

Abstract
Kisspeptin (Kiss1) neurons are essential for reproduction, but their role in the control of energy balance and other homeostatic functions remains unclear. High-frequency firing of hypothalamic arcuate Kiss1 (Kiss1ARH) neurons releases kisspeptin into the median eminence, and neurokinin B (NKB) and dynorphin onto neighboring Kiss1ARH neurons to generate a slow EPSP mediated by TRPC5 channels that entrains intermittent, synchronous firing of Kiss1ARH neurons. High-frequency optogenetic stimulation of Kiss1ARH neurons also releases glutamate to excite the anorexigenic proopiomelanocortin (POMC) neurons and inhibit the orexigenic neuropeptide Y/agouti-related peptide (AgRP) neurons via metabotropic glutamate receptors. At the molecular level, the endoplasmic reticulum (ER) calcium-sensing protein stromal interaction molecule 1 (STIM1) is critically involved in the regulation of neuronal Ca2+ signaling and neuronal excitability through its interaction with plasma membrane (PM) calcium (e.g., TRPC) channels. Therefore, we hypothesized that deletion of Stim1 in Kiss1ARH neurons would increase neuronal excitability and their synchronous firing, which ultimately would affect energy homeostasis. Using optogenetics in combination with whole-cell recording and GCaMP6 imaging in slices, we discovered that deletion of Stim1 in Kiss1 neurons significantly increased the amplitude and duration of the slow EPSP and augmented synchronous [Ca2+]i oscillations in Kiss1ARH neurons. Deletion of Stim1 in Kiss1ARH neurons amplified the actions of NKB and protected ovariectomized female mice from developing obesity and glucose intolerance with high-fat dieting (HFD). Therefore, STIM1 appears to play a critical role in regulating synchronous firing of Kiss1ARH neurons, which ultimately affects the coordination between energy homeostasis and reproduction.
SIGNIFICANCE STATEMENT Hypothalamic arcuate kisspeptin (Kiss1ARH) neurons are essential for stimulating the pulsatile release of gonadotropin-releasing hormone (GnRH) and maintaining fertility. However, Kiss1ARH neurons appear to be a key player in coordinating energy balance with reproduction. The regulation of calcium channels and hence calcium signaling is critically dependent on the endoplasmic reticulum (ER) calcium-sensing protein stromal interaction molecule 1 (STIM1), which interacts with the plasma membrane (PM) calcium channels. We have conditionally deleted Stim1 in Kiss1ARH neurons and found that it significantly increased the excitability of Kiss1ARH neurons and protected ovariectomized female mice from developing obesity and glucose intolerance with high-fat dieting (HFD).
Keywords: calcium, KNDy neurons, neurokinin B, slow EPSP, stromal interaction molecule 1, TRPC5 channel
Introduction
Nutrition and reproduction are inextricably linked across all mammalian species, i.e., high circulating concentrations of 17β-estradiol (E2) during the late follicular phase of the reproductive cycle correlate with reduced food intake (Czaja, 1978; Asarian and Geary, 2006; Roepke et al., 2010). However, we are just beginning to understand the central mechanisms by which E2 feedback coordinates reproduction and energy balance (Castellano and Tena-Sempere, 2013; Nestor et al., 2014; Navarro, 2020). Kisspeptin neurons in the hypothalamic arcuate nucleus (Kiss1ARH neurons) appear to be critical for coordinating these two homeostatic processes. First, Kiss1 and its G-protein-coupled receptor (GPR54) are essential for pubertal development and reproductive function (Kuohung and Kaiser, 2006). Mutations in Kiss1 or GPR54 cause hypogonadotropic hypogonadism in humans (De Roux et al., 2003; Seminara et al., 2003; Topaloglu et al., 2012), and deletion of Kiss1 or GPR54 causes defective sexual development and reproductive failure in mice (Seminara et al., 2003; d'Anglemont de Tassigny et al., 2007). These effects on fertility are directly dependent on Kiss1/GPR54 signaling in gonadotropin-releasing hormone (GnRH) neurons (Han et al., 2005; Pielecka-Fortuna et al., 2008; Zhang et al., 2008). Moreover, Kiss1 signaling appears to be also important for normal metabolism and glucose homeostasis. GPR54 deletion in female, but not male, mice causes severe obesity, reduced metabolism, glucose intolerance and hyperleptinemia (Tolson et al., 2014, 2019). Also, Kiss1ARH neurons are directly depolarized/excited by leptin (Qiu et al., 2011) and insulin (Qiu et al., 2014), so they are quite possibly key neurons involved in conveying metabolic information to GnRH neurons.
High-frequency optogenetic stimulation of Kiss1ARH neurons expressing channelrhodopsin (ChR2) generates pulsatile release of luteinizing hormone (LH; Clarkson et al., 2017). Kiss1ARH neurons co-express neurokinin B (NKB) and dynorphin (Goodman et al., 2007; Navarro et al., 2009) and high-frequency firing (10–20 Hz) of these neurons co-releases NKB and dynorphin to coordinate the synchronous firing of the whole population of Kiss1ARH neurons (Qiu et al., 2016). NKB binds to tachykinin 3 receptor (TacR3) in neighboring Kiss1ARH neurons to activate canonical transient receptor potential 5 (TRPC5) channels to cause a robust depolarization (slow EPSP), whereas co-released dynorphin feeds back to bind to presynaptic κ-opioid receptors to limit the release of NKB to discrete bursts of activity (Qiu et al., 2016). The co-release of the two peptide neurotransmitters coordinates the synchronous firing of Kiss1ARH neurons that drives the pulsatile release of GnRH into the median eminence (Qiu et al., 2016; Clarkson et al., 2017).
The activity of TRPC channels is modulated by stromal interaction molecule 1 (STIM1), which is localized to the endoplasmic reticulum (ER) membrane of cells, and its N-terminal domain contains an EF-hand that senses changes in ER calcium concentrations and maintains intracellular Ca2+ homeostasis through store-operated Ca2+ entry (SOCE; Salido et al., 2011). Upon depletion of ER Ca2+, STIM1 oligomerizes and then interacts with plasma membrane (PM) calcium (TRPC) channels (Yuan et al., 2007; Salido et al., 2011). Phosphorylation of STIM1 is required for oligomerization, and E2 inhibits the phosphorylation of STIM1 and its interaction with PM Orai and TRPC channels and hence SOCE (Yuan et al., 2007; Salido et al., 2011). Under normal physiological conditions, TRPC5 channels are coupled to PM receptors (Qiu et al., 2010, 2014; Gao et al., 2017), but in cellular stressed states (e.g., inflammation, obesity), TRPC5 channels may associate with STIM1 to replete ER Ca2+ stores (Birnbaumer, 2009; Qiu et al., 2018b). E2 maintains the excitatory effects of insulin in proopiomelanocortin (POMC) neurons, mediated by TRPC5 channel opening, by downregulating Stim1 expression in the ARH, thereby protecting against insulin resistance in obese females (Qiu et al., 2018b). E2 also downregulates Stim1 expression in the ARH of female guinea pigs, indicating that E2's effects maybe universal (Qiu et al., 2018b). Therefore, we hypothesized that deletion of Stim1 in Kiss1ARH neurons would augment TacR3 mediated depolarization via TRPC5 channels to ultimately drive synchronous firing of the “pulse-generator” Kiss1ARH neurons.
Materials and Methods
Animals
All animal procedures were conducted at Oregon Health and Science University (OHSU) according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with approval from the OHSU Animal Care and Use Committee.
We used female mice in all of the experiments. Kiss1Cre:GFP (v2) mice (Richard D. Palmiter; University of Washington; Padilla et al., 2018) were housed under constant temperature (21–23°C) and 12/12 h light/dark cycle schedule (lights on at 6 A.M. and lights off at 6 P.M.), with free access to food (Lab Diets 5L0D) and water. Kiss1Cre:GFP mice were used for viral injection to express ChR2 or GCaMP6s in Kiss1ARH neurons or they were crossed with homozygous Ai32 mice (RRID:IMSR_JAX:024109, C57BL/6 background) purchased from The Jackson Laboratory. These Ai32 mice carry the ChR2 (H134R)–EYFP gene in their Gt(ROSA)26Sor locus (Madisen et al., 2012). The gene is separated from its CAG promoter by a loxP-flanked transcriptional STOP cassette, allowing its expression in a Cre-dependent manner. To test for this, we dispersed and harvested EYFP neurons in the ARH from Kiss1Cre:GFP::Ai32 females and used single-cell RT-PCR (scRT-PCR) to determine Kiss1 mRNA expression as described below and according to previous published methods (Bosch et al., 2013). Data from 126 ARHEYFP neurons from 6 Kiss1Cre:GFP::Ai32 females documented that 99% of the EYFP neurons expressed Kiss1.
To generate mice with conditional knock-out of Stim1 in Kiss1 neurons (Stim1kko), we first crossed Kiss1Cre/+ (v2) males (Padilla et al., 2018) with Stim1fl/fl females (The Jackson Laboratory stock #023350, RRID:IMSR_JAX:023350; Oh-hora et al., 2008). This cross knocks out Stim1 through excising exon 2 (Oh-hora et al., 2008) of the floxed Stim1 gene in cells in which Cre is expressed under the control of a promoter specific for the expression of Kiss1 (Padilla et al., 2018; Qiu et al., 2018a). The F1 mice produced were Kiss1Cre/+::Stim1+/lox and Stim1+/lox. The F2 mice were generated by crossing these Kiss1Cre/+::Stim1+/lox males with Stim1lox/lox females. Approximately 25% of the offspring were Kiss1Cre/+::Stim1lox/lox such that Stim1 was deleted in Kiss1 cells (Stim1kko), and all the Stim1 knock-out mice were seen at the expected frequency and viable throughout adulthood. We used Kiss1Cre/+ mice as controls. To increase the yield of Stim1 knock-out mice, we crossed Kiss1Cre/+::Stim1lox/lox males with Stim1 lox/lox females. We maintained not only this strain but also the Kiss1Cre/+ strain at the same time. Genotypes for Stim1 were determined using forward primer JAX#18 885 (5'-CGA TGG TCT CAC GGT CTC TA-3') and reverse primer JAX#18 886 (5'-GCT CTG CTG ACC TGG AAC TA-3'), which distinguished between lox/lox, lox/+, and +/+ genotypes. Cre genotypes were determined using forward primer 5'-GCG GTC TGG CAG TAA AAA CTA TC-3' and reverse primer 5'-TTC CAT GAG TGA ACG AAC CTG G-3', which distinguished between carriers and non-carriers of the Cre allele.
Puberty onset and estrous cyclicity
To determine whether deleting Stim1 in Kiss1-expressing neurons might impact fertility, we evaluated female Stim1kko mice and wild-type (WT) female littermates for pubertal onset and estrous cyclicity. For breeding, male and female mice were mated at 1:1, and the number of pups per litter was counted. The Stim1kko mice showed similar fecundity as control mice. Puberty onset in females was assessed by monitoring for vaginal opening daily between 9 and 10 A.M. starting at three weeks of age. For estrous cycle studies, Stim1kko and Kiss1Cre:GFP female mice were group housed and were habituated to handling for at least one week by the same investigator before estrous cycle monitoring. Vaginal lavage was performed daily for 13 consecutive days between 9 and 10 A.M. Cytology was evaluated using a light microscope and scored as diestrus, proestrus or estrus as previously described (Qiu et al., 2018a). The Number of estrous and diestrous days were counted for each animal and used for statistical analysis (Mann–Whitney U test).
Gonadectomy
At least 7 d before each experiment, ovaries were removed as described previously while under inhalant isofluorane anesthesia (Piramal Enterprises Limited; Qiu et al., 2018a). Each mouse received analgesia (carprofen; 5 mg/kg, s.c.) immediately after a surgery for relief of postoperative pain.
Metabolic studies
For the metabolic studies, Stim1kko and Kiss1 littermate control females were ovariectomized at two to four months of age and put on a high-fat diet (HFD; 45% kcal from fat; Research Diets; D12451) for eight weeks. Mice were group housed (because of COVID-19 restrictions) and individually weighed every week. The evening before the glucose tolerance test (GTT), all mice were assessed for body composition (fat and lean mass) using an EchoMRI 4-in-1-500 Body Composition Analyzer.
For GTT, age matched Kiss1Cre and Stim1kko mice were fasted overnight for 15 h, and baseline glucose levels measured with the aid of an Accu-Check Advantage blood glucose meter (Roche) using blood collected from the tail vein. All mice were then injected intraperitoneally with glucose (1 mg/g lean mass as determined by EchoMRI) in sterile PBS and blood glucose levels were measured 15, 30, 60, 90, and 120 min after injection. The glucose clearance (area under the curve; AUC) was calculated based on the glucose baseline levels at 0 min (Ayala et al., 2010).
