

Abstract
Background
Hereditary gingival fibromatosis (HGF) is a rare condition characterized by slowly progressive overgrowth of the gingiva. The severity of overgrowth may differ from mild causing phonetic and masticatory issues, to severe resulting in diastemas or malposition of teeth. Both, autosomal-dominant and autosomal-recessive forms of HGF are described. The aim of this review is a clinical overview, as well as a summary and discussion of the involvement of candidate chromosomal regions, pathogenic variants of genes, and candidate genes in the pathogenesis of HGF. The loci related to non-syndromic HGF have been identified on chromosome 2 (GINGF, GINGF3), chromosome 5 (GINGF2), chromosome 11 (GINGF4), and 4 (GINGF5). Of these loci, pathogenic variants of the SOS-1 and REST genes inducing HGF have been identified in the GINGF and the GINGF5, respectively. Furthermore, among the top 10 clusters of genes ranked by enrichment score, ATP binding, and fibronectin encoding genes were proposed as related to HGF.
Conclusion
The analysis of clinical reports as well as translational genetic studies published since the late’90s indicate the clinical and genetic heterogeneity of non-syndromic HGF and point out the importance of genetic studies and bioinformatics of more numerous unrelated families to identify novel pathogenic variants potentially inducing HGF. This strategy will help to unravel the molecular mechanisms as well as uncover specific targets for novel and less invasive therapies of this rare, orphan condition.
Keywords: Chromosome, Gene, Hereditary gingival fibromatosis, Linkage analysis, Pathogenic variant , Whole-exome sequencing
Background
Gingival fibromatosis (GF), also known as gingival hyperplasia or gingival overgrowth may be manifested as pathologic, diffuse, or local growth of the gingiva. By definition, it affects the masticatory mucosa (the marginal and attached gingiva and the interdental papilla), but it does not spread beyond the mucogingival junction. The excess gingival tissue can cover part of or the entire crown of a tooth, and can result in diastemas, teeth displacement, retention of primary or permanent teeth, and may also cause masticatory, phonetic, psychological, and esthetic problems [1, 2]. In rare cases, the condition may be associated with hereditary factors and occurs as an isolated (non-syndromic) hereditary gingival fibromatosis (HGF, MIM 135300), otherwise known as hereditary gingival overgrowth affecting exclusively gingiva. It can appear as a complete diffuse condition when it affects both the maxilla and the mandible, however, it can also affect exclusively one jaw (part diffuse condition) or it can appear locally as a nodule-like form [3–5]. Both, autosomal-dominant and autosomal-recessive forms of this condition have been reported [3].
The first clinical signs of HGF manifest in the primary dentition period. Parents and/or close relatives usually have had their own experience with this condition and seek medical advice for the child. At this age, the most prominent feature of HGF is an extremely wide zone of keratinized gingiva. During adolescence, the connective tissue within the gingiva appears thickened, mostly due to overproduction of collagenous proteins. Except for recurrent cases, gingival overgrowth in HGF is characterized by a slow progression rate. GF associated with hereditary factors may also co-exist as a part of rare genetic diseases, syndromes, or chromosomal abnormalities, e.g.,Ramon syndrome (MIM 266270), Jones syndrome (MIM 135550), juvenile hyaline fibromatosis (MIM 228600). In the case of genetic syndromes or diseases, gingival overgrowth co-exists with a variety of systemic symptoms, such as skeletal defects, hypertrichosis, epilepsy, sensorineural hearing loss, joint contractures, and others. For more detailed information, refer to Gawron et al. [3].
To date, five distinct loci related to the non-syndromic variant of HGF have been identified, two on chromosome 2 (GINGF on 2p21-p22, MIM 135300 and GINGF3 on 2p22.3-p23.3, MIM 609955) [6–8], one mapped to chromosome 5 (GINGF2 on 5q13-q22, MIM 605544) [9], one to chromosome 11 (GINGF4 on 11p15, MIM 611010) [10], and one to chromosome 4 (GINGF5 on 4q12, MIM 617626) [11]. Of these loci, HGF-inducing pathogenic variants of the Son-of-Sevenless-1 (SOS-1) gene (MIM 182530) and in the RE1-silencing transcription factor (REST) gene (MIM 600571) have been identified within the GINGF and the GINGF5 loci, respectively. Wild-type human SOS-1 gene encodes a highly conserved protein, Son-of-Sevenless-1 (SOS-1), and the product of the REST gene, is the RE1-silencing transcription factor [11, 12]. Originally, a single-cytosine insertion in the genomic sequence of the SOS-1 gene (GenBank: NC_000002.12 (NM_005633.4): g.126,142-126,143insC), mapped to the 2p21-p22 locus has been identified as a causative factor of non-syndromic HGF type 1 (HGF1) in one family from Brazil [6, 12]. A dominant HGF locus has been also mapped to the region 2p21-p22 in chromosome 2 in four Chinese families [8]. In contrast to the Brazilian family, where suppression of recombination was found, similar characteristics were not observed in the Chinese families. Furthermore, the SOS-1 locus was likely not affected in this and one more study of other Chinese families [6, 8, 13]. Similarly, the presence of single-cytosine insertion in the SOS-1 gene, and the corresponding HGF1 phenotype have not been found in two affected families from the Polish population investigated in our study [14]. Otherwise, whole-exome sequencing (WES) analysis of eleven affected individuals with HGF from three unrelated families, carried out by Bayram et al., led to the identification of three heterozygous pathogenic variants predicted by conceptual translation to result in premature termination of the nascent transcript, two frameshifts, and one nonsense allele in the REST gene [11]. This gene has been mapped to the GINGF5 locus on chromosome 4 (4q12). Likewise, the group from Korea performed WES followed by gene set enrichment analysis (GSEA), and a protein functional network study in two affected donors from a three-generation family and proposed the candidate representative genes of the clusters of related genes potentially associated with non-syndromic HGF. They proposed ATP binding and fibronectin gene clusters as the central candidate that may cause gingival overgrowth [15]. The clinical communications, as well as the reports from translational studies on the genetics of HGF published over the last twenty years, suggest a heterogeneous nature of non-syndromic HGF however, so far, an updated overview of this topic has not been published.
