

Abstract
Alzheimer’s disease (AD) and other neurodegenerative diseases are characterized by chronic neuroinflammation and a reduction in brain energy metabolism. An important role has emerged for small, non-coding RNA molecules known as microRNAs (miRNAs) in the pathophysiology of many neurodegenerative disorders. As epigenetic regulators, miRNAs possess the capacity to regulate and fine tune protein production by inhibiting translation. Several miRNAs, which include miR-146a, are elevated in the brain, CSF, and plasma of AD patients. miR-146a participates in pathways that regulate immune activation and has several mRNA targets which encode for proteins involved in cellular energy metabolism. An additional role for extracellular vesicles (EVs) has also emerged in the progression AD, as EVs can transfer functionally active proteins and RNAs from diseased to healthy cells. In the current study, we exposed various cell types present within the CNS to immunomodulatory molecules and observed significant upregulation of miR-146a expression, both within cells and within their secreted EVs. Further, we assessed the effects of miR-146a overexpression on bioenergetic function in primary rat glial cells and found significant reductions in oxidative phosphorylation and glycolysis. Lastly, we correlated miR-146a expression levels within various regions of the AD brain to disease staging and found significant, positive correlations. These novel results demonstrate that the modulation of miR-146a in response to neuroinflammatory stimuli may mediate the loss of mitochondrial integrity and function in cells, thereby contributing to the progression of beta-amyloid and tau pathology in the AD brain.
Keywords: Neuroinflammation, microRNAs, miR-146a, extracellular vesicle, oxidative phosphorylation, glycolysis
Introduction
Many neurodegenerative diseases, including Alzheimer’s disease (AD), are characterized by chronic neuroinflammation. Prolonged, unregulated inflammation leads to the increased production of various pro-inflammatory molecules, which likely contribute to disease progression. Although the genetics of late-onset Alzheimer’s disease are complex, recent studies have observed significant downregulation of many genes necessary for proper cellular bioenergetics (Liang et al. 2008). Moreover, functional imaging studies of patients suffering from AD reveal that the severity of brain hypometabolism is correlated with dementia (Brier et al. 2012; Yin et al. 2016). It is clear that dysfunctional brain energetics is an important aspect of AD pathology, yet the molecular mechanisms responsible are poorly understood.
Due to their ability to regulate translational repression of multiple mRNA targets, microRNAs (miRNAs) have recently been implicated as major players in the pathogenesis of AD. A deleterious, pro-inflammatory role has emerged for miR-146a in AD (Alexandrov et al. 2014), as well as other neurological (Nguyen et al. 2018) and degenerative diseases (Pogue and Lukiw 2018). Classically, miR-146a has been considered a well-characterized anti-inflammatory miRNA, as several mRNA targets include genes that encode for interleukin 1 receptor associated kinase (IRAK1), TNFR-associated factor 6 (TRAF6), and signal transducer and activator of transcription 1 (STAT1) (Taganov et al. 2006; Cui et al. 2010; Iyer et al. 2012; Wang et al. 2013; Saba et al. 2014). Many of these proteins work as transcription factors to increase production of inflammatory cytokines or induce apoptosis; therefore miR-146a mediated translational-repression of these signaling molecules can reduce inflammatory responses within cells. In contrast, more recent studies also suggest a pro-inflammatory role for miR-146a function and have observed increased expression of miR-146a in response to oxidative stress and inflammation (Lukiw and Alexandrov 2012; Wu et al. 2015; Cardoso et al. 2016). Elevated levels of miR-146a have also been shown to downregulate expression of complement factor H (CFH), an important repressor of inflammation (Lukiw et al. 2008). Thus, a growing body of literature strongly suggests that miR-146a participates in both pro- and anti-inflammatory roles in human health and disease.
In addition to the physical hallmarks of AD such as extracellular Aβ plaques, intracellular neurofibrillary tangles, and neuronal loss, widespread hypometabolism is also observed within the AD brain (de Leon et al. 2001; Mosconi et al. 2006, 2008). Interestingly, several mRNAs encoding for proteins involved in cellular metabolism have recently been identified as targets of miR-146a; these proteins have been demonstrated to play important roles in glycolysis, mitochondrial respiration, mitochondrial transport, and apoptosis (Rippo et al. 2014). Due to its inflammation-induced upregulation, and the mRNAs in which it targets, miR-146a may be a key player in the pathophysiology and progression of AD.
How miR-146a-mediated inflammatory signals accelerate or exacerbate AD progression is unclear. It is well-established that AD typically progresses in a very characteristic manner with initial pathology developing in the entorhinal cortex, then spreading throughout the temporal cortex, and finally into the frontal cortex (Smith 2002). Propagation of disease pathology from one cell to another may be due, in part, to communication between cells via extracellular vesicles (EVs). EVs are small, nano-sized particles shed or excreted from most cell types, which have the capacity to transfer functional RNAs, including miRNAs, from one cell to another (Valadi et al. 2007; van Niel et al. 2018). Thus, EVs have been implicated in the spread of pathological molecules during disease states that include AD and other neurodegenerative diseases (reviewed by Thompson et al. 2016).
Previous work from our laboratory and others, have shown that miR-146a is significantly upregulated in the brains of AD patients, compared to their age-matched controls (AMCs) (Sarkar et al. 2016; Pogue and Lukiw 2018). In the current study, we assessed the effects of various inflammatory mediators on different cell types within the CNS, the functional bioenergetic outcomes of miR-146a overexpression, and the relationship between human brain miR-146a expression and AD pathology. We hypothesized that miR-146a expression would increase after exposure to inflammatory stimuli, and that overexpression of this miRNA would lead to decreased cellular bioenergetics. Further, we hypothesized that AD brains would have significantly higher levels of miR-146a expression when compared to age matched controls (AMCs), and these levels would positively correlate with disease severity, and negatively correlate with total mitochondrial protein expression.
Materials and Methods
Animals
Pregnant rats (Sprague Dawley) were acquired from Hilltop Laboratory (Scottdale, PA) to obtain E18 brain tissue for preparation of mixed glial and cortical neuronal cultures. Three timed-pregnant female rats were used for each primary culture experiment, with 6–10 embryos per dam, and each experiment was repeated three times. Therefore, nine timed pregnant female rats were used for all animal experiments. All animal studies were approved by the Institutional Animal Care and Use Committee at West Virginia University.
Cell culture
Primary culture:
Pregnant rats were euthanized, and fetal brain tissue was harvested from E18 rats. Brains were removed and placed into magnesium (Mg2+) free Hank’s balanced salt solution (HBSS). Cortices and hippocampi were removed under a dissecting microscope, washed, and placed into neurobasal culture media (without phenol red) supplemented with 1X B27 and 1% penicillin-streptomycin (Gibco, Carlsbad, CA). The cortices and hippocampi were triturated using a graded series of fine polished Pasteur pipettes, and then filtered through a 40 μm nylon cell strainer (Becton Dickinson Labware, Franklin Lakes, NJ). Neurons were plated on poly-L-lysine coated 150-mm dishes and cultured in vitro in 95% humidity and 5% CO2 atmosphere at 37°C for 15 days. At day 2, cells were exposed to 5 μM 1-β-D- arabinofuranosyl cytosine (AraC) to inhibit glial cell growth as previously described (Sarkar et al., 2015, 2016). To obtain glial cells, following trituration the remaining cortical and hippocampal cell suspension was grown in DMEM for 4–5 days until a mixed glial population of astrocytes and microglia reached confluency.
Immortalized cell lines:
A hippocampal neuronal cell line, HT-22 cells, was obtained from Dr. David Schubert at the Salk institute. Additionally, a cerebellar microglial cell line, C8-B4 cells, and a brain microvascular endothelial cell line, bEnd.3 cells, were both obtained from ATCC (Manassas, VA). All cell lines were of murine origin and grown in Dulbecco’s Modified Essential Medium (high glucose) (Hyclone) with 10% FBS 1% pen/strep in humidified 95% air/5% CO2 at 37°C.
Human brain tissues
Brain tissue from the temporal cortex, frontal cortex, and cerebellum of confirmed AD patients and AMCs was obtained from the Kathleen Price Bryan Brain Bank, Bryan Alzheimer’s Disease Research Center, Duke University Medical Center, Durham, NC. All tissues were staged for AD pathology with Braak and Braak staging (Braak and Braak 1991) and cerebral amyloid angiopathy (CAA) (Thal et al. 2003) as shown in Table 1. Brain tissues were used in accordance with the institutional review board/ethical guidelines of the donor institution.
Table 1. Human sample demographics and neuropathological correlations.
