

Abstract
Sarcopenia is a common geriatric disorder characterized by decreased muscle strength, low muscle mass and poor physical performance. This aging-related skeletal muscle deterioration leads to adverse outcomes and severely impairs the quality of life of patients. The accumulation of dysfunctional mitochondria with aging is an important factor in the occurrence and progression of sarcopenia. Mitochondrial quality control (MQC) fundamentally ensures the normal mitochondrial functions and is comprised of four main parts: proteostasis, biogenesis, dynamics and autophagy. Therefore, any pathophysiologic factors compromising the quality control of homeostasis in the skeletal muscle may lead to sarcopenia. However, the specific theoretical aspects of these processes have not been fully elucidated. Current therapeutic interventions using nutritional and pharmaceutical treatments show a modest therapeutic efficacy; however, only physical exercise is recommended as the first-line therapy for sarcopenia, which can ameliorate skeletal muscle deficiency by maintaining the homeostatic MQC. In this review, we summarized the known mechanisms that contribute to the pathogenesis of sarcopenia by impairing normal mitochondrial functions and described potential interventions that mitigate sarcopenia through improving MQC.
Keywords: sarcopenia, mitochondria, mitochondrial quality control, therapeutic intervention
1. Introduction
Senescence is a natural process of aging associated with degeneration of physical functions. The gradual loss of muscle mass, strength and function is one of the most important hallmarks of aging. Muscle mass starts to slightly decline from the age between 30 to 40 years of age along with a reduction in muscle function [1]. The reduction of muscle mass is more serious in populations with an inactive or sedentary lifestyle. The loss of muscle mass probably reaches 1 to 2% per year from 50 to 60 years and 3% to 5% per year at older ages [1]. In these people, 30% to 50% of the muscle mass may lose from 40 to 80 years totally [1]. Some of the cases may reach the diagnostic criteria of sarcopenia.
The term sarcopenia was coined by Irwin Rosenberg in the 1980s to describe an age-dependent decline in muscle mass and its adverse effects on human health [2]. In 2019, the European Working Group on Sarcopenia in Older People 2 (EWGSOP2) launched the latest diagnostic criteria for sarcopenia, including low muscle strength, decreased muscle quantity/quality or poor physical performance [3], highlighting the fundamental role of low muscle strength in the pathogenesis of sarcopenia. As a multifaceted geriatric disease characterized by progressive and generalized loss of skeletal muscle mass and function [4-6], the occurrence and progression of sarcopenia is always concomitant with various negative outcomes, including falls [7], fractures [8], loss of locomotion [9] and even mortality [10]. At the cellular and molecular levels, a constellation of latent mechanisms have been shown to participate in the sarcopenia, such as mitochondrial dysfunction[11], insulin resistance [12], inflammation [13], oxidative stress [14], adipose tissue infiltration [15] and neuromuscular impairment [16]. Notably, an increasing number of studies have indicated that dysfunctional mitochondria may play a central role in the pathogenesis of sarcopenia.
Mitochondria have strong impacts on the maintenance of cellular viability, including ATP production, oxidative phosphorylation (OXPHOX) homeostasis, calcium buffering and apoptosis. Therefore, healthy quality control is crucial for the preservation of intracellular homeostasis of muscle cells with aging. The MQC includes mitochondrial proteostasis, biogenesis, dynamics and autophagy [17, 18]. Orchestrated mechanisms contain several cellular factors and signaling pathways to ensure the integrity of mitochondria. Mitochondrial biogenesis is responsible for the generation of new mitochondria through the synergistic interaction of the nuclear and mitochondrial genes [19]; mitochondrial dynamics is achieved by continual transformation between fusion and fission to eliminate the accumulation of unhealthy mitochondria [20]; mitochondrial autophagy (mitophagy) is a process of selective removal of the hypofunctional and damaged mitochondria [21]. Adverse alternations in the quality control mechanisms may lead to mitochondrial dysfunction, which can further contribute to muscle wasting and even sarcopenia [22-25].
The incidence rate of sarcopenia in the mid-life and elderly population varies according to different age, operational definitions, regions and ethnicities [26-31]. A number of epidemiological studies have shown that the prevalence of sarcopenia gradually increases with age. It is conservatively estimated that 5%-13% of elderly individuals aged 60-70 years are suffering from sarcopenia. The numbers increase to 11%-50% among those aged 80 or above [32]. Since the number and proportion of the global aging population is rapidly growing, the socio-economic burden of individuals and society may increase due to higher prevalence of sarcopenia. Sarcopenia has been formally recognized as a disease with an ICD-10-CM (M62.84) code in 2016 [33, 34], which attracted additional attention for this degenerative disease. Physical activity is recommended as the primary treatment for sarcopenia to improve muscle strength and mass [35], although no specific drugs have been developed with therapeutic effects in sarcopenia. In this review, we summarized the potential mechanisms of mitochondrial dysfunction with an emphasis on promising therapeutic interventions to prevent and ameliorate sarcopenia during aging.
2. Mitochondrial Quality Control in Sarcopenia
Mitochondrial quality control is an elaborate and complicated network in eukaryocytes for maintenance of mitochondria homeostasis in eukaryotes by means of four core processes: mitochondrial proteostasis, biogenesis, dynamics and autophagy. Mitochondrial dysfunction amplified by defective quality control processes is gradually regarded as the major pathophysiologic mechanism of sarcopenia (Fig. 1).
Figure 1.
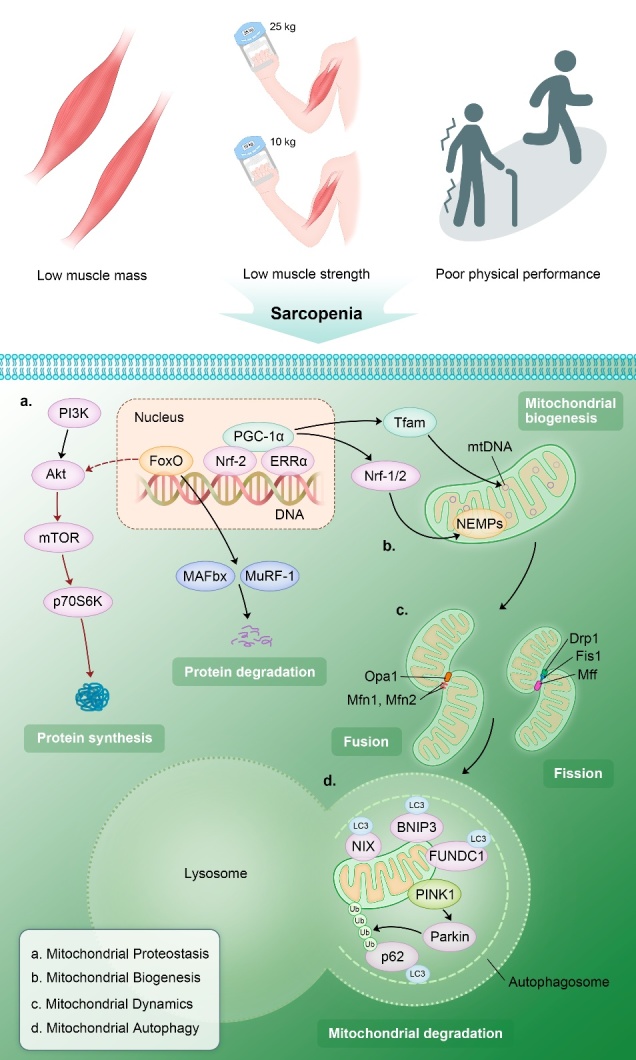
The potential mechanisms of mitochondrial quality control (MQC) dyshomeostasis in sarcopenia. Impaired mitochondrial proteostasis, biogenesis, dynamics and autophagy have been regarded as the major molecular mechanisms in mitochondrial dysfunction, which could lead to the onset and progression of sarcopenia. (PIK3: phosphoinositide 3-kinase; Akt: protein kinase B; mTOR: mechanistic target of rapamycin; FoxO: Forkhead Box O; MAFbx: muscle atrophy F-box; MuRF-1: muscle RING finger protein 1; PGC-1α: peroxisome proliferative activated receptor-γ coactivator-1α; Nrf-1 and 2: nuclear respiratory factor-1 and -2; ERRα: estrogen-related receptor alpha; Tfam: mitochondrial transcription factor A; NEMPs: nuclear-encoded mitochondrial proteins; mtDNA: mitochondrial DNA; Opa1: optic atrophy 1; Mfn1 and 2: mitofusin 1 and 2; Fis1: fission protein 1; Drp1: dynamin-related protein 1; Mff: mitochondrial fission factor; NIX: BCL2 interacting protein 3 like; BNIP3: BCL2 interacting protein 3; FUNDC1: FUN14 domain containing 1; PINK1: PTEN induced putative kinase 1; p62: sequestosome 1; LC 3: microtubule-associated protein light chain 3).
