

Abstract
We investigated the A3AR affinity and selectivity of a series of 2-substituted 3′-azido and 3′-amino adenosine derivatives as well as some 5′-uronamide derivatives thereof. All compounds showed high A3AR selectivity. While the 3′-azides appeared to be A3AR antagonists with moderate A3AR affinity, their 3′-amino congeners exhibit significantly improved A3AR affinity and behave as partial agonists. For both the 3′-azides and the 3′-amines, the 5′-methylcarbamoyl modification improved the overall affinity. Introduction of a 2-phenylethynyl substituent provided high affinity for the A3AR.
Keywords: Adenosine A3 receptor, Selectivity, Affinity, Ribose and purine modifications
1. Introduction
Adenosine receptors (AR) belong to the family of G protein-coupled receptors. They are subdivided into four subtypes designated A1, A2A, A2B and A3, according to the chronological discovery of the receptors.1 The A3ARs are coupled to Gi proteins and, therefore, inhibit adenylate cyclase leading to a decrease in intracellular levels of cAMP.2 The selective activation of the A3AR is both cardioprotective and cerebroprotective in a variety of ischaemic models.3,4 Selective A3AR antagonists promise to be useful in the regulation of cell growth5,6 and as anti-asthmatic,7 cerebroprotective4,8 and anti-inflammatory agents.9 A3AR antagonists appear to lower the intraocular pressure in mice and monkeys and are proposed as new potential therapeutics for the treatment of glaucoma.10,11
Adenosine receptors are ubiquitously distributed throughout the body. As a consequence, ligands need to be highly selective in their action with respect to receptor subtype and tissue to be of therapeutic value.12 Numerous structure–activity studies of adenosine derivatives as receptor agonists conclude that selectivity may be provided by specific substitutions of the adenine ring.13,1 Substitution at the 8-position of the ring is not well tolerated by any AR subtype.14,15 The nitrogen atoms at positions 3 and 7 are required for high affinity of adenosine at all subtypes.1 2-Alkynyl derivatives of NECA possess high affinity at the A3 receptor subtype. Moreover, the presence of 2-alkyne substituents enhanced the A3AR selectivity.16
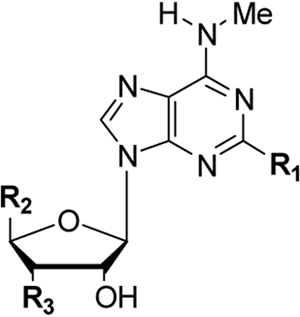
DeNinno et al. discovered that introduction of an amino group at the 3′ position improves the selectivity for the human A3AR, while enhancing the water solubility. The affinity drop caused by this 3′-substitution could be overcome by elaborating the N6-substituents.17 The combination of a large N6-substitituent with a 2-alkynyl group has proven to be unsuccessful because of the steric hindrance caused by the two large substituents, reflected by a decrease in A3AR affinity.16 Therefore, the present study investigated the effect of a 2-alkynyl substituent in concert with a small N6-substituent on the affinity and selectivity of a series of 3′-azido and 3′-amino adenosine derivatives. In addition, we evaluated the effect of the 5′-methylcarbamoyl modification on the overall affinity and efficacy of these compounds (Fig. 1).
Figure 1.
2. Results and discussion
2.1. Chemistry
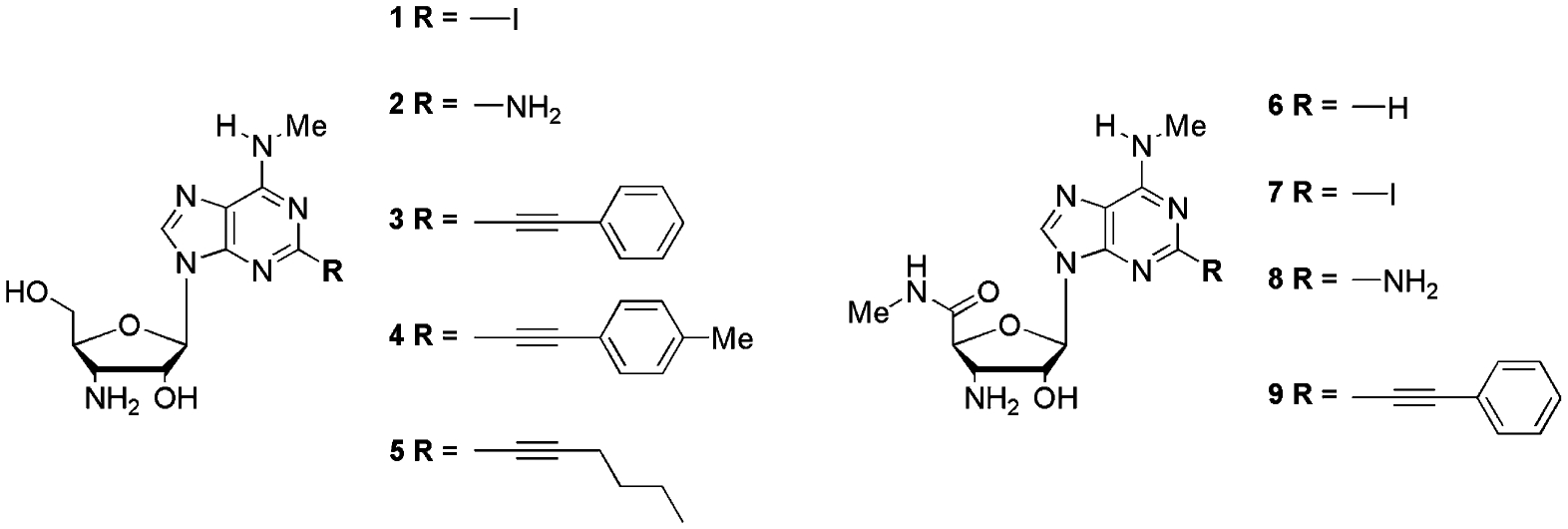
3-Azido-3-deoxy-1,2-di-O-acetyl-α-d-ribofuronamide (10) was prepared from the commercially available 1,2-O-isopropylidene-α-d-xylofuranose as described by us previously.18 It was coupled under Vorbrüggen conditions with silylated 2-amino-6-chloropurine to give 11 in 79% yield. Classical procedures allowed a straightforward conversion of 11 to 13. Triphenylphosphine reduction of the azido moiety yielded the corresponding amine 7.
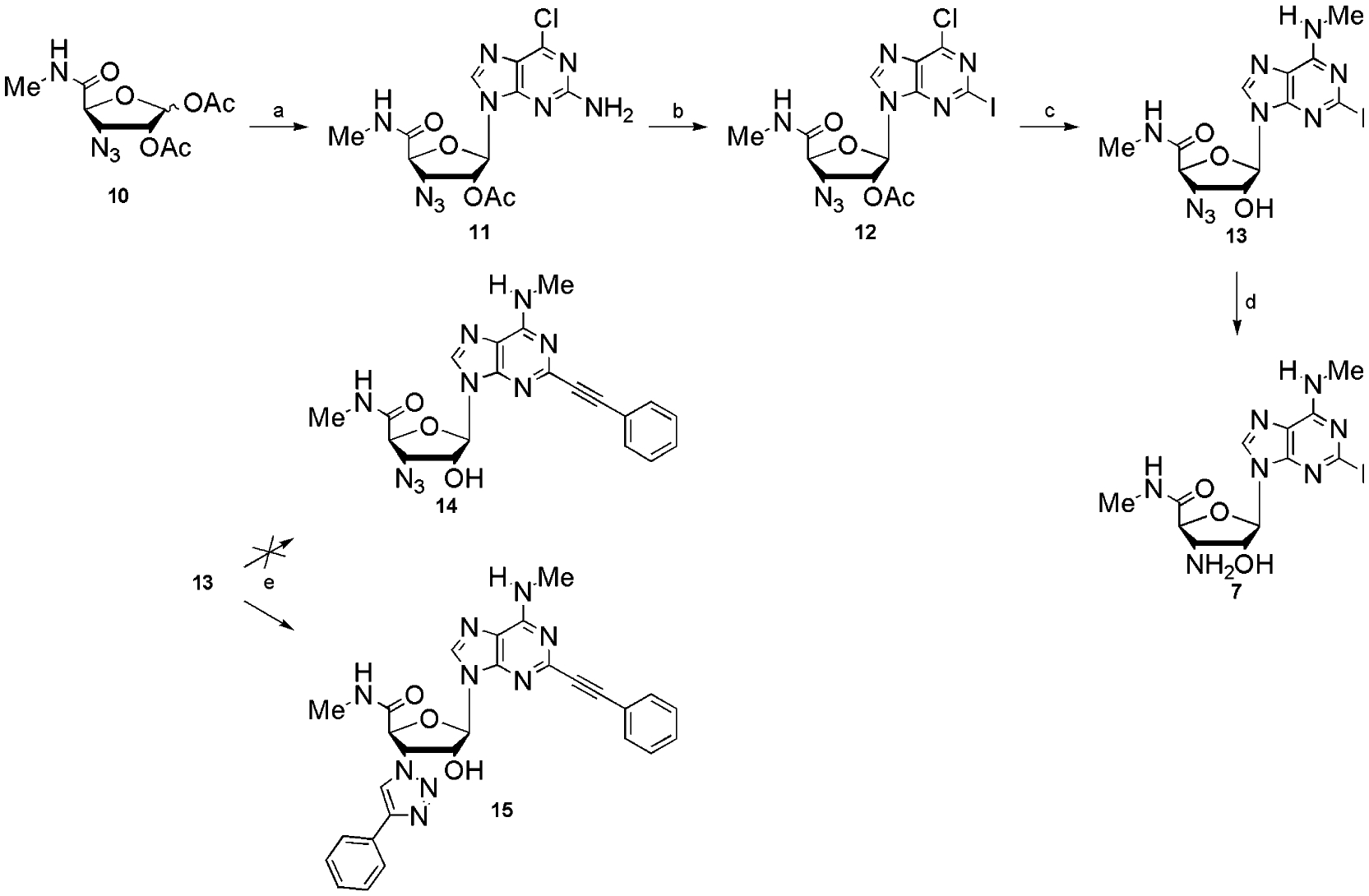
Based on the results of Cristalli et al.19 we have chosen phenylethynyl as the most promising C2-substituent. Reaction conditions used to perform a Sonogashira coupling20 of 13 with phenylacetylene yielded the 3′-(4-phenyl-1,2,3-triazol-1-yl) derivative (15) of the 2-alkynylated compound (Scheme 1). This result was due to a Cu+-catalysed Huisgen dipolar cycloaddition21 of the 3′-azide with phenylacetylene. Consequently, another strategy was used to gain access to compound 14 (Scheme 2), starting from 6-chloro-2-iodo-(9-tetrahydropyran-2-yl)purine (16), obtained from the 2-unsubstituted analogue via a lithiation-mediated stannyl transfer process followed by 2-tributylstannyl-iodine exchange.22 Sonogashira coupling of 17, followed by deprotection, provided 19. Unfortunately, classical Vorbrüggen coupling,23 as described for the synthesis of 11, did not give satisfying results. By using N,O-bis(trimethylsilyl)acetamide (BSA) as silylating agent,24 9-(2-acetyl-3-azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-phenylethynyladenine (20) was obtained in poor yield.