In line with this, the aim of this review is a clinical overview, as well as a summary and discussion of the involvement of candidate chromosomal regions, candidate genes, and pathogenic variants of genes in the pathogenesis of HGF.
Clinical manifestations of HGF
Isolated (non-syndromic) HGF (MIM 135300) is a rare genetic disease with unknown prevalence, manifested by a slowly progressive, non-hemorrhagic, benign, and fibrous overgrowth of the gingiva. The incidence and severity of the disease appear to depend on the penetrance of the mutated gene [6, 7]. Hypertrophic gingiva is usually normal in colour and consists of dense fibrous tissue that feels firm and nodular on palpation. Gingival overgrowth, in addition to being disfiguring, leads to compromised quality of life of the patient due to increased difficulty to maintain oral hygiene, which may lead to caries development, and several periodontal and orthodontic problems [1, 16–18]. Clinically, the onset coincides with the eruption of primary or permanent dentition and is rarely present at birth [19]. Males and females are affected equally. Many reports, including clinical cases or case series present variable clinical aspects of this condition. Whereas the majority of the reports present diffuse (generalized) appearance of gingival overgrowth in both, the mandible and the maxilla or less commonly within only one jaw, others describe exclusively local presence of nodule-like overgrowth around a group of teeth in one or both dental arches [1, 3, 20, 21]. The cases described differ in the severity of the overgrowth. In mild and moderate conditions, phonetic and masticatory problems are usually observed. Severe overgrowth, which covers the major part or entire crowns of the teeth may result in prolonged retention of primary or permanent dentition, diastemas, malposition of teeth and/or may cause psychological problems [3, 18, 19, 21]. In moderate and severe cases periodontal problems also occur, such as bleeding, enhanced risk of caries, and bone loss due to the excess of gingival tissue, presence of pseudopockets, and bacterial plaque accumulation. Sustained oral infections may ultimately impair systemic health due to their effect on the host immune system [3, 22, 23]. Another clinical problem found in patients with HGF is the recurrence of the lesions after surgical removal. In some cases, gingival overgrowth may reoccur within several weeks, months or even years after surgery, whereas in other patients the recurrence is not observed.. The patients with recurrent gingival overgrowth may also present some psychological issues due to frequent surgical interventions or postoperative complications [4, 20, 24–30]. Clinical manifestation, postoperative complications, and management of a highly recurrent, fibrotic gingival overgrowth have been well illustrated in a recently published clinical case report from our laboratory. In brief, an 11-year-old Caucasian female presented for evaluation of a gingival overgrowth that caused functional, hygienic, and psychological problems. Intraoral clinical examination revealed a painless, firm, and pale pink tissue covering 30% of her teeth crowns in the maxilla and the mandible. The recurrence of 25% of the pre-operative tissue volume in the maxilla and 45% of the initial tissue volume in the mandible was observed two and four weeks post-gingivectomy, respectively. Two months later, a gradual decrease in the recurrence was noted in both jaws. In contrast, the follow-up examinations at one, three, and 6 months after gingivectomy carried out in a 13-year-old sister of the patient revealed uneventful healing and no signs of recurrence, whereas complete surgery of diffuse overgrowth within mandibular and maxillary arches was performed in a 32-year-old mother of the patient resulted in the recurrence twelve months post-surgery, particularly in the mandible [18, 25]. These observations showed that the clinical appearance, episodes, and anatomical region of recurrence may vary among members of the same family and stand in accordance with other communications published over the last two decades, indicating that HGF represents a clinically heterogeneous entity. The phenotypic heterogeneity of HGF is presented in Fig. 1.
