

Abstract
Bacterial infections are a common and deadly threat to vulnerable patients. Alternative strategies to fight infection are needed. β-glucan, an immunomodulator derived from the fungal cell wall, provokes resistance to infection by inducing trained immunity, a phenomenon that persists for weeks to months. Given the durability of trained immunity, it is unclear which leukocyte populations sustain this effect. Macrophages have a life span that surpasses the duration of trained immunity. Thus, we sought to define the contribution of differentiated macrophages to trained immunity. Our results show that β-glucan protects mice from Pseudomonas aeruginosa infection by augmenting recruitment of innate leukocytes to the site of infection and facilitating local clearance of bacteria, an effect that persists for more than 7 days. Adoptive transfer of macrophages, trained using β-glucan, into naïve mice conferred a comparable level of protection. Trained mouse bone marrow-derived macrophages assumed an antimicrobial phenotype characterized by enhanced phagocytosis and reactive oxygen species production in parallel with sustained enhancements in glycolytic and oxidative metabolism, increased mitochondrial mass and membrane potential. Beta-glucan induced broad transcriptomic changes in macrophages consistent with early activation of the inflammatory response followed by sustained alterations in transcripts associated with metabolism, cellular differentiation and antimicrobial function. Trained macrophages constitutively secreted CCL chemokines and robustly produced pro-inflammatory cytokines and chemokines in response to LPS challenge. Induction of the trained phenotype was independent of the classic β-glucan receptors Dectin-1 and Toll-like receptor-2. These findings provide evidence that β-glucan induces enhanced protection from infection by driving trained immunity in macrophages.
Keywords: inflammation, chemotaxis, metabolic reprogramming, antimicrobial immunity, mitochondria, phagocytosis, gene transcription, dectin-1, TLR2
INTRODUCTION
Despite nearly a century of antibiotic development and administration, infectious diseases account for significant morbidity and mortality in the modern healthcare system (1). This is particularly true in the hospital setting where aging, immunocompromised, and critically ill patients are exposed to a broad array of pathogens (2). Therefore, new approaches to decrease the burden of infection among vulnerable populations are needed. Prophylactic immunomodulation that bolsters the host response to infection is a promising strategy to combat infection. Classically, vaccines have filled this niche. Vaccines induce highly specific and long-lived (months to years) cellular and humoral immunity that is mediated by memory T and B lymphocytes, respectively, of the adaptive immune system (3, 4). Yet, recent research shows that the innate immune system also retains memory of prior pathogen exposure and becomes armed to elicit more robust responses to subsequent infection (5, 6). This augmented state has been termed trained immunity or innate immune memory (7–9). Trained immunity differs from vaccine-induced immunity in two important ways. The typical duration is shorter and protection from infection is broad-based. Whereas vaccines evoke a memory response that last months to years, the duration of trained immunity is typically weeks to months (10, 11). While vaccines target a specific pathogen, trained immunity provides broad protection against a diverse cohort of pathogens and protection is not specific to the organism from which the training ligand was derived (12–14).
Immunomodulatory compounds that induce trained immunity are a promising tool to augment the host response to infection or serve as an adjunct to antibiotics (15). Beta-glucan, the most abundant polysaccharide of the fungal cell wall, possesses immunomodulatory properties that enhance the host response to diverse bacterial, fungal and viral pathogens (16). Beta-glucan reprograms the metabolic and epigenetic landscape of monocytes to induce enhancement of antimicrobial responses (17–19). Importantly, β-glucan’s protective effects last weeks to months via trained immunity (20). However, monocytes have a life span of a few days, which is incompatible with sustaining the trained phenotype beyond that short timeframe (21). Evidence indicates that epigenetic changes in bone marrow progenitors may serve as a mechanism of providing trained monocytes to sustain the trained phenotype (22). Alternatively, training of differentiated macrophages provides a complementary mechanism to consider. Unlike monocytes, tissue macrophages have a lifespan of months to years and are key regulators of the host response to infection and inflammation (23). With the exception of the small intestinal walls, dermis, and heart, circulating monocytes do not significantly contribute to the population of tissue macrophages in most organs (24). Thus, whether β-glucan trains differentiated macrophages similarly to circulating monocytes is unclear. Additionally, the contribution of trained macrophages to protection from infection has not been adequately explored.
Macrophages are crucial for initiating and coordinating the host immune response to infection. The detection of microbial pathogen-associated molecular patterns (PAMPs) by macrophages initiates a rapid inflammatory response that leads to mobilization of innate immune cells, chiefly monocytes and neutrophils, and their recruitment to the site of infection. Recruited myeloid cells respond by phagocytosing and killing the pathogen locally. Failure of the innate response may lead to bacterial dissemination and systemic infection, which dramatically increases morbidity and mortality (25). Therefore, the development of approaches to augment macrophage antimicrobial functions safely and effectively could serve to prevent and treat infections, especially in vulnerable populations where innate immune function is suppressed or dysregulated (26, 27).
Macrophages detect β-glucan by the pattern recognition receptors (PRRs) Dectin-1 and Toll-like receptor (TLR)-2 (28–30). Genetic deficiency of Dectin-1 leads to recurrent infections with Candida albicans in humans (31). However, some macrophage responses to β-glucan are independent of Dectin-1 and TLR2 (32). Furthermore, the importance of Dectin-1 and TLR2 for inducing trained immunity in macrophages has not been determined. Understanding the signaling cascades that contribute to protective phenotype is important for optimizing the translational potential of β-glucan immunotherapy.
We tested the hypothesis that training of differentiated macrophages with β-glucan will provide resistance against infection with Pseudomonas aeruginosa, a common nosocomial pathogen with a high frequency of antibiotic resistance (33, 34). Furthermore, we hypothesized that β-glucan-induced protection is driven by antimicrobial, metabolic, and biochemical alterations in trained macrophages. Our results show that adoptive transfer of macrophages trained with β-glucan protects mice from P. aeruginosa infection by augmenting innate leukocyte recruitment to sites of infection and enhancing local bacterial clearance. Further, using a combination of transcriptomic, metabolomic, and immunologic techniques, we show that β-glucan-trained macrophages develop distinct metabolic and phenotypic characteristics, and a robust antimicrobial phenotype. Finally, we demonstrate that β-glucan training is independent of Dectin-1 and TLR2 signaling. These findings advance the paradigm of innate immune memory by demonstrating the trained phenotype in differentiated macrophages treated with β-glucan.
MATERIALS AND METHODS
Mice.
Wild type male and female C57BL/6 mice, age 10-12 weeks, were purchased from The Jackson Laboratory (Bar Harbor, ME). Clec7a−/− (Dectin-1 KO) and Tlr2−/− (TLR2 KO) mice were purchased from The Jackson Laboratory. Clec7a−/−Tlr2−/− (double knockout; DKO) mice were generated at Vanderbilt University. The authenticity of knockout strains was validated by routine genotyping. All experiments and procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Beta-glucan preparation and administration.