Scheme 1.
Reagents and conditions: (a) i—HMDS, (NH4)2SO4, reflux, 20 h, ii—2-amino-6-chloropurine, TMSOTf; (b) isoamyl nitrite, I2, CuI, CH2I2 in THF, reflux; (c) CH3NH3Cl, Et3N, EtOH, reflux; (d) Ph3P, H2O, THF, 2 days; (e) phenylacetylene, (PPh3)2PdCl2, CuI, Et3N, DMF.
Scheme 2.
Reagents and conditions: (a) CH3NH3Cl, DMAP, EtOH, reflux; (b) phenylacetylene (PPh3)2PdCl2, CuI, Et3N, DMF; (c) TFA, CH2Cl2; (d) 10, BSA, TMSOTf, CH3CN reflux; (e) 7 N NH3 in MeOH; (f) Ph3P, H2O, THF, 2 days.
During the course of this work it became clear that the 3′-amino-analogues generally exhibit much better A3AR affinities than their 3′-azide precursors. Consequently, we focussed only on the 3′-amino derivatives for further synthesis. This direction permitted us to reduce the 3′-azide to a 3′-amine before the Sonogashira coupling, avoiding the unwanted cycloaddition.
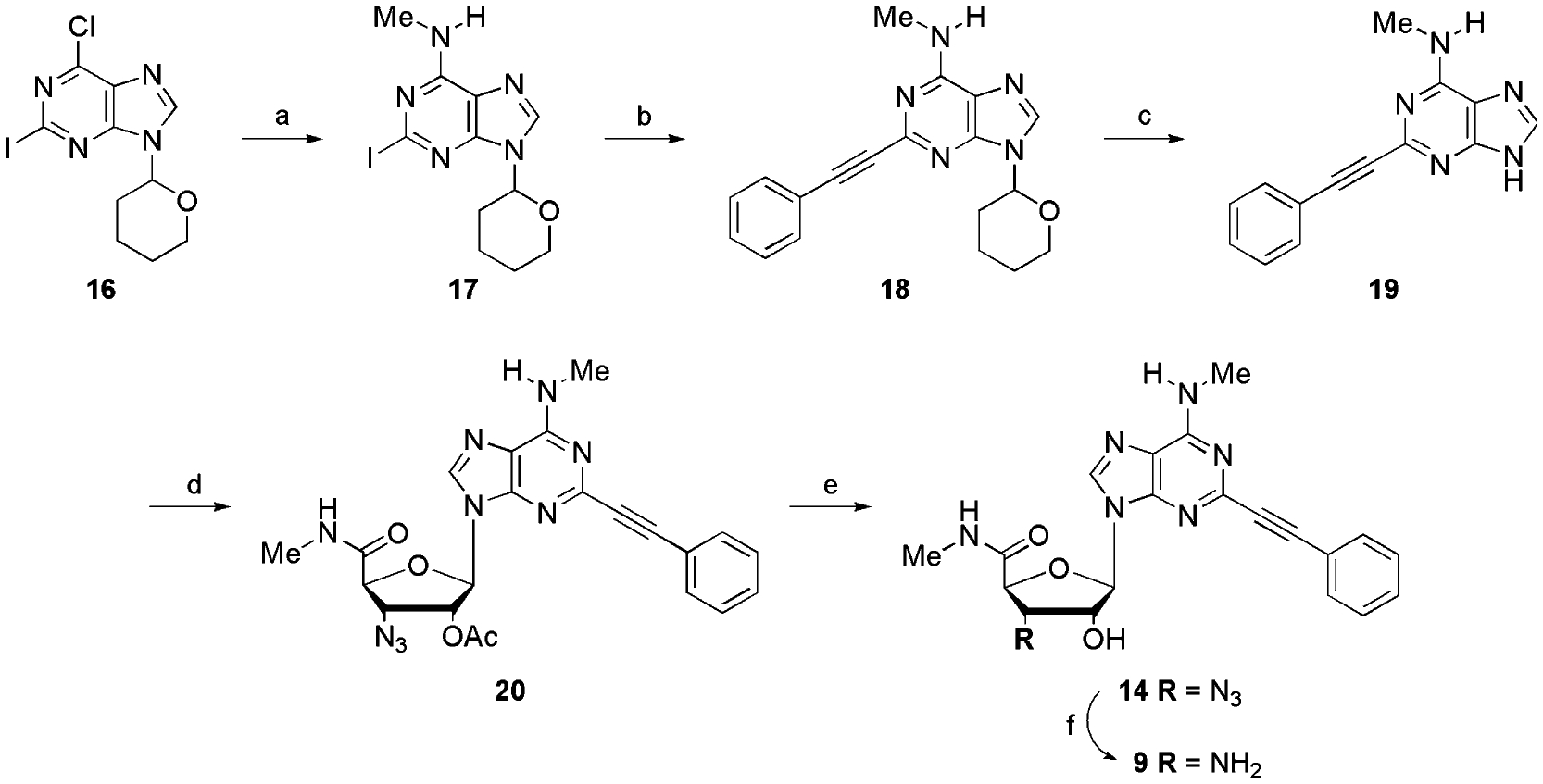
9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-N6-methyl-2-iodopurine (1) served as a suitable synthon for the synthesis of the 2-alkynylated 3′-amino-adenosines 3–5 (Scheme 3). It was obtained by coupling of sugar 21 with 2-amino-6-chloropurine. Elaboration of the base moiety to yield 25 was essentially accomplished as for 13. Staudinger reduction allowed the unmasking of the amine group. Finally, Sonogashira coupling on amine 1 provided the alkynylated analogues 3–5 in 80–82% yield.
Scheme 3.
Reagents and conditions: (a) 2-amino-6-chloropurine, BSA, TMSOTf, CH3CN, reflux; (b) isoamylnitrite, I2, CuI, CH2I2 in THF, reflux; (c) i—CH3NH3Cl, Et3N, EtOH, reflux, ii—7 N NH3 in MeOH; (d) Na° in MeOH; (e) Ph3P, H2O, THF, 2 days; (f) alkyne, (PPh3)2PdCl2, CuI, Et3N, DMF.