2.1 Mitochondrial Proteostasis
Mitochondrial proteostasis plays an essential role in retaining the dynamic balance between new protein synthesis and impaired protein degradation. Imbalanced proteostasis leads to the accumulation of unnecessary and defective proteins, which further adversely impacts multiple physiological functions, including skeletal muscle activity [36].
An intricate mechanism for the removal of misfolded and dysfunctional proteins is present in muscle mitochondria. The ubiquitin-proteasome system (UPS) and autophagy-lysosome system (ALS) are two predominant pathways to selectively eliminate damaged mitochondrial proteins [37, 38]; UPS mainly eliminate single and unfolded proteins under the tight regulation of AMP-activated protein kinase (AMPK) and the Forkhead Box O (FoxO) transcription factor family [37, 39]. FoxO3 can decrease skeletal muscle mass directly by activating the downstream muscle-specific ubiquitin ligases, the Muscle RING Finger 1 (MuRF1) and the Muscle Atrophy F-box (MAFbx), that are considered as the key regulators of protein turnover contributing to muscle atrophy [40, 41]; FoxO3 can also act indirectly through inactivating the mammalian target of rapamycin complex 1 (mTORC1), a pro-anabolic regulator of protein production [42]. On the other hand, diverse mitochondrial stress conditions associated with the aging process stimulate the mitochondrial unfolded protein response (UPRmt) that serves as a central node to restore proteomic homeostasis [43, 44]. When nascent polypeptides are perturbed during the import and fold processes, mitochondria-specific chaperones, mitochondrial proteases (mitoproteases) and the UPRmt are initiated to repair these “errors”. Chaperones assist with protein import into mitochondria and are responsible for folding of unfolded or misfolded proteins [45, 46], while mitoproteases form the first line of defense against cellular stress responses by degrading irreversibly damaged proteins [47]. Intriguingly, numerous stress responses simultaneously activate the UPRmt and mitophagy [48]. In this scenario, UPRmt serves as the basal folding and degradation mechanism to counteract mitochondrial protein dyshomeostasis, and mitophagy can completely eliminate the unsalvageable mitochondria suggesting that UPRmt-dominated protein degradation and mitophagy are complementary processes [48].
The synthesis of new and properly functioning proteins is an important part of protein turnover in mitochondria. An increasing number of extensively studied regulators of protein synthesis in skeletal muscle are likely to be associated with the pathogenesis of sarcopenia. Akt is the key element of the PI3K/Akt/mTOR signaling pathway and acts as a pivotal mediator in the homeostasis of muscle mass through the mTOR/p70S6K and FoxO3/MuRF-1 and MAFbx pathways to regulate protein synthesis and degradation, respectively [49]. Moreover, AMPK can abrogate the protein synthesis in skeletal muscle by suppressing the mTOR signaling pathway and activating the FoxO-dependent degradation pathway [50]. Furthermore, peroxisome proliferative activated receptor-γ coactivator-1α (PGC-1α) and its variants can stimulate protein synthesis and mitigate muscle protein degradation by UPS through repressing the activities of nuclear factor κB (NF-κB) and FoxO3 activity [51-53]. PGC-1α knockout mice manifested a failure of skeletal muscle function partially due to UPRmt dysregulation [54].
Protein dyshomeostasis may induce muscle mass and strength insufficiency in sarcopenia. UPRmt-related genes were significantly decreased in the muscle of sarcopenia patients, including those encoding mitochondrial heat shock proteins (HSP) and proteases [25]. In addition, mTOR signaling pathway, an important hallmark of mitochondrial protein anabolism, was restrained in sarcopenic participants as well [25]. In the skeletal muscle of senescence-accelerated mouse (SAM) prone 8 (SAMP8), a canonical sarcopenia model, protein synthesis-related markers (Akt, mTOR and p70S6K) were reduced; however, the protein degradation-associated markers (FoxO3, MuRF-1 and MAFbx) were elevated indicating during advancing process of aging, protein turnover has a pro-degradation trend that leads to muscle atrophy which contributes to the occurrence of sarcopenia [55]. A study indicated that the activation of FoxO and proteolytic systems was not involved in sarcopenia, but mTORC1 overactivity was found in aged mice [56, 57]. However, these results were not remarkably observed in human individuals. In addition, rapamycin, an inhibitor of mTORC1, ameliorated sarcopenic symptoms, including less age-related loss of muscle mass and improved muscle functions [57]. Therefore, the functional roles of FoxO and mTORC1 in sarcopenia are controversial and need further investigations. Imbalanced mitochondrial proteostasis significantly reduces the muscle mass and compromises mitochondrial biogenesis resulting in the dysfunction of mitochondria and skeletal muscle. However, the extent of associations of imbalanced mitochondrial proteostasis in senescent muscle with sarcopenia requires additional investigations.
2.2 Mitochondrial Biogenesis
Mitochondrial biogenesis is a multistage process that generates new mitochondria [19]. Mitochondrial proteins are encoded by the nuclear and mitochondrial genomes. The nuclear genome encodes most of proteins involved in mitochondria genesis, whereas mitochondrial DNA (mtDNA) encodes a small number of crucial subunits of the electron transport chain (ETC) complexes. Mitochondrial biogenesis mainly comprise three steps: transcription of nuclear genes, import of nuclear-encoded mitochondrial proteins (NEMPs), and transcription and replication of mtDNAs [58]. PGC-1α is the main factor in regulating mitochondrial biogenesis in the cooperation with downstream nuclear transcription cofactors [38, 59], such as nuclear respiratory factor-1 and -2 (Nrf-1 and Nrf-2) and estrogen-related receptor alpha (ERRα) [59]. Once activated, Nrf-1 and Nrf-2 cofactors bind to the target nuclear genes and promote the expression of NEMPs and mitochondrial transcription factor A (Tfam), which can directly bind to target mtDNA and activate the replication and transcription of the corresponding regions of mtDNA [58, 60].
Mitochondrial biogenesis and its regulators collectively participate in the pathophysiologic changes in sarcopenia. In an interethnic study of human sarcopenia, it was demonstrated that expression profiles of PGC-1α, ERRα and other coactivators were reduced in sarcopenic individuals [25]. PGC-1α, Nrf-1 and Tfam are downregulated in SAMP8 mice during the onset and development of sarcopenia [55]. Moreover, a pronounced decline in muscle mass, muscle performance and frailty was observed in old Nrf-2 knockout (Nrf-2 KO) mice with decreased expression levels of PGC-1α, Nrf-1 and Tfam, suggesting that knockout of Nrf-2 may exacerbate skeletal muscle frailty and sarcopenia through compromising mitochondrial biogenesis [61]. In addition to prototypical cofactors of mitochondrial biogenesis, Zhou and colleagues [62] demonstrated that deficiency of high-temperature requirement protein A2 (HtrA2/Omi) protease is involved in the development of sarcopenia by negatively regulating mitochondrial biogenesis. HtrA2/Omi is a quality control protease that mainly localizes in the intermembrane space of the mitochondria [63]. HtrA2 mnd2 (-/-) mice manifested low muscle mass, poor physical function and sarcopenia phenotypes. Intriguingly, HtrA2 apparently regulates mitochondrial biogenesis in sarcopenia via the differential expression of Nrf-1/2, but not PGC-1α.