The β-(1,3)-D-glucan training reagent and linear glucan were isolated from Candida albicans and purified as described previously (35). The purity of β-glucan isolates was confirmed using 1H and 13C NMR spectroscopy. All preparations of β-glucan were determined to be endotoxin-free. It is important to note that the β-glucan training reagent used in this research is the same as that used in the studies of Ifrim (36), Cheng (19), Saeed (17) and Garcia-Valtanen (11). This reagent is prepared exclusively in the Williams laboratory and is recognized worldwide as the “gold standard” immune training agent. Mice received 1 mg of β-glucan by intraperitoneal (i.p.) injection of 0.2 mL vortexed suspension of 5% dextrose in H20 (D5W). Treatment groups in cell culture received 5 mg/mL of β-glucan for the indicated times. Mice and macrophages treated with vehicle served as control.
Intraperitoneal infection model.
Mice were infected with Pseudomonas aeruginosa via the i.p. route as described in our previous studies (37). P. aeruginosa was purchased from American Type Culture and Collection (Manassas, VA; ATCC 19660). Bacterial cultures were grown in tryptic soy broth for 22 hours at 37°C, washed, and diluted in sterile saline. Mice were inoculated i.p. with 1 x 108 colony-forming units (CFUs) P. aeruginosa in 0.5 mL saline. Six hours after inoculation, body temperatures were measured by rectal thermometer and mice were anesthetized. Whole blood was collected by carotid artery laceration under isoflurane anesthesia into heparinized microcentrifuge tubes, centrifuged at 2000 x g for 15 minutes at 4°C and plasma was collected and stored at −80°C for storage until subsequent cytokine analysis. Following cervical dislocation, the peritoneal cavity was lavaged with 5 mL of cold sterile phosphate buffered saline (PBS). A portion of lavage fluid was diluted and plated on trypic soy agar overnight and bacterial colonies were counted to determine CFUs/mL recovered. The remaining peritoneal lavage fluid was centrifuged at 300 x g for 6 minutes at 4°C and diluted appropriately for flow cytometric analyses.
Bone marrow-derived macrophages (BMDM).
Femurs were harvested from mice and flushed with RPMI 1640 containing 2 mM glutamine and 25 mM HEPES (Gibco, Grand Island, New York) supplemented with 10% certified performance plus fetal bovine serum (FBS, Gibco), 1% antibiotic-antimycotic (Gibco), and 10 ng/mL mouse recombinant macrophage-colony stimulating factor (M-CSF, R&D Systems), henceforth referred to complete media. Bone marrow cell suspensions were centrifuged at 300 x g for 6 minutes at 4°C and plated at a concentration of 5 x 104 cells/mL in complete media. After 7 days of differentiation, BMDM received fresh media and were treated with 5 mg/mL β-glucan or vehicle as unstimulated controls for 24 hours. Macrophages were washed and allowed to rest in complete media for 3 days to generate the trained phenotype (3 days-post group; 3dp). Separately, BMDM were maintained in complete media and stimulated with 5 mg/mL β-glucan or vehicle for 4 or 24 hours prior to assessment (4h and 24h groups).
BMDM adoptive transfer.
Trained BMDM were prepared as above, harvested, and resuspended at a concentration of 5 x 106 cells/mL in PBS. One day prior to infection, mice received 1 x 106 control or trained BMDM by i.p. injection. Twenty-four hours later, mice were inoculated i.p. with 1 x 108 CFUs of P. aeruginosa. Core body temperature, whole blood collection, peritoneal lavage, flow cytometry, and cytokine analysis were performed as described above.
Cytokine analysis.
The concentration of IL-6, CCL3, CCL4, and TNFα were measured by DuoSet ELISA kits from R&D Systems (Minneapolis, MN). Cytokines were measured from in vivo infection samples or conditioned cell culture media where indicated.
Flow cytometry.
Cells collected by peritoneal lavage were resuspended in PBS at a concentration of 1 x 107 cells/mL and incubated with 1 mg/mL anti-mouse CD16/32 (eBioscience, San Diego, CA) prior to addition of fluorochrome-conjugated antibodies (0.5 mg/106 cells) and incubation for 15 minutes at room temperature. Antibodies used to differentiate peritoneal leukocytes included anti-F4/80-FITC (clone BM8; eBioscience), anti-Ly6G-PE (clone 1A8; BD Biosciences, San Jose, CA), anti-Ly6C-PE Cy5.5 (clone HK1.4; eBioscience) alongside respective isotype controls. Monocytes were identified as F4/80+Ly6C+, macrophages as F4/80+Ly6C−, and neutrophils as Ly6G+F4/80−. Data were collected using an Accuri C6 flow cytometer and analyzed using Accuri C6 software (BD Biosciences).
Reactive oxygen species measurements.
BMDM were assessed with the Respiratory Burst Assay Kit (Cayman Chemical, Ann Arbor, MI). BMDM were incubated with dihydrorhodamine-123 for 1 hour at 37°C. Rhodamine-123 fluorescence was determined by flow cytometry.
Phagocytosis assay.
Staphylococcus aureus particles (Invitrogen) labelled with pHrodo-red dye were suspended in phenol red-free RPMI 1640 and sonicated for 10 minutes. pHrodo particles were added to BMDM cultures and placed in a Synergy H1 plate reader at 37°C (BioTek, Winooski, VT). pHrodo fluorescence was measured every 15 minutes for the indicated time.
Bacterial killing assay.
One day prior to the assay, BMDM were plated at 1 x 105 cells/well in a 96-well plate. Cells were washed with warm PBS 5 times and placed in RPMI 1640 containing 0.1% FBS (Gibco). P. aeruginosa was prepared as above and diluted to 1 x 106 CFU/mL after passage of stock through a 26g needle. Bacteria were added to BMDM and plates were centrifuged at 515 x g for 4 minutes at 4°C. BMDM and bacteria were co-incubated for 1 hour at 37°C and supernatants were collected and plated for bacterial counting, as above. BMDM were then washed with warm PBS 5 times and placed in RPMI 1640 containing 0.1% FBS (Gibco) and 300 mg/mL gentamicin (Sigma-Aldrich) for 1 hour. BMDM were washed with PBS and a solution of 0.02% triton-X (Sigma-Aldrich) in PBS was added to the 96-well plate and vigorously pipetted to generate cellular lysates. Lysates were collected and plated for bacterial counting, as above.
Seahorse extracellular flux analysis.
One day prior to the assay, BMDM were plated at 5 x 104 cells/well in a 96-well Seahorse assay plate. All measurements were performed on a Seahorse XFe96 Extracellular Flux Analyzer (Agilent). The glycolysis and mitochondrial stress tests were performed using the manufacturer’s protocol. Briefly, extracellular acidification rate was measured at baseline and after the addition of 10 mM glucose (Sigma-Aldrich), 1 mM oligomycin (Agilent), and 50 mM 2-deoxyglucose (2-DG; Sigma-Aldrich). Oxygen consumption rate was measured at baseline and after the addition of 1 mM oligomycin, 1 mM FCCP (Agilent), and 0.5 mM antimycin A and rotenone (Agilent).
