

Abstract
The single-nucleotide polymorphism (SNP), rs3184504, is broadly associated with increased risk for multiple autoimmune and cardiovascular diseases. Although the allele is uniquely enriched in European descent, the mechanism for the widespread selective sweep is not clear. Here, we find the rs3184504*T allele had a strong association with reduced mortality in a human sepsis cohort. The rs3184504*T allele associates with a loss-of-function amino acid change (p.R262W) in the adaptor protein SH2B3, a likely causal variant. To better understand the role of SH2B3 in sepsis, we utilized mouse modeling and challenged SH2B3-deficient mice with a polymicrobial cecal-ligation puncture (CLP) procedure. We found SH2B3 deficiency improved survival and morbidity with less organ damage and earlier bacterial clearance compared to control mice. The peritoneal infiltrating cells exhibited augmented phagocytosis in SH2B3KO mice with enriched recruitment of Ly6Chi inflammatory monocytes despite equivalent or reduced chemokine expression. Rapid cycling of monocytes and progenitors occurred uniquely in the Sh2b3KO mice following CLP suggesting augmented myelopoiesis. To model the hypomorphic autoimmune risk allele, we created a novel knockin mouse harboring a similar point mutation in the murine PH domain of SH2B3. At baseline, phenotypic changes suggested a hypomorphic allele. In the CLP model, homozygous knockin mice displayed improved mortality and morbidity compared to wildtype or heterozygous mice. Collectively, these data suggest that hypomorphic SH2B3 improves the sepsis response and that balancing selection likely contributed to the relative frequency of the autoimmune risk variant.
Keywords: Human, Rodent, Monocytes, Neutrophils, Autoimmunity, Bacterial, Sepsis
INTRODUCTION
Autoimmune risk alleles have been associated in genome-wide association studies (GWAS) for multiple autoimmune diseases, but the underlying molecular mechanisms remain elusive (1). The same loci may improve fitness against a known pathogen(s) and thus be enriched in the population (2). The 12q24.12 region is an example of a large linkage disequilibrium region shown to have undergone a selective sweep in humans of European descent (3). The allele has progressed to become the predominant haplotype in several human populations despite associating with numerous autoimmune diseases, including type 1 diabetes (T1D) (4), celiac disease, and rheumatoid arthritis (RA) (5), as well as inflammatory conditions, such as coronary artery disease (6) and hypertension (7).
The only credible coding variant in the region is the non-synonymous single-nucleotide polymorphism (SNP), rs3184504*T, encoding a hypomorphic variant in the adaptor protein SH2B3 (also termed LNK) (6). SH2B3 is a negative regulator of a range of receptor/non-receptor tyrosine kinases (RTK) and cytokine receptors (including JAK2, c-fms, c-kit, mpl, IL-7R) (9). Loss-of-function or deletion of human SH2B3 associates with acute lymphoblastic leukemia and several myeloid neoplasms (10). Missense somatic and germline PH domain mutations in SH2B3 have previously been identified in subjects with myeloproliferative disorders and in juvenile myelomonocytic leukemia (JMML) (30, 31), but the 262W variant has not been implicated (32). The human rs3184504*T allele did associate with enhanced JAK2-dependent cytokine receptor signaling in vitro (6) suggesting that the degree of augmented signaling is not pathogenic. Furthermore, the rs3184504*T allele associates with multiple blood indices across numerous cell lineages including progenitor cell populations (15–18). SH2B3 variants may alter the immune response by skewing hematopoiesis during infection. However, any fitness variant effect on immune function may be tempered by malignancy risk.
Sh2b3-deficient mice have been extensively studied and demonstrate baseline augmentation of several hematopoietic lineages including hematopoietic stem cells, pre-B to mature B cells, dendritic cells, lymphocytes, monocytes and megakaryocytes (8). Sh2b3−/− animals additionally develop a myeloproliferative-like disease with a progressive phenotype in aged mice showing extramedullary hematopoiesis and splenomegaly (11–13). Mice on a high-fat inflammatory diet demonstrated augmented myelopoiesis, but direct testing of the role of SH2B3 in sepsis and during bacterial infections has not been performed. No hypomorphic Sh2b3 animal studies have been reported.
In this paper, we directly explore the role of the risk variant in bacterial sepsis. We tested the association of the human rs3184504*T allele with human sepsis outcomes and modeled this genetic variant in murine models for mechanistic insight. We used several cohorts of extensively genotyped patients, both healthy controls and sepsis patients, to assess circulating human peripheral blood subsets and human survival in sepsis in relation to the genetic allele. Next, we challenged mice with global deletion of SH2B3 or mice harboring a hypomorphic murine allele with cecal-ligation puncture, a model of polymicrobial sepsis. We tested the hypothesis that reduced function of SH2B3 promotes survival in sepsis.
MATERIALS & METHODS:
Human Subjects.
Healthy control adult subjects provided informed consent and studies were approved by the Institutional Review Board at Benaroya Research Institute (IRB07109–372). Subjects were genotyped and complete blood count data analyzed with gPLINK-2.050. Deidentified sepsis genotypes were obtained from the iSPAAR (identification of SNPs Altering ALI Risk) consortium, a large NHLBI funded multicenter genomics study to study genetic risk factors for Acute Respiratory Distress Syndrome (ARDS). Subjects were limited to Caucasians with sepsis (n=1542). Genomic and clinical variables for these subjects are publicly available (dbGaP Study Accession: phs000631.v1.p1). Subjects were originally genotyped using the Illumina 660Quad BeadChip (San Diego, CA). TagSNPs in the gene for SH2B3 were identified using the University of Washington genome variation server. Primary and secondary outcomes were inpatient mortality and peak 24-hour white blood cell count with select plasma cytokines measured by immunoassay (Meso Scale Discovery), respectively, with a (n=554) obtained within 48 hours of enrollment. Continuous outcome measures were natural log transformed prior to association testing in linear models. We performed multiple logistic regression adjusting for subject age and gender using an additive genetic model performed in Golden Helix (Bozeman, MT) or Stata 15 (College Station, TX).
Mice.
Mice were bred and maintained at the Seattle Children’s Research Institute Animal Facility. All animal care and experimentation occurred with Institutional Animal Care and Use Committee approval. The SH2B3flox/flox and SH2B3tm3Draw mice were two separate lines created (Biocytogen) by inserting LoxP sites engineered into intron 3 and downstream of 3’ UTR using C57BL6 mixed (75% B6/J and 25% B6/N) embryonic stem cells (Biocytogen). SH2B3tm3Draw mice additionally had a single nucleotide change (NM_008507.4:c701C>G, p.P234R) in the coding region. Mice were crossed to B6.C-Tg(CMV-cre)1Cgn/JCMV-Cre mice (JAX # 06054; N11) generated the SH2B3KO (SH2B3−/−) mouse line or the B6.Cg-Tg(ACTFLP3)9205Dym mice (JAX #005703) to remove the FRT-flanked neomycin-resistance cassette and maintained backcrossed to Jackson C57BL/6 mice. Bone marrow chimeric mice were generated by two equal daily 500 rad irradiation doses followed by intravenous injection of 5 × 106 purified bone marrow cells from either Sh2b3WT or Sh2b3KO mice. Murine complete blood counts (CBC) were performed at Pheonix Lab veterinary services facility using a Siemens Advia 2120 flow cytometry device with peroxidase methodology followed by confirmatory manual differentials. Baseline bone marrow and spleen from age-matched (16–18 months) littermate mice were preserved in 10% neutral buffered formalin+/− decalcification prior to paraffin-embedding and preparing 4μm sections, then stained with hematoxylin eosin. Blinded enumeration of megakaryocytes was performed in 5 high-power fields (40X) and summed per animal.
Procedures.
High-grade CLP model was performed as described previously (19) with survival rates in control mice expected to be ~20% similar to published protocols (42–43). All sepsis experiments had a blinded experimental design. Briefly, the distal 2/3 of the cecum from a 12- to 18-week-old male mouse was ligated and punctured with a 22-G needle prior to placing the cecum back into the abdomen. Sterile saline (1mL) was administered into the peritoneal cavity and the incision was closed using sutures and 9-mm steel wound clips. Antibiotics were not administered secondary to experimental design. Mouse rectal temperatures were measured at 24-hours after CLP. Clinical monitoring of mice was performed at least four times daily for the first 3 days, and then twice daily for 7 days. Animals considered to be moribund or below body temperature threshold were euthanized by CO2 inhalation. Tissues from CLP animals were harvested at 24-hours post-procedure. An all-inclusive histologic semi-quantitative severity score was utilized which a blinded comparative pathologist used to score lesions in liver and lung based on a scale of 1+ (minimal) to 4+ (severe). Criteria included the distribution of lesions, the severity of the inflammatory cell accumulation, severity of hepatocellular degeneration (for liver sections) or perivascular edema (for lung sections). The overall severity score is a sum of all scores for each mouse.
