oral prednisone daily and subsequent slight tapering off over several months and until radiologic remission to reduce subsequent recurrences. Immunosuppressive drugs such as azathioprine and cyclophosphamide6,7,9 or brain biopsy should be considered for patients who fail to respond to corticosteroid pulse therapy.
Acknowledgment
This manuscript is dedicated to the loving memory of Prof. Massimo Musicco. The authors thank the patients and their families referred to the iCAβ International Network.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ARIA
amyloid-related imaging abnormalities
- ARIA-E
ARIA edema
- CAA
cerebral amyloid angiopathy
- CAA-ri
CAA-related inflammation
- CI
confidence interval
- CMB
cerebral microbleed
- CRF
case report form
- cSS
cortical superficial siderosis
- GRE
gradient recalled echo
- iCAβ
Inflammatory Cerebral Amyloid Angiopathy and Alzheimer's Disease βiomarkers
- ICH
intracerebral hemorrhage
- SINdem
Italian Society of Neurology–dementia
- VE
vasogenic edema
- WMH
white matter hyperintensity
Appendix. Authors
Footnotes
CME Course: NPub.org/cmelist
Study Funding
This work was supported by Alzheimer's Association (ModelCAA Research Grant AARG-18-561699), Fondazione Cariplo (biomarkARIA Research Project 2015-0820), and SINdem CAA Study Group.
Disclosure
F. Piazza reports grants from the Canadian Institute of Health Research outside the submitted work. F. Piazza received personal and institutional fees from and served as a consultant for Hoffmann La-Roche, outside the submitted work. F. Piazza is the inventor (without ownership) of a patent for a method and kit for the Aβ autoantibody test, outside the submitted work. All the other authors declare no competing interests for the submitted work. Go to Neurology.org/N for full disclosures.
References
- 1.Piazza F, Greenberg SM, Savoiardo M, et al. . Anti-amyloid beta autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol. 2013;73(4):449-458. [DOI] [PubMed] [Google Scholar]
- 2.Auriel E, Charidimou A, Gurol ME, et al. . Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2016;73(2):197-202. [DOI] [PubMed] [Google Scholar]
- 3.Castro Caldas A, Silva C, Albuquerque L, Pimentel J, Silva V, Ferro JM. Cerebral amyloid angiopathy associated with inflammation: report of 3 cases and systematic review. J Stroke Cerebrovasc Dis. 2015;24(9):2039-2048. [DOI] [PubMed] [Google Scholar]
- 4.Corovic A, Kelly S, Markus HS. Cerebral amyloid angiopathy associated with inflammation: a systematic review of clinical and imaging features and outcome. Int J Stroke. 2018;13(3):257-267. [DOI] [PubMed] [Google Scholar]
- 5.Kimura A, Sakurai T, Yoshikura N, et al. . Corticosteroid therapy in a patient with cerebral amyloid angiopathy-related inflammation. J Neuroinflammation 2013;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kloppenborg RP, Richard E, Sprengers ME, Troost D, Eikelenboom P, Nederkoorn PJ. Steroid responsive encephalopathy in cerebral amyloid angiopathy: a case report and review of evidence for immunosuppressive treatment. J Neuroinflammation. 2010;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luppe S, Betmouni S, Scolding N, Wilkins A. Cerebral amyloid angiopathy related vasculitis: successful treatment with azathioprine. J Neurol. 2010;257(12):2103-2105. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi H, Ueda A, Kosaka T, et al. . Cerebral amyloid angiopathy-related inflammation presenting with steroid-responsive higher brain dysfunction: case report and review of the literature. J Neuroinflammation. 2011;8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Regenhardt RW, Thon JM, Das AS, et al. . Association between immunosuppressive treatment and outcomes of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2020;77(10):1261-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boncoraglio GB, Piazza F, Savoiardo M, et al. . Prodromal Alzheimer's disease presenting as cerebral amyloid angiopathy-related inflammation with spontaneous amyloid-related imaging abnormalities and high cerebrospinal fluid anti-A beta autoantibodies. J Alzheimers Dis. 2015;45(2):363-367. [DOI] [PubMed] [Google Scholar]