AAV delivery to Kiss1Cre:GFP and Stim1kko mice
Fourteen to 21 d before each experiment, Kiss1Cre:GFP mice or Stim1kko mice (>60 d old) received bilateral ARH injections of a Cre-dependent adeno-associated viral (AAV; serotype 1) vector encoding ChR2-mCherry (AAV1-Ef1a-DIO-ChR2: mCherry) or ChR2-YFP (AAV1-Ef1a-DIO-ChR2:YFP, Stephanie L. Padilla; University of Washington) or GCaMP6s (AAV9-Syn-Flex-GCaMP6s-WPRE-SV40; Addgene, #100845-AAV9). Using aseptic techniques, anesthetized female mice (1.5% isoflurane/O2) received a medial skin incision to expose the surface of the skull. The glass pipette (Drummond Scientific #3-000-203-G/X) with a beveled tip (diameter = 45 μm) was filled with mineral oil, loaded with an aliquot of AAV using a Nanoject II (Drummond Scientific). ARH injection coordinates were anteroposterior (AP): −1.20 mm, mediolateral (ML): ± 0.30 mm, dorsoventral (DL): −5.80 mm (surface of brain z = 0.0 mm); 500 nl of the AAV (2.0 × 1012 particles/ml) was injected (100 nl/min) into each position, left in place for 10 min postinjection, then the pipette was slowly removed from the brain. The skin incision was closed using skin adhesive, and each mouse received analgesia (carprofen; 5 mg/kg) for 2 d postoperation.
Electrophysiology
Coronal brain slices (250 µm) containing the ARH from gonadectomized females were prepared as previously described (Qiu et al., 2003). Whole-cell, patch recordings were performed in voltage clamp and current clamp using an Olympus BX51W1 upright microscope equipped with video-enhanced, infrared-differential interference contrast (IR-DIC) and an Exfo X-Cite 120 Series fluorescence light source. Electrodes were fabricated from borosilicate glass (1.5-mm outer diameter; World Precision Instruments) and filled with a normal internal solution: 128 mm potassium gluconate, 10 mm NaCl, 1 mm MgCl2, 11 mm EGTA, 10 mm HEPES, 3 mm ATP, and 0.25 mm GTP (pH was adjusted to 7.3–7.4 with 1N KOH, 290–300 mOsm). Pipette resistances ranged from 3 to 5 MΩ. In whole cell configuration, access resistance was <20 MΩ; access resistance was 80% compensated. For some experiments measuring the ramp current–voltage (I–V) relationship, K+-gluconate in the normal internal solution was replaced with Cs+-gluconate (pH 7.35 with CsOH), and the extracellular solution contained Na+, K+, Ih (HCN), Ca2+, and GABAA channel blockers (126 mm NaCl, 5 mm 4-aminopyridine, 2.5 mm KCl, 1.2 mm MgCl2, 2 mm CsCl, 1.4 mm CaCl2, 1 mm CoCl2, 0.01 mm nifedipine, 20 mm HEPES, 8 mm NaOH, 10 mm glucose, 0.001 mm tetrodotoxin (TTX), and 0.1 mm picrotoxin). The slope conductance was measured from −40 to −20 mV in the optimum range for detecting TRPC5 channel activity (Blair et al., 2009). For optogenetic stimulation, a light-induced response was evoked using a light-emitting diode (LED) 470-nm blue light source controlled by a variable 2A driver (ThorLabs) with the light path delivered directly through an Olympus 40 × water-immersion lens. High fidelity response to light (470 nm) stimulation of Kiss1ARH::ChR2-mCherry expressing neurons was observed, and both evoked inward currents (in voltage clamp, Vhold = −60 mV) or depolarization (in current clamp) were measured. To characterize action potential (AP) properties, current-clamp recordings were obtained in the presence of antagonists of ionotropic glutamate and GABA receptors (50 μm d-APV, 10 μm CNQX, and 100 μm picrotoxin). The membrane potential of neurons was subsequently maintained around −70 mV by direct current injection. The excitability of neurons was assessed by injecting depolarizing currents (1 s steps, 5 pA increments). The first current step to display an AP was defined as the rheobase, and the first spike was analyzed in detail. AP properties were analyzed using Clampfit 11.2 software. Full-width at half-maximum (FWHM) of the AP, and afterhyperpolarization (AHP) time and amplitude were measured relative to threshold. Electrophysiological signals were amplified with an Axopatch 200A and digitized with Digidata 1322A (Molecular Devices), and the data were analyzed using p-Clamp software (RRID:SCR_011323, version 9.2, Molecular Devices). The amplitude of the slow EPSP was measured after low pass filtering to eliminate the barrage of APs riding on the depolarization. The liquid junction potential was corrected for all data analysis.
Calcium imaging
For calcium imaging, brain slices were placed in a RC-22C slide recording chamber (Harvard/Warner Instruments) and imaged on an inverted Nikon TiE microscope equipped with a Yokogawa CSU-W1 spinning disk confocal head, integrated under NIS Elements v4.20 (Nikon). The preparation, kept at 32°C via a cage incubator (Okolab), was continuously perfused with oxygenated artificial CSF (aCSF) at a flow rate of 1.25 ml/min. Images were acquired on a Zyla v5.5 sCMOS camera (Andor) at 0.5 Hz. frame-rate, through an 10 × (NA 0.45) or 20 × (NA 0.75) objective, combining 488 nm laser excitation with 500- to 550-nm emission collection. A single focal plane (z-axis) was maintained using the Nikon Perfect Focus System. Minor tissue drift in the x-y-axis was corrected using NIS Elements. Imaging displaying major drift were excluded from final analysis. Changes in Kiss1ARH neuron Ca2+ levels were measured in regions of interest (ROIs) comprising the GCaMP6s-positive cell bodies. In all recordings, background fluorescence measured in an ROI drawn on nearby tissue was subtracted from every ROI. [Ca2+]i variations after drug applications were assessed as changes in fluorescence signals over baseline (ΔF/F0). To normalize the fluorescence value of each cell, we first separated experimental trials into two parts: a baseline period (2 min) corresponding to all the frames recorded before addition of drugs, and a stimulus period, after the onset of the drug (such as bath-applied senktide) application and lasting several minutes. Next, for each ROI we calculated ΔF/F0 for each frame (t), where ΔF/F0 equals (F(t) – F0)/F0, and F0 was the mean fluorescence value for that ROI for all frames in the baseline period for that trial. The AUC was calculated over the time periods of 2 min before and 18 min after drug application. Maximal peak reached after drug application was also measured and used in quantitative analysis. Data were averaged across all Kiss1ARH neurons in a slice (two slices per animal), which were used as the statistical unit over a minimum of three animals per condition.
scRT-PCR
Coronal brain sections from the ARH of three female Stim1kko and three Kiss1Cre:GFP::Ai32 mice were prepared for electrophysiology and scRT-PCR. The 3–4 slices obtained were divided between electrophysiological recording experiments and single-cell harvesting. Single-cell dispersion and harvesting was performed as described previously with some modifcations as described below (Bosch et al., 2013; Zhang et al., 2013b). Briefly, the ARH was dissected and digested in papain (7 mg/ml in aCSF, Sigma-Aldrich). Gentle trituration using varying sizes of flame polished Pasteur pipets were used to disperse the cells and then they were plated onto a glass bottom dish. A constant flow of oxygenated aCSF (125 mm NaCl, 5 mm KCl, 1.44 mm NaH2PO4, 5 mm HEPES, 10 mm D-glucose, 26 mm NaHCO3, 2 mm MgSO4·7H2O, and 2 mm CaCl2) was applied to the dish to keep the cells healthy and to clear debris. Fluorescent neurons were visualized under an inverted microscope. The Xenoworks Microinjection system (Sutter Instruments) was used to manipulate a 10 µm tip size glass capillary tube to approach single neurons, apply gently suction and harvest single cells or pools of 10 cells into a siliconized tube containing a solution of 1× Invitrogen Superscript III Buffer (LifeTech), 15U of RNasin (Promega), 10 mm of dithiothreitol (DTT) and diethylpyrocarbonate (DEPC)-treated water in a total of 5 µl for single cells or 8 µl for pools of 10 cells. Corresponding controls were collected at the same time including single neurons (processed without reverse transcriptase) and aCSF from the surrounding area. Hypothalamic tissue RNA was also processed with and without reverse transcriptase. First strand cDNA synthesis was performed on single cells, pools of cells and controls in a 20 µl (single cells) or 25 µl (10 cell pools) volume containing a final concentration of 1× Invitrogen Superscript III Buffer, 30 U of RNasin, 15 mm DTT, 10 mm dNTP, 100 ng Random Primers (Promega), 400 ng Anchored Oligo (dT)20 Primer (Invitrogen), 100 U Superscript III Reverse Transcriptase (Life Tech), and DEPC-treated water according to manufactures protocol and stored at −20°C. Clone Manager software (Sci Ed Software) was used to design primers that cross at least one intron-exon boundary. In order to confirm that STIM1 was knocked out, STIM1 primers were designed to include part of exon 2 (see Table 1). Single-cell PCR conditions were optimized for primer concentration, magnesium concentration and annealing temperature. Standard curves were generated using hypothalamic cDNA with dilutions from 1:50 to 1:12,800 for primers used for quantitative PCR (qPCR) to determine the efficiency (E = 10(−1/m)−1; Table 1). Primer pairs with efficiencies of 90–100% permit the use of the comparative ΔΔCT method for analysis (Livak and Schmittgen, 2001; Pfaffl, 2001).
Table 1.
Primer table
| Gene name (encodes for) | Accession number | Primer location (nt) | Product length (bp) | Annealing temperature (°C) | Efficiency slope | r 2 | % |
|---|---|---|---|---|---|---|---|
| Kiss1 (Kiss1)a,b | NM_178260 | 64–80 (exon 1) | 120 | 57a, 60b | −3.410 | 0.989 | 97 |
| 167–183 (exon 2) | |||||||
| Stim1 (STIM1)a | NM_009287 | 797–816 (exon 2) | 204 | 59 | |||
| 981–1000 (exon 3) | |||||||
| Stim1 (STIM1)b | NM_009287 | 821–839 (exon 2) | 135 | 60 | −3.311 | 0.977 | 100 |
| 937–955 (exon 3) | |||||||
| Stim2 (STIM2) | NM_001363348 | 620–638 (exon 2) | 172 | 59 | |||
| 773–791 (exon 4) | |||||||
| Stim2 (STIM2)b | NM_001363348 | 1784–1803 (exon 11) | 131 | 60 | −3.439 | 0.993 | 95 |
| 1895–1914 (exon 12) | |||||||
| Gapdh (GAPDH)b | NM_008084 | 689–706 (exon 4) | 93 | 60 | −3.352 | 0.998 | 99 |
| 764–781 (exon 5) | |||||||
| Trpc4 (TRPC4)a,b | NM_016984 | 1888–1907 (exon 6) | 116 | 60 | −3.318 | 0.940 | 100 |
| 1984–2003 (exon 7) | |||||||
| Trpc5 (TRPC5)a | NM_009428 | 2206–2227 (exon 6) | 131 | 63 | |||
| 2315–2336 (exon 7) | |||||||
| Trpc5 (TRPC5)b | NM_009428 | 734–753 (exon 2) | 118 | 60 | −3.161 | 0.953 | 100 |
| 832–851 (exon 3) |
aprimers for scRT-PCR.
bprimers for qPCR.
PCR for Kiss1, Stim1, Trpc4, and Trpc5 mRNAs was performed on 3 µl of cDNA from single cells in a 30-µl reaction volume containing 1× GoTaq Flexi buffer (Promega), 2 mm MgCl2, 10 mm dNTP, 0.33 μm forward and reverse primers, 2 U GoTaq Flexi Polymerase (Promega), and 0.22 µg TaqStart Antibody (Clontech). A total of 45–50 cycles of amplification were performed on a Bio-Rad C1000 thermocycler and the resulting product visualized with ethidium bromide on a 2% agarose gel.
qPCR was performed on 4 µl of cDNA from pools of 10 cells (three to four pools per animal) in duplicate for the target genes (Stim1, Stim2, Trpc4, and Trpc5) and 2 µl in duplicate for the reference gene (Gapdh) in a 20 µl reaction volume containing 1× Power SYBR Green PCR Master Mix (Applied Biosystems) and 0.5 μm forward and reverse primers. Forty cycles of amplification were run on a Quant Studio 7 Flex Real-Time PCR System (Applied Biosystems) and the resulting data were analyzed using the comparative ΔΔCT method (Livak and Schmittgen, 2001; Pfaffl, 2001). The relative linear quantity was determined with the 2-ΔΔCT equation (Bosch et al., 2013). The mean of all of the ΔCT values (ΔCT = CT of the target gene – CT of the reference gene) from the controls was used as the calibrator and the data are expressed as fold change in gene expression.