Fig. 1.
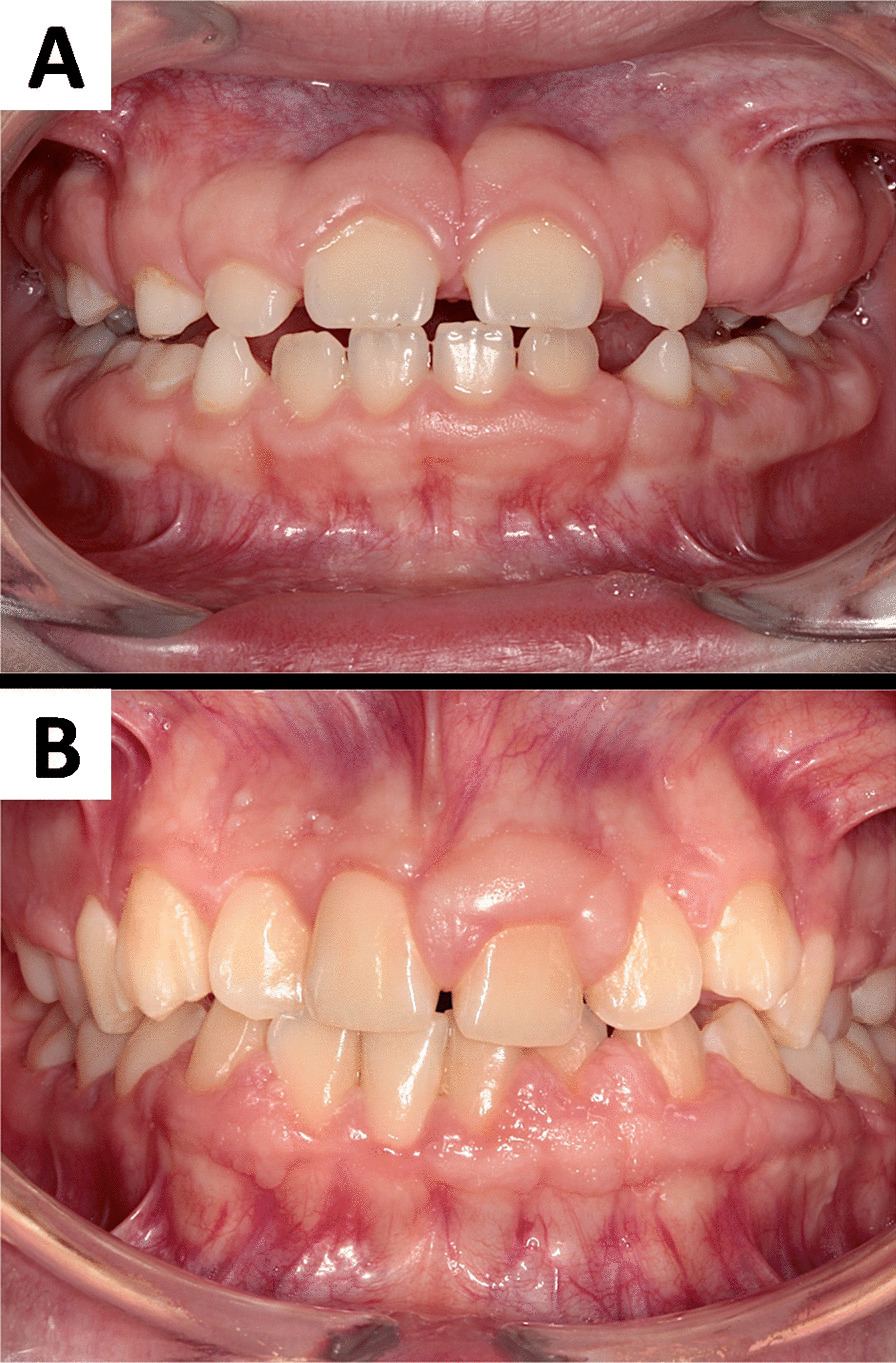
Phenotypic heterogeneity of hereditary gingival fibromatosis. A. the photography shows the gingiva from a 10-year-old Caucasian male patient diagnosed with HGF. Both mother of the patient and her sister are also affected. The surface of the gingiva is almost homogeneous, smooth and with a normal stippling of the attached gingiva. An unusually wide zone of the keratinized gingiva is equally distributed along teeth in both dental arches in the maxilla and in the mandible. The margin of gingiva obscures half of the crowns’ height, which makes teeth appearing not completely erupted; B. a 15-year-old Caucasian female diagnosed with asymmetric gingival overgrowth of hereditary origin in the maxilla and in the mandible. Hypertrophy of gingiva caused spacing between teeth. It also makes clinical crowns appearing to be shorter than their anatomical length. Gingiva around some teeth looks almost normal while in other locations it presents a significant amount of keratinized tissue, which covers the teeth surface. The surface of the gingiva is heterogeneous at different sites of oral cavity. Some areas are smooth with a normal stippling of the gingiva, whereas other resemble multiple verrucous lesions. Some interdental papillae are overgrown, inflamed, and bleed easily during tooth brushing
Besides an isolated form manifested in HGF, a fibrous gingival overgrowth can also occur as part of some genetic syndromes and diseases, causing serious disabilities e.g., mental deficiency and epilepsy in Ramon syndrome (MIM 266270) and Zimmermann-Laband syndrome (MIM 135500), progressive sensorineural hearing loss in Jones syndrome (MIM 135550), and many others [3, 31–36].
Pathogenic variants of genes in HGF
Non-syndromic HGF is transmitted as a Mendelian trait in an autosomal-dominant or, less frequently, an autosomal-recessive mode [1]. The incidence ratio of the condition does not depend on gender. Linkage analysis of families suffering from a non-syndromic autosomal-dominant variant of HGF revealed several regions on chromosomes that may potentially contain pathogenic variants of genes contributing to this condition. The candidate loci have been identified on chromosome 2p21-p22 (GINGF, HGF1, MIM 135300) in a Brazilian family, and chromosomes 5q13-q22 (GINGF2, MIM 605544), 2p22.3-p23.3 (GINGF3, MIM 609955), and 11p15 (GINGF4, MIM 611010) in several Chinese families [6, 7, 9, 10] (Table 1, Fig. 2).
Table 1.
Chromosomal regions with affected or candidate genes contributing to non-syndromic HGF
| Chromosomal region/gene locus | Affected# or candidate## gene | Gene/locus MIM number | Disease type | Phenotype MIM number | Inheritance | References |
|---|---|---|---|---|---|---|
|
2p21-p22 (D2S1788, D2S441) GINGF, GINGF1, GGF1 |
SOS-1# | 182530 |
Hereditary gingival fibromatosis type 1 (HGF1) |
135300 | AD | [6, 8, 12, 65] |
|
2p16-p13 (D2S1788, D2S2298) GINGF |
– | – | Hereditary gingival fibromatosis type 1 (HGF1) | 135300 | AD, AR | [64, 65] |
|
5q13-q22 (D5S1491, D5S1453) GINGF2, GGF2 |
CAMK4## | 114080 |
Hereditary gingival fibromatosis type 2 (HGF2) |
605544 | AD | [9, 66, 67] |
|
2p23.3-p22.3 (D2S2221, D2S1788) GINGF3, GGF3 |
– | – |
Hereditary gingival fibromatosis type 3 (HGF3) |
609955 | AD | [7, 68] |
|
11p15 (D11S1984- D11S1338) GINGF4, GGF4 |
– | – |
Hereditary gingival fibromatosis type 4 (HGF4) |
611010 | MI | [10, 69] |
|
4q12 GINGF5, GGF5 |
REST# | 600571 |
Hereditary gingival fibromatosis type 5 (HGF5) |
617626 | AD | [11, 70] |
O(MIM), Online Mendelian Inheritance in Man; AD, autosomal dominant; AR, autosomal recessive; MI, maternal inheritance; CAMK4, calcium/calmodulin-dependent protein kinase IV; SOS-1, Son-of-Sevenless-1; REST, RE1-silencing transcription factor
# affected gene, ## candidate gene contributing to HGF
Fig. 2.

