

Summary
Microdroplet single-cell ATAC-seq is widely used to measure chromatin accessibility, however highly scalable and simple sample multiplexing procedures are not available. Here, we present a transposome-assisted single nucleus barcoding approach for ATAC-seq (SNuBar-ATAC) that utilizes a single oligonucleotide adapter for multiplexing samples during the existing tagmentation step and does not require a pre-labeling procedure. The accuracy and scalability of SNuBar-ATAC was evaluated using cell line mixture experiments. We applied SNuBar-ATAC to investigate treatment-induced chromatin accessibility dynamics by multiplexing 28 mice with lung tumors that received different combinations of chemo, radiation, targeted and immunotherapy. We also applied SNuBar-ATAC to study spatial epigenetic heterogeneity by multiplexing 32 regions from a human breast tissue. Additionally, we show that SNuBar can multiplex single cell ATAC&RNA multiomic assays in cell lines and human breast tissue samples. Our data show that SNuBar is a highly accurate, easy-to-use and scalable system for multiplexing scATAC-seq and scATAC&RNA-seq experiments.
Graphical Abstract
In brief
Wang et.al reported the development of an accurate, easy-to-use and scalable system (SNuBar) for multiplexing single cell ATAC or multi-omics (ATAC&RNA) assays, and applied this method to multiplex therapy combinations in a lung cancer mouse model and macro-spatial regions of normal breast tissues and samples.
Introduction
Single-cell ATAC-seq (scATAC-seq) methods have emerged as powerful tools to measure the chromatin accessibility landscape and investigate the role of transcription factors at promoters, enhancers, activators and insulators at single cell genomic resolution (Cusanovich et al., 2015)(Buenrostro et al., 2015)(Chen et al., 2018)(Mezger et al., 2018). While the first-generation scATAC-seq methods were generally low-throughput and had substantial technical noise, the development of microdroplet-based technologies (e.g., 10X Genomics and dscATAC-seq) have overcome these technical limitations and enabled the profiling of thousands of single cells in parallel (Lareau et al., 2019; Satpathy et al., 2019). While microdroplet-based methods can generate data on thousands of cells from a single sample, it remains difficult and expensive to run these assays on large sample collections, since each sample must be run in a single microfluidics channel. While methods such as combinatorial indexing can enable sample multiplexing (eg. dscATAC-seq), most of these methods require complex molecular processing to generate a large quantity of custom modified Tn5 transposomes (Lareau et al., 2019). These issues represent a major technical barrier for studies that require the profiling of large-scale sample numbers for scATAC-seq, including combinatorial drug screening, large-scale patient cohort studies, longitudinal time-series experiments and multi-spatial sampling of normal or malignant tissues.
Several different multiplexing methods have been developed for single-cell RNA sequencing (scRNA-seq) that perform indexing using existing genetic information such as Demuxlet (Kang et al., 2018), CellTag indexing (Guo et al., 2019) and Perturb-Seq (Dixit et al., 2016), or by using methods to label cells with antibody-based cell hashing (Stoeckius et al., 2018), lipid based MULTI-seq (McGinnis et al., 2019), transient barcoding (Shin et al., 2019) or by staining with oligonucleotides followed by fixation (sci-Plex) (Srivatsan et al., 2020). In contrast, there are limited methods available for multiplexing large-scale scATAC-seq experiments (Fiskin et al., 2020; Mimitou et al., 2020) and no methods available for multiplexing single cell ATAC&RNA co-assays (10X Genomics) on microdroplet platforms. Furthermore, while several prior methods for multiplexing scRNA-seq experiments have been developed, they have low scalability, a limited scope of applications (McGinnis et al., 2019) or complex requirements to synthesize custom chemical reagents (such as lipid conjugates, modified oligos, protein-conjugated antibodies or formalin fixation). Notably, all of these approaches require additional experimental steps to barcode and label the cells in advance that creates additional lab work for the users.
Here, we report an important technical advance, in the development of a transposome-assisted Single Nucleus Barcoding (SNuBar) approach for scATAC-seq that can easily label and multiplex a large number of samples together for parallel sequencing in a single microdroplet experiment (eg. 10X Genomics). The SNuBar-ATAC approach utilizes a simple workflow by adding a single oligonucleotide barcode during the existing tagmentation step in the scATAC assays to label each sample with a unique identifier that is demultiplexed in the data post-processing steps. We validated the performance, efficiency and scalability of SNuBar-ATAC in cell line experiments and applied it to two different biological applications, including profiling chromatin accessibility changes induced by drug treatment combinations and studying macro-spatial areas of breast tissue regions. Additionally, we applied SNuBar for multiplexing single cell ATAC&RNA co-assays of combined samples from cell lines and human breast tissues.
Results
Design
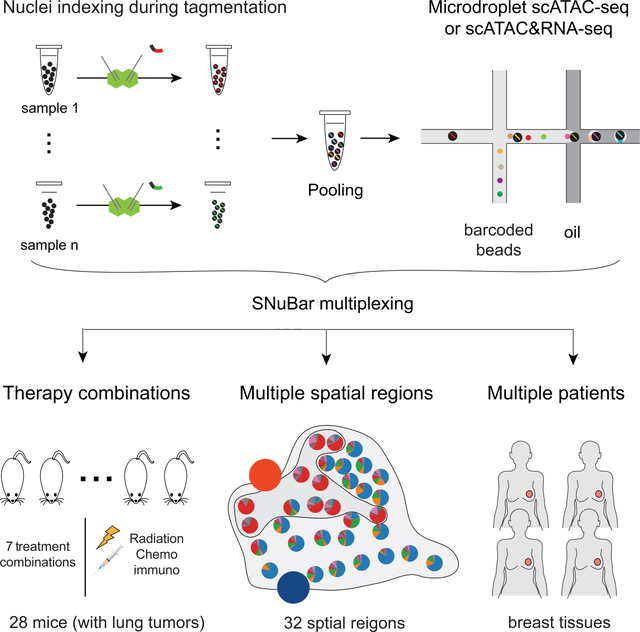
SNuBar uses a simple oligonucleotide adapter to perform single nuclei barcoding by integrating into the universal adaptors of the Tn5 transposome. The SNuBar-ATAC barcode oligonucleotide (SBO) is comprised of three parts: 1) a Complementary Sequence of the Transposome (CST) universal tail, which enables the barcode adapter to hybridize to the universal tails of the transposome, and is amplified by the primer with the cell barcodes, 2) the sample barcode sequence and Unique Molecular Identifier (UMI), which are used to index each sample and remove PCR duplicates from the sample barcodes, and 3) the PCR handle which enables the whole sequence to be amplified (Figure S1A). The SBO is delivered into the nuclei via two simultaneous methods: 1) by hybridizing directly to the transposome, followed by delivery into the nuclei by transporting across the nuclear membrane, and 2) by diffusing into the nucleus, followed by hybridizing to the transposome universal tails that are already present inside of the nucleus and bound to the DNA. These two complimentary delivery processes occur concurrently at the same time, labeling each cell with a unique set of sample barcodes that are encoded in the SBO.
To perform the whole procedure, nuclei are isolated from cell suspensions or macro-dissected from tissue regions and used for tagmentation reactions (Figure 1A, Methods). SBO with unique sets of barcode sequences are then added during the tagmentation reaction to each sample and incubated. The barcoded nuclei from different samples are then pooled together and loaded into a high-throughput droplet-based scATAC-seq platform (eg. 10X Genomics). The scATAC-seq library and the sample barcode library are prepared and mixed for sequencing (Figure S1B). In the same cell, the scATAC and sample barcode libraries have identical cell barcodes, which allows the data to be matched for each cell in the data post-processing steps, to organize groups of cells together with their respective SBO sample barcodes.
Figure 1. SNuBar-ATAC workflow and technical performance.

(A) Overview of the SNuBar-ATAC workflow. Compared to the standard 10X scATAC workflow, SNuBar-ATAC involves adding a unique SBO oligonucleotide to each sample during the tagmentation step to perform sample multiplexing. The barcoded samples are then pooled together and loaded into a microdroplet platform (10X Genomics) to perform scATAC-seq.
(B) SNuBar-ATAC quality control plots for A20 cells from the 10X (10XSTD), SNuBar-HighAK and SNuBar-LowAK experiments, in which each dot represents one droplet with at least 100 fragments.
(C) Comparison of the aggregated counts per million (CPM) fragments within peaks for the A20 cell line in 10XSTD, SNuBar-HighAK, and SNuBar-LowAK.
(D) Comparison of scATAC profiles of the A20 cells from the 10XSTD, SNuBar-HighAK, and SNuBar-LowAK experiments in a region of chromosome 7. Upper panels show the aggregated profiles of all cells and lower panels show fragments present in each of the 100 random cells.
(E-F) Upper panels show a heatmap of cell numbers from different species determined by barnyard analysis (rows) and SNuBar barcode classifications (columns). Lower panels show a t-SNE plot colored by the SNuBar barcode classifications for SNuBar-HighAK (E) and SNuBar-LowAK (F).
(G) Experimental workflow used to assess the scalability of SNuBar-ATAC by multiplexing 96 samples from three cell lines.
(H) UMAP of the scATAC-seq profiles from MDA-MB-231, MDA-MB-436, and K562 cells.
(I) Clustered heatmap showing copy number aberrations inferred from the 96-plex SNuBar-ATAC data.
(J) Histogram showing the frequencies of SBO counts in the singlets from the three cell lines.
(K) Heatmap showing the normalized SBO counts from 1,000 random single cells with the SNuBar classifications and the inferred cell lines indicated.
See also Figures S1, S2 and Table S1.
Evaluation of Technical Performance and Scalability
To evaluate the technical performance of SNuBar-ATAC for multiplexing different samples, we used SNuBar-ATAC to label a mouse B lymphocyte (A20) and a human bone marrow cell line (K562) with two unique SBOs and pooled them together for high-throughput scATAC-seq on a microdroplet (10X Genomics) platform (STAR Methods). Two independent replicate experiments were performed, in which the nuclei from one sample was overloaded (SNuBar-HighAK) and another in which the nuclei were not overloaded (SNuBar-LowAK) during microdroplet generation to evaluate the doublet rate and reproducibility of our method. In the resulting data we removed low quality nuclei using cut-offs of 1000 unique nuclear fragments per cell and a transcription start site (TSS) enrichment score of 8, as previously described (Satpathy et al., 2019). From this data we estimated that 53.2% and 62.3% of the fraction of transposition events were located in peaks of the SNuBar-HighAK and SNuBar-LowAK experiments respectively, suggesting that the accessibility profiles of SNuBar are highly enriched for open chromatin under both conditions. Furthermore, we found that the aggregate chromatin accessibility of SNuBar A20 was highly correlated (Pearson’s R = 0.97, p < 2.2e−16) to standard 10X Genomics A20 data generated without barcoding, and also highly correlated (Pearson’s R = 0.99, p < 2.2e−16) between the two independent experiments (Figure 1B–D, STAR Methods). Similarly, we found that the SNuBar accessibility profiles for K562 were highly correlated (Pearson’s R = 0.84, p < 2.2e−16) between two replicate experiments and correlated with dscATAC-seq data (Figure S1C–E, STAR Methods). We next used the ATAC-seq profiles from A20 and K562 as ground truth profiles to evaluate the efficiency of SNuBar barcoding. This analysis showed that 98.91% (5725/5788) of s1 (barcode for A20) barcoded cells were correctly matched with A20 cells, and 99.97% of s2 (barcode for K562) cells were correctly matched with the K562 in the SNuBar-HighAK data. Similarly, 99.97% and 99.49% of s1 and s2 barcoded cells were correctly matched with A20 and K562 in the SNuBar-LowAK data, respectively (Figure 1E–F). Moreover, SNuBar identified hundreds of cell doublets that cannot easily be distinguished from the chromatin accessibility signals alone in the SNuBar-HighAK and SNuBar-LowAK data (Figure 1E–F, Figure S1F). In addition, we show that the SNuBar-ATAC assay is compatible with the NextGEM microdroplet chemistry for the scATAC-seq assay (10X Genomics) using the A20 and K562 cell lines (Figure S2A–H).
