

Abstract
Precious and rare elements have traditionally dominated inorganic photophysics and photochemistry, but now we are witnessing a paradigm shift toward cheaper and more abundant metals. Even though emissive complexes based on selected first-row transition metals have long been known, recent conceptual breakthroughs revealed that a much broader range of elements in different oxidation states are useable for this purpose. Coordination compounds of V, Cr, Mn, Fe, Co, Ni, and Cu now show electronically excited states with unexpected reactivity and photoluminescence behavior. Aside from providing a compact survey of the recent conceptual key advances in this dynamic field, our Perspective identifies the main design strategies that enabled the discovery of fundamentally new types of 3d-metal-based luminophores and photosensitizers operating in solution at room temperature.
Keywords: Photoluminescence, first-row transition metal complexes, coordination chemistry, metal-to-ligand charge transfer, ligand-to-metal charge transfer, Earth-abundant metals
1. Introduction
Many coordination compounds with platinum group metals have rich photophysical and electrochemical properties,1,2 but their electronic structures are difficult to emulate with first-row transition metals. The key challenge is that a given coordination environment imposes a considerably weaker ligand field on a 3d-metal than on a 4d- or 5d-species, because the more contracted 3d-orbitals have weaker spatial overlaps with the relevant ligand orbitals than the 4d- or 5d-orbitals (Figure 1a).3 Consequently, complexes with partially filled 3d-orbitals typically feature a multitude of metal-centered (MC) excited states, which are energetically close to each other and to the electronic ground state. This scenario is problematic, because it can allow for a cascade of nonradiative relaxation processes from one MC state to the other and finally back to the ground state (Figure 1b).4 Moreover, in some of the MC states, the metal–ligand bonding situation is very different from the ground state, for example, when d-electrons are promoted from nonbonding to antibonding orbitals. The respective excited states are then said to be “distorted”, and this is captured by the displacement of potential wells along a certain nuclear coordinate (Figure 1b/c), for instance, the metal–ligand bond distance.5 From the resulting overall picture, it is quite obvious that the barriers for crossing from one potential well into the next lower one can be undesirably low, causing rapid nonradiative relaxation.
Figure 1.
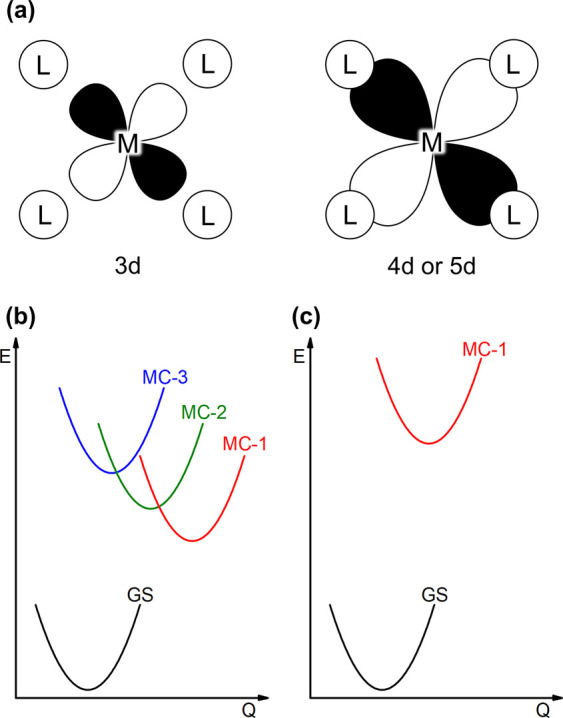
(a) Illustration of the overlap between metal d-orbitals and ligand orbitals. (b) Exemplary single configurational coordinate diagram for a 3d-metal complex (c) Exemplary single configurational coordinate diagram for a 4d- or 5d-metal complex. Q is a nuclear coordinate.
The stronger ligand fields of 4d- and 5d-metal complexes typically do not only lead to greater energy gaps between the individual MC states, but also to a sizable energy gap between the lowest MC state and the electronic ground state (Figure 1c). In such cases, that lowest MC state can become luminescent and photochemically active,5 because nonradiative relaxation becomes less efficient when the excitation energy has to be dissipated by an increasing number of vibrational quanta.6 Energetically high lying MC states furthermore open the possibility for the installment of fundamentally different excited states at lower energies (while maintaining a sufficiently large gap to the ground state), for instance states, in which electron density is shifted from the metal to the ligands or vice versa. Since the lowest excited state commonly governs the photophysical and photochemical properties, that state is usually the most important one, and the aforementioned metal-to-ligand charge transfer (MLCT) and ligand-to-metal charge transfer (LMCT) are very attractive for many applications. The redox properties in these charge-transfer states are drastically altered with respect to the ground state, enabling them to engage in photoredox chemistry with organic substrates7,8 or to act as dyes in inorganic semiconductor solar cells.9 Ligand-to-ligand charge transfer (LLCT), in which electron density is shifted from one coordinated ligand to another, can be similarly useful.10 This is fundamentally different from so-called intraligand (IL) excitations, in which the electron density redistribution typically occurs within the π-conjugation system of a given (individual) ligand.
Contrary to typical organic compounds, many metal complexes facilitate rapid intersystem crossing from initially excited singlet to lower-lying triplet excited states by virtue of strong spin–orbit coupling.11 This is important because triplet states give access to photochemical reactivities that are unattainable from singlets, they can have improved kinetic reactivity due to longer lifetimes,12 and photoinduced electron transfer from triplet states can produce primary photoproducts that undergo less (undesired) charge recombination, leading to inherently greater quantum efficiencies of photoredox reactions.12,13 Numerous applications underscore the important role played by (so far mostly precious) metal-based compounds, including luminophores in organic light emitting diodes (OLEDs)14 and light-emitting electrochemical cells (LEECs),15,16 photocatalysts in synthetic photochemistry and artificial photosynthesis,17,18 light-harvesters in dye-sensitized solar cells,19 and sensitizers in photochemical upconversion or photodynamic therapy.20,21 Thus, there are many good reasons to search for photoactive complexes based on cheap and abundant metals.22−24
Copper(I) emerged as an attractive candidate nearly 40 years ago,25 and furthermore, some chromium(III) complexes had long been known to emit in solution at room temperature (Figure 2).26 With both metal ions, key conceptual progress has been made recently, and complexes with exceptional luminescence quantum yields and excited-state lifetimes were reported.27 For other elements such as cobalt and nickel, there had been a few selected reports on emission from molecular complexes in the older literature,28 but just recently several studies revived interest in these two elements that were long neglected in molecular photophysics and photochemistry. As the most abundant transition metal element in Earth’s crust, iron continuously attracts much attention, but two recent landmark studies29,30 lead to unexpected insight that could revolutionize the field.31 Manganese and vanadium represent two comparatively abundant d-metal elements that only just appeared on the landscape of molecular complexes showing luminescence in solution at room temperature.32−34
Figure 2.
Red: Elements for which luminescent complexes have long been known but important conceptual progress has been made recently. Yellow: A few selected luminescent complexes were known for these elements, and a few more examples have been added recently. Green: New elements on the landscape of emissive molecular complexes in solution at room temperature. Gray: Many luminescent ZnII complexes are known, but they typically feature ligand-centered emission. White: Elements for which room temperature emission in solution still remains a challenge and no recent reports have appeared. Spin states and appertaining oxidation states of so far known examples of luminescent molecular complexes in solution at room temperature are listed below the individual elements.
The focus of our Perspective is on the recent conceptual key discoveries in the field of molecular complexes with first-row transition metals that luminesce in solution at room temperature. When a compound emits under these conditions, this typically implies excited-state lifetimes that are sufficiently long for diffusion-controlled bimolecular reactions; hence, these compounds can in principle become suitable for photochemical applications that go far beyond simple lighting, and we then call those complexes “photoactive”.23 Of particular interest here is the direct involvement of the metal in shaping the photophysical and photochemical properties, whereas complexes in which IL excitations play a dominant role (such as, for example, in many zinc(II) compounds)35 are not considered. Our Perspective is structured into four core sections for the four main types of photoactive excited states (LLCT, MLCT, LMCT, and MC) that are relevant for the various 3dn-electron configurations (n = 2–10). Based on the conceptual insights gained from this survey, we then give a brief outlook into the possible future of luminescent base metal complexes.
2. LLCT Emitters: 3d10 (CuI) Complexes
In the 3d10 electron configuration, there are no energetically low-lying MC states, and therefore, emissive MLCT and LLCT excited states can be installed comparatively easily. Homoleptic bis(α-diimine)copper(I) as well as heteroleptic variants combining α-diimine with diphosphine ligands have attracted much attention in the past,19,36 but recent work demonstrated that such four-coordinate tetrahedral CuI MLCT luminophores are outperformed by two-coordinate linear CuI LLCT emitters.37 The combination of unconventional N-heterocyclic carbenes with N-bound amide ligands leads to charge-neutral CuI compounds featuring luminescence quantum yields approaching 100% in the solid state and in solution at room temperature (Figure 3a).38,39 The (anionic) amide ligands are strong π-electron donors, whereas cyclic (alkyl)(amino)carbene (CAAC) ligands (or related unconventional N-heterocyclic carbenes) act as π-acceptors, leading to luminescence from an LLCT excited state. This is a key conceptual difference to traditional four-coordinate CuI complexes, which have photoactive MLCT states that undergo Jahn–Teller distortion with a consequent increase of nonradiative decay rates.40 In the new linear compounds, the metal remains of key importance, as it mediates significant electronic coupling between the donor and acceptor ligands through its d-orbitals (Figure 3b) and furthermore facilitates intersystem crossing (ISC) between singlet and triplet states. As long as both ligands are coplanar (such as the case in the S0 and T1 states of suitably designed complexes), the LLCT is strongly allowed both in absorption and emission,38 meaning that extinction coefficients are sizable (4000–6000 M–1 cm–1) despite considerable donor–acceptor distances (ca. 3.7 Å) and radiative decay rates (kr) are comparatively high (ca. 105–106 s–1). Nonradiative relaxation is decelerated by steric encumbrance on the carbene ligands (Figure 3a), in part because this prevents undesired bending (Renner–Teller) distortions (Figure 3c).41 The limited metal character associated with the photoactive excited state(s) is beneficial for minimization of the Renner–Teller distortion.39
Figure 3.

(a) Linear molecular structure of two-coordinate CAAC-CuI-amido complexes (at the top) and structures of exemplary CAAC and amido ligands.38 (b) Key frontier orbitals in CAAC-CuI-amido complexes and direction of charge transfer upon LLCT excitation.38 (c) Renner–Teller distortion upon MLCT excitation of linear d10 complexes.41 (d) Energy-level diagram illustrating emission processes of CAAC-CuI-amido complexes. (e) Molecular structures of linear CuI complexes based on cyclic mono- or diamidocarbene ligands.39 (f) Molecular structures of some exemplary CAArC-CuI complexes.45 R′ = H, CN. R″ = H, CN. Ra = CH3, Ph. Rb = H, CH3. CNCz, when R′ = CN and R″ = H.
Since the HOMO and the LUMO are spatially well separated, the singlet–triplet energy gap (ΔES-T) of the emissive LLCT state can become small enough such that S1 can be repopulated from T1 via reverse intersystem crossing (RISC), causing a situation reminiscent of thermally activated delayed fluorescence (TADF), though neither the singlet nor the triplet is spin-pure in these CuI complexes (Figure 3d).38,39 Thus, ΔES-T remains small while kr of the luminescence process is comparatively large, two properties which are usually considered mutually exclusive but highly desirable for TADF emitters. Consequently, light-emitting diodes with outstanding quantum efficiencies become possible.38,42
Variations of the donor and acceptor ligands of the linear CuI complexes permit emission color tuning over the entire visible spectral range (Table 1), for example, when combining cyclic monoamidocarbene (MAC*) or cyclic diamidocarbene (DAC*) ligands with carbazolates (Figure 3e).39 The carbonyl groups of these carbenes enhance their π-acceptor properties, and their bulky 2,6-diisopropylphenyl (dipp) substituents suppress the formation of exciplexes with solvent molecules; one of the most common nonradiative deactivation pathways for traditional CuI complexes.40,43,44 Cyclic (amino)(aryl)carbenes (CAArCs) have a more extended π-system (Figure 3f) and are more electrophilic than their alkyl counterparts (CAACs), leading to a substantial red-shift of the lowest-energy UV–vis absorption bands and providing access to deep red and near-infrared (NIR) luminophores.45 High radiative rate constants and small singlet–triplet energy gaps remain accessible, making this a new subclass of compounds with unusually low triplet energies and promising TADF properties.
Table 1. Complexes Emitting from Charge-Transfer Excited States and Some of Their Key Photophysical Properties in Deaerated Solution at Room Temperature.
| compd | electron configuration | solvent | λem [nm] | τ0 | φ [%] | type of emission | ref |
|---|---|---|---|---|---|---|---|
| [Cu(MAC*)(CN2Cz)] | d10 | 2-MeTHF | 448 | 2.3 μs | 24 | LLCT | (39) |
| [Cu(MAC*)(CNCz)] | d10 | 2-MeTHF | 492 | 1.2 μs | ∼100 | LLCT | (39) |
| [Cu(MAC*)(Cz)] | d10 | 2-MeTHF | 542 | 1.1 μs | 55 | LLCT | (39) |
| [Cu(DAC*)(CNCz)] | d10 | 2-MeTHF | 666 | 52 ns | 2 | LLCT | (39) |
| [Cu(CAAC1)(Cz)] | d10 | 2-MeTHF | 492 | 2.5 μs | ∼100 | LLCT | (38) |
| [Cu(CAAC2)(Cz)] | d10 | 2-MeTHF | 510 | 2.3 μs | 68 | LLCT | (38) |
| [Cu(CAAC1)(OMe2Cz)] | d10 | 2-MeTHF | 558 | 0.28 μs | 25 | LLCT | (38) |
| [Cu(CAAC1)(NPh2)] | d10 | 2-MeTHF | 580 | 0.87 μs | 16 | LLCT | (38) |
| [Cr(LtBu)3] | d6 | THF | 630 | 2.2 ns | 0.001 | MLCT | (46) |
| [Cr(Lpyr)3] | d6 | cyclooctane | 682 | 6.10 nsa | 0.09 | MLCT | (47) |
| [Mn(Lbi)3]+ | d6 | CH3CN | 485 | 0.74 nsb | 0.05 | MLCT | (32) |
| [Mn(Ltri)2]+ | d6 | CH3CN | 525 | 1.73 nsc | 0.03 | MLCT | (32) |
| [Ni(BuImp)(CH3CN)](PF6) | d8 | solid state | 534d | (48) | |||
| [Ni(dpb)(Cz)] | d8 | solid state | 600d | 0.11 μsd,e | IL/MLCT | (49) | |
| [Cp*2ScCl] | d0 | isooctane/methylcyclohexane (1:1) | 520 | 2.0 μs | 0.01 | LMCT | (50) |
| [Fe(btz)3]3+ | d5 | CH3CN | 600 | 0.1 ns | 0.03 | LMCT | (29) |
| [Fe(PhB(MeIm)3)2]+ | d5 | CH3CN | 655 | 2.0 ns | 2.1 | LMCT | (30) |
| [Fe(ImP)2]+ | d5 | CH3CN | 450 | 4.2 nsf | MLCT | (51) | |
| 675 | 0.22 ns | <1 | LMCT | ||||
| [Co(dgpy)2]3+ | d6 | CH3CN | 440 | 5.07 ns | 0.7 | LMCT | (52) |
| [Co(dgpz)2]3+ | d6 | CH3CN | 412 | 6.55 nsg | 0.4 | LMCT | (52) |
Weighted average from biexponential decay fit (4.05 ns (48%), 8.00 ns (52%)).
Weighted average from a triexponential decay fit (0.374 ns (79.2%), 1.84 ns (19.3%), 5.85 ns (1.5%)).