Schematic representation of the loci and affected genes associated with a non-syndromic variant of HGF. Two loci are present on chromosome 2 (GINGF, 2p21-p22 and GINGF3, 2p22.3-p23.3), one on chromosome 4 (GINGF5, 4q12), chromosome 5 (GINGF2, 5q13-q22) and one on chromosome 11 (GINGF4, 11p15). Pathogenic variants of the SOS-1 (Son-of-Sevenless-1) and REST (RE1-silencing transcription factor) genes were associated with GINGF and GINGF5, respectively
Analysis of the SOS-1 gene
Further sequencing analyses revealed an insertion of a single base (cytosine) in exon 21 (g.126,142-126,143insC; c.3248-3249insC) of the SOS-1 gene (MIM 182530) in the 2p21-p22 candidate region, underlying GINGF locus which segregated in a dominant manner over four generations of a large Brazilian family with HGF. The chromosomal region 2p21-p22 has been discovered and described as the first candidate region containing the pathogenic variant, which contributes to HGF, hence the HGF induced by insertion of cytosine in exon 21 of the SOS-1 gene has been designed as “type 1” (HGF1) [12]. Wild type human SOS-1, a bifunctional guanine nucleotide exchange factor (GEF) is a highly conserved 1,333-amino acid protein that is constitutively maintained in a down-regulated state under physiological conditions. The carboxyl-terminal domain of this protein exerts negative allosteric control on the interaction of the SOS-1 catalytic domain with Ras and Rac related proteins in cells. Ras regulates several protein kinases, including members of the mitogen-activated protein kinase (MAPK), phosphoinositide-3 kinase (PI3K), and proteins of the Rho family [37]. Indirect activation of Ras by SOS-1 resulting in phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) of the MAPK signaling pathway or leading to activation of downstream molecules involved in the PI3K pathway and the Ral nucleotide exchange factor (RalGDS) family are the key mechanisms regulating cell proliferation, survival, gene transcription, and differentiation [38]. SOS-1 also acts as a GEF for Rac when it forms a complex with the adaptor molecules E3b1 and Eps8. Activation of Rac regulates the actin remodeling and cytoskeletal organization that is critical for the transport of signaling molecules inside the cell, cell adhesion, and migration. Whether SOS-1 will function as a Ras or Rac exchange factor is dictated by its C-terminal proline-rich region, which contains binding sites for SRC Homology 3 (SH3) domains present in both adaptor proteins, growth factor receptor-bound protein 2 (Grb2), and E3b1 [39, 40]. Therefore, competition between these two adaptor proteins for binding to SOS-1 may determine whether the Ras or the Rac pathway gets activated [41, 42]. Phosphorylation of SOS-1 on tyrosine residues by the MAPK pathway does not affect the stability of the SOS-1/E3b1/Eps8 complex, but it disrupts the SOS-1/Grb2 complex [43]. Hence, phosphorylation of SOS-1 provides a mechanism to balance signaling between the Ras and Rac pathways. In addition to growth factor receptors, certain integrin-type extracellular matrix (ECM) receptors also regulate the Ras and Rac pathways through SOS-1 [44–46]. The single-cytosine insertion causes a frameshift and an early termination of the protein biosynthesis which yields a chimeric 1,105-amino acid protein that consists of 1,083 SOS-1 N-terminal amino acids followed by 22 replaced amino acids. Since the resultant chimeric protein lacks the regulatory carboxyl-terminal domain, it has augmented activity. This suggests that, in HGF1, mutated SOS-1 is constantly in an activated state, which may increase the activity of the MAPK pathway. The consequence is inappropriate transcriptional control of genes required for cell proliferation, differentiation, migration, ECM, and growth factors signaling [12]. Similar to humans, studies in several species, including yeast and Drosophila melanogaster indicate that truncation of the proline-rich carboxyl-terminal region of SOS-1 is associated with a gain-of-function [47]. Additionally, a transgene constructs with an SOS-1 carboxyl-terminal deletion (called “SOS-F”) induces skin tumor development in mice [48]. Furthermore, Lee et al. reported increased proliferation of fibroblasts and significantly increased levels of collagen in the histological studies of gingival tissue of the patients with HGF caused by the g.126,142-126,143insC pathogenic variant of the SOS-1 [49]. Similarly, analyses in monolayer and three-dimensional cultures of gingival fibroblasts harboring the g.126,142-126,143insC in the SOS-1 demonstrated increased cell proliferation rates, an altered ability to attach, an enhanced propensity to form protrusions with lamellipodia and dense staining of the ECM indicating clustering of collagen fibers. These data indicate that the presence of the g.126,142-126,143insC in exon 21 of the SOS-1 gene producing the truncated protein chimera p.K1084fsX1105 induces HGF1 in an extended kindred through a gain-of-function mechanism [6, 12, 49]. Moreover, this pathogenic variant of the SOS-1 has been suggested to be associated with increased fibroblast proliferation and collagen matrix synthesis in vivo and in vitro [49].
On contrary, the analyses carried out in patients from two unrelated, Polish families with a history of non-syndromic autosomal-dominant HGF did not confirm single cytosine insertion (g.126,142-126,143insC) nor other pathogenic variants in exon 21 of the SOS-1 gene. Further analyses of those families also did not reveal other insertions, any deletion, or substitution in the contiguous exons 19, 20, and 22 of the SOS-1 [14]. As previously discussed, the single cytosine insertion in exon 21 of the SOS-1 has been associated with increased synthesis of collagen type I and increased proliferation of gingival fibroblasts [49]. Although the patients from Polish families presented a highly fibrotic phenotype of HGF and increased proliferation rates of gingival fibroblasts, such associations were not found in our studies [14, 17, 50]. Likewise, Ma et al. did not detect a single-cytosine insertion in exon 21 nor any other pathogenic variant in 23 exons of the SOS-1 gene in three multigeneration Chinese families with isolated HGF [13]. These observations suggest that the analyzed families from Poland and China are not affected by HGF1, which indirectly indicates that the pathogenic variant(s) of other genes inducing other type(s) of HGF might be associated.
Pathogenic variants of the REST gene
In fact, in a more recent study performed by Bayram et al., the WES analysis of eleven affected individuals with HGF from three unrelated families led to the identification of three heterozygous pathogenic variants predicted by conceptual translation to result in premature termination of the nascent transcript, two frameshifts, and one nonsense allele in the REST gene that has been mapped in the GINGF5 locus on chromosome 4 (GINGF5 on 4q12, MIM 617626) [11] (Table 1, Fig. 2).