Metabolite, cytokine and chemokine measurements:
Murine TNF and IL-6 levels were measured using ELISA MAX Kits (BioLegend) with detection limits of 4 and 2 pg/ml, respectively. Murine KC amounts were measured with Mini ABST ELISA Kits (Peprotech) with detection limits of 16pg/ml and 4 pg/ml, respectively. Luminex Cytokine & Chemokine Panel 1 (26-plex) (ThermoFisher) analysis was performed. Human cytokines and chemokines were measured by immunoassay (Meso Scale Discovery). ALT and BUN levels were measured using a colorimetric method with detection limits are 0.5 IU/L and 100mg/dl, respectively (Teco Diagnostics).
Bacterial quantification, reactive oxygen species (ROS), and phagocytosis capacity assessment:
Peritoneal fluids or blood were diluted on LB agar (peritoneal fluids) or tryptic soy agar (TSA) with 5% sheep blood plates (blood). Colonies were visually scored after overnight incubation at 37°C. The oxidative stress indicator, CM-H2DCFDA (ThermoFisher) and FITC-labeled Escherichia coli (K-12 strain) BioParticles (ThermoFisher) were used for ROS and phagocytosis assays, respectively. Cells were stained with markers for neutrophils (CD11b+, Ly6G+, Ly6C−), monocytes (CD11b+, Ly6G−, Ly6C+), or macrophages (CD11b+, F4/80+) and evaluated via flow cytometry. As a control for ROS reactivity of each sample, the cells were treated with or without 100ng/mL PMA for 7 minutes at 37 degrees C.
Cell cycle analysis using in vivo EdU labeling.
5-ethynyl-2’-deoxyuridine (EdU, Thermo Fisher) was dissolved in sterile 1x PBS (10% DMSO) and 1mg in a volume of 100ul was injected i.p. 1 hr prior to sacrifice. EdU detection was performed with the Click-iT Plus EdU Pacific Blue Flow Cytometry Assay Kit (ThermoFisher). Cells from total bone marrow were stained for surface markers or for myeloid progenitors in the BM, total BM was subject to lineage depletion prior to surface staining followed by fixation and incubation with the Click-iT Plus reaction cocktail according to manufacturer’s instructions.
Immunoblot analysis.
Whole-cell lysates were analyzed by Western blot using primary antibodies: mouse anti-Lnk (Santa Cruz), rabbit anti-β-actin (SigmaAldrich). Anti-mouse and rabbit secondaries (LI-COR Biosciences) were used and imaged on Odyssey Infrared Imager (LI-COR Biosciences) and quantified by ImageJ software.
Cytokine stimulation and flow cytometry:
Murine cells were stained using Phycoerythrin (PE)-Ly-6G (1A8); eFluor 450-CD11b (M1/70); allophycocyanin (APC)-F4/80 (BM8); PE-Cy7-Ly6C (HK1.4). PacificBlue-CD117 (c-kit) (2B8), PE-CD115 (AFS98), Ly6C PE-Cy7 (HK1.4), Ly6G PE (1A8), CD16/32 PerCP-Cy5.5 (93)(Biolegend). Fixable live dead (near-IR or AF350) (Invitrogen) or 4’, 6-diamidino-2-phenylindole (DAPI) was used to detect dead cells. EdU detection was performed with the Click-iT Plus EdU Pacific Blue Flow Cytometry Assay Kit (Thermo Fisher). Cells were acquired on a BD LSR II with FACSDiva software and analyzed with FlowJo software (version 10.2, Tree Star).
Statistical analysis.
All statistical analysis used GraphPad Prism version 9.0 except where noted above. All specific statistical tests and P-values are indicated in the relevant figures. Data was tested for Normality using either Anderson-Darling, D’Agostino & Pearson, Shapiro-Wilk and Kolmogorov-Smirnov test depending on sample size. To assess statistical significance between two groups with normal distribution data sets, the Student’s t-test was used. Non-normal data was analyzed by Mann-Whitney test. When three groups were analyzed, we used one-way ANOVA with Tukey’s multiple comparisons post-hoc analysis or Kruskal-Wallis test for non-parametric data with post-hoc Dunn’s multiple comparisons analysis.
Data availability.
The data that support the findings of this study are available from the corresponding author upon request.
RESULTS
Human subjects with the rs3184504*T autoimmune risk allele display reduced sepsis mortality.
The rs3184504*C allele encodes for SH2B3 Arg262 (CGG, Arg, R) and represents the ancestral allele or “non-risk” for autoimmune disease. The autoimmune associated “risk allele” (TGG, Trp, W), is a derived allele that is rare in East Asian and African populations (gnomAD maf T=0.001 and T= 0.073 respectively), while approaching the major allele in cohorts of non-Finnish European sub-groups (T= 0.487 in gnomAD). The positive selection of the haplotype has been previously noted (3), but further testing of the host response to infection is lacking. Here, we analyzed the rs3184504 allele frequency in a known sepsis cohort of genotyped patients. We utilized the NHLBI funded iSPAAR (identification of SNPs Altering ALI Risk) consortium of genotyped patients with sepsis defined using the 2001 consensus definition used at the time of enrollment (20). Re-analysis showed these subjects (1527/1542 or 99%) also meet the Sepsis-3 guideline definition for ICU patients with an admission SOFA (Sequential Organ Failure Assessment) score >=2 along with a suspected infection (Table S1) (21). Analysis was limited to Caucasian subjects (n=1050) given the allele frequency bias. Initial analysis revealed subjects with the rs3184505*T/T genotype had 10.42% mortality (39/374) compared to 17.30% (135/780) and 17.26% (67/388) mortality with rs3184504*T/C or C/C genotypes, respectively.
Next, we performed a multivariate logistic regression model to consider variants within 2kb upstream or downstream of the SH2B3 gene, with a minor allele frequency > 0.05 in the HapMap CEU population and chose an R2 threshold of 0.5. This identified four SNPs previously genotyped in this dataset (rs2239196, rs2239194, rs3184504, and rs739496) all with Hardy-Weinberg equilibrium p>0.001 and call rate >0.99 (Figure S1). The primary outcome measure for our analysis was inpatient mortality, while secondary outcomes included white blood cell (WBC) count and plasma cytokine levels. In a multiple logistic regression model adjusted for age and gender, the rs3184504*T allele uniquely associated with improved survival [OR 0.76 (95% CI 0.62–0.92); Adj p=0.0054] (Table I). Given that genome-wide data was available for the subjects in iSPAAR, next we calculated principal components using the genomic data adjusting for the 1st three principal components, in addition to age and gender, to account for unmeasured confounding and further adjustment for APACHE III score, a measure for severity of illness in ICU patients. Similar estimates and degree of significance were found [OR was 0.80 (95% CI 0.64–0.99); Adj p = 0.04]. The rs3184504*T allele also associated with an increased peak absolute WBC in the first 24-hours of sepsis (β= +0.05; Adj p=0.032) (Figure S1). Conversely, IL-8 levels, a known mediator associated with poor outcomes (22), were reduced with the rs3184504*T allele (β= −0.19; p=0.025). IL-6, G-CSF and sTNFR1 were not different between rs3184504 genotype groups. Thus, the rs3184504*T/T genotype associated with reduced sepsis mortality, elevated peak WBC and lower circulating IL-8 levels in this cohort. Collectively, homozygosity of the rs3184504*T risk allele was associated with reduced sepsis mortality in this cohort.
Table I. Multiple logistic regression model shows selective SH2B3 SNP (rs3184504) association with reduced mortality in sepsis cohort.
Single Nucleotide Polymorphisms (SNP) within 2kB of the coding region of SH2B3 were analyzed for association with sepsis mortality. Data from the identification of SNPs Altering ALI Risk (iSPAAR) consortium Caucasian subjects with sepsis (n=1542 total subjects) were analyzed for inpatient sepsis mortality displayed by genotype and error illustrated by the adjusted p value from the multiple logistic regression model for sepsis inpatient mortality adjusted for age and gender.
| SNP | Adj p** | P-Value | OR** | OR 95% low CI** | OR 95% hi CI** | N | Bonferroni P | FDR |
|---|---|---|---|---|---|---|---|---|
| rs2239196 (intron) | 0.5749 | 0.626 | 1.15 | 0.7111 | 1.858 | 1543 | 1 | 0.575 |
| rs2239194 (intron) | 0.4315 | 0.532 | 1.15 | 0.815 | 1.622 | 1543 | 1 | 0.575 |
| rs3184504 (exon) | 0.0054 | 0.01 | 0.755 | 0.618 | 0.921 | 1542 | 0.022 | 0.022 |
| rs739496 (intron) | 0.1195 | 0.16 | 1.201 | 0.956 | 1.51 | 1542 | 0.478 | 0.236 |
adjusted for age, gender (age is associated with mortality)
Hypomorphic SH2B3 promotes survival during polymicrobial CLP model with rapid bacterial clearance.