Weighted average from a triexponential decay fit (0.635 ns (56.4%), 2.07 ns (33.6%), 6.74 ns (10.0%)).
Data obtained in the solid state.
Measured at 77 K. The emission maximum at 77 K is at 468 nm.
Average value, see original reference for details.
Weighted average from a biexponential decay fit (3.21 ns (39%), 8.69 ns (61%)).
Traditional four-coordinate CuI complexes25,53 as well as polynuclear CuI compounds54−56 continue to receive significant attention,57−62 yet much of this work has been reviewed and will not be considered further here.15,19,36,63
3. MLCT Emitters: 3d6 (MnI and Cr0) Complexes and a 3d8 (NiII) Compound
Metal complexes with the 4d6 and 5d6 valence electron configurations, in particular with RuII, ReI, OsII, and IrIII, are among the most widely employed MLCT emitters.1,2,64 The idea of obtaining analogous 3MLCT luminescence from 3d6 complexes is attractive, among other things because the above-mentioned metals are all very precious and rare. Most efforts until now have focused on iron, but no FeII complexes emitting from an MLCT excited state under steady-state irradiation at room temperature have been reported to date.65 The key problem is that the more contracted 3d-orbitals of FeII experience a substantially weaker ligand field than the 4d-orbitals of RuII (Figure 1a),3 and in classical octahedral polypyridine complexes the metal-centered 3T1g and 5T2g excited states (Figure 4a) are energetically below the lowest MLCT state. Consequently, relaxation from the MLCT to these MC states is typically ultrafast (Figure 4b).66,67 The MC states can indeed be exploited for photoredox reactions though they are nonemissive.68−71 Recently, there has been important progress in elongating the lifetimes of dark MLCT states, as well as in understanding their population and deactivation pathways.72−76 Much of this work has been reviewed,65,77−79 and since these compounds are nonluminescent, they are not considered further here.
Figure 4.
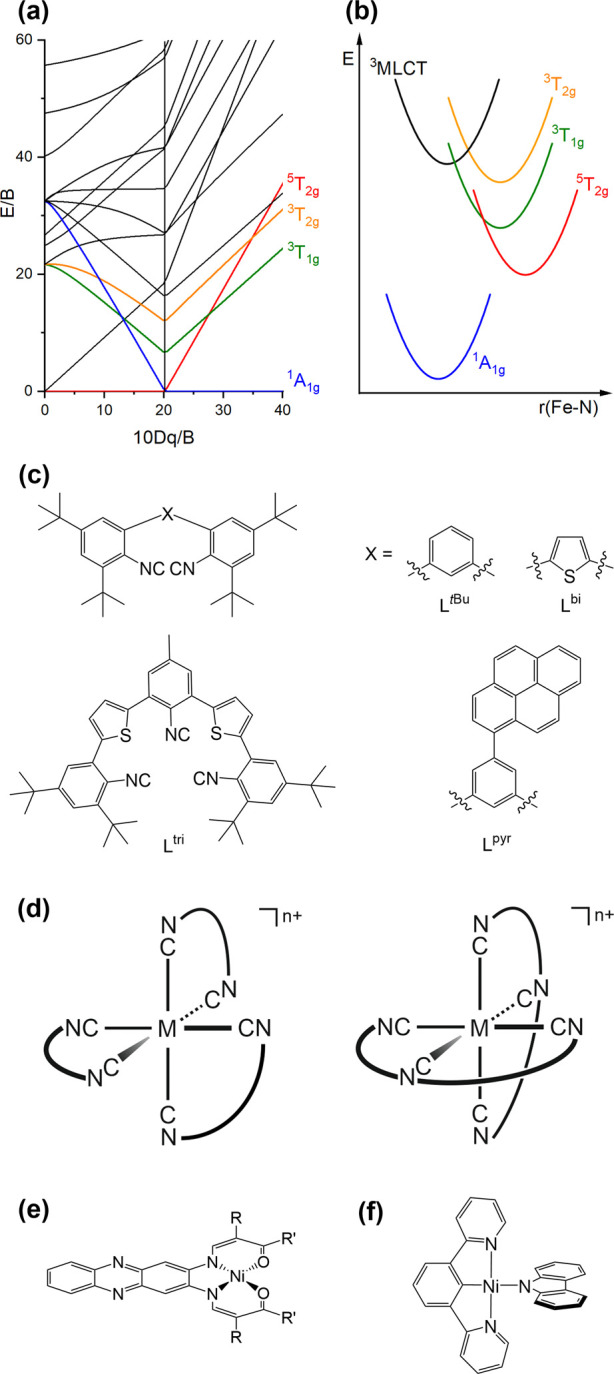
(a) Tanabe–Sugano diagram for the d6-electron configuration in an octahedral ligand field. (b) Single configurational coordinate diagram involving some key electronic states in [Fe(bpy)3]2+.66 (c) Molecular structures of the isocyanide ligands LtBu, Lbi, Lpyr, and Ltri. (d) Generic structures of homoleptic tris(diisocyanide) and bis(triisocyanide) complexes. M = Cr0, n = 0; MnI, n = 1.32,46,47 (e) Molecular structures of NiII complexes showing luminescence that strongly resembles ligand fluorescence (R = COOEt, COOMe; R′ = Me, CF3).80 (f) Molecular structure of [NiII(dpb)(Cz)].49
Following earlier work on W0 arylisocyanides,81,82 we found that chelating diisocyanide ligands give access to luminescent Mo0 complexes.83−85 This chemistry was further extendable to the first row of transition metals and resulted in a Cr0 complex (LtBu ligand in Figure 4c, complex structure in Figure 4d) that emitted from a 3MLCT state in solution at room temperature.46 The emission quantum yield of [Cr(LtBu)3] is very low (ca. 10–5), but the MLCT lifetime of 2.2 ns in deaerated THF at 20 °C is remarkable (Table 1). The sterically demanding tert-butyl substituents in ortho-position to the isocyanide groups protect the metal center from the chemical environment and rigidify the overall complex. Such shielding and rigidification strategies are generally quite successful for improvement of chemical robustness and for the deceleration of nonradiative excited-state relaxation processes in photoactive metal complexes made from Earth-abundant metals.84,86,87 Early studies reported on peculiar dual MLCT emission from several [Cr(α-diimine)(CO)4] complexes in benzene at room temperature.88,89 It is not a priori obvious why two MLCT states, which are energetically relatively close and distorted to different extents relative to each other, can both be emissive.90 Furthermore, photoinduced CO-dissociation can be an important excited-state deactivation pathway in this compound class.91 Be that as it may, the [Cr(LtBu)3] complex is a comparatively clear-cut 3d6 MLCT emitter under continuous-wave irradiation at room temperature.
Attachment of pyrene moieties in the ligand backbone of the diisocyanide ligands in [Cr(LtBu)3] afforded the complex [Cr(Lpyr)3] (Lpyr ligand in Figure 4c, complex structure in 4d).47 [Cr(Lpyr)3] showed an MLCT lifetime of 6.10 ns and an accompanying luminescence quantum yield of 0.09% in deaerated cyclooctane at 20 °C. The largely improved photophysical properties of [Cr(Lpyr)3] relative to those of [Cr(LtBu)3] are due to the delocalization effect.92−94 The delocalization effect is a well-known strategy to improve photophysical properties of ruthenium(II) polypyridines, and [Cr(Lpyr)3] is the first 3d6 emitter to benefit from this concept. Both the luminescence quantum yield and the MLCT lifetime of [Cr(Lpyr)3] showed an unusual bell-shaped solvent dependency, indicative of two counteracting effects controlling the MLCT deactivation giving rise to optimal emissive conditions in cyclooctane. The two effects were identified as predominantly deactivation either through an energetically nearby lying MC excited state in the most apolar solvents such as n-hexane or alternatively via direct nonradiative relaxation to the ground state in accordance with the energy gap law in more polar solvents such as 1,4-dioxane. A tetracarbonyl complex of Cr0 with a bidentate pyridyl-mesoionic carbonyl ligand was recently reported to show near-infrared-II emission, albeit exclusively in the solid state.95
Recently, chelating isocyanide ligands enabled the synthesis and characterization of the first luminescent MnI complexes.32 Isoelectronic with FeII and Cr0, they are 3d6 MLCT emitters, but the higher oxidation state with respect to Cr0 makes the MnI complexes more air-stable. The [Mn(Lbi)3]+ and [Mn(Ltri)2]+ complexes (Lbi and Ltri ligands in Figure 4c, complex structures in 4d) can be considered first-row analogues of the well-known [Ru(bpy)3]2+ and [Ru(tpy)2]2+ compounds, and they luminesce with quantum yields of 0.03–0.05% in acetonitrile at room temperature (Table 1). The strong ligand field created by the π-acceptor isocyanide ligands shifts the detrimental 3T1g and 5T2g MC states (Figure 4a) to sufficiently high energies, such that blue MLCT emission can become competitive with nonradiative relaxation, though the comparatively low quantum yields indicate that dissipation of excitation energy by molecular vibrations remains the major relaxation pathway in this compound class, at least until now. The luminescence behavior of these tris(diisocyanide) and bis(triisocyanide) manganese(I) compounds stands in contrast to long-known MnI tricarbonyl diimines, which undergo efficient CO ligand release (due to the rapid population of the above-mentioned dissociative MC states), but no emission is accompanied by this process.4,96−98 The MLCT lifetimes of [Mn(Lbi)3]+ and [Mn(Ltri)2]+ are on the order of 1–2 ns in CH3CN at 20 °C, and this is long enough for bimolecular electron and (triplet–triplet) energy transfer reactions.32 Thus, the manganese(I) isocyanide compounds exhibit the same type of photoreactivity as RuII polypyridines and cyclometalated IrIII complexes. Though manganese is not as abundant as iron, its natural abundance in Earth’s crust exceeds that of copper by a factor 18,99 which seems noteworthy given the long-standing interest of the photophysics and photochemistry community in copper.
The photophysics and the photochemistry of molecular nickel(II) complexes recently gained considerable attention in the context of light-driven (or at least light-initiated) cross coupling catalysis,100−104 though this typically involves nonemissive NiII compounds with energetically low-lying MC states that deactivate nonradiatively.105 A recent study reported on unexpectedly intense photoluminescence from square-planar NiII compounds, where it seems that the emission is essentially due to a fluorescent ligand (Figure 4e).80 Another very recent study had a clear focus on installing emissive charge-transfer states in NiII complexes and succeeded in identifying some key design principles for obtaining square-planar complexes that luminesce in the solid state at 298 K or in frozen glasses at 77 K.49 The carbazolylnickel(II) complex in Figure 4f has a lowest-lying UV–vis absorption band attributable to a mixed IL (localized on the cyclometalating terdentate ligand) and MLCT excitation according to TD-DFT calculations. Weak luminescence at room temperature is observed, and at 77 K its lifetime is 0.11 μs, suggesting an emissive triplet state. In the 77 K emission spectrum vibrational progressions in a skeletal vibration mode of the ligand framework (ca. 1400 cm–1) point to a significant IL contribution. Nevertheless, it seems clear that the use of strong-field terdentate ligands is a promising route toward NiII complexes exhibiting MLCT luminescence.48,49 So far, this has only been achievable with tetrahedral Ni0 complexes,106,107 which exhibit comparable photophysical properties to four-coordinate CuI diimine complexes; however, tetrahedral Ni0 compounds tend to be very air-sensitive. Other new developments in NiII photochemistry involve the use of macrocyclic ligands, but no emission has been reported so far.108,109
4. LMCT Emitters: 3d0 (ScIII), 3d3 (MnIV), 3d5 (FeIII), and 3d6 (CoIII) Complexes
Energetically low-lying LMCT states call for electron-rich ligands and electron-deficient metals. The most promising valence electron configuration for achieving long-lived LMCT states is d0, because in this specific case there are no MC states through which the LMCT excited state can deactivate nonradiatively. Consequently, group 3 metals in the +III oxidation state and group 4 metals in the +IV oxidation state are very promising candidates. Early on, [Cp*2ScCl] was found to emit in fluid solution at room temperature with a luminescence lifetime of 2.0 μs and the emissive excited state was assigned as LMCT from the p-Cl nonbonding lone pair to the Sc3+ ion.50,110 Many TiIV based metallocenes with the general formula Cp2TiX2 also exhibit LMCT luminescence,111 albeit typically only in the solid state at 77 K.112 More recently, this compound class has become of interest for photoredox catalysis.113,114 A TiIV carbene complex seemed to be nonemissive in solution at room temperatures while its ZrIV and HfIV analogues emitted under these conditions.115 Recent work on related work focused much on ZrIV (and comparatively little on TiIV), suggesting that room temperature emission is much more tricky to obtain for TiIV than for ZrIV.87,116−118
The electron-donating tris(carbene)borate ligand PhB(MeIm)3 stabilizes manganese in its +IV oxidation state,119 and the homoleptic [Mn(PhB(MeIm)3)2]2+ complex (Figure 5a, M = MnIV, n = 2) shows ruby-like red emission in the solid state at room temperature, similar to many isoelectronic CrIII compounds. Unexpectedly, this compound furthermore shows green luminescence from an LMCT excited state (up to room temperature), even though several MC states of the d3 valence electron configuration are energetically lower. The green emission spectrum mirrors a corresponding LMCT absorption band with a high oscillator strength, indicative of a spin-allowed transition, which implies a high radiative decay rate constant that could be crucially important to make luminescence competitive with nonradiative relaxation. An emission lifetime of 50 ns at 85 K (where nonradiative relaxation should be largely suppressed) is compatible with this interpretation, and the vibrational fine structure in the emission spectrum (featuring progressions in the typical range of ligand breathing modes) further supports the LMCT assignment. This MnIV study was the first in a series of recent reports on unexpected LMCT emissions from 3d-metal complexes, in particular among the FeIII compounds considered in the following.
Figure 5.