Drugs
A standard artificial cerebrospinal fluid was used (Qiu et al., 2011). All drugs were purchased from Tocris Bioscience unless otherwise specified. TTX was purchased from Alomone Labs (1 mm), DL-amino-5-phosphonovaleric acid (AP5; 50 mm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 mm) were dissolved in H2O. Thapsigargin (Tg; 2 mm), Picrotoxin (100 mm), TacR3 agonist senktide (1 mm), and TRPC4/5 antagonist, HC070 (from MedChemExpress, 10 mm), and 2-aminoethyl diphenylborinate (2-APB, 100 mm) were prepared in dimethylsulfoxide (DMSO). 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) tetrasodium salt (10 mm) replaced EGTA in the internal solution. Aliquots of the stock solutions were stored as appropriate until needed.
Data analysis
For qPCR four Kiss1 neuronal pools (10 cells/pool) from each animal were run in duplicate for the mRNAs that encode for STIM1, STIM2 and GAPDH and the mean value of each gene from each animal (n = 3 animals) was used for statistical analysis. Data are expressed as mean ± SEM and were analyzed using an unpaired Student's t test. In addition, Kiss1 neuronal pools (10 cells/pool) were used to determine the expression of Trpc4 and Trpc5 in these neurons. For scRT-PCR the number of Kiss1-positive cells harvested from Kiss1Cre:GFP females injected with Cre-dependent ChR2-mCherry or from Kiss1Cre:GFP::Ai32 females were used to qualitatively assess the number of Kiss1 neurons with Stim1, Stim2, Trpc4, Trpc5, and percent expression.
Comparisons between different treatments were performed using a repeated measures, two-way or one-way ANOVA analysis with the post hoc Bonferroni's test. Differences were considered statistically significant if p < 0.05. All data are expressed as mean ± SEM.
Results
Validation of conditional deletion of Stim1 in Kiss1 neurons
STIM1 is involved in the regulation of neuronal firing in cerebellar Purkinje neurons (Hartmann et al., 2014; Ryu et al., 2017), midbrain dopaminergic neurons (Sun et al., 2017) and hypothalamic arcuate nucleus POMC neurons (Qiu et al., 2018b). Initially to determine whether STIM1 regulates Kiss1ARH neuronal excitability, we measured the mRNA expression of Stim1 and its close homolog Stim2 in manually harvested Kiss1ARH neurons by quantitative real-time PCR (Fig. 1A). Based on the qPCR, mRNA levels of Stim1 were greater than those of Stim2 in Kiss1ARH neurons (Fig. 1A1). Likewise, in cerebellar Purkinje neurons, Stim1 is also much more abundant than Stim2 (Hartmann et al., 2014), while in hippocampal (Berna-Erro et al., 2009) and cortical neurons (Gruszczynska-Biegala et al., 2011) Stim2 expression levels exceed those of Stim1. A qualitative, unbiased sampling of Kiss1ARH neurons (n = 60) from ovariectomized Kiss1Cre females (n = 3) revealed that Stim1 mRNA was expressed in 81.7 ± 7.6% and Stim2 mRNA was detected in 81.2 ± 2.7% of Kiss1ARH neurons with 70% of neurons expressing both Stim1 and Stim2.
Figure 1.

Expression patterns of Stim1 and Stim2 in the arcuate Kiss1 neurons. A1–A3, qPCR assay measuring Stim1 and Stim2 mRNA in Kiss1ARH neuronal pools (n = 3 animals, 10 cells in each pool, 4 pools/animal) from Kiss1Cre control and Stim1kko female mice (n = 3 animals per group). A1, Comparison between Stim1 and Stim2 in controls only. Bar graphs represent mean ± SEM (unpaired t test, t(4) = 3.079, *p = 0.0370). A2, Stim1 was non-detectable (ND) in the STIM1KKO neuronal pools (unpaired t test, t(4) = 7.559, **p = 0.0016). A3, The Stim2 expression level of Kiss1ARH neurons was not different between Kiss1Cre control and Stim1kko female mice (unpaired t test, t(4) = 0.7143, p = 0.5145). B, Representative gels illustrating mRNA expression of Stim1 and Stim2 in single Kiss1ARH neurons from Stim1kko mice. The expected base pair (bp) sizes are Kiss1, 120 bp; Stim1, 204 bp; Stim2, 172 bp. A single neuron was processed without reverse transcriptase (–RT) and RNA extracted from hypothalamic tissue was used as positive (+, with RT) and negative (–, without RT) tissue controls. MM, molecular marker. C, left, Schematic of a coronal section showing the bilateral viral injections in the ARH with AAV-DIO-GCaMP6s. Right, Photomicrograph showing a coronal section confirming targeted bilateral injections of DIO-GCaMP6s into the ARH. D, E, Representative traces of GCaMP6s activity based on cytosolic Ca2+ measurements in Kiss1ARH neurons from Kiss1Cre:GCaMP6s mice (D) and Stim1kko:GCaMP6s mice (E). ER Ca2+ stores were depleted with 2 μm Tg, a SERCA inhibitor, after 20 min of perfusion with aCSF containing 0 mm Ca2+. SOCE was evaluated by substituting the extracellular aCSF containing 0 mm Ca2+ with aCSF containing 2 mm Ca2+. F, Averaged traces from D, E revealed that deletion of Stim1 in Kiss1ARH neurons attenuated the SOCE. G, Bar graphs summarizing the effects of depletion of Ca2+ store by Tg and Ca2+ influx (SOCE) in Kiss1ARH neurons from Kiss1Cre:GCaMP6s and Stim1kko:GCaMP6s mice (two-way ANOVA: main effect of treatment (F(1,3) = 13.84, p = 0.0338), main effect of time (F(1,3) = 5.199, p = 0.1069), and interaction (F(1,3) = 52.14, p = 0.0055); n = number of slices; post hoc Bonferroni test, **p < 0.01, for SOCE; ns = not significant for depletion of Ca2+ store.
To elucidate the functional role of STIM1 in Kiss1 neurons, we generated mice that lacked STIM1 selectively in Kiss1 neurons (Stim1kko; detailed in Materials and Methods). We confirmed the Stim1 deletion in Stim1kko mice using single-cell qPCR of pools of harvested Kiss1ARH neurons (n = 3 animals; Fig. 1A2). Consistent with the scRT-PCR results (Fig. 1B), Stim1 mRNA was undetectable in Stim1kko neurons (Fig. 1A2), whereas there was no reduction in Stim2 mRNA expression (Fig. 1A3). In contrast, Stim1 mRNA was still expressed in the majority of adjacent nonfluorescent neurons obtained from both Stim1kko and Kiss1Cre mice.
Stim1 deletion reduces SOCE
SOCE constitutes an important source of calcium entry and signaling in neurons. Depletion of ER Ca2+ stores causes the ER Ca2+ sensor STIM proteins (STIM1 and STIM2) to interact with and activate cell surface Ca2+ release-activated Ca2+ (CRAC) channels, thereby resulting in a second wave of cytoplasmic Ca2+ rise (Moccia et al., 2015). Genetic suppression of Stim1 in neural progenitor cells results in abrogation of this second wave of calcium rise that constitutes SOCE (Somasundaram et al., 2014). We asked whether deletion of Stim1 in Kiss1ARH neurons (Stim1kko) attenuates neuronal SOCE. Kiss1Cre and Stim1kko mice received bilateral ARH injections of GCaMP6 viral vector (Fig. 1C1,C2), and Kiss1ARH neurons expressing GCaMP6s were imaged using spinning disk confocal microscopy (Movie 1). ER Ca2+ stores were released by treatment with 2 μm Tg, a blocker of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) pump. As expected, Tg treatment of neurons bathed in Ca2+-free aCSF generated an initial wave of cytoplasmic Ca2+ release ([Ca2+]i) as measured by an increase in GCaMP6s activity both in control and Stim1-deleted neurons (Fig. 1D–F). As long as neurons were kept in Ca2+-free aCSF, the ER stores remained empty, a situation that was presumably sensed by the Ca2+ sensor STIMs. Upon switching to a normal aCSF containing 2 mm Ca2+, an immediate SOCE response was observed as a second wave of cytoplasmic Ca2+ rise. Consistent with a role for STIM1 regulation, we observed an attenuation of SOCE in Kiss1ARH neurons from Stim1kko mice (ΔF/F0 × 100 = 1274.5 ± 49.4, n = 4, Kiss1Cre mice vs 389.0 ± 86.1, n = 4, Stim1kko mice, which was measured from the 15 min time point to the peak; Fig. 1D–G), indicating that STIM1 plays a major role in SOCE after Tg-induced ER Ca2+ depletion in Kiss1ARH neurons as has been shown in other CNS neurons (Guner et al., 2017; Pavez et al., 2019).
NKB receptor agonist senktide induces [Ca2+]i increase in Kiss1ARH neurons expressing GCaMP6s. Imaging of transient Ca2+ changes in an arcuate slice using spinning disk confocal microscopy. Fluorescence intensity was measured over 20 min, before and after application of senktide (1 µm). The period represented is 20 min.
TacR3-induced increase in [Ca2+]i is augmented by deletion of Stim1
TacR3 classically couples to a Gαq protein-calcium signaling and excites Kiss1ARH neurons (de Croft et al., 2013; Ruka et al., 2013; Qiu et al., 2016). Calcium is of critical importance to neurons as it participates in the transmission of depolarizing signals and contributes to synaptic activity (Brini et al., 2014). Therefore, we tested whether STIM1 can modulate TacR3-mediated calcium responses. We first measured the effects of the TacR3, which is Gq-coupled, agonist senktide on GCaMP6s-expressing Kiss1ARH neurons in arcuate slices from Kiss1Cre mice; senktide (1 μm) rapidly induced an increase in [Ca2+]i (Fig. 2A,C). Next, we investigated whether STIM1 contributes to intracellular rise in [Ca2+]i after senktide activation. Indeed, deletion of Stim1 significantly augmented the peak TacR3-mediated response by ∼3-fold [ΔF/F0 × 100 = 244.0 ± 27.7, n = 7 slices, Kiss1Cre mice vs 622.1 ± 133.2, n = 6 slices, Stim1kko mice; two-way ANOVA: main effect of treatment (F(1,11) = 5.265, p = 0.0424), main effect of time (F(19,209) = 42.69, p < 0.0001), and interaction (F(19,209) = 6.486, p < 0.0001); post hoc Bonferroni test, ****p < 0.001, **p < 0.01, *p < 0.05; Fig. 2B,C]. Likewise, the AUC was significantly increased in the Stim1kko group by 4-fold (Kiss1Cre: 954.8 ± 200.4, n = 7 vs Stim1kko: 3746.0 ± 1227.0, n = 6; Fig. 2D).
Figure 2.

Senktide-induced increase in [Ca2+]i is augmented by deletion of Stim1 in GCaMP6s-expressing Kiss1ARH neurons from Stim1kko mice. A, B, Representative traces of senktide-induced [Ca2+]i in Kiss1ARH neurons from Kiss1Cre (A) and Stim1kko (B) mice. Traces represent individual cells within a single slice. C, Summary of the potentiation of senktide-induced [Ca2+]i by deletion of Stim1. Two-way ANOVA: main effect of treatment (F(1,11) = 5.265, p = 0.0424), main effect of time (F(19,209) = 42.69, p < 0.0001), and interaction (F(19,209) = 6.486, p < 0.0001); n = number of slices; post hoc Bonferroni test, ****p < 0.001, **p < 0.01, *p < 0.05. D, AUC of Kiss1ARH neurons from Kiss1Cre and Stim1kko mice from C. There was a significant difference (unpaired t test, t(11) = 2.430, *p = 0.0334) between the two groups.