To better understand the improved sepsis response in humans, we used a murine model to directly test the role of SH2B3 during a polymicrobial sepsis response. The rs3184504*T allele has been shown to be hypomorphic as a negative regulator (6), thus we first performed studies in SH2B3 deficient mice (Figure 1A). Using a cecal-ligation puncture (CLP) model, we found Sh2b3KO mice exhibited decreased mortality over 7 days (Figure 1B) and demonstrated reduced morbidity measured by drop in body temperature at 24-hours, an accurate surrogate for mortality (Figure 1C) (44). Endothelial cell expression of SH2B3 has been reported (23), thus we tested whether SH2B3 deficiency in hematopoietic cells was sufficient for the protective phenotype. We generated bone marrow (BM) chimeras using Sh2b3WT or Sh2b3KO BM transferred into irradiated wildtype hosts. Chimeras were rested 12-weeks to allow reconstitution and then CLP surgeries were performed. We found SH2B3 deficiency in hematopoietic cells was sufficient for improved morbidity (Figure 1D). For the remainder of our studies, we thus utilized non-irradiated mice. We next analyzed mice for early signs of tissue damage post-sepsis. At 24-hours following CLP, lung and liver tissue showed early signs of tissue damage uniquely in the Sh2b3WT mice including neutrophil infiltration with edema and hepatocellular degeneration and perivascular edema (Figure 1E–F). Average semi-quantitative severity across mice were scored. Additionally, blood urea nitrogen (BUN) was elevated in the Sh2b3WT mice compared to Sh2b3KO mice (Figure 1G). Overall, Sh2b3KO mice lacked significant organ injury at 24-hours.
Figure 1. SH2B3 deficiency promotes rapid bacterial clearance and improved sepsis response.

(A) Sh2b3 knock-out (SH2B3KO) genomic targeting strategy (B–J) Comparison of Sh2b3+/+ (WT) and Sh2b3KO (KO) littermate male mice during experimental sepsis using a cecal ligation and puncture (CLP) model. (B) Survival curve in mice over 7 days following CLP (n=13/group) analyzed with the Kaplan- Meier test. *WT vs KO (p=0.03) via the Chi square test. (C–D) Drop in body temperature measured 24-hours post-CLP in (C) SH2B3WT and SH2B3KO mice (n=22 WT, n=18 KO) or in (D) bone marrow chimeric mice (n=6 WT>WT; n=6 KO>WT) rested 12-weeks post-reconstitution prior to CLP procedure. (E) Colony-forming units (CFU) in the peritoneal fluid (left) and blood (right) at 24-hours post-CLP (WT n=8, KO n=8). (F–G) Comparison of peritoneal myeloid subsets function in WT versus KO mice 24-hours post-CLP. Ex-vivo peritoneal neutrophil (CD11b+Ly6G+Ly6C+) and inflammatory monocytes (CD11b+Ly6G−Ly6C+) were analyzed for (F) fluorescent E.coli phagocytosis via flow cytometry or (G) ex-vivo reactive oxygen species (ROS) by flow cytometry (n=11 WT; n=11KO). (H) Blood urea nitrogen (BUN) was measured at 24-hours post-CLP in Sh2b3WT and Sh2b3KO mice (WT n=13, KO n=14). (I) Histology images shown for comparison of lung injury between WT and KO mice 24hrs post-CLP. Dilated perivascular (PV) space indicates edema and neutrophil influx (*) with erythrocytes present as scattered populations or as dense accumulations within lymphatic sumps (arrow). Lungs from Sh2b3KO mice show normal perivascular tissue with the exception of slightly dilated lymphatics (L). (J) Representative liver sections taken from WT mice show minimal inflammatory cell infiltrates in some sinusoids (*) and marked degree of hepatocellular degeneration recognized histologically as severe cell swelling with occasional loss of individual hepatocytes. Comparison liver sections from Sh2b3KO mice were histologically normal. An average semi-quantitative severity score is displayed below images for each genotype (n=3 mice/genotype). Statistical analysis with unpaired t test (C-D, F-G) or Mann-Whitney (E) (**** p<0.0001, *** p<0.001, **p<0.01, *p<0.05). Data are presented as means (±SEM).
Sh2b3KO mice also demonstrated improved bacterial clearance and less organ damage following CLP. At 24-hours following surgery, Sh2b3KO mice exhibited improved systemic and local bacterial clearance measured by colony forming units from blood and peritoneal cavity, respectively (Figure 1H). Peritoneal granulocytes from Sh2b3KO mice (gated separately on neutrophils (CD11b+Ly6C−Ly6G+) and “classical” inflammatory monocytes (CD11b+Ly6Chi Ly6G−), respectively) demonstrated enhanced ex vivo phagocytosis compared to controls (Figure 1I). Peritoneal cells were also tested for reactive oxygen species (ROS) following PMA stimulation and SH2B3 deficient monocytes produced reduced ROS from each animal compared to control mice, whereas neutrophils demonstrated no difference (Figure 1J). Thus, in the setting of polymicrobial CLP, deficiency of SH2B3 results in a more rapid bacterial clearance, enhanced phagocytosis and less organ damage.
SH2B3 deficiency promotes monocyte influx in response to sepsis.
For most pathogens, monocyte recruitment to the site of inflammation enhances resistance to that infection (24). We next characterized and quantified peritoneal granulocytes ex vivo following the CLP (Figure 2A). Total cellular influx and absolute neutrophils were not significantly different in the peritoneal cavity between genotypes, but Sh2b3KO mice displayed augmented absolute monocyte counts at both 12- and 24-hours compared to Sh2b3WT mice (Figure 2B). There was no significant difference in circulating blood monocytes in Sh2b3KO mice following CLP, while the percentage of neutrophils was reduced (Figure 2C). Thus, in the absence of SH2B3, inflammatory monocytes were increased at sites of peritoneal inflammation.
Figure 2. SH2B3 knockout mice have expanded inflammatory monocytes post-CLP.
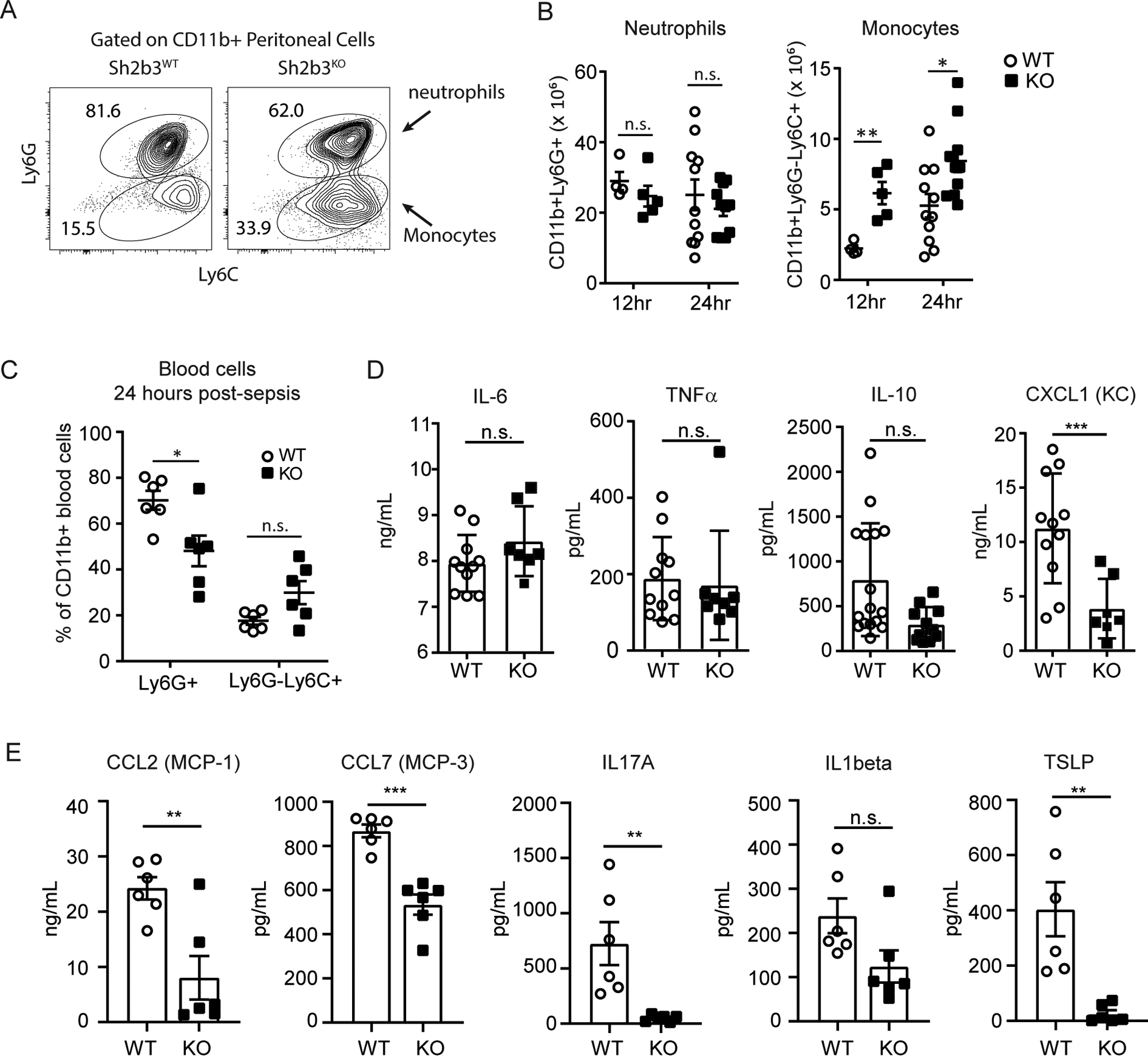
(A–C) Peripheral blood or peritoneal cells from Sh2b3WT and Sh2b3KO littermate male mice were analyzed 24-hours post-CLP. (A) Representative flow cytometry of CD3-B220-CD11b+ cells identifying neutrophil (Ly6G+Ly6C+) and inflammatory monocytes (Ly6G−Ly6C+) and (B) cell numbers quantified at 12 hours (WT n=4, KO n=5) or 24-hours post-CLP (WT n=11, KO n=9). Statistical analysis with unpaired t test (**p<0.01, *p<0.05) (C) Peripheral blood cells were analyzed for percentages of neutrophil or inflammatory monocyte within the CD11b+ myeloid population in circulation. Statistical analysis with Two-way ANOVA (**p<0.01). (D–E) Cytokine and chemokines were quantified in peritoneal fluid from Sh2b3WT and Sh2b3KO mice at 24-hours post-CLP. (D) Cytokine and chemokine levels were quantified via targeted ELISA for CXCL1 (KC), IL-6, TNF-alpha and IL-10 (n=6 per genotype) or (E) using Luminex bead-based capture panel. Statistics performed by grouped multiple comparisons and discovery determined by two-stage linear step-up procedure *** q value <0.001, ** q value <0.01, * q value <0.05). Data are presented as means (±SEM).