Demographic information including age, sex, apolipoprotein E (APOE) alelles, post-mortem intervals (in hours), and neuropathological correlations including diagnosis of Alzheimer’s disease (AD), Braak & Braak staging, and cerebrovascular pathologies were collected for all samples.
| Age | Sex | APOE | PMI | Diagnoses | B&B stage | Atherosclerosis* | Amyloid angiopathy* | Other | |
|---|---|---|---|---|---|---|---|---|---|
| Age-matched control subjects (AMC) | 78 | F | 33 | 5.5 | Normal CERAD 1A | 1 | |||
| 80 | F | 33 | 1.2 | Normal CERAD 1A | 1 | Mild | |||
| 72 | F | 34 | 3.0 | Normal CERAD 1A | 1 | ||||
| 74 | F | 24 | 4.7 | Normal CERAD 1B | 3 | Other | |||
| 72 | F | 33 | 30.0 | Normal CERAD 1B | 2 | ||||
| 78 | M | n/a | 9.8 | Normal CERAD 1A | 1 | ||||
| 75 | M | 22 | 29.0 | Normal CERAD 1A | 1 | Lacunes | |||
| 75 | M | 33 | 18.9 | Normal CERAD 1B | 1 | ||||
| 80 | M | 33 | 4.3 | Normal CERAD 1B | 1 | ||||
| 81 | M | 34 | 7.2 | Normal CERAD 1B | 1 | Lacunes | |||
| Alzheimer’s Disease patient subjects (AD) | 76 | F | 33 | 10.5 | AD | 3 | |||
| 76 | F | 44 | 5.5 | AD | 3 | Mild | Mild | ||
| 75 | F | 34 | 20.1 | AD | 3 | Severe | Mild | ||
| 74 | F | 34 | 20.5 | AD | 6 | ||||
| 75 | F | 33 | 13.3 | AD | 6 | Mild | |||
| 79 | F | n/a | 20.8 | AD | 6 | Moderate | Infarcts | ||
| 75 | F | 34 | 24.0 | AD | 6 | Severe | |||
| 77 | M | 33 | 16.8 | AD | 3 | ||||
| 75 | M | 33 | 3.0 | AD | 3 | Mild | |||
| 78 | M | 34 | 0.8 | AD | 3 | Mild | |||
| 71 | M | 33 | 25.2 | AD | 6 | Mild | |||
| 77 | M | 44 | 25.4 | AD | 6 | Mild | Mild | ||
| 75 | M | 34 | 2.6 | AD | 6 | Moderate |
Pathological scores were analyzed by a pathologist for atherosclerosis and amyloid angiopathy using CERAD guidelines and NIA-Reagan criteria.
Preparation of soluble amyloid-beta (Aβ)
Synthetic Aβ1–42 (Tocris, Ellisville, MO) oligomers were prepared without the fibrillar component according to published methods (Barghorn et al. 2005; Sarkar et al. 2015). Briefly, Aβ1–42 was dissolved in 1,1,1,3,3,3- hexafluoro-2-propanol to 1 mM. The clear solution was then evaporated to dryness. Dried peptide was diluted in DMSO to 5 mM and sonicated for 10 min in a bath sonicator. The peptide solution was resuspended in cold Neurobasal medium and immediately vortexed. The solution was then incubated at 4°C for 24 h. After high speed centrifugation, the supernatant was collected, visualized by polyacrylamide gel electrophoresis and silver staining, and found to be comprised of fibrillar-free oligomers and monomers.
Extracellular vesicle isolation
HT-22 cells were seeded in 100 mm dishes and exposed to 1 ng/ml tumor necrosis factor-alpha (TNF-α) when cells reached 80% confluency. C8-B4 and bEnd.3 cells were seeded in 150 mm dishes, and when cells reached 50% confluency, they were exposed to either 1 μg/ml LPS (Sigma-Aldrich, St. Louis, MO) or 1 ng/ml TNF-α (R&D, Minneapolis, MN). Similarly, rat cortical primary neuronal and glial cells were grown in 150 mm dishes and when cells were 75% confluent, they were exposed to 250 or 500nM of oligomeric Aβ. For all cell types and exposure conditions, after 24 hours, the media was collected for EV isolation as previously performed by our group, and downstream miRNA purification. EVs were isolated from the microglia and brain endothelial cell lines and primary cell culture media by ultracentrifugation (Théry et al. 2006; Sarkar et al. 2015; Dakhlallah et al. 2019) and from neuronal cell line culture media (Russell et al. 2019) with the ExoRNeasy Isolation Kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol. Using the ultracentrifugation method, the conditioned cell culture media was centrifuged (320 × g for 5 minutes) to remove cells, and the supernatant was filtered through a Steriflip filter (0.22 μm pore size). EVs were pelleted from the filtered media by ultracentrifugation at 100,000 g for 60 min at 4°C using Beckman SW 28 rotor running in a Beckman Coulter Optima L-100 XP ultracentrifuge (Beckman Coulter, Brea, CA). EVs were washed once with sterile PBS, collected by ultracentrifugation, and resuspended in 1 ml of sterile PBS.
Overexpression of miR-146a in primary mixed glial cells
To induce overexpression of miR-146a in cells and their secreted EVs, rat primary mixed glial cells were transfected with either vector or miR-146a expression plasmid DNA by Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The miR-146a expression vector was purchased through Addgene (Addgene plasmid #15092, Cambridge, MA), which was deposited from Dr. David Baltimore’s group (Taganov et al. 2006). Forty hours after transfection, EVs in the conditioned media were prepared as described above. The presence or absence of miR-146a was determined by isolating total RNA using the miRNeasy Kit (Qiagen) followed by qRT-PCR as described below.
miRNA expression analysis
Total RNA enriched in miRNAs was isolated from approximately 5 mg of individual frozen brain tissue samples and various cell samples by using the miRNeasy Micro Kit Isolation Kit (Qiagen). Total RNA enriched in miRNAs was also isolated from EVs using the miRNeasy Serum/Plasma Kit. Prior to isolation of EV RNA, 3.5 μl C. elegans miR-39 (at a concentration of 1.6 × 108 copies/μl) was added to each sample as a spike-in control. Total RNA concentrations were measured using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA). Using the miScript II RT Kit (Qiagen), 2μg of total RNA containing miRNA was reverse transcribed in a total volume of 10μl reaction mix to make cDNA. Expression of miR-146a was determined by quantitative RT-PCR, which was performed using target specific miScript primer assays and the miScript SYBR® Green PCR Kit (Qiagen). All reactions were performed in triplicate for each sample using a StepOne Plus PCR system (Applied Biosystems, Foster City, CA) for 40 cycles as follows: 10 sec at 95 °C, 30 sec at 55°C, 30 sec at 70 °C. Negative control reactions were included as wells containing only master mix and nuclease-free water without any template cDNA. All miRNA specific primers were from Qiagen and included: miScript Primer Assay Hs_miR-146a (MS00003535), Hs_RNU6 (MS00033740), and Ce_miR-39_1 (219610). The expression levels of miR-146a were standardized against those of RNU6 (an internal control for brain tissue and cell samples) and miR-39 (a spike-in control for EV samples) detected in identical cDNA samples. Quantification of PCR amplified miRNA specific cDNA was done by the comparative cycle threshold CT method (ΔΔCT) (Livak and Schmittgen 2001).
Measurement of mitochondrial respiration: oxygen consumption rate (OCR)
Approximately 20,000 primary mixed glial cells per well were seeded in a XFe 96 cell culture microplate (Agilent, Santa Clara, CA) in DMEM medium with 10% FBS and 1% pen/strep. The next day, glial cells were transfected with either vehicle vector or miR-146a expression plasmid DNA (1, 2, 3, or 4 μg/ml) by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Forty hours after transfection, OCR was measured by the XFe 96 Extracellular Flux Analyzer using a Mito Stress Kit (Agilent) according to the manufacturer’s protocol. To measure mitochondrial function in real time, oligomycin, carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP), and antimycin A plus rotenone were added separately to a sensor cartridge and injected sequentially through ports in the XF Assay cartridge to final concentrations of 1 μg/ml, 1 μM and 10 μM, respectively. Addition of these reagents allowed for measurement and calculation of the basal level of mitochondrial oxygen consumption, the amount of oxygen consumption linked to ATP production, the level of non-ATP-linked oxygen consumption (proton leak), the maximal respiratory capacity, and the non-mitochondrial oxygen consumption. For each experimental condition, six replicate wells were used.
Measurement of glycolytic function: extracellular acidification rate (ECAR)
Similarly, as performed for OCR, primary mixed glial cells were cultured in a XFe96 cell culture microplate and transfected with vector or miR-146a expression plasmid DNA. Forty hours after transfection, ECAR was measured by XFe 96 Extracellular Flux Analyzer using a Glycolysis Stress Kit (Agilent) which measures three key parameters of glycolytic function: glycolysis, glycolytic capacity, and glycolytic reserve. A saturating concentration of glucose, oligomycin, and 2-deoxy-glucose (2-DG; a glucose analog) were added separately in a sensor cartridge and injected sequentially through ports in the cartridge to final concentrations of 10 mM, 0.6 μM, 125mM, respectively. This assay allows for the calculation of the basal rate of glycolysis/glycolytic flux, maximum glycolytic capacity, and glycolytic reserve. For each experimental condition, six replicate wells were used.
Western blot analysis
To prepare total protein extracts, frozen human brain tissues were homogenized in Pierce® RIPA buffer (Thermo Scientific) with 1x Halt™ Protease Inhibitor Single-Use Cocktail (Thermo Scientific), followed by centrifugation at 13,400 g for 5 min at room temperature. The supernatant was collected and the protein concentrations were determined using Pierce™ BCA Protein Assay (Thermo Scientific). Approximately 15 μg total protein was separated by size in Bolt 4–12% Bis-Tris Plus poly-acrylamide gels (Novex®, Thermo Scientific) through electrophoresis, along with the Chameleon™ Kit Pre-stained Protein Ladder (LI-COR, Lincoln, NE) size marker. Proteins were transferred to iBlot® 2 PVDF membranes (Invitrogen, Thermo Scientific) by iBlot® 2 Gel Transfer Device (Life Technologies). The membranes were blocked in Odyssey blocking buffer in TBS (LI-COR) for 1 hour at room temperature, and then incubated with primary antibodies of target proteins in sequential manner overnight at 4°C: VDAC (MilliporeSigma, Burlington, MA, (MABN504), 1:1,000 dilution), and β-actin (Adipogen (YIF-LF-PA0209) 1:15,000 dilution). The following day, membranes were washed with 1x TBST buffer (prepared with 1x Tris buffered saline with 0.1% Tween 20) 3 times for 5 minutes, and incubated with appropriate anti-rabbit, anti-mouse, secondary antibody conjugated with fluorescence dye (LI-COR, IRDye 800CW anti-Rabbit and anti-Mouse, both 1:15000 diluted in blocking buffer) for one hour, then washed again in 1x TBST 3 times in 5 minute intervals prior to imaging. The specific reaction was visualized by using the Odyssey CLx (LI-COR), and immunoblotting bands were quantified by densitometry using Image Studio 5.1.