Deletion of STIM1 enhances slow EPSP in Kiss1ARH neurons
Kiss1ARH neurons are the chief component of the GnRH pulse generator circuit (Navarro et al., 2009, 2011; Lehman et al., 2010; Okamura et al., 2013), such that they synchronize their activity to trigger the release of peptides to drive pulsatile release of GnRH (Qiu et al., 2016; Clarkson et al., 2017). To investigate whether STIM1 modulates the activity of Kiss1ARH neurons, we bilaterally injected AAV1-Ef1a-DIO-ChR2:mCherry into the arcuate nucleus of Kiss1Cre and Stim1kko mice. To verify that Trpc5 mRNAs is co-localized in these Kiss1ARH neurons, we harvested 50 Kiss1ARH neurons from two females and did scRT-PCR for Trpc5 and Trpc4 expression. The single-cell analysis revealed that Trpc5 transcript was detectable in 82% of Kiss1ARH neurons, but Trpc4 mRNA was not detected in Kiss1ARH neurons (Fig. 3A). Moreover, quantitative single-cell PCR documented that Trpc5 but not Trpc4 mRNA was expressed in Kiss1ARH neurons (Fig. 3B). Initially, whole-cell patch recording in Kiss1ARH neurons from ovariectomized Kiss1Cre or Stim1kko female mice revealed that there was no difference in the resting membrane potential (RMP: Kiss1Cre: −66.0 ± 1.7 mV, n = 38 vs Stim1kko: −68.2 ± 0.9 mV, n = 58) or membrane capacitance (Cm: Kiss1Cre: 25.0 ± 1.0 pF, n = 38 vs Stim1kko: 27.3 ± 0.9 pF, n = 58). However, there was a significant difference in the membrane input resistance (Rin: Kiss1Cre: 524.2 ± 42.4 Ω, n = 38, vs Stim1kko: 417.3 ± 26.9 Ω, n = 58, unpaired two-tailed t test, t(94) = 2.242, p = 0.0273; Table 2), which has also been reported with Stim1 knock-out in cerebellar Purkinje neurons (Ryu et al., 2017). We also measured the rheobase in the presence of synaptic blockers (CNQX, AP5, and picrotoxin) and the frequency-current relationship, and there were no differences between control Kiss1Cre and Stim1kko mice (Fig. 3C,D).
Figure 3.
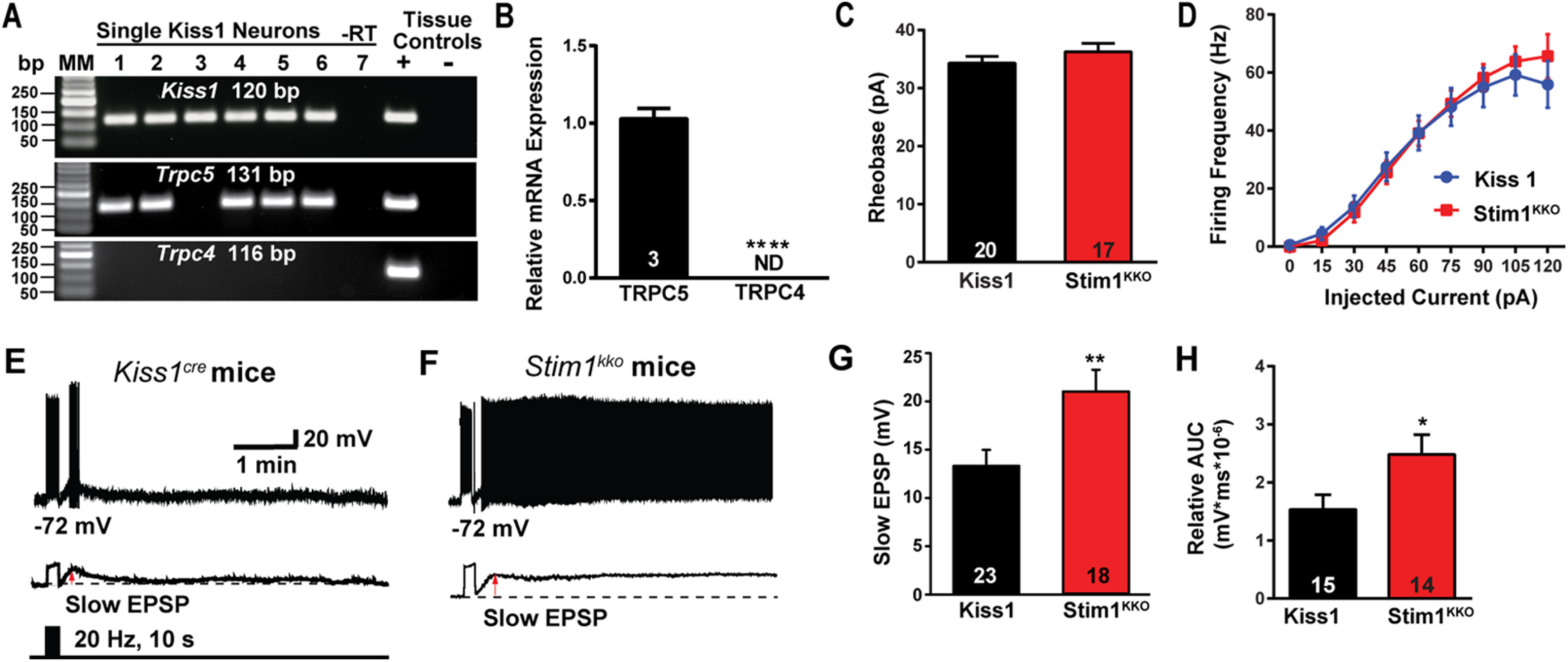
Deletion of Stim1 augments high-frequency optogenetic stimulation-induced slow EPSP depolarization in Kiss1ARH neurons. A, Representative gel image illustrating the mRNA expression of Trpc5 channel subunit in Kiss1ARH neurons harvested from female mice. The expected size of PCR products for Kiss1 and Trpc5 are indicated. Trpc4 mRNA was not detected in Kiss1ARH neurons. MM, molecular marker; –RT indicates a harvested Kiss1 neuron reacted without RT; + indicates positive tissue control (with RT); – indicates negative tissue control (without RT) using cDNA from mouse medial basal hypothalamic tissue; RT, reverse transcriptase. B, Quantitative single-cell PCR (3 × 10 cell pools per animal, n = 3 animals) verified that Trpc5 was expressed in Kiss1ARH neurons, whereas Trpc4 mRNA was not detected (unpaired t test, t(4) = 15.67, ****p < 0.0001). C, The mean rheobase of Kiss1ARH neurons between Kiss1 and Stim1kko groups was not different (unpaired t test, t(35) = 1.042, p = 0.3045). D, There was no difference in the evoked firing rate between Kiss1 and Stim1KKO groups (two-way ANOVA: main effect of treatment, F(1,32) = 0.08594, p = 0.7713, main effect of time, F(8,256) = 125.6, p < 0.0001, and interaction, F(8,256) = 1.359, p = 0.2149; Kiss1Cre, n =19, Stim1kko, n = 15; post hoc Bonferroni test, p > 0.05). E, F, High-frequency optogenetic stimulation (20 Hz, 10 s) generated slow EPSPs in a ChR2-expressing Kiss1ARH neuron from control Kiss1Cre mice (E) and in a ChR2-expressing Kiss1ARH neuron from Stim1kko mice (F). The lower traces show the slow EPSP after low-pass filtering the traces from E, F (red arrow). G, Summary of the effects of Stim1 deletion on the slow EPSP amplitude (unpaired t test, t(39) = 2.802, **p = 0.0079). H, Summary of the effects of Stim1 deletion on the AUC of slow EPSP (unpaired t test, t(27) = 2.246, *p = 0.0331). Bar graphs represent the mean ± SEM. Cell numbers are indicated.
Table 2.
Electrophysiological properties of Kiss1ARH neurons from Kiss1cre and Stim1kko female mice
| RMP (mV)a | Rin (MΩ)b | Cm (pF)c | Rheobase (pA)d | Threshold (mV)e | Overshoot (mV)f | Peak amplitude (mV) g | FWHM (ms)h | AHP amplitude (mV)i | |
|---|---|---|---|---|---|---|---|---|---|
| Kiss1cre | −66.0 ± 1.7 | 524.2 ± 42.4 | 25.0 ± 1.0 | 34.3 ± 1.1 | −46.1 ± 1.0 | 55.8 ± 1.9 | 91.8 ± 2.0 | 0.9 ± 0.0 | 30.5 ± 1.7 |
| n = 38 | n = 38 | n = 38 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | |
| Stim1KKO | −68.2 ± 0.9 | 417.3 ± 26.9* | 27.3 ± 0.9 | 36.2 ± 1.5 | −45.4 ± 0.9 | 56.0 ± 2.7 | 91.4 ± 2.9 | 0.9 ± 0.0 | 32.8 ± 1.7 |
| n = 58 | n = 58 | n = 58 | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 |
RMP, resting membrane potential; Rin, input resistance; Cm, membrane capacitance; FWHM, full-width at half-maximum of the AP; AHP, afterhyperpolarization; n = neurons.
ap = 0.2232
bp = 0.0273, unpaired two-tailed t test, t(94) = 2.242
cp = 0.0997
dp = 0.3045
ep = 0.6296
fp = 0.9507
gp = 0.9116
hp = 0.4476; ip. = 0.3348.
Kiss1ARH neurons expressing ChR2-mCherry in slices were photostimulated at 20 Hz for 10 s (Movie 2) to generate slow EPSPs as previously described (Qiu et al., 2016). As we had hypothesized, deletion of Stim1 increased the amplitude and the duration of the slow EPSP induced by high-frequency optogenetic stimulation (Fig. 3E–H). Therefore, these results indicate that STIM1 expression governs the activity of TRPC5 channels, which ultimately contributes to the synchronous activity of Kiss1ARH neurons.
High-frequency photostimulation induces a slow EPSP. Slow EPSP was induced by a 10-s 20-Hz photostimulation (light intensity 0.9 mW and pulse duration, 10 ms) in a ChR2-expressing Kiss1ARH neuron in a slice from a Kiss1Cre::Ai32 mouse. The period represented is 1 min, 34 s.
NKB agonist activates TRPC5 channels in Kiss1ARH neurons from Kiss1Cre and Stim1kko mice
Based on our previous findings that TPRC5 channel protein is expressed in Kiss1ARH neurons and is activated by the NKB agonist senktide (Qiu et al., 2011; Kelly et al., 2018), we investigated the contribution of TRPC5 channels to generating the inward current underlying the slow EPSP. In the presence of TTX to block voltage-gated Na+ channels, we observed that senktide induced larger inward currents in Kiss1ARH neurons from Stim1kko mice versus Kiss1Cre mice (Fig. 4A,D,F,G). The senktide-induced cation current was increased by ∼2-fold as a result of Stim1 deletion (Stim1kko: 96.4 ± 9.2 pA, n = 6, vs Kiss1Cre: 56.4 ± 9.8 pA, n = 7, unpaired t test, t(11) = 2.929, p = 0.0137). We also antagonized the effects of senktide with the TPRC1,3,4,5,6 channel blocker 2-APB (Clapham et al., 2005) or with the more selective TRPC4,5 channel blocker HC070 (Just et al., 2018). HC070 suppressed the senktide-induced inward current in Kiss1ARH neurons by 45% in Kiss1Cre mice and 42% in Stim1kko mice, respectively (Fig. 4B,E–G), whereas 2APB was more efficacious to inhibit (65%) the senktide-induced inward current in Kiss1ARH neurons (Fig. 4C,F). Since Trpc4 mRNA is not expressed in Kiss1ARH neurons (Fig. 3), we would argue that TRPC5 channels mediate the slow EPSP in these neurons. The I–V plots recorded using an Cs+ internal solution revealed that the slope conductance induced by senktide between −40 to −20 mV was increased (Fig. 4H,I) by 2.5-fold (Kiss1Cre: 0.7 ± 0.2 nS, n = 16, vs Stim1kko: 1.8 ± 0.5 nS, n = 10, unpaired two-tailed t test, t(24) = 2.172, p = 0.0399), but the reversal potential for the current was not different between Kiss1ARH neurons recorded from Stim1kko mice or Kiss1Cre mice (Kiss1Cre: −13.9 ± 1.6 mV, n = 16, vs Stim1kko: −14.7 ± 2.1 mV, n = 10, unpaired two-tailed t test, t(24) = 0.3200, p = 0.7518; Fig. 4J), indicating that another conductance was not contributing to the increased current.
Figure 4.