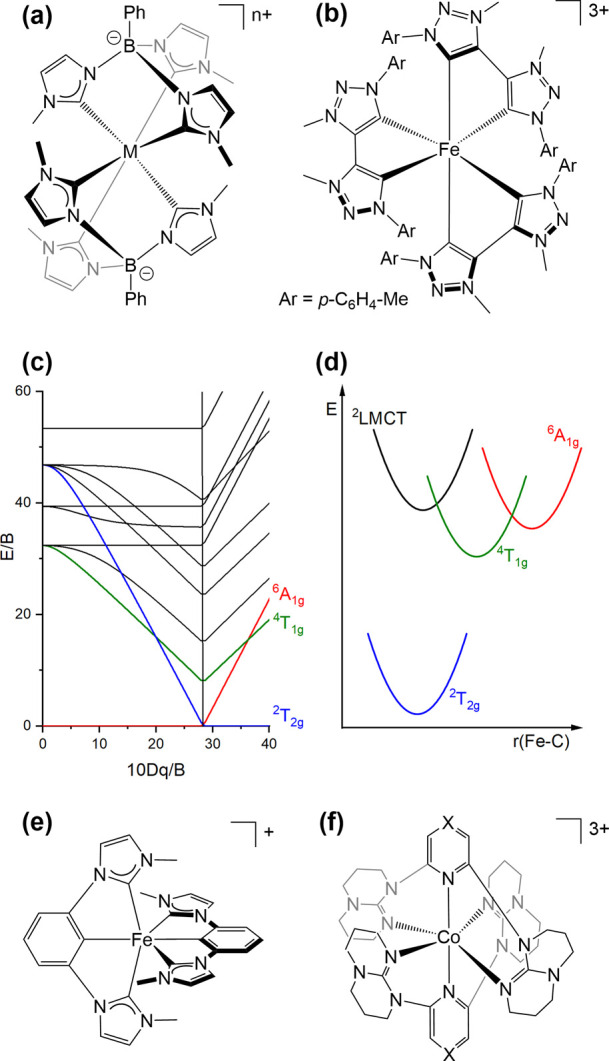
Molecular structures of (a) [M(PhB(MeIm)3)2]n+ (with M = MnIV, n = 2; M = FeIII, n = 1; M = CoIII, n = 1)30,119,149 and (b) [Fe(btz)3]3+.29 (c) Tanabe–Sugano diagram for the d5 electron configuration in an octahedral ligand field. (d) Single configurational coordinate diagram with key electronic states in [Fe(btz)3]3+.29 Molecular structures of (e) [Fe(Imp)2]+ and (f) [Co(dgpy)2]3+ (X = CH) as well as [Co(dgpz)2]3+ (X = N).51,52
N-Heterocyclic carbene (NHC) ligands caught the attention of the iron photophysics research community some time ago,77,78,120,121 yet it was mostly the recent use of a mesoionic carbene (MIC) ligand (Figure 5b) that led to a conceptual breakthrough. While NHCs are merely strong σ-donors, MICs combine good σ-donor with strong π-acceptor properties, causing very strong ligand fields in which nonradiatively deactivating MC states are energetically destabilized and emissive charge-transfer excited states can instead become accessible.65 The original aim with the btz ligand was likely to obtain an FeII compound with a long-lived MLCT state,122 but the resulting homoleptic complex underwent facile oxidation to FeIII (Figure 5b). Strikingly, the resulting [Fe(btz)3]3+ complex turned out to be photoluminescent,29 which was not to be expected for a low-spin d5 complex. Given the presence of energetically low-lying MC states in this valence electron configuration, the observation of LMCT emission at room temperature is very surprising at first glance but can be rationalized on the basis of detailed spectroscopic and computational studies: First of all, the LMCT emission is spin-allowed and therefore has a high radiative rate constant. Second, the small Stokes shift between LMCT absorption and emission bands (0.15 eV) indicates that the LMCT state is only weakly distorted with respect to the ground state (Figure 5d), which limits the efficiency of direct nonradiative relaxation. Third, although low-lying MC states are considerably distorted with respect to the LMCT, the activation barriers for internal conversion are sizable (4–22 kJ/mol), and consequently nonradiative relaxation via the MC states is not ultrafast, but instead permits LMCT luminescence with a lifetime of 100 ps and a quantum yield of 3 × 10–4 at room temperature (Table 1).
In a follow-up study, the energies of the low-lying 4MC and 6MC states were further increased by creating an even stronger ligand field with the more σ-donating (anionic) PhB(MeIm)3 ligand (Figure 5a, M = FeIII, n = 1).30 According to an Arrhenius-type analysis, the crossing frequency from the emissive 2LMCT to the dark 4MC state in the [Fe(PhB(MeIm)3)2]+ complex is reduced by at least an order of magnitude compared to [Fe(btz)3]3+, which is likely of key importance for the observable improved photophysical properties. Though a luminescence quantum yield of 2% and an excited-state lifetime of 2.0 ns may seem low compared to precious metal emitters,123 these figures of merit are spectacular for an open-shell first-row transition metal complex (Table 1).
The spin-allowed nature of the relevant LMCT transition is instrumental to make radiative relaxation competitive with nonradiative excited-state deactivation in these FeIII compounds, but at the same time this has a detrimental effect on the photoreactivity of the luminescent 2LMCT state: Though its reductive quenching by sacrificial electron donors is fast, the photoreduction efficiencies are very low (5%), because photogenerated electron–hole pairs rapidly recombine before they are spatially separated into longer-lived reduction and oxidation products.124 This so-called geminate recombination within the “solvent cage” is a well-known phenomenon, which becomes particularly cumbersome when the electron–hole recombination is spin-allowed (i. e., associated with no net spin change).125 Unfortunately, this is the case for electron transfer from tertiary amine donors to the 2LMCT-excited FeIII complex, because this yields a low-spin FeII complex (a singlet) and an oxidized amine (a doublet), which can undergo spin-allowed reverse electron transfer to form the FeIII complex in its doublet ground state and the neutral amine in its singlet state.124 In RuII complexes, photoreduction efficiencies are in the range of 5–60%, indicating that this is a general problem, which does however diminish when the electron–hole recombination becomes spin-forbidden.125
The [Fe(PhB(MeIm)3)2]+ complex furthermore undergoes “self-quenching” in sufficiently concentrated acetonitrile solution (70 mM). In this reaction, the 2LMCT-excited FeIII compound reacts with one equivalent of itself in the electronic ground state, to afford the FeII and FeIV forms of this complex.126 This is likely the first clear-cut case of photoinduced symmetry-breaking charge separation in a transition metal complex, and it became possible due to the fact that the [Fe(PhB(MeIm)3)2]+ complex features three different metal-centered redox processes (without interfering ligand-based redox events). As a consequence, the symmetry-breaking charge separation reaction becomes strongly exergonic, and although geminate in-cage charge recombination is rapid, the FeII and FeIV photoproducts form in 4% yield.
The [Fe(ImP)2]+ complex (Figure 5e) seems to show even more unusual luminescence behavior than the only other two FeIII emitters known so far.51 UV excitation of [Fe(ImP)2]+ in CH3CN is thought to populate a “hot” 2MLCT excited state, from which a branching into the relaxed 2MLCT and a lower-lying 2LMCT states occurs. Strikingly, both of these states seem to be luminescent (Table 1), one at 450 nm and the other at 675 nm, with lifetimes of 4.2 and 0.2 ns, respectively.
The photophysical properties of the [Co(dgpy)2]3+ and [Co(dgpz)2]3+ complexes (Figure 5f) are comparatively easy to rationalize.52 Both complexes emit blue-green with nanosecond lifetime in acetonitrile at room temperature and luminescence quantum yields of 0.4–0.7% (Table 1). The combination of relatively electron-rich ligands with a metal trication leads to a charge transfer reversal with respect to classical FeII polypyridines,127,128 and the emissive excited state has 3LMCT character. The large bite angle of the dgpy and dgpz ligands (in particular when compared to tpy as a prototypical terdentate ligand in photoactive metal complexes)129 likely plays a key role in establishing a sufficiently strong ligand field to shift MC states to high energies and to decelerate 3LMCT deactivation via such MC states. In this sense, the molecular design of [Co(dgpy)2]3+ and [Co(dgpz)2]3+ takes some of the lessons learned about the importance of bite angle optimization in FeII and CrIII complexes into account.130,131
5. MC Emitters: 3d2 (VIII), 3d3 (CrIII, MnIV), and 3d6 (CoIII) Complexes
Spin-flip transitions within a given dn electron configuration (n = 2–8) typically give rise to excited states that are only weakly distorted relative to the ground state, because they usually do not lead to significant changes of the metal–ligand bonding situation, and this is beneficial to decelerate nonradiative deactivation. For octahedrally coordinated d2 and d3 complexes such spin-flips are expected as lowest excited states in sufficiently strong ligand fields (Figure 6a/b). Doped into inorganic host materials, d2 and d3 ions have long been known to emit from the respective spin-flip excited states,132 and some molecular CrIII complexes were shown to luminesce in solution at room temperature, though until recently mostly with very low quantum yields.133
Figure 6.
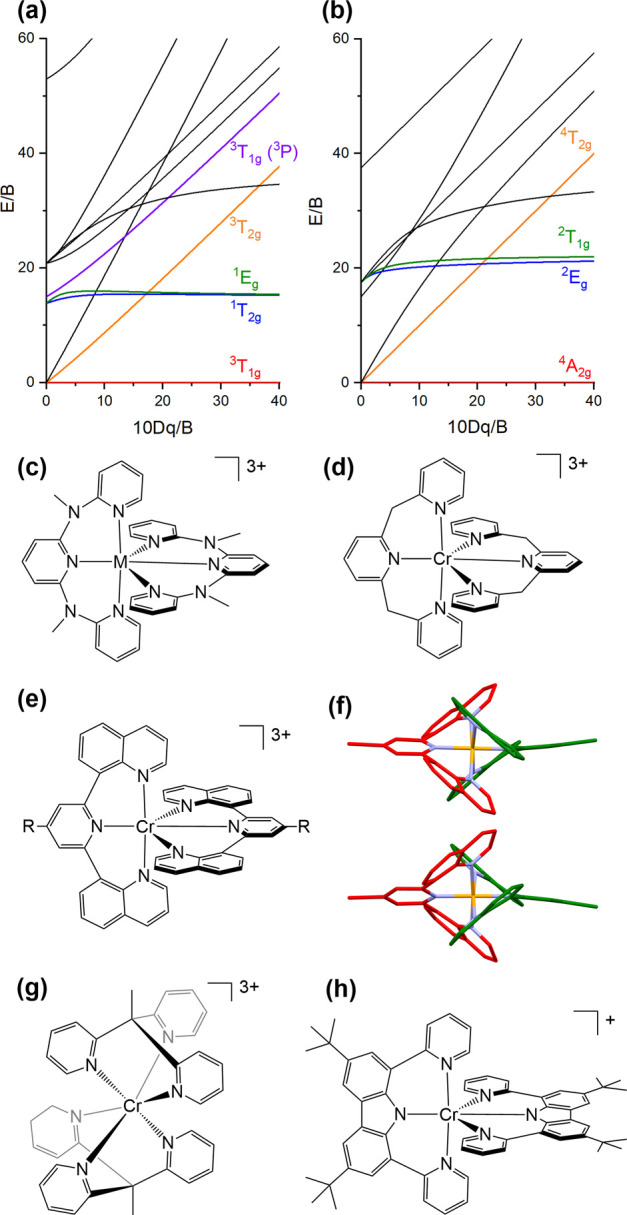
Tanabe–Sugano diagrams for the d2 (a) and the d3 (b) electron configurations in an octahedral ligand field. (c) Molecular structure of [M(ddpd)2]3+; M = CrIII or VIII.34,131,134−137,139 (d) Molecular structure of [Cr(bpmp)2]3+.140 (e) Molecular structure of [Cr(Rdqp)2]3+, where R = H, OMe, Br or C≡CH.142,144,145 (f) Enantiomeric pair PP-[Cr(Brdqp)2]3+ (upper) and MM-[Cr(Brdqp)2]3+ (lower). The CrIII ion is orange, N atoms are blue, and the two Brdqp ligands are in red and green, respectively.145 (g) Molecular structure of [Cr(tpe)2]3+.147 (h) Molecular structure of [Cr(dpc)2]+.148
The field of luminescent CrIII complexes recently experienced a strong revival, initiated by the discovery of the exceptional photophysical properties of [Cr(ddpd)2]3+ (Figure 6c, M = Cr).131 Compared to other tridentate ligands such as tpy, the ddpd ligand offers a larger bite angle that causes a stronger ligand field as a result of better overlap between metal 3d- and ligand-orbitals. This is helpful, because it widens the energy gap between the emissive 2Eg and the higher-lying 4T2 state (blue and orange in Figure 6b, respectively), leading to a smaller thermal population of the latter via reverse intersystem crossing and limiting nonradiative relaxation from this strongly distorted excited state. Further optimizations were possible by ligand deuteration, which decelerates unwanted nonradiative processes and thereby leads to a luminescence quantum yield of 30% paired with a 2E lifetime of 2.3 ms in solution at room temperature (Table 2).134 Applications in temperature135 and pressure sensing were possible,136 and the singlet oxygen sensitization capability of [Cr(ddpd)2]3+ was exploited for photocatalysis.137 Much of this work has been reviewed.22,138,139
Table 2. Complexes Emitting from Metal-Centered Excited States and Some of Their Key Photophysical Properties in Deaerated Solution at Room Temperature.
| compd | electron configuration | solvent | λem [nm] | τ0 | φ [%] | type of emission | ref |
|---|---|---|---|---|---|---|---|
| mer-[V(ddpd)2]3+ | d2 | CD3CN | 396 | 5.4 nsa | 2.1 | MC/LMCT | (34) |
| 1109/1123 | 1.35 μsb | 0.00018 | MC | (34) | |||
| [VCl3(ddpd)] | d2 | solid state | 1102/1219/1256c | 0.5 μsc | MC | (33) | |
| [Cr(CN)6]3– | d3 | H2O | d | 0.12 μs | MC | (146) | |
| fac-[Cr(tpe)2]3+ | d3 | D2O/DClO4 | 748 | 4.5 ms | 8.2 | MC | (147) |
| mer-[Cr([D9]-ddpd)2]3+ | d3 | CD3CN | 738/778 | 2.3 ms | 30.1 | MC | (134) |
| mer-[Cr(bpmp)2]3+ | d3 | D2O/DClO4 | 709 | 1.8 ms | 19.6 | MC | (140) |
| [Cr(dpq)2]3+ | d3 | H2O | 724/747 | 1.2 ms | 5.2 | MC | (144) |
| [Cr(dqp)(tpy)]3+ | d3 | CH3CN | 724/746 | 578 μs | 0.2 | MC | (142) |
| [Cr(ddpd)(dqp)]3+ | d3 | CH3CN | 728/762 | 642 μs | 6.0 | MC | (142) |
| [Cr(dqp)(OMe dqp)]3+ | d3 | CH3CN | 725/752 | 855 μs | 6.5 | MC | (142) |
| [Cr(OMedqp)2]3+ | d3 | H2O | 720/756 | 1.35 ms | 17 | MC | (145) |
| [Cr(dpc)2]+ | d3 | CH3CNe | 1067e | 2.0 μsf | <0.00089e | MC | (148) |
| [Mn(PhB(MeIm)3)2](OTf)2 | d3 | solid state | 815g | 1.5 μsg | MC | (119) | |
| 600 | 50 ns | LMCT | |||||
| [Co(CN)6]3– | d6 | H2O | 726h | 2.6 ns | MC | (28), (146) | |
| [Co(PhB(MeIm)3)2]+ | d6 | CH3CN | 690 | 0.82 μs | 0.01 | MC | (149) |
Weighted average from a biexponential decay fit (3.2 ns (56%), 8.2 ns (44%)).
Weighted average from a biexponential decay fit (790 ns (93%), 8800 ns (7%)), measured at 77 K in nBuCN.
Data obtained in the solid state at 298 K.
No emission maximum was reported in the original work.
Measured at 77 K.
Weighted average from a biexponential decay fit (1.4 μs (88%), 6.3 μs (12%), measured at 77 K.
Data obtained in the solid state at 77 K.
Measured in an ethylene glycol/water mixture (60/40 vol %) at 165 K.
In a very recent study, the NMe bridging units of ddpd were replaced by −CH2– groups (Figure 6d), which induces a blue-shift of the emission band maximum from 775 nm in [Cr(ddpd)2]3+ to 709 nm in the new [Cr(bpmp)2]3+ complex.140 As the 2E energy depends essentially on the repulsion between metal d-electrons, this experimental observation suggests that the extent of metal–ligand bond covalence is different in [Cr(bpmp)2]3+ than in [Cr(ddpd)2]3+. Curiously, analysis of the spectroscopic properties of [Cr(bpmp)2]3+ in terms of ligand field theory yielded a Racah B parameter of 1003 cm–1, which is higher than the B-value for the free CrIII ion in the gas phase (918 cm–1) and substantially larger than the Racah B parameter determined for [Cr(dqp)2]3+ (656 cm–1).141,142 The new complex furthermore has a high luminescence quantum yield (20%), and it was noted that [Cr(bpmp)2]3+ surpasses [Ru(bpy)3]2+, though spin-flip emission from a very weakly distorted MC excited state and luminescence from an MLCT state are two very different things. The comparison to an europium(III)-based emitter also made in the respective study seems more appropriate, because that compound’s emissive MC state is likely more similarly weakly distorted as the luminescent (spin-flip) MC state of [Cr(bpmp)2]3+.