We hypothesized that augmented monocyte-related chemokine expression would be found in the peritoneal fluid of Sh2b3KO mice during these models. We performed a broad cytokine and chemokine multiplex array and targeted ELISA testing on peritoneal fluid 24-hours post-CLP from Sh2b3WT (n=6) and Sh2b3KO mice (n=6) (Figure 2D–E). Overall, compared to controls, Sh2b3KO mice had a broad reduction in the proinflammatory cytokines associated with tissue damage, including IL-6, TNF-alpha, TSLP, IL-1beta, IL-17A; findings that likely reflected earlier bacterial clearance in Sh2b3KO mice. Consistent with reduced levels of IL-8 in septic human subjects with the rs3184504*T autoimmune risk allele, the murine surrogates for IL-8 [CXCL1 (KC) and CXCL2 (MIP-2)], were also decreased in the peritoneum of Sh2b3KO mice in the setting of CLP. Surprisingly, monocyte chemotactic factors measured, including CCL2 (MCP-1) and CCL7 (MCP-3), were also reduced in Sh2b3KO mice. Thus, while the reduction in proinflammatory cytokines, including IL-8 surrogates, correlated with the reduced morbidity post-CLP, altered chemokine levels could not explain the augmented monocytes in SH2B3 deficient mice.
SH2B3 deficiency promotes monocyte and progenitor proliferation in response to sepsis.
We next hypothesized that peripheral monocytes and/or their myeloid progenitors may be proliferating to account for the enhanced monocyte population at the site of infection. In the CLP model, immature myeloid cells and hematopoietic stem and progenitor cells (HSPC) are mobilized from the BM niches and can be measured in the spleen (25–27). Thus, we performed an intraperitoneal injection of EdU (5-ethynyl-2’-deoxyuridine) at 24-hours post-CLP and one hour prior to sacrifice (Figure 3A). EdU positive cycling cells were identified in the BM and spleen. The EdU+B220- fraction was similar between mice indicating equal labeling of the BM (Figure 3B). We focused on the splenic non-B, non-T cell fraction that contains myeloid cells and HSPCs. We analyzed the neutrophils, monocytes and c-kit positive cells and found a significant increase in CD11b−c-kit+ cells in the spleen from Sh2b3KO mice, as well as Ly6Chi monocytes (CD11b+Ly6G−Ly6Chi)(Figure 3C–D). Thus, reduced SH2B3 function correlates with increased peripheral myeloid progenitors and monocytes following CLP in mice. Overall, absence of the SH2B3 adaptor improved the immediate sepsis response in mice.
Figure 3. CLP induces cycling in Ly6C+ monocytes and progenitors in SH2B3 knockout mice.

(A) Sh2b3WT and Sh2b3KO littermate male mice underwent CLP and at 24-hours were injected with EdU+ or DMSO control 1 hour prior to sacrifice. The bone marrow (BM) and spleen were then analyzed via flow cytometry for cycling (EdU+) cells. (B) Equal labeling was demonstrated in the BM with equal staining of B220- cells (EdU n=5/genotype; DMSO n=1/genotype). (C) Non-B, non-T cell (B220−CD3−) cells were further analyzed for splenic progenitors (CD11bneg c-kit+, monocytes (CD11b+Ly6Gneg Ly6C+), and a population negative for all markers. **** p<0.0001, *** p<0.001, ** p<0.001, *p<0.05. Data are presented as means (±SEM). (D) Representative flow cytometry of B220−CD3−Ly6G− splenocytes following CLP and EdU labeling comparing DMSO control to Sh2b3WT and Sh2b3KO mice with gating strategy for CD11b-Ly6C-CD117+ progenitor cells (left) or Ly6Chi monocytes (right). Data representative of two independent experiments each with 2–3 animals per genotype.
Characterization of a novel SH2B3 hypomorphic knock-in mouse model.
Human SH2B3 deficiency associates with malignancy and myelodysplasia. In contrast, the autoimmune risk allele involving SH2B3 is hypomorphic. Thus, we reasoned that a hypomorphic murine SH2B3 variant similar to the human autoimmune risk allele may be sufficient to improve the response to the CLP. The human ancestral arginine at position 262 is highly conserved in mammals outside of rodents with proline at the homologous position 234. Based upon in silico modeling from the murine SH2B2 (APS) PH domain (PDB: 1V5M), we predicted an arginine substitution (P234R) in the murine SH2B3 would partially disrupt the PH domain. We created a novel knockin mouse using homologous recombination and introduced the 234R into the genomic murine locus (Figure 4A). We included flanking loxP sites allowing cre-dependent excision of exons 3–8 as well. Founder mice were established that were healthy and fertile and heterozygous mice were intercrossed to deliver littermate controls (Sh2b3WT) and homozygous knockin (Sh2b3KI) mice for all remaining experiments. We compared protein expression and found the 234R coding change did not impact protein expression (Figure 4B).
Figure 4. Generation and characterization of the SH2B3P234R knock-in mouse model.