Statistical analyses
All experiments were repeated at least 3 times with n=3–6 wells/treatment/experiment. Results from the experiments are reported as means ± SEM. All data were subjected to the Shapiro-Wilk normality test and met the criteria for normality. Quantitative results that satisfied criteria for parametric analyses were assessed for significance using Welch’s t-test for two sample comparisons or Welch’s ANOVA with Tukey’s HSD post hoc test for three or more comparisons. Data in Figures 2, 3, and 4 were analyzed using simple linear regression where miRNA is the independent variable and indices of oxidative phosphorylation or glycolytic function were dependent variables, wherein the slope for all models showed a significant linear trend. Spearman’s rho (ρ) was calculated to determine correlations between miR-146a levels and Braak or CAA staging. Effect sizes were reported for all significant results by the following: t-tests - ηp2, ANOVA- ω2, and simple linear regression – R2; effect sizes follow p values in all figure legends. Additional specific details of the statistical reporting for each experiment are provided in the corresponding figure legend. All analyses were performed using R 3.5.1 (https://www.R-project.org) and GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA). A p value ≤ 0.05 was used to establish significant; significant presented as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.0001.
Figure 2. Effects of Aβ on intracellular and EV miR-146a levels in primary neurons and mixed glial cells.
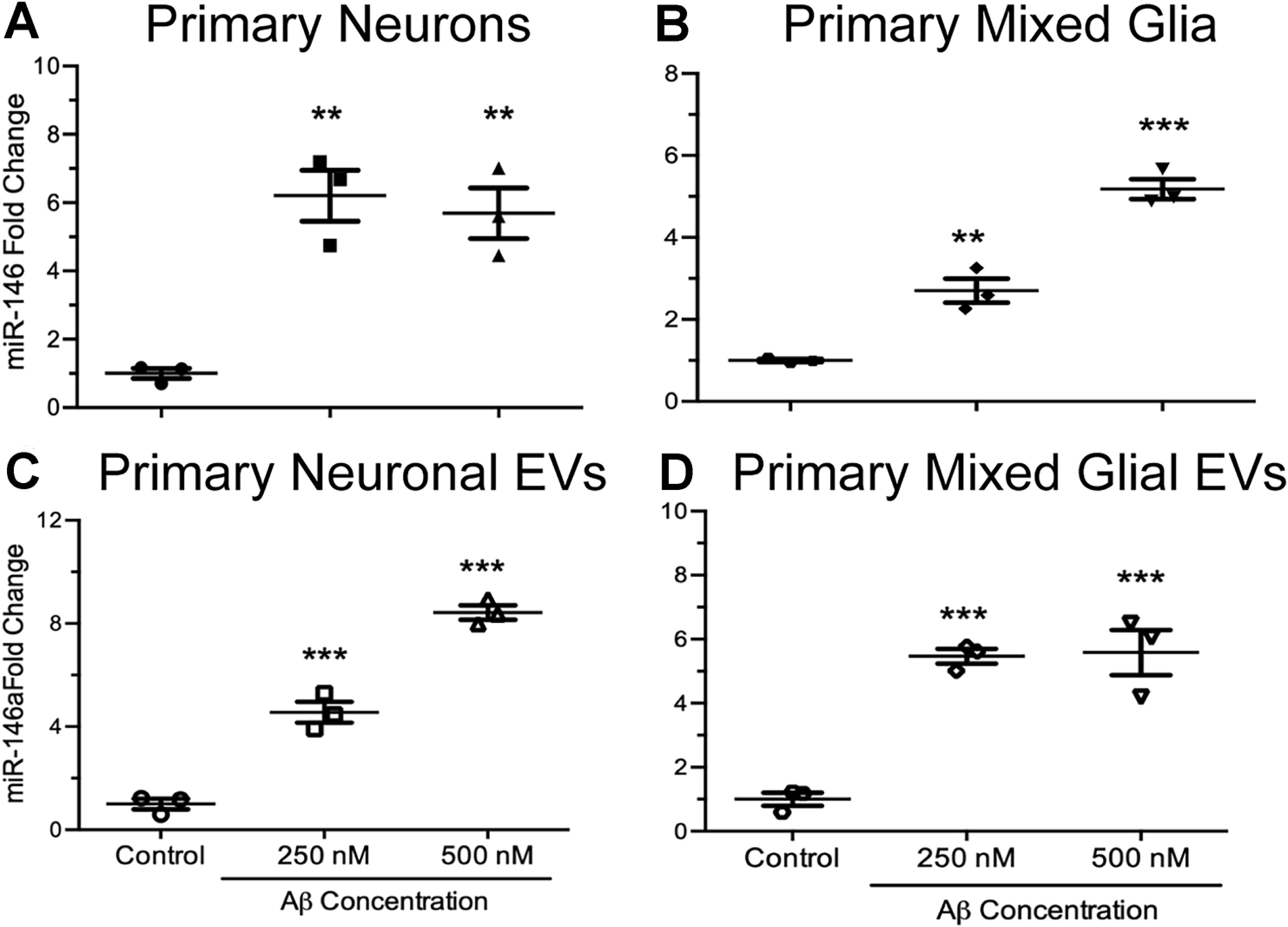
(A & C) Rat primary neurons and (B & D) mixed glial cells were exposed to 0, 250, and 500 nM of Aβ for 24 hours, followed by purification of cellular and EV miRNA. Primary neurons exposed to both concentrations of Aβ expressed significantly higher levels of intracellular miR-146a (A) when compared to unexposed, control cells (t7 = 3.16, p ≤ 0.05, R2 = 0.53). Similarly, EVs secreted from primary neurons exposed to both concentrations of Aβ expressed significantly higher levels of miR-146a (C) when compared to EVs secreted from unexposed, control cells (t7 = 18.28, p ≤ 0.0001, R2 = 0.98). Primary microglia exposed to both concentrations of Aβ expressed significantly higher levels of intracellular miR-146a (B) when compared to unexposed control cells (t7 = 156.0, p ≤ 0.0001, R2 = 0.95). Lastly, EVs secreted from primary microglia exposed to both concentrations of Aβ expressed significantly higher levels of miR-146a (D) when compared to EVs secreted from unexposed, control cells (t7 = 16.96, p ≤ 0.01, R2 = 0.66). Data are presented as means ± SEM with significance as *p≤0.05, **p≤0.01, ***p≤0.0001 compared to control.
Figure 3. Functional effects of ectopically expressed miR-146a on oxidative phosphorylation in rat primary mixed glial cells.
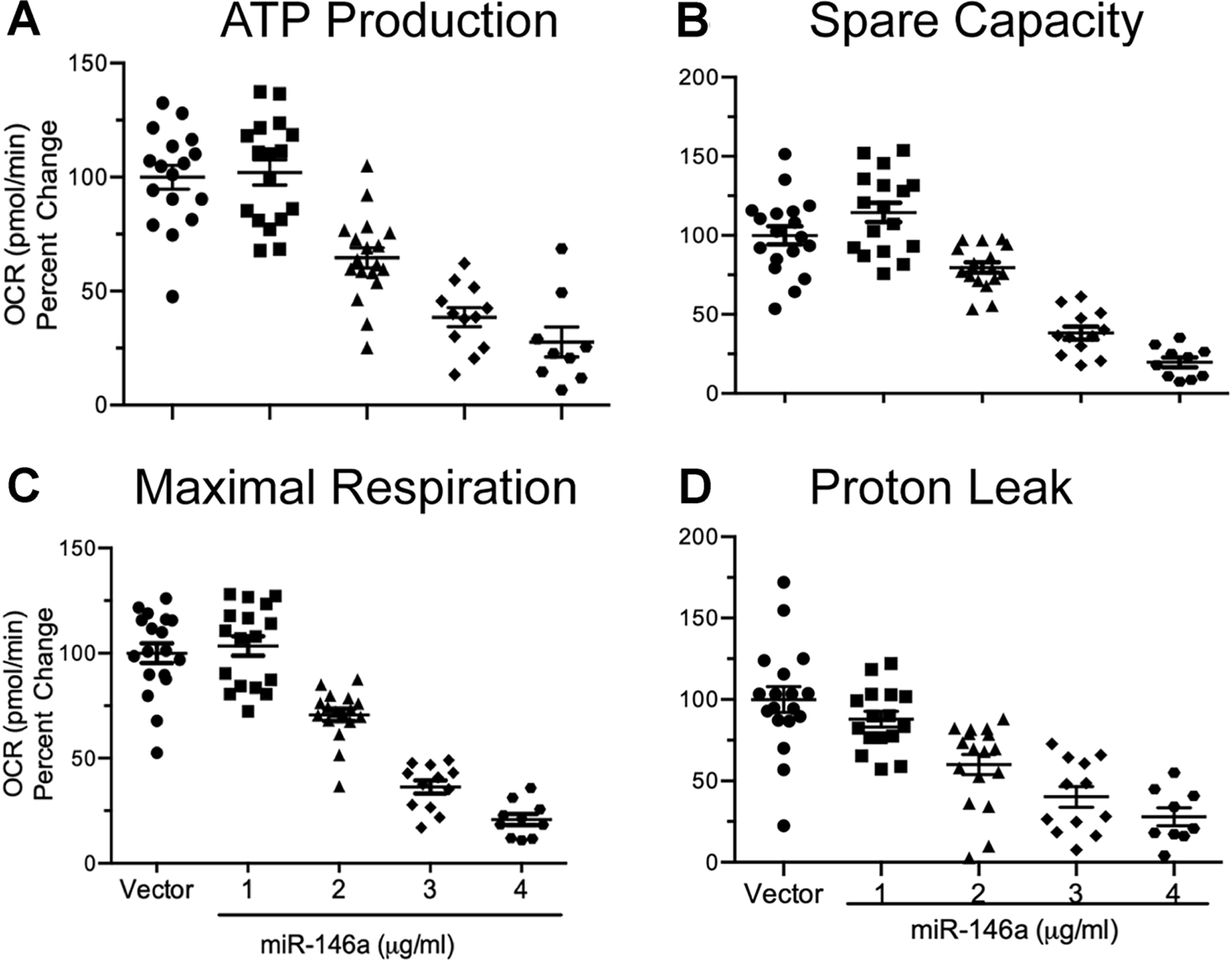
Mixed primary glial cells were transfected with a miR-146a overexpression vector for 40 hours, and mitochondrial function was assessed. ATP production (A) was significantly reduced in a miR-146a dose-dependent fashion (t71 = −10.95, p ≤ 0.0001, R2 = 0.62). Spare respiratory capacity (B) was significantly reduced in a dose-dependent fashion (t72 = −10.98, p ≤ 0.0001, R2 = 0.62). Maximal respiration (C) was significantly reduced in a dose-dependent fashion (t72 = −14.13, p ≤ 0.0001, R2 = 0.73). Proton leak (D) was significantly reduced in a dose-dependent fashion (t70 = −8.81, p ≤ 0.0001, R2 = 0.52). Data were analyzed by using simple linear regression which showed a negative linear trend between the independent variable (miR-146a) and each dependent variable. All data are presented as means ± SEM.