PGC-1α is a critical regulator that maintains muscle homeostasis and has attracted considerable attention due to the studies of its potential effects in aging-associated diseases [17]. In mammalian cells, PGC-1α serves as a nuclear-mitochondrial hub in mitochondrial biogenesis [64] by translocating from the cytosol to the nucleus and mitochondria [64, 65]. Muscle atrophy and poor exercise performance combined with reduced PGC-1α levels in skeletal muscle have been detected in the elderly population [66], and the mRNA and protein expression levels of PGC-1α were considerably decreased in the soleus muscle of old rats, indicating that PGC-1α downregulation may participate in the course of aging [67]. The expression levels of PGC-1α, Tfam and Nrf-1 were decreased in the muscle of old SAMP8 mice [55] and elderly individuals [66, 68]. In contrast, PGC-1α overexpression can counteract the loss of muscle mass [69] and muscle atrophy [52] through regulating the homeostatic mechanisms of MQC, although it has not been found in the process of muscle aging. Notably, three prominent metabolic regulators (AMPK, PGC-1α and SIRT1) apparently cooperate to hinder the progression of sarcopenia. AMPK plays a crucial regulatory role in mitochondrial biogenesis [70, 71] and its biological activity in mitochondrial biogenesis declines with aging [72]. Activation of the AMPK/PGC-1α pathway facilitates mitochondrial biogenesis in skeletal muscle [73-75]. Moreover, silent mating type information regulation 2 homolog sirtuin 1 (SIRT1), an NAD+-dependent deacetylase [63], can directly deacetylate and activate PGC-1α in the cytoplasm. SIRT1 colocalizes with PGC-1α in the mitochondria and serves as a downstream regulator of AMPK in response to exercise and fasting [76]. It has been shown that the AMPK/SIRT1/PCG-1α pathway protects the heart from aging and stress [77]. In sarcopenia patients, the NAD+ levels was reduced [25], while AMPK can elevate NAD+ levels and thus activate NAD+-dependent SIRT1 [25, 78]. Moreover, activated SIRT1 deacetylates PCG-1α and induces its expression to enhance mitochondrial synthesis, assembly, growth and antioxidant capability in the heart [77]. It is thus reasonable to suggest that AMPK, PGC-1α and SIRT1 form an integrated and coordinated pathway to attenuate impaired mitochondrial biogenesis associated with aging. Considering the important role of PGC-1α in MQC, the pathway may become a promising target for the prevention and treatment of sarcopenia.
2.3 Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that constantly fuse with surrounding mitochondria and split into daughter mitochondria [79]. Mitochondrial dynamics involves two processes (fusion and fission) [20] that are indispensable for mitochondrial maintenance [79]. Fusion and fission enable the efficient distribution and exchange of mitochondrial contents to meet the local demands of the cells [80]. Mitochondrial fusion is a complementary process that mixes the contents of damaged mitochondria under multiple stress conditions, and mitochondrial fission is a divisive process that results in the production of new mitochondria and contributes to the homeostasis of MQC by removing malfunctioning mitochondria and promoting apoptosis [81]. A family of conserved large GTPases are essential for the regulation of mitochondrial dynamics, such as mitofusins 1 and 2 (Mfn1, Mfn2) and optic atrophy 1 (Opa1) for mitochondrial fusion, and dynamin-related protein 1 (Drp1), mitochondrial fission factor (Mff) and fission protein 1 (Fis1) for mitochondrial fission [82-84]. Mitochondrial dynamics can repair mild damage to mitochondria and is associated with autophagy to thoroughly eliminate impaired mitochondria, especially through mitochondrial fission [81]. Imbalanced mitochondrial dynamics is a common hallmark of senescence [85] that can lead to mitochondrial swelling, decreased cristae production and defective respiratory function [86] thus severely affecting cellular homeostasis.
Several proteins that mediate mitochondrial dynamics are found to be dysregulated in skeletal muscle. For instance, Drp1 knockdown was found to induce muscle atrophy [87, 88]. Currently, the great majority of researches demonstrated that mediators relevant to mitochondrial dynamics are decreased or deficient in aged muscle. Paucity of Mfn2 [89] and deletion of Opa1 and Drp1 [90] have been shown to lead to skeletal muscle atrophy in mice of advanced age. Mitochondrial fusion- and fission-related proteins are differentially expressed in mice and humans with sarcopenia or sarcopenic symptoms. Decreased mRNA expression levels of Mfn1, Mfn2 and Opa1 were detected in the skeletal muscle of elderly mice with sarcopenic phenotypes [91]. In another study, the expression levels of Mfn2, Fis1 and Opa1 were downregulated in sarcopenic muscle, suggesting that mitochondrial dynamics participates in the pathogenesis of sarcopenia [25]. Similarly, mitochondrial fusion genes (Mfn2 and Opa1) were substantially downregulated in old SAMP8 mice and demonstrated a downward trend during the whole progression of sarcopenia [55]. In sarcopenia patients, Opa1 expression was reduced during senescence [92]. Mfn2 protein expression was also markedly decreased in hip-fractured patients with sarcopenia [93]. It is universally appreciated that mitochondrial fission plays an indispensable role for the elimination of impaired mitochondria by mitophagy, whereas some studies have shown that the mitochondrial dynamics is shifted toward fusion in sarcopenia. Intriguingly, fusion-related proteins (Mfn1/Mfn2) were significantly upregulated in older wild type (WT) mice due to the downregulation of fission protein Fis1 [94], which is consistent with the results obtained in some hip-fractured patients of advanced age with sarcopenia [95]. In addition, Huang et al. [61] identified that mitochondrial fusion-related factors (Mfn1, Mfn2 and Opa1) and mitochondrial fission-related factor (Drp1) were decreased in old Nrf-2 KO mice with sarcopenia, suggesting that loss of Nrf-2 can aggravate sarcopenia by disrupting balanced mitochondrial dynamics and that mitochondrial biogenesis and dynamics may be associated with each other.
These aforementioned studies demonstrated the prominent role of abnormal mitochondrial dynamics in muscle aging and sarcopenia (Table 1); however, the mechanisms of these processes remain poorly understood. The alternations in mitochondrial fusion and fission per se are closely associated with mitophagy and mitobiogenesis to form a finely tuned network that prevents and repairs mitochondrial damage. Therefore, investigation of the molecular mechanism of this process is needed, and effective interventions to delay and even reverse the progression of sarcopenia should be developed.
Table 1.
Mitochondrial dynamics-related factors in sarcopenia.
| Key Regulators | Biofunction | Expression in Sarcopenia | Reference | |
|---|---|---|---|---|
| Mitochondrial Fusion | Mfn1, Mfn, and Opa1 | Mixing the contents of impaired mitochondria | Downregulated | [55, 61, 92, 93] |
| Mitochondrial Fission | Drp1, Mff, and Fis1 | Generating daughter mitochondria, removing damaged mitochondria and promoting apoptosis | Downregulated | [61, 94, 95] |
2.4 Mitochondrial Autophagy
Entire mitochondrion can be degraded by autophagosomes with a fused lysosome through an intricate catabolic process termed autophagy. Mitophagy is an exceptional type of macroautophagy that primarily mediates the selective removal of the damaged or superfluous organelles and protein aggregation to maintain mitochondrial homeostasis [96, 97]. Traditionally, the activation of the ULK1-Atg13-FIP200 complex is considered as the canonical initiator of autophagy that culminates in the generation of a double-membrane autophagosome, engulfment of cellular proteins and organelles and fusion with lysosomes [98, 99]. Recently, the PINK1/Parkin pathway has been recognized as one of the most important signaling pathways that regulates ubiquitin-dependent mitophagy [100]. When the membrane potential of damaged mitochondrion is depolarized, the import of PINK1 into IMM is inhibited and the protein accumulates on the outer mitochondrial membrane (OMM) inducing the recruitment of Parkin from the cytosol to the OMM [96, 101-103]. Parkin is an E3 ubiquitin ligase, and its activity is triggered by the PINK-dependent phosphorylation [104, 105]. Then, activated Parkin ubiquitinates outer membrane proteins, generating ubiquitin (Ub) and poly-ubiquitin (poly-Ub) chains. Poly-Ub chains are subsequently phosphorylated by PINK1 and serve as an autophagic signal. Ubiquitin-binding adaptor proteins, including p62, optineurin (OPTN) and nuclear dot protein 52 (NDP52), recognize phosphorylated poly-Ub chains on mitochondrial proteins and recruit damaged mitochondria to the isolation membrane through their interaction with microtubule-associated protein light chain 3 (LC3) [96, 103]. Finally, the damaged mitochondrion is engulfed by an autophagosome that can further fuse with a lysosome to form an autolysosome thus eliminating the entire mitochondrion. In addition to the PINK1/Parkin pathway, important mitophagy receptors (NIX [106], BNIP3 [107, 108] and FUNDC1 [109]) can localize to OMM and directly bind to LC3 to recruit autophagosomes and facilitate mitochondrial elimination as well [103].