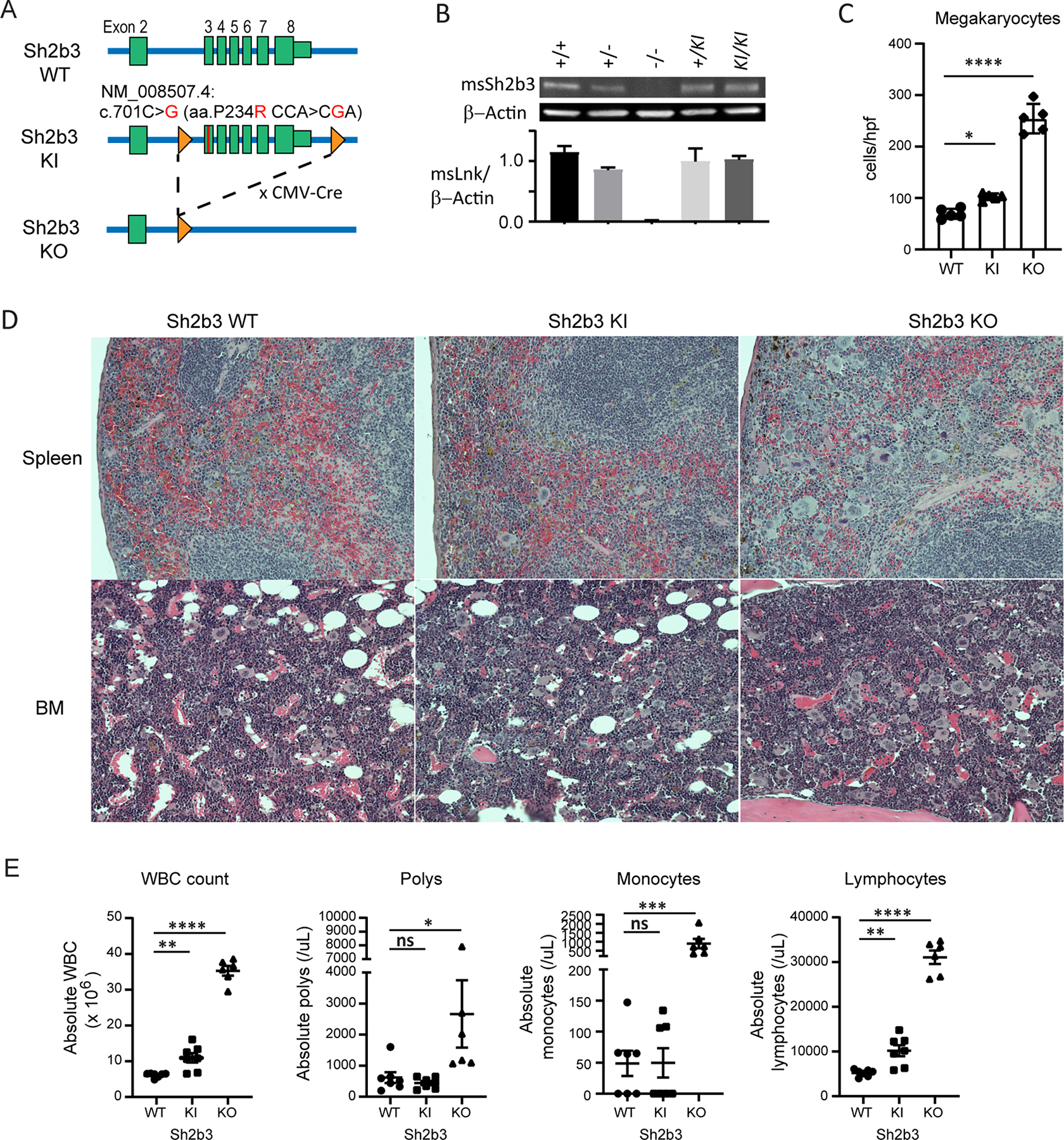
(A) Knock-in (SH2B3KI) mouse targeting strategy with P234R point mutation within exon 3 of Sh2b3 gene introducing a hypomorphic variant into the endogenous locus. Residual FRT site following Flp-induced removal of the neomycin cassette and the location of LoxP sites are indicated. The knockout (SH2B3KO) mouse line was generated by crossing to CMV-Cre strain. (B) Representative western blot using spleen whole cell lysates from Sh2b3+/+(WT), Sh2b3+/−, SH2B3KO or mice with one (Sh2b3+/KI) or two (Sh2b3KI/KI) knock-in alleles analyzed for total SH2B3 and beta-actin with averaged normalized densitometry shown graphically (n=3). (C–E) Sh2b3WT, Sh2b3KI, and Sh2b3KO mice were aged 15–18 months and then peripheral blood counts and tissue pathology compared. (C–D) Hematoxylin and eosin stained histological sections of spleen and bone marrow (BM) from Sh2b3WT, Sh2b3KI, and Sh2b3KO mice (n=5 per genotype). (C) Megakaryocytes were enumerated across 5 random high-powered fields (hpf) per animal (n=5 per genotype) (magnification 40X). Statistical analysis via one-way ANOVA with comparison to wildtype; *p<0.05. (D) Representative hematoxylin and eosin stained histological sections of spleen and BM from Sh2b3WT (left), Sh2b3KI (middle) and Sh2b3KO (right) mice. Megakaryocytes and other myeloid cells are increased within the red pulp of the spleen and within the BM compared to WT mice. Sh2b3KO mice demonstrated numerous large, pale-staining megakaryocytes and marked hypercellularity of the splenic red pulp and crowding of the BM. An intermediate phenotype with fewer, but still increased numbers of megakaryocytes and other myeloid cells is present within the splenic red pulp and BM from Sh2b3KI mice. Original magnification 20X (E) Murine complete blood counts were performed by flow cytometry with peroxidase methodology confirmed by blinded manual differential from aged mice showing total white blood cell count (WBC), polymorphonuclear leukocyte (Polys), monocyte, and lymphocyte counts (Sh2b3WT n=7, Sh2b3KI n=7, Sh2b3KO n=6). Statistical analysis via one-way ANOVA **** p<0.0001, *** p<0.001, ** p<0.001, *p<0.05. Data are presented as means (±SEM).
We next compared littermates for in vivo pathology previously observed in SH2B3 knockout mice (11). Deficiency in SH2B3 augments numerous cytokine receptor signaling pathways (8). Sh2b3−/− mice demonstrate augmented peripheral white blood cell, lymphocyte and monocyte counts and aged mice demonstrate myeloproliferative phenotypes with augmented megakaryocytes and extramedullary hematopoiesis (11). We hypothesized that the Sh2b3KI mice would demonstrate an intermediate phenotype between Sh2b3WT and Sh2b3KO mice, including splenomegaly, leukocytosis, extramedullary hematopoiesis and increased megakaryocytes (11). Thus, we analyzed the blood, BM and spleen of aged mice (Sh2b3WT, Sh2b3KI and Sh2b3KO aged 15–18 months) to compare the phenotype to the published phenotype from Sh2b3-deficient mice (Figure 4C–E). Histologic analysis of bone marrow revealed a higher megakaryocyte cell density in both Sh2b3KI and Sh2b3KO mice per high power field (Figure 4C) and low power showed increasing hypercellularity in Sh2b3KI and Sh2b3KO mice, respectively (Figure 4D). Increases in other myeloid cells were also observed within the red pulp of the spleen. Murine peripheral blood predominantly contains lymphocytes (28), which was also observed in our studies. Peripheral blood analysis from Sh2b3-deficient mice displayed leukocytosis with elevations in lymphocytes, monocytes and neutrophils (Figure 4E), similar to cell counts reported in the literature (11). The Sh2b3KI mice also showed leukocytosis when compared to Sh2b3WT mice, specifically with elevated circulating absolute lymphocyte counts intermediate between Sh2b3WT and Sh2b3KO mice (Figure 4E). Thus, introduction of the hypomorphic variant into the murine PH domain mimicked Sh2b3KO mice, but resulted in a less severe, intermediate phenotype.
SH2B3 hypomorphic knock-in mice demonstrate an improved sepsis response
We next tested the hypothesis that homozygous Sh2b3KI mice would exhibit reduced mortality following induction of CLP. We compared mortality over 7 days post-CLP induction. Homozygous Sh2b3KI mice showed less mortality compared to heterozygous knockin mice (Sh2b3234P/R) and Sh2b3WT littermate control mice (Figure 5A). Morbidity post-CLP, as measured by drop in body temperature in the first 24-hours, revealed an allele effect with more homozygous animals maintaining body temperature compared to the Sh2b3234P/R mice (Figure 5B). Bacterial clearance in the homozygous Sh2b3KI and Sh2b3234P/R mice was not significantly different from the Sh2b3WT littermate control mice, although a trend for lower bacterial burden in the peritoneal cavity and systemically was observed stepwise with allele burden (Figure 5C). Lastly, comparing the local cellular infiltrate in the peritoneal cavity, inflammatory monocytes (CD11b+Ly6C+Ly6G−) were significantly increased in Sh2b3KI mice compared to Sh2b3WT control mice (Figure 5D). In contrast, the peritoneal cells in Sh2b3234P/R mice were not discernibly different from wildtype mice, suggesting again that homozygosity was necessary to improve the sepsis response in the CLP animal model. Overall, our data suggest that reduced SH2B3 function increased inflammatory monocyte recruitment resulting in improved bacterial clearance and reduced mortality in hypomorphic Sh2b3KI mice.
Figure 5. Homozygous Sh2b3 knock-in mice demonstrate improved sepsis response.