Figure 4. Overexpression of miR-146a reduces glycolysis in rat primary mixed glia.
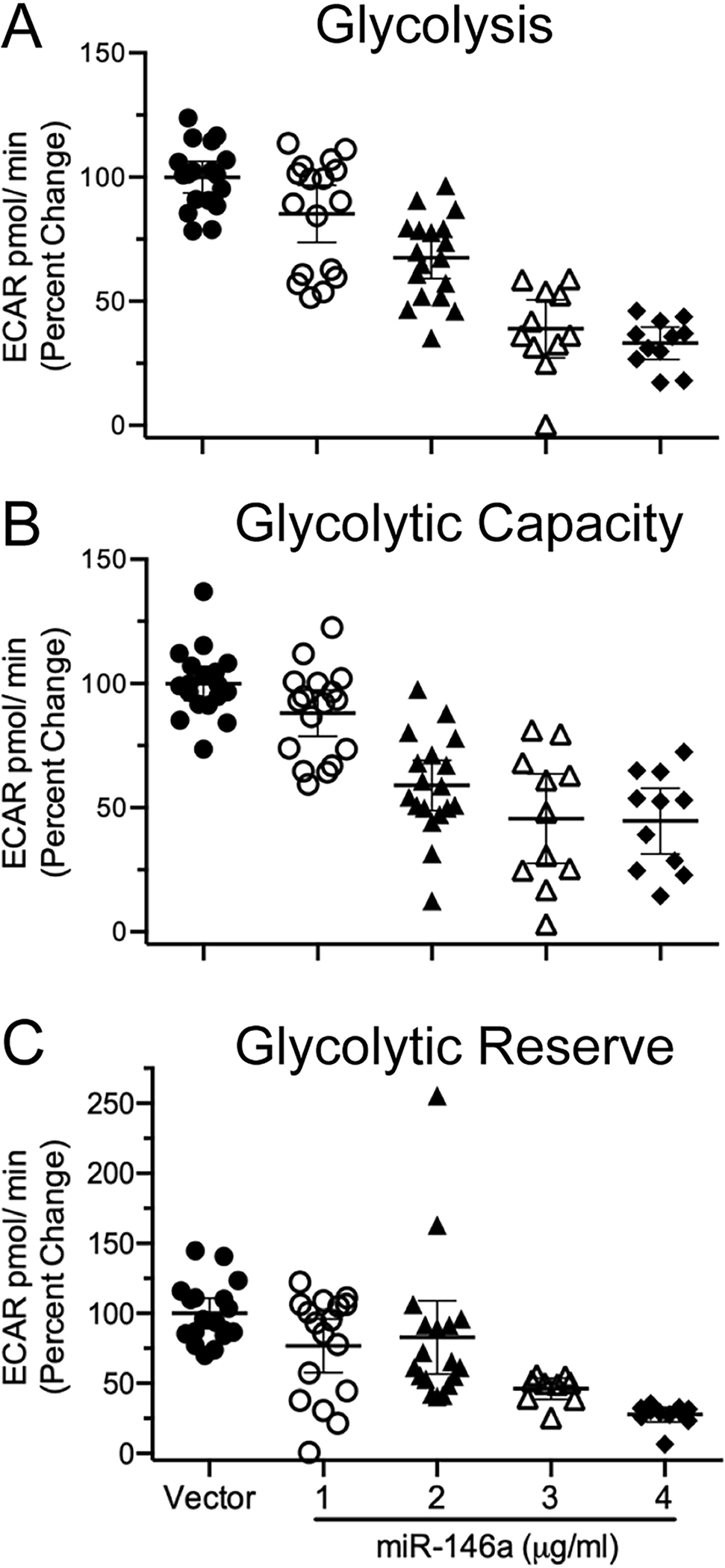
Rat primary mixed glia were transfected with a miR-146a overexpression vector and 40 hours after transfection, glycolysis was assessed. All data were analyzed using an ANOVA linear contrast. Glycolysis (A) was significantly reduced in a miR-146a dose-dependent manner (t74 = −12.28, p ≤ 0.0001, R2 = 0.66). Glycolytic capacity (B) was significantly reduced in a dose-dependent fashion (t73 = −9.23, p ≤ 0.0001, R2 = 0.53). Glycolytic reserve (C) was significantly reduced in a dose-dependent manner (t71 = −5.68, p ≤ 0.0001, R2 = 0.30). Data were analyzed by simple linear regression which showed a negative linear trend between the independent variable (miR-146a) and each dependent variable. All data are presented as means ± SEM.
Results
Neuroinflammatory stimuli induce upregulation of miR-146a in several CNS cell types and their EVs
Due to the heterogeneity of cell types within the CNS, our first goal was to understand how diverse inflammatory stimuli influence miR-146a production in various cells and their released EVs. To this end, we utilized an immortalized neuronal cell line (HT-22), a microglial cell line (C8-B4), and a brain endothelial cell line (bEnd.3). Cells were exposed to either TNF-α (1 ng/ml), a proinflammatory cytokine which is upregulated in AD (Decourt et al. 2016), or lipopolysaccharide (LPS) (1 μg/ml), which may be released from bacteria in the gastrointestinal tract and cross the aged BBB (Zhao and Lukiw 2018), for 24 hours. TNF-α exposure of HT-22 cells significantly increased miR-146a expression both intracellularly (Fig 1A) and in secreted EVs (Fig 1D). Levels of miR-146a in C8-B4 cells were also significantly increased in response to LPS exposure both intracellularly (Fig 1B) and in secreted EVs (Fig 1E). A similar pattern of miR-146a expression was also observed in bEnd.3 cells (Fig 1C) and EVs (Fig 1F) exposed to LPS, however, TNF-α exposure appears to affect these cells in a different manner. Although exposure to this cytokine induced significant upregulation of miR-146a in bEnd.3-derived EVs (Fig 1F), intracellular expression of miR-146a is significantly reduced. These data suggest the possibility of cell-type specific, as well as stimulus specific regulatory mechanisms, which influence miRNA packaging and secretion in EVs.
Figure 1. Effects of pro-inflammatory stimuli on intracellular and EV miR-146a levels in different brain cell types.