MitoTracker and Tetramethylrhodamine, methyl ester (TMRM) staining.
BMDM were plated at 2.4 x 105 cells/well in a 24-well plate one day prior to the assay. 50 mM MitoTracker Green dye (Invitrogen, Carlsbad, CA) or 100 nnM TMRM dye (Invitrogen) was added for 30 minutes at 37°C to stain total and active mitochondria, respectively. BMDM were washed and assessed by flow cytometry using channel FL1 (green) for MitoTracker and FL3 (red) for TMRM.
Lipopolysaccharide (LPS) stimulation.
BMDM were treated with β-glucan as described above. Fresh media containing 100 ng/mL LPS (ultrapure, Invitrogen) derived from Escherichia coli 0111:B4 was added to the cell culture. Four hours after incubation with LPS, cellular lysates were harvested for RNA as above. Separately, conditioned cell culture media was collected six hours after incubation. After 6 and 24 hours of LPS stimulation, the Seahorse extracellular flux assay was performed as above.
RNA sequencing.
Total RNA was isolated under an RNase-free environment, using the RNeasy Mini Kit (Qiagen, Hilden, Germany), and treated with DNase (Qiagen). Total RNA quality and concentration were verified with a Thermo Scientific NanoDrop 2000 spectrophotometer. Purified RNA was assessed with Qubit and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and RNA integrity number (RIN) was determined for each sample (38, 39). All samples had a RIN score of 7 or greater. mRNA Libraries were prepared using NEBNext Poly(A) selection (New England Biolabs). Sequencing was performed at paired-end 150 bp on an Illumina NovaSeq 6000 with at least 50 million reads per sample by Vanderbilt Technologies for Advanced Genomics. RNASeq data were analyzed with Basepair (www.basepairtech.com). Briefly, reads were aligned to the mm10 genome using STAR after trimming and undergoing quality control with QC30 (40). Read counts were measured using featureCounts (41). Differentially expressed genes were identified using DESeq2 (42). Transcripts with log2foldchange of at least 1.0 and p-adjusted < 0.05 were considered significant for individual gene analysis. Gene Set Enrichment Analysis (GSEA) was used to define significantly altered biological processes sorted with Gene Ontology (43–45). Raw data was deposited using the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). Accession Number: GSE174141.
Western blotting.
Cellular lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MI) containing cOmplete protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail (Roche, Basel, Switzerland). Lysate protein concentrations were quantified using the bicinchoninic acid assay for normalization (Pierce, ThermoFisher, Waltham, MA). Samples were separated by gel electrophoresis on Mini-Protean 4-20% Tris-glycine gels (Bio-Rad). Sample proteins were transferred onto nitrocellulose membranes overnight (PerkinsElmer, Boston, MA). Membranes were blocked with 5% fraction V BSA (RPI, Mount Prospect, IL) and incubated with primary antibodies (1:1000 dilution) overnight at 4 °C. Membranes were washed and then placed in HRP-conjugated secondary antibodies (1:2000 dilution) for two hours at room temperature followed by ECL reagent (Bio-Rad). Protein bands were detected by film exposure.
Statistics.
Data were analyzed using GraphPad Prism 8.3.0 (La Jolla, CA) software unless otherwise noted. Data are expressed as mean ± SEM or median where noted. Data from experiments containing multiple groups were compared using one-way ANOVA followed by Tukey’s post hoc multiple comparison test. Body temperature and bacterial counts were compared using the Mann-Whitney test when comparing two groups or Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test when comparing more than two groups. Differences in gene expression for RNASeq data were determined using the DESeq2 protocol. A p value of < 0.05 was considered statistically significant.
RESULTS
Beta-glucan augments innate immune defense against P. aeruginosa in mice
To characterize the innate immune response to a clinically relevant pathogen after β-glucan training, mice were treated i.p. with 1 mg β-glucan or vehicle on two consecutive days and were infected with Pseudomonas aeruginosa at one, three, seven, or fourteen days later. Rectal temperature, bacterial counts and leukocytes numbers were measured 6 hours after infection (Fig. 1A). Vehicle-treated control mice became hypothermic while β-glucan-primed mice maintained normothermia across all time points (Fig. 1B). Beta-glucan-treated mice showed significantly lower P. aeruginosa CFUs in peritoneal lavage as compared to vehicle-treated mice for up to 7 days following β-glucan treatment and returned to control levels on day 14 (Fig. 1C). The number of monocytes in the peritoneal cavity was significantly elevated in mice treated with β-glucan at 1 and 3 days prior to infection and returned to control levels at day 7 (Fig. 1D). Mice treated with β-glucan displayed elevated neutrophils in the peritoneal cavity following infection for up to 14 days, although infection-elicited intraperitoneal neutrophil numbers waned after day 7 (Fig. 1E). The number of macrophages in the peritoneal cavity after infection was significantly elevated for 7 days in mice treated with β-glucan treatment but diminished by day 14 (Fig 1F). Taken together, these results demonstrate that β-glucan treatment augments leukocyte recruitment and bacterial clearance for at least 7 days and confers physiologic protection for at least 14 days in response to infection with the clinically relevant pathogen P. aeruginosa.
Figure 1. Beta-glucan augments innate immune defense against P. aeruginosa in mice.
