

Abstract
Chronic hypoxia is a common cause of pulmonary hypertension, preeclampsia, and intrauterine growth restriction (IUGR). The molecular mechanisms underlying these diseases are not completely understood. Chronic hypoxia may induce the generation of reactive oxygen species (ROS) in mitochondria, promote endoplasmic reticulum (ER) stress, and result in the integrated stress response (ISR) in the pulmonary artery and uteroplacental tissues. Numerous studies have implicated hypoxia-inducible factors (HIFs), oxidative stress, and ER stress/unfolded protein response (UPR) in the development of pulmonary hypertension, preeclampsia and IUGR. This review highlights the roles of HIFs, mitochondria-derived ROS and UPR, as well as their interplay, in the pathogenesis of pulmonary hypertension and preeclampsia, and their implications in drug development.
Keywords: hypoxia, pulmonary hypertension, preeclampsia, reactive oxygen species, mitochondria, endoplasmic reticulum, unfolded protein response, integrated stress response, vascular remodeling
Teaser:—
The integrated stress response (ISR) is a major contributor to pulmonary hypertension and preeclampsia, and a potential therapeutic target for these two diseases.
Introduction
Oxygen (O2) is essential to sustain mammalian life. O2 is primarily utilized for energy generation and for biomolecule synthesis via oxidation-reduction reactions. Many biological processes in mammalian cells are dependent on the ATP produced by the electron transport chain (ETC) in mitochondria, with O2 being the terminal electron acceptor in this chain. O2 is also a pivotal substrate in protein synthesis and protein folding in the endoplasmic reticulum (ER). Both oxidative phosphorylation and oxidative protein folding are coupled to the generation of reactive oxygen species (ROS) [1, 2]. A reduction of O2 supply (i.e. hypoxia) disrupts mitochondrial and ER functions. Hypoxia has been shown to alter the homeostasis of mitochondrial ROS (mitoROS) and to induce ER stress [3]. To respond to hypoxic stress, cells undergo adaptive responses that are primarily mediated by the activation of hypoxia-inducible factors (HIFs), reprogramming of mitochondrial metabolism, increased ROS flux, an unfolded protein response (UPR) in the ER, and the subsequent integrated stress response (ISR). HIFs are transcriptional activators that function as master regulators of hypoxia-activated gene expression, whereas the UPR/ISR attempts to restore proteostasis. Interestingly, HIFs, ROS and UPR/ISR are mutually connected [3, 4]. When the production of HIF and ROS and the UPR/ISR become sustained, they are deleterious and disrupt cell functions.
In mammals, the lung extracts oxygen from the air and transfers it into the bloodstream, and discharges carbon dioxide from the bloodstream into the air. In utero, fetal respiratory function is executed by the placenta where gas exchange occurs between maternal and fetal blood. The pulmonary and uteroplacental circulatory systems share some similarities. Both are low-pressure, high-flow systems [5, 6]. Upon exposure to acute hypoxia, systemic arteries and arterioles dilate, whereas pulmonary and uteroplacental arteries and arterioles constrict, leading to increased vascular resistance and reduced blood flow [7, 8].
Pulmonary hypertension is a disorder of pulmonary arteries, characterized by a mean pulmonary arterial pressure ≥20 mm Hg [9]. The disorder is classified into five groups based on the proceedings of the 6th World Symposium on Pulmonary Hypertension (WSPH), held in 2019 [9]. Among these categories, Group III involves cases of pulmonary hypertension that are caused by lung diseases and/or hypoxia, including obstructive lung disease, restrictive lung disease, other lung disease with mixed restrictive/obstructive pattern, hypoxia without lung disease and developmental lung disorders. In pulmonary hypertension, the pulmonary arteries are constricted and/or obstructed due to vasoconstriction and vascular remodeling, leading to increased pulmonary vascular resistance and pulmonary pressure. The remodeling of pulmonary arteries (a pathological process) primarily involves the proliferation of vascular smooth muscle cells (VSMCs) [10]. Pulmonary hypertension often leads to right ventricular overload, and heart failure is the most common cause of mortality in patients who have pulmonary hypertension[11]. The incidence, prevalence, and mortality of patients who have pulmonary hypertension are increasing[12].
Adequate uteroplacental blood supply to the placenta is pivotal for the development and growth of both the placenta and the fetus. The increase in uteroplacental blood flow during pregnancy is mainly achieved by structural and functional adaptation of uterine arteries[13, 14]. One critical process in uteroplacental vascular adaptation is the remodeling of the uterine radial arteries (a physiological process). During this process, invading extravillous trophoblasts (EVTs) replace endothelial cells and VSMCs from the arterial walls, transforming the arteries into widened, low-resistance vascular channels. Preeclampsia is a common pregnancy complication originated in the placenta, in which there is new onset of hypertension after 20 weeks of pregnancy. It affects ~5% of pregnant women worldwide and is associated with high maternal and fetal morbidity and mortality[15]. This disorder is currently defined as elevated systolic and diastolic blood pressure (≥140 and ≥90 mmHg, respectively) with one or more of following criteria: proteinuria, other maternal organ dysfunction, and/or uteroplacental dysfunction[16]. In preeclampsia, uterine vascular remodeling is incomplete and uterine vascular adaptation is compromised, resulting in increased vascular resistance and reduced blood flow in the uteroplacental circulation[17]. Uteroplacental blood flow is a critical determinant of fetal growth and health [18], and preeclampsia is frequently complicated by intrauterine growth restriction (IUGR) [19].
Preeclampsia presents in heterogeneous forms and is commonly classified into early-onset (delivery before 34 weeks’ gestation) and late-onset (delivery after 34 weeks’ gestation) according to the time of delivery. Early-onset preeclampsia accounts for ≤20% of all preeclampsia cases, and is typically associated with placental dysfunction (i.e., insufficient trophoblast invasion, reduced spiral artery remodeling and placental malperfusion), hypoxia/ischemia-reperfusion injury, and IUGR. By contrast, late-onset preeclampsia, comprising ≥80% preeclampsia cases, is believed to result from maternal factors and is often associated with normal placental and fetal growth [20]. Late-onset preeclampsia frequently occurs in women with pregestational obesity and diabetes who may have existing chronic systematic inflammation [21, 22]. Intriguingly, early-onset preeclamptic placentas exhibit reduced antioxidant capacity, whereas placentas of late-onset preeclampsia display mitochondria-related adaptions and compensatory antioxidant responses [23]. Moreover, placentas of late-onset preeclampsia are unable to respond to hypoxia and display no apparent activation of the UPR when compared to normotensive placentas [24, 25]. Early-onset preeclampsia is the most clinically important form because it contributes to most of the maternal and perinatal morbidity and mortality [26–28]. Given the scope of this review, we primarily discuss early-onset preeclampsia herein.
Pulmonary hypertension and preeclampsia are two distinct diseases. Their phenotypes and biochemistries are compared in Table 1. Nevertheless, hypoxia is a common contributor to the pathogenesis of both diseases. Circulating endothelin-1 and its receptors in VSMCs are increased in pulmonary hypertension, in preeclampsia and in animal models of these diseases [29, 30]. Moreover, prolonged hypoxia impairs pulmonary and uteroplacental function, resulting in pulmonary hypertension and preeclampsia, respectively [14, 31]. HIFs, ROS and UPR are all induced or activated in these two disorders [32–35]. This review summarizes our knowledge of the roles of HIFs, ROS, UPR and their interplay in the ISR and in the pathogenesis of hypoxia-related pulmonary hypertension and preeclampsia. Potential therapeutic approaches for pulmonary hypertension and preeclampsia are also discussed.
Table 1.
Comparison of phenotype and biochemistry between pulmonary hypertension and preeclampsia.
| Pulmonary hypertension | Preeclampsia | |
|---|---|---|
| Local vascular resistance |
|
|
| Systemic blood pressure |
|
|
| Vascular remodeling |
|
|
| Complications |
|
|
| Endothelin-1 |
|
|
| Hypoxia, hypoxia inducible factor (HIFs) and microRNA-210 (miR-210) |
|
|
| Reactive oxygen species (ROS) and oxidative stress |
|
|
| ER stress and the unfolded protein response (UPR) |
|
|
Roles of oxidative stress and stress responses in the mitochondria and ER ROS and oxidative stress
ROS are products of O2 metabolism in cells. Common ROS include the superoxide (O2•−), hydrogen peroxide (H2O2) and the hydroxyl radical (HO•). In general, O2•−, which is the precursor of most other ROS, is produced by the reduction of molecular O2. The major sources of ROS in mammalian cells are mitochondria, ER, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase s (NOXs). Redox hemostasis is maintained by a delicate balance of the generation and destruction of ROS. Oxidative stress occurs when ROS generation overwhelms the antioxidant defense. ROS can be detrimental or beneficial to cellular functions depending on their levels in the cells. ROS may modulate cellular homeostasis through direct oxidative damage and by altering signal transduction pathways. Owing to their extreme reactivity, O2•− and HO• often cause lipid, protein, and DNA damage when present at high levels. H2O2 can alter the conformation and/or activity of target proteins, such as enzymes and transcription factors, by oxidizing cysteine thiols (Cys-SH) to cysteine sulfenic acids (Cys-SOH) [36].
ER stress and the UPR
Only properly folded proteins are allowed to exit the ER. The accumulation of unfolded or misfolded proteins in the ER lumen promotes ER stress, and activates the UPR to mitigate this stress and to restore protein-folding capacity. The UPR can be activated in various cellular compartments or organelles. To date, a UPR has been identified in the ER, mitochondria, and cytoplasm [37, 38], but only the ER UPR is discussed in this review. In the ER, the UPR is mediated by three types of ER transmembrane receptor: protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1α(IRE1α) and activating transcription factor 6 (ATF6) [39]. The luminal domains of these proteins sense the protein folding status of the ER. Under normal conditions, these sensors are kept in the inactive state by forming a complex with the ER chaperone BiP/Grp78 (binding immunoglobulin protein/78-kDa glucose-regulated protein). However, the accumulation of unfolded and/or misfolded proteins in the ER lumen causes the dissociation of BiP/Grp78 from the sensors, leading to changes in the oligomerization state of the sensors and to downstream signaling activities. PERK activation promotes the global inhibition of translation through the phosphorylation and subsequent inactivation of eukaryotic translation initiation factor 2α(eIF2α), which results in reduced protein synthesis and misfolded protein load. Paradoxically, eIF2α phosphorylation also triggers the translation of ATF4, which in turn stimulates the transcription of a variety of genes, including those that boost antioxidant defense mechanisms [40]. During ER stress, the phosphorylation of nuclear factor 2 (Nrf2), mediated by PERK, promotes the dissociation of Nrf2 from Kelch-like ECH-associated protein 1 (KEAP1) in the cytosol, enabling Nrf2 to enter the nucleus where it regulates genes encoding antioxidant proteins [41]. ATF4 also activates the transcription of C/EBP homologous protein (CHOP) during chronic ER stress. The dissociation of BiP/Grp78 promotes IRE1α trans-autophosphorylation and its endoribonuclease activity, generating the stable transcription factor X-box binding protein 1 (XBP1) and thus upregulating UPR-targeted genes including BiP/Grp78 and components of ER-associated degradation (ERAD). Following the release of BiP/Grp78 from its luminal domain, ATF6 transits to the Golgi apparatus where it is cleaved by site-specific proteases S1P and S2P. The released cytosolic fragment, ATF6f, migrates to the nucleus where it activates the transcription of genes encoding various ER chaperones.
ISR
The ISR is a signaling pathway in mammalian cells that regulates gene expression by reprogramming mRNA translation. A variety of extrinsic or intrinsic stresses, such as hypoxia, amino acid deprivation, heme deprivation, oxidative stress, and viral infection, are sensed by eIF2 kinases, which include double-stranded RNA-dependent protein kinase (PKR), PERK, general control nonderepressible 2 (GCN2) and heme-regulated inhibitor (HRI) [42]. PKR is activated primarily by double-stranded RNA during viral infection. The accumulation of unfolded protein in ER triggers PERK activation. GCN2 is activated by amino acid deprivation and by UV light. HRI, which is mainly expressed in erythroid cells, is activated in response to heme deficiency and oxidative stress [42, 43].
The activation of these eIF2 kinases converges on the phosphorylation of serine 51 in eIF2α, leading to the ISR. eIF2 is a heterotrimer composed of an α-, a β-, and a γ-subunit. eIF2 forms a ternary complex (TC) with GTP and Met-tRNA, and the binding of the TC to the 40S ribosomal subunit results in the formation of the 43S preinitiation complex. The recognition of the start codon AUG triggers the hydrolysis of GTP and the subsequent dissociation of eIF2-GDP from the preinitiation complex, initiating the elongation phase of translation [44]. To reactivate eIF2 for the next round of translation initiation, the guanine nucleotide exchange factor eIF2B catalyzes the exchange of GDP with GTP. eIF2α phosphorylation converts eIF2-GDP into a competitive inhibitor of eIF2B, thus limiting the availability of the TC to form the preinitiation complex and inhibiting global protein synthesis, while selectively triggering the translation of a subset of genes with special 5’ untranslated regions that encode transcription factors and other proteins including ATF4 [45].
Both pro-survival and pro-apoptotic pathways are activated in the ISR and the cell fate is determined by the balance of these two pathways, which is largely determined by the magnitude and duration of the ISR. The ISR is terminated by eIF2α dephosphorylation mediated by protein phosphatase 1 (PP1) with the participation of one of the regulatory subunits: PPP1R15A (also known as growth arrest and DNA damage-inducible protein, GADD34) or PPP1R15B (also known as constitutive repressor of eIF2α phosphorylation, CReP) [46, 47]. Where PPP1R15A is induced by the ISR, PPP1R15B is constitutively expressed [43]. Lines of evidence have implicated the ISR in the pathogenesis of various human diseases, including cardiovascular and lung diseases [48, 49].
ROS biogenesis and metabolism in mitochondria and ER
The ETC, composed of complexes I to IV in the inner mitochondrial membrane, transfers electrons from NADH (the reduced form of nicotinamide adenine dinucleotide) or FADH2 (a reduced form of flavin adenine dinucleotide (FAD)) to O2 via a series of oxidation-reduction reactions, leading to the generation of H2O (Figure 1A). During the electron transferring process, protons (H+) are pumped from the mitochondrial matrix into the intermembrane space by complexes I, II and IV. The resultant H+ gradient is then used by the ATP synthase complex (complex V) for the phosphorylation of ADP to ATP. The transfer of electrons by the ETC is not perfect. It is estimated that up to 2% of electrons escape from the chain and interact with O2 to produce superoxide (O2•−) [50]. Complexes I and III are the major sites of ROS production in mitochondria (Figure 1A) [50, 51]. Complex I generates O2•− exclusively in the mitochondrial matrix, whereas Complex III releases O2•− into both the matrix and the intermembrane space. Dismutation of O2•− to H2O2 in mitochondria is fulfilled by superoxide dismutase 1 (SOD1, Cu, Zn-SOD) in the intermembrane space and by SOD2 (Mn-SOD) in the matrix [1] In an antioxidant mechanism that occurs in mitochondria, H2O2 is decomposed into H2O primarily via the glutathione (GSH) redox system, which involves GSH peroxidases 1 and 4 (GPX1 and GPX4), GSH reductase (GR), peroxiredoxins 3 and 5 (PRX3 and PRX5), thioredoxin-2 (TRX2), and TRX reductase 2 (TRXR2) [1, 52].