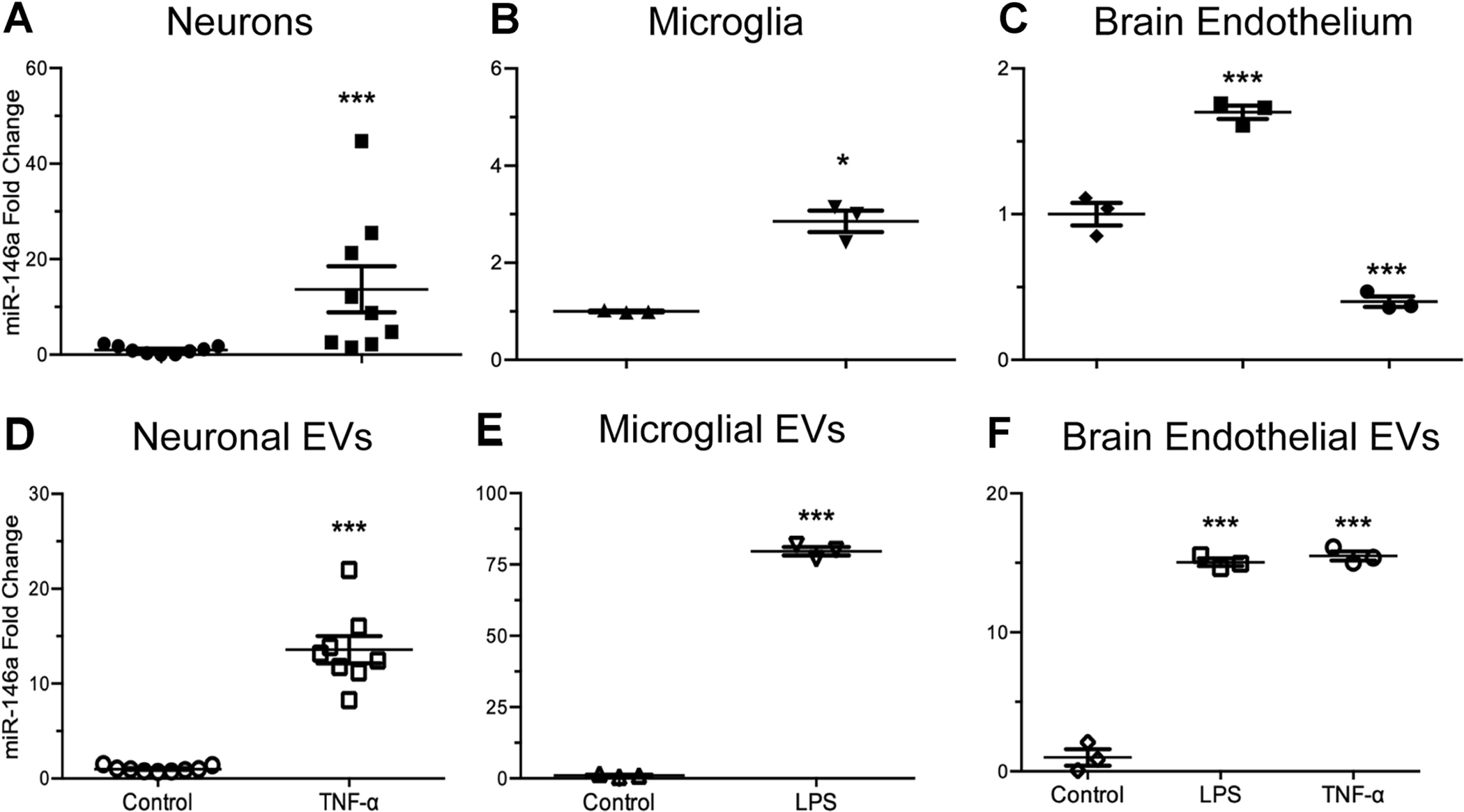
(A & D) Murine hippocampal neurons (HT-22 cells), (B & E) brain microglia (C8-B4 cells), or (C & F) brain endothelial cells (bEnd.3) were exposed to either LPS (1 μg/ml) or TNF-α 1 ng/ml for 24 hours, followed by purification of cellular and EV miRNA. Intracellular miR-146a expression was greater in TNF-α exposed neurons (A) than in untreated, control cells (t8.1 = 2.63, p ≤ 0.05, ηp2 = 0.68). Similarly, TNF-α exposure also significantly increased miR-146a expression in secreted neuronal EVs (D) when compared to EVs secreted from unexposed, control cells (t7.1 = 8.75, p ≤ 0.0001, ηp2 = 0.96). Intracellular miR-146a expression was not significantly altered in microglial cells after exposure to LPS (B) when compared to unexposed, control cells (t2.1 = 8.39, p ≤ 0.05, ηp2 = 0.99). Interestingly however, microglial EVs contained significantly elevated levels of miR-146a after exposure to LPS (E) when compared to unexposed control cells (t2.2 = 53.58, p ≤ 0.001, ηp2 = 0.99). Intracellular miR-146a expression levels in brain endothelial cells exposed to LPS were significantly elevated when compared to control, while TNF-α exposure lead to significantly reduced miR-146a expression (C) (F2,6 = 135.6, p ≤ 0.0001, ω2 = 0.97). EVs secreted from brain endothelial cells had significantly elevated miR-146a expression after LPS or TNF-α exposure, when compared to EVs secreted from unexposed, control cells (F) (F2,6 = 383.7, p ≤ 0.0001, ω2=0.09). Data are presented as means ± SEM with significance as *p≤0.05, **p≤0.01, ***p≤0.0001 compared to control.
Aβ exposure induces upregulation of miR-146a in primary neurons and mixed glial cells and their EVs
To determine if neuroinflammation-induced alterations in miR-146a were species specific, we next used rat primary neurons and primary mixed glial cells. For 24 hours, these cells were exposed to Aβ; a neuroinflammatory component of plaques and one of the hallmarks of AD. We observed a significant upregulation of miR-146a expression in primary neurons (Fig 2A), and a dose-dependent increase in their secreted EVs (Fig 2C). Exposure of primary mixed glia to Aβ induced similar responses, with a dose-dependent increase of miR-146a expression intracellularly (Fig 2B), and a significant upregulation within their secreted EVs (Fig 2D).
miR-146a inhibits oxidative phosphorylation and glycolysis in primary mixed glial cells
Previous work has shown that miR-146a targets several mRNAs which encode for proteins of the mitochondrial electron transport chain (Rippo et al. 2014) and glycolysis (Raschzok et al. 2011). Hence, we hypothesized that elevated levels of miR-146a would lead to significant impairment of cellular bioenergetics. To test this, we overexpressed miR-146a in rat primary mixed glial cells and measured oxidative phosphorylation and glycolysis using the Mito Stress Kit or Glycolysis Stress Test Kit (Agilent Biosciences). To ensure transfection efficiency prior to use of either of the metabolic stress kits, rat primary mixed glial cells were transfected with either vehicle vector or miR-146a overexpression vector, and cultured for 40 hours. We then assessed miR-146a expression levels intracellularly and in secreted EVs qRT-PCR. As shown in Table 2, when compared to vehicle vector, transfection of the miR-146a overexpression vector increased intracellular miR-146a levels more than 2-fold, while the levels in EVs were increased in a dose-dependent manner (53 to 116-fold). In addition, we assessed cellular viability 40 hours after transfection with a calcein AM assay and observed no effects on cellular viability (data not shown).
Table 2. Fold change in miR-146a levels following transfection in primary mixed glial cells.
Forty hours after transfection of vehicle or overexpression vector, total RNA was purified from primary mixed glial cells and their isolated EVs and miR-146a expression levels were assessed.
| miR-146a expression (relative fold change) | |||
|---|---|---|---|
| Transfection | Intracellular | Extracellular Vesicle | |
| 4 μg/ml | 1.0 ± 0.2 | 1.0 ± 0.0 | |
| 2 μg/ml | 2.1 ± 0.1 | 53 ± 19 | |
| 4 μg/ml | 2.3 ± 0.4 | 116 ± 20 * | |
All data were expressed as mean ± SEM;
p<0.05 compared to vector control.
We next examined the effects of miR-146a overexpression on oxidative phosphorylation. After 40 hours of transfection, we observed a dose dependent decrease in all OCR parameters assessed, including ATP production (Fig 3A), spare capacity (Fig 3B), maximal respiration (Fig 3C), and proton leak (Fig 3D) when compared to vector transfected cells. In addition, ECAR values were also measured 40 hours after transfection. Similarly, we observed a dose-dependent decrease in glycolysis (Fig 4A), glycolytic capacity (Fig 4B), and glycolytic reserve (Fig 4C) when compared to vector transfected cells. All slopes were significantly negative, which demonstrates that all indices of cellular bioenergetics are decreased when miR-146a expression is increased. These data show that elevated levels of miR-146a have the capacity to alter cellular bioenergetics by impairing both mitochondrial function and glycolysis.
miR-146a expression is positively correlated with pathological scores in human AD brains
Several groups have demonstrated that miR-146a levels are significantly increased in neocortical brain regions of AD patients, along with multiple biological fluids including serum, plasma, and cerebrospinal fluid (Alexandrov et al. 2012; Lukiw et al. 2012; Denk et al. 2015; Dong et al. 2015; Kumar and Reddy 2016). To test if miR-146a expression exhibits a brain region specific phenotype, we analyzed miR-146a levels from the temporal cortex (TC), frontal cortex (FC), and cerebellum (CB) of 10 control and 13 AD patient brains. In accordance with previous reports by Lukiw et al. (2012), we confirmed that miR-146a levels are significantly increased in the TC of AD patients (3.6-fold) when compared to AMCs (Fig 5A). We also observed increased levels of miR-146a in the FC (5.3-fold; Fig 5D) and CB (3.7-fold; Fig 5G), but this trend did not reach statistical significance due to high variation among samples.
Figure 5. miR-146a profile and correlation with pathology in human Alzheimer’s disease (AD) regions compared to age-matched controls (AMC) in temporal cortex, frontal cortex, and cerebellum.