The dqp ligand was known from previous work with RuII143 and recently turned out to be applicable in CrIII coordination chemistry as well.142 The development of a new synthetic strategy gave access to heteroleptic CrIII compounds, in which ddpd, dpq, and tpy (as well as substituted variants thereof) were combined (Table 2). In the homoleptic [Cr(dqp)2]3+ complex (Figure 6e), the two meridionally coordinating dqp ligands wrap around each other to result in a helical conformation around a pseudo-C2 axis, stabilized by intramolecular π-stacking interactions (Figure 6f).144 The complexation reaction leads exclusively to the PP-[Cr(dqp)2]3+ and MM-[Cr(dqp)2]3+ pair of enantiomers, while the PM diastereomer is not formed. No racemization was observed over several months and the absolute configurations of the two enantiomers were determined. PP-[Cr(dqp)2]3+ and MM-[Cr(dqp)2]3+ show circularly polarized luminescence (CPL) with dissymmetry factors (glum) that surpass those for previously reported d-metal complexes by at least an order of magnitude,144 owing to the fact that the two relevant emission spin-flip transitions (2E → 4A2 and 2T1 → 4A2) have magnetic-dipole allowed and electric-dipole forbidden character. For transitions of this type, glum theoretically maximizes to values of −2 or +2 when either pure right- or left-handed polarized light is emitted.145 [Cr(dqp)2]3+ in CH3CN gave |glum| = 0.2 for the 2E emission at 749 nm,144 and nearly equally high values were obtained for three congeners with modified dqp ligands (Table 2).145 The functionalization of dqp with a methoxy-group at the central pyridine ring improved the chiral resolution and allowed for a larger scale preparation of the [Cr(OMedqp)2]3+ luminophore with an emission quantum yield of 17% and an excited-state lifetime of up to 1.35 ms at room temperature in aqueous solution.145 Similar CPL studies were performed with [Cr(ddpd)2]3+, for which the chiral resolution of PP- and MM-enantiomers proved more difficult, and a somewhat lower glum value of −0.093 was found, yet this performance factor remains competitive with many lanthanoid-based CPL emitters.150
The ddpd and dqp ligands preferentially coordinate CrIII in a meridional fashion (though facially coordinated dqp complexes of RuII are known);151 hence, no inversion center is present in these complexes and the parity selection rule for d–d transitions does not hold true in a strict sense. In the [Cr(tpe)2]3+ complex (Figure 6g), the tridentate ligand is forced to coordinate facially, which installs an inversion center and renders the 2Eg → 4A2g luminescence transition Laporte-forbidden.147 The radiative 2E decay rate is therefore only on the order of 20 s–1, similarly low as in centrosymmetric [Cr(CN)6]3–.146 Since nonradiative relaxation pathways remain moderately efficient also in [Cr(tpe)2]3+, the new design principle resulted in an exceptionally long luminescence lifetime of 4.5 ms in aqueous solution at room temperature.
The CrIII polypyridine complexes known until very recently all had their emission band maxima in the rather narrow spectral range between 709 and 778 nm. The [Cr(dpc)2]+ complex (Figure 6h) is the first near-infrared-II emitter in this compound family, featuring an emission band maximum at 1067 nm in frozen CH3CN at 77 K.148 This drastic red-shift seems to be the result of increased metal–ligand bond covalence achievable by the mixed amido and pyridine coordination environment. This design principle of exploiting the nephelauxetic effect, which operates particularly well on intraconfigurational (spin-flip) states, is radically different from the common approach of enhancing the ligand field strength, which is typically applied to modulate the energies of interconfigurational states.
While many molecular CrIII complexes are emissive from their 2E state,152 currently it seems that there exists only one example of an isoelectronic molecular MnIV compound showing the same type of photoluminescence.119 Specifically, the [Mn(PhB(MeIm)3)2](OTf)2 complex (Figure 5a, M = MnIV, n = 2) reported in section 4 shows visible LMCT emission alongside NIR 2E → 4A2 luminescence with a band maximum at 814 nm in the solid state (Table 2). This is significantly red-shifted compared to most of the molecular CrIII complexes and likely reflects a high degree of metal–ligand bond covalence in this MnIV complex.119 Presumably, the higher metal oxidation state leads to a stronger nephelauxetic effect, decreasing the mutual repulsion between d-electrons and lowering the energy of the emissive 2E state.
Taking a leftward step from CrIII in the periodic table, the d3 valence electron configuration is achievable with VII, for example in [V(bpy)3]2+ and [V(phen)3]2+. These compounds have low-lying 2MLCT states that mix with the lowest metal-centered doublet states.153,154 This produces geometric distortion and energetic stabilization, causing efficient nonradiative relaxation. The finding that a 2MLCT state is at lower energy than the corresponding 4MLCT implies that Hund’s rule is not necessarily obeyed in cases with substantial overlap between singly occupied ligand- and metal-centered orbitals. It seems plausible that the use of bpy and phen ligands with electron-donating ligands will elevate the MLCT energies to the extent that 2E emission will become observable.153
As noted above, aside from the d3 valence electron configuration, octahedral d2 complexes are obvious candidates for a spin-flip as lowest-lying excited state (Figure 6a), but until recently no molecular complexes of this type have been shown to emit in solution. The [V(ddpd)2]3+ complex (Figure 6c, M = V) features 1Eg/1T2g → 3T1g emission around 1100 nm with 1.8 × 10–4 % quantum yield in CH3CN at room temperature, representing the first case of NIR-II emission by a 3d-metal complex in fluid solution.34 The second overtone of aromatic C–H vibrations has significant spectral overlap with this emission, yet 17-fold deuteration of the ddpd ligand had no clear effect on the luminescence quantum yield, hence the nonradiative decay path could not be unraveled on this basis. The most striking observation was the appearance of blue fluorescence with a quantum yield of 2.1% upon UV excitation. Higher excited-state emission from MC states had been previously reported for d2 species, but only at cryogenic temperatures.155 In [V(ddpd)2]3+ this emission was tentatively attributed to the 3T1g (3P) → 3T1g transition (purple and red in Figure 6a, respectively) or to 3LMCT transitions.34 Though there is a large energy gap of ca. 2 eV between the two emissive excited states, there are several other excited states in between, and it seems that intersystem crossing from the higher-lying triplet states to the singlets is comparatively inefficient.
The complex [VCl3(dppd)] likewise emits from the 1Eg/1T2g manifold but seemingly only in the solid state.33 Unlike in the homoleptic [V(ddpd)2]3+ congener, excited-state relaxation is strongly susceptible to ligand deuteration, confirming that multiphonon relaxation involving C–H oscillators is a major decay pathway. The large splitting of the electronic ground state, manifesting in the appearance of multiple 1Eg/1T2g → 3T1g emission band maxima, likely further facilitates nonradiative excited-state relaxation by reducing the relevant energy gap. Though vanadium has a very low spin–orbit coupling constant, the rate of intersystem crossing following excitation into singlet states is very high (kISC = 6.7 × 1011 s–1), and trajectory surface hopping simulations rationalized that finding.156 This resonates with a previous study, which found no direct correlation between the magnitude of the spin–orbit coupling constant and the rate for intersystem crossing.157 Complexation of the V3+ ion to the ligands (C6F5)3tren and CNtBu resulted in the trigonal bipyramidal complex [V[(C6F5)3tren](CNtBu)] with a 3A ground state, and the lowest lying excited state is a 1E state due to the strong ligand field.158 The 1E → 3A transition is an intraconfigurational, spin-flip transition, which radiates in the near-infrared, however only in frozen matrices at 77 K. Cocrystallization of the V3+ ion with the diamagnetic analogue Ga3+ ion afforded [V0.023Ga0.977[(C6F5)3tren](CNtBu)], for which single crystals were luminescent at room temperature.
[Co(CN)6]3– is a rare (and long known) example of a 3d6 complex that emits from a strongly distorted MC state in solution at room temperature.28,146 With the PhB(MeIm)3 ligand, it was now possible to create a similarly strong ligand field as with CN– (10 Dq = 38 600 cm–1), making the [Co(PhB(MeIm)3)2]+ complex (Figure 5a, M = CoIII, n = 1) an MC emitter with a microsecond lifetime in CH3CN at room temperature (Table 2).149 Excitation has to occur into charge-transfer bands in the UV, from which the emissive 3T1g state is populated via internal conversion and intersystem crossing. While that 3T1g state is highly problematic for obtaining MLCT emission from FeII complexes (because it depopulates the MLCT and rapidly decays via nonradiative relaxation, Figure 4a/b),66 in the cobalt complex, here this state maintains a sizable energy gap to the ground state even in its relaxed geometry, owing to the strong ligand field.149 The emission is spin-forbidden and therefore associated with a comparatively low radiative rate constant, which makes nonradiative relaxation very competitive despite the large energy gap. Consequently, the emission quantum yield at room temperature is only 0.01%. Since the σ- and π-bonding properties of the PhB(MeIm)3 scorpionate ligand can in principle be tuned, this opens the perspective to further enhance the photophysical properties of CoIII-based 3T1g emitters.
6. Summary and Outlook
Traditional luminophores made from precious d6 or d8 metals such as RuII, OsII, ReI, IrIII, PtII, and AuIII typically function on the basis of MLCT (or the closely related MMLCT) transitions.63,64 Using the less noble element copper in its +I oxidation state with its completely filled d10 subshell, this photophysical behavior was emulated already nearly 40 years ago.25 The absence of low-lying MC states in the d10 valence electron configuration greatly facilitated this early discovery, and nowadays four-coordinate CuI MLCT emitters are a highly developed compound class.19,36,57 With the open-shell 3d6 and 3d8 configurations, emissive MLCT excited states are much harder to install, because nonradiative relaxation via low-lying MC states tends to be ultrafast. Until now, most efforts to obtain 3d6 MLCT emitters have concentrated on FeII complexes, but despite considerable recent progress in enhancing their MLCT lifetimes and in understanding their electronic structures no FeII based MLCT luminophores are yet known.66−68,72−74,77,78,120−122 Very recently, complexes of isoelectronic Cr0 and MnI have been identified as 3d6 MLCT emitters showing analogous photophysical and photochemical behavior as RuII polypyridines or cyclometalated IrIII complexes (Figure 7a).32,46,47,159,160 In parallel, considerable progress has been made toward 3d8 MLCT luminophores based on NiII, though in this case the breakthrough has not yet been made in the sense that room-temperature MLCT emission has not been achievable until now.49,109
Figure 7.

Recent key developments summarized graphically.
A paradigm change occurred with the recent emergence of highly emissive two-coordinate CuI complexes which are LLCT luminophores (Figure 7b),37−39,42,45 and with the discovery of MnIV and FeIII LMCT emitters (Figure 7a).29,30,119 The reversal of the charge transfer direction when going from MLCT to LMCT has direct consequences for possible applications in dye-sensitized solar cells;161 yet as far as the luminescence properties are concerned, LMCT or LLCT emitters seem highly competitive with MLCT luminophores.87,127 This is particularly true for the new class of linear CuI LLCT emitters,37−39,42,45 which represents a conceptual breakthrough in the photophysics of copper(I) after a long period with a dominant focus on tetrahedral CuI MLCT luminophores.36 The FeIII LMCT emitters reported so far exhibit spin-allowed luminescence,29,30,51,124 which is helpful to boost luminescence quantum yields because it makes radiative relaxation kinetically more competitive with nonradiative processes. On the other hand, the spin-allowed character of the photoactive excited state is likely disadvantageous for some photochemical applications, because spin-allowed geminate charge recombination is expected to hamper cage-escape yields,13,124 thereby perhaps limiting the effectiveness of these sensitizers for photoinduced electron transfer.
Analogously to the LMCT emitters made from FeIII and MnIV, new MC luminophores based on CrIII and VIII do not have diamagnetic ground states, which represents another key difference to the traditional d6 and d10 MLCT luminophores. In the case of CrIII this leads to the well-known ruby-like emission that has been optimized and brought to new applications in recent years (Figure 7c),131,134−139,142,144,145,147,148,150,162 whereas in the case of VIII an analogous spin-flip transition has now become emissive in a molecular complex.34 Similarly to the CrIII emitters reported recently, a new CoIII based MC luminophore outperforms an earlier investigated analogue and benefits from new insights concerning the design of coordination environments to maximize ligand field strength and minimize unwanted nonradiative relaxation processes.149 The spin-forbidden nature of the MC emissions of these d3 and d6 compounds is similar to the traditional 3MLCT luminescence in d6 or d10 compounds, though different spin selection rules will apply for energy transfer depending on the exact electron configuration of the reactive excited states.163 This could have important implications in energy transfer catalysis,164 because not all photocatalyst/substrate combinations will represent equally good donor–acceptor pairs in which energy transfer is equally spin-allowed.
One particularly striking new finding is that several of the newly reported first-row transition metal complexes emit from higher excited states. In the FeIII carbene complexes, there are two MC states with different spin multiplicities below the emissive 2LMCT state.29,30 In the MnIV and VIII complexes, it seems possible that their higher excited-state emissions even originate from states that have the same multiplicity as lower-lying (MC) states34,119 and as such would represent exceptions to Kasha’s rule, according to which typically only the lowest electronically excited state of a given multiplicity emits with appreciable quantum yields.165 In the solid state at cryogenic temperatures, such behavior has been known for some first-row transition metals (including Ti2+ and Ni2+ when doped into halide host matrices),166 but for molecular compounds at room temperature this is very uncommon behavior.
Decelerating nonradiative excited-state relaxation remains the most challenging part in the development of new emissive metal complexes, particularly in the first row of transition metals, where the ligand field is typically comparatively weak and low-lying MC states can represent effective deactivation channels (Figure 1b).3,22,23,65,77 There is no single recipe that is uniformly applicable to address this issue in different metals and oxidation states, because each dn valence electron configuration has its own characteristics. Fairly general guidelines include however the design of rigid coordination environments to limit vibrational degrees of freedom,38,39,84,86,87 the enhancement of the ligand field strength to shift MC states to higher energies,73,77,78,120−122,167 the optimization of coordination geometries to minimize splitting of degenerate states into several sublevels and to maximize energy gaps between individual states,130,131,138,162 the installment of an inversion center to influence radiative excited-state decay rates,147 and the introduction of push- and/or pull-character to control the energies of charge-transfer states.73,168 Perhaps less obvious but potentially interesting could be strategies based on optimizing metal–ligand bond covalence to minimize repulsion between spectroscopically relevant d-orbitals148 as well as the exploitation of triplet reservoir and/or delocalization effects47 in first-row transition metal-based compounds with covalently attached organic chromophores.92−94 Ligand deuteration, a well-known strategy in the field of lanthanide luminophores, is now successfully applied to first-row transition metal emitters.134
Aside from the broadened perspectives with regard to the accessible emission types (including LMCT and LLCT in addition to the traditionally explored MLCT and MC transitions), there are very important recent developments beyond the first-row of transition metals. On the one hand, this includes comparatively abundant second- and third-row transition metals (for example Zr,87,116 Mo,83,84,95,160 or W82,95,160,169,170) or f-elements (for example Ce171,172). On the other hand, molecular complexes of abundant main group elements are now attracting increasing attention as luminophores and provide additional insight into how nonradiative relaxation can be tamed.173−175 At the same time, further developments of precious metal-based complexes (for example, Ru,167,176,177 Rh,178,179 Re,180−182 Ir,183−185 or Pt186−188) have significantly advanced the field of inorganic photophysics and photochemistry. Regardless of whether complexes of d-, f-, or main group elements are considered, the interplay between synthetic, spectroscopic, and computational work189 seems crucial for the development of new designer luminophores.
Acknowledgments
This work was supported by the Swiss National Science Foundation through grant number 200021_178760. C. W. thanks the Independent Research Fund Denmark for an international postdoctoral grant (9059-00003B).