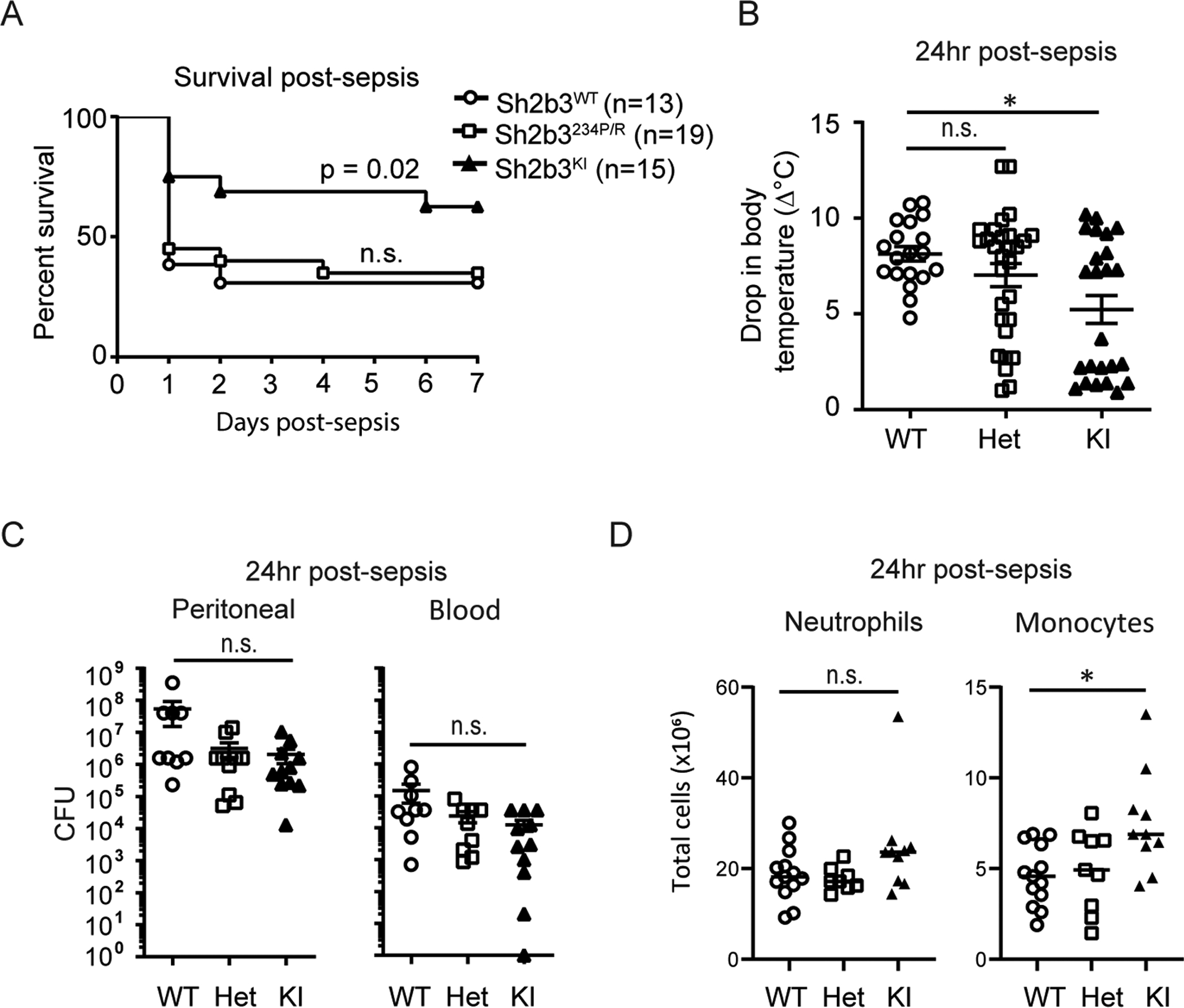
Comparison of Sh2b3WT, heterozygote (Sh2b3234P/R), and Sh2b3KI mice using a similar high-grade CLP model. (A) Survival curve in mice over 7 days following CLP (n=13–19/group, WT n=13, HET n=19, KI n=15) analyzed with the Kaplan-Meier test. * WT vs KI (p=0.02) via the Chi square test. (B) Body temperature measured 24-hours post-CLP (n = 18–30/group; WT n=30, HET n=25, KI n=24) (**WT vs KI (p=0.0134). Analyzed via Kruskal-Wallis test; post-hoc analysis by Dunn’s multiple comparisons test. (C) Colony-forming units (CFU) in the peritoneal fluid (left) and blood (right) at 24-hours post-CLP (WT n=11, HET n=9, KI n=13). Statistical analysis by one-way ANOVA. * p<0.05. (D) Comparison of peritoneal myeloid subsets at 24-hours post-CLP. Ex-vivo peritoneal neutrophil (CD11b+Ly6G+Ly6C+) and inflammatory monocytes (CD11b+Ly6G−Ly6C+) were quantified and characterized by flow cytometry (WT n=13; HET n=9; KI n=10). Statistical analysis by one-way ANOVA. * p<0.05. Data are presented as means (±SEM).
DISCUSSION
Here, we show that reduced function of the SH2B3 adaptor protein results in an improved host response to bacterial sepsis in both humans and mice. First, we found in human subjects with sepsis, homozygosity for the rs3184504*T autoimmune risk allele significantly associated with reduced mortality and higher peak leukocyte counts. Previous studies using human cord blood cells suggested that expression of the rs3184504*T allele correlated with increased TPO signaling, consistent with a hypomorphic or reduced function of this inhibitory adaptor protein (6). Given the linkage disequilibrium at this locus, we transitioned to murine modeling to examine the role of the SH2B3 adaptor protein in sepsis. We found that SH2B3 deficiency decreased mortality, morbidity and demonstrated less organ damage in a CLP model. The SH2B3 deficient mice achieved pathogen control with enhanced phagocytosis and increased monocytes at the inflammatory site. Finally, we created a knockin Sh2b3 mouse model with a variant in the PH domain demonstrating similar features as the Sh2b3−/− mice including increased megakaryocytes, augmented baseline leukocytes and tissue pathology findings all suggestive of a hypomorphic allele. To model the human variant, we challenged the Sh2b3 knockin mice with the CLP model showing decreased mortality, increased bacterial clearance, and increased monocytes at the site of inflammation. Taken together, these findings suggest that the SH2B3262W autoimmune risk variant provides protection during acute bacterial challenge in both mice and humans.
Negative regulators of cytokine signaling have emerged as targets of positive natural selection often with improved fitness against infections (1). Simultaneously, these alleles are also important potential autoimmune risk variants (29). Our data suggest that SH2B3 fits within this growing paradigm. Previous work, and ours, have consistently found rs3184504*T allele associates with many increased hematopoietic traits including elevated monocytes (Figure S1) (15,16, 40). Consistent with these findings, human monocyte progenitors and terminal monocytes highly express SH2B3 transcripts (34, 35). Given the plasticity of the myeloid lineages and short neutrophil life span, genetic risk factors that regulate aspects of myelopoiesis are likely impactful and only starting to be explored.
Hematopoietic progenitors are rapidly mobilized following CLP. During the early phases post-CLP in murine sepsis, distinct neutrophil progenitors are expanded in the bone marrow at the expense of monocytes (47). Our data showed increased cell cycling of c-kit+ as well as the CD11b+Ly6C+ fraction in the spleen uniquely in the Sh2b3 deficient animals as evidence for altered myelopoiesis. Emergency or demand-adapted myelopoiesis has been a well described observation of shift in bone marrow hematopoiesis favoring myelopoiesis at the expense of lymphopoiesis (45). However, growing evidence suggests that the neutrophil versus monocyte-dendritic cells lineage decision is equally critical in the modulation of the immune response (36). In human studies, pooled genomic signatures of sepsis mortality revealed whole blood gene expression of several neutrophil specific genes (DEFA4, MPO, CTSG, BPI) increased with high mortality and several monocyte specific genes (CCR2, EIF5A, CX3CR1, EMR3), decreased with high mortality (46). While it is likely that the ratio of myeloid subpopulations plays an important role in the sepsis response, further studies are needed to assess potential differences in these progenitors in the absence of SH2B3 and whether expanded progenitor populations are sufficient for an improved response.
Pathogen-imposed selection pressures have also been linked to altered genetic alleles involved in innate sensing pathways (37, 38). Previous work using PBMCs from human subjects stimulated with muramyl dipeptide, a known NOD2 ligand, demonstrated increased IL-8 secretion in association with the rs3184504*T/T autoimmune risk genotype (3). In contrast, we found, similar to studies in macrophages from Sh2b3 deficient mice (14), that reduced human SH2B3 function based on the rs3184504-T allele did not significantly alter TLR signaling on a per cell basis (Figure S2–S3). Augmented monocyte absolute numbers may explain the previously reported findings as we found consistently higher absolute monocyte numbers associating with the rs3184504 allele in a healthy control cohort (Figure S1). Unfortunately, our sepsis cohort only collected peak total white blood counts and not further differential data to determine if the monocytosis was augmented during human sepsis.
Here, we also demonstrated that reduced function of Sh2b3 associated with rapid bacterial clearance and enhanced phagocytic abilities. Previous work with bone marrow derived macrophages from Sh2b3 deficient mice showed no significant difference in zymosan-labeled particle phagocytosis (14), but minimal follow up has been performed with primary cells from Sh2b3−/− mice. In vivo studies of mice with augmented JAK2 signaling (JAK2V617F mice) showed increased erythrophagocytosis in the macrophages at baseline suggesting that a JAK2-dependent cytokine receptor may augment this function (48). Hematopoietic myeloid progenitor cells and the developmental cues have been found to produce functionally distinct monocytes based upon gene signature, but direct assessment of phagocytic function was not performed (36). In the early phase following CLP, we found the microbicidal function of granulocytes augmented with enhanced phagocytosis, but not altered ROS. The rapid bacterial clearance was in the absence of antibiotic prophylaxis in our animal model and thus represents a possible gain-of-function effect that may explain the selective sweep that occurred with the rs3184504*T genetic allele (3). Further studies will be needed to understand the detailed mechanism of the enhanced phagocytic function following a bacterial challenge.