To further evaluate the scalability of SNuBar-ATAC, we multiplexed 96 different samples from three cell lines, including two breast cancer cell lines (MDA-MB-231, MDA-MB-436) and the K562 leukemia cell line (32 aliquots of each cell line) in a single microdroplet experiment (Figure 1G, Table S1). We obtained scATAC profiles from 8,702 single cells passing quality control (QC) (Figure S2I, STAR Methods). To evaluate the barcoding efficiency and crossover contamination of SNuBar-ATAC, we first determined the ground-truth identities of each nucleus by combining the results from two different approaches: 1) copy number profiles inferred from ATAC fragments, and 2) SNP-based sample identification. To determine the sample identity from the copy number profiles, we computed aneuploid copy number profiles of each nucleus from the ATAC-seq read counts and compared the CNA markers to previous studies. For cluster-1, chromosomal gains of chr6p, chr7q and a loss of chr9 were detected and were consistent with previous data from K562 (Zhou et al., 2019; Dixon et al., 2018), while cluster-2 had gains of chr3p, chr5, chr14 and chr20q as previously reported for MDA-MB-231 (Johnstone et al., 2018), and cluster-3 showed gains of chr8q, chr15p and a loss of chr6p. For the SNP based approach, we deconvoluted the nuclei identities by single nucleotide polymorphisms (SNPs) using “demuxlet” (Kang et al., 2018) (Figures 1H–I, STAR Methods). The identities of the three clusters were further supported by the inferred RNA expressions of the cell line specific genes (Figure S2J). Next, we investigated the barcode crossover contamination by evaluating sample barcode distributions across different cells. The mean on-target barcode for singlets was 594 counts, while the most abundant off-target number was 10 counts on average, indicating that each cell was exclusively enriched for a single barcode (Figure 1J). Importantly, the SNuBar barcodes were able to identify all 32 samples from each cell line and matched 100% of the barcodes assigned to each cell line (Figure 1K). Furthermore, SNuBar-ATAC identified 985 additional multiplets distributed across different samples from the “singlets” classified by chromatin accessibility signals alone (Figure 1K). Collectively, these data show that SNuBar-ATAC is a highly accurate and scalable method for multiplexing microdroplet scATAC-seq experiments for parallel analysis.
Multiplexing drug treatment combinations
To investigate the chromatin accessibility dynamics induced by therapeutic treatment combinations, we applied SNuBar-ATAC to multiplex 28 mice with syngeneic lung tumors derived from the LKR13 lung cancer cell line. The mice were treated with 7 different treatment combinations that included different doses of radiation, chemotherapy, PD-L1 checkpoint inhibitors and a targeted PARP inhibitor (Talazoparib) (Figure 2A, STAR Methods). SNuBar-ATAC barcoded 28 samples were pooled together for analysis using a microdroplet platform (10X Genomics) resulting in 9,280 single nuclei that passed QC (STAR Methods). In average, 1,325 cells from each treatment group (331 cells per mouse) were analyzed. For each single nucleus, we obtained an average of 6,893 ATAC fragments, of which 34% were located within peaks and an average of 803 counts with on-target SNuBar barcodes (Figure 2B, Figure S3A–B). High-dimensional clustering of the scATAC-seq profiles identified 5 major clusters that corresponded to one epithelial population of tumor cells, one fibroblast cluster, and three immune clusters consisting of T-cells, B-cells, myeloid cells based on the TSS-flanking scATAC signals and the inferred expression of cell type-specific marker genes (Figure 2C–D, Figure S3C).
Figure 2. Multiplexing of therapeutic treatment conditions from a lung cancer model.

(A) Experimental workflow for the treatment combinations used to treat syngeneic mice with LKR13 lung cancer cell line injections.
(B) SNuBar-ATAC barcode distribution for single cells.
(C) UMAP projection of scATAC profiles of tumor and non-malignant cell types from all mice samples across all treatment conditions.
(D) Track plots showing the normalized fragment counts per cell type within the TSS-flanking regions of the known genes for specific cell types.
(E) UMAP projection of scATAC profiles showing cells per treatment group colored by cell densities, with star symbols highlighting differences in cell clusters between the treatment and control groups. The total cell number of each group are indicated in brackets.
(F) Barplot showing the microenvironment cell type percentages in each treatment group.
(G) Volcano plot showing the DAPs in the tumor cells between 4Gy+PDL1+Tala and the control group.
(H) NES (normalized enrichment score) of significantly enriched pathways reported by the module-based GSEA using the up-regulated DAPs in 4Gy+PDL1+Tala compared with the control group.
(I) Volcano plot showing the DAPs within the tumor cells in a comparison between Chemo+2Gy+Tala and the control group.
(J) NES of significantly enriched pathways reported by the module-based GSEA using the up-regulated DAPs in the Chemo+2Gy+Tala group compared with the control group.
See also Figures S3.
We next investigated differences in chromatin accessibility across the cell types that were induced by the various treatment conditions. Compared to the control group, the cells from 4Gy, 4Gy+PDL1, Chemo+2Gy and Chemo+2Gy+PDL1 treatment groups showed similar distributions in high-dimensional space, suggesting that treatment did not have a large impact on their chromatin accessibility profiles. Furthermore, none of the significant differential accessible peaks (DAPs) were detected in the tumor cells in these treatment groups (adjusted p ≥ 0.05, logistic regression). In contrast, the tumor microenvironment of both the 4Gy+PDL1+Tala and Chemo+2Gy+Tala treatment groups showed large decreases in the tumor-infiltrating myeloid cells after treatment (Figure 2E–F). Additionally, many of the tumor cells in these two treatment groups showed large shifts in high-dimensional space, indicating a change of their epigenetic landscapes in response treatment (Figure 2E, Figure S3D–E).
To further investigate significant DAPs in the tumor cells that were affected by treatment, we compared the two treatment groups to the control samples. We identified 43 DAPs that were significantly enriched (fold-change > 1.5, adjusted p < 0.05) and 15 DAPs that were significantly down-regulated (fold-change < −1.5, adjusted p < 0.05) in the 4Gy+PDL1+Tala group (Figure 2G). Several of the top down-regulated DAPs were near genes in cancer-related pathways, such as Hif1α which is involved in hypoxia and angiogenesis (Semenza, 2003) and Dlx4 which promotes tumor migration, invasion, and metastasis (Zhang et al., 2012). In contrast, several of the top up-regulated DAPs were cancer-suppressive, such as Jarid2 (Manceau et al., 2013), Casp2(Puccini et al., 2013) and Sin3a (Suzuki et al., 2008). To further investigate the regulation of DAPs in tumor cells, we performed module-based gene set enrichment analysis (GSEA) using the MSigDB hallmark pathways (STAR Methods). Consistent with the DAP results, this data showed that many oncogenic hallmark pathways were significantly down-regulated (GSEA FDR q < 0.05), such as hypoxia, angiogenesis, and immune-suppressive pathways such as IL6/JAK/STAT3 and TGFβ signaling pathways, suggesting a strong efficacy in combining radiotherapy, immunotherapy, and target therapy to modulate the tumor-immune microenvironment towards a more tumor-suppressive state (Figure 2H).
A similar DAPs analysis of the Chemo+2Gy+Tala treatment group showed that the chromatin accessibility of 175 genes were significantly enriched (fold-change > 1.5, adjusted p < 0.05) in the tumor cells after treatment (Figure 2I). In contrast to the 4Gy+PDL1+Tala group, the top highly-enriched DAPs-nearest genes included several oncogenes, such as KDM1A that was reported to promote proliferation, migration and invasion in non-small cell lung cancer (Lv et al., 2012), MAP4K4 which plays a central role in cell migration and invasion (Collins et al., 2006), and PMEPA1 that was shown to promote tumorigenesis in lung cancer cells (Vo Nguyen et al., 2014). The pathway enrichment analysis showed that most of the cancer-related hallmark pathways were positively enriched in the Chemo+2Gy+Tala group, including DNA repair, UV response, G2M checkpoints and TP53 pathways, suggesting an anti-tumor effect from combining PARP inhibitors with conventional chemoradiation (Figure 2J). Overall, these data show that SNuBar-ATAC can be used to multiplex large combinations of treatment-induced samples to identify epigenetic changes in both the tumor cells and microenvironment.
Resolving macro-spatial epigenetic heterogeneity in tissues
Another application of SNuBar that we investigated was the macro-spatial profiling of different topographic regions of a complex tissue, to delineate the spatial epigenomic heterogeneity of cell types and cell states. We selected a fresh human breast tissue from a reduction mammoplasty and applied SNuBar-ATAC to profile 32 macro-dissected spatial regions that were barcoded and pooled together for a single microdroplet experiment (10X Genomics) (Figure 3A, Figure S3F–G, STAR Methods). High-dimensional analysis identified 8 major clusters corresponding to luminal secretory epithelial cells (LS), luminal hormone-responsive epithelial cells (LHr), basal epithelial cells (Bs), adipocytes (Adp), T-cells (T), myeloid cells (Myl), endothelial cells (Endo) and fibroblasts (Fb) based on the TSS-flanking scATAC signals and the inferred RNA expression of known cell type-specific marker genes (Figure 3B–C, Figure S3H). These cell types showed different frequencies across the 32 spatial regions, with some areas having high levels of adipocytes, while other areas having high levels of LS epithelial cells (Figure 3D). Further unbiased hierarchical clustering of the cell type frequencies from each spatial region identified two distinct clusters that corresponded to high levels of adipocytes (cluster-1) or LS epithelial cells (cluster-2), and one intermixed cluster (cluster-3) (Figure 3E). The single cells from different cell types were mapped to their original spatial locations using the SNuBar barcoding information (Figure 3F). Based on the high cell percentages for adipocytes (> 33%), the spatial regions were assigned into two major topographic areas: fatty areas (A) that included 10 consecutive spatial areas and epithelial areas (E) that included 22 consecutive spatial areas, which were consistent with the hierarchical clustering results (Figure 3E–F). Independent of the SNuBar data, the histological staining supported the topographic designations of these areas as either fatty, or epithelial (ductal or lobular regions) (Figure 3G).
Figure 3. Multiplexing of macro-spatial areas from a normal breast tissue.

(A) Macro-dissection plot of 32 spatial regions from a normal breast tissue labeled by white identifiers (barcoded by SNuBar) and adjacent regions labelled in blue (histology staining).
(B) t-SNE of scATAC-seq profiles of cells obtained from the 32 spatial regions.
(C) Track plots of known breast cell type markers showing the normalized fragments counts aggregated by the cell types at TSS-flanking regions.