Figure 1. Reactive oxygen species (ROS) biogenesis and metabolism in mitochondria and the endoplasmic reticulum (ER).
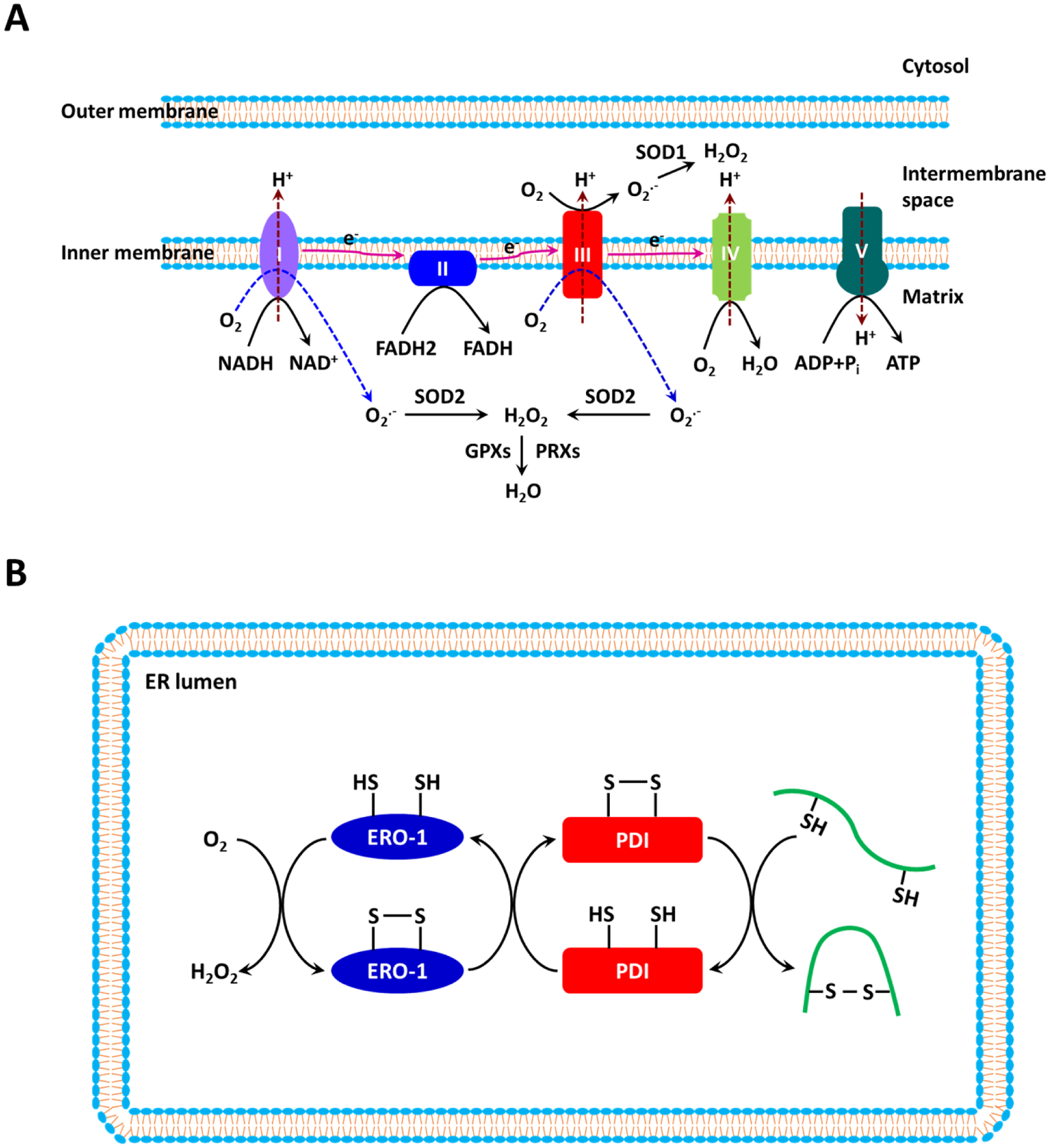
(a) The mitochondrial electron transport chain (ETC), which is composed of four complexes (I–IV) in the inner mitochondrial membrane, transfers electrons from electron donors to electron acceptors. Electrons from reduced nicotinamide adenine dinucleotide (NADH) enter the ETC at Complex I, whereas electrons from reduced flavin adenine dinucleotide (FADH2) enter the ETC at Complex II. Molecular O2 serves as the final electron acceptor. The chief function of the ETC is to synthesize ATP by coupling oxidative phosphorylation with the ATP synthase. Superoxide (O2•−) is produced primarily at Complexes I and III as a result of the incomplete reduction of O2. At Complex I, O2•− is produced within the matrix, whereas at Complex III, O2•− is released into both the matrix and the intermembrane space. O2•− is dismutated to H2O2 by superoxide dismutase 1 (SOD1) in the intermembrane space and by SOD2 in the matrix. H2O2 is subsequently reduced to H2O by glutathione peroxidases (GPXs) and peroxiredoxins (PRXs). (b) The formation of disulfide bonds in nascent proteins in the ER is driven by protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductin-1 (ERO-1). H2O2 is generated as the result of electron transfer between PDI and ERO-1 during the oxidative protein folding process.
Oxidative protein folding in the ER is also driven by redox reactions [53]. Protein disulfide isomerase (PDI) catalyzes the formation of disulfide in the nascent unfolded proteins via oxidation of two adjacent cysteines. To be reactivated, PDI is re-oxidized by ER oxidoreductin 1 (ERO1). ERO1 is then re-oxidized by transferring electrons to molecular O2 in the presence of FAD. The activity of ERO1 produces stoichiometric amounts of H2O2 for every disulfide bond generated. H2O2 is eliminated by PRX4 and GPX7/8 to prevent its build-up inside the ER (Figure 1B) [54]. In addition, ER transmembrane NOX4 also contributes to the ER ROS pool by constitutively producing H2O2 [55].
Interplay between oxidative stress and ER stress
Oxidative stress and ER stress are interconnecting processes and often form a vicious cycle. As mentioned earlier, the activation of the PERK pathway is associated with the increased transcription of genes that are involved in antioxidant defense, implicating ER stress in redox regulation. PERK-induced activation of Nrf2 contributes to the maintenance of intracellular GSH levels following ER stress [56]. PERK deficiency increases ROS generation in mouse embryonic fibroblasts following ER stress [57]. ATF4 deletion impairs the expression of genes that are involved in glutathione biosynthesis [57]. Mitochondria and the ER appear to be important sites of ROS generation during ER stress and the UPR. Tunicamycin-induced oxidative stress is attenuated in ρ0 fibroblasts devoid of endogenous mitochondrial DNA (mitoDNA) and a functional ETC. [56]. Activation of CHOP in the PERK pathway upregulates ERO1α, promoting H2O2 generation in the ER [58].
Alternatively, ER stress and the UPR could be the downstream of oxidative stress. The PERK pathway appears to be the major UPR component targeted by oxidative stress. Exogenous H2O2 increased both PERK-dependent and -independent phosphorylation of eIF2α [59] Interestingly, NOX4-derived ER H2O2 apparently activates the PERK pathway as knockdown of NOX4 or expression of ER-targeted catalase prevented tunicamycin-induced activation of this pathway [60]. Overexpression of ERO1β also induces ER stress, as evidenced by increased expression of BiP/Grp78 and CHOP [61]. MitoROS appear to play a critical role in ER stress and the UPR. Hypoxia-induced eIF2α phosphorylation and ATF4 accumulation, as well as the induction of BiP/Grp78 and CHOP, were attenuated by the expression of catalase and in cytochrome-c null cells [59], implicating a role for mitoROS in promoting ER stress.
ER–mitochondria coupling and ROS generation
Electron microscopy reveals a physical connection between mitochondri a and ER [62]. The contact sites are termed mitochondria-associated endoplasmic reticulum membranes (MAMs). This coupling has functional importance, allowing lipid and Ca2+ transfer [63]. ER inositol trisphosphate receptor (IP3R) and mitochondrial voltage-dependent anion-selective channel protein 1 (VDAC1), which are proteins that occur on the outer membrane of the mitochondria (OMM), are enriched in MAMs. IP3R and VDAC1 form a conduit for Ca2+ transfer, and Grp75 structurally links these two channels [64]. Ca2+ that is released from the ER lumen through IP3Rs is funneled via VDAC1 into the intermembrane space of mitochondria, and is subsequently transported into the mitochondrial matrix via mitochondrial Ca2+ uniporter (MCU) in the inner mitochondrial membrane (IMM) [65–67]. The increased Ca2+ load in the matrix stimulates the activity of various enzymes of the tricarboxylic acid (TCA) cycle, ATP synthesis, and ROS generation [66, 68]. ER stress increases ER–mitochondria coupling, resulting in increased ATP levels and mitochondrial Ca²⁺ uptake [69]. Moreover, abundant ERO1α is also found in MAMs and enhances Ca2+ shuttling from the ER to the mitochondria by stimulating IP3Rs [70].
Impacts of hypoxia on mitochondrial and ER function
Hypoxia and HIFs
To cope with hypoxia, mammals have evolved adaptive mechanisms. Central to this adaptation are HIFs that reprogram the expression of a broad range of genes, including those involved in energetic metabolism, angiogenesis, and proliferation [3, 71]. HIFs are heterodimers consisting of an oxygen-regulated α-subunit (HIF-1α, HIF-2α or HIF-3α) and a constitutively expressed nuclear β-subunit (HIF-1β). Under normoxic conditions, the HIF-α subunit is hydroxylated at conserved proline residue(s) by prolyl hydroxylase domain proteins (PHDs), which utilize O2 and α-ketoglutarate (also known as 2-oxoglutarate) as substrates. The hydroxylated HIF-α subunit is recognized and targeted for proteasomal degradation by the von Hippel-Lindau (VHL) E3 ubiquitin ligase complex. In hypoxic conditions, PHD activity is inhibited. The HIF-α subunit is then stabilized and translocated to the nucleus, where the α and β subunits form a heterodimer that binds to hypoxia response elements (HREs) in the promoter regions of HIF-regulated genes, triggering their transcription.
Hypoxia, HIFs and mitochondrial function
Mitochondrial oxidative phosphorylation depends on substrate availability. Hypoxia has been shown to disrupt substrate supply to the TCA and oxidative phosphorylation. A variety of glycolytic enzymes and glucose transporters are induced or activated by hypoxia through HIF-1. Among them are glucose transporter 1 (GLUT1), hexokinase 2 (HK2), phosphoglycerate kinase 1 (PGK1), pyruvate dehydrogenase kinase 1 (PDK1), and lactate dehydrogenase A (LDH-A) [72]. These adaptive changes that occur in response to hypoxia shift ATP generation from oxidative phosphorylation towards glycolysis. A similar phenomenon of glycolytic shift can also occur even under an adequate supply of O2, and is termed ‘the Warburg effect’.
ETC activity is also altered during hypoxia, in part due to the remodeling of ETC components exerted by HIFs [3]. HIF-1-mediated downregulation of NADH dehydrogenase (ubiquinone) 1α subcomplex, 4-like 2 (NDUFA4L2) is found to inhibit Complex I activity [73]. In addition, HIF-1 upregulates the expression of cytochrome c oxidase 4–2 (COX4–2) of Complex IV and the mitochondrial protease LON [74]. LON degrades COX4–1 and facilitates the swapping of COX4–1 for COX4–2, thereby enhancing the efficiency of mitochondrial respiration. The overall impacts of these changes are reduced ETC activity and ROS generation [74]. Moreover, hypoxia also impacts the ETC via HIF-1-responsive microRNA-210 (miR-210). MiR-210 downregulates the iron sulfur cluster protein (ISCU), succinate dehydrogenase complex subunit D (SDHD), and the cytochrome c oxidase (COX) assembly protein COX10, promoting ROS generation [75, 76]. Therefore, HIF-1 causes a paradoxical regulation of ROS production in mitochondria during hypoxia.
Both decrease and increase in ROS generation in mitochondria during hypoxia have been reported. Hypoxia reduces ROS generation in isolated liver mitochondria [77]. However, many studies have demonstrated an increase in mitoROS generation in response to hypoxia [78, 79]. Complexes I and III are the major sites of hypoxia-stimulated ROS production [79, 80]. Thus, the effect of hypoxia on ROS production is likely to be context-dependent and is regulated by the levels and durations of hypoxia.
Hypoxia, HIFs and ER function
The accumulation of unfolded or misfolded proteins in the ER that is induced by hypoxia triggers ER stress and the UPR [81]. Prolyl 4-hydroxylase subunit beta (P4HB), a PDI and a target of miR-210, is downregulated by miR-210 [82]. Hence, hypoxia may disrupt protein folding via the HIF-1α–miR-210 axis. ERO1α expression is increased by hypoxia in an HIF-1α-dependent way, thereby elevating H2O2 and triggering ER stress [83]. Among the three branches of the UPR, the PERK pathway appears to be the branch that is predominantly affected by hypoxia. Hypoxia activates the PERK pathway by causing PERK autophosphorylation and subsequent eIF2α phosphorylation [84]. Interestingly, the PERK pathway is a major component of the ISR [42]. Activation of the PERK–eIF2α–ATF4 axis appears to be a major mechanism to inhibit the translation of mRNAs and protein synthesis in hypoxia [85–87]. Hypoxia-induced eIF2α phosphorylation is reduced by catalase and cytochrome c deficiency and imitated by exogenous H2O2 [59], thus implicating a role of mitochondrial oxidative stress–ER stress coupling in regulating the UPR during hypoxia. The IRE1–XBP1 and ATF6 pathways are also targeted by hypoxia. Hypoxia induces HIF-1α-dependent XBP1 mRNA expression and splicing [88]. The ATF6α/ATF6 ratio is increased during hypoxia [89].
Regulation of HIFs by ROS and the UPR
Intriguingly, HIF expression and/or stabilization is also regulated by mitochondrial ROS and the UPR. Studies showing that exogenous H2O2 stimulated the accumulation of HIFs under normoxia and that scavenging of ROS prevents hypoxic HIF induction suggest a critical role for ROS in regulating HIF signaling [79, 90]. The loss of HIF-1α stabilization in hypoxia in ρ0 cells implicates the mitochondrial ETC as the HIF-inducing source of ROS [79, 90]. ROS produced at Complex III are found to be the primary contributors to HIF-1α stabilization [79, 90]. Inhibition of the hypoxic stabilization of HIF-1α by the overexpression of catalase, but not of SOD1 or SOD2, suggests that mitochondria-derived H2O2, but not O2•−, is required for hypoxic stabilization of HIFα [79] Inhibition of ER stress with tauroursodeoxycholic acid (TUDCA) also abolishes the increase in HIF-1 expression and activity induced by hypoxia, implicating ER stress as a HIF-1 activator during hypoxia [91]. XBP1 enhances the HIF-1α-activated gene expression in hypoxia by assembling a transcriptional complex with HIF-1α and recruiting RNA polymerase II [88]. These observations suggest that HIFs and ROS or ER stress are interdependent. Depending on the severity and/or duration of hypoxia, their integration may lead to beneficial or deleterious outcomes.