Biological aging adversely affects mitophagy homeostasis in various organs and systems [110-112] and impaired mitophagy plays a crucial role in the loss of normal mitochondrial functions [111]. Certain alternations in numerous mitophagy regulators have been reported in senescent animals and humans [94, 113] to lead to mitophagy deficiency and subsequent accumulation of malfunctional mitochondria in skeletal muscle with aging [114, 115]. The accumulation of damaged mitochondria causes the dysfunction of skeletal muscle cells accompanied by muscle wasting and muscle strength reduction [116, 117]. Therefore, insufficient mitophagy potentially plays a causative role in sarcopenia. During the course of sarcopenia progression in SAMP8 mice, elevated Atg13 and LC3-II levels were associated with the accumulation of p62 and lysosome-associated membrane protein 1 (LAMP1), suggesting that poor fusion between autophagosomes and lysosomes and impaired mitophagy in sarcopenic mice [55]. In another premature aging model with sarcopenia, old WT mice had lower expression levels of beclin-1 and p62 than those in young mice, indicating an autophagic dysfunction in aging muscle [94]. The downregulated expression of the autophagy mediator, LC3B, has also been detected in muscle from elderly hip-fractured patients with sarcopenia as well [93]. Recently, many studies have demonstrated that Parkin deletion results in inadequate muscle mass and poor physical performance in old individuals and mice [114, 118], whereas Parkin overexpression has a protective effect on skeletal muscle [118]. Similarly, Leduc-Gaudet el al. [119] have demonstrated that Parkin overexpression can ameliorate the reduction in muscle mass and strength during senescence. The authors demonstrated that after intramuscular injection of adeno-associated virus (AAV) vectors for encoding Parkin induced the upregulation of Parkin expression and attenuated the loss of skeletal muscle mass and strength in old mice versus those in the control group. Moreover, mitochondrial derived vesicles (MDVs) function as important mediators in the vesicle transport between mitochondria and lysosomes, which was regarded as an Drp1-independent mitophagy pathway [120]. And MDV-derived nicotinamide adenine dinucleotide may become a novel biomarker for sarcopenia [121].
Additionally, mitochondrial autophagy, dynamics and biogenesis are closely associated with each other. Mitophagy and mitochondrial dynamics are interrelated because the conversion between fusion and fission is a prerequisite for mitophagy [79]. PINK1 can indirectly activate Drp1 to promote the degradation of defective mitochondria [122]. Parkin induces the proteasomal degradation of mitofusins thus shifting the mitochondrial fusion/fission balance towards fission to suppress mitochondrial fusion and induce segregation of malfunctioning mitochondria from the healthy mitochondrial network [123]. Besides, Parkin is positively associated with mitochondrial biogenesis due to proteasomal degradation of Parkin-interacting substrate (PARIS), a zinc-finger protein that inhibits the synthesis and secretion of PGC1α and blunts the activation of its target cofactors and genes [124]. These findings indicate that mitophagy may contribute to sarcopenia through a complex MQC network. Considering multiple roles of mitophagy in quality control, it is deserved to expect that Parkin or other important regulators of mitophagy may become novel targets for the prevention and attenuation of sarcopenia during aging.
In summary, the maintenance of homeostatic MQC is attributed to the synergistic regulation of mitochondrial proteostasis, biogenesis, dynamics and autophagy in senescence. Disruption of quality control homeostasis results in accunulation of dysfunctional mitochondria that negatively affects skeletal muscle health in older individuals and may even lead to sarcopenia.
3. Potential Interventions for Sarcopenia
3.1 Exercise Therapy
Physical activity, especially resistance exercise training, has been recommended as the first-line intervention to manage sarcopenia according to the evidence-based clinical practice guidelines [35].
3.1.1 Resistance exercise for sarcopenia
Physical activities that set off skeletal muscle contraction against resistance can be defined as resistance exercise. Resistance exercise increases muscle mass, elevates muscle strength and improves the performance of physical exercise in elderly sarcopenia patients [35, 125, 126]. Several studies have shown that resistance exercise promotes mitochondrial protein synthesis and thus plays a role in amelioration of mitochondrial dysfunction [127, 128] to improve muscle mitochondrial biofunctions in human skeletal muscle [129]. Resistance exercise has been shown to optimize mitochondrial functions, particularly by improving mitochondrial biogenesis. Specific molecular mechanisms mainly include the AMPK/SIRT1/PGC-1α or FoxO1 axis as regulators of mitochondrial biogenesis in response to physical exercise of skeletal muscle [76]. In such a scenario, exercise training activates AMPK due to an increase in the AMP/ATP ratio [130] which directly phosphorylates PGC-1α. Subsequently, activated PGC-1 translocates from cytosol into the nucleus and coactivates the transcription factors and nuclear receptors to enhance mitochondrial biogenesis [65, 131]. Besides, long-term moderate exercise positively regulates mitochondrial biogenesis through coordinated interactions of AMPK, SIRT1 and PGC-1α in the skeletal muscle of older mice [132]. In addition to the effects on mitochondrial biogenesis, several studies have demonstrated that exercise training also contributes to mitochondrial homeostasis by preserving mitophagy in skeletal muscle, which may potentially benefit patients with sarcopenia [133, 134]. Interestingly, voluntary resistance wheel exercise (RWE) has been warranted as an effective way to prevent sarcopenia in old C57BL/6J mice [135]. Exercised sarcopenic mice manifested improved mitophagy (increased LC3II/I ratios) and mitochondrial functions (higher mitochondrial density and better oxidative capacity) compared to those in 23-month-old sedentary controls, and the differences were not sex-specific.
3.1.2 Endurance training for sarcopenia
In addition to resistance-based exercise, endurance training is involved in the maintenance of MQC in skeletal muscle as well, including mitochondrial protein synthesis [127, 136, 137], mitophagy [138, 139] and also general functions [137], which likely contribute to the prevention and management of sarcopenia during aging. Endurance exercise promoted the clearance of impaired proteins in skeletal muscle in addition to the prosynthetic effects of physical activity [140]. But the clinical significance of endurance exercise on managing sarcopenia requires further investigation.
Current studies indicate that physical exercises enhance muscle performance and relieve sarcopenic manifestations, in part due to optimized MQC. Currently, consistent recommendation of specific physical exercise for sarcopenia patients is not available; however, exercise plans should correspond to patient abilities and rehabilitation goals to achieve the best therapeutic effect.
3.2 Nutritional and Pharmacological Interventions
Certain limitations for physical exercises in patients with sarcopenia include increased risk of fractures and falls in older adults and poor adherence to long-term training programs [141]. Therefore, valid nutritional and pharmacological interventions have to be identified to ameliorate sarcopenia in a more easily available fashion. Currently, clinically approved drugs for specific treatment of sarcopenia are unavailable; however, a few medicines are undergoing phase I and II clinical trials [4]. The supplementary nutrients mainly include protein, vitamin D, antioxidants, myostatin inhibitors and anabolic hormones [4, 35, 142]. Nutritional manipulations demonstrated certain efficacy in the improvement of the symptoms of sarcopenia; however, none of these manipulations are recommended as conventional methods for therapy of sarcopenia, except protein intake, which is conditionally recommended [35].