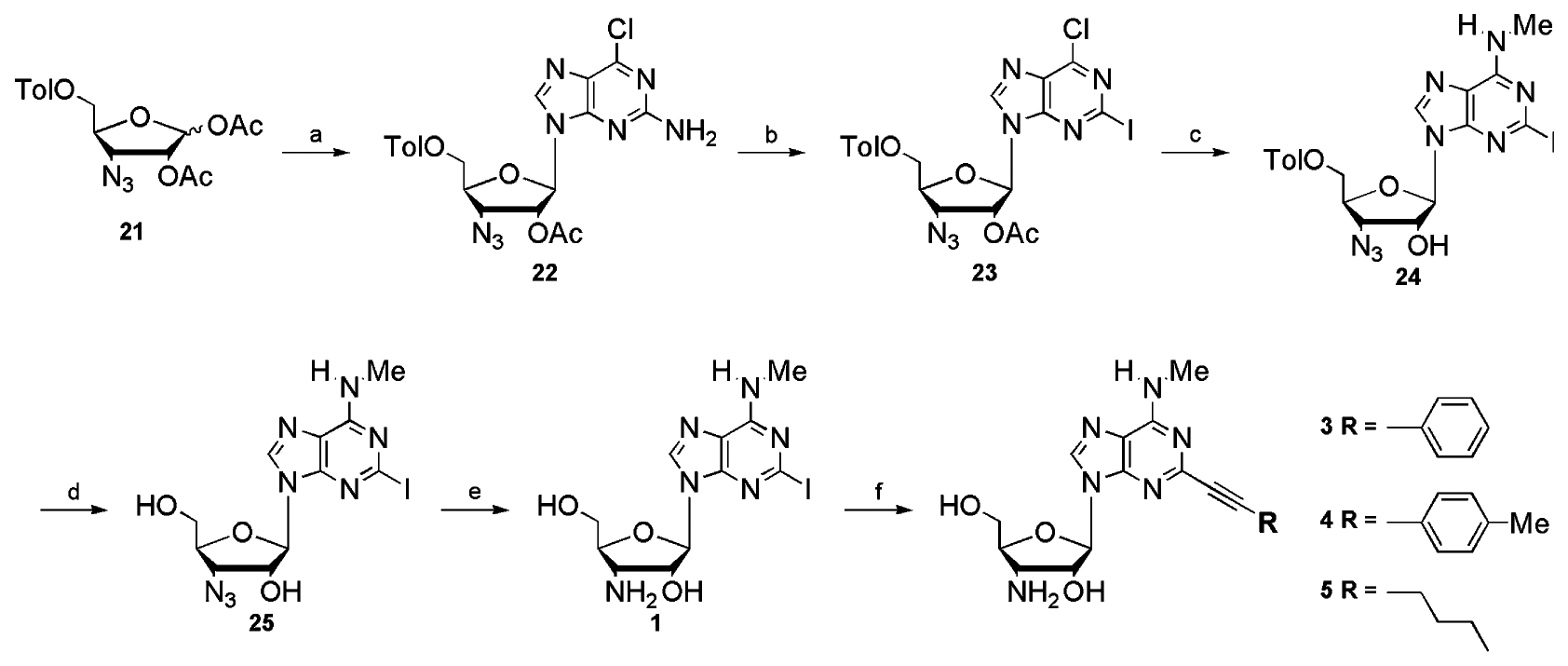
To continue the exploration of the 2-position, we synthesized the 2-I and the 2-NH2 derivatives of the 5′-OH and the 2-H and the 2-I derivatives of the 5′-methylcarbamoyl 3′-amino-N6-aminomethyl adenosine analogues (Scheme 4).
Scheme 4.
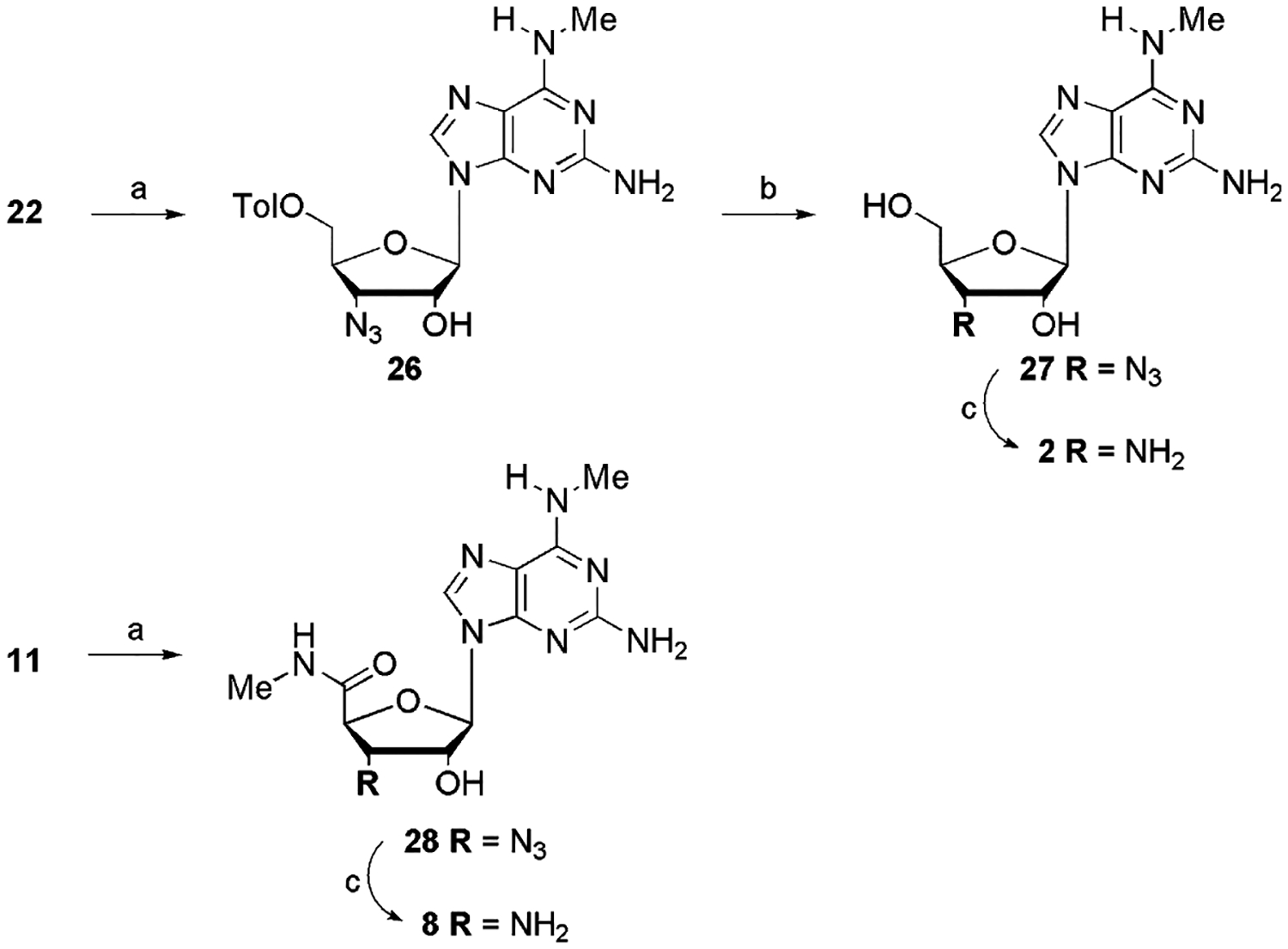
Reagents and conditions: (a) i—CH3NH3Cl, Et3N, EtOH, reflux, ii—7 N NH3 in MeOH; (b) Na° in MeOH; (c) Ph3P, H2O, THF, 2 days.
3′-Amine 6 (Fig. 1) was prepared by catalytic hydrogenation of the 3′-azide precursor which has been described.18
2.2. Biological evaluation
For the adenosine derivatives prepared in this study (1–9, 13, 14, 15, 25, 27 and 29) we measured both the binding affinities at the hA1, hA2A and hA3AR and their degrees of activation of the A3AR subtype. The results are reported in Table 1. The ability of each of these adenosine derivatives to compete for radioligand binding at each of these hARs was evaluated at a fixed concentration of 10 μM, and full competition curves were determined at the A3AR. Six different 2-substituents were included: H, I, NH2, Ph-C≡C, pMePh-C≡C and nBu-C≡C. The choice of the methyl group as a small N6 substituent was based on the results of Cristalli et al., who demonstrated that it increased the affinity for the human A3AR and significantly enhanced the A3AR selectivity.19
Table 1.