Glossary
Abbreviations
- bpmp
2,6-bis(2-pyridinmethyl)pyridine
- bpy
2,2′-bipyridine
- btz
3,3′-dimethyl-1,1′-bis(p-tolyl)-4,4′-bis(1,2,3-triazol-5-ylidene))
- CAAC
cyclic (alkyl)(amino)carbene
- Cp
cyclopentadienyl anion
- Cp*
pentamethylcyclopentadienyl anion
- CPL
circularly polarized luminescence
- Cz
carbazolyl
- DAC*
diamidocarbene
- ddpd
N,N′-dimethyl-N,N′-dipyridine-2-ylpyridine-2,6-diamine)
- dgpy
1,3-bis(3,4,7,8-tetrahydro-2H-pyrimido[1,2-a]pyrimidin-1(6H)-yl)benzene
- dgpz
3,5-bis(3,4,7,8-tetrahydro-2H-pyrimido[1,2-a]pyrimidin-1(6H)-yl)pyridine
- dipp
2,6-diisopropylphenyl
- dpb
1,3-di(pyridine-2-yl)benzene
- dqp
2,6-di(quinolin-8-yl)pyridine
- dpc
3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazole
- OMedqp
2,6-di(quinolin-8-yl)-4-methoxy-pyridine
- GS
ground state
- HBuImP
1,1′-(1,3-phenylene)bis(3-butyl-1-imidazol-2-ylidene)
- HImP
1,1′-(1,3-phenylene)bis(3-methyl-1-imidazol-2-ylidene)
- HOMO
highest occupied molecular orbital
- ISC
intersystem crossing
- Lbi
2,5-bis(3,5-di-tert-butyl-2-isocyanophenyl)thiophene
- LLCT
ligand-to-ligand charge transfer
- LMCT
ligand-to-metal charge transfer
- LtBu
3,3″,5,5″-tetra-tert-butyl-2,2″-diisocyano-1,1′:3′,1″-terphenyl
- Ltri
5,5′-(2-isocyano-5-methyl-1,3-phenylene)bis(2-(3,5-di-tert-butyl-2-isocyanophenyl)thiophene)
- LUMO
lowest unoccupied molecular orbital
- MAC*
monoamido-aminocarbene
- MC
metal-centered
- MLCT
metal-to-ligand charge transfer
- MMLCT
metal–metal-to-ligand charge transfer
- OTf
triflate
- PhB(MeIm)3
tris(3-methylimidazolin-2-ylidene)(phenyl)borate
- phen
1,10-phenanthroline)
- RISC
reverse intersystem crossing
- TADF
thermally activated delayed fluorescence
- TD-DFT
time-dependent density functional theory
- tpe
1,1,1-tris(pyrid-2-yl)ethane)
- tpy
2,2′;6′,2″-terpyridine; tren = tris(2-aminoethyl)amine
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
References
- Balzani V.; Campagna S.. Photochemistry and Photophysics of Coordination Compounds I. Springer: Berlin, Heidelberg, New York, 2007. [Google Scholar]
- Balzani V.; Campagna S.. Photochemistry and Photophysics of Coordination Compounds II. Springer: Berlin, Heidelberg, New York, 2007. [Google Scholar]
- McCusker J. K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Ford P. C. From curiosity to applications. A personal perspective on inorganic photochemistry. Chem. Sci. 2016, 7, 2964–2986. 10.1039/C6SC00188B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A.; Reber C. Spectroscopy and Chemical Bonding in Transition Metal Complexes. Struct. Bonding (Berlin, Ger.) 2016, 172, 291–312. 10.1007/430_2015_195. [DOI] [Google Scholar]
- Caspar J. V.; Kober E. M.; Sullivan B. P.; Meyer T. J. Application of the energy gap law to the decay of charge-transfer excited states. J. Am. Chem. Soc. 1982, 104, 630–632. 10.1021/ja00366a051. [DOI] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T. P.; Ischay M. A.; Du J. N. Visible Light Photocatalysis as a Greener Approach to Photochemical Synthesis. Nat. Chem. 2010, 2, 527–532. 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- Ardo S.; Meyer G. J. Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces. Chem. Soc. Rev. 2009, 38, 115–164. 10.1039/B804321N. [DOI] [PubMed] [Google Scholar]
- Li L.-K.; Tang M.-C.; Lai S.-L.; Ng M.; Kwok W.-K.; Chan M.-Y.; Yam V. W.-W. Strategies towards rational design of gold(III) complexes for high-performance organic light-emitting devices. Nat. Photonics 2019, 13, 185–191. 10.1038/s41566-018-0332-z. [DOI] [Google Scholar]
- Yersin H.; Rausch A. F.; Czerwieniec R.; Hofbeck T.; Fischer T. The Triplet State of Organo-Transition Metal Compounds. Triplet Harvesting and Singlet Harvesting for Efficient OLEDs. Coord. Chem. Rev. 2011, 255, 2622–2652. 10.1016/j.ccr.2011.01.042. [DOI] [Google Scholar]
- Montalti M.; Credi A.; Prodi L.; Gandolfi M. T.. Handbook of Photochemistry; CRC Taylor & Francis: Boca Raton, FL, 2006. [Google Scholar]
- Olmsted J.; Meyer T. J. Factors Affecting Cage Escape Yields Following Electron-Transfer Quenching. J. Phys. Chem. 1987, 91, 1649–1655. 10.1021/j100290a071. [DOI] [Google Scholar]
- Bizzarri C.; Spuling E.; Knoll D. M.; Volz D.; Bräse S. Sustainable metal complexes for organic light-emitting diodes (OLEDs). Coord. Chem. Rev. 2018, 373, 49–82. 10.1016/j.ccr.2017.09.011. [DOI] [Google Scholar]
- Costa R. D.; Orti E.; Bolink H. J.; Monti F.; Accorsi G.; Armaroli N. Luminescent ionic transition-metal complexes for light-emitting electrochemical cells. Angew. Chem., Int. Ed. 2012, 51, 8178–8211. 10.1002/anie.201201471. [DOI] [PubMed] [Google Scholar]
- Henwood A. F.; Zysman-Colman E. Luminescent Iridium Complexes Used in Light-Emitting Electrochemical Cells (LEECs). Top. Curr. Chem. 2016, 374, 36. 10.1007/s41061-016-0036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S.; Boixel J.; Pellegrin Y.; Blart E.; Becker H. C.; Odobel F.; Hammarström L. Accumulative electron transfer: Multiple charge separation in artificial photosynthesis. Faraday Discuss. 2012, 155, 233–252. 10.1039/C1FD00089F. [DOI] [PubMed] [Google Scholar]
- Arias-Rotondo D. M.; McCusker J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- Housecroft C. E.; Constable E. C. The emergence of copper(I)-based dye sensitized solar cells. Chem. Soc. Rev. 2015, 44, 8386–8398. 10.1039/C5CS00215J. [DOI] [PubMed] [Google Scholar]
- Bharmoria P.; Bildirir H.; Moth-Poulsen K. Triplet-triplet annihilation based near infrared to visible molecular photon upconversion. Chem. Soc. Rev. 2020, 49, 6529–6554. 10.1039/D0CS00257G. [DOI] [PubMed] [Google Scholar]
- Heinemann F.; Karges J.; Gasser G. Critical overview of the use of Ru(II) polypyridyl complexes as photosensitizers in one-photon and two-photon photodynamic therapy. Acc. Chem. Res. 2017, 50, 2727–2736. 10.1021/acs.accounts.7b00180. [DOI] [PubMed] [Google Scholar]
- Förster C.; Heinze K. Photophysics and photochemistry with earth-abundant metals – fundamentals and concepts. Chem. Soc. Rev. 2020, 49, 1057–1070. 10.1039/C9CS00573K. [DOI] [PubMed] [Google Scholar]
- Wenger O. S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. 10.1021/jacs.8b08822. [DOI] [PubMed] [Google Scholar]
- Hockin B. M.; Li C. F.; Robertson N.; Zysman-Colman E. Photoredox Catalysts Based on Earth-Abundant Metal Complexes. Catal. Sci. Technol. 2019, 9, 889–915. 10.1039/C8CY02336K. [DOI] [Google Scholar]
- Dietrich-Buchecker C. O.; Marnot P. A.; Sauvage J. P.; Kirchhoff J. R.; McMillin D. R. Bis(2,9-Diphenyl-1,10-phenanthroline)copper(I) - A Copper Complex With a Long-Lived Charge Transfer Excited State. J. Chem. Soc., Chem. Commun. 1983, 513–515. 10.1039/c39830000513. [DOI] [Google Scholar]
- Maestri M.; Bolletta F.; Moggi L.; Balzani V.; Henry M. S.; Hoffman M. Z. Mechanism of Photochemistry and Photophysics of Tris(2,2′-Bipyridine)chromium(III) ion in Aqueous Solution. J. Am. Chem. Soc. 1978, 100, 2694–2701. 10.1021/ja00477a021. [DOI] [Google Scholar]
- Heinze K.; Förster C.; Vöhringer P.; Sarkar B. Licht und Leuchten bei 3d-Metallen. Nachr. Chem. 2019, 67, 54–59. 10.1002/nadc.20194089001. [DOI] [Google Scholar]
- Viaene L.; D’Olieslager J. Luminescence from and absorption by the 3T1g Level of the Hexacyanocobaltate(III) Ion. Inorg. Chem. 1987, 26, 960–962. 10.1021/ic00253a039. [DOI] [Google Scholar]
- Chábera P.; Liu Y.; Prakash O.; Thyrhaug E.; El Nahhas A.; Honarfar A.; Essén S.; Fredin L. A.; Harlang T. C. B.; Kjaer K. S.; Handrup K.; Ericsson F.; Tatsuno Y.; Morgan K.; Schnadt J.; Häggström L.; Ericsson T.; Sobkowiak A.; Lidin S.; Huang P.; Styring S.; Uhlig J.; Bendix J.; Lomoth R.; Sundström V.; Persson P.; Wärnmark K. A low-spin FeIII complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. 10.1038/nature21430. [DOI] [PubMed] [Google Scholar]
- Kjær K. S.; Kaul N.; Prakash O.; Chabera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K. E.; Haggstrom L.; Ericsson T.; Lindh L.; Yartsev A.; Styring S.; Huang P.; Uhlug J.; Bendix J.; Strand D.; Sundström V.; Persson P.; Lomoth R.; Wärnmark K. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Young E. R.; Oldacre A. Iron Hits the Mark. Science 2019, 363, 225–226. 10.1126/science.aav9866. [DOI] [PubMed] [Google Scholar]
- Herr P.; Kerzig C.; Larsen C. B.; Häussinger D.; Wenger O. S. Manganese(I) complexes with metal-to-ligand charge transfer luminescence and photoreactivity. Nat. Chem. 2021, 10.1038/s41557-021-00744-9. [DOI] [PubMed] [Google Scholar]
- Dorn M.; Kalmbach J.; Boden P.; Kruse A.; Dab C.; Reber C.; Niedner-Schatteburg G.; Lochbrunner S.; Gerhards M.; Seitz M.; Heinze K. Ultrafast and long-time excited state kinetics of an NIR-emissive vanadium(III) complex I: Synthesis, spectroscopy and static quantum chemistry. Chem. Sci. 2021, 12, 10780–10790. 10.1039/D1SC02137K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn M.; Kalmbach J.; Boden P.; Päpcke A.; Gómez S.; Förster C.; Kuczelinis F.; Carrella L. M.; Büldt L. A.; Bings N. H.; Rentschler E.; Lochbrunner S.; González L.; Gerhards M.; Seitz M.; Heinze K. A Vanadium(III) Complex with Blue and NIR-II Spin-Flip Luminescence in Solution. J. Am. Chem. Soc. 2020, 142, 7947–7955. 10.1021/jacs.0c02122. [DOI] [PubMed] [Google Scholar]
- Tungulin D.; Leier J.; Carter A. B.; Powell A. K.; Albuquerque R. Q.; Unterreiner A. N.; Bizzarri C. Chasing BODIPY: Enhancement of Luminescence in Homoleptic Bis(dipyrrinato) ZnII Complexes Utilizing Symmetric and Unsymmetrical Dipyrrins. Chem. - Eur. J. 2019, 25, 3816–3827. 10.1002/chem.201806330. [DOI] [PubMed] [Google Scholar]
- Lazorski M. S.; Castellano F. N. Advances in the Light Conversion Properties of Cu(I)-Based Photosensitizers. Polyhedron 2014, 82, 57–70. 10.1016/j.poly.2014.04.060. [DOI] [Google Scholar]
- Gernert M.; Müller U.; Haehnel M.; Pflaum J.; Steffen A. A Cyclic Alkyl(amino)carbene as Two-Atom π-Chromophore Leading to the First Phosphorescent Linear CuI Complexes. Chem. - Eur. J. 2017, 23, 2206–2216. 10.1002/chem.201605412. [DOI] [PubMed] [Google Scholar]
- Hamze R.; Peltier J. L.; Sylvinson D.; Jung M.; Cardenas J.; Haiges R.; Soleilhavoup M.; Jazzar R.; Djurovich P. I.; Bertrand G.; Thompson M. E. Eliminating nonradiative decay in CuI emitters: > 99% quantum efficiency and microsecond lifetime. Science 2019, 363, 601–606. 10.1126/science.aav2865. [DOI] [PubMed] [Google Scholar]
- Shi S.; Jung M. C.; Coburn C.; Tadle A.; Sylvinson M. R. D.; Djurovich P. I.; Forrest S. R.; Thompson M. E. Highly Efficient Photo- and Electroluminescence from Two-Coordinate Cu(I) Complexes Featuring Nonconventional N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2019, 141, 3576–3588. 10.1021/jacs.8b12397. [DOI] [PubMed] [Google Scholar]
- Chen L. X.; Shaw G. B.; Novozhilova I.; Liu T.; Jennings G.; Attenkofer K.; Meyer G. J.; Coppens P. MLCT state structure and dynamics of a copper(I) diimine complex characterized by pump-probe X-ray and laser spectroscopies and DFT calculations. J. Am. Chem. Soc. 2003, 125, 7022–7034. 10.1021/ja0294663. [DOI] [PubMed] [Google Scholar]
- Li T.-y.; Muthiah Ravinson D. S.; Haiges R.; Djurovich P. I.; Thompson M. E. Enhancement of the Luminescent Efficiency in Carbene-Au(I)-Aryl Complexes by the Restriction of Renner–Teller Distortion and Bond Rotation. J. Am. Chem. Soc. 2020, 142, 6158–6172. 10.1021/jacs.9b13755. [DOI] [PubMed] [Google Scholar]
- Di D. W.; Romanov A. S.; Yang L.; Richter J. M.; Rivett J. P. H.; Jones S.; Thomas T. H.; Jalebi M. A.; Friend R. H.; Linnolahti M.; Bochmann M.; Credgington D. High-performance light-emitting diodes based on carbene-metal-amides. Science 2017, 356, 159–163. 10.1126/science.aah4345. [DOI] [PubMed] [Google Scholar]
- Shaw G. B.; Grant C. D.; Shirota H.; Castner E. W.; Meyer G. J.; Chen L. X. Ultrafast Structural Rearrangements in the MLCT Excited State for Copper(I) bis-Phenanthrolines in Solution. J. Am. Chem. Soc. 2007, 129, 2147–2160. 10.1021/ja067271f. [DOI] [PubMed] [Google Scholar]
- Chen L. X.; Jennings G.; Liu T.; Gosztola D. J.; Hessler J. P.; Scaltrito D. V.; Meyer G. J. Rapid excited-state structural reorganization captured by pulsed X-rays. J. Am. Chem. Soc. 2002, 124, 10861–10867. 10.1021/ja017214g. [DOI] [PubMed] [Google Scholar]