Recently, several bioactive compounds or drugs have been identified as latent and potent options to prevent and delay the progress of sarcopenia through ameliorating mitochondrial dysfunction. Melatonin (N-acetyl-5-methoxytryptamin, aMT) is a pineal hormone that is ubiquitously present in most organs and tissues, including skeletal muscle [143]. Melatonin coordinates physiological adaptations to the light/dark cycle and seasonal alterations and has a number of other biofunctions, such as recovery of aging-related mitochondrial dysfunction of muscle in senescence-accelerated mice [143-145]. Notably, a protective effect of melatonin in sarcopenia is mediated by the modulation of mitochondrial changes in aging. Sayed et al. [146, 147] demonstrated that oral melatonin treatment of early-aged (12 months) mice with sarcopenia resulted in relatively normal muscle structures, increased number of muscle fibers, decreased frailty index (FI) and improved physical performance. Additionally, melatonin supplementation promoted lactate production and diminished tubular aggregate formation and nuclear apoptosis. These results indicated that melatonin plays the prophylactic role in sarcopenia during aging, which should probably be used in clinical therapy in the near future. In addition to endogenous hormones, 5,7-dimethoxy?avone (DMF), a major ?avone detected in Kaempferia parvi?ora, was demonstrated to serve as a natural ingredient to delay sarcopenia [148]. In this study, oral administration of DMF considerably increased muscle mass, strength, and physical ability and three basal evaluation indexes of sarcopenia compared to those in the aged controls. At the molecular level, DMF regulated protein synthesis by stimulating the PI3K-Akt axis and mTOR pathway and restraining the FoxO pathways and enhanced mitochondrial biogenesis by upregulating the mRNA expression of PGC-1α, Nrf-1, and Tfam. Another natural substance, oligonol, has been demonstrated to increase muscle mass and strength by optimizing the quality control of mitochondria in SAMP8 mice [149]. Oligonol is extracted from ?avanol-rich lychee and can regulate metabolism [150, 151]. After 8 weeks of oligonol administration, SAMP8 mice in the experimental group had higher skeletal muscle mass and strength versus those in the control animals. At the molecular level, oligonol positively regulated mitochondrial proteostasis (stimulation of Akt/mTOR/p70S6K and inhibition of FoxO3a/MuRF1 and MAFbx signaling), mitochondrial biogenesis (elevated PGC-1α and Tfam), mitochondrial dynamics (increased Mfn2 and Opa1) and mitophagy (upregulation of PINK1, a reduction in Atg13, LC3-II, and p62, and a decrease in autophagosomes and lysosomes), indicating that oligonol can play a role in the correction of mitochondrial dysfunctions thus preventing sarcopenia. Notwithstanding, the underlying mechanisms derived from aging-accelerated mice may differ from normally senescent mice. Additionally, 5-aminolevulinic acid (ALA), a basic compound in porphyrin synthesis, was demonstrated to reduce the loss of muscle mass and improve physical performance in old mice through facilitating protein synthesis in mitochondria [152]. The decreased level of NAD+ was detected in sarcopenia, cardiovascular aging and neurodegenerative diseases [25, 78, 153]. Nicotinamide, an NAD+ precursor, has been shown to promote mitophagy and suppress cardiac aging through activating Sirtuins [154, 155]. Therefore, targeting NAD+ may become a novel strategy for alleviating sarcopenia. Moreover, urolithin A, as one of the end-products of ellagitannins (ETs) and ellagic acid (EA), was also found to induce mitophagy and improve muscle functions in aged mouse, which probably becomes a promising nutritional supplementation for sarcopenia [156]. However, all of these bioactive compounds or drugs are carried out in animal models, and none of them are tested in clinical trials.
Overall, the combination of physical exercise and protein supplementation is the most effective countermeasure for sarcopenia. However, nutritional and pharmacological interventions are more applicable for the majority of sarcopenia patients. Several potential therapeutic agents have been demonstrated to mitigate mitochondrial malfunction for the prevention and treatment of sarcopenia. Furthermore, gene therapy probably become a novel strategy for alleviating sarcopenia. METTL21c acts as a bone-muscle pleiotropic gene for sarcopenia. Although METTL21c’s biofunction is achieved by NF-κB signaling pathway, its potential role in quality control mechanisms of mitochondria deserves more investigation [157]. Additionally, some mitokines potentially function as biomarkers for the diagnosis of sarcopenia, such as growth differentiation factor 15 (GDF15) and fibroblast growth factor 21 (FGF21) [158, 159]. Therefore, an increasing number of studies are needed to confirm the effectiveness of the present findings and to explore novel countermeasures and diagnostic methods for sarcopenia, which may greatly improve the quality of life of elderly patients.
4. Conclusion
Sarcopenia is a prevalent and degenerative skeletal muscle disease that leads to poor quality of life in patients, particularly elderly individuals. Numerous pathophysiological alterations contribute to a progressive decline in muscle strength, muscle mass and physical performance, which are the predominant hallmarks of sarcopenia. Dyshomeostasis of mitochondrial quality control is one of the primary factors for the initiation and progression of sarcopenia. MQC requires coordination of mitochondrial proteostasis, biogenesis, dynamics and autophagy. Alterations in mitochondrial quality control exacerbate muscle atrophy and reduce muscle strength during aging concomitant with restricted locomotive function. The important regulators and signaling pathways in MQC may be involved in the etiology of sarcopenia and are comprehensively summarized in this review. Physical exercise, the most recommended therapeutic intervention for sarcopenia, improves skeletal muscle quantity and quality partially by facilitating the restoration of malfunctional mitochondria. Additionally, nutritional and pharmaceutical treatments ameliorate sarcopenia to a certain degree, although compelling evidence and mechanisms remain to be identified. Overall, dysfunctional MQC plays a causative role in sarcopenia and revealing latent mechanisms may shed light on efficient preventive and intervention strategies for patients with sarcopenia.
Acknowledgements
This work was supported by National Key R&D Program of China (2019YFA0111900), National Natural Science Foundation of China (No. 81874030, 82072506), Provincial Natural Science Foundation of Hunan (No. 2020JJ3060), Provincial Clinical Medical Technology Innovation Project of Hunan (No. 2020SK53709), the Administration of Traditional Chinese Medicine of Hunan Province (No.2021075), Innovation-Driven Project of Central South university (No. 2020CX045), Wu Jieping Medical Foundation (No. 320.6750.2020-03-14), CMA?Young and Middle-aged Doctors Outstanding Development Program--Osteoporosis Specialized Scientific Research Fund Project (No. G-X-2019-1107-12), the Clinical and Rehabilitation Research Foundation of Xiangya Hospital and Weiming of Peking University (No. xywm2015II04), and the Key program of Health Commission of Hunan Province (No. 20201902).
Footnotes
Conflicts of interest
The authors disclose no potential conflicts of interest.
References
- [1].Denison HJ, Cooper C, Sayer AA, Robinson SM (2015). Prevention and optimal management of sarcopenia: a review of combined exercise and nutrition interventions to improve muscle outcomes in older people. Clin Interv Aging, 10:859-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rosenberg IH (1997). Sarcopenia: origins and clinical relevance. J Nutr, 127:990s-991s. [DOI] [PubMed] [Google Scholar]
- [3].Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, et al. (2019). Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing, 48:16-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cruz-Jentoft AJ, Sayer AA (2019). Sarcopenia. Lancet, 393:2636-2646. [DOI] [PubMed] [Google Scholar]
- [5].Borsch A, Ham DJ, Mittal N, Tintignac LA, Migliavacca E, Feige JN, et al. (2021). Molecular and phenotypic analysis of rodent models reveals conserved and species-specific modulators of human sarcopenia. Commun Biol, 4:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Börsch A, Ham DJ, Mittal N, Tintignac LA, Migliavacca E, Feige JN, et al. (2021). Molecular and phenotypic analysis of rodent models reveals conserved and species-specific modulators of human sarcopenia. Commun Biol, 4:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bischoff-Ferrari HA, Orav JE, Kanis JA, Rizzoli R, Schlögl M, Staehelin HB, et al. (2015). Comparative performance of current definitions of sarcopenia against the prospective incidence of falls among community-dwelling seniors age 65 and older. Osteoporos Int, 26:2793-2802. [DOI] [PubMed] [Google Scholar]
- [8].Yu R, Leung J, Woo J (2014). Incremental predictive value of sarcopenia for incident fracture in an elderly Chinese cohort: results from the Osteoporotic Fractures in Men (MrOs) Study. J Am Med Dir Assoc, 15:551-558. [DOI] [PubMed] [Google Scholar]