Two different heterozygous truncating variants in the REST gene including a frameshift deletion (GenBank: NM_005612.4; exon 4; c.2865_2866delAA [p.Asn958Serfs*9]; chr4:57,797,888_CAA>C were identified in family 1 and a nonsense variant (GenBank: NM_005612.4; exon 4; c.1310T>A [p.Leu437*]; chr4:57,796,334_T>A was detected in family 2. Both affected siblings from family 1 were found to be heterozygous for the identified frameshift c.2865_2866delAA (p.Asn958Serfs*9) variant allele, the mother was found to be wild-type, while the mildly affected father also appeared to contain two wild-type alleles. However, the observed mild HGF phenotype in the father suggested possible tissue-specific mosaicism and transmission of the frameshift variant to the affected siblings from the father. In family 2, all five affected individuals, including the proband, two siblings, mother, and a maternal uncle were heterozygous carriers for the WES-identified stop-gain variant (c.1310T>A [p.Leu437*]) and the unaffected maternal aunt were wild-type at this locus. In family 3 the de novo REST variant (GenBank: NM_005612.4; exon 4; c.2413delC [p.Leu805Phefs*38]; chr4:57,797,436_CC>C) was identified in the proband. The REST gene is located on chromosome 4q12 and encodes a 1097-amino acid zinc finger protein. REST protein has a critical function as a transcriptional repressor during embryogenesis and neurogenesis and exerts an important role in several cellular mechanisms, such as tumor-suppressor as well as osteoblast, cardiac and hematopoietic differentiation [51–55]. The data from computational algorithm performed by Bayram et al. suggest that truncating variants may act through either a dominant-negative (antimorphic) or gain-of-function (neomorphic) effect, rather than by a haploinsufficiency mechanism, indicating that mutant transcripts may reduce the repressor function of REST in gingival fibroblasts from HGF-affected donors [11]. Although the pathophysiologic mechanisms underlying HGF remain not fully understood, it is considered that the pathologic manifestation of gingival overgrowth is driven by the excessive production of ECM components, particularly collagen type I by gingival fibroblasts [17, 18, 56–59]. Production of ECM is controlled by cytokines and growth factors that initiate and/or modulate signaling cascades mediated by specific receptors expressed by fibroblastic cells. The most potent cytokines involved in many biological processes, including regulation of collagen metabolism in connective tissues are transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF), and IL-6 [58–60]. Kong et al. showed that putative REST target genes were widely involved in TGF-β signaling, whereas the inhibition of REST upregulated the TGF-β signaling pathway [61]. Another study implemented on neuroendocrine differentiation in prostate cancer cells revealed that knockdown of REST activates the IL-6-induced autophagy, while IL-6-induced neuronal cell morphology changes in prostate cancer cells were significantly inhibited due to the REST overexpression [62]. Taken together, it has been speculated that the truncating variants of the REST gene may reduce the repressor function of REST that would induce overexpression of the TGF-β gene and upregulation of TGF-β signaling pathways, indirectly leading to increased synthesis and accumulation of collagen type I in HGF.
Bayram et al. also reported the availability of some truncating variants in the last exon of the REST gene in the public databases, i.e., ExAC [11]. However, since they did not have access to the samples and lack clinical information from that database, they could not validate these putative truncating variants nor check whether the individuals with truncating variants were affected by HGF or not. Importantly, the calling of indels in genome-wide sequencing remains a challenge because of poor mappability [63]. Therefore, the frameshift variants identified in exome/genome sequencing need to be confirmed with DNA (Sanger) sequencing or allele-specific cloning.
Overview of other candidate genes
Xiao et al. identified a four-generation Chinese family in which the gingival overgrowth manifests one year after birth, without the combined phenotypes of mental retardation, deafness, and hypertrichosis, thereby indicating non-syndromic HGF. The genome-wide search enabled to exclude the loci identified erenow for HGF and map the GINGF2 locus on the chromosome 5q13-q22 (MIM 605544) in this family. A strong candidate gene located within this region is calcium/calmodulin-dependent protein kinase IV (CAMK4) (MIM 114080) [9]. It is a multifunctional serine/threonine protein kinase, that mediates Ca2+-signaling pathways and is expressed in lymphocytes, neurons, male germ cells, and gingival tissue. It is, therefore hypothesized, that gingival overgrowth induced by cyclosporine A in genetically susceptible individuals may result from increased phosphorylation and activity of CAMK4 [9, 66, 67].
In the study of Hwang et al., instead of identification of specific pathogenic variants of genes responsible for HGF, the researchers proposed the candidate representative genes of the clusters of related genes potentially associated with non-syndromic HGF using WES followed by GSEA, and a protein functional network study [15]. A three-generation family whose grandparents and parents have normal gingiva while the children have gingival fibromatosis was enrolled in the study. A 13-year-old male showed gingival and alveolar overgrowth in the maxillary posterior area. His nine-year-old sister reported delayed eruption of teeth and was unable to close her mouth completely due to generalized gingival overgrowth. Her panoramic radiograph revealed delayed root development of primary second maxillary molars and mandibular canines. These two affected individuals had no history of systemic disease in pediatrics. Histological examinations of tissues obtained from the patients showed gingival fibrosis with myxoid changes. Protein functional network analysis was performed to see how genes from diverse functions were associated with each other in biological coordination and how the interactions between them might be associated with the pathogenesis of HGF. To better understand the function of genetic variants from HGF patients, the investigators searched for significantly enriched functional clusters. Among the top 10 clusters ranked by GSEA, multiple genes sets related to fibronectin, autoimmunity/immunity, C2 domain, myosin, microtubule, epidermal growth factor (EGF), GTPase, ATP binding, SH3 domain, and immunoglobulin were discovered [15]. In brief, ATP binding and fibronectin clusters were located in the center of a functional association network, which implies that fibronectin and ATP binding might be associated with the central cause of gingival hyperplasia. In turn, the immunoglobulin and EGF clusters were linked with fibronectin, while C2 domain, GTPase, myosin, and microtubule clusters were linked with ATP binding.
Conclusions
Although the studies on the genetic background of HGF have been initiated in the late’90s, the pathogenic variants inducing HGF were discovered only in the SOS-1 and the REST genes. The CAMK4 was identified as a strong candidate gene, while fibronectin and ATP binding clusters were found as candidate representatives of the clusters of related genes potentially associated with non-syndromic HGF. Altogether, these observations clearly indicate the genetic heterogeneity of non-syndromic HGF and point out the importance of further experimental as well as bioinformatic studies of the SOS-1, REST, and other genes located within previously reported candidate chromosomal regions as well as initially described candidate genes and/or associated clusters of genes, such as fibronectin, employing numerous unrelated multi-generation families probing to identify novel pathogenic variants of genes associated with HGF. This strategy will help to unravel the molecular mechanisms underlying gingival overgrowth, followed by the identification of specific targets for less invasive therapies into the dental practice.