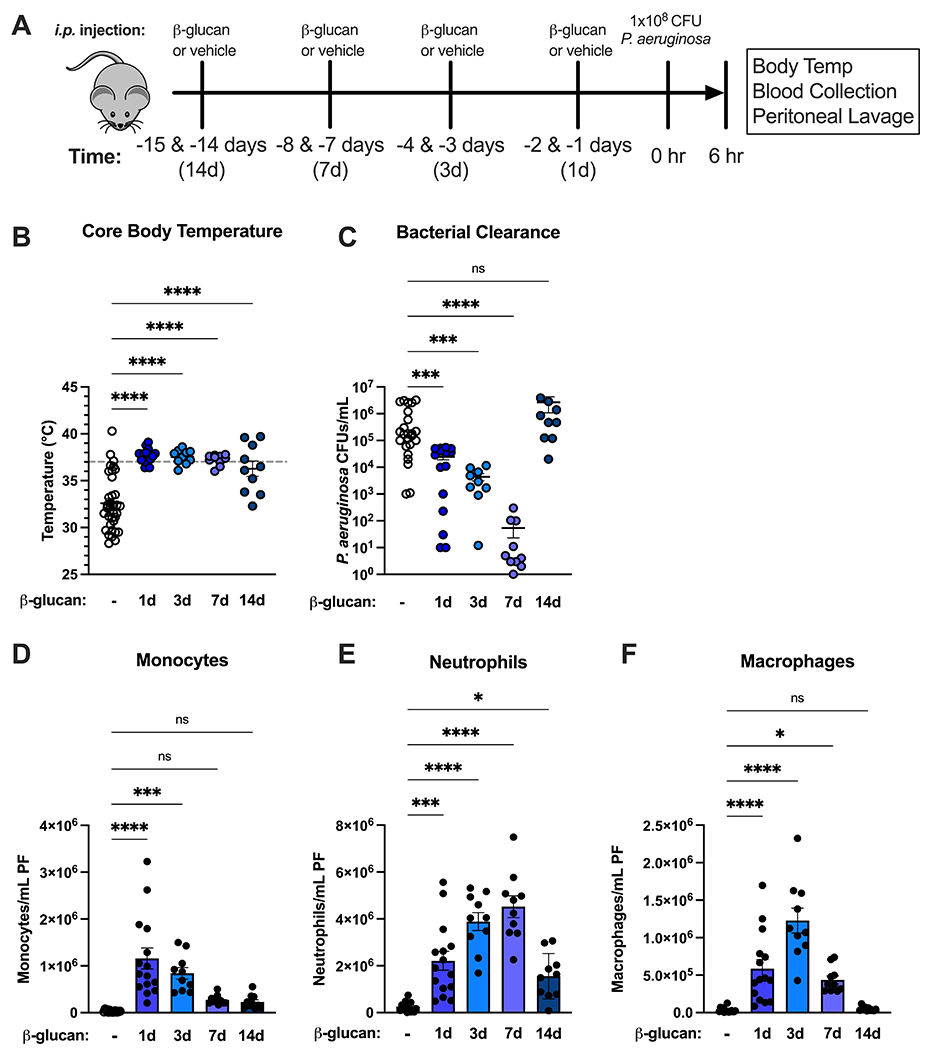
(A) C57Bl/6 mice were injected i.p. with β-glucan (1 mg) or vehicle on 2 consecutive days at 1, 3, 7 and 14 days prior to i.p. inoculation with 108 CFU P. aeruginosa with harvest of plasma and peritoneal lavage fluid 6 hours after infection. (B) Core (rectal) body temperature in vehicle- or β-glucan-treated mice. (C) CFUs of P. aeruginosa per mL of peritoneal fluid. (D-F) Number of monocytes (D), neutrophils (E), or macrophages (F) in infected vehicle- or β-glucan-treated mice. Body temperature and clearance data shown as median. All other data shown as mean ± SEM. N = 10-20 mice per group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by Mann-Whitney U test (B, C) or ANOVA with Tukey’s post-hoc multiple comparison test (D-F).
Adoptive transfer of β-glucan-trained macrophages protects against P. aeruginosa infection
Trained or control BMDM were adoptively transferred i.p. 24 hours prior to infection with P. aeruginosa (Fig. 2A). Mice treated with vehicle or control BMDM developed hypothermia 6 hours after infection, while mice that received β-glucan-treated BMDM maintained normothermia (Fig. 2B). Similar to in vivo systemic training with β-glucan, mice that received trained BMDM had lower bacterial counts in the peritoneal cavity than mice treated with vehicle or control BMDM (Fig. 2C). Mice treated with β-glucan-trained BMDM had significantly more neutrophils (Fig. 2D), monocytes (Fig. 2E) and macrophages (Fig. 2F) at the site of infection compared to mice receiving vehicle or control BMDM. These data demonstrate that adoptive transfer of trained macrophages is sufficient to reproduce the protective benefit of β-glucan treatment, supporting the premise that macrophages contribute to β-glucan-induced trained immunity and resistance to infection.
Figure 2. Adoptive transfer of β-glucan-trained macrophages protects against P. aeruginosa infection.
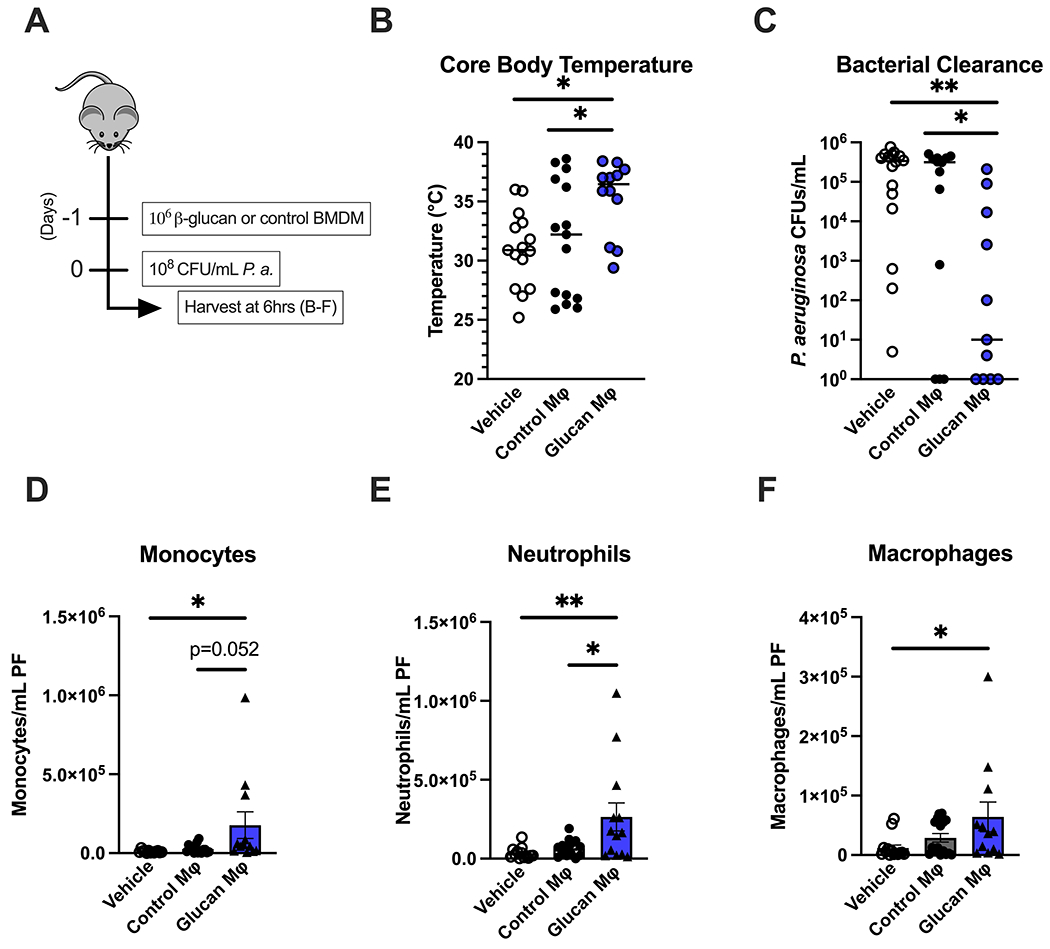
(A) BMDM were treated with β-glucan (5 μg) or vehicle for 24 hours, washed and allowed to rest for 3 days. C57Bl/6 mice were injected i.p. with vehicle (PBS), control BMDM, or β-glucan-treated BMDM 24 hours prior to i.p. inoculation with 108 CFU P. aeruginosa with subsequent harvest of plasma and peritoneal lavage fluid 6 hours later. (B) Core (rectal) body temperature in vehicle, control-BMDM, or β-glucan-BMDM-treated mice after P. aeruginosa challenge. (C) CFU of P. aeruginosa per mL of peritoneal fluid. (D-F) Number of monocytes (D), neutrophils (E), or macrophages (F) in vehicle, control- or β-glucan-BMDM-treated mice. Body temperature and clearance data shown as median. All other data shown as mean ± SEM. N = 10-15 mice per group. * p<0.05, ** p<0.01 by Kruskal Wallis test with Dunn’s post-hoc multiple comparison test (B, C) or ANOVA with Tukey’s post-hoc multiple comparison test (D-F).