- 11.Bouthour W, Sveikata L, Vargas MI, Lobrinus JA, Carrera E. Clinical reasoning: rapid progression of reversible cognitive impairment in an 80-year-old man. Neurology. 2018;91(24):1109-1113. [DOI] [PubMed] [Google Scholar]
- 12.Gera A, Witek N, Bailey M. Pearls & oy-sters: CAA-related inflammation presents as subacute cognitive decline in a patient with Parkinson disease. Neurology. 2019;92(23):1116-1118. [DOI] [PubMed] [Google Scholar]
- 13.Poli L, De Giuli V, Piazza F, et al. . A challenging diagnosis of reversible "vascular" dementia: cerebral amyloid angiopathy-related inflammation. J Neuroimmunol. 2020;338:577109. [DOI] [PubMed] [Google Scholar]
- 14.Van Ommeren R, Izenberg A, Shadowitz S, Aviv R, Keith J. Clinical reasoning: an 81-year-old woman with decreased consciousness and fluctuating right facial droop. Neurology. 2020;94(19):843-848. [DOI] [PubMed] [Google Scholar]
- 15.Carmona-Iragui M, Fernandez-Arcos A, Alcolea D, et al. . Cerebrospinal fluid anti-amyloid-beta autoantibodies and amyloid PET in cerebral amyloid angiopathy-related inflammation. J Alzheimers Dis. 2016;50(1):1-7. [DOI] [PubMed] [Google Scholar]
- 16.Aisen PS, Cummings J, Doody R, et al. . The future of anti-amyloid trials. J Prev Alzheimers Dis. 2020;7(3):146-151. [DOI] [PubMed] [Google Scholar]
- 17.Joseph-Mathurin N, Wang G, Kantarci K, et al. . Longitudinal accumulation of cerebral microhemorrhages in dominantly inherited Alzheimer disease. Neurology. 2021;96(12):e1632-e1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenberg SM, Frosch MP. Life imitates art: anti-amyloid antibodies and inflammatory cerebral amyloid angiopathy. Neurology. 2011;76(9):772-773. [DOI] [PubMed] [Google Scholar]
- 19.Piazza F, Winblad B. Amyloid-related imaging abnormalities (ARIA) in immunotherapy trials for Alzheimer's disease: need for prognostic biomarkers?. J Alzheimers Dis. 2016;52(2):417-420. [DOI] [PubMed] [Google Scholar]
- 20.Sperling R, Salloway S, Brooks DJ, et al. . Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werring DJ, Sperling R. Inflammatory cerebral amyloid angiopathy and amyloid-modifying therapies: variations on the same ARIA?. Ann Neurol. 2013;73(4):439-441. [DOI] [PubMed] [Google Scholar]
- 22.Mintun MA, Lo AC, Duggan Evans C, et al. . Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691-1704. [DOI] [PubMed] [Google Scholar]
- 23.DiFrancesco JC, Longoni M, Piazza F. Anti-abeta autoantibodies in amyloid related imaging abnormalities (ARIA): candidate biomarker for immunotherapy in Alzheimer's disease and cerebral amyloid angiopathy. Front Neurol. 2015;6:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperling RA, Jack CR Jr., Black SE, et al. . Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inflammatory Cerebral Amyloid Angiopathy and Alzheimer's Disease Biomarkers International Network (iCAβ). Accessed May 8, 2021. Available at: sites.google.com/site/icabinternationalnetwork. [Google Scholar]
- 26.Chung KK, Anderson NE, Hutchinson D, Synek B, Barber PA. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry. 2011;82(1):20-26. [DOI] [PubMed] [Google Scholar]
- 27.Conti E, Galimberti G, Tremolizzo L, et al. . Cholinesterase inhibitor use is associated with increased plasma levels of anti-Abeta 1-42 antibodies in Alzheimer's disease patients. Neurosci Lett. 2010;486(3):193-196. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee G, Carare R, Cordonnier C, et al. . The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry. 2017;88(11):982-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellis RJ, Olichney JM, Thal LJ, et al. . Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology. 1996;46(6):1592-1596. [DOI] [PubMed] [Google Scholar]
- 30.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm (Vienna). 2002;109(5-6):813-836. [DOI] [PubMed] [Google Scholar]