Acknowledgements
We are grateful to Professor Aleksander L. Sieron, PhD from the Department of Molecular Biology and Genetics, Faculty of Medical Sciences in Katowice, Medical University of Silesia, Poland for critical comments to this review.
Abbreviations
- AD
Autosomal dominant
- AR
Autosomal recessive
- CAMK4
Calcium/calmodulin-dependent protein kinase IV
- CTGF
Connective tissue growth factor
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- ERK1/2
Extracellular signal-regulated kinase 1/2
- GEF
Guanine nucleotide exchange factor
- GF
Gingival fibromatosis
- Grb2
Growth factor receptor-bound protein 2
- GSEA
Gene set enrichment analysis
- HGF
Hereditary gingival fibromatosis
- HGF1
Non-syndromic hereditary gingival fibromatosis type 1
- MAPK
Mitogen-activated protein kinase
- MI
Maternal inheritance
- O(MIM)
Online Mendelian Inheritance in Man
- PI3K
Phosphoinositide-3 kinase
- RalGDS
Ral nucleotide exchange factor
- REST
RE1-silencing transcription factor
- SH3
SRC Homology 3 domains
- SOS-1
Son-of-Sevenless-1
- TGF-β
Transforming growth factor-β
- WES
Whole-exome sequencing
Authors' contributions
KS: Literature Search, Methodology, Data Acquisition, Data Analysis. AD: Literature Search, Methodology, Visualisation. KŁB: Conceptualization, Data Acquisition, Data Analysis. AMG: Data Analysis, Visualisation, Writing-Reviewing and Editing. EG: Literature Search, Methodology, Data Analysis. TK: Conceptualization, Writing-Reviewing and Editing. PP: Writing- Reviewing and Editing, Supervision. KG: Conceptualization and Design, Methodology, Writing-Original Draft Preparation, Supervision. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study. All articles reviewed for this study are mentioned in this article.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Häkkinen L, Csiszar A. Hereditary gingival fibromatosis: characteristics and novel putative pathogenic mechanisms. J Dent Res. 2007;86(1):25–34. doi: 10.1177/154405910708600104. [DOI] [PubMed] [Google Scholar]
- 2.Millet C, Rodier P, Farges JC, Labert N, Duprez JP. Surgical and prosthetic treatment in an elderly patient affected by unilateral idiopathic gingival fibromatosis: a case report. Gerodontology. 2012;29:e1185–1189. doi: 10.1111/j.1741-2358.2011.00564.x. [DOI] [PubMed] [Google Scholar]
- 3.Gawron K, Łazarz-Bartyzel K, Potempa J, Chomyszyn-Gajewska M. Gingival fibromatosis: clinical, molecular and therapeutic issues. Orphanet J Rare Dis. 2016;11:9. doi: 10.1186/s13023-016-0395-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorlin R, Cohen MM, Levis LS. Syndromes of the head and neck (Oxford monographs on medical genetics) 3. New York: Oxford University Press; 1990. [Google Scholar]
- 5.Seymour RA, Thomason JM, Ellis JS. The pathogenesis of drug-induced gingival overgrowth. J Clin Periodontol. 1996;23(3 Pt 1):165–175. doi: 10.1111/j.1600-051x.1996.tb02072.x. [DOI] [PubMed] [Google Scholar]
- 6.Hart TC, Pallos D, Bowden DW, Bolyard J, Pettenati MJ, Cortelli JR. Genetic linkage of hereditary gingival fibromatosis to chromosome 2p21. Am J Hum Genet. 1998;62(4):876–883. doi: 10.1086/301797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye X, Shi L, Cheng Y, Peng Q, Huang S, Liu J, et al. A novel locus for autosomal dominant hereditary gingival fibromatosis, GINGF3, maps to chromosome 2p22.3-p23.3. Clin Genet. 2005;68(3):239–244. doi: 10.1111/j.1399-0004.2005.00488.x. [DOI] [PubMed] [Google Scholar]
- 8.Xiao S, Wang X, Qu B, Yang M, Liu G, Bu L, et al. Refinement of the locus for autosomal dominant hereditary gingival fibromatosis (GINGF) to a 3.8-cM region on 2p21. Genomics. 2000;68(3):247–252. doi: 10.1006/geno.2000.6285. [DOI] [PubMed] [Google Scholar]
- 9.Xiao S, Bu L, Zhu L, Zheng G, Yang M, Qian M, et al. A new locus for hereditary gingival fibromatosis (GINGF2) maps to 5q13-q22. Genomics. 2001;74(2):180–185. doi: 10.1006/geno.2001.6542. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Zhang W, Huo Z, Zhang Y, Xia Y, Li B, et al. A novel locus for maternally inherited human gingival fibromatosis at chromosome 11p15. Hum Genet. 2007;121(1):113–123. doi: 10.1007/s00439-006-0283-1. [DOI] [PubMed] [Google Scholar]
- 11.Bayram Y, White JJ, Elcioglu N, Cho MT, Zadeh N, Gedikbasi A, et al. REST final-exon-truncating mutations cause hereditary gingival fibromatosis. Am J Hum Genet. 2017;101(1):149–156. doi: 10.1016/j.ajhg.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet. 2002;70(4):943–954. doi: 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Sun Z, Hu Y, Liu Y, Jin L, Zhang F. Non-syndromic hereditary gingival fibromatosis in three Chinese families is not due to SOS1 gene mutations. Cell Biochem Biophys. 2014;70:1869–1873. doi: 10.1007/s12013-014-0144-9. [DOI] [PubMed] [Google Scholar]
- 14.Gawron K, Bereta G, Nowakowska Z, Łazarz-Bartyzel K, Potempa J, Chomyszyn-Gajewska M, et al. Analysis of mutations in the SOS-1 gene in two Polish families with hereditary gingival fibromatosis. Oral Dis. 2017;23:983–989. doi: 10.1111/odi.12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang J, Kim YL, Kang S, Kim S, Kim SO, Lee JH, et al. Genetic analysis of hereditary gingival fibromatosis using whole exome sequencing and bioinformatics. Oral Dis. 2017;23(1):102–109. doi: 10.1111/odi.12583. [DOI] [PubMed] [Google Scholar]
- 16.Shi J, Lin W, Li X, Zhang F, Hong X. Hereditary gingival fibromatosis: a three-generation case and pathogenic mechanism research on progress of the disease. J Periodontol. 2011;82(7):1089–1095. doi: 10.1902/jop.2010.100599. [DOI] [PubMed] [Google Scholar]