- Gernert M.; Balles-Wolf L.; Kerner F.; Müller U.; Schmiedel A.; Holzapfel M.; Marian C. M.; Pflaum J.; Lambert C.; Steffen A. Cyclic (Amino)(aryl)carbenes Enter the Field of Chromophore Ligands: Expanded π System Leads to Unusually Deep Red Emitting CuI Compounds. J. Am. Chem. Soc. 2020, 142, 8897–8909. 10.1021/jacs.0c02234. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Guo X.; Vogel R.; Prescimone A.; Wenger O. S. A tris(diisocyanide)chromium(0) complex is a luminescent analog of Fe(2,2′-bipyridine)32+. J. Am. Chem. Soc. 2017, 139, 985–992. 10.1021/jacs.6b11803. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Häussinger D.; Wenger O. S. Pyrene-Decoration of a Chromium(0) Tris(Diisocyanide) Enhances Excited State Delocalization: A Strategy to Improve the Photoluminescence of 3d6 Metal Complexes. J. Am. Chem. Soc. 2021, 10.1021/jacs.1c07345. [DOI] [PubMed] [Google Scholar]
- Cope J. D.; Denny J. A.; Lamb R. W.; McNamara L. E.; Hammer N. I.; Webster C. E.; Hollis T. K. Synthesis, characterization, photophysics, and a ligand rearrangement of CCC-NHC pincer nickel complexes: Colors, polymorphs, emission, and Raman spectra. J. Organomet. Chem. 2017, 845, 258–265. 10.1016/j.jorganchem.2017.05.046. [DOI] [Google Scholar]
- Wong Y. S.; Tang M. C.; Ng M. G.; Yam V. W. W. Toward the Design of Phosphorescent Emitters of Cyclometalated Earth-Abundant Nickel(II) and Their Supramolecular Study. J. Am. Chem. Soc. 2020, 142, 7638–7646. 10.1021/jacs.0c02172. [DOI] [PubMed] [Google Scholar]
- Pfennig B. W.; Thompson M. E.; Bocarsly A. B. A new class of room temperature luminescent organometallic complexes: luminescence and photophysical properties of permethylscandocene chloride in fluid solution. J. Am. Chem. Soc. 1989, 111, 8947–8948. 10.1021/ja00206a044. [DOI] [Google Scholar]
- Bauer M.; Steube J.; Päpcke A.; Bokareva O.; Reuter T.; Demeshko S.; Schoch R.; Hohloch S.; Meyer F.; Heinze K.; Kühn O.; Lochbrunner S., Janus-type dual emission of a Cyclometalated Iron(III) complex. Research Square, September 4, 2020, ver. 1. 10.21203/rs.3.rs-64316/v1. [DOI] [Google Scholar]
- Pal A. K.; Li C. F.; Hanan G. S.; Zysman-Colman E. Blue-emissive cobalt(III) complexes and their use in the photocatalytic trifluoromethylation of polycyclic aromatic hydrocarbons. Angew. Chem., Int. Ed. 2018, 57, 8027–8031. 10.1002/anie.201802532. [DOI] [PubMed] [Google Scholar]
- Cuttell D. G.; Kuang S. M.; Fanwick P. E.; McMillin D. R.; Walton R. A. Simple Cu(I) Complexes with Unprecedented Excited-State Lifetimes. J. Am. Chem. Soc. 2002, 124, 6–7. 10.1021/ja012247h. [DOI] [PubMed] [Google Scholar]
- Ford P. C.; Cariati E.; Bourassa J. Photoluminescence properties of multinuclear copper(I) compounds. Chem. Rev. 1999, 99, 3625–3647. 10.1021/cr960109i. [DOI] [PubMed] [Google Scholar]
- Dias H. V. R.; Diyabalanage H. V. K.; Eldabaja M. G.; Elbjeirami O.; Rawashdeh-Omary M. A.; Omary M. A. Brightly Phosphorescent Trinuclear Copper(I) Complexes of Pyrazolates: Substituent Effects on the Supramolecular Structure and Photophysics. J. Am. Chem. Soc. 2005, 127, 7489–7501. 10.1021/ja0427146. [DOI] [PubMed] [Google Scholar]
- Yam V. W. W.; Lo K. K. W. Luminescent polynuclear d10 metal complexes. Chem. Soc. Rev. 1999, 28, 323–334. 10.1039/a804249g. [DOI] [Google Scholar]
- Hossain A.; Bhattacharyya A.; Reiser O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713. 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- Heberle M.; Tschierlei S.; Rockstroh N.; Ringenberg M.; Frey W.; Junge H.; Beller M.; Lochbrunner S.; Karnahl M. Heteroleptic Copper Photosensitizers: Why an Extended π-System Does Not Automatically Lead to Enhanced Hydrogen Production. Chem. - Eur. J. 2017, 23, 312–319. 10.1002/chem.201604005. [DOI] [PubMed] [Google Scholar]
- Larsen C. B.; Wenger O. S. Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. - Eur. J. 2018, 24, 2039–2058. 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- Hernandez-Perez A. C.; Collins S. K. Heteroleptic Cu-Based Sensitizers in Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1557–1565. 10.1021/acs.accounts.6b00250. [DOI] [PubMed] [Google Scholar]
- Rentschler M.; Schmid M. A.; Frey W.; Tschierlei S.; Karnahl M. Multidentate Phenanthroline Ligands Containing Additional Donor Moieties and Their Resulting Cu(I) and Ru(II) Photosensitizers: A Comparative Study. Inorg. Chem. 2020, 59, 14762–14771. 10.1021/acs.inorgchem.9b03687. [DOI] [PubMed] [Google Scholar]
- Rosko M. C.; Wells K. A.; Hauke C. E.; Castellano F. N. Next Generation Cuprous Phenanthroline MLCT Photosensitizer Featuring Cyclohexyl Substituents. Inorg. Chem. 2021, 60, 8394–8403. 10.1021/acs.inorgchem.1c01242. [DOI] [PubMed] [Google Scholar]
- Yam V. W. W.; Wong K. M. C. Luminescent metal complexes of d6, d8 and d10 transition metal centres. Chem. Commun. 2011, 47, 11579–11592. 10.1039/c1cc13767k. [DOI] [PubMed] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P.; Von Zelewsky A. Ru(II) Polypyridine Complexes - Photophysics, Photochemistry, Electrochemistry, and Chemi-Luminescence. Coord. Chem. Rev. 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- Wenger O. S. Is Iron the New Ruthenium?. Chem. - Eur. J. 2019, 25, 6043–6052. 10.1002/chem.201806148. [DOI] [PubMed] [Google Scholar]
- Zhang W. K.; Gaffney K. J. Mechanistic Studies of Photoinduced Spin Crossover and Electron Transfer in Inorganic Complexes. Acc. Chem. Res. 2015, 48, 1140–1148. 10.1021/ar500407p. [DOI] [PubMed] [Google Scholar]
- Auböck G.; Chergui M. Sub-50-fs photoinduced spin crossover in Fe(bpy3)2+. Nat. Chem. 2015, 7, 629–633. 10.1038/nchem.2305. [DOI] [PubMed] [Google Scholar]
- Woodhouse M. D.; McCusker J. K. Mechanistic Origin of Photoredox Catalysis Involving Iron(II) Polypyridyl Chromophores. J. Am. Chem. Soc. 2020, 142, 16229–16233. 10.1021/jacs.0c08389. [DOI] [PubMed] [Google Scholar]
- Gualandi A.; Marchini M.; Mengozzi L.; Natali M.; Lucarini M.; Ceroni P.; Cozzi P. G. Organocatalytic Enantioselective Alkylation of Aldehydes with [Fe(bpy)3]Br2 Catalyst and Visible Light. ACS Catal. 2015, 5, 5927–5931. 10.1021/acscatal.5b01573. [DOI] [Google Scholar]
- Parisien-Collette S.; Hernandez-Perez A. C.; Collins S. K. Photochemical Synthesis of Carbazoles Using an [Fe(phen)3](NTf2)2/O2 Catalyst System: Catalysis toward Sustainability. Org. Lett. 2016, 18, 4994–4997. 10.1021/acs.orglett.6b02456. [DOI] [PubMed] [Google Scholar]
- Zhou W.-J.; Wu X.-D.; Miao M.; Wang Z.-H.; Chen L.; Shan S.-Y.; Cao G.-M.; Yu D.-G. Light Runs Across Iron Catalysts in Organic Transformations. Chem. - Eur. J. 2020, 26, 15052–15064. 10.1002/chem.202000508. [DOI] [PubMed] [Google Scholar]
- Tatsuno H.; Kjær K. S.; Kunnus K.; Harlang T. C. B.; Timm C.; Guo M.; Chàbera P.; Fredin L. A.; Hartsock R. W.; Reinhard M. E.; Koroidov S.; Li L.; Cordones A. A.; Gordivska O.; Prakash O.; Liu Y.; Laursen M. G.; Biasin E.; Hansen F. B.; Vester P.; Christensen M.; Haldrup K.; Németh Z.; Sárosiné Szemes D.; Bajnóczi É.; Vankó G.; Van Driel T. B.; Alonso-Mori R.; Glownia J. M.; Nelson S.; Sikorski M.; Lemke H. T.; Sokaras D.; Canton S. E.; Dohn A. O.; Møller K. B.; Nielsen M. M.; Gaffney K. J.; Wärnmark K.; Sundström V.; Persson P.; Uhlig J. Hot Branching Dynamics in a Light-Harvesting Iron Carbene Complex Revealed by Ultrafast X-ray Emission Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 364–372. 10.1002/anie.201908065. [DOI] [PubMed] [Google Scholar]
- Braun J. D.; Lozada I. B.; Kolodziej C.; Burda C.; Newman K. M. E.; van Lierop J.; Davis R. L.; Herbert D. E. Iron(II) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes. Nat. Chem. 2019, 11, 1144–1150. 10.1038/s41557-019-0357-z. [DOI] [PubMed] [Google Scholar]
- Paulus B. C.; Adelman S. L.; Jamula L. L.; McCusker J. K. Leveraging excited-state coherence for synthetic control of ultrafast dynamics. Nature 2020, 582, 214–218. 10.1038/s41586-020-2353-2. [DOI] [PubMed] [Google Scholar]
- Vittardi S. B.; Magar R. T.; Schrage B. R.; Ziegler C. J.; Jakubikova E.; Rack J. J. Evidence for a lowest energy 3MLCT excited state in [Fe(tpy)(CN)3]−. Chem. Commun. 2021, 57, 4658–4661. 10.1039/D1CC01090E. [DOI] [PubMed] [Google Scholar]
- Reuter T.; Kruse A.; Schoch R.; Lochbrunner S.; Bauer M.; Heinze K. Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion. Chem. Commun. 2021, 57, 7541–7544. 10.1039/D1CC02173G. [DOI] [PubMed] [Google Scholar]
- Liu Y. Z.; Persson P.; Sundström V.; Wärnmark K. Fe N-heterocyclic carbene complexes as promising photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. 10.1021/acs.accounts.6b00186. [DOI] [PubMed] [Google Scholar]
- Duchanois T.; Liu L.; Pastore M.; Monari A.; Cebrian C.; Trolez Y.; Darari M.; Magra K.; Frances-Monerris A.; Domenichini E.; Beley M.; Assfeld X.; Haacke S.; Gros P. C. NHC-based iron sensitizers for DSSCs. Inorganics 2018, 6, 63. 10.3390/inorganics6020063. [DOI] [Google Scholar]
- Ashley D. C.; Jakubikova E. Ironing out the Photochemical and Spin-Crossover Behavior of Fe(II) Coordination Compounds with Computational Chemistry. Coord. Chem. Rev. 2017, 337, 97–111. 10.1016/j.ccr.2017.02.005. [DOI] [Google Scholar]
- Kurz H.; Schötz K.; Papadopoulos I.; Heinemann F. W.; Maid H.; Guldi D. M.; Köhler A.; Hörner G.; Weber B. A Fluorescence-Detected Coordination-Induced Spin State Switch. J. Am. Chem. Soc. 2021, 143, 3466–3480. 10.1021/jacs.0c12568. [DOI] [PubMed] [Google Scholar]
- Mann K. R.; Gray H. B.; Hammond G. S. Excited-state reactivity patterns of hexakisarylisocyano complexes of chromium(0), molybdenum(0), and tungsten(0). J. Am. Chem. Soc. 1977, 99, 306–307. 10.1021/ja00443a083. [DOI] [Google Scholar]
- Sattler W.; Henling L. M.; Winkler J. R.; Gray H. B. Bespoke photoreductants: tungsten arylisocyanides. J. Am. Chem. Soc. 2015, 137, 1198–1205. 10.1021/ja510973h. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Guo X.; Prescimone A.; Wenger O. S. A Molybdenum(0) Isocyanide Analogue of Ru(2,2′-Bipyridine)32+: A Strong Reductant for Photoredox Catalysis. Angew. Chem., Int. Ed. 2016, 55, 11247–11250. 10.1002/anie.201605571. [DOI] [PubMed] [Google Scholar]
- Herr P.; Glaser F.; Büldt L. A.; Larsen C. B.; Wenger O. S. Long-lived, strongly emissive, and highly reducing excited states in Mo(0) complexes with chelating isocyanides. J. Am. Chem. Soc. 2019, 141, 14394–14402. 10.1021/jacs.9b07373. [DOI] [PubMed] [Google Scholar]
- Bilger J. B.; Kerzig C.; Larsen C. B.; Wenger O. S. A Photorobust Mo(0) Complex Mimicking [Os(2,2′-bipyridine)3]2+ and Its Application in Red-to-Blue Upconversion. J. Am. Chem. Soc. 2021, 143, 1651–1663. 10.1021/jacs.0c12805. [DOI] [PubMed] [Google Scholar]
- McCusker C. E.; Castellano F. N. Design of a Long-Lifetime, Earth-Abundant, Aqueous Compatible Cu(I) Photosensitizer Using Cooperative Steric Effects. Inorg. Chem. 2013, 52, 8114–8120. 10.1021/ic401213p. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lee T. S.; Favale J. M.; Leary D. C.; Petersen J. L.; Scholes G. D.; Castellano F. N.; Milsmann C. Delayed fluorescence from a zirconium(IV) photosensitizer with ligand-to-metal charge-transfer excited states. Nat. Chem. 2020, 12, 345–352. 10.1038/s41557-020-0430-7. [DOI] [PubMed] [Google Scholar]
- Manuta D. M.; Lees A. J. Emission and photochemistry of M(CO)4(diimine) (M = chromium, molybdenum, tungsten) complexes in room-temperature solution. Inorg. Chem. 1986, 25, 1354–1359. 10.1021/ic00229a012. [DOI] [Google Scholar]
- Lees A. J. Luminescence properties of organometallic complexes. Chem. Rev. 1987, 87, 711–743. 10.1021/cr00080a003. [DOI] [Google Scholar]
- Röhrs M.; Escudero D. Multiple Anti-Kasha Emissions in Transition-Metal Complexes. J. Phys. Chem. Lett. 2019, 10, 5798–5804. 10.1021/acs.jpclett.9b02477. [DOI] [PubMed] [Google Scholar]
- Farrell I. R.; Matousek P.; Towrie M.; Parker A. W.; Grills D. C.; George M. W.; Vlček A. Direct Observation of Competitive Ultrafast CO Dissociation and Relaxation of an MLCT Excited State: Picosecond Time-Resolved Infrared Spectroscopic Study of [Cr(CO)4(2,2‘-bipyridine)]. Inorg. Chem. 2002, 41, 4318–4323. 10.1021/ic020218h. [DOI] [PubMed] [Google Scholar]
- Howarth A. J.; Majewski M. B.; Wolf M. O. Photophysical properties and applications of coordination complexes incorporating pyrene. Coord. Chem. Rev. 2015, 282, 139–149. 10.1016/j.ccr.2014.03.024. [DOI] [Google Scholar]
- McClenaghan N. D.; Barigelletti F.; Maubert B.; Campagna S. Towards ruthenium(II) polypyridine complexes with prolonged and predetermined excited state lifetimes. Chem. Commun. 2002, 602–603. 10.1039/b110291e. [DOI] [PubMed] [Google Scholar]
- Dierks P.; Papcke A.; Bokareva O. S.; Altenburger B.; Reuter T.; Heinze K.; Kuhn O.; Lochbrunner S.; Bauer M. Ground- and Excited-State Properties of Iron(II) Complexes Linked to Organic Chromophores. Inorg. Chem. 2020, 59, 14746–14761. 10.1021/acs.inorgchem.0c02039. [DOI] [PubMed] [Google Scholar]