Roles of HIFs, mitoROS and UPR in the pathogenesis of pulmonary hypertension and preeclampsia
Hypoxia is central to the pathogenesis of hypoxia-related pulmonary hypertension [92, 93] and preeclampsia [14, 94]. In humans, prolonged hypoxia due to chronic obstructive pulmonary disease and cystic fibrosis contributes to the development of pulmonary hypertension [92]. Placental hypoxia is a principal factor initiating preeclampsia [95]. Further evidence comes from observations in human and animal models under both hypobaric and normobaric hypoxia. Residing at high altitude is associated with elevated pulmonary artery pressure and pulmonary vascular remodeling in humans [96, 97]. Similarly, the incidence of preeclampsia is ~ 3-fold higher in pregnant women living at high altitude than in those living at low altitude [98, 99]. Rodent models that are exposed to normobaric hypoxia recapitulate many of the features of pulmonary hypertension and preeclampsia seen in humans [100–106].
Roles of HIFs in pulmonary hypertension and preeclampsia
HIFs in pulmonary hypertension
HIFs are major regulators of cellular responses to hypoxia. HIF-1α plays a major role in the proliferation of pulmonary artery smooth muscle cells (PASMCs), whereas HIF-2α primarily promotes endothelial growth pertinent to pulmonary vascular remodeling [107]. Hypoxia induces HIF-1α in human PASMCs and HIF-2α in human lung vascular endothelial cells, as well as HIF-1α in pulmonary arteries or PASMCs in animal models of hypoxia-induced pulmonary hypertension [103, 108–110]. Genetic manipulation has demonstrated that HIFs are mediators of hypoxia-induced pulmonary hypertension. HIF-1α knockdown suppresses the proliferation of PASMCs [103, 111–113]. Mice with a partial HIF-1α deletion (Hif-1α+/−) and a smooth muscle-specific HIF-1α deletion have attenuated hypoxia-induced pulmonary vascular remodeling and pulmonary hypertension [114–116]. Endothelial HIF-2α also contributes to the development of pulmonary hypertension in hypoxia. Hif-2α+/− mice are protected from pulmonary vascular remodeling and pulmonary hypertension [117–119]. Moreover, mice expressing a human HIF-2α gain-of-function mutation (G537W) develop pulmonary hypertension [120].
HIF-1α-induced hypoxia inhibits the expression and activities of voltage-gated potassium channels Kv1.5 and Kv2.1 [108, 115] and upregulates the expression of transient receptor potential channels TRPC1 and TRPC6 [121] in PASMCs. Downregulation of the Kv channel and upregulation of the TRPCs leads to an increase in intracellular Ca2+ concentrations ([Ca2+]i), which contributes to increased PASMC proliferation and pulmonary vascular tone in pulmonary hypertension [115, 122, 123]. Treatment with HIF-1α short interfering RNA (siRNA) prevents PASMC proliferation by suppressing dynamin-related protein 1 (Drp1) expression and phosphorylation [103, 109], suggesting that HIF-1α also promotes PASMC proliferation by regulating mitochondrial dynamics.
HIFs in preeclampsia
Adequate blood supply to the placenta during pregnancy depends in part on the successful completion of physiological remodeling in the uteroplacental spiral arteries. Failure to transform these vessels is a common feature of preeclampsia and IUGR [17, 124]. High altitude pregnancies also display a decrease in remodeling of the uteroplacental spiral arteries [125]. Following implantation, cytotrophoblasts (CTB) are differentiated to EVT s within anchoring villi. EVTs at the tips of the anchoring villi then invade the spiral arteries, forming plugs that occlude the lumen of these vessels [126]. Thus, placental development in the first trimester of pregnancy occurs in a low O2 environment, resulting from a lack of uteroplacental blood flow caused by trophoblastic plugs, which is physiological at this stage. At the beginning of second trimester, the trophoblast plugs dislodge from spiral arteries, enabling the onset of uteroplacental blood flow and allowing high flow, low resistance, low velocity maternal blood to enter the intervillous space [127, 128]. Trophoblast invasion and spiral artery remodeling continue until the middle of the second trimester. The regulation of CTB biology by oxygen in the first trimester remains controversial. Some studies have found that hypoxia promotes trophoblast proliferation and inhibits trophoblast differentiation [129, 130], others demonstrate that low oxygen promotes the differentiation of trophoblasts into cells that have an invasive phenotype [131, 132]. It has been proposed that hypoxia promotes the formation of immature EVTs, and that the maturation of these cells into EVT that have invasive potential requires rising oxygen tension [133].
The expression of HIF-1α and HIF-2α in the placenta is consistently high in the first trimester and declines thereafter [129, 134]. This pattern of HIF expression is important for uterine spiral artery remodeling. Antisense-induced inhibition of HIF-1α expression in placental explants at 5–8 weeks of gestation results in inhibition of EVT proliferation and triggers a switch from a proliferative to an invasive trophoblast phenotype [129]. However, Cindrova-Davies and colleagues [135] report undetectable HIF-1α and HIF-2α in the first trimester placenta, a discrepancy that probably results from tissues being collected by different modes of delivery. Placental HIF-1α and HIF-2α are overexpressed in preeclampsia [136–138] and in trophoblast cell lines under hypoxic conditions [139, 140]. High-altitude pregnancy also exhibits high levels of HIF-1α in the placental and uterine arteries [141, 142]. Similarly, elevated placental HIF-1α is also observed in a rodent hypoxic model of preeclampsia [104]. The upregulation of HIFs in uteroplacental tissues plays a crucial role in the pathogenesis of preeclampsia. In an in vitro study, constitutive expression of an O2-insensitive form of HIF-1α (CA-HIF-1α) suppresses the differentiation of rat trophoblast giant cells, which are analogous to human EVTs [143]. Pregnant mice with global overexpression of HIF-1α are hypertensive with proteinuria [144]. Prolonged expression of CA-HIF-1α in the mouse, specifically in trophoblasts using lentiviral blastocyst transduction and non-surgical embryo transfer, also leads to failure to remodel spiral arteries, maternal hypertension, proteinuria, and fetal growth restriction [145]. Hypoxia-induced HIF-2α upregulates the expression of soluble fms-like tyrosine kinase-1 (sFlt-1) in trophoblasts [139]. Overexpression of sFlt-1 in animal models causes endothelial dysfunction, maternal hypertension and proteinuria, recapitulating preeclampsia phenotypes [146]. Elevated mitoROS induced by sFlT-1 probably contributes to endothelial dysfunction in preeclampsia [147].
HIF-sensitive miR-210, pulmonary hypertension and preeclampsia
miR-210 expression is upregulated in the pulmonary arteries of patients with pulmonary hypertension and is induced in the pulmonary arteries of mice by chronic hypoxia [148, 149]. miR-210 targets ISCU in pulmonary artery endothelial cells (PAECs) to disrupt mitochondria function, contributing to the development of pulmonary hypertension. [149]. In addition, miR-210 exhibits an antiapoptotic role in PASMCs [148]. HIF-1α may also indirectly impair uteroplacental functions by inducing miR-210 expression [142, 150, 151]. Overexpression of miR-210 is observed in preeclamptic placenta and in the uterine arteries in high-altitude pregnancies [150, 152, 153]. miR-210 inhibits trophoblast invasion, in part by promoting mitochondrial dysfunction [150, 154]. Moreover, miR-210 suppresses the expression and function of the large-conductance Ca2+-activated K+ channel (BKCa) in uterine arteries [153, 155], leading to increased uterine arterial myogenic tone and uterine vascular resistance.
mitoROS in the pathogenesis of pulmonary hypertension and preeclampsia
mito ROS and pulmonary hypertension
Disruption of mitochondrial ETC is a major contributor to ROS dysregulation. PAECs from patients with pulmonary arterial hypertension (PAH) exhibit elevated levels of mitochondrial O2•− and H2O2 [156]. Mice overexpressing mitochondria-targeted catalase (MCAT) have an attenuated hypoxia-induced increase in pulmonary vessel muscularization, whereas mice overexpressing SOD2 (TghSOD2) have exacerbated hypoxia-induced pulmonary hypertension [157]. These findings suggest that H2O2 is an important mediator of pulmonary hypertension. Fe-S clusters are important cofactors in Complexes I, II, and III that participate in electron transfer. The NFU1 Fe-S cluster scaffold protein is involved in the biogenesis of Fe-S clusters. NFU1 mutations impair ETC function and are associated with pulmonary hypertension [158, 159]; for example, introduction of the NFU1G206C point mutation in the rat, the equivalent of human G208C, replicates the human pulmonary hypertension phenotype [160]. Like NFU1, ISCU facilitates the assembly of Fe-S clusters. miR-210 is upregulated and its target ISCU is downregulated both in the pulmonary arteries of humans with pulmonary hypertension and in mouse models of pulmonary hypertension induced by chronic hypoxia, SU5416/chronic hypoxia and VHL deficiency (VHL−/−) [149]. In human PAECs, miR-210 is induced by prolonged hypoxia exposure [161]. ISCU suppression reduces the activity of mitochondrial Complex I, resulting in increased mitoROS generation in PASMCs and PAECs under hypoxic conditions [75, 161, 162]. Hypoxia also boosts the generation of mitoROS by activating the mitochondrial ATP-dependent K+ channel (mitoKATP) in PASMCs [163], which subsequently stimulates the HIF–miR-210–ISCU axis to produce more mitoROS [164].
The expression levels and activities of the ion channels and enzymes that regulate Ca2+ movement in pulmonary arteries are impacted by chronic hypoxia. Reduced expression of Kv1.5 in pulmonary arteries and in PASMCs has been documented in human patients and in animal models of pulmonary hypertension [108, 165, 166], as well as in PASMCs exposed to chronic hypoxia [167]. By contrast, the expression of TRPC1, TRPC6, transient receptor potential cation channel subfamily V 4 (TRPV4), and ryanodine receptor 2 (RyR2) in PASMCs is increased by chronic hypoxia [121, 168–170]. Moreover, PASMCs from mice exposed to chronic hypoxia display higher phospholipase Cγ 1 (PLCγ 1) expression and activity [171]. ROS, including mitoROS, apparently contribute to the altered expression and activity of ion channels and phospholipase C. Studies from Archer’s group show that a reduction in mitoROS causes downregulation of Kv1.5 by activating HIF-1α and nuclear factor of activated T cells cytoplasmic 2 (NFATc2) in PASMCs [108, 166]. Other studies suggest that chronic hypoxia activates mitoKATP to produce H2O2 in mitochondria, resulting in Kv1.5 downregulation in PASMCs [163, 172]. H2O2 also appears to regulate the expression of TRPC1 and TRPC6 in PASMCs. The abundance of TRPC1 protein in PASMCs is increased by polyethylene glycol (PEG)-conjugated superoxide dismutase (PEG-SOD) under normoxia and is decreased by PEG-catalase under hypoxia [173]. Chronic hypoxia-promoted dissociation of FKBP12.6 (FK506 binding protein, 12.6 kDa molecular weight) from RyR2 in PASMCs from a chronic-hypoxia-induced pulmonary hypertension mouse model is mediated by ROS generated in Complex III, as evidenced by the suppression of this dissociation by deletion of the Rieske iron-sulfur protein (RISP) [174]. The increased activity of phospholipase C in PASMCs in hypoxia is mediated by H2O2 [171]. Overall, the altered expression levels or activities of these channels and enzymes result in increased basal [Ca2+]i in PASMCs and in increased vascular tone [169, 171, 175, 176].
In response to chronic hypoxia, PASMCs and PAECs display increased proliferation and/or reduced apoptosis [103, 157]. Pulmonary vascular remodeling is exacerbated and mitigated in mice overexpressing SOD2 and mitochondria-targeted catalase, respectively [157]. TEMPOL and mitochondrial-targeted coenzyme Q (MitoQ) reduce the migration and proliferation of Sugen 5416/hypoxia-induced microvascular endothelial cells (MVECs) [177]. Hypoxia promotes PASMC proliferation and suppresses apoptosis by activating mitoKATP and increasing the production of mitoROS and H2O2 [162, 164]. Hypoxia also increases the migration and proliferation of Sugen 5416/hypoxia-induced MVECs through mitoROS-stimulated TRPV4 activation [177]. However, Archer et al. [178] demonstrated that the reduction of mitochondrial H2O2 due to SOD2 downregulation promotes the proliferation and suppresses the apoptosis of PASMCs in fawn-hooded rats.
Ca2+ plays a pivotal role in cell proliferation [179]. Nuclear factor kappa B (NF-κB) and NFAT are Ca2+-regulated transcription factors. NF-κB promotes cell proliferation through transcriptional regulation of cyclin D1, whereas NFAT regulates genes involved in the cell cycle and apoptosis. NF-κB activation is elevated in the PASMCs and PAECs of patients with pulmonary hypertension [180]. Inhibition of NF-κB decreases pulmonary artery hypertrophy in vivo, suppresses proliferation, and promotes the apoptosis of PASMCs in vitro, establishing a role for NF-κB in pulmonary vascular remodeling [181]. Hypoxia increases the nuclear translocation of NF-κB in rodent lungs and in cultured PAECs [182, 183]. Similarly, the expression and nuclear translocation of NFATc, which is activated in the cytoplasm by the Ca2+-dependent phosphatase calcineurin, is increased in the PASMCs of patients with pulmonary hypertension and of monocrotaline-induced pulmonary artery hypertensive rats, as well as in PASMCs that are exposed to hypoxia [166, 184, 185]. Increased [Ca2+]i promotes the proliferation and migration, and reduces the apoptosis, of PASMCs by activating NFATc and increasing the nuclear translocation of NFATc [166, 184, 185].
MitoROS and preeclampsia
The placenta, being metabolically active in order to meet the requirements of placental and fetal development and growth, consumes significant amounts of O2, nutrients, and energy. Perturbations in mitochondrial function in the placenta may lead to excessive generation of ROS, contributing to the pathogenesis of preeclampsia and IUGR [186, 187]. Oxidative stress is commonly detected in placentas from pregnant women with preeclampsia [34], and women with mitochondrial dysfunction have a high incidence of pre-eclampsia [188]. Preeclamptic placentas display damaged mitochondria, as evidenced by swollen mitochondria and broken cristae [150, 189]. Proteomic analysis reveals that a number of proteins that are involved in fatty acid oxidation, TCA, ETC, and ROS homeostasis are altered in preeclamptic placentas [189. 190]. Among them, 2-oxoglutarate dehydrogenase (OGDH), LDH-A, PRXs, NADH dehydrogenase (ubiquinone) iron-sulfur protein 3 (NDUFS3), ubiquinol-cytochrome c reductase core protein 2 (UQCRC2), and ATP synthase are downregulated, whereas NDUFS7, NUDFB8, and NDUFB7 are upregulated.