- 17.Gawron K, Ochała-Kłos A, Nowakowska Z, Bereta G, Łazarz-Bartyzel K, Grabiec AM, et al. TIMP-1 association with collagen type I overproduction in hereditary gingival fibromatosis. Oral Dis. 2018;24(8):1581–1590. doi: 10.1111/odi.12938. [DOI] [PubMed] [Google Scholar]
- 18.Gawron K, Łazarz-Bartyzel K, Fertala A, Plakwicz P, Potempa J, Chomyszyn-Gajewska M. Gingival fibromatosis with significant de novo formation of fibrotic tissue and a high rate of recurrence Version 2. Am J Case Rep. 2016;17:655–659. doi: 10.12659/AJCR.899997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breen GH, Addante R, Black CC. Early onset of hereditary gingival fibromatosis in a 28-month-old. Pediatr Dent. 2009;31:286–288. [PubMed] [Google Scholar]
- 20.Goyal L, Bey A, Gupta ND, Varshney A. Diagnosis and management of nonsyndromic hereditary gingival fibromatosis in a 13 year old girl: report of a rare case. Contemp Clin Dent. 2012;3(Suppl 2):S210–S213. doi: 10.4103/0976-237X.101097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhadse PV, Yeltiwar RK, Pandilwar PK, Gosavi SR. Hereditary gingival fibromatosis. J Indian Soc Periodontol. 2012;16:606–609. doi: 10.4103/0972-124X.106930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandhu SP, Kakar V, Gogia G, Narula SC. Unilateral gingival fibromatosis with localized aggressive periodontitis (involving first molars): an unusual case report. J Indian Soc Periodontol. 2009;13:109–113. doi: 10.4103/0972-124X.55834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casavecchia P, Uzel MI, Kantarci A, Hasturk H, Dibart S, Hart TC, et al. Hereditary gingival fibromatosis associated with generalized aggressive periodontitis: a case report. J Periodontol. 2004;75(5):770–778. doi: 10.1902/jop.2004.75.5.770. [DOI] [PubMed] [Google Scholar]
- 24.Baptista IP. Hereditary gingival fibromatosis: a case report. J Clin Periodontol. 2002;29:871–874. doi: 10.1034/j.1600-051x.2002.290913.x. [DOI] [PubMed] [Google Scholar]
- 25.Gawron K, Lazarz-Bartyzel K, Lazarz M, Steplewska K, Pyrc K, Potempa J, et al. In vitro testing the potential of a novel chimeric IgG variant for inhibiting collagen fibrils formation in recurrent hereditary gingival fibromatosis: chimeric antibody in a gingival model. J Physiol Pharmacol. 2014;65:585–591. [PubMed] [Google Scholar]
- 26.Zhou M, Xu L, Meng HX. Diagnosis and treatment of a hereditary gingival fibromatosis case. Chin J Dent Res. 2011;14:155–158. [PubMed] [Google Scholar]
- 27.Tripathi AK, Dete G, Saimbi CS, Kumar V. Management of hereditary gingival fibromatosis: a 2 years follow-up case report. J Indian Soc Periodontol. 2015;19:342–344. doi: 10.4103/0972-124X.148643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavvadia K, Pepelassi E, Alexandridis C, Arkadopoulou A, Polyzois G, Tossios K. Gingival fibromatosis and significant tooth eruption delay in an 11-year-old male: a 30-month follow-up. Int J Paediatr Dent. 2005;15:294–302. doi: 10.1111/j.1365-263X.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- 29.Kelekis-Cholakis A, Wiltshire WA, Birek C. Treatment and long-term follow-up of a patient with hereditary gingival fibromatosis: a case report. J Can Dent Assoc. 2002;68:290–294. [PubMed] [Google Scholar]
- 30.Salinas CF. Orodental findings and genetic disorders. Birth Defects Orig Artic Ser. 1982;18(1):79–120. [PubMed] [Google Scholar]
- 31.Ramon Y, Berman W, Bubis JJ. Gingival fibromatosis combined with cherubism. Oral Surg. 1967;24:436–448. doi: 10.1016/0030-4220(67)90416-1. [DOI] [PubMed] [Google Scholar]
- 32.Suhanya J, Aggarwal C, Mohideen K, Jayachandran S, Ponniah I. Cherubism combined with epilepsy, mental retardation and gingival fibromatosis (Ramon syndrome): a case report. Head Neck Pathol. 2010;4:126–131. doi: 10.1007/s12105-009-0155-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones G, Wilroy RS, Jr, McHaney V. Familial gingival fibromatosis associated with progressive deafness in five generations of a family. Birth Defects Orig Art Ser. 1977;13:195–201. [PubMed] [Google Scholar]
- 34.Hartsfield JK, Jr, Bixler D, Hazen RH. Gingival fibromatosis with sensorineural hearing loss: an autosomal dominant trait. Am J Med Genet. 1985;22:623–627. doi: 10.1002/ajmg.1320220323. [DOI] [PubMed] [Google Scholar]
- 35.Stefanova M, Atanassov D, Krastev T, Fuchs S, Kutsche K. Zimmermann-Laband syndrome associated with a balanced reciprocal translocation t(3;8) (p21.2;q24.3) in mother and daughter: molecular cytogenetic characterization of the breakpoint regions. Am J Med Genet. 2003;117:289–294. doi: 10.1002/ajmg.a.10174. [DOI] [PubMed] [Google Scholar]
- 36.Kim HG, Higgins AW, Herrick SR, Kishikawa S, Nicholson L, Kutsche K, et al. Candidate loci for Zimmermann–Laband syndrome at 3p14.3. Am J Med Genet A. 2007;143A:107–111. doi: 10.1002/ajmg.a.31544. [DOI] [PubMed] [Google Scholar]
- 37.Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Regulation of Sos activity by intramolecular interactions. Mol Cell Biol. 1998;18(2):880–886. doi: 10.1128/mcb.18.2.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 39.Nimnual A, Bar-Sagi D. The two hats of SOS. Sci STKE. 2002;202(145):pe36. doi: 10.1126/stke.2002.145.pe36. [DOI] [PubMed] [Google Scholar]