Beta-glucan-trained macrophages display a robust antimicrobial phenotype
Based on our in vivo data, we hypothesized that β-glucan enhances macrophage antimicrobial capacity, which we assessed using several approaches. BMDM trained with β-glucan (3dp) were significantly larger (Fig. 3A) and more granular (Fig. 3B) than control and 24h BMDM, as measured by forward and side scatter using flow cytometry. Rhodamine-123 staining showed increased reactive oxygen species (ROS) production in 24h and 3dp BMDM as compared to the control BMDM (Fig. 3C). Trained BMDM phagocytosed significantly more pHrodo-labelled bacteria particles over time than control BMDM (Fig. 3D). Increased numbers of viable P. aeruginosa were also present in trained BMDM as compared to controls, confirming augmented phagocytosis (Fig. 3E).
Figure 3. Beta-glucan-trained macrophages display a robust antimicrobial phenotype.
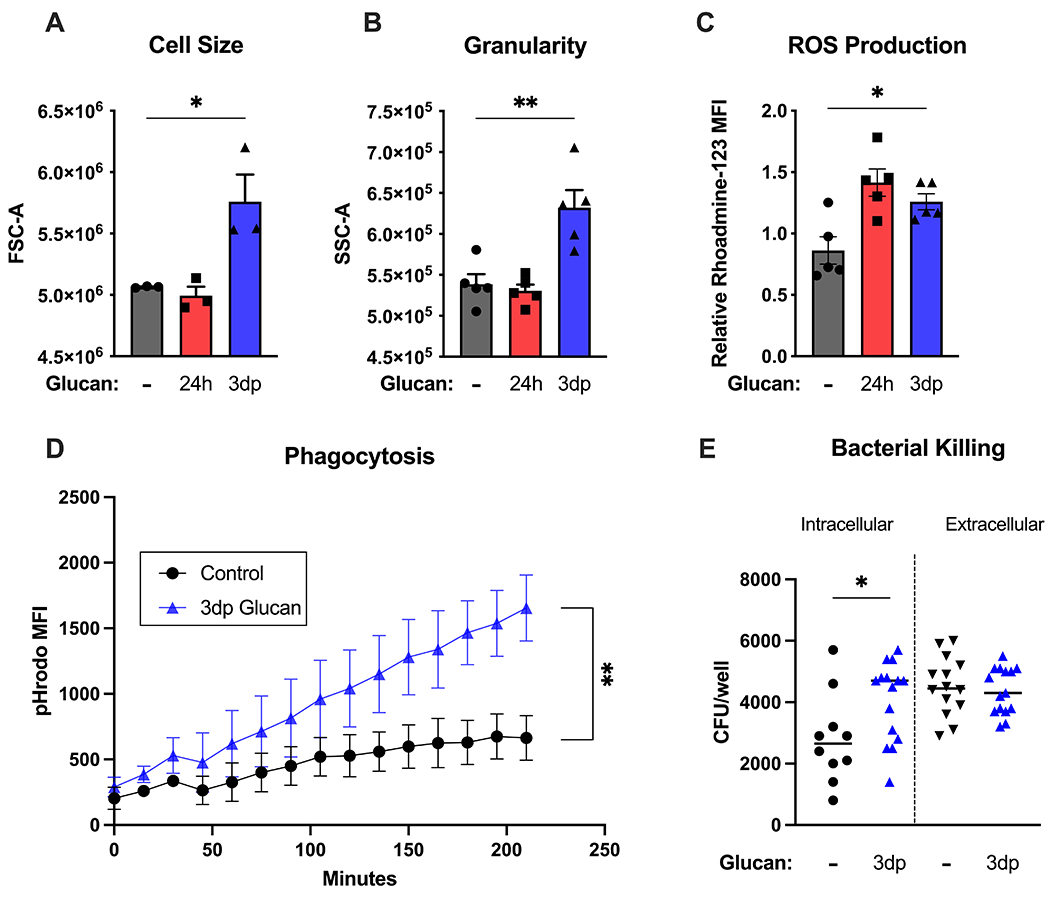
BMDM were treated with β-glucan (5 μg) or vehicle for 24 hours (24h), washed and allowed to rest for 3 days (3dp) followed by assessment of macrophage phenotype. (A) Cell size was measured by forward scatter. (B) Cell granularity as measured by side scatter. (C) Rhodamine-123 fluorescence was measured after a 15-minute incubation period using flow cytometry. (D) Control or trained BMDM were incubated with pHrodo S. aureus particles. pHrodo MFI was measured every 15 minutes for 5 hours. (E) BMDM were incubated with P. aeruginosa. Intracellular and extracellular CFU were quantified as described in Methods. Data shown as mean ± SEM. Experiments were performed with 3-5 biological replicates. * p<0.05, ** p<0.01 by ANOVA with Tukey’s post-hoc multiple comparison test (A-C) or repeated two-way ANOVA (D).
Beta-glucan training augments metabolism and increases mitochondrial content and membrane potential in macrophages
We next sought to evaluate the metabolic phenotype of macrophages trained with β-glucan (Fig. 4). BMDM treated with β-glucan for 24 hours had elevated baseline and maximal extracellular acidification rate (ECAR), an indirect measure of lactate production and glycolysis., compared to control (Fig. 4A–C). Trained (3dp) BMDM displayed significantly higher ECAR levels than control and those treated for 24 hours (Fig. 4A–C). Both 24h and 3dp BMDM displayed elevated baseline and maximal oxygen consumption rate (OCR) compared to control, indicating a sustained increase in oxidative phosphorylation (Fig. 4D–F).
Figure 4. Beta-glucan training augments metabolism and increases mitochondrial content and membrane potential in macrophages.