Binding affinities of adenine derivatives at human A1, A2A and A3ARs expressed in CHO cellsa
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | % Inhibition (hA3AR) | Ki (nM) | % Activationd (hA3AR) | |
| (hA1AR) (%) | (hA2AAR) (%) | ||||||
| 1 | I | CH2OH | NH2 | 39 | 12 | 879 ± 346 | 67 ± 6 |
| 2 | NH2 | CH2OH | NH2 | 16 | 4 | 654 ± 42 | 57 ± 2 |
| 3 | Ph-C≡C | CH2OH | NH2 | 50 | 14 | 126 ± 4 | 36 ± 6 |
| 4 | pMePh-C≡C | CH2OH | NH2 | 33 | 0 | 145 ± 35 | 23 ± 3 |
| 5 | nBu-C≡C | CH2OH | NH2 | 4 | 3 | 389 ± 112 | 66 ±11 |
| 6 | H | CONHMe | NH2 | 18 | 12 | 32.3 ± 3.9 | 72 ± 10 |
| 7 | I | CONHMe | NH2 | 16 | 24 | 71.4 ± 12.8 | 18 ± 10 |
| 8 | NH2 | CONHMe | NH2 | −14 | 10 | 536 ± 247 | 92 ± 3 |
| 9 | Ph-C≡C | CONHMe | NH2 | 9 | 9 | 15.6 ± 3.6 | 67 ± 11 |
| 13 | I | CONHMe | N3 | 16 | 0 | 2530b | −8 ± 5 |
| 14 | Ph-C≡C | CONHMe | N3 | 31 | 16 | 78.9 ± 12.4 | −11 ± 8 |
| 15 | Ph-C≡C | CONHMe | 4-Ph-1,2,3-triazol-1-yl | 14 | 2 | 1820 ± 770 | 0 |
| 25 | I | CH2OH | N3 | 85b,c | 27% | 6540 ± 320 | −3 ± 2 |
| 27 | NH2 | CH2OH | N3 | 49 | 16 | 28,800b | 0 |
| 29 18 | H | CONHMe | N3 | 12 | 10 | 1140 ± 300 | 38 ± 4 |
All A3AR experiments were performed using adherent CHO cells stably transfected with cDNA encoding one of the human adenosine receptors. Binding at human A1, A2A and A3ARs in this study was carried out as described in methods using [3H]PIA, [3H]CGS 21680 or [125I]AB-MECA as radioligand. Values from the present study are expressed as Ki values (means ± SEM, n = 3, unless noted) or as percent displacement of radioligand at 10 μM.
n = 1.
Ki (A1 AR) = 1850 nM.
% Inhibition at 100 μM of forskolin-stimulated cAMP production at 10 μM, in CHO cells expressing the hA3AR, as a percentage of the response of the full angonist CI-IB-MECA (n = 3).
Results from the competition experiments showed that the A3AR affinities of the 3′-amines were much higher than those of the 3′-azides. For all derivatives studied, the 5′-methylcarbamoyl modification, in general, enhanced the affinity at the A3AR in comparison to 5′-CH2OH. Except for 25, all evaluated compounds showed a very high selectivity for the A3AR compared to the other ARs. The most potent compound (9) displayed a Ki value of 16 nM at the A3AR. The C-2 substituent of this compound, a phenylethynyl moiety, was previously shown to enhance A3AR affinity and selectivity,19 and proved to have a superior contribution to A3AR affinity than a p-methyl-phenylethynyl (4) or a 1-hexynyl (5) moiety. Furthermore, the 2-phenylethynyl modification appeared to overcome the reduction of affinity caused by the 3′-azide (cf. Ki = 78.9 nM for 14 vs 1140 nM for 29). Consequently, this C-2 substituent was selected to be combined with a 3′-amino and a 5′-methylcarbamoyl modification.
Previous studies showed that 3′-amino derivatives exhibit a decreased affinity at the A3AR compared to their 3′-hydroxy analogues. The affinity reduction associated with this 3′ modification could be overcome by elaborating the N6-substituents, for example with a substituted benzyl group.17 The high affinity of compound 9 (Ki = 15.6 nM) demonstrated that a 2-phenylethynyl modification in concert with a small N6-substituent was likewise capable of overcoming this reduction in affinity. Note that in our experiments the affinity of derivative 6 for the A3AR (Ki = 32.3 nM) was 4-fold higher than that reported by DeNinno et al.15 The 2-I analogue 7 also showed appreciable A3AR affinity (Ki = 71.4 nM). Conversely, the 2-NH2 analogue 8 exhibited weak A3AR affinity (Ki = 536 nM).
The results of the cyclic cAMP-assay (Table 1) indicated that all 3′-azides were A3AR antagonists, except for compound 29 which showed partial agonist activity. Also the 3′-(4-phenyl-1,2,3-triazol-1-yl) derivative 15 appeared to be an A3AR antagonist. All other compounds were partial agonists, except for compound 8, which manifested full agonist activity.
3. Conclusions
The 2,3′,5′-trisubstituted and 2,3′-disubstituted N6-methyl adenosine derivatives described in the present study were synthesized in good overall yields. All the compounds had A3AR affinities in the low micromolar or nanomolar range and showed very high A3AR selectivity. The 3′-azides appeared to be A3AR antagonists with a moderate A3AR affinity. The 3′-amino modification significantly improved the A3AR affinity and resulted in partial A3AR agonists. For both the 3′-azido and the 3′-amino derivatives, the 5′-methylcarbamoyl modification improved the overall affinity. Curiously, the presence of a 5′-uronamide did not restore full A3AR efficacy in 2-position derivatives, as was demonstrated in the case of N6-substituents that reduced efficacy.26 The 2-phenylethynyl derivative 9 demonstrated high A3AR receptor affinity with a Ki value of 15.6 nM and >1000-fold selectivity. Previous studies revealed that 3′-amines exhibit a decreased affinity compared to their 3′-hydroxy analogues. This study demonstrated that introduction of a 2-phenylethynyl substituent in concert with the N6-methyl group is capable of overcoming this affinity drop.
4. Experimental
4.1. Chemicals and solvents
All reagents were from standard commercial sources and of analytic grade.
4.2. Chromatography
Precoated Merck silica gel F254 plates were used for TLC and spots were examined under UV light at 254 nm and further visualized by sulfuric acid-anisalde-hyde spray. Column chromatography was performed on Uetikon silica (0.2–0.06 mm).
4.3. Instruments and analyses
NMR spectra were obtained with a Varian Mercury 300 MHz spectrometer. Chemical shifts are given in ppm (δ) relative to the residual solvent signal, in the case of DMSO-d6 2.54 ppm for 1H and in the case of CDCl3 7.26 ppm for 1H. All signals assigned to amino, amide hydrogen and hydroxyl groups were exchangeable with D2O. Exact mass measurements were performed on a quadrupole/orthogonal-acceleration time-of-flight (Q/oaTOF) tandem mass spectrometer (qToF 2, Micromass, Manchester, UK) equipped with a standard electrospray ionization (ESI) interface. Samples were infused in a 2-propanol/water (1:1) mixture at 3 μL/min.
4.4. 9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-2-iodo-N6-methyladenine (1)
This compound was synthesized from 30 mg (0.069 mmol) of 25 by the procedure described for the synthesis of 7; yield: 26 mg (92%). 1H NMR (300 MHz, DMSO-d6): δ 1.66 (br s, 2H, NH2), 2.87 (d, 3H, J = 4 Hz, N6-CH3), 3.41 (app t, 1H, J = 6.0 Hz, H3′), 3.54–3.71 (m, 3H, H4′ and H5′A and H5′B), 4.10 (br s, 1H, 2′-OH), 4.19 (dd, 1H, J = 2.6 and 4.4 Hz, H2′), 4.98 (br s, 1H, 5′-OH), 5.82 (d, 1H, J = 2.6 Hz, H1′), 8.11 (d, 1H, J = 4 Hz, N6H), 8.30 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C11H16N6O3I[M+H]+: 407.0330. Found: 407.0332.
4.5. 9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-2-amino-N6-methyladenine (2)
This compound was synthesized from 27 (35 mg, 0.11 mmol) by the procedure described for the synthesis of 7; yield: 30 mg (93%). 1H NMR (300 MHz, DMSO-d6): δ 1.70 (br s, 2H, 3′-NH2), 2.86 (s, 3H, N6-CH3), 3.34 (app t, 1H, J = 6.0 Hz, H3′), 3.52–3.70 (m, 3H, H4′ and H5′A and H5′B), 4.16 (br s, 2H, 2′-OH and H2′), 5.16 (br s, 1H, 5′-OH), 5.74 (d, 1H, J = 2.8 Hz, H1′), 5.83 (s, 2H, 2-NH2), 7.24 (br s, 1H, N6H), 7.91 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C12H18N7O3 [M+H]+: 296.1471. Found: 296.1470.
4.6. General procedure for the synthesis of alkynes 3, 4 and 5 from 1
Compound 1 (50 mg, 0.12 mmol) was dissolved in Et3N (1.5 mL) and DMF (1 mL). After purging the solution with N2, (PPh3)2PdCl2 (8.6 mg, 0.012 mmol) and CuI (2.3 mg, 0.012 mmol) were added. The appropriate alkyne (2 equiv) was subsequently added dropwise and the mixture was stirred at room temperature for 3 h. The solvents were removed under reduced pressure, the residue was taken up in ethyl acetate and the solution was filtered over a Celite pad. The residue remaining after solvent evaporation was purified on silica gel column (CH2Cl2/MeOH, 90:10).