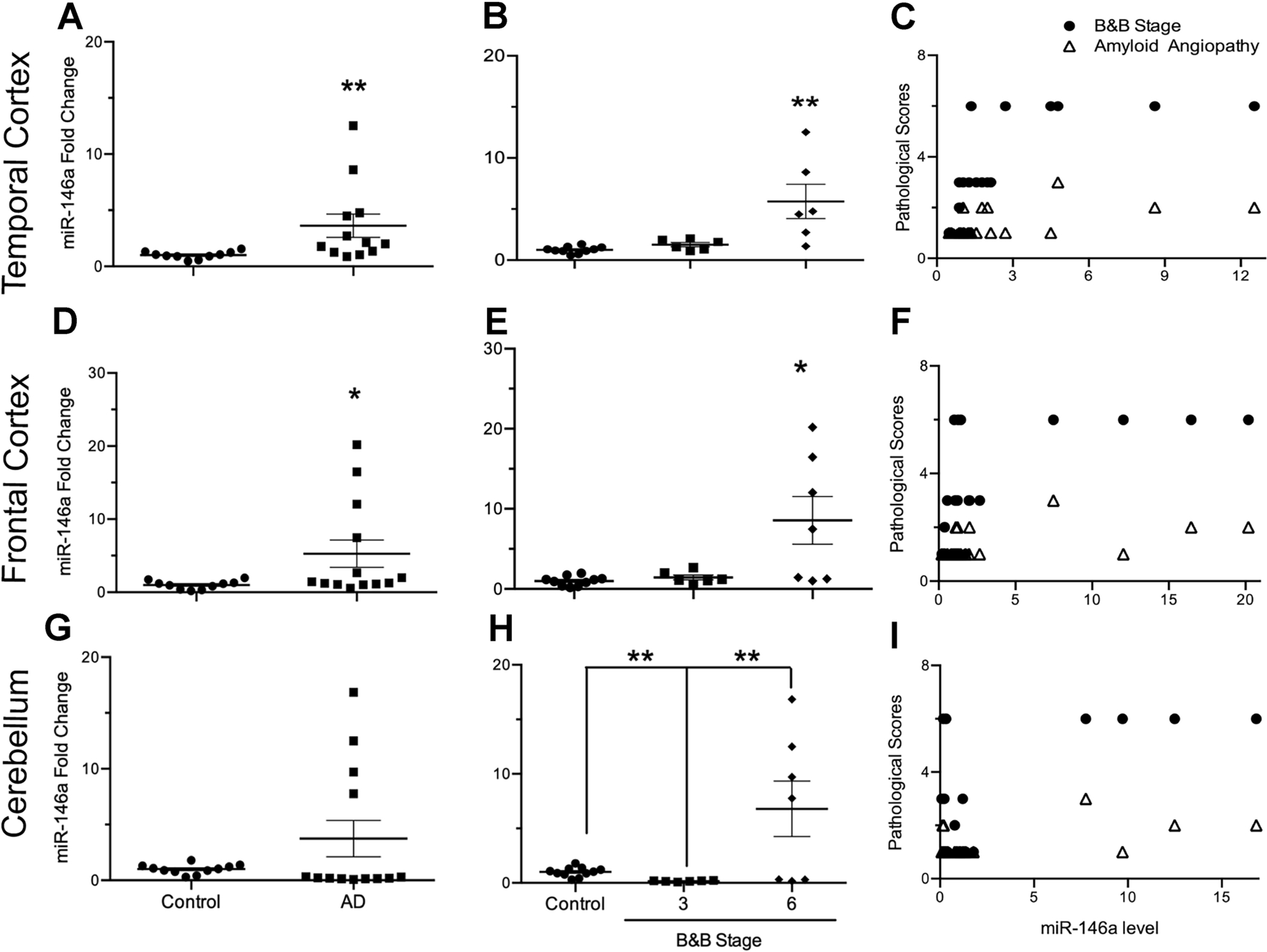
Relative fold change of miR-146a levels in AD cases (n=13) compared with AMCs (n=10) shows significant increases in the temporal cortex (A; t11.2 = 2.54, p ≤ 0.05, ηp2 = 0.60) and frontal cortex (D; t12.2 = 2.29, p ≤ 0.05, ηp2 = 0.55) of AD brains when compared to AMCs, while there were no significant changes in the cerebellum (G; t12.2 = 1.67, p = 0.12, ηp2 = 0.43). Of the AD brains, seven AD cases were diagnosed as Braak & Braak (B&B) stage VI, and six were diagnosed as stage III. The relationship of miRNA expression with B&B stage (control, 0 vs. 3 vs. 6) was also assessed to determine B&B stage specific miR-146a expression in human brains. Significant differences were identified within the temporal cortex (B; F2,19 = 9.91, p ≤ 0.01, ω2 = 0.45), frontal cortex (E; F2,20 = 7.05, p ≤ 0.01, ω2 = 0.35) and cerebellum (H; F2,21 = 7.19, p ≤ 0.01, ω2 = 0.34). Correlation of miR-146a expression with B&B stage or cerebral amyloid angiopathy (CAA) was calculated by using Spearman’s rho (ρ). Significant correlations between miR-146a with B&B stage were observed in the temporal (C; ρ = 0.77, p ≤ 0.001) and frontal cortices (F; ρ = 0.58, p ≤ 0.01), but not in the cerebellum (I: ρ = −0.01, p = 0.96). Similarly, miR-146a expression was significantly correlated with CAA in the temporal (C; ρ = 0.53, p ≤ 0.05) and frontal cortices (F; ρ = 0.48, p ≤ 0.05), but not in the cerebellum (I; ρ = 0.08, p = 0.70). Data are presented as means ± SEM with significance as *p≤0.05, **p≤0.01, ***p≤0.0001 compared to control.
To test if the variation was due to the severity of AD pathology, we further analyzed miR-146a levels in brains when stratified by Braak stage (3 and 6). The increase of miR-146a in AD brains is most highly correlated with Braak stage 6, which designates late-stage AD, regardless of which brain region we assessed; 5.7-fold in TC (Fig 5B), 8.6-fold in FC (Fig 5E), and 6.8-fold in CB (Fig 5H). We observed a significant positive correlation between increased levels of miR-146a and the severity of Braak staging of AD patients for the temporal and frontal cortices (Figs 5C and 5F), but not in the cerebellum (5I). Further correlational analyses also indicated that levels of miR-146a showed a similar pattern of correlation for CAA. These data suggest that both tau pathology (as assessed with Braak staging) and amyloid burden on cerebral vasculature (as assessed with CAA) are correlated with increased levels of miR-146a in the frontal and temporal cortices.
Due to our finding that overexpression of miR-146a in primary mixed glial cells significantly reduces cellular bioenergetics, and miR-146a expression is significantly elevated in AD brains, we next probed for changes in total mitochondrial protein (VDAC1) levels within the temporal cortices of AD and AMC brains (Fig 6A). We found a significant negative correlation between increased expression levels of miR-146a and decreased mitochondrial protein (Fig 6B). Together, our findings suggest that as AD progresses, expression of miR-146a increases and is correlated with decreased expression of mitochondrial protein.
Fig 6. Mitochondrial protein levels are inversely correlated with miR-146a gene expression.
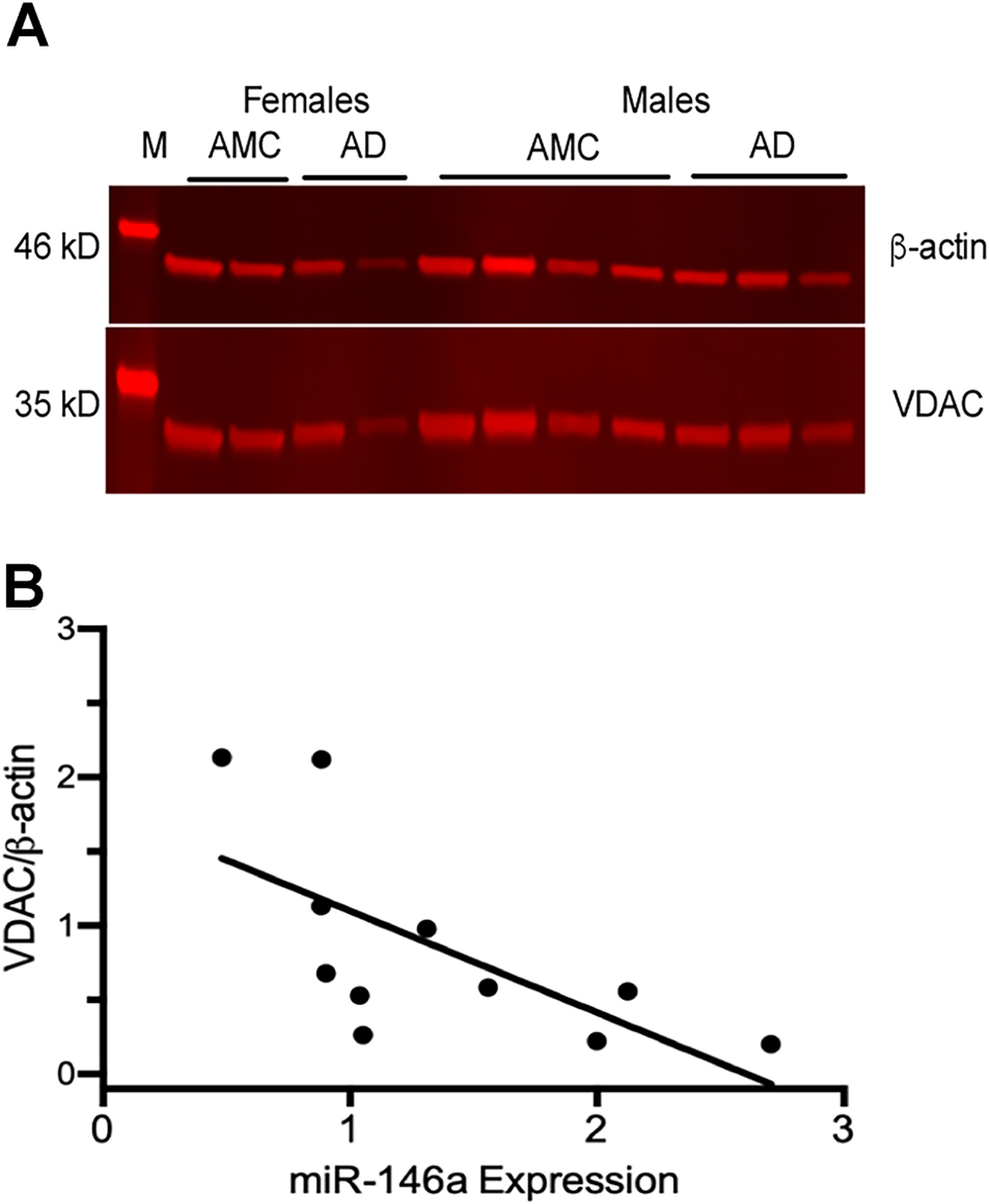
(A) Western blot of representative AD and AMC temporal cortex shows levels of the mitochondrial protein, voltage-dependent anion channel (VDAC, 35 kD), and the cytosolic housekeeping protein, β-actin (46 kD). (B) VDAC levels were normalized to β -actin for each sample (VDAC/β-actin). The correlation between the VDAC/β-actin ratio and miR-146a expression revealed an inverse relationship between total mitochondrial proteins and miR146a expression (Spearman’s ρ = −0.80, p ≤ 0.01).
Discussion
In the current study, we have described a novel role for miR-146a in the pathophysiology of AD. We demonstrated that various inflammatory stimuli induce upregulation of miR-146a in several cell types of both mouse and rat origins. Our assessment of the functional impacts of elevated levels of miR-146a on cellular bioenergetics reveal novel and very interesting findings which indicate that further mechanistic probing is warranted. In addition to our in vitro studies, we utilized human brain samples to demonstrate that patients suffering from AD express significantly elevated levels of miR-146a when compared to AMCs. From these data, we propose that miR-146a plays a key role in the progression and pathophysiology of AD.