(D) Barplots showing the percentages of the 8 cell types across the 32 spatial regions.
(E) Clustered heatmap showing the percentage of cell types for each spatial region.
(F) Pie charts of cell types across the spatial regions of the breast tissue that were sampled, showing two major spatial areas annotated as adipocyte areas (A) or epithelial areas (E).
(G) H&E images at 10X magnification of the spatial regions indicated in panel F.
(H-K) UMAP of scATAC profiles of two fibroblasts cell states (H). Volcano plot of DAPs between the two fibroblast cell states (I). Pie chart maps of the two fibroblast cell state frequencies, showing only regions with more than 5 cells (J). Cell state percentages in the A and E areas with Chi-square test for significance (K).
(L-O) UMAP of scATAC profiles of two luminal epithelial secretory cell states (L). Volcano plot of DAPs between two luminal secretory cell states (M). Pie chart maps of luminal secretory cell state frequencies across the spatial regions in the breast tissue, showing only regions with more than 5 cells (N). Hierarchical clustering of cell state percentages across the spatial regions, in which dendrograms are colored by clusters (O).
See also Figures S3.
We next investigated whether the two major topographic areas (A, E) had an influence on the epigenomic programs of the same cell types, resulting in distinct cell states. Unbiased clustering of scATAC peaks identified two different clusters (F1, F2) of fibroblasts (Figure 3H). DAPs analysis identified significant chromatin accessibility changes for 9 genes between F1 and F2 (fold-change > 1.5, adjusted p-value < 0.05, logistic regression) (Figure 3I). The F1 cell state was significantly enriched (p = 0.002, Chi-square test) in the A area after we mapped the two fibroblast cell states back to their original spatial locations by their spatial barcodes (Figure 3J–K). Several top genes in the F1 cell state were associated with adipocyte functions, such as FTO that acts as a regulator of fat mass, adipogenesis and energy homeostasis (Jia et al., 2011), LRRC8C that regulates fat oil droplets (Tominaga et al., 2004), and WNT2 and WNT10 which have been shown to regulate lipogenic genes to protect against diet-induced metabolic dysfunction (Bagchi et al., 2020). Based on this data, we conclude that the F1 fibroblasts are ‘lipofibroblasts’ that represent a distinct cell state in the adipocyte regions of the breast tissue.
Re-clustering analysis also identified two distinct cell states (LS1, LS2) of luminal secretory epithelial cells (Figure 3L). DAPs analysis identified 47 genes that were significantly enriched in LS1, while 10 genes were significantly enriched in LS2 (fold-change > 1.5, adjusted p-value < 0.05, logistic regression) (Figure 3M). As expected, the LS cells were mainly distributed to the E areas (22/25 regions). However, we found that the two LS cell states were not distributed evenly in the E area, when mapped to their spatial locations (Figure 3N). Instead, hierarchical clustering of the cell states frequencies across the spatial regions identified two major clusters, with high LS1 regions represented in cluster 1, while high LS2 regions were found in cluster 2B (Figure 3O). Moreover, we found that the regions of cluster 1 were located together in the same spatial regions (assigned as area D), while most of regions in cluster 2B were also located together (assigned as area L) (Figure 3N). Histopathological analysis of the D and L epithelial areas showed different macro-epithelial structures, in which the D area corresponded to having more ductal structures, while the L area consisted mainly of lobules (Figure 3G). This data suggests that the LS cells in the ducts and lobules may have different epigenetic profiles and biological functions.
Re-clustering of the endothelial cells also identified two distinct cell states in which Endo1 had significantly enriched chromatin accessibility for 4 genes, while Endo2 had enrichment for 53 genes (fold-change > 1.5, adjusted p-value < 0.05, logistic regression) (Figure S3I–J). The Endo1 cell state enriched genes were related to vascular endothelial cell functions, such as ENG, which is required for normal structure and integrity of adult vasculature(Lee and Blobe, 2007) and regulation of vascular endothelial cell migration (McAllister et al., 1994). In contrast, the Endo2 cell state was enriched for genes that were related with lymphatic endothelial cell functions, including TBX1 which is required for the growth and maintenances of lymphatic vessels(Chen et al., 2010), and FLT4 which is a marker for lymphatic vessels (Kaipainen et al., 1995). These data suggest that Endo1 represents a vascular endothelial cell state, while Endo2 represents a lymphatic endothelial cell state in the human breast tissue. Notably, the two endothelial cell states also showed different spatial distributions (LM, VS) but did not correspond to the A and E areas (Figures S3K–L). Taken together, our data suggests that normal breast tissues have epigenetic heterogeneity in both cell types and cell states, that is influenced by their macro-spatial topography and local cellular neighborhoods.
Multiplexing of single cell ATAC&RNA co-assays with SNuBar
Droplet-based high-throughput single cell ATAC&RNA co-assay sequencing technologies have been developed that can profile both the epigenome and transcriptome simultaneously in thousands of single cells, such as SNARE-seq (Chen et al., 2019) and Chromium Single Cell Multiome ATAC + Gene Expression (10X Genomics). While these methods are capable of running single samples, running large numbers of samples on microdroplet platforms remains difficult due to the high cost and lack of available multiplexing methods. To overcome these technical barriers, we modified the SNuBar oligonucleotide barcode for compatibility with the scATAC&RNA co-assay (SBOC), which consists of a CST sequence that can hybridize to the universal tails of the Tn5 transposome, along with a PCR handle, sample barcode and an additional polyA tail sequence that enables the full sequence to be captured by the poly-T probes on the microdroplet platform (eg. 10X Genomics) cell barcode beads (Figure S4A). Similar to the SNuBar-ATAC approach, the barcodes are introduced during the existing tagmentation step of the ATAC&RNA co-assay experiment (Figure 4A). To evaluate the performance of SNuBar in multiplexing chromatin accessibility and RNA expression co-profiling experiments (SNuBar-ARC), we first barcoded nuclei from the K562 and A20 cell lines during the tagmentation step using SNuBar, followed by loading the barcoded nuclei for high-throughput scATAC&RNA multi-omic sequencing using the microdroplet platform (10X Genomics). In total, we detected 4,277 cells from K562 and 4,435 cells from A20 that passed the QC filters for ATAC-seq. We found that 59.9% of the transposition events were located in ATAC peaks, which is similar to the 53.2% and 62.3% estimated in the SNuBar-HighAK and SNuBar-LowAK experiments, respectively. Furthermore, this data showed that the aggregate chromatin accessibility profiles for SNuBar-ARC were highly consistent with SNuBar-HighAK and SNuBar-LowAK in both the K562 and A20 cells (Figure 4B–D, Figure S4B–D). From the gene expression data, we identified two clusters with median gene counts of 4,723 and 2,034 that corresponding to the K562 and A20 cells, respectively (Figure 4E). The expression profiling of A20 and K562 from SNuBar-ARC were highly consistent (R=0.89 and 0.79 for A20 and K562, respectively) with publicly available bulk (A20 cell lines) datasets (Sl et al., 2020) and single cell (K562) datasets (Dixit et al., 2016) (Figure 4F). Moreover, the high-dimensional clustering results for the scRNA-seq data were highly similar to the chromatin accessibility clustering results (Figure 4G). By matching the cell barcodes between the ATAC and RNA assays, we found that 9,592 cells (80.80%) had both ATAC-seq and RNA expression signals present in the same cells, while 365 cells (3.07%) showed only ATAC signal, and 1,915 cells (16.13%) showed only gene expression signal (Figure 4H). Collectively, these data suggest that the SNuBar barcoding procedure had minimum impact on the chromatin accessibility profiles and gene expression signals that were measured on the microdroplet platform. We also evaluated SNuBar barcoding efficiency by using the cells that were identified as singlets (9,014) by both ATAC-seq signal and gene expression and found that 99.13% of s1 (barcode for A20) barcoded cells were correctly matched with A20 cells, while 99.36% of s2 (barcode for K562) were correctly matched with K562 in the SNuBar-ARC data, suggesting that SNuBar achieved high barcoding efficiency for multiplexing single cell multi-omics experiments (Figure 4I).
Figure 4. Multiplexing of cell lines by SNuBar-ARC.

(A) Overview of scATAC&RNA co-assay in which a single SBOC oligonucleotide is added during the tagmentation step to perform sample multiplexing in SNuBar-ARC. After barcoding, single nuclei are pooled together, and the libraries are loaded into the microdroplet-based scATAC&RNA co-assay.
(B) Quality control for the scATAC data from the A20 cells in SNuBar-ARC.
(C) Comparison of log-normalized fragments within peaks for aggregated A20 cells in SNuBar-ARC, SNuBar-HighAK and SNuBar-LowAK. Each dot denotes a peak.
(D) Comparison of scATAC profiles of the A20 cells from the SNuBar-ARC, SNuBar-HighAK, and SNuBar-LowAK experiments in a genomic region on chromosome 7. Upper panels show aggregated fragments of all cells and lower panels show fragment present in each of the 100 random cells.
(E) Detected gene counts of each single nucleus from A20, K562 and doublet cells using the RNA assay of SNuBar-ARC.
(F) Comparison between the aggregated scRNA data of A20, K562 cells and the public datasets with Pearson’s R and p-values indicated. Each dot represents a gene.
(G) t-SNE projection of chromatin accessibility and gene expression profiles from A20 and K562 cells in the SNuBar-ARC experiments.
(H) Venn diagram showing number of cells detected by one or both assays in SNuBar-ARC.
(I) Heatmap of cell numbers from different species determined by barnyard analysis (rows) and SNuBar barcode classifications (columns).
See also Figure S4.
We next applied SNuBar-ARC to multiplex 4 normal human breast tissue samples (reduction mammoplasties) from 4 different women (Figure 5A). In total, we profiled 10,331 cells that passed the quality control metrics (Figure S5A–B, STAR Methods, Table S2). To further determine the performance of SNuBar-ARC, we used “souporcell”(Heaton et al., 2020), a SNPs based method to determine the sample identity of each cell, which we considered as ground truth (Figure 5B, Figure S5C). This analysis showed that 96.47% (± 2.26%) of the singlets identified by SNuBar-ARC were correctly matched to the “souporcell” sample labels, suggesting a high sample barcoding accuracy of SNuBar-ARC. SNuBar-ARC also identified an additional 10.53% (± 1.69%) of nuclei doublets from the “souporcell” singlet results. High-dimensional plots identified 9 different clusters, which represent three different epithelial cell types (LHr, LS, Bs), three immune cell types (T, Myl, B) and three stromal cell types (Fb, Endo and Pc - pericytes) according to the RNA expression and TSS-flanking ATAC fragments of the known gene markers (Figure 5C, Figure S5D–E). The cell type clusters were represented well in all of the 4 patients (Figure S5F). The top 5 differential expressed genes (DEGs) identified in the RNA profiles were concordant with the gene activity (cis-regulatory interaction) inferred from the chromatin accessible TSS-flanking ATAC fragments (Figure 5D). By performing a more detailed correlation analysis using 850 variable genes in the RNA expression levels and the ATAC gene activity, we found a high average Pearson correlation value of 0.67 (Figure 5E). Next, we measured chromatin accessibility sharing a TF binding motif using chromVAR (Schep et al., 2017) and identified TFs that regulated the cell type identities (Table S3). For example, we found FOXA1 and TP63 had the highest chromVAR scores and specific gene expressions in the LHr and Bs cells respectively (Figure 5F), which had also been previously reported as the lineage TFs in LHr and Bs cells (Pellacani et al., 2016). Additionally, both TF profiles showed cell-type specific ‘footprints’ surrounding the binding motif, suggesting their active roles in regulating the cell type identities (Figure 5G).