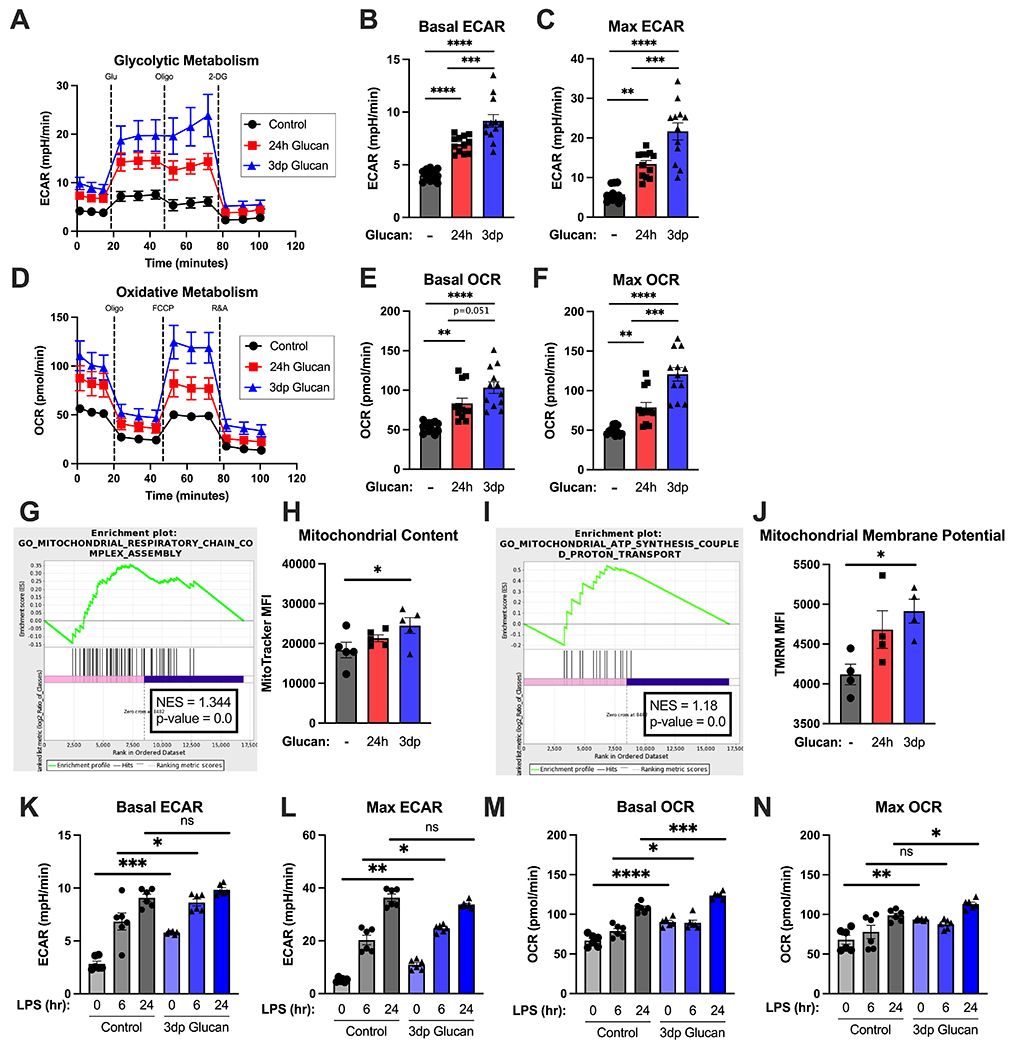
BMDM were treated with β-glucan (5 μg) or vehicle for 24 hours (24h), washed and allowed to rest for 3 days (3dp) followed by assessment of macrophage metabolic phenotype. (A) Glycolysis stress test of control, 24h, and trained (3dp) BMDM on the Seahorse XFe96. Extracellular acidification rate was measured over time at baseline and after glucose, oligomycin, and 2-deoxyglucose addition. (B) Basal ECAR as determined from 3 separate runs. (C) Maximum ECAR as determined from 3 separate runs. (D) Oxidative stress test of control, 24h, and trained BMDM on the Seahorse XFe96. Oxygen consumption rate was measured over time at baseline and after oligomycin, FCCP, and R&A administration. (E) Basal OCR as determined from 3 separate runs. (F) Maximum OCR as determined from 3 separate runs. (G) Enrichment plot for GO term “Mitochondrial Respiratory Chain Complex Assembly” for control vs. trained (3dp) BMDM. (H) MitoTracker Green MFI was measured in BMDM by flow cytometry after 30 minutes incubation. (I) Enrichment plot for GO term “Mitochondrial ATP Synthesis-Coupled Proton Transport” for control vs. Trained (3dp) BMDM. (J) TMRM MFI was measured in BMDM by flow cytometry after 30 minutes incubation. (K-L) Basal and maximal ECAR as determined by Seahorse XFe96 in control and trained BMDM stimulated with vehicle or 100 ng/mL LPS for 4hr and 24hr. (M-N) Basal and maximal OCR as determined by Seahorse XFe96 in control and trained BMDM stimulated with vehicle or 100 ng/mL LPS for 4hr and 24hr. Data shown as mean ± SEM. Experiments were performed with 3-5 biological replicates. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by ANOVA with Tukey’s post-hoc multiple comparison test.
Because β-glucan increased oxidative metabolism in macrophages, we assessed mitochondrial mass and activity. First, we asked whether trained BMDM increased the available mitochondrial pool. We performed an unbiased transcriptomic analysis of macrophages trained with β-glucan and assessed metabolic pathways using gene ontology (GO) to identify enriched pathways. BMDM trained with β-glucan (3dp) displayed increases in transcriptomic signatures associated with oxidative phosphorylation (GO:0033108, NES = 1.344, Fig. 4G). In concert with this finding, 3dp BMDM had significantly higher mitochondrial content as compared to control or 24h BMDM as demonstrated by increased MitoTracker Green staining (Fig. 4H). These changes were mirrored by an increase in transcription related to ATP biosynthesis (GO: 0042775, NES = 1.18, Fig. 4I) and mitochondrial membrane potential, as measured by tetramethylrhodamine (TMRM) staining (Fig. 4J). Thus, β-glucan treatment induces a unique metabolic phenotype in differentiated macrophages characterized by elevated ECAR and OCR in association with increases in mitochondrial content and function.
Next, we sought to determine the effect of β-glucan training on the macrophage metabolic response to LPS challenge (Fig. 4K–N). Control and β-glucan-trained BMDM were stimulated with 100 ng/mL LPS for 6 and 24 hours and ECAR and OCR were measured. Trained BMDM displayed a significant increase in basal and maximal glycolytic rate at baseline and after 6 hours of LPS stimulation as compared to control BMDM (Fig. 4K–L). After 24 hours of LPS stimulation, both control and trained BMDM showed similar glycolytic rates. Trained BMDM showed increased basal and maximal OCR in the absence of LPS stimulation compared to control BMDM. Both control and trained BMDM showed no or modest increases in basal and maximal OCR, respectively, at 6 hours after LPS challenge (Fig. 4M–N). However, trained BMDM showed more robust increases in basal and maximal OCR in response to 24 hours of LPS stimulation compared to control BMDM.