Stim1 deletion augments senktide-induced Kiss1ARH neuronal excitability through TRPC channel activation. A–C, Representative traces of senktide (1 μm)-induced inward current in the absence (A) or presence of TRPC4/5 channel blocker HC070 (B, 100 nm) or 2APB (C, 100 μm) from Kiss1Cre mice in the presence of fast sodium channel blockade (TTX, 1 μm). Vhold = –60 mV. D–E, Stim1 deletion augments senktide-induced inward current (D), which is antagonized by the TRPC4/5 channel blocker HC070 (E, 100 nm). F, Summary of the effects of HC070 and 2APB on senktide-induced inward current in Kiss1ARH neurons from Kiss1Cre mice. Comparisons between different treatments were performed using a one-way ANOVA analysis (F(2,19) = 7.737, p = 0.0035 with the Newman–Keuls's post hoc test; *p < 0.05 or **p < 0.01 vs control). G, Summary of the effects of HC070 on senktide-induced inward current in Kiss1ARH neurons from Stim1kko mice (unpaired t test, t(10) = 3.457, *p = 0.0062). H, I, The I–V relationships before and during the peak response to senktide (Senk) in Kiss1ARH neurons from Kiss1Cre (H) and Stim1kko (I) mice indicated that although the slope conductance increased by 2.5-fold, the reversal potential of the nonselective cation current (∼−14 mV) did not change. Note, the TRPC channel blocker 2APB clearly abrogated the senktide response (H). J, Summary of the reversal potentials of the senktide-induced cation current recorded in Kiss1ARH neurons from Kiss1Cre and Stim1kko mice. Bar graphs represent the mean ± SEM (unpaired two-tailed t test, t(24) = 0.3200, p = 0.7518). Cell numbers are indicated.
Chelating [Ca2+]i with BAPTA abolishes the slow EPSP and persistent firing
Similar to a number of cortical neurons (Zylberberg and Strowbridge, 2017), Kiss1ARH neurons appear to express the biophysical properties that allow them to continue to persistently fire even after a triggering synaptic event has subsided. The “intrinsic bi-stability” of a neuron that generates the persistent firing activity has been linked to a calcium-activated, non-specific cation current (ICAN; Zylberberg and Strowbridge, 2017). TRPC channels, specifically TRPC5 channels, are thought to be responsible for the ICAN current in cortical neurons (Zhang et al., 2011). Therefore, we hypothesized that with TacR3 activation there is an influx of Ca2+ through TRPC5 channels leading to greater build-up of [Ca2+]i (Fig. 2) that facilitates the opening of more TRPC5 channels in a self-sustaining manner, and buffering with 10 mm BAPTA should attenuate the response (Blair et al., 2009). We used our standard optogenetic protocol to activate the slow EPSP and persistent firing of Kiss1ARH neurons. Notably, 10 mm BAPTA inhibited the slow EPSP by ∼80% in both control (13.3 ± 1.7 vs 3.1 ± 0.5 mV) and Stim1kko (21.0 ± 2.2 vs 5.6 ± 2.4 mV), ovariectomized females (Fig. 5A–F). This would indicate that Ican through TRPC5 channels generates a slow EPSP that causes the persistent firing in Kiss1ARH neurons.
Figure 5.

Stim1 deletion augmentation of senktide-induced Kiss1ARH neuronal excitability is dependent on intracellular calcium. A, B, High-frequency photostimulation-induced slow EPSP recorded using normal internal solution (A, EGTA 11 mm) and BAPTA internal solution (B, EGTA was replaced by 10 mm BAPTA) in Kiss1ARH neurons from Kiss1Cre mice. C, Summary of the effects of buffering intracellular calcium on the slow EPSP in A, B (unpaired t test, t(30) = 3.741, ***p = 0.0008). D, E, High-frequency photostimulation-induced slow EPSP recorded using normal internal solution (D, EGTA 11 mm) and BAPTA internal solution (E) in Kiss1ARH neurons from Stim1KKO mice. F, Summary of the effects of buffering intracellular calcium on the slow EPSP in D, E (unpaired t test, t(21) = 3.425, **p = 0.0025). Data points represent the mean ± SEM. Cell numbers are indicated.
Stim1 deletion in Kiss1ARH neurons has minimal effects on estrous cycle
Stim1kko mice on the C57BL/6 background were viable at the expected Mendelian ratio and did not show any difference in the time to vaginal opening (Stim1kko mice: postnatal day 30.2 ± 0.8, n = 21 vs Kiss1Cre mice: postnatal day 29.1 ± 0.8, n = 19, unpaired t test, t(38) = 1.003, p = 0.3222). However, since kisspeptin neurons are responsible for the maintenance of the reproductive cycle (Seminara et al., 2003; d'Anglemont de Tassigny et al., 2007; Mayer et al., 2010), and Stim1 deletion facilitated the synchronous firing of Kiss1ARH neurons, we measured the effects of Stim1 deletion in Kiss1 neurons on the estrous cycle. We monitored the estrous cycle of Stim1kko and Kiss1Cre female mice with vaginal lavage for two weeks before ovariectomy for the metabolic studies (see below). Stim1kko female mice exhibited prolonged estrous cycles versus the Kiss1Cre females (Fig. 6A–C vs D–F) with a slight decrease in diestrous days and a prolongation of estrous days (Fig. 6G,H). Although more in depth analysis is warranted (i.e., measurement of pulsatile LH), the results were not unexpected since augmented synchronous activity of Kiss1ARH neurons, as we documented at the cellular level, should still drive LH pulses in these female mice (Qiu et al., 2016; Clarkson et al., 2017).
Figure 6.
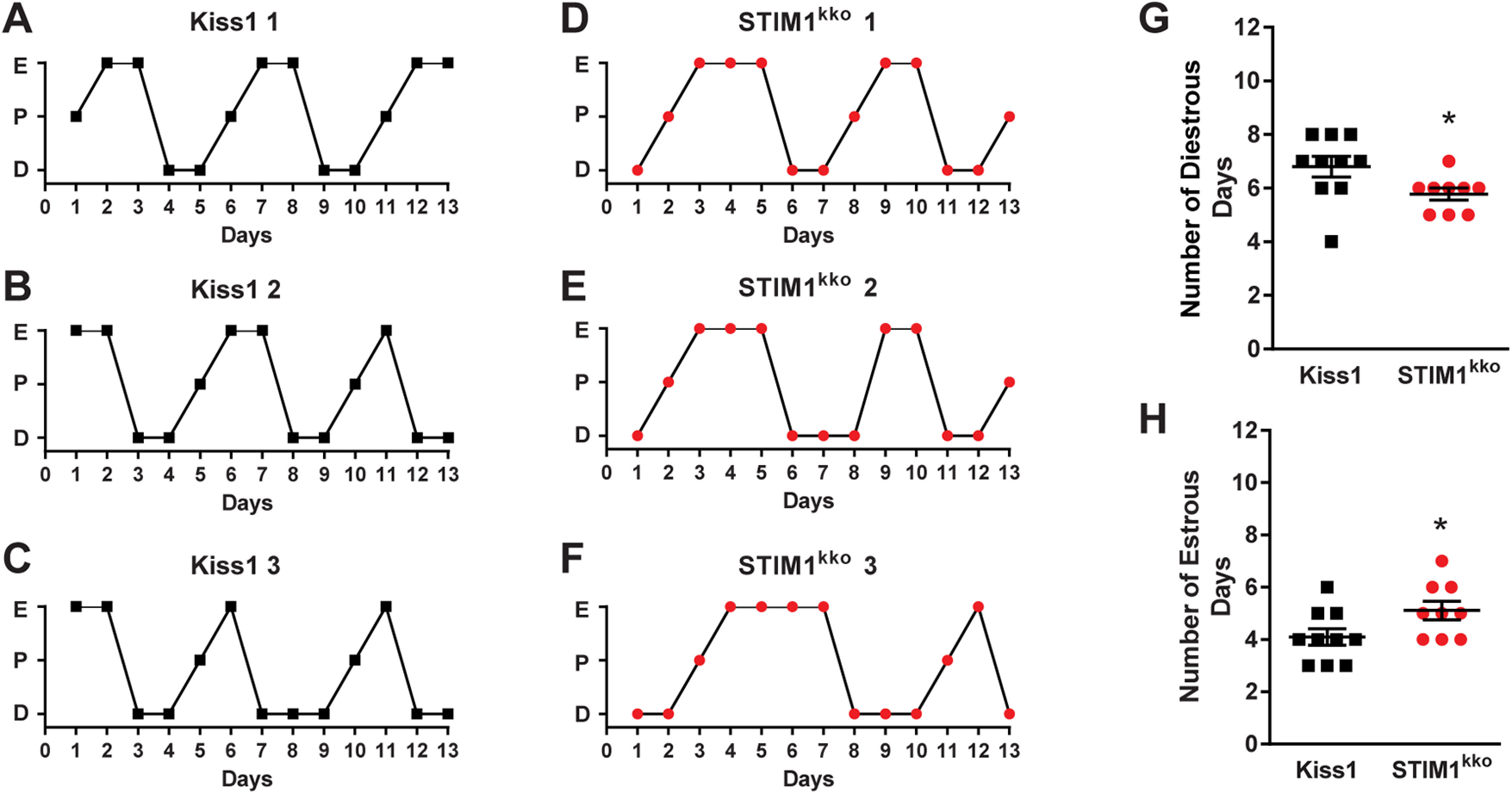
Stim1kko mice exhibit more estrous days. A–F, Representative estrous cycle data from three representative control Kiss1Cre and three Stim1 kko mice over a 13-d period. Vaginal lavage was done daily at 9:30 A.M., and cell cytology was observed and recorded as diestrus (D), proestrus (P), or estrus (E). Summary data for the number of diestrous days (G) and estrous days (H) during the 13-d period was compared between Kiss1Cre (n = 10) and Stim1 kko mice (n = 9; unpaired, two-tailed t test for G, t(17) = 2.215, *p = 0.0407; unpaired two-tailed t test for H, t(17) = 2.151, *p = 0.0461).
Stim1 deletion in Kiss1ARH neurons protects ovariectomized females against diet-induced obesity
Subsequently, the same two cohorts of female mice, Stim1kko (n = 10) and the littermate control Kiss1Cre (n = 10) mice, were ovariectomized at around three months of age and put on a HFD for eight weeks (see Materials and Methods). Over this time period, there was significantly less gain in body weight in the Stim1kko versus the Kiss1Cre mice (Fig. 7A,B). Moreover, the average fat mass of Stim1kko mice was significantly lighter than that of Kiss1Cre controls by week 6 (Stim1kko vs Kiss1Cre mice fat mass: 7.6 ± 0.9 g, n = 10 vs 11.4 ± 1.1 g, n = 10; Fig. 7C). The lean mass of Stim1kko mice was also significantly less versus the Kiss1Cre mice (Stim1kko vs the Kiss1Cre mice lean mass: 16.9 ± 0.4 g, n = 10 vs 18.9 ± 0.4 g, n = 10; Fig. 7D). After six weeks, both Stim1kko and Kiss1Cre controls were assessed for glucose tolerance using an intraperitoneal GTT (see Materials and Methods). Both Stim1kko and Kiss1Cre females started at relatively the same blood glucose levels after an overnight fast (Fig. 7E, time 0), suggesting similar whole-body homeostatic conditions after fasting. However, Stim1kko female mice had significantly lower glucose levels after intraperitoneal glucose compared with Kiss1Cre females, indicating that Stim1kko females were more glucose tolerant compared with Kiss1Cre controls. Stim1kko females had a significantly higher glucose clearance rate than controls based on the integrated AUC (Stim1kko vs the Kiss1Cre controls AUC: 20,232 ± 868 mg/dl/min, n = 6 vs 22,622 ± 624 mg/dl/min, n = 6). Finally, when both groups were euthanized after eight weeks on HFD and the tissues harvested, both the intrascapular brown adipose tissue (iBAT) and perigonadal adipose tissue (GAT) were dissected from each mouse and weighed. Both iBAT and GAT masses were significantly lighter in the Stim1kko versus the Kiss1Cre females (Stim1kko vs the Kiss1Cre iBAT: 73.3 ± 6.0 mg, n = 10 vs 97.3 ± 9.6 mg, n = 10; Stim1kko vs the Kiss1Cre GAT: 1.5 ± 0.2 g, n = 10 vs 2.3 ± 0.2 g, n = 10; Fig. 7F,G). Overall, these results suggest that conditional deletion of Stim1 in Kiss1ARH neurons affords some protection against diet-induced obesity. However, we cannot overlook the possibility that deletion of Stim1 in Kiss1-expressing hepatocytes might contribute to this metabolic phenotype (Song et al., 2014).
Figure 7.