- [9].Morley JE, Abbatecola AM, Argiles JM, Baracos V, Bauer J, Bhasin S, et al. (2011). Sarcopenia with limited mobility: an international consensus. J Am Med Dir Assoc, 12:403-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].De Buyser SL, Petrovic M, Taes YE, Toye KR, Kaufman JM, Lapauw B, et al. (2016). Validation of the FNIH sarcopenia criteria and SOF frailty index as predictors of long-term mortality in ambulatory older men. Age Ageing, 45:602-608. [DOI] [PubMed] [Google Scholar]
- [11].Huang JH, Hood DA (2009). Age-associated mitochondrial dysfunction in skeletal muscle: Contributing factors and suggestions for long-term interventions. IUBMB Life, 61:201-214. [DOI] [PubMed] [Google Scholar]
- [12].Cleasby ME, Jamieson PM, Atherton PJ (2016). Insulin resistance and sarcopenia: mechanistic links between common co-morbidities. J Endocrinol, 229:R67-81. [DOI] [PubMed] [Google Scholar]
- [13].Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. (2017). Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol, 8:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Meng SJ, Yu LJ (2010). Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci, 11:1509-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pagano AF, Brioche T, Arc-Chagnaud C, Demangel R, Chopard A, Py G (2018). Short-term disuse promotes fatty acid infiltration into skeletal muscle. J Cachexia Sarcopenia Muscle, 9:335-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, et al. (2013). Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol, 33:194-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Picca A, Calvani R, Bossola M, Allocca E, Menghi A, Pesce V, et al. (2018). Update on mitochondria and muscle aging: all wrong roads lead to sarcopenia. Biol Chem, 399:421-436. [DOI] [PubMed] [Google Scholar]
- [18].Green DR, Van Houten B (2011). SnapShot: Mitochondrial quality control. Cell, 147:950, 950.e951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kelly DP, Scarpulla RC (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev, 18:357-368. [DOI] [PubMed] [Google Scholar]
- [20].Archer SL (2013). Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med, 369:2236-2251. [DOI] [PubMed] [Google Scholar]
- [21].Nunnari J, Suomalainen A (2012). Mitochondria: in sickness and in health. Cell, 148:1145-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Calvani R, Joseph AM, Adhihetty PJ, Miccheli A, Bossola M, Leeuwenburgh C, et al. (2013). Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem, 394:393-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alway SE, Mohamed JS, Myers MJ (2017). Mitochondria Initiate and Regulate Sarcopenia. Exerc Sport Sci Rev, 45:58-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zane AC, Reiter DA, Shardell M, Cameron D, Simonsick EM, Fishbein KW, et al. (2017). Muscle strength mediates the relationship between mitochondrial energetics and walking performance. Aging Cell, 16:461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Migliavacca E, Tay SKH, Patel HP, Sonntag T, Civiletto G, McFarlane C, et al. (2019). Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun, 10:5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim H, Suzuki T, Kim M, Kojima N, Yoshida Y, Hirano H, et al. (2015). Incidence and predictors of sarcopenia onset in community-dwelling elderly Japanese women: 4-year follow-up study. J Am Med Dir Assoc, 16:85.e81-88. [DOI] [PubMed] [Google Scholar]
- [27].Gielen E, O'Neill TW, Pye SR, Adams JE, Wu FC, Laurent MR, et al. (2015). Endocrine determinants of incident sarcopenia in middle-aged and elderly European men. J Cachexia Sarcopenia Muscle, 6:242-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu R, Wong M, Leung J, Lee J, Auyeung TW, Woo J (2014). Incidence, reversibility, risk factors and the protective effect of high body mass index against sarcopenia in community-dwelling older Chinese adults. Geriatr Gerontol Int, 14 Suppl 1:15-28. [DOI] [PubMed] [Google Scholar]
- [29].Dodds RM, Granic A, Davies K, Kirkwood TB, Jagger C, Sayer AA (2017). Prevalence and incidence of sarcopenia in the very old: findings from the Newcastle 85+ Study. J Cachexia Sarcopenia Muscle, 8:229-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cruz-Jentoft AJ, Landi F, Schneider SM, Zúñiga C, Arai H, Boirie Y, et al. (2014). Prevalence of and interventions for sarcopenia in ageing adults: a systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing, 43:748-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sánchez-Rodríguez D, Marco E, Ronquillo-Moreno N, Miralles R, Vázquez-Ibar O, Escalada F, et al. (2017). Prevalence of malnutrition and sarcopenia in a post-acute care geriatric unit: Applying the new ESPEN definition and EWGSOP criteria. Clin Nutr, 36:1339-1344. [DOI] [PubMed] [Google Scholar]
- [32].von Haehling S, Morley JE, Anker SD (2010). An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J Cachexia Sarcopenia Muscle, 1:129-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anker SD, Morley JE, von Haehling S (2016). Welcome to the ICD-10 code for sarcopenia. J Cachexia Sarcopenia Muscle, 7:512-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vellas B, Fielding RA, Bens C, Bernabei R, Cawthon PM, Cederholm T, et al. (2018). Implications of ICD-10 for Sarcopenia Clinical Practice and Clinical Trials: Report by the International Conference on Frailty and Sarcopenia Research Task Force. J Frailty Aging, 7:2-9. [DOI] [PubMed] [Google Scholar]
- [35].Dent E, Morley JE, Cruz-Jentoft AJ, Arai H, Kritchevsky SB, Guralnik J, et al. (2018). International Clinical Practice Guidelines for Sarcopenia (ICFSR): Screening, Diagnosis and Management. J Nutr Health Aging, 22:1148-1161. [DOI] [PubMed] [Google Scholar]
- [36].Ayyadevara S, Balasubramaniam M, Suri P, Mackintosh SG, Tackett AJ, Sullivan DH, et al. (2016). Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging (Albany NY), 8:3486-3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. Embo J, 29:1774-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson ML, Robinson MM, Nair KS (2013). Skeletal muscle aging and the mitochondrion. Trends Endocrinol Metab, 24:247-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pohl C, Dikic I (2019). Cellular quality control by the ubiquitin-proteasome system and autophagy. Science, 366:818-822. [DOI] [PubMed] [Google Scholar]
- [40].Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell, 117:399-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Biedasek K, Andres J, Mai K, Adams S, Spuler S, Fielitz J, et al. (2011). Skeletal muscle 11beta-HSD1 controls glucocorticoid-induced proteolysis and expression of E3 ubiquitin ligases atrogin-1 and MuRF-1. PLoS One, 6:e16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rom O, Reznick AZ (2016). The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic Biol Med, 98:218-230. [DOI] [PubMed] [Google Scholar]
- [43].Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ (2002). A mitochondrial specific stress response in mammalian cells. Embo J, 21:4411-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Seli E, Wang T, Horvath TL (2019). Mitochondrial unfolded protein response: a stress response with implications for fertility and reproductive aging. Fertil Steril, 111:197-204. [DOI] [PubMed] [Google Scholar]
- [45].Moehle EA, Shen K, Dillin A (2019). Mitochondrial proteostasis in the context of cellular and organismal health and aging. J Biol Chem, 294:5396-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, et al. (1994). Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature, 371:768-774. [DOI] [PubMed] [Google Scholar]
- [47].Quirós PM, Langer T, López-Otín C (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol, 16:345-359. [DOI] [PubMed] [Google Scholar]
- [48].Pellegrino MW, Haynes CM (2015). Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol, 13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Drummond MJ, Dreyer HC, Fry CS, Glynn EL, Rasmussen BB (2009). Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J Appl Physiol (1985), 106:1374-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Schiaffino S, Mammucari C (2011). Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle, 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, et al. (2012). A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell, 151:1319-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A, 103:16260-16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martínez-Redondo V, Jannig PR, Correia JC, Ferreira DM, Cervenka I, Lindvall JM, et al. (2016). Peroxisome Proliferator-activated Receptor γ Coactivator-1 α Isoforms Selectively Regulate Multiple Splicing Events on Target Genes. J Biol Chem, 291:15169-15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, et al. (2011). The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab, 13:160-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu HW, Chang YC, Chan YC, Hu SH, Liu MY, Chang SJ (2020). Dysregulations of mitochondrial quality control and autophagic flux at an early age lead to progression of sarcopenia in SAMP8 mice. Biogerontology, 21:367-380. [DOI] [PubMed] [Google Scholar]
- [56].Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, et al. (2013). Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology, 14:303-323. [DOI] [PubMed] [Google Scholar]