4.7. 9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-N6-methyl-2-phenylethynyladenine (3)
The reaction of 1 (50 mg, 0.12 mmol) with phenylacetylene (27 μL, 0.24 mmol) gave compound 3 in 81% yield. 1H NMR (300 MHz, DMSO-d6) δ 2.07 (br s, 2H, 3′-NH2), 2.94 (s, 3H, N6-CH3), 3.44 (app t, 1H, J = 5.3 Hz, H3′), 3.56–3.75 (m, 3H, H4′ and H5′A and H5′B), 4.09 (br s, 1H, 2′-OH), 4.24 (dd, 1H, J = 2.6 and 4.4 Hz, H2′), 5.11 (br s, 1H, 5′-OH), 5.94 (d, 1H, J = 2.3 Hz, H1′), 7.46 (m, 3H, Ph), 7.61 (m, 2H, Ph), 7.95 (br s, 1H, N6H), 8.47 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C19H21N6O3[M+H]+: 381.1675. Found: 381.1675.
4.8. 9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-N6-methyl-2-(4-methyl-phenyl)ethynyladenine (4)
The reaction of 1 (50 mg, 0.12 mmol) with 4-methylphenylacetylene (31 μL, 0.24 mmol) gave compound 4 in 82% yield. 1H NMR (300 MHz, DMSO-d6) δ 2.33 (s, 3H, CH3Ph), 2.94 (s, 3H, N6-CH3), 3.50–3.79 (m, 4H, H3′, H4′, H5′A and H5′B), 4.31 (dd, 1H, J = 2.4 and 4.5 Hz, H2′), 5.15 (br s, 1H, 5′-OH), 5.74 (s, 1H, 2′-OH), 5.96 (d, 1H, J = 2.4 Hz, H1′), 7.25 (m, 2H, Ph), 7.50 (m, 2H, Ph), 7.95 (br s, 1H, N6H), 8.46 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C20H23N6O3[M+H]+: 395.1831. Found: 395.1823.
4.9. 9-(3-Amino-3-deoxy-β-d-ribofuranosyl)-N6-methyl-2-(1-hexyn-1-yl)adenine (5)
The reaction of 1 (50mg, 0.12 mmol) with 1-hexyn (28 μL, 0.24 mmol) gave compound 5 in 80% yield. 1H NMR (300 MHz, DMSO-d6) δ 0.90 (t, 3H, J = 7.03 Hz, CH2CH3), 1.37–1.54 (m, 4H, CH2CH2CH3), 2.41 (t, 2H, J = 7.04 Hz, C≡CCH2), 2.88 (s, 3H, N6-CH3), 3.48–3.73 (m, 4H, H3′, H4′, H5′A and H5′B), 4.10–4.23 (m, 2H, H2′ and 2′-OH), 5.10 (br s, 1H, 5′-OH), 5.90 (d, 1H, J = 2.5 Hz, H1′), 7.83 (br s, 1H, N6H), 8.41 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C17H25N6O3[M+H]+: 361.1988. Found: 361.1982.
4.10. 9-(3-Amino-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-iodoadenine (7)
Compound 13 (50 mg, 0.11 mmol) and PPh3 (57 mg, 0.21 mmol) were dissolved in THF (2 mL). After stirring for 10 min, H2O was added (270 μL, 15 μmol) and the reaction mixture was stirred for 2 days. The residue obtained after solvent evaporation was purified by column chromatography (CH2Cl2/MeOH, 95:5) to yield 82% of compound 7. 1H NMR (300 MHz, DMSO-d6): δ 1.79 (s, 2H, 3′-NH2), 2.69 (d, 3H, J = 4.7 Hz, CH3NHCO), 2.88 (d, 3H, J = 4.1 Hz, N6-CH3), 3.54 (t, 1H, J = 11.1 Hz, H3′), 4.10 (d, 1H, J = 6.2 Hz, H4′), 4.30 (app t, 1H, J = 8.8 Hz, H2′), 5.91 (br s, 1H, 2′-OH), 5.93 (d, 1H, J = 3.81 Hz, H1′), 8.03 (d, 1H, J = 4,7 Hz, NHCO), 8.13 (d, 1H, J = 4.1 Hz, N6H), 8.48 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C12H17ClIN7O3[M+H]+ : 434.0439. Found: 434.0445.
4.11. 9-(3-Amino-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-2-amino-N6-methylpurine (8)
Compound 28 (40 mg, 0.12 mmol) and PPh3 (66 mg, 0.25 mmol) were dissolved in THF (2 mL). After stirring for 10 min, H2O was added (310 μL, 17 mmol) and the mixture was stirred for 2 days. The residue obtained after solvent evaporation was purified by column chromatography (CH2Cl2/MeOH, 80:20) to give compound 8 in 80% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.95 (br s, 2H, 3′-NH2), 2.66 (d, 3H, J = 4.7 Hz, CH3NHCO), 2.86 (s, 3H, N6-CH3), 3.54 (app t, 1H, J = 5.4 Hz, H3′), 4.05 (d, 1H, J = 5.6 Hz, H4′), 4.37 (app t, 1H, J = 4.7 Hz, H2′), 5.82 (d, 1H, J = 4.1 Hz, H1′), 5.88 (s, 2H, 2-NH2), 7.31 (s, 1H, N6H), 8.04 (s, 1H, H8), 8.27 (d, 1H, J = 4.4 Hz, NHCO); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C12H19N8O3[M+H]+: 323.1580. Found: 323.1579.
4.12. 9-(3-Amino-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-phenylethynyladenine (9)
This compound was synthesized by the procedure described for the synthesis of 7 from 20 mg (0.046 mmol) of 14 in 96% yield (18 mg). 1H NMR (300 MHz, DMSO-d6): δ 1.78 (s, 2H, 3′-NH2), 2.71 (d, 3H, J = 4.4 Hz, CH3NHCO), 2.94 (s, 3H, N6-CH3), 4.33 (dd, 1H, J = 5.0 and 5.3 Hz, H3′), 4.13 (d, 1H, J = 5.3 Hz, H4′), 4.36 (br s, 1H, H2′), 5.94 (s, 1H, 2′-OH), 6.02 (d, 1H, J = 4.40, H1′), 7.46 (m, 3H, Ph), 7.61 (m, 2H, Ph), 8.04 (d, 1H, J = 4.4 Hz, N6H), 8.41 (d, 1H, J = 4.7 Hz, NHCO), 8.62 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C20H22N7O3[M+H]+: 408.1784. Found: 408.1787.
4.13. 9-(2-O-Acetyl-3-azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-2-amino-6-chloropurine (11)
4.13.1. Silylation of the base.
2-Amino-6-chloropurine (462 mg, 2.7 mmol) was treated with 1,1,1,3,3,3-hexamethyldisilazane (HMDS, 40 mL) and (NH4)2SO4 (0.27 mmol, 36 mg) and refluxed for 20 h. The silylated compound was concentrated and used without further purification.
4.13.2. Vorbrüggen coupling.
3-Azido-3-deoxy-1,2-di-O-acetyl-α-d-ribofuronamide (10, 600 mg, 2.09 mmol) in dry 1,2-dichloroethane (25 mL) was added to the silylated 2-amino-6-chloropurine (462 mg, 2.7 mmol). The solution was gently refluxed, and after 5 min TMSOTf (417μL, 2.3 mmol) was added dropwise. After 4 h, the mixture was cooled to room temperature, quenched with a cold saturated NaHCO3 solution (80 mL) and extracted with CH2Cl2 (40 mL). The organic layer was dried with MgSO4, filtered and evaporated to dryness. The residue was purified by column chromatography (CH2Cl2/MeOH, 98:2) to give 650 mg (79%) of compound 11. 1H NMR (300 MHz, CDCl3): δ 2.15 (s, 3H, CH3CO), 2.90 (d, 3H, J = 5.0 Hz, CH3N), 4.53 (d, 1H, J = 3.2 Hz, H4′), 4.90 (dd, 1H, J = 3.5 and 5.0 Hz, H3′), 5.14 (s, 2H, NH2), 5.94 (t, 1H, J = 5.3 Hz, H2′), 5.97 (d, 1H, J = 6.2 Hz, H1′), 7.09 (br s, 1H, NHCO), 7.82 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C13H15ClN9O4[M+H]+: 396.0935. Found: 396.0932.