Beta-glucan induces a distinct transcriptomic profile in macrophages
To understand the evolution of the macrophage response to β-glucan over time, we looked further at the transcriptomic analysis of macrophages treated with β-glucan for 4 or 24 hours or after completion of our training protocol (i.e., 3dp; Fig. 5). Principal component analysis showed that β-glucan induced significant alterations in macrophage gene expression. This representation demonstrates that acute stimulation (4hr) with β-glucan caused the largest variance in PC1 compared to control, with 24h and 3dp BMDM more closely resembling control BMDM in PC1, although 24h and 3dp BMDM sustained distinct differences in PC2 (Fig. 5A). After 4 hours of β-glucan stimulation, the expression of 257 (211 up/ 46 down) genes were altered compared to vehicle-treated controls (Fig. 5B). Twenty-four hours after stimulation, BMDM were less transcriptionally active than after acute 4hr exposure, with 104 (67 up/ 37 down) transcripts significantly altered (Fig. 5B). After completion of the trained immunity protocol, 109 transcripts were altered, with most being downregulated rather than upregulated (29 up/ 80 down) (Fig. 5B). Evaluation of gene ontology (GO) terms sorted by enrichment score (ES) showed that macrophages treated with β-glucan for 4 hours triggered pathways consistent with acute inflammation and activation of the innate immune system (Figure 5C). At 24 hours and in 3dp BMDMs, gene pathways associated with housekeeping and cellular differentiation predominated (Figures 5D and E). These results point toward alterations in gene transcription that underpin the antimicrobial phenotype seen in trained BMDM.
Figure 5. Beta-glucan induces a distinct transcriptomic profile in macrophages.
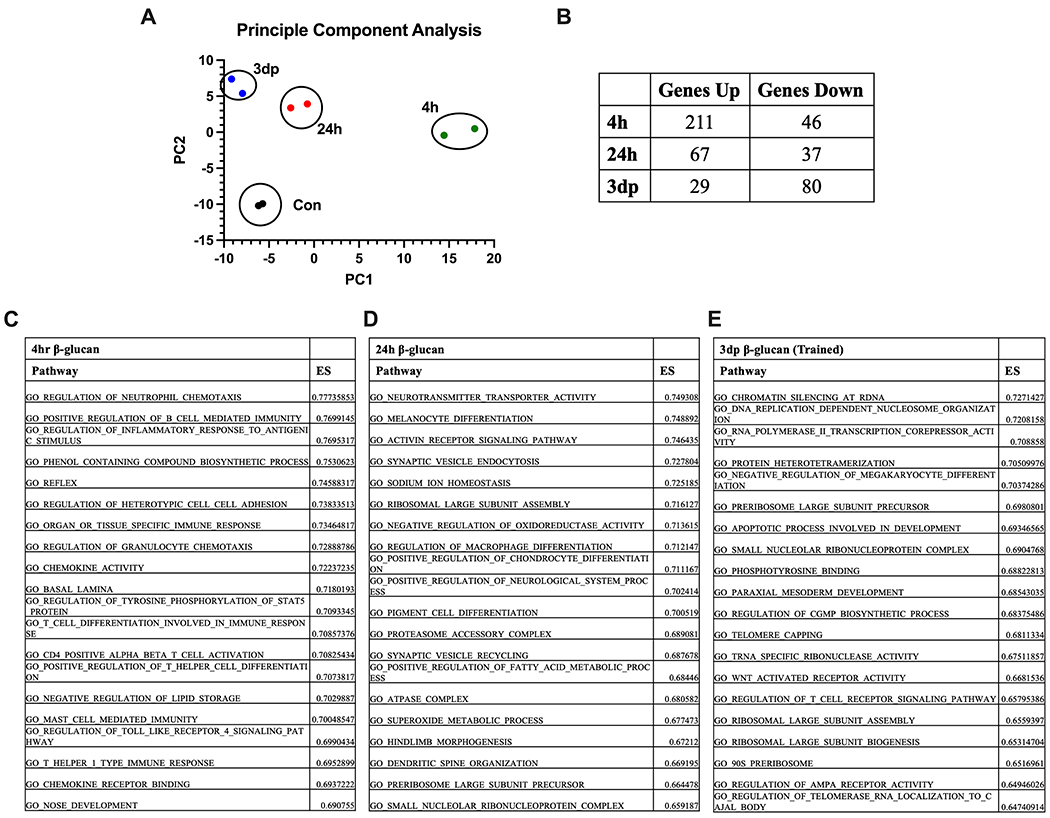
BMBM were treated with β-glucan (5 μg) or vehicle for 4 (4h) or 24 (24h) hours. A subset of 24h BMDM were washed and allowed to rest for 3 days (3dp). Gene expression was measured using RNAseq. (A) Principal Component Analysis of BMDM harvested at specified time points. (B) Relative gene expression by BMDM treated with β-glucan relative to control at the specified time points. ES = enrichment score (C) Top 20 pathways identified by gene ontology analysis at 4 hours after β-glucan treatment relative to control. (D) Top 20 pathways identified by gene ontology analysis at 24 hours after β-glucan treatment relative to control. (E) Top 20 pathways identified by gene ontology analysis at 3dp after β-glucan treatment relative to control. RNA quantification performed in duplicate.
Impact of β-glucan on macrophage cytokine and chemokine production
We analyzed the impact of β-glucan treatment on cytokine production at the mRNA and protein levels (Fig. 6). Transcriptomic data revealed that treatment of BMDM with β-glucan for 4 hours induced transcription of numerous proinflammatory cytokines and reduced transcription of others, such as transforming growth factor-β (Fig. 6A). In 24h and 3dp BMDM, transcription of pro-inflammatory cytokines returned to near or below baseline levels and suppressed cytokines remained suppressed (Fig. 6A). Challenge of control and 3dp BMDM with LPS potently induced cytokine production with no discernable differences among groups (Fig 6A). Measurement of TNFα and IL-6 concentrations in culture supernatants from control and 3dp BMDM also showed no significant difference in LPS-induced cytokine production among groups (Fig. 6B–C). Because β-glucan training in vivo leads to increased leukocyte recruitment, we next sought to determine if training impacted the ability of BMDM to produce and secrete chemokines. Transcripts of several chemokines that facilitate neutrophil and monocyte recruitment were upregulated 4 hours after β-glucan stimulation in BMDM (Fig. 6D). Many of these transcripts remained upregulated in the 24h and 3dp BMDM groups. Challenge of control and 3dp BMDM with LPS potently induced chemokine production with no discernable differences among the groups (Fig 6D). Measurement of CXCL1 and CXCL2 concentrations in culture supernatants from control and 3dp BMDM showed potent induction of chemokine production with small differences among groups, although concentrations of CXCL2 were significantly higher in 3dp BMDM compared to control (Fig. 6E–F). Similar patterns of cytokine and chemokine secretion by control and 3dp BMDM were observed after challenge with heat-killed P. aeruginosa (Suppl Fig 1). Thus, LPS-induced production of pro-inflammatory cytokines and chemokines is sustained in trained BMDM.
Figure 6. Beta-glucan alters macrophage cytokine and chemokine production.