During periods of neuroinflammation, microglia and astrocytes act as the first responders and initiate an immune response (Dong and Benveniste 2001; Yang et al. 2010). This response can be beneficial in the short term, as invading pathogens or aberrantly expressed molecules are cleared to protect and preserve neuronal function (Sochocka et al. 2017). The negative consequences of neuroinflammation arise when the inflammatory response becomes prolonged, or chronic, such as the case for patients suffering from AD (Sochocka et al. 2017). A noteworthy hypothesis by which long-term, chronic neuroinflammation may be induced is via the gut microbiome. Various gut microbes secrete metabolic byproducts that can cause barriers, like the gastrointestinal barrier and blood-brain barrier to become leaky; these neurotoxic byproducts can then enter the brain and induce chronic inflammation (Lukiw 2020). As AD progresses, the cells of the CNS are exposed to many inflammatory stimuli, including cytokines, chemokines, and Aβ aggregates (Akiyama et al. 2000). Here, we have shown that with prolonged exposure to these immunomodulatory molecules, miR-146a expression is significantly increased. In line with our results, other recent studies have also shown that miR-146a expression occurs within cells of the CNS, including choroid plexus epithelium (Balusu et al. 2016), human neural cells (Lukiw et al. 2008), and brain endothelial cells (Wu et al. 2015). Similar to our findings, another group recently showed that exposure of glial cells to Aβ activates nuclear factor kappa-B (NF-κB) and induces miR-146a expression (Denk et al. 2015). Interestingly, inflammation-induced miR-146a expression has been reported in various other tissues/cell types, including immune cells (macrophage, dendritic, neutrophil, T cells and B cells) (Taganov et al. 2006; Lu et al. 2010; Boldin et al. 2011; Sun et al. 2015), hepatocytes (Sun et al. 2015), cardiomyocytes, bronchial epithelial cells, endothelial cells (HUVEC), orbital adipose tissue, as well as mesenchymal stem cells (Song et al. 2017; Fu et al. 2017). As reviewed by Saba et al., (2014) miR-146a has been demonstrated to be a key regulator of the immune response, both systemically and within the central nervous system. Together, these findings support the notion that inflammatory stimuli induce upregulation of miR-146a in a wide variety of cell types, and that this mechanism is likely involved in the immune response.
In addition to intracellular increases of miR-146a, we also found significant increases in miR-146a expression in the EVs secreted from cells exposed to various inflammatory stimuli. EV-mediated spread of disease may exacerbate the neuroinflammatory response (Rajendran et al. 2006) due to their ability to transfer functionally active proteins and RNAs to recipient cells (Ratajczak et al. 2006; Valadi et al. 2007). As inflammation induced EVs are taken up by neighboring cells, their contents can be transferred into recipient cells and exert functional effects (i.e., miRNAs contained within EVs can repress protein translation in recipient cells) (Valadi et al. 2007). We observed a substantial upregulation of miR-146a in isolated EVs induced from exposure neuroinflammatory stimuli at levels much greater than control, and conclude that this is indicative of an increase in EVs derived from cultured cells. Taken together, our results are in agreement with others (Alexander et al. 2015; Song et al. 2017), and support the concept that EVs play a role in disease progression due to their ability to transfer aberrant proteins and RNAs to recipient cells.
In the AD brain, Aβ plaque deposition is considered a pathological hallmark of the disease and has been shown to induce inflammation. Previous work has observed the accumulation of Aβ aggregates within microglia, as these brain immune cells attempt to degrade the insoluble protein aggregates to no avail (Paresce et al. 1997; Lee and Landreth 2010; Krabbe et al. 2013; Baik et al. 2016). Additionally, complement factors and other inflammatory molecules have been observed within plaques themselves (Eikelenboom et al. 1989). It is clear that while the brain is trying to rid itself of these extracellular aggregates, an inflammatory response is being initiated within the surrounding tissue. Evidence from in vitro studies has shown that in response to Aβ exposure, cultured human neuronal and glial cells upregulate expression of miR-146a due to the activation of NF-κB (Lukiw et al. 2008; Cui et al. 2010). Increased expression of miR-146a has been associated with another major characteristic of AD, tau hyperphosphorylation, via the repression of rho-associated, coiled-coil containing protein kinase 1 (ROCK1) (Wang et al. 2016).
To better understand the interplay between inflammatory stimuli and decreased cellular bioenergetics, we investigated the potential effects of elevated levels of miR-146a on mitochondrial function and glycolysis. We show for the first time, that overexpression of miR-146a leads to profound reductions in several parameters of mitochondrial function and glycolytic capabilities of primary mixed glial cells, which supports the notion that miR-146a exerts pro-inflammatory effects when it is significantly upregulated. Although in the current experiment we did not test the direct effects of inflammatory stimuli on cellular bioenergetics, previous work has demonstrated that exposure to pro-inflammatory cytokines such as TNF-α and IL-1β leads to alterations in glucose metabolism and oxidative stress in astrocytes (Gavillet et al. 2008) and in a neuronal cell line (Russell et al. 2019). In addition to classical inflammatory mediators, chronic exposure to soluble Aβ peptides also results in impairment of energy homeostasis due to the decreased respiratory capacity of the mitochondrial electron transport chain; this may accelerate neuronal death (Rhein et al. 2009) as neurons are extremely dependent on aerobic metabolism and oxygen use. Despite a large reservoir of ATP, neurons are heavily dependent on aerobic metabolism and oxygen use, so reductions in ATP availability due to impaired glycolytic and/or mitochondrial function results in neuronal cell death (Goldberg and Choi 1993; Lee et al. 2012).
Lukiw (2007) first demonstrated differential miRNA expression patterns in AD hippocampi, and later Cogswell et al. (2008) observed this phenomenon in specific brain regions and cerebrospinal fluid (CSF). Another group also found that miR-146a expression levels were significantly upregulated in both the CSF and hippocampi of AD patients (Denk et al. 2015). In addition to measuring miR-146a expression, we also correlated levels within specific brain regions (frontal and temporal cortices, and cerebellum) to Braak & Braak staging (Braak and Braak 1991), and found significant positive correlations between these two factors. Additionally, expression levels of miR-146a were also positively correlated with CAA, which is characterized by deposition of Aβ in the media and adventitia of cortical and leptomeningeal vessels (Salvarani et al. 2016). Aging and AD are established risk factors for developing CAA (Yamada 2015), suggesting that not only parenchymal deposition of Aβ, but also intravascular accumulation, affects the expression of miR-146a during disease progression. Further experimentation is necessary to evaluate the effects of Aβ exposure on miR-146a expression in primary endothelial cells and their released EVs. These data indicate that multiple cell types may mediate the increase in miR-146a levels in AD and other neurological disease states. Brain hypometabolism is also significantly correlated with increased dementia in AD (Liang et al. 2008). Here, we have shown that within the temporal cortex of AD patients, as miR-146a expression increases, total mitochondrial protein decreases. Although VDAC is not a direct target of miR-146a, several other glycolytic and mitochondrial proteins are (www.targetscan.org); downregulation of these proteins may lead to impaired mitochondrial function and reduced total mitochondria within the AD brain. Together these data support the hypothesis that inflammation-induced miR-146a overexpression contributes to brain hypometabolism in AD.
In summary, we have shown that inflammatory stimuli expressed within the AD brain induce upregulation of miR-146a in a variety of cell types. Parallel findings in multiple brain cell types (neurons, astrocytes, microglia, and endothelial cells) and species (mouse, rat, and human) demonstrate that our observations may represent a common CNS inflammatory response mediated, in part, by miR-146a. Our functional data indicate that the overexpression of miR-146a significantly impairs cellular bioenergetic function and may lead to the hypometabolic phenotype seen in clinical AD. These results suggest a novel pro-inflammatory mechanism of action for miR-146a in the neuroinflammatory etiology of AD and related dementias.
Funding:
The project was supported by NIH Grants K01NS081014 (C.M.B.), U54GM104942, P20 GM109098 (J.W.S and C.M.B.), and T32AG052375 (A.E.R.).
Footnotes
Conflicts of interest/Competing interests: Not applicable
Ethics approval (include appropriate approvals or waivers): All animal experiments were approved by the West Virginia University Institutional Animal Care and Use Committee.