In summary, our data show that reduced function of the SH2B3 adaptor improves the early sepsis response and suggest an important role for the SH2B3262W coding variant in immune regulation. Our findings support the hypothesis that the SH2B3262W risk protein is a causative variant that underwent positive selection to promote improved monocyte function and accumulation in the absence of severe tissue damage. Future work using the models described here will help to comprehensively assess the impact of the SH2B3262W risk in autoimmune pathogenesis including whether regulating lifespan or function of innate myeloid responses may contribute to the initiation and/or acceleration of human autoimmunity.
Supplementary Material
Key Points:
Hypomorphic SH2B3 improves the sepsis response in humans and mice.
SH2B3 deficiency results in enhanced phagocytosis and rapid bacterial clearance.
Lack of SH2B3 augments myelopoiesis in sepsis promoting monocytes.
ACKNOWLEDGEMENTS
We thank S.Khim for animal husbandry; the University of Washington Histology and Imaging Core and Benaroya Research Institute (BRI) Translational Research Core.
Supported by the US National Institutes of Health (DP3-DK097672 to J.H.B.; DP3-DK111802 to D.J.R.; 1K08DK114568 to E.J.A.), Rheumatology Research Foundation (ACR ID# 104242 to E.J.A.), the Children’s Guild Association Endowed Chair in Pediatric Immunology and the Benaroya Family Gift Fund (D.J.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DISCLOSURE OF CONFLICTS OF INTERESTS
The authors declare no conflicts of interests.
REFERENCES
- 1.Gutierrez-Arcelus M, Rich SS, and Raychaudhuri S. 2016. Autoimmune diseases - connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet 17: 160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman SJ, and Hill AVS. 2012. Human genetic susceptibility to infectious disease. Nat. Rev. Genet 13: 175–188. [DOI] [PubMed] [Google Scholar]
- 3.Zhernakova A, Elbers CC, Ferwerda B, Romanos J, Trynka G, Dubois PC, de Kovel CGF, Franke L, Oosting M, Barisani D, Bardella MT, Finnish Celiac Disease Study Group, Joosten LAB, Saavalainen P, van Heel DA, Catassi C, Netea MG, and Wijmenga C. 2010. Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am. J. Hum. Genet 86: 970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steck AK, Xu P, Geyer S, Redondo MJ, Antinozzi P, Wentworth JM, Sosenko J, Onengut-Gumuscu S, Chen W-M, Rich SS, Pugliese A, and Type 1 Diabetes TrialNet Study Group. 2017. Can Non-HLA Single Nucleotide Polymorphisms Help Stratify Risk in TrialNet Relatives at Risk for Type 1 Diabetes? J. Clin. Endocrinol. Metab 102: 2873–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su D-F, Yang J, Xie G, Keystone E, Westra H-J, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJH, van Riel PLCM, van de Laar MAFJ, Guchelaar H-J, Huizinga TWJ, Dieudé P, Mariette X, Bridges SL Jr, Zhernakova A, Toes REM, Tak PP, Miceli-Richard C, Bang S-Y, Lee H-S, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapää-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M, RACI consortium, GARNET consortium, Eyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae S-C, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, and Plenge RM. 2014. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506: 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Tang Y, Wang Y, Tascau L, Balcerek J, Tong W, Levine RL, Welch C, Tall AR, and Wang N. 2016. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ. Res 119: e91–e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huan T, Esko T, Peters MJ, Pilling LC, Schramm K, Schurmann C, Chen BH, Liu C, Joehanes R, Johnson AD, Yao C, Ying S-X, Courchesne P, Milani L, Raghavachari N, Wang R, Liu P, Reinmaa E, Dehghan A, Hofman A, Uitterlinden AG, Hernandez DG, Bandinelli S, Singleton A, Melzer D, Metspalu A, Carstensen M, Grallert H, Herder C, Meitinger T, Peters A, Roden M, Waldenberger M, Dörr M, Felix SB, Zeller T, International Consortium for Blood Pressure GWAS (ICBP), Vasan R, O’Donnell CJ, Munson PJ, Yang X, Prokisch H, Völker U, van Meurs JBJ, Ferrucci L, and Levy D. 2015. A meta-analysis of gene expression signatures of blood pressure and hypertension. PLoS Genet. 11: e1005035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gery S, and Koeffler HP. 2013. Role of the adaptor protein LNK in normal and malignant hematopoiesis. Oncogene 32: 3111–3118. [DOI] [PubMed] [Google Scholar]
- 9.Tong W, Zhang J, and Lodish HF. 2005. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 105: 4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maslah N, Cassinat B, Verger E, Kiladjian J-J, and Velazquez L. 2017. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 31: 1661–1670. [DOI] [PubMed] [Google Scholar]
- 11.Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, Chikwava KR, and Tong W. 2010. Lnk constrains myeloproliferative diseases in mice. J. Clin. Invest 120: 2058–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balcerek J, Jiang J, Li Y, Jiang Q, Holdreith N, Singh B, Chandra V, Lv K, Ren J-G, Rozenova K, Li W, Greenberg RA, and Tong W. 2018. Lnk/Sh2b3 deficiency restores hematopoietic stem cell function and genome integrity in Fancd2 deficient Fanconi anemia. Nat. Commun 9: 3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, Paige CJ, and Pawson T. 2002. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med 195: 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gueller S, Goodridge HS, Niebuhr B, Xing H, Koren-Michowitz M, Serve H, Underhill DM, Brandts CH, and Koeffler HP. 2010. Adaptor protein Lnk inhibits c-Fms-mediated macrophage function. J. Leukoc. Biol 88: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, Lambourne JJ, Sivapalaratnam S, Downes K, Kundu K, Bomba L, Berentsen K, Bradley JR, Daugherty LC, Delaneau O, Freson K, Garner SF, Grassi L, Guerrero J, Haimel M, Janssen-Megens EM, Kaan A, Kamat M, Kim B, Mandoli A, Marchini J, Martens JHA, Meacham S, Megy K, O’Connell J, Petersen R, Sharifi N, Sheard SM, Staley JR, Tuna S, van der Ent M, Walter K, Wang S-Y, Wheeler E, Wilder SP, Iotchkova V, Moore C, Sambrook J, Stunnenberg HG, Di Angelantonio E, Kaptoge S, Kuijpers TW, Carrillo-de-Santa-Pau E, Juan D, Rico D, Valencia A, Chen L, Ge B, Vasquez L, Kwan T, Garrido-Martín D, Watt S, Yang Y, Guigo R, Beck S, Paul DS, Pastinen T, Bujold D, Bourque G, Frontini M, Danesh J, Roberts DJ, Ouwehand WH, Butterworth AS, and Soranzo N. 2016. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167: 1415–1429. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, Finucane HK, Aryee MJ, Buenrostro JD, and Sankaran VG. 2019. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet 51: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMullin MF, Wu C, Percy MJ, and Tong W. 2011. A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis. Am. J. Hematol 86: 962–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Harst P, Zhang W, Mateo Leach I, Rendon A, Verweij N, Sehmi J, Paul DS, Elling U, Allayee H, Li X, Radhakrishnan A, Tan S-T, Voss K, Weichenberger CX, Albers CA, Al-Hussani A, Asselbergs FW, Ciullo M, Danjou F, Dina C, Esko T, Evans DM, Franke L, Gögele M, Hartiala J, Hersch M, Holm H, Hottenga J-J, Kanoni S, Kleber ME, Lagou V, Langenberg C, Lopez LM, Lyytikäinen L-P, Melander O, Murgia F, Nolte IM, O’Reilly PF, Padmanabhan S, Parsa A, Pirastu N, Porcu E, Portas L, Prokopenko I, Ried JS, Shin S-Y, Tang CS, Teumer A, Traglia M, Ulivi S, Westra H-J, Yang J, Zhao JH, Anni F, Abdellaoui A, Attwood A, Balkau B, Bandinelli S, Bastardot F, Benyamin B, Boehm BO, Cookson WO, Das D, de Bakker PIW, de Boer RA, de Geus EJC, de Moor MH, Dimitriou M, Domingues FS, Döring A, Engström G, Eyjolfsson GI, Ferrucci L, Fischer K, Galanello R, Garner SF, Genser B, Gibson QD, Girotto G, Gudbjartsson DF, Harris SE, Hartikainen A-L, Hastie CE, Hedblad B, Illig T, Jolley J, Kähönen M, Kema IP, Kemp JP, Liang L, Lloyd-Jones H, Loos RJF, Meacham S, Medland SE, Meisinger C, Memari Y, Mihailov E, Miller K, Moffatt MF, Nauck M, Novatchkova M, Nutile T, Olafsson I, Onundarson PT, Parracciani D, Penninx BW, Perseu L, Piga A, Pistis G, Pouta A, Puc U, Raitakari O, Ring SM, Robino A, Ruggiero D, Ruokonen A, Saint-Pierre A, Sala C, Salumets A, Sambrook J, Schepers H, Schmidt CO, Silljé HHW, Sladek R, Smit JH, Starr JM, Stephens J, Sulem P, Tanaka T, Thorsteinsdottir U, Tragante V, van Gilst WH, van Pelt LJ, van Veldhuisen DJ, Völker U, Whitfield JB, Willemsen G, Winkelmann BR, Wirnsberger G, Algra A, Cucca F, d’Adamo AP, Danesh J, Deary IJ, Dominiczak AF, Elliott P, Fortina P, Froguel P, Gasparini P, Greinacher A, Hazen SL, Jarvelin M-R, Khaw KT, Lehtimäki T, Maerz W, Martin NG, Metspalu A, Mitchell BD, Montgomery GW, Moore C, Navis G, Pirastu M, Pramstaller PP, Ramirez-Solis R, Schadt E, Scott J, Shuldiner AR, Smith GD, Smith JG, Snieder H, Sorice R, Spector TD, Stefansson K, Stumvoll M, Tang WHW, Toniolo D, Tönjes A, Visscher PM, Vollenweider P, Wareham NJ, Wolffenbuttel BHR, Boomsma DI, Beckmann JS, Dedoussis GV, Deloukas P, Ferreira MA, Sanna S, Uda M, Hicks AA, Penninger JM, Gieger C, Kooner JS, Ouwehand WH, Soranzo N, and Chambers JC. 2012. Seventy-five genetic loci influencing the human red blood cell. Nature 492: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shubin NJ, Clauson M, Niino K, Kasprzak V, Tsuha A, Guga E, Bhise G, Acharya M, Snyder JM, Debley JS, Ziegler SF, and Piliponsky AM. 2020. Thymic stromal lymphopoietin protects in a model of airway damage and inflammation via regulation of caspase-1 activity and apoptosis inhibition. Mucosal Immunol. 