Figure 5. Multiplexing of human tissue samples by SNuBar-ARC.

(A) Overview of SNuBar-ARC experimental workflow for multiplexing 4 human breast tissues.
(B) Heatmap of cell numbers from the 4 samples determined by “souporcell” (rows) and SNuBar barcode classifications (columns).
(C) UMAP projection of all single cells based on the weighted-nearest neighbor (wnn) graph integrating the RNA and ATAC modalities. Cells (dots) are colored by the cell types.
(D) Dotplot showing the RNA expression and gene activity (inferred from ATAC) of top 5 DEGs per cell type. The dot color represents the scaled average values. The dot size indicates the percentage of cells in which the gene is detected.
(E) Heatmap showing Pearson correlation of RNA expression and gene activity across different cell types.
(F) UMAP projection of all single cells colored by the RNA expression, gene activity and TF binding potential (measured by chromVar) of FOXA1 and TP63.
(G) Footprints of the TFs FOXA1 and TP63 in three different epithelial cell types (LHr, LS, Bs). The bottom panels showing the Tn5 insertion bias track.
See also Figure S5.
Discussion
Here, we developed a transposome-assisted single nucleus barcoding method, SNuBar, to perform multiplexing of high-throughput scATAC and multi-omic (scATAC&RNA) sequencing experiments. SNuBar performs barcoding through the addition of a simple oligonucleotide barcode adapter during the existing tagmentation step, which minimizes the experimental steps required for multiplexing and avoids challenges involved in generating customized or in-house assembled pre-indexed transposomes, as previously reported (Cusanovich et al., 2015; Lareau et al., 2019). We show that SNuBar-ATAC has both high scalability and high accuracy (>99%) for delivering barcodes into single cells as demonstrated by cross-species cell lines mixture experiments and 96 sample multiplexing experiments. SNuBar also allows overloading of single cells into microdroplets during the encapsulation step, by using the sample barcodes to distinguish cell doublets. In our experiments, we successfully loaded and sequenced 4,000 – 40,000 single cells in a single droplet encapsulation reaction (Table S2). We further showed that a small modification of the SNuBar adapters enables multiplexing multi-omic ATAC&RNA co-assays (SNuBar-ARC) of single cells (10X Genomics), which we expect will be widely used, due to the high cost and inability to multiplex samples when running these assays.
The application of SNuBar to multiplex a large collection of 28 syngeneic mice treated by 7 different treatment combinations in a pre-clinical screen for lung cancer allowed us to identify two treatment therapeutic combinations that altered epigenetic chromatin accessibility states in treated mice compared to the control group. Our data identified immune-suppressive hallmark pathways that were significantly down-regulated in the 4Gy+PDL1+Tala group, while DNA damage pathways were significantly up-regulated in the Chemo+2Gy+Tala treated group. In both groups we identified treatment-specific epigenetic responses for different genes, including Hif1α, Dlx4, Jarid2, Casp2, Setd1a and Pmepa1. Targeting these genes may lead to re-sensitizing the tumor cells to treatment, or improving the efficacy of immunotherapies by adding a target therapy agent (eg. Talazobarib) to treat lung cancer patients.
Another application of SNuBar involved the macro-spatial profiling of normal breast tissues to investigate the epigenomic heterogeneity of cell types and cell states across different spatial tissue areas. SNuBar profiling of 32 macro-spatial regions in a single microdroplet scATAC-seq experiment that revealed 8 cell types and multiple cell states that were organized across three major topographic areas: one fatty area and two epithelial areas that corresponded to ductal and lobular regions by histopathology. Our data showed that the adipocyte areas were correlated with a lipofibroblast cell state that was enriched for lipid related pathways and metabolism and two distinct luminal secretory epithelial cell states that may have different biological roles in the lobular and ductal areas. These data suggest that the epigenomic profiles of cell types correspond to their spatial organization in distinct topographic areas of the breast tissue.
We also applied SNuBar-ARC to profile normal breast tissues from 4 different women and identified many TFs that regulate cell type identities. Additionally, to generalize the tagmentation reaction in scATAC&RNA-seq coassay experiments, we also tested the Illumina Tn5 transposome and the in-house assembled Tn5 transposome for SNuBar-ARC experiments. Our results show that the Illumina Tn5 is comparable in read usage efficiency to the Tn5 transposome from 10X Genomics (0.6 versus 0.4 efficiency, STAR Methods). While the in-house assembled Tn5 had lower read usage efficiency (0.12), this may be explained by the free mosaic end sequences in the reaction, which can potentially enter the droplets and consume the cell barcodes.
In summary, we expect that SNuBar will have numerous applications for sample multiplexing of single cell ATAC-seq and multi-omics experiments. While we demonstrated multiplexing of therapeutic combinations and macro-spatial regions, other applications may include running large numbers of time-series experiments, the analysis of large patient cohorts or perturbation experiments that require a large number of samples to be analyzed. In the near future, we anticipate that SNuBar will make it possible to multiplex and sequence hundreds to thousands of samples or spatial regions in one experiment, due to recent developments in microdroplet platforms that have enabled sequencing up to 1M cells in one experimental run (eg. 10X Genomics Chromium X) compared to 10K cells which is the maximum on current platforms. Finally, we expect that SNuBar will provide an important tool for projects such as the Human Cell Atlas (HCA), HubMap and the Human Tumor Atlas Network (HTAN) that aim to understand the spatial organization of cell states and cell types across many normal organ systems and diseases such as cancer.
Limitations of the Study
One limitation of SNuBar is that unlike methods that use genetic information to multiplex samples (Kang et al., 2018; Heaton et al., 2020), SNuBar uses a separate barcode library for recording sample/spatial information, consumes about 5–20% of the total sequencing reads. Additionally, we found that the mapping rate of SNuBar-ATAC (63.80%−81.00%) was slightly lower than standard 10X Genomics scATAC-seq (82.8%−84.4%) due to the chimeric reads that are generated from the sample barcodes. This issue can be improved with one additional purification step in the last step of the library construction, as we found in the SNuBar-lowAK library, which increased the mapping rate from 73.4% to 78.5% after the additional purification. However, the SNuBar-ARC approach showed a comparable mapping rate (82.8–84.4%) when running the 10X Genomics ATAC&RNA co-assay. Another limitation is that SNuBar has the capacity to perform splitting and pooling combinatorial indexing at the nuclei level, but not at the molecular level, because SNuBar was designed to barcode each nucleus instead of barcoding each molecule in methods (eg. dscATAC-seq). We also noted that SNuBar-ARC failed to barcode some of the nuclei in the human breast tissue experiments, which may relate with the quality of the tissue samples. Despite this issue, SNuBar-ARC was still able to profiled a high number of barcoded cells, since this method allows overloading of very high number of cells during the microdroplet runs. These limitations can likely be improved in future developments of the SNuBar methods.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicholas Navin (nnavin@mdanderson.org).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact, Nicholas Navin (nnavin@mdanderson.org).
Data and Code availability
The data was deposited in the NCBI Gene Expression Omnibus under accession: GSE162798. The code for the SNuBar method is available on the following GitHub repository: https://github.com/navinlabcode/snubar_atac_scripts
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Cell lines.
The mouse A20 and human K562 cell lines were purchased from the American Type Culture Collection (ATCC TIB-208 and CCL-243 respectively) and cultured based on ATCC guidelines. The MDA-MB-231 and MDA-MB-436 human breast cancer cell lines were obtained from the MD Anderson Characterized Cell Line Core Facility and were cultured in Dulbecco’s Modified Eagle’s Medium-high glucose (DMEM, Sigma, D5976) medium supplemented with 10% FBS (Sigma, F0926) and 1X penicillin-streptomycin. The mouse lung cancer cell line LKR13 was cultured in RPMI-1640 medium supplemented with 10% FBS and 1X penicillin-streptomycin. All cell lines were maintained at 37°C with 5% CO2. Short tandem repeat (STR) profiling and mycoplasma tests were performed by ATCC or the MD Anderson Characterized Cell Line Core Facility.
Patient samples.
The human normal breast tissue (HBCA) was obtained from Baylor College of Medicine. This study was approved by the Institutional Review Board (IRB) and the patient was consented through an informed consent process.
Mouse drug treatment conditions.
5-week old male and female 129S mice were purchased from the Jackson Laboratory. When the mice grew to 7-weeks old, 0.75 million LKR13 tumor cells were subcutaneously implanted into the right thigh. After the implanted tumors reached the tumor volume of 150 mm3, four mice were randomly assigned to each of the following 7 groups: 1) Control; 2) Chemotherapy + 2 Gy Radiotherapy; 3) 4 Gy Radiotherapy; 4) Chemotherapy + 2 Gy Radiotherapy + Talazoparib; 5) Chemotherapy + 2 Gy Radiotherapy + PD-L1 Blockade; 6) 4Gy Radiotherapy + PD-L1 Blockade; 7) 4Gy Radiotherapy + Talazoparib + PD-L1 Blockade. Chemotherapy (16mg/kg paclitaxel and 30 mg/kg carboplatin) was administered by i.p. injection on day 1. The PXI (Precision X-RAY) X-RAD 225Cx irradiator was used to irradiate the leg tumors using an anterior-posterior appositional field. 2 Gy radiotherapy was delivered in the dose of 2Gy daily to the mice for consecutive 6 days. 4 Gy radiotherapy was delivered in the dose of 4 Gy daily to the mice for consecutive 3 days. Talazoparib was purchased from Selleck Chemicals and administered by oral gavage at a dose of 0.3mg/kg daily for 3 weeks. Anti-mouse PD-L1 (B7-H1) was purchased from Bio X Cell. PD-L1 blockade was administered by i.p. injection at dose of 100 μg/kg three times in the first week and 200 μg/kg weekly for the following two weeks. The tumors were collected one week after the treatment was completed.
Mouse tissue dissociation.
Tumor tissues were minced in 1X dissociation buffer containing DMEM F12/HEPES (Gibco, 113300), 2.5% BSA fraction V (Gibco, 15260037) and 0.25U/ul Collagenase IV (Fisher, 17104019). The dissociating tissues were incubated at 37 °C for 30 minutes, trypsinized using TrypLE express enzyme (Fisher, 12605010) for 5 min, and filtered through 70 μm filters. Cells were centrifuged at 400 rcf for 5 minutes. Red blood cells were removed by incubating the cell pellet in 1x RBC lysis buffer (MACS Miltenyi Biotec, 130–094-183) for 10 minutes at room temperature. Cells were then centrifuged at 400 rcf for 5 minutes and washed once with DMEM. Lastly, the cells were centrifuged at 400 rcf for 5 minutes and resuspended in PBS containing 0.04% BSA for downstream SNuBar experiment procedures.
Human breast tissue dissociation.