Availability of data and material (data transparency):
Data can be made available upon request
References
- Akiyama H, Barger S, Barnum S, et al. (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander M, Hu R, Runtsch MC, et al. (2015) Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun 6:7321. 10.1038/ncomms8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov PN, Dua P, Hill JM, et al. (2012) microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int J Biochem Mol Biol 3:365–73 [PMC free article] [PubMed] [Google Scholar]
- Alexandrov PN, Dua P, Lukiw WJ (2014) Up-Regulation of miRNA-146a in Progressive, Age-Related Inflammatory Neurodegenerative Disorders of the Human CNS. Front Neurol 5:181. 10.3389/fneur.2014.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Son SM, Mook-Jung I (2016) Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 64:2274–2290. 10.1002/glia.23074 [DOI] [PubMed] [Google Scholar]
- Balusu S, Van Wonterghem E, De Rycke R, et al. (2016) Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus‐derived extracellular vesicles. EMBO Mol Med 8:1162–1183. 10.15252/emmm.201606271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, et al. (2005) Globular amyloid beta-peptide1–42 oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem 95:834–847. 10.1111/j.1471-4159.2005.03407.x [DOI] [PubMed] [Google Scholar]
- Boldin MP, Taganov KD, Rao DS, et al. (2011) miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 208:1189–201. 10.1084/jem.20101823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–59 [DOI] [PubMed] [Google Scholar]
- Brier MR, Thomas JB, Snyder AZ, et al. (2012) Loss of Intranetwork and Internetwork Resting State Functional Connections with Alzheimer’s Disease Progression. J Neurosci 32:8890–8899. 10.1523/JNEUROSCI.5698-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AL, Guedes JR, de Lima MCP (2016) Role of microRNAs in the regulation of innate immune cells under neuroinflammatory conditions. Curr Opin Pharmacol 26:1–9. 10.1016/J.COPH.2015.09.001 [DOI] [PubMed] [Google Scholar]
- Cogswell JP, Ward J, Taylor IA, et al. (2008) Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimer’s Dis 14:27–41. 10.3233/JAD-2008-14103 [DOI] [PubMed] [Google Scholar]
- Cui JG, Li YY, Zhao Y, et al. (2010) Differential Regulation of Interleukin-1 Receptor-associated Kinase-1 (IRAK-1) and IRAK-2 by MicroRNA-146a and NF-κB in Stressed Human Astroglial Cells and in Alzheimer Disease. J Biol Chem 285:38951–38960. 10.1074/jbc.M110.178848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakhlallah DA, Wisler J, Gencheva M, et al. (2019) Circulating extracellular vesicle content reveals de novo DNA methyltransferase expression as a molecular method to predict septic shock. J Extracell Vesicles 8:1669881. 10.1080/20013078.2019.1669881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, Convit A, Wolf OT, et al. (2001) Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-D-glucose/positron-emission tomography (FDG/PET). Proc Natl Acad Sci 98:10966–10971. 10.1073/pnas.191044198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourt B, Lahiri D, Sabbagh M (2016) Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr Alzheimer Res 13:1–1. 10.2174/1567205013666160930110551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk J, Boelmans K, Siegismund C, et al. (2015) MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer`s Disease. PLoS One 10:e0126423. 10.1371/journal.pone.0126423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Li J, Huang L, et al. (2015) Serum MicroRNA Profiles Serve as Novel Biomarkers for the Diagnosis of Alzheimer’s Disease. Dis Markers 2015:625659. 10.1155/2015/625659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–90 [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Hack CE, Rozemuller JM, Stam FC (1989) Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol 56:259–62 [DOI] [PubMed] [Google Scholar]
- Fu Y, Zhang L, Zhang F, et al. (2017) Exosome-mediated miR-146a transfer suppresses type I interferon response and facilitates EV71 infection. PLOS Pathog 13:e1006611. 10.1371/journal.ppat.1006611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavillet M, Allaman I, Magistretti PJ (2008) Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 56:975–989. 10.1002/glia.20671 [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13:3510–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, Zurolo E, Prabowo A, et al. (2012) MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLoS One 7:e44789. 10.1371/journal.pone.0044789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, et al. (2013) Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS One 8:e60921. 10.1371/journal.pone.0060921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Reddy PH (2016) Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta - Mol Basis Dis 1862:1617–1627. 10.1016/j.bbadis.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CYD, Landreth GE (2010) The role of microglia in amyloid clearance from the AD brain. J Neural Transm 117:949–60. 10.1007/s00702-010-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, et al. (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J, et al. (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci 105:4441–4446. 10.1073/pnas.0709259105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lu L-F, Boldin MP, Chaudhry A, et al. (2010) Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell 142:914–929. 10.1016/j.cell.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ (2020) Gastrointestinal (GI) Tract Microbiome-Derived Neurotoxins—Potent Neuro-Inflammatory Signals From the GI Tract via the Systemic Circulation Into the Brain. Front Cell Infect Microbiol 10:. 10.3389/fcimb.2020.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ (2007) Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 18:297–300. 10.1097/WNR.0b013e3280148e8b [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Alexandrov PN (2012) Regulation of Complement Factor H (CFH) by Multiple miRNAs in Alzheimer’s Disease (AD) Brain. Mol Neurobiol 46:11–19. 10.1007/s12035-012-8234-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN (2012) Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer’s disease (AD) and in age-related macular degeneration (AMD). Int J Biochem Mol Biol 3:105–16 [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Zhao Y, Cui JG (2008) An NF-κB-sensitive Micro RNA-146a-mediated Inflammatory Circuit in Alzheimer Disease and in Stressed Human Brain Cells. J Biol Chem 283:31315–31322. 10.1074/jbc.M805371200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ (2008) Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann N Y Acad Sci 1147:180–195. 10.1196/annals.1427.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Sorbi S, de Leon MJ, et al. (2006) Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med 47:1778–86 [PubMed] [Google Scholar]
- Nguyen LS, Fregeac J, Bole-Feysot C, et al. (2018) Role of miR-146a in neural stem cell differentiation and neural lineage determination: relevance for neurodevelopmental disorders. Mol Autism 9:38. 10.1186/s13229-018-0219-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem 272:29390–7 [DOI] [PubMed] [Google Scholar]
- Pogue AI, Lukiw WJ (2018) Up-regulated Pro-inflammatory MicroRNAs (miRNAs) in Alzheimer’s disease (AD) and Age-Related Macular Degeneration (AMD). Cell Mol Neurobiol 38:1021–1031. 10.1007/s10571-017-0572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, et al. (2006) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci 103:11172–11177. 10.1073/pnas.0603838103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschzok N, Werner W, Sallmon H, et al. (2011) Temporal expression profiles indicate a primary function for microRNA during the peak of DNA replication after rat partial hepatectomy. Am J Physiol Regul Integr Comp Physiol 300:1363–1372. 10.1152/ajpregu.00632.2010 [DOI] [PubMed] [Google Scholar]
- Ratajczak J, Wysoczynski M, Hayek F, et al. (2006) Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20:1487–1495. 10.1038/sj.leu.2404296 [DOI] [PubMed] [Google Scholar]
- Rhein V, Baysang G, Rao S, et al. (2009) Amyloid-beta Leads to Impaired Cellular Respiration, Energy Production and Mitochondrial Electron Chain Complex Activities in Human Neuroblastoma Cells. Cell Mol Neurobiol 29:1063–1071. 10.1007/s10571-009-9398-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippo MR, Olivieri F, Monsurrò V, et al. (2014) MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp Gerontol 56:154–163. 10.1016/j.exger.2014.03.002 [DOI] [PubMed] [Google Scholar]
- Russell AE, Jun S, Sarkar S, et al. (2019) Extracellular vesicles secreted in response to cytokine exposure increase mitochondrial oxygen consumption in recipient cells. Front Cell Neurosci 13:. 10.3389/fncel.2019.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Sorensen DL, Booth SA (2014) MicroRNA-146a: A dominant, negative regulator of the innate immune response. Front. Immunol 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvarani C, Morris JM, Giannini C, et al. (2016) Imaging Findings of Cerebral Amyloid Angiopathy, Aβ-Related Angiitis (ABRA), and Cerebral Amyloid Angiopathy-Related Inflammation: A Single-Institution 25-Year Experience. Medicine (Baltimore) 95:e3613. 10.1097/MD.0000000000003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Jun S, Rellick S, et al. (2016) Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res 1646:139–151. 10.1016/J.BRAINRES.2016.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Jun S, Simpkins JW (2015) Estrogen amelioration of Aβ-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERβ, AKAP and Drp1. Brain Res 1616:101–111. 10.1016/j.brainres.2015.04.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD (2002) Imaging the progression of Alzheimer pathology through the brain. Proc Natl Acad Sci U S A 99:4135–7. 10.1073/pnas.082107399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochocka M, Diniz BS, Leszek J (2017) Inflammatory Response in the CNS: Friend or Foe? Mol Neurobiol 54:8071–8089. 10.1007/s12035-016-0297-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Dou H, Li X, et al. (2017) Exosomal miR-146a Contributes to the Enhanced Therapeutic Efficacy of Interleukin-1β-Primed Mesenchymal Stem Cells Against Sepsis. Stem Cells 35:1208–1221. 10.1002/stem.2564 [DOI] [PubMed] [Google Scholar]
- Sun X, Zhang J, Hou Z, et al. (2015) miR-146a is directly regulated by STAT3 in human hepatocellular carcinoma cells and involved in anti-tumor immune suppression. Cell Cycle 14:243–252. 10.4161/15384101.2014.977112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang K-J, Baltimore D (2006) NF-B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses [DOI] [PMC free article] [PubMed]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–301 [DOI] [PubMed] [Google Scholar]
- Théry C, Amigorena S, Raposo G, Clayton A (2006) Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. In: Current Protocols in Cell Biology. John Wiley & Sons, Inc., Hoboken, NJ, USA, p Unit 3.22 [DOI] [PubMed] [Google Scholar]
- Thompson AG, Gray E, Heman-Ackah SM, et al. (2016) Extracellular vesicles in neurodegenerative disease — pathogenesis to biomarkers. Nat Rev Neurol 12:346–357. 10.1038/nrneurol.2016.68 [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, et al. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19:213–228. 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- Wang G, Huang Y, Wang L-L, et al. (2016) MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci Rep 6:26697. 10.1038/srep26697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang X, Ju Y, et al. (2013) MicroRNA-146a Feedback Suppresses T Cell Immune Function by Targeting Stat1 in Patients with Chronic Hepatitis B. J Immunol 191:293–301. 10.4049/jimmunol.1202100 [DOI] [PubMed] [Google Scholar]
- Wu D, Cerutti C, Lopez-Ramirez MA, et al. (2015) Brain endothelial miR-146a negatively modulates T-cell adhesion through repressing multiple targets to inhibit NF-κB activation. J Cereb Blood Flow Metab 35:412–23. 10.1038/jcbfm.2014.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M (2015) Cerebral amyloid angiopathy: emerging concepts. J stroke 17:17–30. 10.5853/jos.2015.17.1.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang I, Han SJ, Kaur G, et al. (2010) The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci 17:6–10. 10.1016/j.jocn.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Sancheti H, Patil I (2016) Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med 100:108–122. 10.1016/J.FREERADBIOMED.2016.04.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Lukiw WJ (2018) Microbiome-Mediated Upregulation of MicroRNA-146a in Sporadic Alzheimer’s Disease. Front Neurol 9:145. 10.3389/fneur.2018.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data can be made available upon request