13: 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent J-L, Ramsay G, and SCCM/ESICM/ACCP/ATS/SIS. 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit. Care Med 31: 1250–1256. [DOI] [PubMed] [Google Scholar]
- 21.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J-D, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent J-L, and Angus DC. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong HR, Cvijanovich N, Wheeler DS, Bigham MT, Monaco M, Odoms K, Macias WL, and Williams MD. 2008. Interleukin-8 as a stratification tool for interventional trials involving pediatric septic shock. Am. J. Respir. Crit. Care Med 178: 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devallière J, Chatelais M, Fitau J, Gérard N, Hulin P, Velazquez L, Turner CE, and Charreau B. 2012. LNK (SH2B3) is a key regulator of integrin signaling in endothelial cells and targets α-parvin to control cell adhesion and migration. FASEB J. 26: 2592–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi C, and Pamer EG. 2011. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol 11: 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delano MJ, Kelly-Scumpia KM, Thayer TC, Winfield RD, Scumpia PO, Cuenca AG, Harrington PB, O’Malley KA, Warner E, Gabrilovich S, Mathews CE, Laface D, Heyworth PG, Ramphal R, Strieter RM, Moldawer LL, and Efron PA. 2011. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J. Immunol 187: 911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takizawa H, Boettcher S, and Manz MG. 2012. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 119: 2991–3002. [DOI] [PubMed] [Google Scholar]
- 27.Ziegler P, Boettcher S, Takizawa H, Manz MG, and Brümmendorf TH. 2016. LPS-stimulated human bone marrow stroma cells support myeloid cell development and progenitor cell maintenance. Ann. Hematol 95: 173–178. [DOI] [PubMed] [Google Scholar]
- 28.O’Connell KE, Mikkola AM, Stepanek AM, Vernet A, Hall CD, Sun CC, Yildirim E, Staropoli JF, Lee JT, and Brown DE. 2015. Practical murine hematopathology: a comparative review and implications for research. Comp. Med 65: 96–113. [PMC free article] [PubMed] [Google Scholar]
- 29.Housley WJ, Fernandez SD, Vera K, Murikinati SR, Grutzendler J, Cuerdon N, Glick L, De Jager PL, Mitrovic M, Cotsapas C, and Hafler DA. 2015. Genetic variants associated with autoimmunity drive NFκB signaling and responses to inflammatory stimuli. Sci. Transl. Med 7: 291ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baran-Marszak F, Magdoud H, Desterke C, Alvarado A, Roger C, Harel S, Mazoyer E, Cassinat B, Chevret S, Tonetti C, Giraudier S, Fenaux P, Cymbalista F, Varin-Blank N, Le Bousse-Kerdilès M-C, Kiladjian J-J, and Velazquez L. 2010. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood 116: 5961–5971. [DOI] [PubMed] [Google Scholar]
- 31.Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang Y-D, Mazor T, Esquivel E, Yu A, Seepo S, Olsen S, Rosenberg M, Archambeault SL, Abusin G, Beckman K, Brown PA, Briones M, Carcamo B, Cooper T, Dahl GV, Emanuel PD, Fluchel MN, Goyal RK, Hayashi RJ, Hitzler J, Hugge C, Liu YL, Messinger YH, Mahoney DH Jr, Monteleone P, Nemecek ER, Roehrs PA, Schore RJ, Stine KC, Takemoto CM, Toretsky JA, Costello JF, Olshen AB, Stewart C, Li Y, Ma J, Gerbing RB, Alonzo TA, Getz G, Gruber T, Golub T, Stegmaier K, and Loh ML. 2015. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet 47: 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMullin MF, and Cario H. 2016. LNK mutations and myeloproliferative disorders. Am. J. Hematol 91: 248–251. [DOI] [PubMed] [Google Scholar]
- 33.Auer PL, Teumer A, Schick U, O’Shaughnessy A, Lo KS, Chami N, Carlson C, de Denus S, Dubé M-P, Haessler J, Jackson RD, Kooperberg C, Perreault L-PL, Nauck M, Peters U, Rioux JD, Schmidt F, Turcot V, Völker U, Völzke H, Greinacher A, Hsu L, Tardif J-C, Diaz GA, Reiner AP, and Lettre G. 2014. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat. Genet 46: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sønderby CK, Winther O, Rapin N, and Porse BT. 2016. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 44: D917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hay SB, Ferchen K, Chetal K, Grimes HL, and Salomonis N. 2018. The Human Cell Atlas bone marrow single-cell interactive web portal. Exp. Hematol 68: 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yáñez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, Salomonis N, Grimes HL, and Goodridge HS. 2017. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 47: 890–902. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quintana-Murci L 2019. Human Immunology through the Lens of Evolutionary Genetics. Cell 177: 184–199. [DOI] [PubMed] [Google Scholar]
- 38.Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, Arkatkar T, Clough C, Dai X, Khim S, Pestal K, Liggitt D, Cerosaletti K, Stetson DB, James RG, Oukka M, Concannon P, Gale M Jr, Buckner JH, and Rawlings DJ. 2017. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat. Immunol 18: 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Netea MG, Nold-Petry CA, Nold MF, Joosten LAB, Opitz B, van der Meer JHM, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JWM, and Dinarello CA. 2009. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113: 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, Chen M, Raffield LM, et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell. 2020. September 3;182(5):1214–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moldawer LL, Efron PA. Cecal ligation and puncture. Curr Protoc Immunol. 2010. November; Chapter 19:Unit 19.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mai SHC, Sharma N, Kwong AC, Dwivedi DJ, Khan M, Grin PM, Fox-Robichaud AE, Liaw PC. Body temperature and mouse scoring systems as surrogate markers of death in cecal ligation and puncture sepsis. Intensive Care Med Exp. 2018. July 27;6(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sweeney TE, Perumal TM, Henao R, Nichols M, Howrylak JA, Choi AM, Bermejo-Martin JF, Almansa R, Tamayo E, Davenport EE, Burnham KL, Hinds CJ, Knight JC, Woods CW, Kingsmore SF, Ginsburg GS, Wong HR, Parnell GP, Tang B, Moldawer LL, Moore FE, Omberg L, Khatri P, Tsalik EL, Mangravite LM, Langley RJ. A community approach to mortality prediction in sepsis via gene expression analysis. Nat Commun. 2018. February 15;9(1):694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012. March 29;119(13):2991–3002. [DOI] [PubMed] [Google Scholar]
- 47.Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, Evrard M, Li JLY, Tran HTN, Tan Y, Liu D, Mishra A, Liong KH, Leong K, Zhang Y, Olsson A, Mantri CK, Shyamsunder P, Liu Z, Piot C, Dutertre C, Cheng H, Bari S, Ang N, Biswas SK, Koeffler HP, Tey HL, Larbi A, Su I, Lee B, St John A, Chan JKY, Hwang WYK, Chen J, Salomonis N, Chong SZ, Grimes LH, Liu B, Hidalgo A, Newell EW, Cheng T, Ginhoux F, Ng LG. Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity. 2020. August 18;53(2):303–318. [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, Welch C, Cai B, Silvestre-Roig C, Ai D, Yang Y, Hidalgo A, Soehnlein O, Tabas I, Levine RL, Tall AR, Wang N. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 V617F Mice. Circ Res. 2018. November 9;123(11):e35–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.