- 40.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401:290–293. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- 42.Innocenti M, Tenca P, Frittoli E, Faretta M, Tocchetti A, Di Fiore PP, et al. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol. 2002;156:125–136. doi: 10.1083/jcb.200108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sini P, Cannas A, Koleske AJ, Di Fiore PP, Scita G. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat Cell Biol. 2004;6:268–274. doi: 10.1038/ncb1096. [DOI] [PubMed] [Google Scholar]
- 44.Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- 45.Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- 46.Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, et al. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell. 2001;8:115–127. doi: 10.1016/s1097-2765(01)00285-4. [DOI] [PubMed] [Google Scholar]
- 47.McCollam L, Bonfini L, Karlovich CA, Conway BR, Kozma LM, Banerjee U, et al. Functional roles for the pleckstrin and Dbl homology regions in the Ras exchange factor Son-of-sevenless. J Biol Chem. 1995;270(27):15954–15957. doi: 10.1074/jbc.270.27.15954. [DOI] [PubMed] [Google Scholar]
- 48.Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell. 2000;102(2):211–220. doi: 10.1016/s0092-8674(00)00026-x. [DOI] [PubMed] [Google Scholar]
- 49.Lee EJ, Jang SI, Pallos D, Kather J, Hart TC. Characterization of fibroblasts with Son-of Sevenless-1 mutation. J Dent Res. 2006;85(11):1050–1055. doi: 10.1177/154405910608501115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gawron K, Łazarz-Bartyzel K, Kowalska A, Bereta G, Nowakowska Z, Plakwicz P, et al. Fibroblasts from recurrent fibrotic overgrowths reveal high rate of proliferation in vitro-findings from the study of hereditary and idiopathic gingival fibromatosis. Connect Tissue Res. 2019;60(1):29–39. doi: 10.1080/03008207.2018.1517758. [DOI] [PubMed] [Google Scholar]
- 51.Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 52.Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 53.Thakore-Shah K, Koleilat T, Jan M, John A, Pyle AD. REST/NRSF knockdown alters survival, lineage differentiation and signaling in human embryonic stem cells. PLoS ONE. 2015;10:e0145280. doi: 10.1371/journal.pone.0145280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwahara K. Role of NRSF/REST in the regulation of cardiac gene expression and function. Circ J. 2013;77:2682–2686. doi: 10.1253/circj.cj-13-1210. [DOI] [PubMed] [Google Scholar]
- 55.Liu B, Cheng S, Xing W, Pourteymoor S, Mohan S. RE1-silencing transcription factor (Rest) is a novel regulator of osteoblast differentiation. J Cell Biochem. 2015;116:1932–1938. doi: 10.1002/jcb.25148. [DOI] [PubMed] [Google Scholar]
- 56.Tipton DA, Howell KJ, Dabbous MK. Increased proliferation, collagen, and fibronectin production by hereditary gingival fibromatosis fibroblasts. J Periodontol. 1997;68:524–530. doi: 10.1902/jop.1997.68.6.524. [DOI] [PubMed] [Google Scholar]
- 57.Coletta RD, Almeida OP, Ferreira LR, Reynolds MA, Sauk JJ. Increase in expression of Hsp47 and collagen in hereditary gingival fibromatosis is modulated by stress and terminal procollagen N-propeptides. Connect Tissue Res. 1999;40:237–249. doi: 10.3109/03008209909000702. [DOI] [PubMed] [Google Scholar]
- 58.Tipton DA, Dabbous MK. Autocrine transforming growth factor beta stimulation of extracellular matrix production by fibroblasts from fibrotic human gingiva. J Periodontol. 1998;69(6):609–619. doi: 10.1902/jop.1998.69.6.609. [DOI] [PubMed] [Google Scholar]
- 59.Martelli-Junior H, Cotrim P, Graner E, Sauk JJ, Coletta RD. Effect of transforming growth factor-beta1, interleukin-6, and interferon-gamma on the expression of type I collagen, heat shock protein 47, matrix metalloproteinase (MMP)-1 and MMP-2 by fibroblasts from normal gingiva and hereditary gingival fibromatosis. J Periodontol. 2003;74:296–306. doi: 10.1902/jop.2003.74.3.296. [DOI] [PubMed] [Google Scholar]
- 60.Trackman PC, Kantarci A. Connective tissue metabolism and gingival overgrowth. Crit Rev Oral Biol Med. 2004;15(3):165–175. doi: 10.1177/154411130401500305. [DOI] [PubMed] [Google Scholar]
- 61.Kong QR, Xie BT, Zhang H, Li JY, Huang TQ, Wei RY, et al. RE1-silencing transcription factor (REST) is required for nuclear reprogramming by inhibiting transforming growth factor β signaling pathway. J Biol Chem. 2016;291:27334–27342. doi: 10.1074/jbc.M116.743849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang PC, Wang TY, Chang YT, Chu CY, Lee CL, Hsu HW, et al. Autophagy pathway is required for IL-6 induced neuroendocrine differentiation and chemoresistance of prostate cancer LNCaP cells. PLoS ONE. 2014;9:e88556. doi: 10.1371/journal.pone.0088556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Z, Liu X, Yang BZ, Gelernter J. The role and challenges of exome sequencing in studies of human diseases. Front Genet. 2013;4:60. doi: 10.3389/fgene.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shashi V, Pallos D, Pettenati MJ, Cortelli JR, Fryns JP, von Kap-Herr C, et al. Genetic heterogeneity of gingival fibromatosis on chromosome 2p. J Med Genet. 1999;36:683–686. [PMC free article] [PubMed] [Google Scholar]
- 65.https://omim.org/entry/135300.
- 66.https://omim.org/entry/605544.
- 67.https://omim.org/entry/114080.
- 68.https://omim.org/entry/609955.
- 69.https://omim.org/entry/611010.
- 70.https://omim.org/entry/617626.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study. All articles reviewed for this study are mentioned in this article.