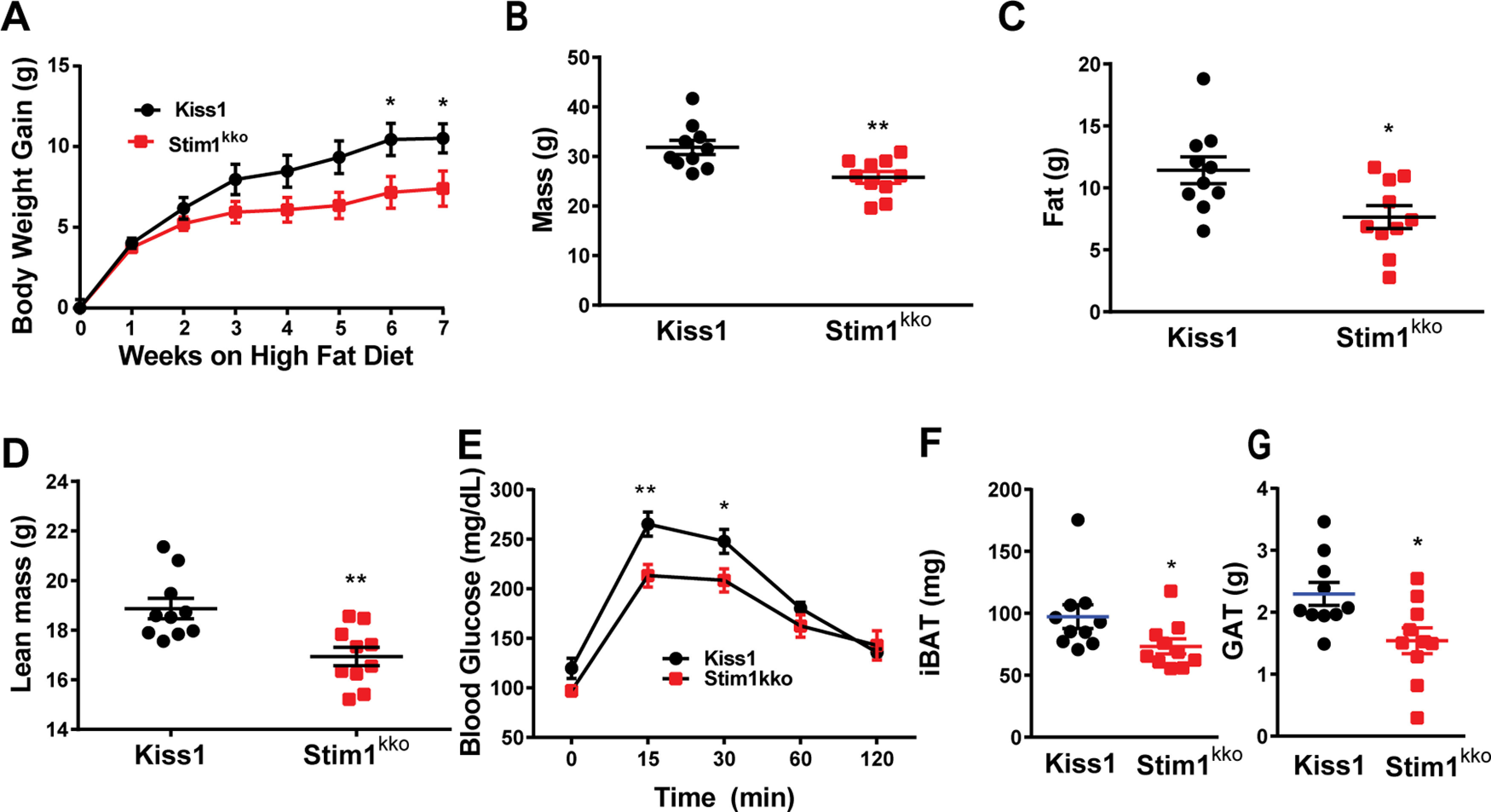
Ablation of Stim1 in Kiss1 neurons attenuates body mass, fat, and lean in mice on a HFD. Stim1kko and Kiss1Cre littermate control females were ovariectomized and fed a HFD (45% kcal from fat) for seven weeks. A, Body-weight gain measured once a week for seven weeks. The HFD caused significant weight gain in both groups relative to their baseline with the Kiss1Cre females gaining significantly more weight by six weeks [two-way ANOVA: main effect of treatment (F(1,18) = 3.839, p = 0.0657), main effect of time (F(7,126) = 98.07, p < 0.0001), and interaction (F(7,126) = 4.645, p = 0.0001); Kiss1 control, n = 10, Stim1kko, n = 10; post hoc Bonferroni test, *p < 0.05]. B–D, Mass (B), total body fat (C), and lean mass (D) measured by an EchoMRI whole-body composition analyzer. Lean mass did not include bone and fluids within organs. The difference in mass (B), body fat (C), and lean mass (D) between the groups was significantly different by six weeks on HFD (unpaired, two-tailed t test for B, t(18) = 3.222, **p = 0.0047; unpaired two-tailed t test for C, t(18) = 2.662, *p = 0.0159; unpaired, two-tailed t test for D, t(18) = 3.489, **p = 0.0026). E, Six weeks after HFD, there was a significant difference in GTTs between the two groups [two-way ANOVA: main effect of treatment (F(1,9) = 6.282, p = 0.0335), main effect of time (F(4,36) = 88.01, p < 0.0001) and interaction (F(4,36) = 3.527, p = 0.0158); Kiss1Cre, n = 6; Stim1kko, n = 5; post hoc Bonferroni test, **p < 0.01, *p < 0.05]. F, G, Both iBAT and GAT mass of Stim1kko were lighter than that of Kiss1Cre mice on a high fat diet after eight weeks (unpaired, two-tailed t test for iBAT, t(18) = 2.127, *p = 0.0475; unpaired two-tailed t test for GAT, t(18) = 2.711, *p = 0.0143).
Discussion
For the first time, we show that conditional knock-out of Stim1 significantly reduces SOCE in Kiss1ARH neurons following Tg-mediated depletion of Ca2+ stores. Based on single-cell qPCR analysis, Stim1 mRNA was expressed at approximately 2-fold higher levels than Stim2 in Kiss1ARH neurons, and deletion of Stim1 did not alter expression of Stim2 in Kiss1ARH neurons, i.e., there was no developmental compensation. Selective deletion of Stim1 in Kiss1ARH neurons augmented the TacR3-mediated increase in [Ca2+]i and synchronous activity of Kiss1ARH neurons by almost 4-fold. Whole-cell recording revealed that both the amplitude and the duration of the slow EPSP induced by high-frequency optogenetic stimulation of Kiss1ARH-ChR2 neurons were significantly enhanced by Stim1 deletion. This augmentation of the slow EPSP was mediated by TacR3 coupling to TRPC5 channel activation since the senktide-induced inward current was equally enhanced. Moreover, the inward current exhibited the signature double rectifying I–V plot of TRPC5 channels and was antagonized by both 2APB and the more selective TRPC 4/5 channel blocker HC070. Furthermore, chelating [Ca2+]i with BAPTA abolished the slow EPSP and persistent firing of Kiss1ARH neurons. Finally, the enhanced TacR3 signaling in Stim1kko female mice translated into more neuroprotection against diet-induced obesity and glucose intolerance.
Mammalian TRPC channels can be activated by G-protein-coupled receptors and receptor tyrosine kinases (Clapham, 2003; Ambudkar and Ong, 2007) and are one of the major targets for Group I metabotropic glutamate receptor (mGluR1) signaling in CNS neurons (Tozzi et al., 2003; Bengtson et al., 2004; Faber et al., 2006; Berg et al., 2007). In substantia nigra dopamine neurons TRPC5 channels are highly expressed, and mGluR1 agonists induce a current that exhibits a double-rectifying I–V plot (Tozzi et al., 2003) similar to the effects of the NKB agonist senktide in Kiss1ARH neurons (Fig. 4). Both mGluR1 and TacR3 are Gq-coupled to phospholipase C (PLC) activation which leads to hydrolyzis of phosphatidylinositol 4,5-biphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3). TRPC channels are cation selective and can associate with Orai calcium channels to form CRAC channels (Birnbaumer, 2009). TRPC5 channels are highly permeable to calcium (PCa/PNa = 9:1; Venkatachalam and Montell, 2007), and a unique feature of TRPC5 (and TRPC 4) channels is that they are potentiated by micromolar concentrations of lanthanum (La3+; Clapham et al., 2005), which we have exploited to characterize TPRC5 signaling in POMC neurons (Qiu et al., 2010, 2014).
Both leptin and insulin excite/depolarize Kiss1ARH and POMC neurons through activation of TRPC5 channels (Qiu et al., 2010, 2011, 2014; Kelly et al., 2018). More recently, we documented a critical role of STIM1 in the insulin signaling cascade in POMC neurons (Qiu et al., 2018b). Stim1 mRNA is highly expressed in POMC (Qiu et al., 2018b) and Kiss1ARH neurons (Fig. 1), and E2 downregulates Stim1 mRNA expression in microdissected arcuate nuclei that encompasses these two populations of neurons. Downregulation of Stim1 is critical for maintaining insulin excitability in POMC neurons with diet-induced obesity (Qiu et al., 2018b). In ovariectomized females that are relatively refractory to insulin excitation, pharmacological blockade of the SOCE complex quickly increases the insulin-mediated excitation of POMC neurons (i.e., activation of the TRPC5-mediated inward current), which supports the concept that TRPC5 channels play a role both in SOCE and receptor operated calcium entry (Birnbaumer, 2009; Salido et al., 2011). Therefore, selective deletion of Stim1 in Kiss1 neurons would ensure that TRPC5 channels function as receptor-operated channels to couple TacR3s and transmit the excitatory effects of NKB to induce synchronous firing of Kiss1ARH neurons as demonstrated in the present findings.
Downregulating STIM1 decreases the SERCA-dependent cytosolic Ca2+ clearance and elevates intracellular Ca2+ levels (Fig. 2; Ryu et al., 2017), which could also contribute to activation of TRPC5 channels in Kiss1ARH neurons (Blair et al., 2009). Indeed, we have found that Ca2+ significantly potentiates the leptin-induced TRPC5 current in POMC neurons (Qiu et al., 2010), which has recently been corroborated in primary cultures of POMC neurons (Perissinotti et al., 2021). What is impressive is that Stim1 deletion not only increased the amplitude, but it also prolonged the duration of the slow EPSP (Fig. 3). The slow EPSP is reminiscent of the “plateau potential” that has been described in hippocampal and cortical neurons (Zhang et al., 2011; Arboit et al., 2020). We know that electrical stimulation (20 Hz) cannot induce the slow EPSP, and the duration is not dependent on the AP firing per se since blockade of sodium channels with QX314, and hence abrogation of AP firing, does little to affect the duration of the depolarization (Qiu et al., 2016). Rather, many neurons express biophysical properties that allow them to continue to persistently fire even after a triggering synaptic event has subsided (Zylberberg and Strowbridge, 2017). Moreover, the intrinsic bi-stability of neurons that generates persistent firing activity has been linked to a calcium-activated, non-specific cation current (Zylberberg and Strowbridge, 2017), and TRPC channels, specifically TRPC5 channels, are thought to be responsible for the ICAN in cortical neurons (Zhang et al., 2011). Therefore, we hypothesized that with TacR3 activation there is an influx of Ca+2 through TRPC5 channels leading to greater build-up of [Ca2+]i (Fig. 2) that facilitates the opening of more TRPC5 channels in a self-sustaining manner. We tested this directly by using the fast intracellular calcium chelator BAPTA, which has been shown to robustly inhibit TRPC5 channel activation (Blair et al., 2009). Indeed, BAPTA essentially abolished the slow EPSP and persistent firing in Kiss1ARH neurons following optogenetic stimulation in both control and Stim1kko female mice (Fig. 5). Moreover, similar to cerebellar Purkinje neurons (Ryu et al., 2017), we measured a significant decrease in the input resistance with Stim1 deletion, which we believe reflects an increase in distribution of TRPC5 channels across the PM and coupling to Tac3 receptors (Fig. 8; Birnbaumer, 2009). A similar scenario occurs in cortical neurons and in heterologous cells expressing Cav1.2 (L-type calcium) channels and Stim1, where inhibition of STIM1 augments Ca2+ influx through L-type calcium channels (Park et al., 2010; Wang et al., 2010). Furthermore, deletion of Stim1 in cardiomyocyte-derived (HL-1) cells increases the peak amplitude and current density of T-type calcium channels and shifts the activation curve toward more negative membrane potentials (Nguyen et al., 2013). Biotinylation assays have revealed that deletion of Stim1 increases T-type calcium channel surface expression, and co-immunoprecipitation assays suggest that STIM1 directly regulates T-type channel activity (Nguyen et al., 2013). Thus, STIM1 appears to dampen the activity of voltage-gated calcium channels. Importantly, estradiol treatment downregulates Stim1 expression in the ARH of ovariectomized female mice and guinea pigs (Qiu et al., 2018b). In contrast, E2 upregulates Cav3.1 channel expression by 3-fold and whole cell currents by 10-fold in Kiss1ARH neurons, which greatly enhances the excitability and contributes to the synchronous firing of Kiss1ARH neurons (Qiu et al., 2018a). The T-type calcium channel Cav3.1 underlies burst firing in anteroventral periventricular preoptic (AVPV) kisspeptin neurons (Zhang et al., 2013b; Wang et al., 2016) and facilitates TRPC 4 channel activation in GnRH neurons (Zhang et al., 2008, 2013a). Calcium influx via Cav3.1 channels may also facilitate TRPC5 channel opening in Kiss1ARH neurons (Fig. 8), but this remains to be determined.