- [57].Ham DJ, Börsch A, Lin S, Thürkauf M, Weihrauch M, Reinhard JR, et al. (2020). The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat Commun, 11:4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ji LL, Kang C (2015). Role of PGC-1α in sarcopenia: etiology and potential intervention - a mini-review. Gerontology, 61:139-148. [DOI] [PubMed] [Google Scholar]
- [59].Picca A, Lezza AM (2015). Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion, 25:67-75. [DOI] [PubMed] [Google Scholar]
- [60].Rebelo AP, Dillon LM, Moraes CT (2011). Mitochondrial DNA transcription regulation and nucleoid organization. J Inherit Metab Dis, 34:941-951. [DOI] [PubMed] [Google Scholar]
- [61].Huang DD, Fan SD, Chen XY, Yan XL, Zhang XZ, Ma BW, et al. (2019). Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp Gerontol, 119:61-73. [DOI] [PubMed] [Google Scholar]
- [62].Zhou H, Yuan D, Gao W, Tian J, Sun H, Yu S, et al. (2020). Loss of high-temperature requirement protein A2 protease activity induces mitonuclear imbalance via differential regulation of mitochondrial biogenesis in sarcopenia. IUBMB Life, 72:1659-1679. [DOI] [PubMed] [Google Scholar]
- [63].Chang HC, Guarente L (2014). SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab, 25:138-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, et al. (2011). Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A, 108:4135-4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO (2007). Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J Biol Chem, 282:194-199. [DOI] [PubMed] [Google Scholar]
- [66].Joseph AM, Adhihetty PJ, Buford TW, Wohlgemuth SE, Lees HA, Nguyen LM, et al. (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell, 11:801-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kang C, Chung E, Diffee G, Ji LL (2013). Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1α. Exp Gerontol, 48:1343-1350. [DOI] [PubMed] [Google Scholar]
- [68].Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB (2005). Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem, 280:32485-32492. [DOI] [PubMed] [Google Scholar]
- [69].Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA (2015). The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. J Physiol, 593:1981-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, et al. (2001). Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab, 281:E1340-1346. [DOI] [PubMed] [Google Scholar]
- [71].Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, et al. (2002). AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A, 99:15983-15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, et al. (2007). Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab, 5:151-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen J, Wong HS, Leong PK, Leung HY, Chan WM, Ko KM (2017). Ursolic acid induces mitochondrial biogenesis through the activation of AMPK and PGC-1 in C2C12 myotubes: a possible mechanism underlying its beneficial effect on exercise endurance. Food Funct, 8:2425-2436. [DOI] [PubMed] [Google Scholar]
- [74].Kou G, Li Z, Wu C, Liu Y, Hu Y, Guo L, et al. (2018). Citrus Tangeretin Improves Skeletal Muscle Mitochondrial Biogenesis via Activating the AMPK-PGC1-α Pathway In Vitro and In Vivo: A Possible Mechanism for Its Beneficial Effect on Physical Performance. J Agric Food Chem, 66:11917-11925. [DOI] [PubMed] [Google Scholar]
- [75].Jiao W, Hu F, Li J, Song J, Liang J, Li L, et al. (2020). Qiangji Jianli Decoction promotes mitochondrial biogenesis in skeletal muscle of myasthenia gravis rats via AMPK/PGC-1α signaling pathway. Biomed Pharmacother, 129:110482. [DOI] [PubMed] [Google Scholar]
- [76].Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, et al. (2010). Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab, 11:213-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hsu CP, Odewale I, Alcendor RR, Sadoshima J (2008). Sirt1 protects the heart from aging and stress. Biol Chem, 389:221-231. [DOI] [PubMed] [Google Scholar]
- [78].Ren J, Zhang Y (2018). Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol Sci, 39:1064-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Westermann B (2010). Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol, 11:872-884. [DOI] [PubMed] [Google Scholar]
- [80].Hoppins S, Lackner L, Nunnari J (2007). The machines that divide and fuse mitochondria. Annu Rev Biochem, 76:751-780. [DOI] [PubMed] [Google Scholar]
- [81].Youle RJ, van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science, 337:1062-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Tilokani L, Nagashima S, Paupe V, Prudent J (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem, 62:341-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. (2019). CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell, 177:1536-1552.e1523. [DOI] [PubMed] [Google Scholar]
- [84].Yu R, Jin SB, Lendahl U, Nistér M, Zhao J (2019). Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. Embo J, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013). The hallmarks of aging. Cell, 153:1194-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Jang JY, Blum A, Liu J, Finkel T (2018). The role of mitochondria in aging. J Clin Invest, 128:3662-3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun, 10:2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Dulac M, Leduc-Gaudet JP, Reynaud O, Ayoub MB, Guérin A, Finkelchtein M, et al. (2020). Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. J Physiol, 598:3691-3710. [DOI] [PubMed] [Google Scholar]
- [89].Wang L, Gao J, Liu J, Siedlak SL, Torres S, Fujioka H, et al. (2018). Mitofusin 2 Regulates Axonal Transport of Calpastatin to Prevent Neuromuscular Synaptic Elimination in Skeletal Muscles. Cell Metab, 28:400-414.e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Romanello V, Scalabrin M, Albiero M, Blaauw B, Scorrano L, Sandri M (2019). Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Del Campo A, Contreras-Hernández I, Castro-Sepúlveda M, Campos CA, Figueroa R, Tevy MF, et al. (2018). Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging (Albany NY), 10:34-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. (2017). Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab, 25:1374-1389.e1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Marzetti E, Calvani R, Lorenzi M, Tanganelli F, Picca A, Bossola M, et al. (2016). Association between myocyte quality control signaling and sarcopenia in old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp Gerontol, 80:1-5. [DOI] [PubMed] [Google Scholar]
- [94].Joseph AM, Adhihetty PJ, Wawrzyniak NR, Wohlgemuth SE, Picca A, Kujoth GC, et al. (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One, 8:e69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Picca A, Calvani R, Lorenzi M, Menghi A, Galli M, Vitiello R, et al. (2017). Mitochondrial dynamics signaling is shifted toward fusion in muscles of very old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp Gerontol, 96:63-67. [DOI] [PubMed] [Google Scholar]
- [96].Ashrafi G, Schwarz TL (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ, 20:31-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mizushima N, Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell, 147:728-741. [DOI] [PubMed] [Google Scholar]
- [98].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell, 20:1981-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Levy JMM, Towers CG, Thorburn A (2017). Targeting autophagy in cancer. Nat Rev Cancer, 17:528-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Pickles S, Vigié P, Youle RJ (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol, 28:R170-r185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sekine S, Youle RJ (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol, 16:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Harper JW, Ordureau A, Heo JM (2018). Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol, 19:93-108. [DOI] [PubMed] [Google Scholar]
- [103].Palikaras K, Lionaki E, Tavernarakis N (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol, 20:1013-1022. [DOI] [PubMed] [Google Scholar]
- [104].Aguirre JD, Dunkerley KM, Mercier P, Shaw GS (2017). Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc Natl Acad Sci U S A, 114:298-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Youle RJ, Narendra DP (2011). Mechanisms of mitophagy. Nat Rev Mol Cell Biol, 12:9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature, 454:232-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Quinsay MN, Thomas RL, Lee Y, Gustafsson AB (2010). Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy, 6:855-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wrighton PJ, Shwartz A, Heo JM, Quenzer ED, LaBella KA, Harper JW, et al. (2021). Quantitative intravital imaging in zebrafish reveals in vivo dynamics of physiological-stress-induced mitophagy. J Cell Sci, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol, 14:177-185. [DOI] [PubMed] [Google Scholar]
- [110].Ajoolabady A, Aslkhodapasandhokmabad H, Aghanejad A, Zhang Y, Ren J (2020). Mitophagy Receptors and Mediators: Therapeutic Targets in the Management of Cardiovascular Ageing. Ageing Res Rev, 62:101129. [DOI] [PubMed] [Google Scholar]
- [111].Gustafsson Å B, Dorn GW 2nd (2019). Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol Rev, 99:853-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Tian H, Chen P, Ren J (2019). Physical exercise, autophagy and cardiometabolic stress in aging. Aging (Albany NY), 11:5287-5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Russ DW, Krause J, Wills A, Arreguin R (2012). "SR stress" in mixed hindlimb muscles of aging male rats. Biogerontology, 13:547-555. [DOI] [PubMed] [Google Scholar]