Tissues were minced in 1X dissociation buffer (DMEM F12/HEPES, 2.5% BSA fraction V and 1g/ul Collagenase A (Sigma, 11088793001)) followed by incubated at 37 °C for 2–3hrs, trypsinized using trypsin containing 0.25% EDTA (Corning, 25053CI) for 5 min, and filtered through 70 μm filters. Cells were centrifuged at 500 rcf for 5 minutes. Red blood cells were removed by incubating the cell pellet in 1x RBC lysis buffer for 10–20 minutes at room temperature. Cells were then centrifuged at 500 rcf for 5 minutes and washed once with DMEM. Lastly, the cells were centrifuged at 500 rcf for 5 minutes and resuspended in FBS containing 10% DMSO to be stored at −80°C until SNuBar-ARC experiment procedures.
Nuclei isolation from cell suspension for SNuBar-ATAC.
Cell nuclei were isolated, washed, and counted according to the Nuclei Isolation for Single Cell ATAC Sequencing Protocol (CG000169 Rev D, 10X Genomics). Briefly, cell lines were washed with PBS containing 0.04% BSA and passed through a 40 μm Flowmi Cell Strainer. 100,000 to 1,000,000 cells were collected in a 2-ml microcentrifuge tube by centrifugation at 300 rcf for 5 min at 4 °C. 100 μl chilled Lysis buffer (10mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 Substitute, 0.01% Digitonin, and 1% BSA) was added to the cell pellet after carefully removing all the supernatant and pipette mixed 10X. Cells were incubated on ice for 5 min (optimized), followed by adding 1 ml chilled Wash Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, and 1% BSA) and pipette mixed 5X. Nuclei were centrifuged at 500 rcf for 5 min at 4°C, followed by removal of the supernatant without disrupting the nuclei pellet. Nuclei were resuspended in 50 μl chilled PBS and counted using a Countess II FL Automated Cell Counter (Life technologies, AMQAX1000), then immediately used to perform Tn5 tagmentation and oligonucleotide barcoding as described below.
Nuclei isolation from tissues.
Tissues were cut into small pieces and cell nuclei were isolated, washed, and counted according to the Nuclei Isolation from Mouse Brain Tissue for Single Cell ATAC Sequencing Protocol (CG000212 Rev B, 10X Genomics). In brief, tissues were cut into small pieces and transferred to a 1.5 ml microcentrifuge tube. 500 μl chilled 0.1X lysis buffer (10mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.01% Tween-20, 0.01% Nonidet P40 Substitute, 0.001% Digitonin, and 1% BSA) was added and tissues were homogenized 15X using a pellet pestle and then incubated on ice. After 5 min incubation, tissues were pipette mixed 10X and incubated for an additional 10 min. 500 μl chilled Wash Buffer was added to the lysed cells and pipette mixed 5X. Nuclei were passed through 40 μm filter (pluriStrainer Mini) and centrifuged at 500 rcf for 5 min at 4°C. Resulting supernatant was removed without disrupting the nuclei pellet. Nuclei were resuspended in 50 μl chilled PBS and counted using a Countess II FL Automated Cell Counter. Nuclei were used immediately to perform Tn5 tagmentation and oligonucleotide barcoding as described below.
Tn5 tagmentation and oligonucleotide barcoding.
Approximately 35K nuclei from cell suspensions (or macro-dissected tissue pieces) were incubated with the Tn5 transposomes in a 50 μl reaction [25 μl 2 × TD buffer (Illumina 20034198), 1 μl 0.5% Digitonin, 5 μl TDE1 (Illumina, 20034198), 5 μl nuclease-free water, 14 μl cells in PBS]. Reactions were incubated at 37 °C for 35 min while mixing at 850 rpm in a program cycling between 15 sec mixing and 15 sec pausing. Then 1μl of 2 μM HPLC purified barcode oligonucleotide adapters 5-’CCTTGGCACCCGAGAATTCCANNNNNNNN-(N)18-CTACTCTGCGTTGATACCAGACGCTGCCGACGA-3’ [NNNNNNNN represents 8bp UMI sequence while -(N)18- sequence represents the 18bp spatial/sample barcode described in further detail on Figure S1A and Table S1] were added, mixed, and incubated with the nuclei at 37 °C for another 35 min while mixing at 850 rpm (15 sec mixing and 15 sec pause cycle to help cell and barcode be well mixed). Nuclei were gently washed with 500 μl chilled PBS containing 1% BSA and centrifuged at 500 rcf for 5 min at 4°C. After the supernatant removal, 1ml chilled PBS containing 1% BSA was slowly added without disrupting the pellet, followed by incubation on ice for 5–10 min. Nuclei were centrifuged at 500 g for 5 min at 4°C and the pellet was resuspended in ~20 μl chilled PBS containing 1% BSA. Nuclei from different cell lines or tissues were then pooled together and passed through the 20 μm filter. After centrifuging at 500 rcf for 5 min at 4°C and discarding the supernatant, nuclei were gently washed with 1ml Diluted Nuclei Buffer (PN-2000153, 10X Genomics) without disrupting the pellet, followed by incubation on ice for 5 min. Nuclei were centrifuged at 500 rcf for 5 min at 4 °C, resuspended in Diluted Nuclei Buffer, and counted using the Countess™ II Automated Cell Counter. Nuclei concentration was adjusted with the Diluted Nuclei Buffer to obtain the desired capture number, based on the Recovery efficiency factor.
Single nuclei ATAC-seq library construction.
8 μl of the adjusted nuclei solution was combined with 7 μl ATAC buffer (10X Genomics, 2000122/2000193) and loaded into the 10X Genomics system for single cell ATAC sequencing according to the manufacturer’s instructions (CG000168 Rev A for Chromium Single Cell ATAC Reagent Kits, or CG000209 Rev D for Chromium Next GEM Single Cell ATAC Library Kit v1.1). The barcode and ATAC templates were separated during Ampure XP beads purification step following GEM generation and Dynabead cleanup. In detail, 48 μl vortexed Ampure XP beads(1.2X) was added to the Dynabeads cleanup product and pipette mixed thoroughly. After incubation at room temperature for 5 min, the solution is centrifuged briefly and placed on the magnet high until the solution cleared. The supernatant containing barcodes was transferred to another tube and added additional 0.6X Ampure XP beads (total 1.8X) for purification. The barcode and ATAC templates were washed twice with 80% ethanol and then eluted with Buffer EB and nuclease-free H2O, respectively.
The ATAC Indexed sequencing library was constructed by mixing Sample Index PCR Mix composed of Amp Mix and SI-PCR Primer B, ATAC template sample, and an individual Chromium i7 Sample Index N, Set A as recommended and further purified with 0.4X and 1.2X double sided size selection using Ampure XP beads, in here we recommend to do one additional 1.2X purification to further enrich target library. The barcode sequencing library was prepared with the following PCR reaction setup: 25 μl of 2X KAPA HiFi HotStart ReadyMix, 10 μl purified barcodes, 1.5 μl 10 μM TruSeq RPIX primer (5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’), 1.5 μl 10 μM P5 Adaptor (5’-AATGATACGGCGACCACCGAGATCTACAC-3’) and 12 μl nuclease-free H2O. The PCR was run at 98 °C for 30 s, 8–10 cycles of (98 °C 15 s, 60 °C 30 s, 72 °C 30 s), 72 °C 1 min, and 4°C hold. The barcode products were further purified with 1.5X Ampure XP beads. Final ATAC and barcode libraries were then mixed at a ratio of 8:2 and sequenced on the Illumina NextSeq instrument. Read 1 (5’-TCGTCGGCAGCGTCTGGTATCAACGCAGAGTAG-3’) and I5 Index Primer (5’-CTACTCTGCGTTGATACCAGACGCTGCCGACGA-3’) were spiked into the sequencing step.
SNuBar-ARC.
Nuclei were isolated, washed and counted according to the Nuclei Isolation for Single Cell Multiome ATAC + Gene Expression Sequencing Protocol (10X Genomics, CG000365 Rev A). Approximately 16 K nuclei from each sample were utilized for tagmentation following the manufacturer’s instructions (10X Genomics, CG000338 Rev A for Chromium Next GEM Single Cell Multiome ATAC + Gene Expression). Tagmentation reactions were incubated at 37 °C for 35 min in a thermal cycler. Then 0.5 μl of 0.5 μM HPLC purified barcode oligonucleotides were added to two different samples respectively, mixed, and incubated at 37 °C for another 35 min with pipetting mixing every 10 mins. Barcode oligonucleotides s1: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCAAGTATGCTCCTTCCGTCCAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’, s2: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCAGCGACGCAGATAAACCCTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’, s3: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCACCATCTGAGGTGTCAGCTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’, s4: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCACGTTGTACTCAGATCTGTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’ were utilized in the cell line and HBCA multiomics experiments. Nuclei were then gently washed with 500 μl chilled PBS containing 1% BSA and centrifuged at 500 rcf for 5 min at 4°C. After supernatant removal, 1ml chilled Diluted Nuclei Suspension Buffer [50 μl 20X Nuclei Buffer (10X Genomics, PN-2000207), 0.625 μl 1M DTT, 25 μl 40U/ul RNase Inhibitor (NEB,M0314L), and 924.4 μl Nuclease-free water] was slowly added without disrupting the pellet, followed by incubation on ice for 5–10 min. Nuclei were centrifuged at 500 rcf for 5 min at 4°C and the pellet was resuspended in 5 μl Diluted Nuclei Suspension Buffer. Nuclei from two cell lines were then pooled together and counted using the Countess™ II Automated Cell Counter. After counting, nuclei concentration was adjusted using Diluted Nuclei Buffer to the desired capture number. Then 8 μl of the adjusted nuclei solution was combined with 7 μl ATAC buffer and loaded into 10X Genomics system for single cell ATAC + Gene Expression sequencing according to the manufacturer’s instructions (10X Genomics CG000338 Rev A for Chromium Next GEM Single Cell Multiome ATAC + Gene Expression). It should be noted that in the cDNA Amplification step, 1 μl 2.5 μM barcode primer bcP: 5’-CCTTGGCACCCGAGAATTCCA-3’ was added into cDNA Amplification Mix. The barcode and cDNA were separated during cDNA Ampure XP Cleanup. Next, 60 μl Ampure XP beads(0.6X) was added to the PCR product and mixed thoroughly. After incubation at room temperature for 5 min, the solution was centrifuged briefly and placed on the magnet until the solution cleared. The supernatant containing barcodes was transferred to another tube and incubated with additional 1.2X Ampure XP beads (total 1.8X) for purification. The cDNA and barcode were washed twice with 80% ethanol and eluted with nuclease-free H2O and EB Buffer respectively. The Gene Expression Library Construction was performed following the CG000338 Rev A instructions, while the barcode sequencing library was prepared with the following PCR reaction setup: 25 μl of 2X KAPA HiFi HotStart ReadyMix, 10 μl purified barcodes, 2 μl 10 μM TruSeq RPIX primer (5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’) and 2 μl 10 μM InPE 1 (5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’), and 11 μl nuclease-free H2O. The PCR was run at 98 °C for 30 s, 6 cycles of (98 °C 15 s, 60 °C 30 s, 72 °C 30 s), 72 °C 1 min, and 4°C hold. The barcode library was then purified with 1.5X Ampure XP beads. RNA and barcode libraries were then mixed at a ratio of 9:1 and sequenced on the Illumina NextSeq2000 instrument. The ATAC library was sequenced separately on the Illumina NextSeq2000 instrument as well.