- Boden P.; Di Martino-Fumo P.; Bens T.; Steiger S.; Albold U.; Niedner-Schatteburg G.; Gerhards M.; Sarkar B. NIR-Emissive Chromium(0), Molybdenum(0), and Tungsten(0) Complexes in the Solid State at Room Temperature. Chem. - Eur. J. 2021, 27, 12959. 10.1002/chem.202102208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke W. C.; Otolski C. J.; Moore W. N. G.; Elles C. G.; Blakemore J. D. Ultrafast Spectroscopy of [Mn(CO)3] Complexes: Tuning the Kinetics of Light-Driven CO Release and Solvent Binding. Inorg. Chem. 2020, 59, 2178–2187. 10.1021/acs.inorgchem.9b02758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottelat E.; Lucarini F.; Crochet A.; Ruggi A.; Zobi F. Correlation of MLCTs of Group 7 fac-M(CO)3+ Complexes (M = Mn, Re) with Bipyridine, Pyridinylpyrazine, Azopyridine, and Pyridin-2-ylmethanimine Type Ligands for Rational photoCORM Design. Eur. J. Inorg. Chem. 2019, 2019, 3758–3768. 10.1002/ejic.201900568. [DOI] [Google Scholar]
- Schatzschneider U. PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications. Inorg. Chim. Acta 2011, 374, 19–23. 10.1016/j.ica.2011.02.068. [DOI] [Google Scholar]
- Holleman A. F.; Wiberg N.. Lehrbuch der Anorganischen Chemie, 102nd ed.; Walter de Gruyter: Berlin, 2007. [Google Scholar]
- Ting S. I.; Garakyaraghi S.; Taliaferro C. M.; Shields B. J.; Scholes G. D.; Castellano F. N.; Doyle A. G. 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc. 2020, 142, 5800–5810. 10.1021/jacs.0c00781. [DOI] [PubMed] [Google Scholar]
- Tian L.; Till N. A.; Kudisch B.; MacMillan D. W. C.; Scholes G. D. Transient Absorption Spectroscopy Offers Mechanistic Insights for an Iridium/Nickel-Catalyzed C-O Coupling. J. Am. Chem. Soc. 2020, 142, 4555–4559. 10.1021/jacs.9b12835. [DOI] [PubMed] [Google Scholar]
- Welin E. R.; Le C.; Arias-Rotondo D. M.; McCusker J. K.; MacMillan D. W. C. Photosensitized, Energy Transfer-Mediated Organometallic Catalysis through Electronically Excited Nickel(II). Science 2017, 355, 380–384. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y. Z.; Sun R.; Gianoulis N. P.; Nocera D. G. Photoredox Nickel-Catalyzed C-S Cross-Coupling: Mechanism, Kinetics, and Generalization. J. Am. Chem. Soc. 2021, 143, 2005–2015. 10.1021/jacs.0c11937. [DOI] [PubMed] [Google Scholar]
- Wenger O. S. Photoactive Nickel Complexes in Cross-Coupling Catalysis. Chem. - Eur. J. 2021, 27, 2270–2278. 10.1002/chem.202003974. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Larsen C. B.; Wenger O. S. Luminescent Ni0 Diisocyanide Chelates as Analogues of CuI Diimine Complexes. Chem. - Eur. J. 2017, 23, 8577–8580. 10.1002/chem.201700103. [DOI] [PubMed] [Google Scholar]
- Malzkuhn S.; Wenger O. S. Luminescent Ni(0) complexes. Coord. Chem. Rev. 2018, 359, 52–56. 10.1016/j.ccr.2018.01.003. [DOI] [Google Scholar]
- Grübel M.; Bosque I.; Altmann P. J.; Bach T.; Hess C. R. Redox and Photocatalytic Properties of a NiII Complex with a Macrocyclic Biquinazoline (Mabiq) Ligand. Chem. Sci. 2018, 9, 3313–3317. 10.1039/C7SC05320G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauenstein R.; Mader S. L.; Derondeau H.; Esezobor O. Z.; Block M.; Römer A. J.; Jandl C.; Riedle E.; Kaila V.; Hauer J.; Thyrhaug E.; Hess C. R. The central role of the metal ion for photoactivity: Zn- vs. Ni-Mabiq. Chem. Sci. 2021, 12, 7521–7532. 10.1039/D0SC06096H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfennig B. W.; Thompson M. E.; Bocarsly A. B. Luminescent d0 scandocene complexes: photophysical studies and electronic structure calculations on Cp*2ScX (X = Cl, I, Me). Organometallics 1993, 12, 649–655. 10.1021/om00027a014. [DOI] [Google Scholar]
- Kenney J. W.; Boone D. R.; Striplin D. R.; Chen Y. H.; Hamar K. B. Electronic luminescence spectra of charge transfer states of titanium(IV) metallocenes. Organometallics 1993, 12, 3671–3676. 10.1021/om00033a045. [DOI] [Google Scholar]
- London H. C.; Whittemore T. J.; Gale A. G.; McMillen C. D.; Pritchett D. Y.; Myers A. R.; Thomas H. D.; Shields G. C.; Wagenknecht P. S. Ligand-to-Metal Charge-Transfer Photophysics and Photochemistry of Emissive d0 Titanocenes: A Spectroscopic and Computational Investigation. Inorg. Chem. 2021, 60, 14399–14409. 10.1021/acs.inorgchem.1c02182. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Hilche T.; Slak D.; Rietdijk N. R.; Oloyede U. N.; Flowers II R. A.; Gansäuer A. Titanocenes as Photoredox Catalysts Using Green-Light Irradiation. Angew. Chem., Int. Ed. 2020, 59, 9355–9359. 10.1002/anie.202001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermi A.; Gualandi A.; Bergamini G.; Cozzi P. G. Shining Light on TiIV Complexes: Exceptional Tools for Metallaphotoredox Catalysis. Eur. J. Org. Chem. 2020, 2020, 6955–6965. 10.1002/ejoc.202000966. [DOI] [Google Scholar]
- Romain C.; Choua S.; Collin J. P.; Heinrich M.; Bailly C.; Karmazin-Brelot L.; Bellemin-Laponnaz S.; Dagorne S. Redox and Luminescent Properties of Robust and Air-Stable N-Heterocyclic Carbene Group 4 Metal Complexes. Inorg. Chem. 2014, 53, 7371–7376. 10.1021/ic500718y. [DOI] [PubMed] [Google Scholar]
- Yang M.; Sheykhi S.; Zhang Y.; Milsmann C.; Castellano F. N. Low power threshold photochemical upconversion using a zirconium(IV) LMCT photosensitizer. Chem. Sci. 2021, 12, 9069–9077. 10.1039/D1SC01662H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Lee T. S.; Petersen J. L.; Milsmann C. A Zirconium Photosensitizer with a Long-Lived Excited State: Mechanistic Insight into Photoinduced Single-Electron Transfer. J. Am. Chem. Soc. 2018, 140, 5934–5947. 10.1021/jacs.8b00742. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Petersen J. L.; Milsmann C. A Luminescent Zirconium(IV) Complex as a Molecular Photosensitizer for Visible Light Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 13115–13118. 10.1021/jacs.6b05934. [DOI] [PubMed] [Google Scholar]
- Harris J. P.; Reber C.; Colmer H. E.; Jackson T. A.; Forshaw A. P.; Smith J. M.; Kinney R. A.; Telser J. Near-infrared 2Eg→4A2g and visible LMCT luminescence from a molecular bis-(tris(carbene)borate) manganese(IV) complex. Can. J. Chem. 2017, 95, 547–552. 10.1139/cjc-2016-0607. [DOI] [Google Scholar]
- Duchanois T.; Etienne T.; Beley M.; Assfeld X.; Perpete E. A.; Monari A.; Gros P. C. Heteroleptic Pyridyl-Carbene Iron Complexes with Tuneable Electronic Properties. Eur. J. Inorg. Chem. 2014, 2014, 3747–3753. 10.1002/ejic.201402356. [DOI] [Google Scholar]
- Liu Y. Z.; Harlang T.; Canton S. E.; Chábera P.; Suarez-Alcantara K.; Fleckhaus A.; Vithanage D. A.; Goransson E.; Corani A.; Lomoth R.; Sundström V.; Wärnmark K. Towards Longer-Lived Metal-to-Ligand Charge Transfer States of Iron(II) Complexes: An N-Heterocyclic Carbene Approach. Chem. Commun. 2013, 49, 6412–6414. 10.1039/c3cc43833c. [DOI] [PubMed] [Google Scholar]
- Chábera P.; Kjaer K. S.; Prakash O.; Honarfar A.; Liu Y. Z.; Fredin L. A.; Harlang T. C. B.; Lidin S.; Uhlig J.; Sundström V.; Lomoth R.; Persson P.; Wärnmark K. FeII hexa N-heterocyclic carbene complex with a 528 ps metal-to-ligand charge-transfer excited-state lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]
- Mills I. N.; Porras J. A.; Bernhard S. Judicious Design of Cationic, Cyclometalated IrIII Complexes for Photochemical Energy Conversion and Optoelectronics. Acc. Chem. Res. 2018, 51, 352–364. 10.1021/acs.accounts.7b00375. [DOI] [PubMed] [Google Scholar]
- Rosemann N. W.; Chábera P.; Prakash O.; Kaufhold S.; Wärnmark K.; Yartsev A.; Persson P. Tracing the Full Bimolecular Photocycle of Iron(III)-Carbene Light Harvesters in Electron-Donating Solvents. J. Am. Chem. Soc. 2020, 142, 8565–8569. 10.1021/jacs.0c00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S.; Wenger O. S.; Kerzig C. Controlling Spin-Correlated Radical Pairs with Donor-Acceptor Dyads: A New Concept to Generate Reduced Metal Complexes for More Efficient Photocatalysis. Chem. - Eur. J. 2021, 27, 4115–4123. 10.1002/chem.202004638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul N.; Lomoth R. The Carbene Cannibal: Photoinduced Symmetry-Breaking Charge Separation in an Fe(III) N-Heterocyclic Carbene. J. Am. Chem. Soc. 2021, 143, 10816–10821. 10.1021/jacs.1c03770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger O. S. A bright future for photosensitizers. Nat. Chem. 2020, 12, 323–324. 10.1038/s41557-020-0448-x. [DOI] [PubMed] [Google Scholar]
- Carey M. C.; Adelman S. L.; McCusker J. K. Insights into the excited state dynamics of Fe(II) polypyridyl complexes from variable-temperature ultrafast spectroscopy. Chem. Sci. 2019, 10, 134–144. 10.1039/C8SC04025G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlycott E. A.; Hanan G. S. Designing Tridentate Ligands for Ruthenium(II) Complexes with Prolonged Room Temperature Luminescence Lifetimes. Chem. Soc. Rev. 2005, 34, 133–142. 10.1039/b316486c. [DOI] [PubMed] [Google Scholar]
- Jamula L. L.; Brown A. M.; Guo D.; McCusker J. K. Synthesis and Characterization of a High-Symmetry Ferrous Polypyridyl Complex: Approaching the 5T2/3T1 Crossing Point for FeII. Inorg. Chem. 2014, 53, 15–17. 10.1021/ic402407k. [DOI] [PubMed] [Google Scholar]
- Otto S.; Grabolle M.; Förster C.; Kreitner C.; Resch-Genger U.; Heinze K. Cr(ddpd)23+: A Molecular, Water-Soluble, Highly NIR-Emissive Ruby Analogue. Angew. Chem., Int. Ed. 2015, 54, 11572–11576. 10.1002/anie.201504894. [DOI] [PubMed] [Google Scholar]
- Beaulac R.; Tremblay J. C.; Bussière G.; Reber C. Application of near-infrared luminescence spectroscopy to vanadium(III) complexes. Characterization of their electronic ground state. Can. J. Anal. Sci. Spectrosc. 2001, 46, 152–161. [Google Scholar]
- Büldt L. A.; Wenger O. S. Chromium Complexes for Luminescence, Solar Cells, Photoredox Catalysis, Upconversion, and Phototriggered NO Release. Chem. Sci. 2017, 8, 7359–7367. 10.1039/C7SC03372A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Otto S.; Dorn M.; Kreidt E.; Lebon J.; Srsan L.; Di Martino-Fumo P.; Gerhards M.; Resch-Genger U.; Seitz M.; Heinze K. Deuterated Molecular Ruby with Record Luminescence Quantum Yield. Angew. Chem., Int. Ed. 2018, 57, 1112–1116. 10.1002/anie.201711350. [DOI] [PubMed] [Google Scholar]
- Otto S.; Scholz N.; Behnke T.; Resch-Genger U.; Heinze K. Thermo-Chromium: A Contactless Optical Molecular Thermometer. Chem. - Eur. J. 2017, 23, 12131–12135. 10.1002/chem.201701726. [DOI] [PubMed] [Google Scholar]
- Otto S.; Harris J. P.; Heinze K.; Reber C. Molecular ruby under pressure. Angew. Chem., Int. Ed. 2018, 57, 11069–11073. 10.1002/anie.201806755. [DOI] [PubMed] [Google Scholar]
- Otto S.; Nauth A. M.; Emilov E.; Scholz N.; Friedrich A.; Resch-Genger U.; Lochbrunner S.; Opatz T.; Heinze K. Photo-Chromium: Sensitizer for Visible Light-Induced Oxidative C-H Bond Functionalization - Electron or Energy Transfer?. ChemPhotoChem 2017, 1, 344–349. 10.1002/cptc.201700077. [DOI] [Google Scholar]
- Otto S.; Dorn M.; Förster C.; Bauer M.; Seitz M.; Heinze K. Understanding and Exploiting Long-Lived Near-Infrared Emission of a Molecular Ruby. Coord. Chem. Rev. 2018, 359, 102–111. 10.1016/j.ccr.2018.01.004. [DOI] [Google Scholar]
- Förster C.; Dorn M.; Reuter T.; Otto S.; Davarci G.; Reich T.; Carrella L.; Rentschler E.; Heinze K. Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes. Inorganics 2018, 6, 86. 10.3390/inorganics6030086. [DOI] [Google Scholar]
- Reichenauer F.; Wang C.; Förster C.; Boden P.; Ugur N.; Báez-Cruz R.; Kalmbach J.; Carrella L.; Rentschler E.; Ramanan C.; Niedner-Schatteburg G.; Gerhards M.; Seitz M.; Resch-Genger U.; Heinze K. Strongly Red-Emissive Molecular Ruby [Cr(bpmp)2]3+ surpasses [Ru(bpy)3]2+. J. Am. Chem. Soc. 2021, 143, 11843–11855. 10.1021/jacs.1c05971. [DOI] [PubMed] [Google Scholar]
- Tanabe Y.; Sugano S. On the Absorption Spectra of Complex Ions II. J. Phys. Soc. Jpn. 1954, 9, 766–779. 10.1143/JPSJ.9.766. [DOI] [Google Scholar]
- Jiménez J. R.; Poncet M.; Doistau B.; Besnard C.; Piguet C. Luminescent polypyridyl heteroleptic CrIII complexes with high quantum yields and long excited state lifetimes. Dalton Trans. 2020, 49, 13528–13532. 10.1039/D0DT02872J. [DOI] [PubMed] [Google Scholar]
- Abrahamsson M.; Jäger M.; Osterman T.; Eriksson L.; Persson P.; Becker H. C.; Johansson O.; Hammarström L. A 3.0 ms Room Temperature Excited State Lifetime of a Bistridentate RuII-polypyridine Complex for Rod-Like Molecular Arrays. J. Am. Chem. Soc. 2006, 128, 12616–12617. 10.1021/ja064262y. [DOI] [PubMed] [Google Scholar]