Figure 8.

A cellular model of STIM1 affecting NKB activation of TRPC5 channels in Kiss1ARH neurons. SOCE is a conserved mechanism by which the depletion of the ER calcium store is conveyed to calcium-permeable channels at the PM, triggering calcium influx from the extracellular space and into the cell cytosol. A physiological mechanism responsible for the activation of SOCE results from the stimulation of G-protein-coupled receptors associated with the IP3 and phospholipase C cascade, resulting in the release of calcium from ER, via the IP3 receptor (IP3R). A, Under physiological stress and in the absence of E2, STIM1 interacts with TPRC5 channels thereby engaging these Ca2+ channels as store-operated channels, which are activated with ER depletion of Ca2+. B, However, under physiological conditions in reproductively active females, in which E2 downregulates the expression of STIM1, TRPC5 channels are converted to receptor-operated channels in Kiss1ARH neurons. NKB binds to its receptor (TacR3) to activate Gαq–PLCβ signaling cascade to facilitate TPRC5 channel opening, generating a robust inward Ca2+/Na+ current to depolarize Kiss1ARH neurons, activating T-type calcium (Cav3.1) channels to greatly increase Kiss1ARH neuronal excitability.
Presumably with conditional knock-out, Stim1 was deleted in all cells expressing kisspeptin, which includes arcuate, AVPV and amygdala kisspeptin neurons, and non-neural kisspeptin cells in the gonads, pancreas and liver (Dudek et al., 2019). Currently, we found that the deletion of Stim1 in kisspeptin neurons had a minor effect on the estrous cycle. Stim1kko mice exhibited more estrous-type vaginal cytology, which may be indicative of higher levels of circulating estrogens because of increased synchronous firing of kisspeptin neurons and excitatory drive to GnRH neurons (Qiu et al., 2016; Clarkson et al., 2017). It is important to note that synchronous firing of “pulse-generator” Kiss1ARH neurons is a failsafe system for maintaining gonadotropin pulses and folliculogenesis in female rodents (Nagae et al., 2021).
Because of the well-documented anorexigenic actions of E2 on POMC and Agouti-related peptide (AgRP) neurons controlling energy homeostasis (Qiu et al., 2006; Roepke et al., 2010; Clegg, 2012; Kelly and Rønnekleiv, 2012; Smith et al., 2013), we ovariectomized the females before feeding them a HFD. After seven weeks on a HFD, Stim1kko females gained less body weight and showed significantly less body fat and lean mass than ovariectomized Kiss1Cre females on a HFD. Most importantly, Stim1kko females exhibited improved glucose tolerance. Kiss1ARH neurons probably mediate these protective effects via their input onto POMC and AgRP neurons. Besides the peptides Kiss1ARH neurons also co-express the vesicular glutamate transporter 2 (vGluT2; Cravo et al., 2011; Nestor et al., 2016), and we have documented that optogenetic stimulation of Kiss1ARH neurons expressing ChR2 releases glutamate, which is dependent on the estrogenic state of females (Qiu et al., 2018a). Although the mRNA expression of Kiss1, Tac2 and Pdyn mRNA in Kiss1ARH neurons are all downregulated by E2 (Navarro et al., 2009; Lehman et al., 2010), Vglut2 mRNA expression is upregulated together with increased probability of glutamate release in E2-treated, ovariectomized females (Qiu et al., 2018a). Low-frequency (1–2 Hz) optogenetic stimulation of Kiss1ARH neurons evokes fast ionotropic glutamatergic EPSCs in POMC and AgRP neurons, but high-frequency (20 Hz) optogenetic stimulation releases enough glutamate to induce a slow excitatory response in POMC neurons but a slow inhibitory response in AgRP neurons (Nestor et al., 2016; Qiu et al., 2016, 2018a). Indeed, the Group I mGluR agonist DHPG depolarizes POMC neurons, while Group II/III mGluR agonists (DCG-IV; AMN082) hyperpolarize AgRP neurons (Qiu et al., 2018a). Group I mGluRs (mGluR1 and mGluR5) are Gq/G11-coupled, while Group II/III mGluRs (mGluR2 and mGluR7) are Gi/Go-coupled (Niswender and Conn, 2010). Hence, the output of Kiss1ARH neurons excites the anorexigenic POMC neurons and inhibits the orexigenic AgRP neurons. Therefore, Kiss1ARH neurons appear to be an integral part of an anorexigenic circuit in the hypothalamus (Qiu et al., 2018a; Rønnekleiv et al., 2019; Navarro, 2020).
Presently, there is compelling evidence that Kiss1ARH neurons are a critical “command” neuron for coordinating energy states with reproductive functions (for review, see Rønnekleiv et al., 2019; Navarro, 2020). We have now documented that conditional knock-out of Stim1 in Kiss1ARH neurons, which augments the NKB-mediated depolarization of these neurons via TRPC5 channels, helps protect ovariectomized, female mice from diet-induced obesity and glucose intolerance. In addition, in preliminary experiments we have found that insulin treatment in vitro increases the synchronous firing (GCaMP6 activity) of Kiss1ARH neurons, which further emphasizes its role as a command neuron. Clearly, Kiss1ARH neurons are part of a hypothalamic circuit for coordinating reproduction with energy balance, but additional experiments are needed to elucidate the cellular mechanisms by which steroid and metabolic hormonal signaling synergize to govern their activity.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS043330 (to O.K.R.), R01-NS038809 (to M.J.K.), and R01-DK068098 (to O.K.R. and M.J.K.). Confocal microscopy was supported by the NIH Grant P30 NS061800 (PI, S. Aicher). We thank Mr. Daniel Johnson for his technical support.
The authors declare no competing financial interests.
References
- Ambudkar IS, Ong HL (2007) Organization and function of TRPC channelosomes. Pflugers Arch 455:187–200. 10.1007/s00424-007-0252-0 [DOI] [PubMed] [Google Scholar]
- Arboit A, Reboreda A, Yoshida M (2020) Involvement of TRPC4 and 5 channels in persistent firing in hippocampal CA1 pyramidal cells. Cells 9:365. 10.3390/cells9020365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asarian L, Geary N (2006) Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci 361:1251–1263. 10.1098/rstb.2006.1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP; NIH Mouse Metabolic Phenotyping Center Consortium (2010) Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3:525–534. 10.1242/dmm.006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Tozzi A, Bernardi G, Mercuri NB (2004) Transient receptor potential-like channels mediate metabotropic glutamate receptor EPSCs in rat dopamine neurones. J Physiol 555:323–330. 10.1113/jphysiol.2003.060061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AP, Sen N, Bayliss DA (2007) TrpC3/C7 and Slo2.1 are molecular targets for metabotropic glutamate receptor signaling in rat striatal cholinergic interneurons. J Neurosci 27:8845–8856. 10.1523/JNEUROSCI.0551-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B (2009) STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal 2:ra67. 10.1126/scisignal.2000522 [DOI] [PubMed] [Google Scholar]
- Birnbaumer L (2009) The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol 49:395–426. 10.1146/annurev.pharmtox.48.113006.094928 [DOI] [PubMed] [Google Scholar]
- Blair NT, Kaczmarek JS, Clapham DE (2009) Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J Gen Physiol 133:525–546. 10.1085/jgp.200810153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch MA, Tonsfeldt KJ, Rønnekleiv OK (2013) mRNA expression of ion channels in GnRH neurons: subtype-specific regulation by 17β-estradiol. Mol Cell Endocrinol 367:85–97. 10.1016/j.mce.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Calì T, Ottolini D, Carafoli E (2014) Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci 71:2787–2814. 10.1007/s00018-013-1550-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Tena-Sempere M (2013) Metabolic regulation of kisspeptin. Adv Exp Med Biol 784:363–383. 10.1007/978-1-4614-6199-9_17 [DOI] [PubMed] [Google Scholar]
- Clapham DE (2003) TRP channels as cellular sensors. Nature 426:517–524. 10.1038/nature02196 [DOI] [PubMed] [Google Scholar]
- Clapham DE, Julius D, Montell C, Schultz G (2005) International union of pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev 57:427–450. 10.1124/pr.57.4.6 [DOI] [PubMed] [Google Scholar]
- Clarkson J, Han SY, Piet R, McLennan T, Kane GM, Ng J, Porteous RW, Kim JS, Colledge WH, Iremonger KJ, Herbison AE (2017) Definition of the hypothalamic GnRH pulse generator in mice. Proc Natl Acad Sci USA 114:E10216–E10223. 10.1073/pnas.1713897114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg DJ (2012) Minireview: the year in review of estrogen regulation of metabolism. Mol Endocrinol 26:1957–1960. 10.1210/me.2012-1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravo RM, Margatho LO, Osborne-Lawrence S, Donato JJ, Atkin S, Bookout AL, Rovinsky S, Frazão R, Lee CE, Gautron L, Zigman JM, Elias CF (2011) Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience 173:37–56. 10.1016/j.neuroscience.2010.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja JA (1978) Ovarian influences on primate food intake: assessment of progesterone actions. Physiol Behav 21:923–928. 10.1016/0031-9384(78)90167-1 [DOI] [PubMed] [Google Scholar]
- d'Anglemont de Tassigny X, Fagg LA, Dixon JPC, Day K, Leitch HG, Hendrick AG, Zahn D, Franceschini I, Caraty A, Carlton MBL, Aparicio SAJR, Colledge WH (2007) Hypogonadotropic hypogonadism in mice lacking a functional KiSS 1 gene. Proc Natl Acad Sci USA 104:10714–10719. 10.1073/pnas.0704114104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Croft S, Boehm U, Herbison AE (2013) Neurokinin B activates arcuate kisspeptin neurons through multiple tachykinin receptors in the male mouse. Endocrinology 154:2750–2760. 10.1210/en.2013-1231 [DOI] [PubMed] [Google Scholar]
- De Roux N, Genin E, Carel J-C, Matsuda F, Chaussain JL, Milgrom E (2003) Hypogonadotropic hypogonadism due to loss of function of the KiSS 1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972–10976. 10.1073/pnas.1834399100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek M, Ziarniak K, Cateau M-L, Dufourny L, Sliwowska JH (2019) Diabetes type 2 and kisspeptin: central and peripheral sex-specific actions. Trends Endocrinol Metab 30:833–843. 10.1016/j.tem.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Faber ES, Sedlak P, Vidovic M, Sah P (2006) Synaptic activation of transient receptor potential channels by metabotropic glutamate receptors in the lateral amygdala. Neuroscience 137:781–794. 10.1016/j.neuroscience.2005.09.027 [DOI] [PubMed] [Google Scholar]
- Gao Y, Yao T, Deng Z, Sohn JW, Sun J, Huang Y, Kong X, Yu KJ, Wang RT, Chen H, Guo H, Yan J, Cunningham KA, Chang Y, Liu T, Williams KW (2017) TrpC5 mediates acute leptin and serotonin effects via Pomc neurons. Cell Rep 18:583–592. 10.1016/j.celrep.2016.12.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CVR, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, Clarke IJ (2007) Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 148:5752–5760. 10.1210/en.2007-0961 [DOI] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J, Pomorski P, Wisniewska MB, Kuznicki J (2011) Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS One 6:e19285. 10.1371/journal.pone.0019285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guner G, Guzelsoy G, Isleyen FS, Sahin GS, Akkaya C, Bayam E, Kotan EI, Kabakcioglu A, Ince-Dunn G (2017) NEUROD2 regulates Stim1 expression and store-operated calcium entry in cortical neurons. eNeuro 4:ENEURO.0255-0216.2017. 10.1523/ENEURO.0255-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE (2005) Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci 25:11349–11356. 10.1523/JNEUROSCI.3328-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Rühlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T, Konnerth A (2014) STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82:635–644. 10.1016/j.neuron.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Just S, Chenard BL, Ceci A, Strassmaier T, Chong JA, Blair NT, Gallaschun RJ, Del Camino D, Cantin S, D'Amours M, Eickmeier C, Fanger CM, Hecker C, Hessler DP, Hengerer B, Kroker KS, Malekiani S, Mihalek R, McLaughlin J, Rast G, et al. (2018) Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLoS One 13:e0191225. 10.1371/journal.pone.0191225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Rønnekleiv OK (2012) Membrane-initiated actions of estradiol that regulate reproduction, energy balance and body temperature. Front Neuroendocrinol 33:376–387. 10.1016/j.yfrne.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Qiu J, Rønnekleiv OK (2018) TRPCing around the hypothalamus. Front Neuroendocrinol 51:116–124. 10.1016/j.yfrne.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuohung W, Kaiser UB (2006) GPR54 and KiSS-1: role in the regulation of puberty and reproduction. Rev Endocr Metab Disord 7:257–263. 10.1007/s11154-006-9020-2 [DOI] [PubMed] [Google Scholar]