To test whether other Tn5 transposomes are compatible with the 10X Genomics ATAC&RNA coassay experiments, Illumina Tn5 (#20034198) and in-house assembled Tn5 were utilized to perform SNuBar-ARC. To assemble the in-house Tn5 transposome, (1) mix 20 μl 100p Tn5-ME-Ap (/5phos/TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’), 20 μl 100p Tn5-ME-rev (/5phos/CTGTCTCTTATACACATCT) and 4 μl 10X Anneal buffer (100 mM Tris-HCl, 10mM EDTA, 250mM NaCl, pH8.0) (2) mix 20 μl 100p Tn5-ME-B (5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’), 20 μl 100p Tn5-ME-rev and 4 μl 10X Annealing buffer. (1) and (2) were incubated at 95 °C for 3min, followed by decreasing 0.1°C/s to 25°C, individually. Annealed products from (1) and (2) were mixed equally, and followed by making an 8-fold dilution. Then 7 μl diluted products were mixed with 5 μl Creative Enzyme Tn5 (#NATE-1629) and incubated at 23 °C for 30 min, after which 6ul glycerol was added. Approximately 35K nuclei from A20 and K562 cell suspensions were incubated with the Tn5 transposomes (Illumina or in-house) in a 50 μl reaction [25 μl 2 × TD buffer (Illumina 20034198), 1 μl 0.5% Digitonin, 5 μl TDE1 (Illumina, 20034198), 5 μl nuclease-free water, 14 μl cells in PBS]. Reactions were incubated at 37 °C for 30 min. Then 1.5 μl of 1 μM HPLC purified barcode oligonucleotide adapters were added and incubated for another 30min at 37 °C. All of the other steps followed the standard SNuBar-ARC protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data pre-processing.
The 10X Genomics CellRanger ATAC (v1.2.0) was utilized to pre-process the scATAC-seq data. The cellranger-atac mkfastq was used to demultiplex raw base call (BCL) files generated by Illumina sequencers and convert the barcode and expression data into FASTQ files. The cellranger-atac count was utilized to take FASTQ files and perform ATAC analysis. Mixing experiment data was aligned to the combined reference genomes of human (hg19 reference) and mouse (mm10 reference). All other experimental data was aligned to human hg19 or mouse mm10 genome. When pre-processing sample/spatial barcodes data, we first regenerated two new FASTQ files including 1) Read1 FASTQ file that composes of 16bp cell barcodes (extracted from original Index2 FASTQ file) and 8bp UMI (extracted from original Read1 files), and 2) Read2 FASTQ file that composes of 18bp sample barcodes sequences (extracted from the first 18bp of original Read1 FASTQ file) using custom scripts (Data and Code availability). The new converted FASTQ files were then processed using CITE-seq-Count (v1.4.3) (Patrick Roelli et al., 2019) (-cbf 1 -cbl 16 -umif 17 -umil 24 --max-error 2 --umi_collapsing_dist 1) to generate the cell barcode and sample/spatial barcode count matrix. The cells called by CellRanger were used for CITE-seq-Counts input. For Snubar-ARC, 10X Genomics CellRanger ARC was used to demultiplex the raw BCL files. Demultiplexed ATAC data was further mapped to the hg19 and mm10 combined reference genome using CellRanger ATAC (v1.2.0) pipelines, while the gene expression data was aligned to hg19 and mm10 combined reference genome using CellRanger (v4.0.0) with parameter --chemistry=JAG-v1. The sample barcode library was converted into the cell barcode and sample barcode matrix using CITE-seq-Count (-cbf 1 -cbl 16 -umif 17 -umil 28 --max-error 2 --umi_collapsing_dist 1) using the orginal Read1 and Read2 FASTQ files as input. The cells called by CellRanger were used for CITE-seq-Counts. For species-mixture experiments, the barnyard analysis was performed to decide the human-mouse multiplets according to the reference genome they mapped.
Binarizing scATAC fragment count matrix.
For datasets without cancer cells involved, we binarized the non-negative fragment count matrix by converting any non-zero value into 1 and maintaining the zero as 0, which was described previously (Satpathy et al., 2019). This procedure was used for the A20 cell line and the HBCA dataset. Binarization was not performed in datasets involving cancer cells, because of possible copy number alterations.
Quality control (QC) for scATAC-seq.
We used unique nuclear fragments and TSS enrichment scores to estimate the signal strength and signal-to-background ratio, respectively. These calculations were performed using the package ‘ArchR’ (Granja et al., 2020). By default, we retained cells having 1) the unique nuclear fragment number no smaller than 1000 and no greater than 10,000, and 2) the TSS enrichment score no smaller than 8 (human cells) and 10 (mouse cells). In the 2-plex experiments, we used 8 as the TSS enrichment threshold regardless of species as done in previous study (Satpathy et al., 2019).
In-silico doublet removal procedure.
The package ‘ArchR’ was utilized to remove doublets in silico. It first synthesizes artificial doublets by mixing reads from the observed cells and quantifies how similar the cells are to synthetic doublets with the function ‘addDoubletScores’. Then it removes cells identified as doublets with the function ‘filterDoublets’. Specifically, we ran ‘addDoubletScores’ with default parameters and then ran ‘filterDoublets’ with different ‘doublet_filter_ratio’ parameter depending on the data. The parameter ‘doublet_filter_ratio’ was set 1.8 for the 96-plex experiment and 1.2 in the mouse drug and human breast tissue experiments. For datasets involving cancer cells, we removed the outlier cell clusters with higher fragment numbers (double or higher) than the others.
SNuBar barcode classification for non-mixed species experiments.
Using cell that passed QC from SNuBar-ATAC or SNuBar-ARC, we attached the SNuBar barcode count matrix and created Seurat object using the package ‘Signac’ (Stuart et al., 2019). We first normalized the SNuBar barcode count matrix with the function ‘NormalizeData’ using the centered log-ratio (CLR) normalization method. Next, we ran the ‘MULTIseqDemux’ function so that a cell was identified as ‘doublet’, ‘singlet’ or ‘negative’ (insufficient SNuBar counts). Finally, each singlet cell was assigned to its best predicted SNuBar barcode and retained for analysis.
Evaluating the aggregated ATAC signal consistency between SNuBar-ATAC, SNuBar-ARC and other technologies.
To compare the signal consistency between multiple datasets for a cell line, datasets were first integrated. Specifically, peaks from multiple datasets were merged if they had 1-bp overlaps to recreate a unified peak set, which resulted in a new fragment count matrix for each dataset. Next, each recreated fragment count matrix was summed up across singe cells per dataset, normalized to counts per million fragments (CPM), and log2-scaled to create a pseudo-bulk ATAC-seq dataset. Finally, the Pearson’s R and p-value of ATAC signals at peaks between two datasets were computed to quantify consistency using the “cor.test” function of R.
Evaluating the aggregated RNA signal consistency between SNuBar-ARC and other technologies.
For each scRNA-seq dataset (SNuBar-ARC RNA (K562 and A20), public K562), we created a pseudo-bulk UMI value vector by summing up the UMI counts of genes across single cells. For K562, the pseudo-bulk UMI value of SNuBar-ARC and the pseudo-bulk public K562 was normalized to counts per million (CPM) and log2-scaled to quantify gene expressions. For A20, the pseudo-bulk UMI value of SNuBar-ARC A20 and the public bulk A20 was also normalized to counts per million (CPM) and log2-scaled to quantify gene expressions. For each species (K562 or A20), only the gene symbols shared by both datasets were used. Finally, the Pearson’s R and p-value of gene expressions between two datasets were computed to quantify the consistency using “cor.test” function of R.
Evaluating the aggregated RNA signal consistency between SNuBar-ARC and other technologies.
For scRNA-seq data, we created a pseudo-bulk UMI count matrix by summing up the UMI values of genes across single cells. Then all pseudo-bulk UMI count and bulk RNA-seq reads matrices were normalized to CPM, log2-scaled for comparison. Only the gene symbols shared by all datasets were used.
scATAC data clustering and visualization.
We used the package ‘Signac’ to make a visualization-ready object. First, we ran the ‘RunTFIDF’ function to perform term frequency-inverse document frequency (TF-IDF) normalization. Then we selected the variable features by running the function ‘FindTopFeatures’ with the parameter min.cutoff=‘q5’ (top 95% most common peaks). To perform dimension reduction and generate the latent semantic indexing (LSI) components, the function ‘RunSVD’ with the parameter n=30 was used to run singular valued decomposition (SVD) on the TD-IDF matrix using selected variable features. The LSI component having a strong correlation (greater than 0.75) with the unique fragment number (assessed by the function ‘DepthCor’) was excluded (only the first LSI component) before performing non-linear dimension reduction and clustering. We applied the functions ‘RunUMAP’ and ‘RunTSNE’ with the default parameters to create UMAP and tSNE dimension reduction spaces, the function ‘FindNeighbors’ with default parameters to build cell-to-cell similarity graph and ‘FindClusters’ with various resolutions to determine the clusters.
Coverage and tile plots.
The normalized aggregated ATAC fragments along genomic positions were visualized using the function ‘CoveragePlot’ of the package ‘Signac’ with scaling fragments number per group if applicable to 1 million (i.e., the parameter scale.factor=1e6). The aligned peaks and genes tracks were added by specifying the parameter annotation=TRUE and peaks=TRUE. The tile plot to visualize the presence of fragments was done with the function ‘TilePlot’ where 100 cells per group if applicable were randomly picked with the parameter order.by=‘random’. All those plots were aligned using the function ‘CombineTracks’.
Deriving inferred copy number alterations (iCNA) from scATAC data.
To infer the copy number alterations from scATAC data, we utilized the same method and code previously described (Satpathy et al., 2019). When visualizing as a heatmap, we used the log2 fold change of signals per genomic bin compared with its 100 nearest neighbors in terms of GC-content.
Deriving inferred RNA (iRNA) expression from scATAC data.
We used the function ‘GeneActivity’ of the package ‘Signac’ to infer the RNA expression derived from the fragments within 2-kb upstream per gene. Then we used function ‘NormalizData’ to log-normalized the inferred RNA expression following scaling to the median of cell sizes (defined as the sum of the iRNA values per cell). We used the gene annotation package ‘EnsDb.Hsapiens.v75’ and ‘EnsDb.Mmusculus.v79’ for human (hg19) and mouse (mm10) datasets, respectively.
Demultiplexing mixed human cell line data using ‘Demuxlet’.
To create the reference SNP profiles of the three human cell lines (MDA-MB-231, MDA-MB-436, and K562), we downloaded the public bulk ATAC-seq data, mapped them to the human genome (hg19) using bowtie2 (Langmead and Salzberg, 2012) with default parameters, called peaks using macs2 with default parameters, and ran the pipeline as previously described. With the reference SNP profile, we then performed ‘Demuxlet’ on the BAM file with the default parameters to determine the cell line identity of each cell based on SNP profiles.
Differentially accessible peaks (DAPs) analysis.
We used the function ‘FindAllMarkers’ to find DAPs across clusters. The parameter ‘latent.vars’ was set to specify the number of fragments. The test method in use was a logistic regression model specifying the parameter test.use=‘LR’. Other parameters remained the default. We used the same parameter setting to run the function ‘FindAllMarkers’ when comparing two specific clusters. Only the resulting peaks with adjusted p-val less than 0.05 and absolute average fold change greater than 1.5 were reported as DAPs and kept for analysis. We also used the function ‘ClosestFeature’ to pair the closet gene to each DAP if the distance was shorter than 100,000 bp. The gene annotation packages were the same as the ones specified for deriving iRNA.