Preeclampsia reduces both ETC components and activity in the placenta [38, 150, 191, 192]. Similar findings are also seen in placentas from high-altitude pregnancies, in the reduced uterine perfusion pressure (RUPP) rat model of preeclampsia, and in trophoblasts exposed to chronic hypoxia [191, 193]. As expected, mitochondrial O2•− and H2O2 are increased in preeclamptic placentas and in placentas of the hypoxic pregnancy model of preeclampsia [194–197]. Decreases in the expression and activities of SOD, TRXR and GPX are also observed in preeclamptic and high-altitude placentas [197–199]. Preeclampsia is thus associated with heightened oxidative stress in the placenta. Hypoxia plays an important role in mitochondrial dysfunction in preeclampsia [200, 201]. mitoROS production in the placenta could be regulated by HIF-dependent pathways [23, 201, 202]. Furthermore, overexpression of sFlt-1 in the placenta in preeclampsia [146] is secondary to hypoxia and is mediated by HIF-1 [203]. Exposure to serum from preeclamptic women, which is rich in sFlt-1, dissipates the mitochondrial membrane potential (Ψm) and increases mitoROS production in first-trimester trophoblast HTR-8/SVneo cells [147]. Mammalian target of rapamycin (mTOR) activity is also regulated by hypoxia [204]. Hypoxia-induced inhibition of the protein kinase B (AKT)/mTOR pathway has been shown to contribute to the development of preeclampsia [205]. Moreover, placental mTOR is downregulated in IUGR [206]. Inhibition of mTORC1 reduces both the protein expression of Complexes I–IV and mitochondrial respiration, mimicking the phenotype of IUGR placenta [207]. An in vitro study revealed that HIF-1α stabilization inhibits first-trimester primary trophoblast invasion by upregulating miR-210 [151]. Thus, if overexpression of HIF occurs in the first trimester (as in early-onset preeclampsia, its inhibitory effect on trophoblast invasion could be executed in part by miR-210. HIF-1α-dependent miR-210 expression is increased in preeclamptic and high-altitude placentas as well as in hypoxic trophoblasts [150, 191, 208]. MiR-210 downregulates ETC components such as ISCU, COX10, NDUFA4, and SDHD and reduces mitochondrial respiration in trophoblasts [150, 151, 209]. Mitochondrial dysfunction and overproduction of mitoROS impair trophoblast invasion [154, 209] and induce trophoblast apoptosis [210, 211], thereby impairing spiral artery remodeling. The increase in mitoROS production can be imitated by exposing placental explants and trophoblasts to chronic hypoxia [23, 202]. The damaged mitochondria, impaired ETC activity and increased mitoROS in preeclamptic placentas are also replicated in rodent models of preeclampsia (RUPP and hypoxia models) [193, 197].
The BKCa channel β1 subunit is upregulated in the placenta and the BKCa channel plays an essential role in reducing uterine vascular resistance and in increasing uteroplacental blood flow in pregnancy [69, 153, 212]. This adaptation was disrupted by upregulated miR-210 in ovine uterine arteries of high-altitude pregnancy [153, 155]. As the expression and activity levels of BKCa channels are regulated by ROS in the uterine arteries [142, 213, 214], miR-210-induced mitochondrial dysfunction and mitoROS may contribute to the uterine vascular maladaptation seen at high altitude.
ER stress and the UPR in the pathogenesis of pulmonary hypertension and preeclampsia
ER stress and the UPR in pulmonary hypertension
The expression of ATF6 and/or CHOP in pulmonary arteries is increased in patients with PAH, idiopathic pulmonary artery hypertension (IPAH) and systemic sclerosis-associated PAH [215, 216]. In the monocrotaline rat model and in the chronic hypoxic mouse model of pulmonary hypertension, all three UPR pathways are activated in the lung [102, 216, 217]. Chronic hypoxia also activates the UPR in vitro, as evidenced by the increased expression of p-eIF2α, IRE1α and ATF6 in cultured PASMCs [215, 218, 219]. Both the PERK and IRE1α pathways are important mediators of hypoxia-induced proliferation of PASMCs. Knocking down eIF2α with siRNA ablates the proliferation of PASMCs under hypoxia [218]. Inhibiting the IRE1 pathway decreases the migration and proliferation of PASMCs and promotes their apoptosis [219].
ER stress and the UPR in preeclampsia and IUGR
Lines of evidence suggest the presence of misfolded proteins in the preeclamptic placenta [220], and a ctivation of the UPR is a common feature of early-onset preeclampsia and IUGR. Significantly elevated ER stress markers, such as BiP/GPR78, pPERK, p-eIF2α, ATF4, IRE1α, ATF6, and CHOP, are observed in preeclamptic and/or IUGR placentas [25, 138, 221–223]. In addition, placentas from high-altitude pregnancy also exhibit ER stress and activation of the PERK pathway [224], which are recapitulated by gestational hypoxia in a rat model of preeclampsia [225]. Hypoxic treatment of placental explants, primary trophoblasts and trophoblast cell lines similarly induces ER stress and activates the UPR [25, 223, 224, 226].
The concurrent increase in HIF-1α and CHOP in preeclamptic placentas suggests that HIF-1α could be the link between hypoxia and the UPR [138]. Induction of ER stress with tunicamycin in pregnant mice reduces both placental and fetal weights [227]. Increased phosphorylation of eIF2α in preeclamptic, IUGR, and high-altitude placentas suppresses the AKT/mTOR pathway, contributing to fetal growth restriction [186, 221, 224]. Thus, it is not surprising that early-onset preeclampsia and high-altitude pregnancy are associated with increased incidence of IUGR. Placental growth factor (PlGF) is important in regulating placental angiogenesis and trophoblast invasion. In preeclampsia, the circulating level of PlGF is decreased [146]. PlGF expression is negatively regulated by ATF4 and ATF6b in trophoblasts [222], impairing the development and maturation of the placental vascular system. Activation of the PERK pathway is also observed in uterine arteries of pregnant sheep at high altitudes and contributes to an increase in uterine vascular tone [214].
ER–mitochondria interplay in pulmonary hypertension and preeclampsia
The ER and mitochondria are two cellular organelles that are structurally and functionally interconnected. Ca2+ flux from the ER to the mitochondria is the most important function of ER-mitochondria crosstalk [228]. Perturbations in ER-mitochondria connections may result in the progression of pulmonary hypertension and preeclampsia. Nogo-B, a member of the reticulon protein family that is primarily localized in the ER, participates in maintaining ER morphology [229]. Patients with pulmonary hypertension and the rat hypoxic model of pulmonary hypertension both have high levels of Nogo-B in their pulmonary arteries [215]. Hypoxia increases the distance between the ER and the mitochondria, reduces mitochondrial Ca2+ and mitoROS, and inhibits Ca2+-dependent mitochondrial enzymes in PASMCs through ATF6-mediated upregulation of Nogo-B expression [215]. Furthermore, Nogo-B knockout mice are resistant to chronic hypoxia-induced pulmonary hypertension [215]. These findings suggest that Nogo-B functions as a link between ER stress and mitochondrial stress under hypoxia that promotes the development of pulmonary hypertension.
Uncoupling protein 2 (UCP2), which functions as a Ca2+ channel, also participates in translocating Ca2+ from the ER to mitochondria [230]. The expression of UCP2 is reduced in the pulmonary microvessels of patients with IPAH and of chronically hypoxic mice [231, 232]. Two independent studies have revealed that UCP2 deletion promotes PASMC proliferation and that UCP2-knockout mice undergo pulmonary vascular remodeling and develop pulmonary hypertension [231, 233]. However, contrasting data on mitoROS generation have been reported from PASMCs of UCP2-knockout mice. Pak et al. [231] observed an increase in mitoROS, whereas Dromparis et al. [233] detected a decrease in mitoROS. The disruption of Ca2+ transport between the ER and mitochondria probably contributes to mitochondrial dysfunction [233]. Placental expression of UCP2 is positively correlated with fetal weight and is reduced in IUGR pregnancies [234]. The downregulation of placental UCP2 is also observed in mice that are exposed to chronic hypoxia [234]. It remains to be determined whether and how ER–mitochondria crosstalk is impacted by reduced UCP2 expression in preeclamptic and IUGR placentas. In rodent models of IUGR, both chronic hypoxia and cadmium promote ER and/or mitochondrial stress in the placenta [225, 235]. Intriguingly, activation of the PERK pathway is prevented by the antioxidants MitoQ and α-phenyl-N-t-butylnitrone, suggesting that communication between the mitochondria and the ER plays an important role in impairing fetal growth in IUGR.
Caution is needed when interpreting data on placental HIFs, mitoROS, ER stress and the UPR because the mode of delivery can greatly impact the generation and activation of stress [236]. During vaginal delivery, unlike caesarean section, the placenta is exposed to uterine contractions and the accompanying intermittent reduction in maternal blood supply, which are potent inducers of these cellular stress [236–238]. Therefore, comparisons of cellular stresses should be made using the samples from the same mode of delivery. The placenta is highly metabolic active, and therefore rapidly becomes hypoxic soon after disconnecting from the maternal blood supply. Ideally, placental tissues that are suitable for research on cellular stress should be collected from non-labored elective caesarean sections, and snap-frozen in liquid nitrogen or fixed within 10–20 min of detachment from the uterus [236]. Yung et al. [221] used samples from elective caesarean sections to demonstrate that ER stress is increased in IUGR placentas, supporting the notion that pregnancy complications heighten cellular stress in the placenta.
Targeting cellular stress as a potential therapy for pulmonary hypertension and preeclampsia
Despite significant progress in elucidating the mechanisms that underlie pulmonary hypertension and preeclampsia, there is still no cure for either disease. The current therapies mainly aim to improve symptoms. Given the high morbidity and mortality of these two diseases, novel therapies are urgently needed. As already discussed, it is apparent that hypoxia, which induces mitochondrial ROS, ER stress and the ISR, is the primary contributor to the pathogenesis of pulmonary hypertension and preeclampsia. Therefore, HIFs, mitochondrial ROS, ER stress, the ISR, and their constituent signaling elements could be promising targets for the development of therapeutics to treat both diseases (Figure 2). Various compounds have been developed to inhibit or lessen HIF signaling, mitochondrial oxidative stress and ER stress or the UPR, and some of them have been evaluated in animal models for the treatment of pulmonary hypertension and preeclampsia (Table 2).
Figure 2. Hypoxia and the integrated stress response (ISR): potential drug targets.
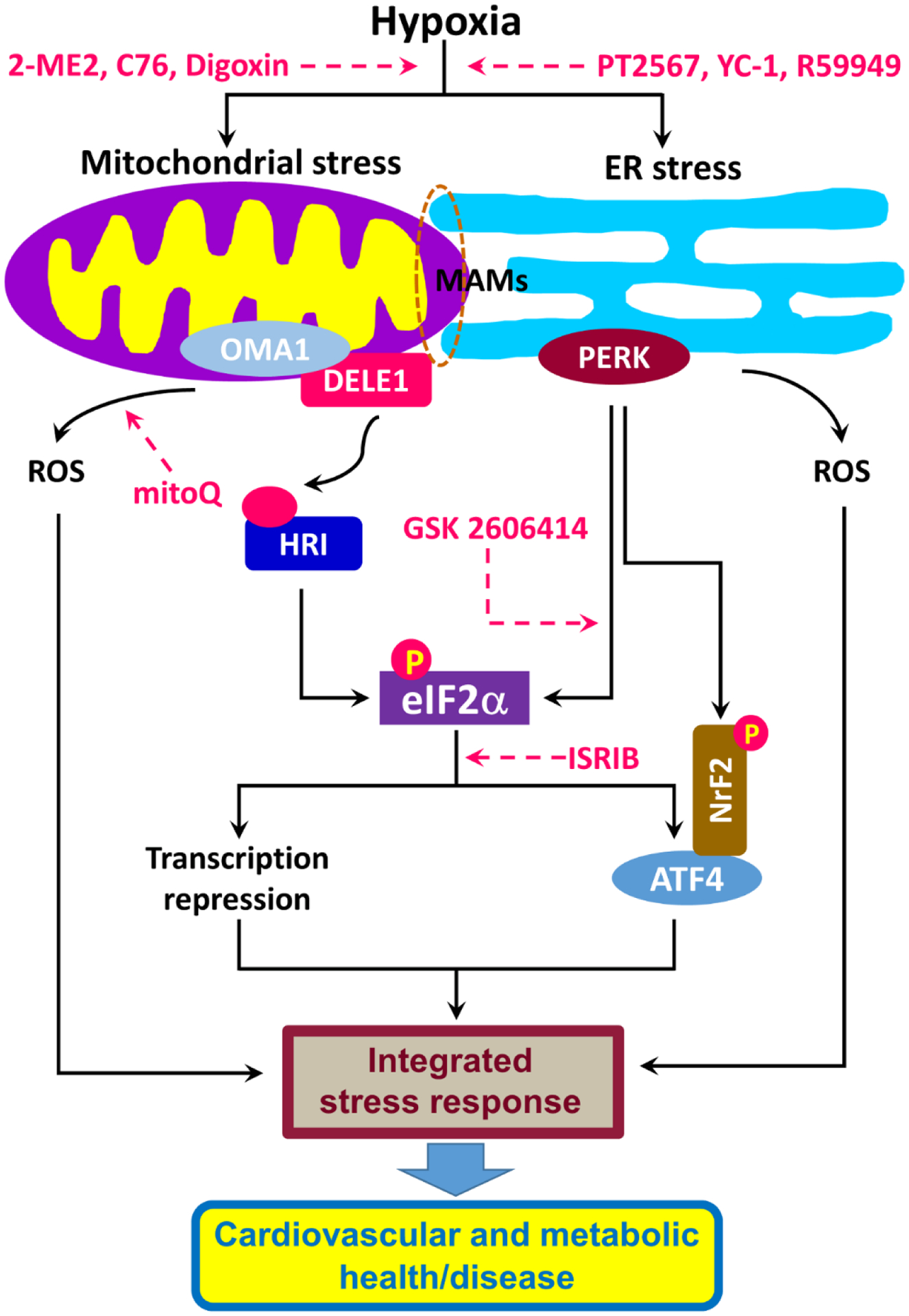
Hypoxia induces mitochondrial stress and endoplasmic reticulum (ER) stress. Mitochondrial stress produced via the OMA1-DELE1-HRI pathway and ER stress resulting from the PERK pathway converge at the phosphorylation of eIF2α, leading to changes in the transcription of genes and subsequent cardiovascular and metabolic (mal)adaptations. The ISR can also be activated by oxidative stress resulting from the accumulation of reactive oxygen species (ROS). Potential targets for drug development are indicated by dashed pink arrows.
Abbreviations: ATF4, activating transcription factor 4; C76, compound 76; DELE1, DAP3-binding cell death enhancer 1; eIF2α, eukaryotic initiation factor 2α; HRI, heme-regulated inhibitor; ISRIB, integrated stress response inhibitor; MAM, mitochondria-associated membrane; ME, 2-methoxyestradiol; mitoQ, mitochondrial-targeted coenzyme Q; NrF2, Nuclear factor erythroid 2-related factor 2; OMA1, a mitochondrial metallopeptidase encoded by the OMA1 gene; PERK, PKR-like ER kinase 2; YC-1: 3-(5’-hydroxymethyl-2’-furyl)-1-benzylindazole.
Table 2.