- 31.Hachinski V, Einhaupl K, Ganten D, et al. . Special topic section: linkages among cerebrovascular, cardiovascular, and cognitive disorders: preventing dementia by preventing stroke: the Berlin Manifesto. Int J Stroke. 2019:1747493019871915. [DOI] [PubMed] [Google Scholar]
- 32.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011;69(2):320-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heffernan AL, Chidgey C, Peng P, Masters CL, Roberts BR. The neurobiology and age-related prevalence of the epsilon4 allele of apolipoprotein E in Alzheimer's disease cohorts. J Mol Neurosci. 2016;60(3):316-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Haj M, Antoine P, Amouyel P, Lambert JC, Pasquier F, Kapogiannis D. Apolipoprotein E (APOE) epsilon4 and episodic memory decline in Alzheimer's disease: a review. Ageing Res Rev. 2016;27:15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Yoon H, Basak J, Kim J. Apolipoprotein E in synaptic plasticity and Alzheimer's disease: potential cellular and molecular mechanisms. Mol Cell. 2014;37(11):767-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piazza F, Galimberti G, Conti E, et al. . Increased tissue factor pathway inhibitor and homocysteine in Alzheimer's disease. Neurobiol Aging. 2012;33(2):226-233. [DOI] [PubMed] [Google Scholar]
- 38.Tibolla G, Norata GD, Meda C, et al. . Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis. 2010;210(1):78-87. [DOI] [PubMed] [Google Scholar]
- 39.Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol. 2015;25(1):63-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee G, Alvares D, Bowen J, Adams ME, Werring DJ. Minimally symptomatic cerebral amyloid angiopathy-related inflammation: three descriptive case reports. J Neurol Neurosurg Psychiatry. 2019;90(1):113-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrowitzki S, Deptula D, Thurfjell L, et al. . Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198-207. [DOI] [PubMed] [Google Scholar]
- 42.Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu E, Wang D, Sperling R, et al. . Biomarker pattern of ARIA-E participants in phase 3 randomized clinical trials with bapineuzumab. Neurology. 2018;90(10):e877–e886. [DOI] [PubMed] [Google Scholar]
- 44.Carare RO, Aldea R, Agarwal N, et al. . Clearance of interstitial fluid (ISF) and CSF (CLIC) group: part of Vascular Professional Interest Area (PIA): cerebrovascular disease and the failure of elimination of amyloid-beta from the brain and retina with age and Alzheimer's disease: opportunities for therapy. Alzheimers Dement (Amst). 2020;12(1):e12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurol ME, Becker JA, Fotiadis P, et al. . Florbetapir-PET to diagnose cerebral amyloid angiopathy: a prospective study. Neurology. 2016;87(19):2043-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson KA, Gregas M, Becker JA, et al. . Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62(3):229-234. [DOI] [PubMed] [Google Scholar]
- 47.Klein G, Delmar P, Rehal S, et al. . Consistently large amyloid reductions in patients with and without ARIA-E in the gantenerumab SCarlet RoAD and Marguerite RoAD open-label extension studies (S9.007). Neurology. 2019;92:S9.007. [Google Scholar]
- 48.Klein G, Delmar P, Voyle N, et al. . Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer's disease: a PET substudy interim analysis. Alzheimers Res Ther. 2019;11(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiFrancesco JC, Touat M, Caulo M, et al. . Recurrence of cerebral amyloid angiopathy-related inflammation: a report of two cases from the iCA beta International Network. J Alzheimers Dis. 2015;46(4):1071-1077. [DOI] [PubMed] [Google Scholar]
- 50.Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta-analysis of phase III RCTs in Alzheimer's disease. Ageing Res Rev. 2021;68:101339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available on reasonable request and prior permission from the iCAβ Network.