Module-based GSEA analysis.
In the mouse drug experiment, DAPs may affect nearby genes in-cis and also in-trans. To understand the effect of DAPs, we used the 3-step module-based GSEA as previously described (LaFave et al., 2020). Briefly, we first defined a module as a set of DAPs, and used the function ‘AddChromatinModule’ in the package ‘Signac’ to compute the module scores representing the enrichment of ATAC signals per module across cells (Schep et al., 2017; Stuart et al., 2019). Next, for the module we sought the gene relevance by iterating each gene and calculating the Pearson correlation coefficient between the module scores and the iRNA values. Thus, the module received a gene-ranked vector decreasingly ordered by the correlation coefficients. Finally, the regular GSEA was performed by using the function ‘GSEA’ in the package ‘clusterProfiler’ (Yu et al., 2012). Only the pathways having FDR q <0.05 were reported as significantly enriched and analyzed.
Estimate the barcoding efficiency of Tn5 transposome in SNuBar-ARC.
We defined the reads usage efficiency as the usable fragments over sequenced read number per cell. The average sequenced reads per cell was calculated by dividing the total sequenced read number with the detected cell number and usable fragment was the median fragments per cell of specific cell line detected by CellRanger ATAC.
QC for the RNA assay of SNuBar-ARC.
Cells that pass all the following three criterions were retained for analysis: 1) the number of detected genes is greater than 100, 2) the log2-scaled number of UMIs (log2nUMI) are less than the median plus 3 times the median absolute deviation (MAD) of log2nUMI across all cells, and 3) the proportion of total UMIs composed by mitochondrial genes (pMT) are less than the median plus 3 times the MAD of pMT across all cells.
scRNA-seq data clustering.
We first normalized the UMI count matrix using the function ‘NormalizeData’ with the default parameters. Having determined the top 3000 highly variable genes using the function ‘FindVariableFeatures’ with the default parameters, we scaled the normalized gene matrix and regressed out the number of total UMIs using the function ‘ScaleData’. Then we performed PCA using the function ‘RunPCA’ with the top 30 PC components stored, after which we applied the function ‘RunUMAP’ and ‘FindNeighbors’ to generate the UMAP projection and cell-cell distance graph, respectively. Next, we applied the function ‘FindClusters’ using the smart local moving (SLM) algorithm to determine the cell clusters.
DEGs detection in scRNA-seq data.
We used the wilcoxon rank sum test of function ‘FindAllMarkers’ to identify DEG genes. To alleviate the difference driven by cell numbers, we down-sampled each cell type by setting the parameter ‘max.cells.per.ident’ to the lowest number of cells for the cell types. Only genes with at least 1.5-fold change, a Bonferroni-adjusted p-value less than 0.05, and a minimum difference in the fraction of detection between two groups greater than 50% were reported as DEGs.
Demultiplexing the SNuBar-ARC data using souporcell.
We ran ‘souporcell_pipeline.py’ on the BAM alignment file of the RNA assay of SNuBar-ARC. The running parameter ‘--fasta’ was set as a hg19 genome FASTA file and the parameter ‘--clusters’ was set as 4 because the SNuBar-ARC data in the normal breast experiment involved 4 samples. Other parameters were set as default.
QC and determining breast cell types in the SNuBar-ARC data.
Cells were tentatively considered if they simultaneously passed the QC for scATAC and scRNA. The gene expression data (RNA) and ATAC data of those cells were clustered separately as described in the above Methods sections. To identify cell types, we annotated each RNA cluster using the known cell type specific marker genes. In parallel, we labelled each ATAC cluster based on the inferred RNA expression and the TSS-flanking fragments of the same marker genes. To remove potential doublets, we only consider cells with the consistent cell type identity in the RNA and ATAC assays and then iterated each cell type to perform scRNA-seq QC. The resulted cells were used for downstream analysis. The RNA assay was processed and clustered again as described in the above Methods section. To capture the possible chromatin accessible peaks specific to certain cell types, we used the function ‘CallPeaks’ of Signac to determine peaks and used the function ‘FeatureMatrix’ of Signac to generate a new fragment count matrix for cells. The resulted ATAC assay was processed and clustered again as described in the above Methods section.
Integrated clustering of SNuBar-ARC breast data.
Before the clusters from the RNA and ATAC modalities were integrated, the sample-specific effect on the clustering of each of the modalities were corrected using the tool Harmony (Korsunsky et al., 2019). With the sample identities of cells determined by SNuBar demultiplexing, we applied the function ‘RunHarmony’ of Seurat on the RNA and ATAC assay using the top 30 PCA components and the top 30 (but excluding the first) LSI components, respectively, resulting in new cell embeddings for each of the modalities (referred as Harmony-RNA and Harmony-ATAC components). To perform the integrated clustering, we used the function ‘FindMultiModalNeighbors’ of Seurat with the top 20 Harmony-RNA and top 20 (but excluding the first) Harmony-ATAC components and then applied ‘RunUMAP’ to generate the UMAP projection for visualization.
ChromVAR analysis and visualization.
To measure the TF regulatory potential per cell, we measured the chromatin accessible regions sharing an in-cis TF binding motif using the tool chromVAR (Schep et al., 2017). To initiate the chromVAR analysis, we applied the function ‘AddMotifs’ of Signac which used the curated motif database ‘human_pwms_v2’ (R package: chromVARmotifs) and the genome FASTA information (R package: BSgenome.Hsapiens.UCSC.hg19). Then we ran the function ‘RunChromVAR’ of Signac to compute chromVAR scores of TF binding motifs across cells. To identify cell type specific motif, we used the function ‘FindAllMarkers’ of Seurat on the chromVAR modality. In details, we used the logistic regression-based test by setting the parameter ‘test.use’ as ‘LR’. The parameter ‘max.cells.per.ident’ was set the lowest number of cells of all cell types to alleviate detecting difference driven by cell number. Because chromVAR scores are not non-negative gene expression values but z-scores, the parameter ‘mean.fxn’ was set as ‘rowMeans’ of R language to aggregate chromVAR score during group-wise comparisons. We also set the parameter ‘latent.vars’ as the total number of fragments per cell to regress out the difference driven by cell total fragments. A motif with its TF names available in RNA assay, a Bonferroni-adjusted p-value lower than 0.05, and an average difference great than 0 was considered cell type specific. As the chromVAR scores of a TF’s motif across cells tend to have a long-tail distribution, the min and max cutoff were set as the 0.05 and 0.95 quantile of a TF’s chromVAR scores when visualized on the UMAP.
TF footprint analysis.
To infer if a TF binds to its motif in a particular cell type, we applied the ‘Footprint’ function of Signac on the ATAC modality which measures the normalized observed versus expected Tn5 insertion frequency for each position surrounding a set of motif instances. In detail, we set the parameter ‘in.peaks=TRUE’ to search motif instances that located within ATAC peaks. Other parameters were default. To visualize the footprint profile, we used the function ‘PlotFootprint’ grouped by all cell types with the parameter set as ‘divide’. For visualization purposes, only the three epithelial cell types were shown.
Downloading of public RNA-seq datasets.
The K562 cell line data was downloaded from SRA: SRX5124174. The A20 cell line data was downloaded from the 10X Genomics website (https://support.10xgenomics.com/single-cell-atac/datasets/1.2.0/atac_v1_hgmm_10k). The bulk RNA-seq data from the A20 cell line was downloaded from NCBI Gene Expression Omnibus (GSM4143267). The scRNA-seq for K562 cell line was downloaded from GEO with the accession number GSE90063.
Downloading of the public bulk ATAC-seq datasets.
The K562 data was downloaded from the ENCODE portal with the experiment number ENCSR868FGK and the accession number ENCFF261UXP for read-1 and ENCFF123TMX for read-2. The MDA-MB-231 and MDA-MB-436 data were downloaded with the SRA accession IDs SRR7225841 and SRR7225843, respectively.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human normal breast tissue samples | Baylor College of Medicine | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Anti-mouse PD-L1 | Bio X Cell | BE0101 |
| Talazoparib | Selleck | S7048; CAS: 1207456-01-6 |
| Carboplatin | Auromedics | 55150-386-01 |
| Paclitaxel | MDACC pharmacy | PremierProRX |
| Tris-HCl (pH7.4) | Sigma | T2194 |
| Tween-20 | Andwin Scientific | NC9022994 |
| Nonidet P40 Substitute | Sigma | 74385 |
| NaCl | Invitrogen | AM9760G |
| MgCl2 | Invitrogen | AM9530G |
| Digitonin | Invitrogen | BN2006 |
| 2-mercaptoethanol | Gibco | 21985023 |
| RPMI-1640 | ATCC | 30–2001 |
| IMDM | ATCC | 30–2005 |
| DMEM | Sigma | D5796 |
| FBS | Sigma | F0926 |
| Penicillin-streptomycin | Corning | 30-002-Cl |
| Collagenase A | Sigma | 11088793001 |
| TrypLE Express Enzyme (1X), phenol red | Fisher | 12605010 |
| Trypsin-EDTA (0.25%) | Corning | 25053CI |
| Ultrapure BSA | Ambion | AM2616 |
| 10x MACS RBC lysis buffer | MACS Miltenyi Biotec | 130-094-183 |
| DMEM F12/HEPES | Gibco | 113300 |
| BSA fraction V | Gibco | 15260037 |
| Creative Enzyme Tn5 | Creative Enzyme | NATE-1629 |
| RNase Inhibitor | NEB | M0314L |
| Critical commercial assays | ||
| Chromium Single Cell ATAC Library & Gel Bead Kit | 10X Genomics | PN-1000110 |
| Chromium Chip E Single Cell ATAC Kit | 10X Genomics | PN-1000155 |
| Chromium i7 Multiplex Kit N, Set A | 10X Genomics | PN-1000084 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit | 10X Genomics | PN-1000175 |
| Chromium Next GEM Chip H Single Cell Kit | 10X Genomics | PN-1000161 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10X Genomics | PN-1000283 |
| Chromium Next GEM Chip J Single Cell Kit | 10X Genomics | PN-1000234 |
| Dual Index Kit TT Set A | 10X Genomics | PN-1000215 |
| Illumina Tagment DNA TDE1 Enzyme and Buffer Kit | Illumina | 20034198 |
| Deposited data | ||
| Raw data files and processed data files | This manuscript | GEO: GSE162798 |
| Experimental models: cell lines | ||
| Human: K562 | ATCC | ATCC: CCL-243 |
| Mouse: A20 | ATCC | ATCC: TIB-208 |
| Human: MDA-MB-231 | MD Anderson Characterized Cell Line Core Facility | N/A |
| Human: MDA-MB-436 | MD Anderson Characterized Cell Line Core Facility | N/A |
| Mouse: LKR13 | MD Anderson Characterized Cell Line Core Facility | N/A |
| Experimental models: organisms/strains | ||
| Mouse: 129S | Jackson Laboratory | 002448 |
| Oligonucleotides | ||
| SBOs, see Table S1 | This manuscript | N/A |
| TruSeq RPIX primer: 5’-CAAGCAGAAGACGGCATACGAGAT-NNNNNN-GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’ | IDT | N/A |
| P5 Adaptor: 5’-AATGATACGGCGACCACCGAGATCTACAC-3’ | IDT | N/A |
| Read 1 Primer: 5’-TCGTCGGCAGCGTCTGGTATCAACGCAGAGTAG-3’ | IDT | N/A |
| I5 Index Primer: 5’-CTACTCTGCGTTGATACCAGACGCTGCCGACGA-3’ | IDT | N/A |
| SNuBar_RNA-I7–1bc: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCAAGTATGCTCCTTCCGTCCAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’ | IDT | N/A |