Promising compounds targeting hypoxia and the mitochondrial ROS and ER stress signaling pathways in animal models of pulmonary hypertension and preeclampsia.
| Compound | Mode of action | Animal model | Pulmonary hypertension | Preeclampsia | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Pulmonary artery pressure (PAP)/right ventricular systolic pressure (RVSP) | Right ventricle hypertrophy | Pulmonary artery remodeling | Mean arterial pressure | Proteinuria | Fetal growth | ||||
| Hypoxia inducible factor (HIF) pathway | |||||||||
| 2-methoxyestradiol | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [239] | |||
| 2-methoxyestradiol | HIF-1α inhibition | COMT−/− mice | ↓ | ↓ | [254] | ||||
| Apigenin | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [240] | |||
| Baicalin | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [241] | |||
| C76 | HIF-2α inhibition | Egln1Tie2Cre mice, monocrotaline (MCT) rats, Sugen/hypoxic rats | ↓ | ↓ | ↓ | [242] | |||
| Caffeic acid phenethyl ester | HIF-1α inhibition | MCT rats | ↓ | ↓ | ↓ | [251] | |||
| Celastramycin | HIF-1α inhibition | Hypoxic mice, MCT rats, Sugen/hypoxic rats | ↓ | ↓ | ↓ | [243] | |||
| Cyclosporin A | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [244] | |||
| Digoxin | HIF-1α inhibition | Hypoxic mice | ↓ | ↓ | ↓ | [245] | |||
| Emetine | HIF-1α and HIF-2α inhibition | MCT rats, Sugen/hypoxic rats | ↓ | ↓ | ↓ | [246] | |||
| Melatonin | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [247] | |||
| PT2567 | HIF-2α inhibition | Hypoxic rats | ↓ | ↓ | [248] | ||||
| Topotecan | HIF-1α inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [249] | |||
| YC-1 | HIF-1α inhibition | Hypoxic mice | ↓ | ↓ | ↓ | [250] | |||
| R59949 | PHD2 activation | Hypoxic mice | ↓ | ↓ | ↓ | [252] | |||
| Anti-CD146 antibody | HIF-1α inhibition | Hypoxic mice | ↓ | ↓ | ↓ | [253] | |||
| HIF-1α small hairpin RNA (shRNA) | HIF-1α silencing | Hypoxic rats | ↓ | ↓ | [112] | ||||
| Anti-miR-210 oligonucleotide | microRNA-210 (miR-210) inhibition | Sugen/hypoxic mice | ↓ | ↓ | [149] | ||||
| mitoROS | |||||||||
| Melatonin | mitoROS scavenger | RUPP rats | ↓ | [257] | |||||
| MitoQ | Mitochondria-targeted antioxidant | Hypoxic mice | ↔ | ↓ | ↔ | [259] | |||
| MitoQ | Mitochondria-targeted antioxidant | RUPP rats | ↓ | ↑ | [263] | ||||
| MitoQ | Mitochondria-targeted antioxidant | RUPP mice | ↓ | ↑ | [197] | ||||
| MitoQ | Mitochondria-targeted antioxidant | Hypoxic sheep | ↑ | [262] | |||||
| MitoQ-nanoparticles (NPs) | Mitochondria-targeted antioxidant | Hypoxic rats | ↑ | [260, 261] | |||||
| Mito-Tempo | Mitochondria-targeted antioxidant | RUPP rats | ↓ | ↑ | [263] | ||||
| ER stress and the unfolded protein response (UPR) | |||||||||
| 4-PBA | ER stress inhibition | Hypoxic mice MCT rats | ↓ | ↓ | ↓ | ↓ | [101] | ||
| 4-PBA | ER stress inhibition | Hypoxic mice | ↓ | ↓ | ↓ | ↓ | [102] | ||
| 4-PBA | ER stress inhibition | MCT rats | ↓ | ↓ | ↓ | ↓ | [217, 266] | ||
| GSK2606414 | PERK inhibition | Sugen/hypoxic rats | ↓ | ↓ | ↓ | ↓ | [267] | ||
| H2S | ER stress inhibition | Hypoxic rats | ↓ | ↓ | ↓ | [268] | |||
Abbreviations: 4-PBA, 4-phenylbutyric acid; C76, compound 76; COMT, catechol-O-methyltransferase; H2S, hydrogen sulfide; MitoQ, Mitochondrial-targeted coenzyme Q; RUPP, reduced uterine perfusion pressure; YC-1, 3-(5’-hydroxymethyl-2’-furyl)-1-benzylindazole.
Given the important role of HIFs in the pathogenesis of pulmonary hypertension and preeclampsia, the suppression of HIF signaling is of great therapeutic interest. A variety of pharmacological HIF inhibitors, including 2-methoxyestradiol, apigenin, baicalin, caffeic acid phenethyl ester, celastramycin, cyclosporin a, digoxin, emetine, melatonin, topotecan, and 3-(5’-hydroxymethyl-2’-furyl)-1-benzylindazole (YC-1), have been found to have beneficial effects in rodent models of pulmonary hypertension. In the hypoxic rodent models of pulmonary hypertension, HIF1α/HIF2α inhibitors prevent or reverse hypoxia-induced pulmonary vascular remodeling, right ventricle hypertrophy, and elevated right ventricular systolic pressure (RVSP) [239–250]. Comparable findings were seen with HIF1α/HIF2α inhibition in Egln1Tie2Cre mice and with monocrotaline-induced PAH rats [251, 252]. R59949, a PHD2 activator that inhibits HIF, also ameliorates pulmonary hypertension in hypoxic mice by reversing hypoxia-induced pulmonary vascular remodeling, right ventricle hypertrophy, and elevated RVSP [252]. Similarly, other therapeutic approaches, such as delivery of anti-CD146 to disrupt the CD146–HIF-1α axis, delivery of small hairpin RNA (shRNA) to silence HIF1α, and delivery of anti-miR-210 oligonucleotide to inhibit HIF-1-responsive miR-210, have been shown to attenuate experimental pulmonary hypertension in hypoxic rodents [112, 149, 253]. 2-Methoxyestradiol is a metabolite of the endogenous 17β-estradiol, formed through the enzymatic actions of cytochrome P-450 and catechol-O-methyltransferase (COMT). The expression of COMT in the placenta and levels of circulating 2-methoxyestradiol are low in pregnant women with preeclampsia [254]. Furthermore, the administration of 2-methoxyestradiol amends hypertension and proteinuria in a COMT−/− mouse model of preeclampsia [254].
The reduction of mitoROS is also an attractive approach in the treatment of pulmonary hypertension and preeclampsia. Mice overexpressing mitochondria-targeted catalase exhibit attenuated chronic-hypoxia-induced increases in RVSP and pulmonary vascular remodeling [157]. Melatonin can be transported into mitochondria via peptide transporters PEPT1 and PEPT2 [255]. and protects mitochondria from stress by scavenging ROS [256]. When administered, melatonin reduced the mean arterial pressure in RUPP rats [257]. MitoQ, which harbors the antioxidant quinone moiety covalently attached to a lipophilic triphenylphosphonium cation, has been used in preclinical studies in rats and mice and in human trials [258], but it fails to prevent pulmonary hypertension in mice exposed to chronic hypoxia [259], probably due to insignificant changes in the lung mitoROS of this animal model. In a rat model of IUGR produced by prenatal hypoxia, maternal injection of MitoQ-loaded nanoparticles prevented placental oxidative stress and rescued fetal growth [260, 261]. MitoQ has also been seen to restore fetal growth in hypoxic sheep [262]. Similarly, the mitochondria-specific antioxidants MitoQ and MitoTEMPO reduced maternal mean arterial pressure and improved fetal growth in the RUPP rat model of preeclampsia [263]. It appears that timing is a critical factor for the actions of MitoQ. MitoQ treatment in early pregnancy exacerbated maternal blood pressure, proteinuria, and fetal growth restriction, whereas MitoQ administration in late gestation alleviated the preeclampsia phenotype in RUPP rats [197].
ER stress and the UPR may also be targeted in the development of treatments for pulmonary hypertension and preeclampsia. Two chemical chaperones, 4-phenylbutyric acid (4-PBA) and TUDCA, have been approved by the US Food and Drug Administration (FDA) for the treatment of urea-cycle disorders and primary biliary cirrhosis, respectively [264, 265]. Interestingly, 4-PBA effectively prevents and reverses the elevat ed pulmonary artery pressure and pulmonary vascular remodeling seen in chronic-hypoxia-exposed mice and/or monocrotaline-induced PAH rats [101, 102, 217, 266]. Pharmacological inhibition of the PERK pathway with GSK2606414 reduced pulmonary vascular remodeling, right ventricle hypertrophy, and elevated RVSP in Sugen 5416/hypoxia PAH mice [267]. H2S also alleviates pulmonary hypertension by inhibiting ER stress in hypoxic rats [268]. Furthermore, TUDCA and GSK2606414 inhibit the effect of gestational hypoxia, reduce uterine arterial myogenic tone and decrease placental protein levels of endothelin-1 and sFlt-1 [213, 214, 269]. A recently developed drug, ISRIB (integrated stress response inhibitor), potently inhibits the ISR and is bioavailable in vivo with no overt toxicity, owing to its bi-mode of action and bell-shaped response to the ISR [42, 265]. It has been found to improve inflammation and cognitive disorders in preclinical studies in rodents [270], and its therapeutic potential in pulmonary hypertension and preeclampsia is yet to be determined.
Although promising preclinical results have been obtained in animal models, the translation of these findings into clinical applications is challenging. As components of HIF, ROS, and the UPR and ISR signaling pathways are broadly expressed, the specific delivery of the therapeutic agent to the target site(s), such as organ, tissue, cell, or organelle undergoing cellular stress, is preferable in order to avoid unintended effects or toxicity. Some progress have been made; for example, mitochondria-targeted antioxidants that conjugate the lipophilic triphenylphosphonium cation to an antioxidant moiety allow these compounds to pass through biological membranes easily and thus to accumulate to levels that are increased several-hundred-fold within mitochondria [271]. The selective delivery of drugs to the placenta has been reviewed recently [201]. Therapeutic molecules conjugated with specific peptide sequences that are designed to recognize cell surface macromolecules in uteroplacental cells have been successfully and selectively delivered to the placenta [201]. It is also important to note that HIFs, mitochondrial ROS and the UPR are also pivotal for physiological signaling. We anticipate the development of therapeutic approaches that can selectively inhibit only the disease-relevant HIF, ROS, UPR or ISR with minimal impact on stress-mediated physiological signaling. Greater effort in future research and drug development will be needed to meet this expectation.
Conclusions
Oxygen is vital sustaining the activities of mitochondria and the ER, and this for cellular functions. Intriguingly, the activities of these two organelles are closely interlinked. Hypoxia has been shown to reprogram or perturb functions performed in the mitochondria and ER, resulting in the activation of HIFs, change in mitoROS production, and induction of the UPR or ISR. Substantial evidence suggests the involvement of HIFs, mitoROS and the UPR in the development and progression of pulmonary hypertension and preeclampsia. HIFs, mitoROS and the UPR or ISR could form a complex signaling network, and could act independently or interdependently to promote these two disorders. Significant progress has been made in revealing the mechanistic links between hypoxia and pulmonary hypertension or preeclampsia. Nevertheless, we are still far away from a comprehensive understanding of the molecular mechanisms underlying these two diseases. F urther research is undoubtedly needed so that we can comprehend the pathogenesis of pulmonary hypertension and preeclampsia and can develop more specific and effective therapeutic interventions.
Highlights.
Hypoxia is a common cause of pulmonary hypertension and preeclampsia.
Mitochondria and endoplasmic reticulum (ER) are the main targets of the hypoxic response.
Oxidative and ER stress converge in the ISR.
The ISR contributes to pulmonary hypertension and preeclampsia.
Potential drug development may target the ISR.
Acknowledgements:
This work was funded in part by National Institutes of Health Grants HD083132, HL128209, HL137649, and HL149608 (all to L. Zhang).
Abbreviations:
- AKT
protein kinase B
- ATF6
activating transcription factor 6
- BiP
binding immunoglobulin protein
- BKCa
large-conductance Ca2+-activated K+ channel
- CHOP
C/EBP homologous protein
- COMT
catechol-O-methyltransferase
- COX4–2
cytochrome c oxidase 4–2
- CTB
cytotrophoblasts
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERO1
ER oxidoreductin 1
- ETC
electron transport chain
- EVT
extravillous trophoblast
- FAD
flavin adenine dinucleotide
- GCN2
general control nonderepressible protein 2
- GPX1
glutathione peroxidase 1
- Grp78
78-kDa glucose-regulated protein
- GSH
glutathione
- HIF
hypoxia inducible factor
- HO•
hydroxyl radical
- HRI
heme-regulated inhibitor
- IP3R
inositol trisphosphate receptor
- IPAH
idiopathic pulmonary artery hypertension
- IRE1α
inositol-requiring enzyme 1α
- ISCU
iron sulfur cluster protein
- ISR
integrated stress response
- IUGR
intrauterine growth restriction
- LDH-A
lactate dehydrogenase A
- mitoKATP
mitochondrial ATP-dependent K+ channel
- mitoQ
mitochondrial-targeted coenzyme Q
- mitoROS
mitochondrial ROS
- MAM
mitochondria-associated endoplasmic reticulum membrane
- mTOR
mammalian target of rapamycin
- NADH
reduced form of nicotinamide adenine dinucleotide
- NDUFS3
NADH dehydrogenase (ubiquinone) iron-sulfur protein 3
- NFATc2
nuclear factor of activated T cells cytoplasmic 2
- NF-κB
nuclear factor kappa B
- NOX
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase
- Nrf2
nuclear factor2
- O2•−
superoxide
- PAEC
pulmonary artery endothelial cell
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary artery smooth muscle cell
- PDI
protein disulfide isomerase
- PEPT1
peptide transporter 1
- PERK
protein kinase RNA-like ER kinase
- PHD
prolyl hydroxylase domain protein
- PKR
double-stranded RNA-dependent protein kinase
- PlGF
placental growth factor
- PRX3
peroxiredoxin 3
- ROS
reactive oxygen species
- RUPP
reduced uterine perfusion pressure
- RVSP
right ventricular systolic pressure
- RyR2
ryanodine receptor 2
- SDHD
succinate dehydrogenase complex subunit D
- sFlt
soluble fms-like tyrosine kinase
- siRNA
short interfering RNA
- SOD1
superoxide dismutase 1
- TC
ternary complex
- TCA
tricarboxylic acid
- TRPC1
transient receptor potential channel 1
- TRPV4
transient receptor potential cation channel subfamily V 4
- TRX2
thioredoxin 2
- TRXR2
thioredoxin reductase 2
- TUDCA
tauroursodeoxycholic acid
- UCP2
uncoupling protein 2
- UPR
unfolded protein response
- VDAC1
voltage-dependent anion-selective channel protein 1
- VHL
von Hippel-Lindau complex
- VSMCs
vascular smooth muscle cells
- XBP1
X-box binding protein 1
Biographies
Xiang-Qun Hu

Dr Hu received his PhD in Pharmacology from Iowa State University in 1994 and is Associate Research Professor at the Lawrence D. Longo, MD Center for Perinatal Biology in the Loma Linda University School of Medicine. His work has focused on the regulation of ion channels including voltage-gated ion channels in smooth muscle and ligand-gated ion channels in the central nervous system. His current research examines the roles of ion channels in uterine vascular adaptation and maladaptation in normal and pathological pregnancies.