- Jiménez J. R.; Doistau B.; Cruz C. M.; Besnard C.; Cuerva J. M.; Campagna A. G.; Piguet C. Chiral Molecular Ruby [Cr(dqp)2]3+ with Long-Lived Circularly Polarized Luminescence. J. Am. Chem. Soc. 2019, 141, 13244–13252. 10.1021/jacs.9b06524. [DOI] [PubMed] [Google Scholar]
- Jiménez J.-R.; Poncet M.; Míguez-Lago S.; Grass S.; Lacour J.; Besnard C.; Cuerva J. M.; Campaña A. G.; Piguet C. Bright Long-Lived Circularly Polarized Luminescence in Chiral Chromium(III) Complexes. Angew. Chem., Int. Ed. 2021, 60, 10095–10102. 10.1002/anie.202101158. [DOI] [PubMed] [Google Scholar]
- Conti C.; Castelli F.; Forster L. S. Photophysics of Cr(CN)63- and Co(CN)63- in Poly-Alcohol-Water Solutions at Room-Temperature. J. Phys. Chem. 1979, 83, 2371–2376. 10.1021/j100481a013. [DOI] [Google Scholar]
- Treiling S.; Wang C.; Förster C.; Reichenauer F.; Kalmbach J.; Boden P.; Harris J.; Carrella L.; Rentschler E.; Resch-Genger U.; Reber C.; Seitz M.; Gerhards M.; Heinze K. Luminescence and Light-Driven Energy and Electron Transfer from an Exceptionally Long-Lived Excited State of a Non-Innocent Chromium(III) Complex. Angew. Chem., Int. Ed. 2019, 58, 18075–18085. 10.1002/anie.201909325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N.; Jiménez J. R.; Pfund B.; Prescimone A.; Piguet C.; Wenger O. S. A near-infrared-II emissive chromium(III) complex. Angew. Chem., Int. Ed. 2021, 10.1002/anie.202106398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufhold S.; Rosemann N. W.; Chábera P.; Lindh L.; Losada I. B.; Uhlig J.; Pascher T.; Strand D.; Wärnmark K.; Yartsev A.; Persson P. Microsecond Photoluminescence and Photoreactivity of a Metal-Centered Excited State in a Hexacarbene-Co(III) Complex. J. Am. Chem. Soc. 2021, 143, 1307–1312. 10.1021/jacs.0c12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dee C.; Zinna F.; Kitzmann W. R.; Pescitelli G.; Heinze K.; Di Bari L.; Seitz M. Strong circularly polarized luminescence of an octahedral chromium(III) complex. Chem. Commun. 2019, 55, 13078–13081. 10.1039/C9CC06909G. [DOI] [PubMed] [Google Scholar]
- Jäger M.; Kumar R. J.; Görls H.; Bergquist J.; Johansson O. Facile Synthesis of Bistridentate RuII Complexes Based on 2,6-Di(quinolin-8-yl)pyridyl Ligands: Sensitizers with Microsecond 3MLCT Excited State Lifetimes. Inorg. Chem. 2009, 48, 3228–3238. 10.1021/ic802342t. [DOI] [PubMed] [Google Scholar]
- Boden P.; Di Martino-Fumo P.; Niedner-Schatteburg G.; Seidel W.; Heinze K.; Gerhards M. Transient FTIR spectroscopy after one- and two-colour excitation on a highly luminescent chromium(III) complex. Phys. Chem. Chem. Phys. 2021, 23, 13808–13818. 10.1039/D1CP01077H. [DOI] [PubMed] [Google Scholar]
- Dill R. D.; Portillo R. I.; Shepard S. G.; Shores M. P.; Rappé A. K.; Damrauer N. H. Long-Lived Mixed 2MLCT/MC States in Antiferromagnetically Coupled d3 Vanadium(II) Bipyridine and Phenanthroline Complexes. Inorg. Chem. 2020, 59, 14706–14715. 10.1021/acs.inorgchem.0c01950. [DOI] [PubMed] [Google Scholar]
- Joyce J. P.; Portillo R. I.; Nite C. M.; Nite J. M.; Nguyen M. P.; Rappé A. K.; Shores M. P. Electronic Structures of Cr(III) and V(II) Polypyridyl Systems: Undertones in an Isoelectronic Analogy. Inorg. Chem. 2021, 60, 12823. 10.1021/acs.inorgchem.1c01129. [DOI] [PubMed] [Google Scholar]
- Wenger O. S.; Güdel H. U. Chemical tuning of the photon upconversion properties in Ti2+-doped chloride host lattices. Inorg. Chem. 2001, 40, 5747–5753. 10.1021/ic010255t. [DOI] [PubMed] [Google Scholar]
- Zobel P.; Knoll T.; González L. Ultrafast and long-time excited state kinetics of an NIR-emissive vanadium(III) complex II. Elucidating Triplet-to-Singlet Excited-State Dynamics. Chem. Sci. 2021, 12, 10791–10801. 10.1039/D1SC02149D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannizzo A.; Blanco-Rodriguez A. M.; El Nahhas A.; Šebera J.; Záliš S.; Vlček A.; Chergui M. Femtosecond fluorescence and intersystem crossing in rhenium(I) carbonyl-bipyridine complexes. J. Am. Chem. Soc. 2008, 130, 8967–8974. 10.1021/ja710763w. [DOI] [PubMed] [Google Scholar]
- Fataftah M. S.; Bayliss S. L.; Laorenza D. W.; Wang X.; Phelan B. T.; Wilson C. B.; Mintun P. J.; Kovos B. D.; Wasielewski M. R.; Han S.; Sherwin M. S.; Awschalom D. D.; Freedman D. E. Trigonal Bipyramidal V3+ Complex as an Optically Addressable Molecular Qubit Candidate. J. Am. Chem. Soc. 2020, 142, 20400–20408. 10.1021/jacs.0c08986. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Wenger O. S. Luminescent Complexes Made from Chelating Isocyanide Ligands and Earth-Abundant Metals. Dalton Trans. 2017, 46, 15175–15177. 10.1039/C7DT03620E. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Wenger O. S. Chromium(0), Molybdenum(0), and Tungsten(0) Isocyanide Complexes as Luminophores and Photosensitizers with Long-Lived Excited States. Angew. Chem., Int. Ed. 2017, 56, 5676–5682. 10.1002/anie.201701210. [DOI] [PubMed] [Google Scholar]
- Wood C. J.; Summers G. H.; Clark C. A.; Kaeffer N.; Braeutigam M.; Carbone L. R.; D’Amario L.; Fan K.; Farré Y.; Narbey S.; Oswald F.; Stevens L. A.; Parmenter C. D. J.; Fay M. W.; La Torre A.; Snape C. E.; Dietzek B.; Dini D.; Hammarström L.; Pellegrin Y.; Odobel F.; Sun L.; Artero V.; Gibson E. A. A comprehensive comparison of dye-sensitized NiO photocathodes for solar energy conversion. Phys. Chem. Chem. Phys. 2016, 18, 10727–10738. 10.1039/C5CP05326A. [DOI] [PubMed] [Google Scholar]
- Jiménez J. R.; Doistau B.; Besnard C.; Piguet C. Versatile Heteroleptic Bis-Terdentate CrIII Chromophores Displaying Room Temperature Millisecond Excited State Lifetimes. Chem. Commun. 2018, 54, 13228–13231. 10.1039/C8CC07671E. [DOI] [PubMed] [Google Scholar]
- Guo D.; Knight T. E.; McCusker J. K. Angular Momentum Conservation in Dipolar Energy Transfer. Science 2011, 334, 1684–1687. 10.1126/science.1211459. [DOI] [PubMed] [Google Scholar]
- Strieth-Kalthoff F.; Glorius F. Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem. 2020, 6, 1888–1903. 10.1016/j.chempr.2020.07.010. [DOI] [Google Scholar]
- Kasha M. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc. 1950, 9, 14–19. 10.1039/df9500900014. [DOI] [Google Scholar]
- Wenger O. S.; Güdel H. U. Influence of crystal field parameters on near-infrared to visible photon upconversion in Ti2+ and Ni2+ doped halide lattices. Struct. Bonding (Berlin, Ger.) 2004, 106, 59–70. 10.1007/b11305. [DOI] [Google Scholar]
- Schmid L.; Kerzig C.; Prescimone A.; Wenger O. S. Photostable Ruthenium(II) Isocyanoborato Luminophores and Their Use in Energy Transfer and Photoredox Catalysis. JACS Au 2021, 1, 819–832. 10.1021/jacsau.1c00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel A. K. C.; Förster C.; Breivogel A.; Mack K.; Ochsmann J. R.; Laquai F.; Ksenofontov V.; Heinze K. A Heteroleptic Push-Pull Substituted Iron(II) Bis(tridentate) Complex with Low-Energy Charge-Transfer States. Chem. - Eur. J. 2015, 21, 704–714. 10.1002/chem.201404955. [DOI] [PubMed] [Google Scholar]
- Fajardo J.; Schwan J.; Kramer W. W.; Takase M. K.; Winkler J. R.; Gray H. B. Third-Generation W(CNAr)6 Photoreductants (CNAr = Fused-Ring and Alkynyl-Bridged Arylisocyanides). Inorg. Chem. 2021, 60, 3481–3491. 10.1021/acs.inorgchem.0c02912. [DOI] [PubMed] [Google Scholar]
- Chan K. T.; Lam T. L.; Yu D. H.; Du L.; Phillips D. L.; Kwong C. L.; Tong G. S. M.; Cheng G.; Che C. M. Strongly Luminescent Tungsten Emitters with Emission Quantum Yields of up to 84%: TADF and High-Efficiency Molecular Tungsten OLEDs. Angew. Chem., Int. Ed. 2019, 58, 14896–14900. 10.1002/anie.201906698. [DOI] [PubMed] [Google Scholar]
- Qiao Y. S.; Schelter E. J. Lanthanide Photocatalysis. Acc. Chem. Res. 2018, 51, 2926–2936. 10.1021/acs.accounts.8b00336. [DOI] [PubMed] [Google Scholar]
- Hu A.; Guo J.-J.; Pan H.; Zuo Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 361, 668–672. 10.1126/science.aat9750. [DOI] [PubMed] [Google Scholar]
- Bestgen S.; Schoo C.; Neumeier B. L.; Feuerstein T. J.; Zovko C.; Köppe R.; Feldmann C.; Roesky H. Intensely Photoluminescent Diamidophosphines of the Alkaline-Earth Metals, Aluminum and Zinc. Angew. Chem., Int. Ed. 2018, 57, 14265–14269. 10.1002/anie.201806943. [DOI] [PubMed] [Google Scholar]
- Pinter P.; Schüßlbauer C. M.; Watt F. A.; Dickmann N.; Herbst-Irmer R.; Morgenstern B.; Grünwald A.; Ullrich T.; Zimmer M.; Hohloch S.; Guldi D. M.; Munz D. Bright luminescent lithium and magnesium carbene complexes. Chem. Sci. 2021, 12, 7401–7410. 10.1039/D1SC00846C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer L. A.; Pearce O. M.; Maharaj F. D. R.; Brown N. L.; Amador C. K.; Damrauer N. H.; Marshak M. P. Open for Bismuth: Main Group Metal-to-Ligand Charge Transfer. Inorg. Chem. 2021, 60, 10137–10146. 10.1021/acs.inorgchem.0c03818. [DOI] [PubMed] [Google Scholar]
- Cerfontaine S.; Wehlin S. A. M.; Elias B.; Troian-Gautier L. Photostable Polynuclear Ruthenium(II) Photosensitizers Competent for Dehalogenation Photoredox Catalysis at 590 nm. J. Am. Chem. Soc. 2020, 142, 5549–5555. 10.1021/jacs.0c01503. [DOI] [PubMed] [Google Scholar]
- Vittardi S. B.; Thapa Magar R.; Breen D. J.; Rack J. J. A Future Perspective on Phototriggered Isomerizations of Transition Metal Sulfoxides and Related Complexes. J. Am. Chem. Soc. 2021, 143, 526–537. 10.1021/jacs.0c08820. [DOI] [PubMed] [Google Scholar]
- Wei F.; Lai S.-L.; Zhao S.; Ng M.; Chan M.-Y.; Yam V. W.-W.; Wong K. M.-C. Ligand Mediated Luminescence Enhancement in Cyclometalated Rhodium(III) Complexes and Their Applications in Efficient Organic Light-Emitting Devices. J. Am. Chem. Soc. 2019, 141, 12863–12871. 10.1021/jacs.9b06308. [DOI] [PubMed] [Google Scholar]
- Whittemore T. J.; Xue C.; Huang J.; Gallucci J. C.; Turro C. Single-chromophore single-molecule photocatalyst for the production of dihydrogen using low-energy light. Nat. Chem. 2020, 12, 180–185. 10.1038/s41557-019-0397-4. [DOI] [PubMed] [Google Scholar]
- Nicholls T. P.; Burt L. K.; Simpson P. V.; Massi M.; Bissember A. C. Tricarbonyl Rhenium(I) Tetrazolato and N-Heterocyclic Carbene Complexes: Versatile Visible-Light-Mediated Photoredox Catalysts. Dalton Trans. 2019, 48, 12749–12754. 10.1039/C9DT02533B. [DOI] [PubMed] [Google Scholar]
- Scattergood P. A.; Sinopoli A.; Elliott P. I. P. Photophysics and photochemistry of 1,2,3-triazole-based complexes. Coord. Chem. Rev. 2017, 350, 136–154. 10.1016/j.ccr.2017.06.017. [DOI] [Google Scholar]
- Rodriguez T. M.; Deegbey M.; Jakubikova E.; Dempsey J. L. The ligand-to-metal charge transfer excited state of [Re(dmpe)3]2+. Photosynth. Res. 2021, 10.1007/s11120-021-00859-7. [DOI] [PubMed] [Google Scholar]
- Shon J. H.; Teets T. S. Molecular Photosensitizers in Energy Research and Catalysis: Design Principles and Recent Developments. ACS Energy Lett. 2019, 4, 558–566. 10.1021/acsenergylett.8b02388. [DOI] [Google Scholar]
- Bevernaegie R.; Wehlin S. A. M.; Piechota E. J.; Abraham M.; Philouze C.; Meyer G. J.; Elias B.; Troian-Gautier L. Improved Visible Light Absorption of Potent Iridium(III) Photo-oxidants for Excited-State Electron Transfer Chemistry. J. Am. Chem. Soc. 2020, 142, 2732–2737. 10.1021/jacs.9b12108. [DOI] [PubMed] [Google Scholar]
- Shon J. H.; Kim D.; Rathnayake M. D.; Sittel S.; Weaver J.; Teets T. S. Photoredox catalysis on unactivated substrates with strongly reducing iridium photosensitizers. Chem. Sci. 2021, 12, 4069–4078. 10.1039/D0SC06306A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J. J.; To W. P.; Liu Y. G.; Lu W.; Che C. M. Efficient Acceptorless Photo-Dehydrogenation of Alcohols and N-Heterocycles with Binuclear Platinum(II) Diphosphite Complexes. Chem. Sci. 2019, 10, 4883–4889. 10.1039/C8SC05600E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrara S.; Aliprandi A.; Hogan C. F.; De Cola L. Aggregation-Induced Electrochemiluminescence of Platinum(II) Complexes. J. Am. Chem. Soc. 2017, 139, 14605–14610. 10.1021/jacs.7b07710. [DOI] [PubMed] [Google Scholar]
- Cebrian C.; Mauro M. Recent advances in phosphorescent platinum complexes for organic light-emitting diodes. Beilstein J. Org. Chem. 2018, 14, 1459–1481. 10.3762/bjoc.14.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel J. P.; González L. The Quest to Simulate Excited-State Dynamics of Transition Metal Complexes. JACS Au 2021, 1, 1116–1140. 10.1021/jacsau.1c00252. [DOI] [PMC free article] [PubMed] [Google Scholar]