| SNuBar_RNA-I7–2bc: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCAGCGACGCAGATAAACCCTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’ | IDT | N/A |
| SNuBar_RNA-I7–3bc: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCACCATCTGAGGTGTCAGCTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’ | IDT | N/A |
| SNuBar_RNA-I7–4bc: 5’-CGAGCCCACGAGACCCTTGGCACCCGAGAATTCCACGTTGTACTCAGATCTGTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’ | IDT | N/A |
| bcP: 5’-CCTTGGCACCCGAGAATTCCA-3’ | IDT | N/A |
| InPE 1: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’ | IDT | N/A |
| Tn5-ME-Ap: /5phos/TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’ | IDT | NA |
| Tn5-ME-rev: /5phos/CTGTCTCTTATACACATCT | IDT | NA |
| Tn5-ME-B: 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’ | IDT | NA |
| Software and algorithms | ||
| CellRanger ATAC (v1.2.0) | 10X Genomics | https://github.com/10XGenomics/cellranger-atac |
| CellRanger ARC (v1.0.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| CellRanger (v4.0.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| CITE-seq-Count (v1.4.3) | DOI: 10.5281/zenodo.2590196 | https://github.com/Hoohm/CITE-seq-Count |
| ArchR (v0.9.5) | Granja et al., 2021 | https://github.com/GreenleafLab/ArchR/tree/cadc5844259aee51a8e42afcf8ab72c5fc32632d |
| Signac (v1.1.1) | Stuart et al., 2020 | https://github.com/timoast/signac/ |
| Demuxlet (implemented in ‘popscle’) | Kang et al., 2017 | https://github.com/statgen/popscle/tree/4b6a88a7b38da04dd7c3544df29240748f54c602 |
| souporcell | Heaton et al., 2020 | https://github.com/wheaton5/souporcell |
| chromVAR (v1.12.0) | Schep, A., Wu, B., Buenrostro, J. et al., 2017 | https://github.com/GreenleafLab/chromVAR |
| Harmony (v1.0) | Korsunsky, I., Millard, N., Fan, J. et al., 2019 | https://github.com/immunogenomics/harmony |
| clusterProfiler | Yu et al., 2012 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| EnsDb.Hsapiens.v75 | NA | https://bioconductor.org/packages/release/data/annotation/html/EnsDb.Hsapiens.v75.html |
| EnsDb.Mmusculus.v79 | NA | http://www.bioconductor.org/packages//release/data/annotation/html/EnsDb.Mmusculus.v79.html |
| chromVARmotifs | NA | https://github.com/GreenleafLab/chromVARmotifs |
| MACS2 | Zhang, Y., Liu, T., Meyer, C.A. et al., 2008 | https://pypi.org/project/MACS2/ |
| Seurat (v4.0.0) | Hao*, Hao*, et al., Cell 2021 | https://github.com/satijalab/seurat/ |
| Ggrastr (v0.2.1) | NA | https://github.com/VPetukhov/ggrastr |
| ComplexHeatmap (v2.6.2) | Zuguang Gu, et al., 2016 | https://github.com/jokergoo/ComplexHeatmap |
| Other | ||
| Detailed protocols (SNuBar-ATAC and SNuBar-ARC) | This paper | Methods S1 |
Highlights.
SNuBar multiplexes scATAC-seq and scATAC&RNA-seq with unmodified oligonucleotides
Study identified treatment-induced chromatin dynamics in 28 mice with lung tumors
Profiled epigenetic heterogeneity of 32 spatial regions in normal breast tissues
SNuBar-ARC identified TFs that regulated breast cell type identities in 4 individuals
Acknowledgements
This study was supported by grants to N.E.N. from the American Cancer Society (129098-RSG-16-092-01-TBG), the NIH National Cancer Institute (RO1CA240526, RO1CA236864, UO1 CA216468), the Emerson Collective Cancer Research Fund and the CPRIT Single Cell Genomics Center (RP180684). N.E.N. is an AAAS Fellow, AAAS Wachtel Scholar, Andrew Sabin Family Fellow and Jack & Beverly Randall Innovator. This study was supported by the MD Anderson Sequencing Core Facility Grant (CA016672). We thank Hongli Tang, Louis Ramagli, and Erika Thompson for their help with next-generation sequencing.
Footnotes
Declaration of Interests declarations
A US patent (UTSC.P1153US.P1) has been filed describing the SNuBar method.
ADDITIONAL RESOURCES
Detailed Protocol. A detailed bench protocol is available as Methods S1.
Table S3. ChromVAR identified transcription factors that regulate cell type identities (related to Figure 5).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bagchi DP, Li Z, Corsa CA, Hardij J, Mori H, Learman BS, Lewis KT, Schill RL, Romanelli SM, and MacDougald OA (2020). Wntless regulates lipogenic gene expression in adipocytes and protects against diet-induced metabolic dysfunction. Molecular Metabolism 39, 100992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Mupo A, Huynh T, Cioffi S, Woods M, Jin C, McKeehan W, Thompson-Snipes L, Baldini A, and Illingworth E (2010). Tbx1 regulates Vegfr3 and is required for lymphatic vessel development. J Cell Biol 189, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lake BB, and Zhang K (2019). High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nature Biotechnology 37, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Miragaia RJ, Natarajan KN, and Teichmann SA (2018). A rapid and robust method for single cell chromatin accessibility profiling. Nature Communications 9, 5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CS, Hong J, Sapinoso L, Zhou Y, Liu Z, Micklash K, Schultz PG, and Hampton GM (2006). A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. PNAS 103, 3775–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, and Shendure J (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al. (2018). Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 50, 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskin E, Lareau CA, Eraslan G, Ludwig LS, and Regev A (2020). Single-cell multimodal profiling of proteins and chromatin accessibility using PHAGE-ATAC. BioRxiv 2020.10.01.322420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2020). ArchR: An integrative and scalable software package for single-cell chromatin accessibility analysis. BioRxiv 2020.04.28.066498. [Google Scholar]
- Guo C, Kong W, Kamimoto K, Rivera-Gonzalez GC, Yang X, Kirita Y, and Morris SA (2019). CellTag Indexing: genetic barcode-based sample multiplexing for single-cell genomics. Genome Biol. 20, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, Durbin R, Hemberg M, and Lawniczak MKN (2020). Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods 17, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, et al. (2011). N 6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone CN, Pattison AD, Gorringe KL, Harrison PF, Powell DR, Lock P, Baloyan D, Ernst M, Stewart AG, Beilharz TH, et al. (2018). Functional and genomic characterisation of a xenograft model system for the study of metastasis in triple-negative breast cancer. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, Breitman M, and Alitalo K (1995). Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A 92, 3566–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nature Biotechnology 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, Naranjo S, Westcott PMK, Duarte FM, Sankar V, et al. (2020). Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 38, 212–228.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, Steemers FJ, Lebofsky R, et al. (2019). Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nature Biotechnology 37, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NY, and Blobe GC (2007). The interaction of endoglin with beta-arrestin2 regulates transforming growth factor-beta-mediated ERK activation and migration in endothelial cells. J Biol Chem 282, 21507–21517. [DOI] [PubMed] [Google Scholar]
- Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, and Song Y (2012). Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLOS ONE 7, e35065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau G, Letouzé E, Guichard C, Didelot A, Cazes A, Corté H, Fabre E, Pallier K, Imbeaud S, Pimpec-Barthes FL, et al. (2013). Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. International Journal of Cancer 132, 2217–2221. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, and Murrell J (1994). Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8, 345–351. [DOI] [PubMed] [Google Scholar]
- McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z, et al. (2019). MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods 16, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, Farmer A, Fordyce P, Linnarsson S, and Greenleaf W (2018). High-throughput chromatin accessibility profiling at single-cell resolution. Nature Communications 9, 3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Lareau CA, Chen KY, Zorzetto-Fernandes AL, Takeshima Y, Luo W, Huang T-S, Yeung B, Thakore PI, Wing JB, et al. (2020). Scalable, multimodal profiling of chromatin accessibility and protein levels in single cells. BioRxiv 2020.09.08.286914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelli Patrick, bbimber Bill Flynn, santiagorevale, and Gege Gui (2019). Hoohm/CITE-seq-Count: 1.4.2 (Zenodo). [Google Scholar]
- Pellacani D, Bilenky M, Kannan N, Heravi-Moussavi A, Knapp DJHF, Gakkhar S, Moksa M, Carles A, Moore R, Mungall AJ, et al. (2016). Analysis of Normal Human Mammary Epigenomes Reveals Cell-Specific Active Enhancer States and Associated Transcription Factor Networks. Cell Reports 17, 2060–2074. [DOI] [PubMed] [Google Scholar]
- Puccini J, Dorstyn L, and Kumar S (2013). Caspase-2 as a tumour suppressor. Cell Death Differ 20, 1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nature Biotechnology 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2003). Targeting HIF-1 for cancer therapy. Nature Reviews Cancer 3, 721–732. [DOI] [PubMed] [Google Scholar]
- Shin D, Lee W, Lee JH, and Bang D (2019). Multiplexed single-cell RNA-seq via transient barcoding for simultaneous expression profiling of various drug perturbations. Science Advances 5, eaav2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sl F, Ap A, S M, Dm G, Rn A, Kk W, D L, and Ah B (2020). CRISPR/Cas9-Mediated Foxp1 Silencing Restores Immune Surveillance in an Immunocompetent A20 Lymphoma Model (Front Oncol). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan SR, McFaline-Figueroa JL, Ramani V, Saunders L, Cao J, Packer J, Pliner HA, Jackson DL, Daza RM, Christiansen L, et al. (2020). Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Ouchida M, Yamamoto H, Yano M, Toyooka S, Aoe M, Shimizu N, Date H, and Shimizu K (2008). Decreased expression of the SIN3A gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer 59, 24–31. [DOI] [PubMed] [Google Scholar]
- Tominaga K, Kondo C, Kagata T, Hishida T, Nishizuka M, and Imagawa M (2004). The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. J Biol Chem 279, 34840–34848. [DOI] [PubMed] [Google Scholar]
- Vo Nguyen TT, Watanabe Y, Shiba A, Noguchi M, Itoh S, and Kato M (2014). TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci 105, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang M, Gan L, He T, Xiao X, Stewart MD, Liu X, Yang L, Zhang T, Zhao Y, et al. (2012). DLX4 upregulates TWIST and enhances tumor migration, invasion and metastasis. Int J Biol Sci 8, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Ho SS, Greer SU, Zhu X, Bell JM, Arthur JG, Spies N, Zhang X, Byeon S, Pattni R, et al. (2019). Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562. Genome Res. 29, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data was deposited in the NCBI Gene Expression Omnibus under accession: GSE162798. The code for the SNuBar method is available on the following GitHub repository: https://github.com/navinlabcode/snubar_atac_scripts
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.