Lubo Zhang

Dr Zhang is Professor of Pharmacology and Physiology and Director of the Lawrence D. Longo, MD Center for Perinatal Biology in the Loma Linda University School of Medicine. He was the President of the Western Pharmacology Society in the US in 2008. He has been a member of various grant review boards for the US National Institutes of Health and the American Heart Association for more than 20 years. Dr. Zhang is the author or coauthor of over 600 scientific articles, book chapters and abstracts. His research interests focus on molecular and epigenetic mechanisms in uteroplacental circulation and on the developmental programming of adult cardiovascular and neurological disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009; 417: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal 2009; 11: 2317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 2020; 21: 268–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schonenberger MJ, Kovacs WJ. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol 2015; 3: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain V, Bordes S, Bhardwaj A. Physiology, pulmonary circulatory system. Treasure Island, FL: StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 6.Wang Y, Zhao S. Placental blood circulation. Vascular biology of the placenta. San Rafael, CA: Morgan & Claypool Life Sciences; 2010. [PubMed] [Google Scholar]
- 7.Weissmann N, Grimminger F, Walmrath D, Seeger W. Hypoxic vasoconstriction in buffer-perfused rabbit lungs. Respir Physiol 1995; 100: 159–69. [DOI] [PubMed] [Google Scholar]
- 8.Makowski EL, Hertz RH, Meschia G. Effects of acute maternal hypoxia and hyperoxia on the blood flow to the pregnant uterus. Am J Obstet Gynecol 1973; 115: 624–31. [DOI] [PubMed] [Google Scholar]
- 9.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, et al. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res 2007; 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zelt JGE, Chaudhary KR, Cadete VJ, Mielniczuk LM, Stewart DJ. Medical therapy for heart failure associated with pulmonary hypertension. Circ Res 2019; 124: 1551–67. [DOI] [PubMed] [Google Scholar]
- 12.Wijeratne DT, Lajkosz K, Brogly SB, Lougheed MD, Jiang L, Housin A, et al. Increasing incidence and prevalence of world health organization groups 1 to 4 pulmonary hypertension: a population-based cohort study in Ontario, Canada. Circ Cardiovasc Qual Outcomes 2018; 11: e003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chi AY, Waypa GB, Mungai PT, Schumacker PT. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal 2010; 12: 603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ducsay CA, Goyal R, Pearce WJ, Wilson S, Hu XQ, Zhang L. Gestational hypoxia and developmental plasticity. Physiol Rev 2018; 98: 1241–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rana S, Lemoine E, Granger JP, Karumanchi SA. Preeclampsia: pathophysiology, challenges, and perspectives. Circ Res 2019; 124: 1094–112. [DOI] [PubMed] [Google Scholar]
- 16.Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018; 72: 24–43. [DOI] [PubMed] [Google Scholar]
- 17.Lyall F, Robson SC, Bulmer JN. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: relationship to clinical outcome. Hypertension 2013; 62: 1046–54. [DOI] [PubMed] [Google Scholar]
- 18.Lang U, Baker RS, Braems G, Zygmunt M, Kunzel W, Clark KE. Uterine blood flow—a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol 2003; 110 Suppl 1: S55–61. [DOI] [PubMed] [Google Scholar]
- 19.Weiler J, Tong S, Palmer KR. Is fetal growth restriction associated with a more severe maternal phenotype in the setting of early onset pre-eclampsia? A retrospective study. PLoS One 2011; 6: e26937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huppertz B The critical role of abnormal trophoblast development in the etiology of preeclampsia. Curr Pharm Biotechnol 2018; 19: 771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung TH, Hsieh TT, Chen SF. Risk of abnormal fetal growth in women with early- and late-onset preeclampsia. Pregnancy Hypertens 2018; 12: 201–6. [DOI] [PubMed] [Google Scholar]
- 22.Robillard PY, Dekker G, Scioscia M, Bonsante F, Iacobelli S, Boukerrou M, et al. Increased BMI has a linear association with late-onset preeclampsia: a population-based study. PLoS One 2019; 14: e0223888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland OJ, Cuffe JSM, Dekker Nitert M, Callaway L, Kwan Cheung KA, Radenkovic F, et al. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis 2018; 9: 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolfo A, Many A, Racano A, Tal R, Tagliaferro A, Ietta F, et al. Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS One 2010; 5: e13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yung HW, Atkinson D, Campion-Smith T, Olovsson M, Charnock-Jones DS, Burton GJ. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J Pathol 2014; 234: 262–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lisonkova S, Joseph KS. Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. Am J Obstet Gynecol 2013; 209: 544. e1–544. e12. [DOI] [PubMed] [Google Scholar]
- 27.Lisonkova S, Sabr Y, Mayer C, Young C, Skoll A, Joseph KS. Maternal morbidity associated with early-onset and late-onset preeclampsia. Obstet Gynecol 2014; 124: 771–81. [DOI] [PubMed] [Google Scholar]
- 28.Veerbeek JH, Hermes W, Breimer AY, van Rijn BB, Koenen SV, Mol BW, et al. Cardiovascular disease risk factors after early-onset preeclampsia, late-onset preeclampsia, and pregnancy-induced hypertension. Hypertension 2015; 65: 600–6. [DOI] [PubMed] [Google Scholar]
- 29.George EM, Granger JP. Endothelin: key mediator of hypertension in preeclampsia. Am J Hypertens 2011; 24: 964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kylhammar D, Radegran G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol (Oxf) 2017; 219: 728–56. [DOI] [PubMed] [Google Scholar]
- 31.Nathan SD, Hassoun PM. Pulmonary hypertension due to lung disease and/or hypoxia. Clin Chest Med 2013; 34: 695–705. [DOI] [PubMed] [Google Scholar]
- 32.Burton GJ, Yung HW. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens 2011; 1: 72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fulton DJR, Li X, Bordan Z, Haigh S, Bentley A, Chen F, et al. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants (Basel) 2017; 6: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci 2018; 19: 1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu Y, Yang W, Xie L, Liu T, Liu H, Liu B. Endoplasmic reticulum stress and pulmonary hypertension. Pulm Circ 2020; 10: 2045894019900121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J 2013; 280: 1944–65. [DOI] [PubMed] [Google Scholar]
- 37.Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol 2019; 11: a033944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yung HW, Colleoni F, Dommett E, Cindrova-Davies T, Kingdom J, Murray AJ, et al. Noncanonical mitochondrial unfolded protein response impairs placental oxidative phosphorylation in early-onset preeclampsia. Proc Natl Acad Sci U S A 2019; 116: 18109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 2015; 10: 173–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santos CX, Nabeebaccus AA, Shah AM, Camargo LL, Filho SV, Lopes LR. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 2014; 20: 121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23: 7198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science 2020; 368: eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep 2016; 17: 1374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wek RC. Role of eIF2α kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol 2018; 10: a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 46.Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol 2003; 23: 1292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, et al. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol 2003; 163: 767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van ‘t Wout EF, Hiemstra PS, Marciniak SJ. The integrated stress response in lung disease. Am J Respir Cell Mol Biol 2014; 50: 1005–9. [DOI] [PubMed] [Google Scholar]
- 49.Santos-Ribeiro D, Godinas L, Pilette C, Perros F. The integrated stress response system in cardiovascular disease. Drug Discov Today 2018; 23: 920–9. [DOI] [PubMed] [Google Scholar]
- 50.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552: 335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med 2009; 47: 333–43. [DOI] [PubMed] [Google Scholar]
- 52.Mailloux RJ, McBride SL, Harper ME. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci 2013; 38: 592–602. [DOI] [PubMed] [Google Scholar]
- 53.Tavender TJ, Bulleid NJ. Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid Redox Signal 2010; 13: 1177–87. [DOI] [PubMed] [Google Scholar]
- 54.Ramming T, Appenzeller-Herzog C. Destroy and exploit: catalyzed removal of hydroperoxides from the endoplasmic reticulum. Int J Cell Biol 2013; 2013: 180906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, et al. Nox4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 2007; 406: 105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 2004; 279: 20108–17. [DOI] [PubMed] [Google Scholar]
- 57.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003; 11: 619–33. [DOI] [PubMed] [Google Scholar]
- 58.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem 2008; 283: 31153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 2010; 30: 3553–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Awazawa M, Futami T, Sakada M, Kaneko K, Ohsugi M, Nakaya K, et al. Deregulation of pancreas-specific oxidoreductin ERO1β in the pathogenesis of diabetes mellitus. Mol Cell Biol 2014; 34: 1290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Copeland DE, Dalton AJ. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 1959; 5: 393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 2017; 62: 1–15. [DOI] [PubMed] [Google Scholar]
- 64.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006; 175: 901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998; 280: 1763–6. [DOI] [PubMed] [Google Scholar]
- 66.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142: 270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011; 476: 341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Booth DM, Enyedi B, Geiszt M, Varnai P, Hajnoczky G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell 2016; 63: 240–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 2011; 124: 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, et al. Ero1α regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid Redox Signal 2012; 16: 1077–87. [DOI] [PubMed] [Google Scholar]
- 71.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol 2014; 9: 47–71. [DOI] [PubMed] [Google Scholar]
- 72.Goda N, Kanai M. Hypoxia-inducible factors and their roles in energy metabolism. Int J Hematol 2012; 95: 457–63. [DOI] [PubMed] [Google Scholar]
- 73.Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordonez A, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting complex I activity. Cell Metab 2011; 14: 768–79. [DOI] [PubMed] [Google Scholar]
- 74.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007; 129: 111–22. [DOI] [PubMed] [Google Scholar]
- 75.Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, et al. Microrna-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein iscu. PLoS One 2010; 5: e10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol 2008; 28: 718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol 2007; 292: H101–8. [DOI] [PubMed] [Google Scholar]
- 78.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 1998; 95: 11715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 2005; 1: 401–8. [DOI] [PubMed] [Google Scholar]
- 80.Wang QS, Zheng YM, Dong L, Ho YS, Guo Z, Wang YX. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic Biol Med 2007; 42: 642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koumenis C, Bi M, Ye J, Feldman D, Koong AC. Hypoxia and the unfolded protein response. Methods Enzymol 2007; 435: 275–93. [DOI] [PubMed] [Google Scholar]
- 82.Lee D, Sun S, Zhang XQ, Zhang PD, Ho AS, Kiang KM, et al. Microrna-210 and endoplasmic reticulum chaperones in the regulation of chemoresistance in glioblastoma. J Cancer 2015; 6: 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takei N, Yoneda A, Sakai-Sawada K, Kosaka M, Minomi K, Tamura Y. Hypoxia-inducible ERO1α promotes cancer progression through modulation of integrin-β1 modification and signalling in HCT116 colorectal cancer cells. Sci Rep 2017; 7: 9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 2002; 22: 7405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012; 486: 126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chee NT, Lohse I, Brothers SP. mRNA-to-protein translation in hypoxia. Mol Cancer 2019; 18: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol 2006; 26: 3955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014; 508: 103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci Rep 2018; 8: 17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275: 25130–8. [DOI] [PubMed] [Google Scholar]
- 91.Belaidi E, Thomas A, Bourdier G, Moulin S, Lemarie E, Levy P, et al. Endoplasmic reticulum stress as a novel inducer of hypoxia inducible factor-1 activity: its role in the susceptibility to myocardial ischemia-reperfusion induced by chronic intermittent hypoxia. Int J Cardiol 2016; 210: 45–53. [DOI] [PubMed] [Google Scholar]
- 92.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006; 99: 675–91. [DOI] [PubMed] [Google Scholar]
- 93.Shimoda LA, Laurie SS. HIF and pulmonary vascular responses to hypoxia. J Appl Physiol (1985). 2014; 116: 867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tal R The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol Reprod 2012; 87: 134. [DOI] [PubMed] [Google Scholar]
- 95.Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension 2001; 38: 718–22. [DOI] [PubMed] [Google Scholar]
- 96.Wilkins MR, Ghofrani HA, Weissmann N, Aldashev A, Zhao L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation 2015; 131: 582–90. [DOI] [PubMed] [Google Scholar]
- 97.Robinson JC, Abbott C, Meadows CA, Roach RC, Honigman B, Bull TM. Long-term health outcomes in high-altitude pulmonary hypertension. High Alt Med Biol 2017; 18: 61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keyes LE, Armaza JF, Niermeyer S, Vargas E, Young DA, Moore LG. Intrauterine growth restriction, preeclampsia, and intrauterine mortality at high altitude in Bolivia. Pediatr Res 2003; 54: 20–5. [DOI] [PubMed] [Google Scholar]
- 99.Zamudio S High-altitude hypoxia and preeclampsia. Front Biosci 2007; 12: 2967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 2009; 297: L1013–32. [DOI] [PubMed] [Google Scholar]
- 101.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013; 127: 115–25. [DOI] [PubMed] [Google Scholar]
- 102.Koyama M, Furuhashi M, Ishimura S, Mita T, Fuseya T, Okazaki Y, et al. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2014; 306: H1314–23. [DOI] [PubMed] [Google Scholar]
- 103.Chen X, Yao JM, Fang X, Zhang C, Yang YS, Hu CP, et al. Hypoxia promotes pulmonary vascular remodeling via HIF-1α to regulate mitochondrial dynamics. J Geriatr Cardiol 2019; 16: 855–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou J, Xiao D, Hu Y, Wang Z, Paradis A, Mata-Greenwood E, et al. Gestational hypoxia induces preeclampsia-like symptoms via heightened endothelin-1 signaling in pregnant rats. Hypertension 2013; 62: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aljunaidy MM, Morton JS, Cooke CL, Davidge ST. Maternal vascular responses to hypoxia in a rat model of intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol 2016; 311: R1068–75. [DOI] [PubMed] [Google Scholar]
- 106.Turan S, Aberdeen GW, Thompson LP. Chronic hypoxia alters maternal uterine and fetal hemodynamics in the full-term pregnant guinea pig. Am J Physiol Regul Integr Comp Physiol 2017; 313: R330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ahmad A, Ahmad S, Malcolm KC, Miller SM, Hendry-Hofer T, Schaack JB, et al. Differential regulation of pulmonary vascular cell growth by hypoxia-inducible transcription factor-1α and hypoxia-inducible transcription factor-2α. Am J Respir Cell Mol Biol 2013; 49: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006; 113: 2630–41. [DOI] [PubMed] [Google Scholar]