- [114].Gouspillou G, Sgarioto N, Kapchinsky S, Purves-Smith F, Norris B, Pion CH, et al. (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. Faseb J, 28:1621-1633. [DOI] [PubMed] [Google Scholar]
- [115].O'Leary MF, Vainshtein A, Iqbal S, Ostojic O, Hood DA (2013). Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol, 304:C422-430. [DOI] [PubMed] [Google Scholar]
- [116].Masiero E, Sandri M (2010). Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy, 6:307-309. [DOI] [PubMed] [Google Scholar]
- [117].Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, et al. (2013). Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol, 45:2288-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Gouspillou G, Godin R, Piquereau J, Picard M, Mofarrahi M, Mathew J, et al. (2018). Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J Physiol, 596:2565-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Leduc-Gaudet JP, Reynaud O, Hussain SN, Gouspillou G (2019). Parkin overexpression protects from ageing-related loss of muscle mass and strength. J Physiol, 597:1975-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol, 22:135-141. [DOI] [PubMed] [Google Scholar]
- [121].Marzetti E, Guerra F, Calvani R, Marini F, Biancolillo A, Gervasoni J, et al. (2020). Circulating Mitochondrial-Derived Vesicles, Inflammatory Biomarkers and Amino Acids in Older Adults With Physical Frailty and Sarcopenia: A Preliminary BIOSPHERE Multi-Marker Study Using Sequential and Orthogonalized Covariance Selection - Linear Discriminant Analysis. Front Cell Dev Biol, 8:564417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Pryde KR, Smith HL, Chau KY, Schapira AH (2016). PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol, 213:163-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol, 191:1367-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, et al. (2011). PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell, 144:689-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Schoenfeld BJ, Ogborn D, Krieger JW (2016). Effects of Resistance Training Frequency on Measures of Muscle Hypertrophy: A Systematic Review and Meta-Analysis. Sports Med, 46:1689-1697. [DOI] [PubMed] [Google Scholar]
- [126].Kim HK, Suzuki T, Saito K, Yoshida H, Kobayashi H, Kato H, et al. (2012). Effects of exercise and amino acid supplementation on body composition and physical function in community-dwelling elderly Japanese sarcopenic women: a randomized controlled trial. J Am Geriatr Soc, 60:16-23. [DOI] [PubMed] [Google Scholar]
- [127].Wilkinson SB, Phillips SM, Atherton PJ, Patel R, Yarasheski KE, Tarnopolsky MA, et al. (2008). Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol, 586:3701-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Robinson MM, Dasari S, Konopka AR, Johnson ML, Manjunatha S, Esponda RR, et al. (2017). Enhanced Protein Translation Underlies Improved Metabolic and Physical Adaptations to Different Exercise Training Modes in Young and Old Humans. Cell Metab, 25:581-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Porter C, Reidy PT, Bhattarai N, Sidossis LS, Rasmussen BB (2015). Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med Sci Sports Exerc, 47:1922-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Winder WW, Hardie DG (1996). Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol, 270:E299-304. [DOI] [PubMed] [Google Scholar]
- [131].Jäger S, Handschin C, St-Pierre J, Spiegelman BM (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A, 104:12017-12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Bayod S, Del Valle J, Lalanza JF, Sanchez-Roige S, de Luxán-Delgado B, Coto-Montes A, et al. (2012). Long-term physical exercise induces changes in sirtuin 1 pathway and oxidative parameters in adult rat tissues. Exp Gerontol, 47:925-935. [DOI] [PubMed] [Google Scholar]
- [133].Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, et al. (2013). Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. Faseb J, 27:4184-4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature, 481:511-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].White Z, Terrill J, White RB, McMahon C, Sheard P, Grounds MD, et al. (2016). Voluntary resistance wheel exercise from mid-life prevents sarcopenia and increases markers of mitochondrial function and autophagy in muscles of old male and female C57BL/6J mice. Skelet Muscle, 6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Scalzo RL, Peltonen GL, Binns SE, Shankaran M, Giordano GR, Hartley DA, et al. (2014). Greater muscle protein synthesis and mitochondrial biogenesis in males compared with females during sprint interval training. Faseb J, 28:2705-2714. [DOI] [PubMed] [Google Scholar]
- [137].Russell AP, Foletta VC, Snow RJ, Wadley GD (2014). Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochim Biophys Acta, 1840:1276-1284. [DOI] [PubMed] [Google Scholar]
- [138].Drake JC, Wilson RJ, Yan Z (2016). Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. Faseb J, 30:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wu NN, Tian H, Chen P, Wang D, Ren J, Zhang Y (2019). Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Pagano AF, Py G, Bernardi H, Candau RB, Sanchez AM (2014). Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med Sci Sports Exerc, 46:1314-1325. [DOI] [PubMed] [Google Scholar]
- [141].Melouane A, Yoshioka M, St-Amand J (2020). Extracellular matrix/mitochondria pathway: A novel potential target for sarcopenia. Mitochondrion, 50:63-70. [DOI] [PubMed] [Google Scholar]
- [142].Coen PM, Musci RV, Hinkley JM, Miller BF (2018). Mitochondria as a Target for Mitigating Sarcopenia. Front Physiol, 9:1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cipolla-Neto J, Amaral FGD (2018). Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr Rev, 39:990-1028. [DOI] [PubMed] [Google Scholar]
- [144].Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (2016). Melatonin as an antioxidant: under promises but over delivers. J Pineal Res, 61:253-278. [DOI] [PubMed] [Google Scholar]
- [145].Rodríguez MI, Escames G, López LC, López A, García JA, Ortiz F, et al. (2008). Improved mitochondrial function and increased life span after chronic melatonin treatment in senescent prone mice. Exp Gerontol, 43:749-756. [DOI] [PubMed] [Google Scholar]
- [146].Sayed RKA, Fernández-Ortiz M, Diaz-Casado ME, Aranda-Martínez P, Fernández-Martínez J, Guerra-Librero A, et al. (2019). Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J Gerontol A Biol Sci Med Sci, 74:1699-1708. [DOI] [PubMed] [Google Scholar]
- [147].Sayed RKA, Fernández-Ortiz M, Diaz-Casado ME, Rusanova I, Rahim I, Escames G, et al. (2018). The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J Gerontol A Biol Sci Med Sci, 73:1330-1338. [DOI] [PubMed] [Google Scholar]
- [148].Kim C, Hwang JK (2020). The 5,7-Dimethoxyflavone Suppresses Sarcopenia by Regulating Protein Turnover and Mitochondria Biogenesis-Related Pathways. Nutrients, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chang YC, Chen YT, Liu HW, Chan YC, Liu MY, Hu SH, et al. (2019). Oligonol Alleviates Sarcopenia by Regulation of Signaling Pathways Involved in Protein Turnover and Mitochondrial Quality. Mol Nutr Food Res, 63:e1801102. [DOI] [PubMed] [Google Scholar]
- [150].Liu HW, Wei CC, Chang SJ (2016). Low-molecular-weight polyphenols protect kidney damage through suppressing NF-κB and modulating mitochondrial biogenesis in diabetic db/db mice. Food Funct, 7:1941-1949. [DOI] [PubMed] [Google Scholar]
- [151].Liu HW, Wei CC, Chen YJ, Chen YA, Chang SJ (2016). Flavanol-rich lychee fruit extract alleviates diet-induced insulin resistance via suppressing mTOR/SREBP-1 mediated lipogenesis in liver and restoring insulin signaling in skeletal muscle. Mol Nutr Food Res, 60:2288-2296. [DOI] [PubMed] [Google Scholar]
- [152].Fujii C, Miyashita K, Mitsuishi M, Sato M, Fujii K, Inoue H, et al. (2017). Treatment of sarcopenia and glucose intolerance through mitochondrial activation by 5-aminolevulinic acid. Sci Rep, 7:4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, et al. (2017). NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol Med, 23:899-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Mitchell SJ, Bernier M, Aon MA, Cortassa S, Kim EY, Fang EF, et al. (2018). Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab, 27:667-676.e664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J (2016). Aging and Autophagy in the Heart. Circ Res, 118:1563-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med, 22:879-888. [DOI] [PubMed] [Google Scholar]
- [157].Huang J, Hsu YH, Mo C, Abreu E, Kiel DP, Bonewald LF, et al. (2014). METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the NF-κB signaling pathway. J Bone Miner Res, 29:1531-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Ji X, Zhao L, Ji K, Zhao Y, Li W, Zhang R, et al. (2017). Growth Differentiation Factor 15 Is a Novel Diagnostic Biomarker of Mitochondrial Diseases. Mol Neurobiol, 54:8110-8116. [DOI] [PubMed] [Google Scholar]
- [159].Kim H, Kim KM, Kang MJ, Lim S (2020). Growth differentiation factor-15 as a biomarker for sarcopenia in aging humans and mice. Exp Gerontol, 142:111115. [DOI] [PubMed] [Google Scholar]