- Lehman MN, Coolen LM, Goodman RL (2010) Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 151:3479–3489. 10.1210/en.2010-0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta CT) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YWA, Garcia AJ, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsáki G, Ramirez JM, Jones AR, Svoboda K, Han X, et al. (2012) A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15:793–802. 10.1038/nn.3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, Levine JE (2010) Timing and completion of puberty in female mice depend on estrogen receptor α-signaling in kisspeptin neurons. Proc Natl Acad Sci USA 107:22693–22698. 10.1073/pnas.1012406108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moccia F, Zuccolo E, Soda T, Tanzi F, Guerra G, Mapelli L, Lodola F, D'Angelo E (2015) Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front Cell Neurosci 9:153. 10.3389/fncel.2015.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Uenoyama Y, Okamoto S, Tsuchida H, Ikegami K, Goto T, Majarune S, Nakamura S, Sanbo M, Hirabayashi M, Kobayashi K, Inoue N, Tsukamura H (2021) Direct evidence that KNDy neurons maintain gonadotropin pulses and folliculogenesis as the GnRH pulse generator. Proc Natl Acad Sci USA 118:e2009156118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM (2020) Metabolic regulation of kisspeptin — the link between energy balance and reproduction. Nat Rev Endocrinol 16:407–420. 10.1038/s41574-020-0363-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA (2009) Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci 29:11859–11866. 10.1523/JNEUROSCI.1569-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM, Castellano JM, McConkey SM, Pineda R, Ruiz-Pino F, Pinilla L, Clifton DK, Tena-Sempere M, Steiner RA (2011) Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab 300:E202–E210. 10.1152/ajpendo.00517.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor CC, Kelly MJ, Rønnekleiv OK (2014) Cross-talk between reproduction and energy homeostasis: central impact of estrogens, leptin and kisspeptin signaling. Horm Mol Biol Clin Investig 17:109–128. 10.1515/hmbci-2013-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor CC, Qiu J, Padilla SL, Zhang C, Bosch MA, Fan W, Aicher SA, Palmiter RD, Rønnekleiv OK, Kelly MJ (2016) Optogenetic stimulation of arcuate nucleus Kiss1 neurons reveals a steroid-dependent glutamatergic input to POMC and AgRP neurons in male mice. Mol Endocrinol 30:630–644. 10.1210/me.2016-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N, Biet M, Simard E, Béliveau E, Francoeur N, Guillemette G, Dumaine R, Grandbois M, Boulay G (2013) STIM1 participates in the contractile rhythmicity of HL-1 cells by moderating T-type Ca(2+) channel activity. Biochimica et Biophysica Acta 1833:1294–1303. 10.1016/j.bbamcr.2013.02.027 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A (2008) Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9:432–443. 10.1038/ni1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H, Tsukamura H, Ohkura S, Uenoyama Y, Wakabayashi Y, Maeda K (2013) Kisspeptin and GnRH pulse generation. Adv Exp Med Biol 784:297–323. 10.1007/978-1-4614-6199-9_14 [DOI] [PubMed] [Google Scholar]
- Padilla SL, Johnson CW, Barker FD, Patterson MA, Palmiter RD (2018) A neural circuit underlying the generation of hot flushes. Cell Rep 24:271–277. 10.1016/j.celrep.2018.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Shcheglovitov A, Dolmetsch R (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330:101–105. 10.1126/science.1191027 [DOI] [PubMed] [Google Scholar]
- Pavez M, Thompson AC, Arnott HJ, Mitchell CB, D'Atri I, Don EK, Chilton JK, Scott EK, Lin JY, Young KM, Gasperini RJ, Foa L (2019) STIM1 is required for remodeling of the endoplasmic reticulum and microtubule cytoskeleton in steering growth cones. J Neurosci 39:5095–5114. 10.1523/JNEUROSCI.2496-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissinotti PP, Martínez-Hernández E, Piedras-Rentería ES (2021) TRPC1/5-Cav3 complex mediates leptin-induced excitability in hypothalamic neurons. Front Neurosci 15:679078. 10.3389/fnins.2021.679078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielecka-Fortuna J, Chu Z, Moenter SM (2008) Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology 149:1979–1986. 10.1210/en.2007-1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, Kelly MJ (2003) Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci 23:9529–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Krust A, Graham S, Murphy S, Korach KS, Chambon P, Scanlan TS, Rønnekleiv OK, Kelly MJ (2006) A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci 26:5649–5655. 10.1523/JNEUROSCI.0327-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ (2010) Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci 30:1560–1565. 10.1523/JNEUROSCI.4816-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Fang Y, Bosch MA, Rønnekleiv OK, Kelly MJ (2011) Guinea pig kisspeptin neurons are depolarized by leptin via activation of TRPC channels. Endocrinology 152:1503–1514. 10.1210/en.2010-1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Zhang C, Borgquist A, Nestor CC, Smith AW, Bosch MA, Ku S, Wagner EJ, Rønnekleiv OK, Kelly MJ (2014) Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab 19:682–693. 10.1016/j.cmet.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Nestor CC, Zhang C, Padilla SL, Palmiter RD, Kelly MJ, Rønnekleiv OK (2016) High-frequency stimulation-induced peptide release synchronizes arcuate kisspeptin neurons and excites GnRH neurons. Elife 5:e16246. 10.7554/eLife.16246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Rivera HM, Bosch MA, Padilla SL, Stincic TL, Palmiter RD, Kelly MJ, Rønnekleiv OK (2018a) Estrogenic-dependent glutamatergic neurotransmission from kisspeptin neurons governs feeding circuits in females. Elife 7:e35656. 10.7554/eLife.35656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Meza C, Navarro UV, Nestor CC, Wagner EJ, Rønnekleiv OK, Kelly MJ (2018b) Estradiol protects proopiomelanocortin neurons against insulin resistance. Endocrinology 159:647–664. 10.1210/en.2017-00793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TA, Bosch MA, Rick EA, Lee B, Wagner EJ, Seidlova-Wuttke D, Wuttke W, Scanlan TS, Rønnekleiv OK, Kelly MJ (2010) Contribution of a membrane estrogen receptor to the estrogenic regulation of body temperature and energy homeostasis. Endocrinology 151:4926–4937. 10.1210/en.2010-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rønnekleiv OK, Qiu J, Kelly MJ (2019) Arcuate kisspeptin neurons coordinate reproductive activities with metabolism. Semin Reprod Med 37:131–140. 10.1055/s-0039-3400251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruka KA, Burger LL, Moenter SM (2013) Regulation of arcuate neurons coexpressing kisspeptin, neurokinin B, and dynorphin by modulators of neurokinin 3 and κ-opioid receptors in adult male mice. Endocrinology 154:2761–2771. 10.1210/en.2013-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu C, Jang DC, Jung D, Kim YG, Shim HG, Ryu HH, Lee YS, Linden DJ, Worley PF, Kim SJ (2017) STIM1 regulates somatic Ca2+ signals and intrinsic firing properties of cerebellar Purkinje neurons. J Neurosci 37:8876–8894. 10.1523/JNEUROSCI.3973-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salido GM, Jardín I, Rosado JA (2011) The TRPC ion channels: association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv Exp Med Biol 704:413–433. 10.1007/978-94-007-0265-3_23 [DOI] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MBL, Crowley WF, Aparicio SAJR, Colledge WH (2003) The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627. 10.1056/NEJMoa035322 [DOI] [PubMed] [Google Scholar]
- Smith AW, Bosch MA, Wagner EJ, Rønnekleiv OK, Kelly MJ (2013) The membrane estrogen receptor ligand STX rapidly enhances GABAergic signaling in NPY/AgRP neurons: role in mediating the anorexigenic effects of 17β-estradiol. Am J Physiol Endocrinol Metab 305:E632–E640. 10.1152/ajpendo.00281.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram A, Shum AK, McBride HJ, Kessler JA, Feske S, Miller RJ, Prakriya M (2014) Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J Neurosci 34:9107–9123. 10.1523/JNEUROSCI.0263-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Mondal P, Wolfe A, Alonso LC, Stamateris R, Ong BW, Lim OC, Yang KS, Radovick S, Novaira HJ, Farber EA, Farber CR, Turner SD, Hussain MA (2014) Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab 19:667–681. 10.1016/j.cmet.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhang H, Selvaraj S, Sukumaran P, Lei S, Birnbaumer L, Singh BB (2017) Inhibition of L-Type Ca2+ channels by TRPC1-STIM1 complex is essential for the protection of dopaminergic neurons. J Neurosci 37:3364–3377. 10.1523/JNEUROSCI.3010-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson KP, Garcia C, Yen S, Simonds S, Stefanidis A, Lawrence A, Smith JT, Kauffman AS (2014) Impaired kisspeptin signaling decreases metabolism and promotes glucose intolerance and obesity. J Clin Invest 124:3075–3079. 10.1172/JCI71075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson KP, Marooki N, Wolfe A, Smith JT, Kauffman AS (2019) Cre/lox generation of a novel whole-body Kiss1r KO mouse line recapitulates a hypogonadal, obese, and metabolically-impaired phenotype. Mol Cell Endocrinol 498:110559. 10.1016/j.mce.2019.110559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, Gurbuz F, Temiz F, Millar RP, Yuksel B (2012) Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med 366:629–635. 10.1056/NEJMoa1111184 [DOI] [PubMed] [Google Scholar]
- Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB (2003) Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci 18:2133–2145. 10.1046/j.1460-9568.2003.02936.x [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387–417. 10.1146/annurev.biochem.75.103004.142819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, DeFazio RA, Moenter SM (2016) Excitability and burst generation of AVPV kisspeptin neurons are regulated by the estrous cycle via multiple conductances modulated by estradiol action. eNeuro 3:ENEURO.0094-16.2016. 10.1523/ENEURO.0094-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330:105–109. 10.1126/science.1191086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9:636–645. 10.1038/ncb1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK (2008) Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci 28:4423–4434. 10.1523/JNEUROSCI.5352-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reboreda A, Alonso A, Barker PA, Séguéla P (2011) TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 21:386–397. 10.1002/hipo.20755 [DOI] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ (2013a) Kisspeptin activation of TRPC4 channels in female GnRH neurons requires PIP2 depletion and cSrc kinase activation. Endocrinology 154:2772–2783. 10.1210/en.2013-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Tonsfeldt KJ, Qiu J, Bosch MA, Kobayashi K, Steiner RA, Kelly MJ, Rønnekleiv OK (2013b) Molecular mechanisms that drive estradiol-dependent burst firing of Kiss1 neurons in the rostral periventricular preoptic area. Am J Physiol Endocrinol Metab 305:E1384–E1397. 10.1152/ajpendo.00406.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberberg J, Strowbridge BW (2017) Mechanisms of persistent activity in cortical circuits: possible neural substrates for working memory. Annu Rev Neurosci 40:603–627. 10.1146/annurev-neuro-070815-014006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NKB receptor agonist senktide induces [Ca2+]i increase in Kiss1ARH neurons expressing GCaMP6s. Imaging of transient Ca2+ changes in an arcuate slice using spinning disk confocal microscopy. Fluorescence intensity was measured over 20 min, before and after application of senktide (1 µm). The period represented is 20 min.
High-frequency photostimulation induces a slow EPSP. Slow EPSP was induced by a 10-s 20-Hz photostimulation (light intensity 0.9 mW and pulse duration, 10 ms) in a ChR2-expressing Kiss1ARH neuron in a slice from a Kiss1Cre::Ai32 mouse. The period represented is 1 min, 34 s.