4.14. 9-(2-O-Acetyl-3-azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-6-chloro-2-iodopurine (12)
Isoamylnitrite (681 μL, 4.98 mmol) was added to a mixture of 11 (650 mg, 1.65 mmol), I2 (418 mg, 1.65 mmol), CH2I2 (1.37 mL, 16.5 mmol) and CuI (330 mg, 1.72 mmol) in 15 mL THF. The mixture was refluxed for 45 min and then cooled to room temperature. Insoluble materials were removed by filtration, and the filtrate was concentrated to dryness. The residue was purified by means of a silica gel column, which was washed with CH2Cl2 until the iodine colour disappeared and then eluted with CH2Cl2/MeOH, 98:2. Compound 12 was obtained in 79% yield. 1H NMR (300 MHz, CDCl3): δ 2.13 (s, 3H, CH3CO), 3.06 (d, 3H, J = 4.98 Hz, CH3N), 4.56 (d, 1H, J = 2.9 Hz, H4′), 4.80 (dd, 1H, J = 2.9 and 5.6 Hz, H3′), 5.75 (dd, 1H, J = 5.9 and 7.0 Hz, H2′), 6.06 (d, 1H, J = 7.3 Hz, H1′), 7.09 (br s, 1H, NHCO), 8.11 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C13H13ClIN8O4[M+H]+: 506.9794. Found: 506.9822.
4.15. 9-(3-Azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-iodoadenine (13)
Compound 12 (460 mg, 0.91 mmol) was dissolved in EtOH (10 mL). Methylammonium chloride (92 mg, 1.36 mmol) and Et3N (158 μL, 1.13 mmol) were added, and the solution was refluxed overnight. The mixture was concentrated to dryness, dissolved in 7 N NH3 in methanol and stirred at room temperature for 2 h to deprotect the 2′-hydroxyl group. The volatiles were removed under reduced pressure, and the residue was purified by silica gel column (CH2Cl2/MeOH, 98:2). The product, 13, was realized, in 77% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.71 (d, 3H, J = 4.7 Hz, CH3NHCO), 2.89 (d, 3H, J = 4.4 Hz, N6-CH3), 4.33 (d, 1H, J = 3.5 Hz, H4′), 4.47 (dd, 1H, J = 3.5 and 5.0 Hz, H3′), 4.92 (dd, 1H, J = 5.6 and 11.4 Hz, H2′), 5.89 (d, 1H, J = 6.5 Hz, H1′), 6.33 (d, 1H, J = 5.6 Hz, 2′-OH), 8.10 (d, 1H, J = 4,7 Hz, NHCO), 8.17 (d, 1H, J = 4.7 Hz, N6H), 8.36 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C12H15IN9O3[M+H]+: 460.0344. Found: 460.0350.
4.16. 9-(3-Azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-phenylethynyladenine (14)
A solution of compound 20 (30 mg, 0.06 mmol) and 10 mL of 7 N NH3 in methanol was kept at room temperature for 2 h to allow deprotection of the 2′-hydroxyl group. The mixture was concentrated to dryness and purified on a silica gel column (CH2Cl2/MeOH, 97:3). A 91% yield of compound 14 was obtained. 1H NMR (300 MHz, DMSO-d6): δ 2.73 (d, 3H, J = 3.5 Hz, CH3NHCO), 2.96 (s, 3H, N6-CH3), 4.33 (d, 1H, J = 2.6 Hz, H4′), 4.51 (dd, 1H, J = 3.0 and 5.27 Hz, H3′), 4.96 (app t, 1H, J = 5.3 Hz, H2′), 5.98 (d, 1H, J = 6.7 Hz, H1′), 7.46 (m, 3H, Ph), 7.64 (m, 2H, Ph), 8.12 (s, 1H, N6H), 8.51 (s, 1H, H8), 8.53 (d, 1H, J = 4.4 Hz, NHCO); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C20H20N9O3[M+H]+: 434.1688. Found: 434.1686.
4.17. Attempted synthesis of compound 14
Attempted conversion of 13 to 14 using the procedure as described for 3, 4 and 5 failed to give 14, but resulted in the formation of triazole 15 as the sole reaction product. 1H NMR (300 MHz, DMSO-d6): δ 2.78 (d, 3H, J = 4.6 Hz, N6-CH3), 3.0 (br s, 3H, CH3NHCO), 5.15 (app q, 1H, J = 6.3 Hz, H2′), 5.17 (d, 1H, J = 3.4 Hz, H4′), 5.61 (dd, 1H, J = 3.6 and 6.3 Hz, H3′), 6.26 (d, 1H, J = 5.4 Hz, 2′OH), 6.27 (d, 1H, J = 6.3 Hz, H1′), 7.36 (t, 1H, J = 7.3 Hz, 4″-Ph), 7.46–7.51 (m, 2H, 4″-Ph and 3H, C≡CPh), 7.65 (br d, 2H, J = 6.8 Hz, C≡CPh), 7.90 (d, 2H, J = 7.6 Hz, 4″-Ph), 8.59 (s, 1H, H8), 8.71 (m, 2H, H5″ and NHCO); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C28H25N9O3[M+H]+: 536.2158. Found: 536.2166.
4.18. 2-Iodo-N6-methyl-(9-tetrahydropyran-2-yl)adenine (17)
Methylammonium chloride (28 mg, 0.41 mmol) and DMAP (67 mg, 0.54 mmol) were added to 100 mg (0.27 mmol) of compound 16 in EtOH (6 mL), and the solution was refluxed overnight. The mixture was concentrated to dryness and the residue was purified on a silica gel column (pentane/ethyl acetate, 50:50). The title compound was obtained in 81% yield. 1H NMR (300 MHz, DMSO-d6): δ 1.52 (m, 6H, H2A′ and H2B′, H3A′ and H3B′, H4A′ and H4B′), 2.87 (s, 3H, NH CH3), 3.68 (t, 1H, J = 11.4 Hz, H5′A), 3.96 (app d, 1H, J = 11.4 Hz, H5′B), 5.43 (dd, 1H, J = 2.3 and 10.9 Hz, H1′), 7.26 (br s, 1H, NHCH3), 7.87 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C11H15N5O3I[M+H]+: 360.0323. Found: 360.0333.
4.19. N6-Methyl-2-phenylethynyl-tetrahydropyranyladenine (18)
Compound 17 (200 mg, 0.557 mmol) was dissolved in a mixture of Et3N (3 mL) and DMF (1 mL) and the solution was purged with N2. (PPh3)2PdCl2 (39 mg, 0.056 mmol) and CuI (10.6 mg, 0.056 mmol) were added. Phenyl acetylene (112 μL, 1.11 mmol) was subsequently added dropwise, and the mixture was stirred at room temperature for 3 h. The solvents were removed under reduced pressure, the residue was taken up in ethyl acetate and the resulting solution was filtered over a pad of Celite. After solvent evaporation, the residue was purified on a silica gel column (pentane/ethyl acetate, 50:50) to give compound 18 in 91% yield. 1H NMR (300 MHz, CDCl3): δ 1.18–2.25 (m, 6H, H2A′ and H2B′, H3A′ and H3B′, H4A′ and H4B′), 3.21 (s, 3H, NH CH3), 3.72 (app t, 1H, J = 11.4 Hz, H5′A), 3.96 (app d, 1H, J = 10.3 Hz, H5′B), 5.79 (d, 1H, J = 9.1 Hz, H1′), 7.20 (s, 1H, H8), 7.30 (d, 3H, J = 5.3 Hz, H-Ph), 7.61 (m, 2H, H-Ph); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C19H20N5O[M+H]+: 334.1667. Found: 334.1671.
4.20. N6-Methyl-2-phenylethynylpurine (19)
To a solution of 18 (170 mg, 0.510 mmol) in 10 mL of CH2Cl2 was added slowly a solution of 0.78 mL TFA (10.2 mmol) and 0.78 mL CH2Cl2. After stirring at room temperature for 1 h, the solvent was evaporated, and the residue was taken up in ethyl acetate, and the solution was washed with 7% NaHCO3. After silica gel chromatography (CH2Cl2/MeOH, 97:3), pure 19 was obtained in a 72% yield. 1H NMR (300 MHz, DMSO): δ 2.97 (s, 3H, NH CH3), 7.30 (m, 3H, H-Ph), 7.61 (m, 2H, H-Ph), 7.88 (br s, 1H, NHCH3), 8.24 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C14H12N5[M+H]+: 250.1092. Found: 250.1073.
4.21. 9-(2-Acetyl-3-azido-3-deoxy-5-methylcarbamoyl-β-d-ribofuranosyl)-N6-methyl-2-phenylethynyladenine (20)
To a mixture of 19 (150 mg, 0.602 mmol) and methyl 3-azido-3-deoxy-1,2-di-O-acetyl-α-d-ribofuronamide (10) (207 mg, 0.722 mmol) in 3 mL CH3CN were successively added 223 μL (0.903 mmol) of N,O-bis(trimethylsilyl)-acetamide (BSA) and 131 μL (0.722 mmol) TMSOTf. The suspension was refluxed for 10 h. After being cooled to room temperature, the reaction was quenched with 7% NaHCO3 and extracted with CH2Cl2. The organic layer was washed with brine, filtered through a short pad of Celite and evaporated to dryness. The crude material was purified by column chromatography (CH2Cl2/MeOH, 99:1), and compound 20 was obtained in 24.4% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.12 (s, 3H, CH3CO), 3.02 (d, 3H, J = 4.69 Hz, CH3NHCO), 3.25 (s, 3H, N6-CH3), 4.58 (d, 1H, J = 2.6 Hz, H4′), 4.78 (dd, 1H, J = 2.1 and 5.3 Hz, H3′), 5.78 (dd, 1H, J = 7.3 and 12.9 Hz, H2′), 5.92 (s, 1H, N6H) 6.02 (d, 1H, J = 7.6 Hz, H1′), 7.40 (app d, 3H, J = 7.0 Hz H-Ph), 7.63 (app d, 2H, J = 9.1 Hz, H-Ph), 7.81 (s, 1H, H8), 8.83 (s, 1H, NHCO); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C22H22N9O4[M+H]+: 476.1794. Found: 476.1800.