- 109.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012; 110: 1484–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-tomesenchymal transition. Am J Physiol Lung Cell Mol Physiol 2018; 314: L256–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Veith C, Zakrzewicz D, Dahal BK, Balint Z, Murmann K, Wygrecka M, et al. Hypoxia- or PDGF-BB-dependent paxillin tyrosine phosphorylation in pulmonary hypertension is reversed by HIF-1α depletion or imatinib treatment. Thromb Haemost 2014; 112: 1288–303. [DOI] [PubMed] [Google Scholar]
- 112.Li Y, Shi B, Huang L, Wang X, Yu X, Guo B, et al. Suppression of the expression of hypoxia-inducible factor-1α by RNA interference alleviates hypoxia-induced pulmonary hypertension in adult rats. Int J Mol Med 2016; 38: 1786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 2018; 23: 1152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 1999; 103: 691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 2001; 281: L202–8. [DOI] [PubMed] [Google Scholar]
- 116.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 2014; 189: 314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003; 111: 1519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest 2010; 120: 827–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation 2016; 133: 2447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, et al. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem 2013; 288: 17134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 2006; 98: 1528–37. [DOI] [PubMed] [Google Scholar]
- 122.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 2002; 283: L144–55. [DOI] [PubMed] [Google Scholar]
- 123.Park WS, Firth AL, Han J, Ko EA. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J Smooth Muscle Res 2010; 46: 89–105. [DOI] [PubMed] [Google Scholar]
- 124.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 2006; 27: 939–58. [DOI] [PubMed] [Google Scholar]
- 125.Tissot van Patot M, Grilli A, Chapman P, Broad E, Tyson W, Heller DS, et al. Remodelling of uteroplacental arteries is decreased in high altitude placentae. Placenta 2003; 24: 326–35. [DOI] [PubMed] [Google Scholar]
- 126.Moser G, Windsperger K, Pollheimer J, de Sousa Lopes SC, Huppertz B. Human trophoblast invasion: new and unexpected routes and functions. Histochem Cell Biol 2018; 150: 361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.James JL, Stone PR, Chamley LW. The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy. Hum Reprod Update 2006; 12: 137–44. [DOI] [PubMed] [Google Scholar]
- 128.Saghian R, Bogle G, James JL, Clark AR. Establishment of maternal blood supply to the placenta: insights into plugging, unplugging and trophoblast behaviour from an agent-based model. Interface Focus 2019; 9: 20190019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 2000; 105: 577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 1997; 277: 1669–72. [DOI] [PubMed] [Google Scholar]
- 131.Wakeland AK, Soncin F, Moretto-Zita M, Chang CW, Horii M, Pizzo D, et al. Hypoxia directs human extravillous trophoblast differentiation in a hypoxia-inducible factor-dependent manner. Am J Pathol 2017; 187: 767–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Horii M, Li Y, Wakeland AK, Pizzo DP, Nelson KK, Sabatini K, et al. Human pluripotent stem cells as a model of trophoblast differentiation in both normal development and disease. Proc Natl Acad Sci U S A 2016; 113: E3882–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chang CW, Wakeland AK, Parast MM. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J Endocrinol 2018; 236: R43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rajakumar A, Conrad KP. Expression, ontogeny, and regulation of hypoxia-inducible transcription factors in the human placenta. Biol Reprod 2000; 63: 559–69. [DOI] [PubMed] [Google Scholar]
- 135.Cindrova-Davies T, van Patot MT, Gardner L, Jauniaux E, Burton GJ, Charnock-Jones DS. Energy status and HIF signalling in chorionic villi show no evidence of hypoxic stress during human early placental development. Mol Hum Reprod 2015; 21: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rajakumar A, Brandon HM, Daftary A, Ness R, Conrad KP. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 2004; 25: 763–9. [DOI] [PubMed] [Google Scholar]
- 137.Kimura C, Watanabe K, Iwasaki A, Mori T, Matsushita H, Shinohara K, et al. The severity of hypoxic changes and oxidative DNA damage in the placenta of early-onset preeclamptic women and fetal growth restriction. J Matern Fetal Neonatal Med 2013; 26: 491–6. [DOI] [PubMed] [Google Scholar]
- 138.Verma S, Pillay P, Naicker T, Moodley J, Mackraj I. Placental hypoxia inducible factor - 1α & CHOP immuno-histochemical expression relative to maternal circulatory syncytiotrophoblast micro-vesicles in preeclamptic and normotensive pregnancies. Eur J Obstet Gynecol Reprod Biol 2018; 220: 18–24. [DOI] [PubMed] [Google Scholar]
- 139.Sasagawa T, Nagamatsu T, Morita K, Mimura N, Iriyama T, Fujii T, et al. HIF-2α, but not HIF-1α, mediates hypoxia-induced up-regulation of Flt-1 gene expression in placental trophoblasts. Sci Rep 2018; 8: 17375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang Z, Huang C, Wang P, Gao J, Liu X, Li Y, et al. HIF-1α affects trophoblastic apoptosis involved in the onset of preeclampsia by regulating FOXO3a under hypoxic conditions. Mol Med Rep 2020; 21: 2484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zamudio S, Wu Y, Ietta F, Rolfo A, Cross A, Wheeler T, et al. Human placental hypoxia-inducible factor-1alpha expression correlates with clinical outcomes in chronic hypoxia in vivo. Am J Pathol 2007; 170: 2171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xiao D, Hu XQ, Huang X, Zhou J, Wilson SM, Yang S, et al. Chronic hypoxia during gestation enhances uterine arterial myogenic tone via heightened oxidative stress. PLoS One 2013; 8: e73731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gultice AD, Kulkarni-Datar K, Brown TL. Hypoxia-inducible factor 1alpha (HIF1A) mediates distinct steps of rat trophoblast differentiation in gradient oxygen. Biol Reprod 2009; 80: 184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tal R, Shaish A, Barshack I, Polak-Charcon S, Afek A, Volkov A, et al. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol 2010; 177: 2950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Albers RE, Kaufman MR, Natale BV, Keoni C, Kulkarni-Datar K, Min S, et al. Trophoblast-specific expression of Hif-1α results in preeclampsia-like symptoms and fetal growth restriction. Sci Rep 2019; 9: 2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003; 111: 649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sanchez-Aranguren LC, Espinosa-Gonzalez CT, Gonzalez-Ortiz LM, Sanabria-Barrera SM, Riano-Medina CE, Nunez AF, et al. Soluble Fms-like tyrosine kinase-1 alters cellular metabolism and mitochondrial bioenergetics in preeclampsia. Front Physiol 2018; 9: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gou D, Ramchandran R, Peng X, Yao L, Kang K, Sarkar J, et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol 2012; 303: L682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, et al. Genetic and hypoxic alterations of the microrna-210-iscu1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med 2015; 7: 695–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Muralimanoharan S, Maloyan A, Mele J, Guo C, Myatt LG, Myatt L. miR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012; 33: 816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Anton L, DeVine A, Polyak E, Olarerin-George A, Brown AG, Falk MJ, et al. HiIF 1α stabilization increases miR-210 eliciting first trimester extravillous trophoblast mitochondrial dysfunction. Front Physiol 2019; 10: 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Luo R, Shao X, Xu P, Liu Y, Wang Y, Zhao Y, et al. MicroRNA-210 contributes to preeclampsia by downregulating potassium channel modulatory factor 1. Hypertension 2014; 64: 839–45. [DOI] [PubMed] [Google Scholar]
- 153.Hu XQ, Dasgupta C, Xiao D, Huang X, Yang S, Zhang L. MicroRNA-210 targets ten-eleven translocation methylcytosine dioxygenase 1 and suppresses pregnancy-mediated adaptation of large conductance Ca2+-activated K+ channel expression and function in ovine uterine arteries. Hypertension 2017; 70: 601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Anton L, Olarerin-George AO, Schwartz N, Srinivas S, Bastek J, Hogenesch JB, et al. MiR-210 inhibits trophoblast invasion and is a serum biomarker for preeclampsia. Am J Pathol 2013; 183: 1437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hu XQ, Dasgupta C, Xiao J, Yang S, Zhang L. Long-term high altitude hypoxia during gestation suppresses large conductance Ca2+-activated K+ channel function in uterine arteries: a causal role for microRNA-210. J Physiol 2018; 596: 5891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Federici C, Drake KM, Rigelsky CM, McNelly LN, Meade SL, Comhair SA, et al. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015; 192: 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adesina SE, Kang BY, Bijli KM, Ma J, Cheng J, Murphy TC, et al. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med 2015; 87: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet 2015; 6: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Birjiniuk A, Glinton KE, Villafranco N, Boyer S, Laufman J, Mizerik E, et al. Multiple mitochondrial dysfunctions syndrome 1: an unusual cause of developmental pulmonary hypertension. Am J Med Genet A 2020; 182: 755–61. [DOI] [PubMed] [Google Scholar]
- 160.Niihori M, Eccles CA, Kurdyukov S, Zemskova M, Varghese MV, Stepanova AA, et al. Rats with a human mutation of NFU1 develop pulmonary hypertension. Am J Respir Cell Mol Biol 2020; 62: 231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hale A, Lee C, Annis S, Min PK, Pande R, Creager MA, et al. An Argonaute 2 switch regulates circulating miR-210 to coordinate hypoxic adaptation across cells. Biochim Biophys Acta 2014; 1843: 2528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hu H, Ding Y, Wang Y, Geng S, Liu J, He J, et al. MitoKATP channels promote the proliferation of hypoxic human pulmonary artery smooth muscle cells via the ROS/HIF/miR-210/ISCU signaling pathway. Exp Ther Med 2017; 14: 6105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu HL, Zhang ZX, Chen CS, Cai C, Zhao JP, Wang X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 2010; 42: 661–6. [DOI] [PubMed] [Google Scholar]
- 164.Lu Y, Huang J, Geng S, Chen H, Song C, Zhu S, et al. MitoKATP regulating HIF/miR-210/ISCU signaling axis and formation of a positive feedback loop in chronic hypoxia-induced PAH rat model. Exp Ther Med 2017; 13: 1697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 1998; 351: 726–7. [DOI] [PubMed] [Google Scholar]
- 166.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A 2007; 104: 11418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 1997; 100: 2347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 2004; 95: 496–505. [DOI] [PubMed] [Google Scholar]
- 169.Yang XR, Lin AH, Hughes JM, Flavahan NA, Cao YN, Liedtke W, et al. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2012; 302: L555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dahan D, Ducret T, Quignard JF, Marthan R, Savineau JP, Esteve E. Implication of the ryanodine receptor in TRPV 4-induced calcium response in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2012; 303: L824–33. [DOI] [PubMed] [Google Scholar]
- 171.Yadav VR, Song T, Mei L, Joseph L, Zheng YM, Wang YX. PLCγ1-PKCε-IP3R1 signaling plays an important role in hypoxia-induced calcium response in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2018; 314: L724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang T, Zhang ZX, Xu YJ. Effect of mitochondrial KATP channel on voltage-gated K+ channel in 24 hour-hypoxic human pulmonary artery smooth muscle cells. Chin Med J (Engl) 2005; 118: 12–9. [PubMed] [Google Scholar]
- 173.Chen TX, Xu XY, Zhao Z, Zhao FY, Gao YM, Yan XH, et al. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca2+ entry into rat pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2017; 312: L477–87. [DOI] [PubMed] [Google Scholar]
- 174.Mei L, Zheng YM, Song T, Yadav VR, Joseph LC, Truong L, et al. Rieske iron-sulfur protein induces FKBP12.6/RyR 2 complex remodeling and subsequent pulmonary hypertension through NF-κ B/cyclin D 1 pathway. Nat Commun 2020; 11: 3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lu W, Ran P, Zhang D, Peng G, Li B, Zhong N, et al. Sildenafil inhibits chronically hypoxic upregulation of canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am J Physiol Cell Physiol 2010; 298: C114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sheak JR, Yan S, Weise-Cross L, Ahmadian R, Walker BR, Jernigan NL, et al. PKCβ and reactive oxygen species mediate enhanced pulmonary vasoconstrictor reactivity following chronic hypoxia in neonatal rats. Am J Physiol Heart Circ Physiol 2020; 318: H470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Suresh K, Servinsky L, Jiang H, Bigham Z, Yun X, Kliment C, et al. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2018; 314: L893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010; 121: 2661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pinto MC, Kihara AH, Goulart VA, Tonelli FM, Gomes KN, Ulrich H, et al. Calcium signaling and cell proliferation. Cell Signal 2015; 27: 2139–49. [DOI] [PubMed] [Google Scholar]
- 180.Price LC, Caramori G, Perros F, Meng C, Gambaryan N, Dorfmuller P, et al. Nuclear factor κ-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS One 2013; 8: e75415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A, et al. Pathophysiological roles of nuclear factor kappaB (NF-κB) in pulmonary arterial hypertension: effects of synthetic selective NF-κB inhibitor IMD-0354. Cardiovasc Res 2013; 99: 35–43. [DOI] [PubMed] [Google Scholar]
- 182.Fan J, Fan X, Li Y, Ding L, Zheng Q, Guo J, et al. Chronic normobaric hypoxia induces pulmonary hypertension in rats: role of NF-κB. High Alt Med Biol 2016; 17: 43–9. [DOI] [PubMed] [Google Scholar]
- 183.Patel H, Zaghloul N, Lin K, Liu SF, Miller EJ, Ahmed M. Hypoxia-induced activation of specific members of the NF-κB family and its relevance to pulmonary vascular remodeling. Int J Biochem Cell Biol 2017; 92: 141–7. [DOI] [PubMed] [Google Scholar]
- 184.Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, et al. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res 2009; 10: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.He RL, Wu ZJ, Liu XR, Gui LX, Wang RX, Lin MJ. Calcineurin/NFAT signaling modulates pulmonary artery smooth muscle cell proliferation, migration and apoptosis in monocrotaline-induced pulmonary arterial hypertension rats. Cell Physiol Biochem 2018; 49: 172–89. [DOI] [PubMed] [Google Scholar]
- 186.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009; 30 Suppl A: S43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Murray AJ. Oxygen delivery and fetal-placental growth: beyond a question of supply and demand? Placenta 2012; 33 Suppl 2: e16–22. [DOI] [PubMed] [Google Scholar]
- 188.Torbergsen T, Oian P, Mathiesen E, Borud O. Pre-eclampsia—a mitochondrial disease? Acta Obstet Gynecol Scand 1989; 68: 145–8. [DOI] [PubMed] [Google Scholar]
- 189.Xu Z, Jin X, Cai W, Zhou M, Shao P, Yang Z, et al. Proteomics analysis reveals abnormal electron transport and excessive oxidative stress cause mitochondrial dysfunction in placental tissues of early-onset preeclampsia. Proteomics Clin Appl 2018; 12: e1700165. [DOI] [PubMed] [Google Scholar]
- 190.Shi Z, Long W, Zhao C, Guo X, Shen R, Ding H. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of preeclampsia. PLoS One. 2013; 8: e64351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot MC, et al. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: a role for miRNA-210 and protein synthesis inhibition. PLoS One 2013; 8: e55194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Beyramzadeh M, Dikmen ZG, Erturk NK, Tuncer ZS, Akbiyik F. Placental respiratory chain complex activities in high risk pregnancies. J Matern Fetal Neonatal Med 2017; 30: 2911–7. [DOI] [PubMed] [Google Scholar]
- 193.Vaka VR, McMaster KM, Cornelius DC, Ibrahim T, Jayaram A, Usry N, et al. Natural killer cells contribute to mitochondrial dysfunction in response to placental ischemia in reduced uterine perfusion pressure rats. Am J Physiol Regul Integr Comp Physiol 2019; 316: R441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang Y, Walsh SW. Placental mitochondria as a source of oxidative stress in preeclampsia. Placenta 1998; 19: 581–6. [DOI] [PubMed] [Google Scholar]