4.22. 9-(2-Acetyl-3-azido-3-deoxy-5-O-toluoyl-β-d-ribofuranosyl)-2-amino-6-chloropurine (22)
To a mixture of 2-amino-6-chloropurine (90 mg, 0.53 mmol) and 3-azido-3-deoxy-1,2-di-O-acetyl-5-O-toluoyl-α-d-ribofuranose (21) (240 mg, 0.64 mmol) in 3 mL CH3CN were successively added 196 μL (0.79 mmol) BSA and 115 μL (0.64 mmol) TMSOTf. The suspension was heated at 80 °C for 3 h. After being cooled to room temperature, the reaction was quenched with 7% NaHCO3 and extracted with CH2Cl2. The organic layer was washed with brine, filtered through a short Celite pad and evaporated to dryness. The residue was purified by column chromatography (CH2Cl2/MeOH, 99:1) to yield 200 mg (65%) of compound 22. 1H NMR (300 MHz, CDCl3): δ 2.19 (s, 3H, CH3CO), 2.41(s, 3H, CH3-Ph), 4.40 (m, 1H, H4′), 4.52–4.58 (m, 1H, H5′A′), 4.76–4.82 (m, 2H, H3′ and H5′B), 5.13 (br s, 2H, NH2), 5.94 (d, 1H, J = 3.8 Hz, H1′), 5.80 (dd, 1H, J = 3.8 and 5.6 Hz, H2′), 7.23 (d, 2H, J = 7.63 Hz, Ph), 7.79 (s, 1H, H8), 7.86 (d, 2H, J = 8.2 Hz, Ph); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C20H19N8O5Cl[M+H]+: 487.1245. Found: 487.1246.
4.23. 9-(2-Acetyl-3-azido-3-deoxy-5-O-toluoyl-β-d-ribofuranosyl)-6-chloro-2-iodopurine (23)
This compound was prepared by the procedure described for the synthesis of 12 from 22 (200 mg, 0.41 mmol); yield: 200 mg (81%). 1H NMR (300 MHz, DMSO-d6): δ 2.15 (s, 3H, CH3CO), 2.35 (s, 3H, CH3-Ph), 4.37 (dd, 1H, J = 4.1 and 7.6 Hz, H4′), 4.52 (dd, 1H, J = 4.4 and 12.3 Hz, H5′A′), 4.65 (dd, 1H, J = 3.2 and 12.3 Hz, H5′B), 4.96 (dd, 1H, J = 5.8 and 7.6 Hz, H3′), 6.03 (dd, 1H, J = 2.6 and 5.28 Hz, H2′), 6.29 (d, 1H, J = 2.9 Hz, H1′), 7.24 (d, 2H, J = 7.9 Hz, Ph), 7.68 (d, 2H, J = 8.2 Hz, Ph), 8.74 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C20H17N7O5ClINa[M+Na]+: 619.9924. Found: 619.9920.
4.24. 9-(3-Azido-3-deoxy-2-hydroxyl-5-O-toluoyl-β-d-ribofuranosyl)-2-iodo-N6-methyladenine (24)
The title compound was prepared as described for the synthesis of 13 from 23 (200 mg, 0.335 mmol); yield: 127 mg (69%). 1H NMR (300 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3-Ph), 2.87 (d, 3H, J = 3.5 Hz, N6-CH3), 4.29 (dd, J = 5.3 and 9.7 Hz, H4′), 4.46–4.60 (m, 3H, H3′, H5′A and H5′B), 5.01 (t, 1H, J = 4.7 Hz, H2′), 5.87 (d, 1H, J = 4.7 Hz, H1′), 6.43 (d, 1H, J = 5.3 Hz, 2′-OH), 7.29 (d, 2H, J = 8.2 Hz, Ph), 7.78 (d, 2H, J = 8.2 Hz, Ph), 8.17 (d, 1H, J = 4.4 Hz, N6H), 8.21 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C19H20N8O4I [M+H]+: 551.0653. Found: 551.0649.
4.25. 9-(3-Azido-3-deoxy-β-d-ribofuranosyl)-2-iodo-N6-methyl-adenine (25)
Ester 24 (127 mg, 0.23 mmol) was dissolved in 2.5 mL MeOH. Na° (11.28 mg, 0.32 mmol) was added and the mixture was stirred at room temperature for 1 h. The reaction was quenched by adding a mixture of CH3COOH/H2O (9:1, v/v) to pH 7. The solution was concentrated to dryness, and the residue was purified by column chromatography (CH2Cl2/MeOH, 98:2) to yield 100 mg (95%) of compound 25. 1H NMR (300 MHz, DMSO-d6): δ 2.88 (d, 3H, J = 4 Hz, N6-CH3), 3.52–3.67 (m, 2H, H5′A and H5′B), 3.94 (dd, 1H, J = 7.26 and 3.81 Hz, H4′), 4.28 (app t, 1H, J = 4.5 Hz, H3′), 4.89 (app t, 1H, J = 5.4 Hz, H2′), 5.20 (br s, 1H, 5′-OH), 5.80 (d, 1H, J = 6.16 Hz, H1′), 6.25 (br s, 1H, 2′-OH), 8.18 (d, 1H, J = 4 Hz, N6H), 8.28 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C11H14N8O3I [M+H]+: 433.0235. Found: 433.0237.
4.26. 9-(3-Azido-3-deoxy-5-O-toluoyl-β-d-ribofuranosyl)-2-amino-N6-methyl-adenine (26)
Derivative 22 (100 mg, 0.21 mmol) was solubilized in EtOH (5 mL). Methylammonium chloride (35 mg, 0.515 mmol) and Et3N (72 μL, 0.515 mmol) were added, and the solution was refluxed overnight. The mixture was concentrated to dryness, the residue redissolved in methanolic NH3 and the solution stirred at room temperature for 2 h to allow deprotection of the 2′-hydroxyl group. The mixture was concentrated to dryness and purified on a silica gel column (CH2Cl2/MeOH, 97:3). Compound 26 was obtained in 68% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3-Ph), 2.84 (br s, 3H, N6-CH3), 4.23 (dd, J = 5.27 and 9.38 Hz, H4′), 4.43 (dd, 1H, J = 5.6 and 11.6, H5′A), 4.55 (m, 2H, H3 and H5′B), 4.48 (app t, 1H, 4.7 Hz, H2′), 5.78 (d, 1H, J = 4.7 Hz, H1′), 5.95 (br s, 2H, 2-NH2), 6.34 (d, 1H, J = 7.9 Hz, 2′-OH), 7.28 (br s, 1H, N6H), 7.29 (d, 2H, J = 7.9 Hz, Ph), 7.79 (s, 1H, H8), 7.82 (d, 2H, J = 8.2 Hz, Ph); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C19H22N9O4 [M+H]+: 440.1794. Found: 440.1789.
4.27. 9-(3-Azido-3-deoxy-β-d-ribofuranosyl)-2-amino-N6-methyladenine (27)
The title compound was synthesized from 26 (60 mg, 0.013 mmol) by the procedure described for the synthesis of 25; yield: 40 mg (91%). 1H NMR (300 MHz, DMSO-d6): δ 2.85 (br s, 3H, N6-CH3), 3.48–3.64 (m, 2H, H5′A and H5′B), 3.89 (dd, 1H, J = 3.2 and 7.0 Hz, H4′), 4.25 (dd, 1H, J = 3.2 and 5.6 Hz, H3′), 4.88 (app t, 1H, J = 5.3 Hz, H2′), 5.58 (t, J = 6.8 Hz, 1H, 5′-OH), 5.71 (d, 1H, J = 6.2 Hz, H1′), 5.85 (s, 2H, NH2), 6.16 (s, 1H, 2′-OH), 7.31 (br s, 1H, N6H), 7.89 (s, 1H, H8); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C11H16N9O3[M+H]+: 322.1376. Found: 322.1365.