- 195.Shibata E, Nanri H, Ejima K, Araki M, Fukuda J, Yoshimura K, et al. Enhancement of mitochondrial oxidative stress and up-regulation of antioxidant protein peroxiredoxin III/SP-22 in the mitochondria of human pre-eclamptic placentae. Placenta 2003; 24: 698–705. [DOI] [PubMed] [Google Scholar]
- 196.Richter HG, Camm EJ, Modi BN, Naeem F, Cross CM, Cindrova-Davies T, et al. Ascorbate prevents placental oxidative stress and enhances birth weight in hypoxic pregnancy in rats. J Physiol 2012; 590: 1377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yang Y, Xu P, Zhu F, Liao J, Wu Y, Hu M, et al. The potent antioxidant mitoq protects against preeclampsia during late gestation but increases the risk of preeclampsia when administered in early pregnancy. Antioxid Redox Signal 2020; 34: 118–36. [DOI] [PubMed] [Google Scholar]
- 198.Wang Y, Walsh SW. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta 2001; 22: 206–12. [DOI] [PubMed] [Google Scholar]
- 199.Zamudio S, Kovalenko O, Vanderlelie J, Illsley NP, Heller D, Belliappa S, et al. Chronic hypoxia in vivo reduces placental oxidative stress. Placenta 2007; 28: 846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Smith AN, Wang X, Thomas DG, Tatum RE, Booz GW, Cunningham MW Jr. The role of mitochondrial dysfunction in preeclampsia: causative factor or collateral damage? Am J Hypertens 2021; 34:442–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hu X-Q, Zhang L. Hypoxia and mitochondrial dysfunction in pregnancy complications. Antioxidants 2021; 10: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Covarrubias AE, Lecarpentier E, Lo A, Salahuddin S, Gray KJ, Karumanchi SA, et al. Ap39, a modulator of mitochondrial bioenergetics, reduces antiangiogenic response and oxidative stress in hypoxia-exposed trophoblasts: relevance for preeclampsia pathogenesis. Am J Pathol 2019; 189: 104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, et al. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 2006; 291: R1085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vadysirisack DD, Ellisen LW. mTOR activity under hypoxia. Methods Mol Biol 2012; 821: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.He C, Shan N, Xu P, Ge H, Yuan Y, Liu Y, et al. Hypoxia-induced downregulation of SRC-3 suppresses trophoblastic invasion and migration through inhibition of the AKT/mTOR pathway: implications for the pathogenesis of preeclampsia. Sci Rep 2019; 9: 10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol 2007; 582: 449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, et al. Mechanistic target of rapamycin complex 1 promotes the expression of genes encoding electron transport chain proteins and stimulates oxidative phosphorylation in primary human trophoblast cells by regulating mitochondrial biogenesis. Sci Rep 2019; 9: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Li L, Huang X, He Z, Xiong Y, Fang Q. miRNA-210–3p regulates trophoblast proliferation and invasiveness through fibroblast growth factor 1 in selective intrauterine growth restriction. J Cell Mol Med 2019; 23: 4422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lee DC, Romero R, Kim JS, Tarca AL, Montenegro D, Pineles BL, et al. Mir-210 targets iron-sulfur cluster scaffold homologue in human trophoblast cell lines: siderosis of interstitial trophoblasts as a novel pathology of preterm preeclampsia and small-for-gestational-age pregnancies. Am J Pathol 2011; 179: 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tang C, Liang J, Qian J, Jin L, Du M, Li M, et al. Opposing role of JNK-p38 kinase and ERK1/2 in hydrogen peroxide-induced oxidative damage of human trophoblast-like JEG-3 cells. Int J Clin Exp Pathol 2014; 7: 959–68. [PMC free article] [PubMed] [Google Scholar]
- 211.Khera A, Vanderlelie JJ, Holland O, Perkins AV. Overexpression of endogenous antioxidants with selenium supplementation protects trophoblast cells from reactive oxygen species-induced apoptosis in a Bcl-2-dependent manner. Biol Trace Elem Res 2017; 177: 394–403. [DOI] [PubMed] [Google Scholar]
- 212.Rosenfeld CR, Cornfield DN, Roy T. Ca(2+)-activated K(+) channels modulate basal and E(2)beta-induced rises in uterine blood flow in ovine pregnancy. Am J Physiol Heart Circ Physiol 2001; 281: H422–31. [DOI] [PubMed] [Google Scholar]
- 213.Hu XQ, Huang X, Xiao D, Zhang L. Direct effect of chronic hypoxia in suppressing large conductance Ca(2+)-activated K(+) channel activity in ovine uterine arteries via increasing oxidative stress. J Physiol 2016; 594: 343–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hu XQ, Song R, Romero M, Dasgupta C, Min J, Hatcher D, et al. Gestational hypoxia inhibits pregnancy-induced upregulation of Ca2+ sparks and spontaneous transient outward currents in uterine arteries via heightened endoplasmic reticulum/oxidative stress. Hypertension 2020; 76: 930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 2011; 3: 88ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yeager ME, Reddy MB, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Activation of the unfolded protein response is associated with pulmonary hypertension. Pulm Circ 2012; 2: 229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wu Y, Adi D, Long M, Wang J, Liu F, Gai MT, et al. 4-phenylbutyric acid induces protection against pulmonary arterial hypertension in rats. PLoS One 2016; 11: e0157538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang AP, Li XH, Yang YM, Li WQ, Zhang W, Hu CP, et al. A critical role of the mTOR/eIF2α pathway in hypoxia-induced pulmonary hypertension. PLoS One 2015; 10: e0130806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cao X, He Y, Li X, Xu Y, Liu X. The IRE1α-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am J Transl Res 2019; 11: 641–54. [PMC free article] [PubMed] [Google Scholar]
- 220.Gerasimova EM, Fedotov SA, Kachkin DV, Vashukova ES, Glotov AS, Chernoff YO, et al. Protein misfolding during pregnancy: new approaches to preeclampsia diagnostics. Int J Mol Sci 2019; 20: 6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol 2008; 173: 451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mizuuchi M, Cindrova-Davies T, Olovsson M, Charnock-Jones DS, Burton GJ, Yung HW. Placental endoplasmic reticulum stress negatively regulates transcription of placental growth factor via ATF4 and ATF6β: implications for the pathophysiology of human pregnancy complications. J Pathol 2016; 238: 550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis 2019; 10: 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yung HW, Cox M, Tissot van Patot M, Burton GJ. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB J 2012; 26: 1970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nuzzo AM, Camm EJ, Sferruzzi-Perri AN, Ashmore TJ, Yung HW, Cindrova-Davies T, et al. Placental adaptation to early-onset hypoxic pregnancy and mitochondria-targeted antioxidant therapy in a rodent model. Am J Pathol 2018; 188: 2704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yang Y, Li J, Han TL, Zhou X, Qi H, Baker PN, et al. Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia. Cell Tissue Res 2020; 379: 589–99. [DOI] [PubMed] [Google Scholar]
- 227.Kawakami T, Yoshimi M, Kadota Y, Inoue M, Sato M, Suzuki S. Prolonged endoplasmic reticulum stress alters placental morphology and causes low birth weight. Toxicol Appl Pharmacol 2014; 275: 134–44. [DOI] [PubMed] [Google Scholar]
- 228.Lombardi AA, Elrod JW. Mediating ER-mitochondrial cross-talk. Science 2017; 358: 591–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ramo O, Kumar D, Gucciardo E, Joensuu M, Saarekas M, Vihinen H, et al. NOGO-A-/RTN4a and NOGO-b/RTN4b are simultaneously expressed in epithelial, fibroblast and neuronal cells and maintain ER morphology. Sci Rep 2016; 6: 35969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 2007; 9: 445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pak O, Sommer N, Hoeres T, Bakr A, Waisbrod S, Sydykov A, et al. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am J Respir Cell Mol Biol 2013; 49: 358–67. [DOI] [PubMed] [Google Scholar]
- 232.Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, et al. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol 2015; 35: 1166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 2013; 113: 126–36. [DOI] [PubMed] [Google Scholar]
- 234.Stark MJ, Hodyl NA, Butler M, Clifton VL. Localisation and characterisation of uncoupling protein-2 (UCP2) in the human preterm placenta. Placenta 2012; 33: 1020–5. [DOI] [PubMed] [Google Scholar]
- 235.Wang Z, Wang H, Xu ZM, Ji YL, Chen YH, Zhang ZH, et al. Cadmium-induced teratogenicity: association with ROS-mediated endoplasmic reticulum stress in placenta. Toxicol Appl Pharmacol 2012; 259: 236–47. [DOI] [PubMed] [Google Scholar]
- 236.Burton GJ, Sebire NJ, Myatt L, Tannetta D, Wang YL, Sadovsky Y, et al. Optimising sample collection for placental research. Placenta 2014; 35: 9–22. [DOI] [PubMed] [Google Scholar]
- 237.Cindrova-Davies T, Yung HW, Johns J, Spasic-Boskovic O, Korolchuk S, Jauniaux E, et al. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am J Pathol 2007; 171: 1168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Veerbeek JH, Tissot Van Patot MC, Burton GJ, Yung HW. Endoplasmic reticulum stress is induced in the human placenta during labour. Placenta 2015; 36: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Docherty CK, Nilsen M, MacLean MR. Influence of 2-methoxyestradiol and sex on hypoxia-induced pulmonary hypertension and hypoxia-inducible factor-1-alpha. J Am Heart Assoc 2019; 8: e011628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.He Y, Fang X, Shi J, Li X, Xie M, Liu X. Apigenin attenuates pulmonary hypertension by inducing mitochondria-dependent apoptosis of PASMCs via inhibiting the hypoxia inducible factor 1α-KV1.5 channel pathway. Chem Biol Interact 2020; 317: 108942. [DOI] [PubMed] [Google Scholar]
- 241.Zhang L, Pu Z, Wang J, Zhang Z, Hu D, Wang J. Baicalin inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via the AKT/HIF-1α/p27-associated pathway. Int J Mol Sci 2014; 15: 8153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, et al. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am J Respir Crit Care Med 2018; 198: 1423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kurosawa R, Satoh K, Kikuchi N, Kikuchi H, Saigusa D, Al-Mamun ME, et al. Identification of celastramycin as a novel therapeutic agent for pulmonary arterial hypertension. Circ Res 2019; 125: 309–27. [DOI] [PubMed] [Google Scholar]
- 244.Koulmann N, Novel-Chate V, Peinnequin A, Chapot R, Serrurier B, Simler N, et al. Cyclosporin A inhibits hypoxia-induced pulmonary hypertension and right ventricle hypertrophy. Am J Respir Crit Care Med 2006; 174: 699–705. [DOI] [PubMed] [Google Scholar]
- 245.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, et al. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci U S A 2012; 109: 1239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Siddique MAH, Satoh K, Kurosawa R, Kikuchi N, Elias-Al-Mamun M, Omura J, et al. Identification of emetine as a therapeutic agent for pulmonary arterial hypertension: novel effects of an old drug. Arterioscler Thromb Vasc Biol 2019; 39: 2367–85. [DOI] [PubMed] [Google Scholar]
- 247.Jin H, Wang Y, Zhou L, Liu L, Zhang P, Deng W, et al. Melatonin attenuates hypoxic pulmonary hypertension by inhibiting the inflammation and the proliferation of pulmonary arterial smooth muscle cells. J Pineal Res 2014; 57: 442–50. [DOI] [PubMed] [Google Scholar]
- 248.Hu CJ, Poth JM, Zhang H, Flockton A, Laux A, Kumar S, et al. Suppression of HIF2 signalling attenuates the initation of hypoxia-induced pulmonary hypertension. Eur Respir J 2019; 54: 1900378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Jiang Y, Zhou Y, Peng G, Liu N, Tian H, Pan D, et al. Topotecan prevents hypoxia-induced pulmonary arterial hypertension and inhibits hypoxia-inducible factor-1α and TRPC channels. Int J Biochem Cell Biol 2018; 104: 161–70. [DOI] [PubMed] [Google Scholar]
- 250.Huh JW, Kim SY, Lee JH, Lee YS. YC −1 attenuates hypoxia-induced pulmonary arterial hypertension in mice. Pulm Pharmacol Ther 2011; 24: 638–46. [DOI] [PubMed] [Google Scholar]
- 251.Cheng CC, Chi PL, Shen MC, Shu CW, Wann SR, Liu CP, et al. Caffeic acid phenethyl ester rescues pulmonary arterial hypertension through the inhibition of AKT/ERK-dependent PDGF/HIF-1α in vitro and in vivo. Int J Mol Sci 2019; 20: 1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chen T, Zhou Q, Tang H, Bozkanat M, Yuan JX, Raj JU, et al. miR-17/20 controls prolyl hydroxylase 2 (PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary artery smooth muscle cell proliferation. J Am Heart Assoc 2016; 5: e004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Luo Y, Teng X, Zhang L, Chen J, Liu Z, Chen X, et al. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun 2019; 10: 3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, et al. Deficiency in catechol-o-methyltransferase and 2-methoxyoestradiol is associated with preeclampsia. Nature 2008; 453: 1117–21. [DOI] [PubMed] [Google Scholar]
- 255.Huo X, Wang C, Yu Z, Peng Y, Wang S, Feng S, et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: an implication of the therapeutic potential. J Pineal Res 2017; 62: e12390. [DOI] [PubMed] [Google Scholar]
- 256.Tan DX, Manchester LC, Qin L, Reiter RJ. Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci 2016; 17: 2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Uzun M, Gencer M, Turkon H, Oztopuz RO, Demir U, Ovali MA. Effects of melatonin on blood pressure, oxidative stress and placental expressions of TNFα, IL-6, VEGF and sFlt-1 in RUPP rat model of preeclampsia. Arch Med Res 2017; 48: 592–8. [DOI] [PubMed] [Google Scholar]
- 258.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 2010; 1201: 96–103. [DOI] [PubMed] [Google Scholar]
- 259.Pak O, Scheibe S, Esfandiary A, Gierhardt M, Sydykov A, Logan A, et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur Respir J 2018; 51: 1701024. [DOI] [PubMed] [Google Scholar]
- 260.Phillips TJ, Scott H, Menassa DA, Bignell AL, Sood A, Morton JS, et al. Treating the placenta to prevent adverse effects of gestational hypoxia on fetal brain development. Sci Rep 2017; 7: 9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Aljunaidy MM, Morton JS, Kirschenman R, Phillips T, Case CP, Cooke CM, et al. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol Res 2018; 134: 332–42. [DOI] [PubMed] [Google Scholar]
- 262.Botting KJ, Skeffington KL, Niu Y, Allison BJ, Brain KL, Itani N, et al. Translatable mitochondria-targeted protection against programmed cardiovascular dysfunction. Sci Adv 2020; 6: eabb1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Vaka VR, McMaster KM, Cunningham MW Jr, Ibrahim T, Hazlewood R, Usry N, et al. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 2018; 72: 703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lee B, Rhead W, Diaz GA, Scharschmidt BF, Mian A, Shchelochkov O, et al. Phase 2 comparison of a novel ammonia scavenging agent with sodium phenylbutyrate in patients with urea cycle disorders: safety, pharmacokinetics and ammonia control. Mol Genet Metab 2010; 100: 221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Crosignani A, Battezzati PM, Setchell KD, Invernizzi P, Covini G, Zuin M, et al. Tauroursodeoxycholic acid for treatment of primary biliary cirrhosis. A dose–response study. Dig Dis Sci 1996; 41: 809–15. [DOI] [PubMed] [Google Scholar]
- 266.Wang JJ, Zuo XR, Xu J, Zhou JY, Kong H, Zeng XN, et al. Evaluation and treatment of endoplasmic reticulum (ER) stress in right ventricular dysfunction during monocrotaline-induced rat pulmonary arterial hypertension. Cardiovasc Drugs Ther 2016; 30: 587–98. [DOI] [PubMed] [Google Scholar]
- 267.Shimizu T, Higashijima Y, Kanki Y, Nakaki R, Kawamura T, Urade Y, et al. PERK inhibition attenuates vascular remodeling in pulmonary arterial hypertension caused by BMPR2 mutation. Sci Signal 2021; 14: eabb3616. [DOI] [PubMed] [Google Scholar]
- 268.Wu J, Pan W, Wang C, Dong H, Xing L, Hou J, et al. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci Rep 2019; 39: BSR20190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Cindrova-Davies T The therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front Pharmacol 2014; 5: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013; 2: e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J 2015; 29: 4766–71. [DOI] [PubMed] [Google Scholar]