4.28. 9-[3-Azido-3-deoxy-5-(methylcarbamoyl)-β-d-ribofuranosyl]-2-amino-6-chloropurine (28)
Derivative 11 (120 mg, 0.30 mmol) was dissolved in EtOH (6 mL). Methylammonium chloride (30 mg, 0.46 mmol) and Et3N (53 μL, 0.38 mmol) were added, and the solution was refluxed overnight. The mixture was concentrated to dryness, the remaining solid dissolved in 7 N NH3 in methanol and the solution stirred at room temperature for 2 h to deprotect the 2′-hydroxyl group. The mixture was concentrated to dryness and the residue was purified on a silica gel column (CH2Cl2/MeOH, 95:5). Compound 28 was obtained in 79% yield. 1H NMR (300 MHz, DMSO-d6): δ 2.68 (d, 3H, J = 4.7 Hz, CH3NHCO), 2.87 (s, 3H, N6-CH3), 4.24 (d, 1H, J = 2.9 Hz, H-4′), 4.42 (dd, 1H, J = 2.9 and 5.3 Hz, H3′), 4.97 (dd, 1H, J = 5.6 and 11.7 Hz, H2′), 5.81 (d, 1H, J = 6.8 Hz, H1′), 5.91 (s, 2H, NH2), 6.26 (d, 1H, J = 5.3 Hz, 2′-OH), 7.31 (s, 1H, N6H), 7.96 (s, 1H, H8), 8.35 (d, 1H, J = 4.7 Hz, NHCO); Exact Mass (ESI-MS, i-PrOH/H2O): Calcd for C12H17N10O3[M+H]+: 349.1484. Found: 349.1491.
4.29. Biological assays
4.29.1. Cell culture and membrane preparation.
CHO cells expressing recombinant human A3ARs were cultured in DMEM (Dulbecco’s modified Eagle’s medium) and F12 (1:1) supplemented with 10% foetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 μmol/mL glutamine and 800 μL geneticin. After harvest and homogenization, the cells were centrifuged at for 10 min. The pellet was resuspended in 50 mM Tris–HCl buffer (pH 8.0) containing 10 mM MgCl2 and 1 mM EDTA. The suspension was homogenized with an electric homogenizer for 10 s and was then recentrifuged at 20,000g for 20 min at 4 °C. The resulting pellets were resuspended in buffer containing 3 U/mL of adenosine deaminase, and the suspension was stored at −80 °C prior to the binding experiments. The protein concentration was measured using the Bradford assay.25
4.29.2. Radioligand binding studies.
For the A3AR binding experiments, the procedures were similar to those previously described.26 Briefly, each tube contained 100 μL of membrane suspension, 50 μL [125I]I-AB-MECA (final concentration 0.5 nM) and 50 μL of increasing concentrations of compounds in Tris–HCl buffer (50 mM, pH 7.4) containing 10 mM MgCl2, 1 mM EDTA. Non-specific binding was determined using 10 μM NECA. The mixtures were incubated at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. The binding of [3H]R-PIA to the recombinant hA1AR and the binding of [3H]CGS21680 to the recombinant hA2AAR were performed as previously described.27,28
4.29.3. Cyclic AMP accumulation assay.
Intracellular cyclic AMP levels were measured by the competitive protein binding method.29 CHO cells expressing recombinant human30 ARs were harvested by trypsinization. After resus-pension in the medium, cells were plated in 24-well plates in 0.5 mL medium/well. After 24 h, the medium was removed and cells were washed three times with 1 mL/well DMEM containing 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 U/mL) and incubated at 37 °C. For A3AR, after 45 min forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated upon removal of the medium, and the cells were lysed with 200 μL/well of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20 °C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate and 30 μL of 0.1 M HCl. Bound radioactivity was separated by rapid filtration through Whatman GF/C filters under reduced pressure and washed once with cold buffer. Bound radioactivity was subsequently measured by scintillation spectrometry.
References and notes
- 1.Müller C Curr. Top. Med. Chem 2000, 7, 1269. [DOI] [PubMed] [Google Scholar]
- 2.Poulsen SA; Quinn RJ Bioorg. Med. Chem 1998, 6, 619. [DOI] [PubMed] [Google Scholar]
- 3.Liang BT; Jacobson KA Proc. Natl. Acad. Sci. U.S.A 1998, 95, 6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Lubitz DK; Lin RC; Popik P; Carter MF; Jacobson KA Eur. J. Pharmacol 1994, 263, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson KA; Moro S; Kim YC; Li AH Drug Dev. Res 1998, 45, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brambilla R; Cattabeni F; Ceruti S; Barbieri D; Franceshi C; Kim Y; Jacobson KA; Klotz KN; Lohse MJ; Abbracchio MP Naunyn-Schmiedeberg’s Arch. Pharmacol 2000, 361, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beaven MA; Ramkumar V; Ali H Trends Pharmacol. Sci 1994, 15, 13. [DOI] [PubMed] [Google Scholar]
- 8.von Lubitz DK; Carter MF; Deutsch SI; Lin RC; Mastropaolo J; Meshulam Y; Jacobson KA Eur. J. Pharmacol 1995, 275, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramkumar V; Stiles G; Beaven MA; Ali H J. Biol. Chem 1993, 268, 16887. [PubMed] [Google Scholar]
- 10.Okamura T; Kurogi Y; Hashimoto K; Sato S; Nishikawa H; Kiryu K; Nagao Y Bioorg. Med. Chem. Lett 2004, 3775. [DOI] [PubMed] [Google Scholar]
- 11.Civan MM; Macknight ADC. Exp. Eye Res 2004, 78, 625. [DOI] [PubMed] [Google Scholar]
- 12.Van Tilburg E; von Frijtag Drabbe Künzel J; de Groote M J. Med. Chem 2002, 45, 420. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson KA; Knutsen L In Handbook of Experimental Pharmacology; Abbracchio M, Williams M, Eds.; Springer, A.: Berlin, 2001; Vol. 151/I, p 302. [Google Scholar]
- 14.Volpini R; Costanzi S; Lambertucci C; Vittori S; Lorenzen A; Klotz K-N; Cristalli G Bioorg. Med. Chem. Lett 2001, 11, 1931. [DOI] [PubMed] [Google Scholar]
- 15.Costanzi S; Lambertucci C; Vittori S; Volpini R; Cristalli G J. Mol. Graph. Model 2003, 21, 253. [DOI] [PubMed] [Google Scholar]
- 16.Volpini R; Constanzi S; Lambertucci C; Vittori S; Cristalli G Nucleosides, Nucleotides Nucleic Acids 2001, 20, 775. [DOI] [PubMed] [Google Scholar]
- 17.DeNinno MP; Masamune H; Chenard LK; DiRico KJ; Eller C; Etienne JB; Tickner JE; Kennedy SP; Knight DR; Kong J; Oleynek JJ; Tracey WR; Hill RJ J. Med. Chem 2003, 46, 353. [DOI] [PubMed] [Google Scholar]
- 18.Jeong LS; Kim MJ; Gao Z-G; Kim S-K; Jacobson KA; Chun MW Bioorg. Med. Chem. Lett 2004, 14, 4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volpini R; Constanzi S; Lambertucci C; Taffi S; Vittori S; Klotz K-N; Cristalli G J. Med. Chem 2002, 45, 3271. [DOI] [PubMed] [Google Scholar]
- 20.Mathieu R; Baurand A; Schmitt M; Gachet C; Bourguignon J-J Bioorg. Med. Chem 2004, 12, 1769. [DOI] [PubMed] [Google Scholar]
- 21.Huisgen R In 1,3—Dipolar Cycloaddition Chemistry; Padwa A, Ed.; Wiley: New York, 1984; pp 1–176. [Google Scholar]
- 22.Brun V; Legraverend M; Grierson DS Tetrahedron Lett. 2001, 42, 8161. [Google Scholar]
- 23.Vorbrüggen H; Krolikiewicz K; Bennua B Chem. Ber 1981, 114, 1234. [DOI] [PubMed] [Google Scholar]
- 24.Caddel J; Chapman A; Cooley B; Downey B; LeBlanc M; Jackson M; O’Connell T; Roper T; Xie SJ Org. Chem 2004, 69, 3212. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM Anal. Biochem 1976, 72, 248. [DOI] [PubMed] [Google Scholar]
- 26.Gao Z-G; Kim S-K; Biadatti T; Chen W; Lee K; Barak D; Kim SG; Johnson CR; Jacobson KA J. Med. Chem 2002, 45, 4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Z-G; Blaustein JB; Gross AS; Melman N; Jacobson KA Biochem. Pharmacol 2003, 65, 1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchison AJ; Williams M; de-Jesus R; Yokoyama R; Oei HH; Ghai GR; Webb RL; Zoganas HC; Stone GA; Jarvis MF J. Med. Chem 1990, 33, 1919. [DOI] [PubMed] [Google Scholar]
- 29.Nordstedt C; Fredholm BB Anal. Biochem 1990, 189, 231. [DOI] [PubMed] [Google Scholar]
- 30.Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10365. [DOI] [PMC free article] [PubMed] [Google Scholar]