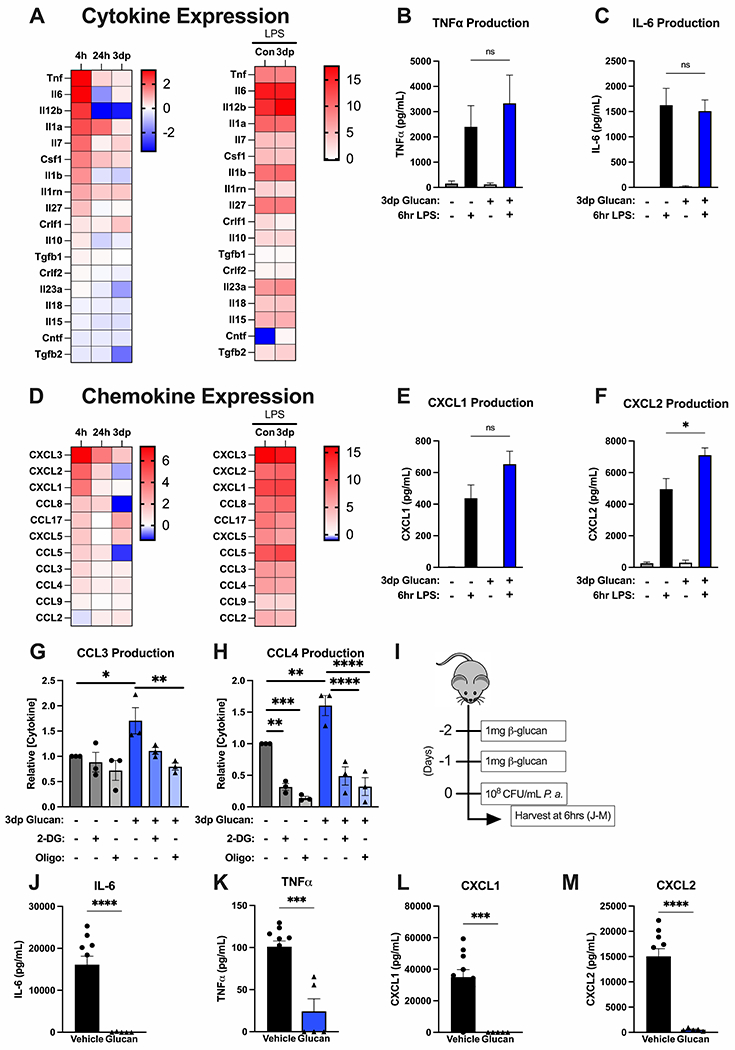
BMBM were treated with β-glucan (5 μg) or vehicle for 4 (4h) or 24 (24h) hours. A subset of 24h BMDM were washed and allowed to rest for 3 days (3dp). Cytokine mRNA expression was measured by RNAseq, protein was measured by ELISA. (A) Heatmaps of cytokine mRNA expression by BMDM at 4h, 24h, or 3dp relative to control and by control and 3dp BMDM at 4h after LPS challenge. (B-C) Concentrations of TNFα and IL-6 in conditioned media from control and 3dp BMDM before and after LPS challenge. (D) Heatmaps of chemokine mRNA expression by BMDM at 4h, 24h, or 3dp relative to control and by control and 3dp BMDM at 4h after LPS challenge. (E-F) Concentrations of CXCL1 and CXCL2 in conditioned media from control and 3dp BMDM before and after LPS challenge. (G-H) Concentrations of CCL3 and CCL4 in conditioned media from control and 3dp BMDM after treatment with 2-DG or oligomycin for 6 hours. Data normalized to untreated control BMDM. (I) C57Bl/6 mice were injected i.p. with β-glucan (1 mg) or vehicle on 2 consecutive days prior to i.p. inoculation with 108 CFU P. aeruginosa with subsequent harvest of plasma 6 hours later. (J-M) Concentrations of IL-6, TNFα, CXCL1 and CXCL2 in peritoneal lavage at 6 hours after P. aeruginosa challenge. RNASeq experiments were performed in duplicate. All other in vitro experiments were performed with 3-5 biological replicates. N=5 mice per group for in vivo experiments. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by ANOVA with Tukey’s post-hoc multiple comparison test.
Consistent with constitutive expression of some chemokine transcripts by 3dp BMDM, conditioned media from 3dp BMDM contained higher levels of CCL3 (Fig. 6G) and CCL4 (Fig. 6H) than untreated controls. Addition of 500 mM 2-DG or 1 mM oligomycin, to block glycolysis or oxidative phosphorylation, respectively, blunted the secretion of both of these chemokines in control and trained macrophages (Fig. 6G–H). Thus, β-glucan facilitates constitutive production of some chemokines in BMDM, which is dependent on β-glucan-induced augmentation of metabolism.
We next undertook experiments to determine whether cytokine and chemokine production after β-glucan training and infection in vivo mirrored that of BMDM ex vivo. Mice were treated with β-glucan for 2 consecutive days followed by challenge with P. aeruginosa 24 hours later. Peritoneal lavage was obtained for cytokine measurements 6 hours after infectious challenge (Fig. 6I). P. aeruginosa challenge potently induced production of IL-6, TNFα, CXCL1 and CXCL2 in control mice (Fig. 6J–M). All of these cytokines were significantly lower in peritoneal lavage from β-glucan-trained mice (Fig. 6J–M). These finding show that local cytokine production is diminished in trained mice, compared to controls, and may be influenced by the greatly augmented clearance of bacteria observed in trained mice (see Fig. 1).
Beta-glucan-induced trained immunity is independent of Dectin-1 and Toll-like receptor-2 activation
We aimed to define the PRR pathways necessary for β-glucan-induced trained immunity. Because Dectin-1 and Toll-like receptor (TLR)-2 are the two major surface receptors for β-glucan on innate leukocytes, we hypothesized that deficiency of these receptors would ablate β-glucan-induced trained immunity. To test this hypothesis, we treated wildtype, Dectin-1 (Dec1 KO), TLR-2 (TLR2 KO), and Dectin-1/TLR-2 double knockout (DKO) mice with β-glucan 48h and 24h prior to i.p. inoculation with P. aeruginosa (Fig. 7A). In all genotypes, vehicle-treated controls developed hypothermia, but β-glucan-treated mice maintained normothermia (Fig. 7B). There were no statistically significant differences observed in core body temperature when comparing β-glucan-treated mice across genotypes. Beta-glucan-treated mice of all genotypes showed lower P. aeruginosa CFUs in peritoneal lavage than vehicle-treated controls (Fig. 7C). There were no differences in P. aeruginosa CFUs between genotypes in the β-glucan-treated mice. Beta-glucan treatment increased the numbers of monocytes (Fig. 7D), neutrophils (Fig. 7E), and macrophages (Fig. 7F) in the peritoneal lavage after infection. Dec1 KO mice had significantly higher numbers of monocytes and neutrophils in the lavage than wild type mice. No other differences were seen between genotypes in the β-glucan-treated groups.
Figure 7. Beta-glucan-induced protection against P. aeruginosa is independent of Dectin-1 and Toll-like receptor (TLR)-2.
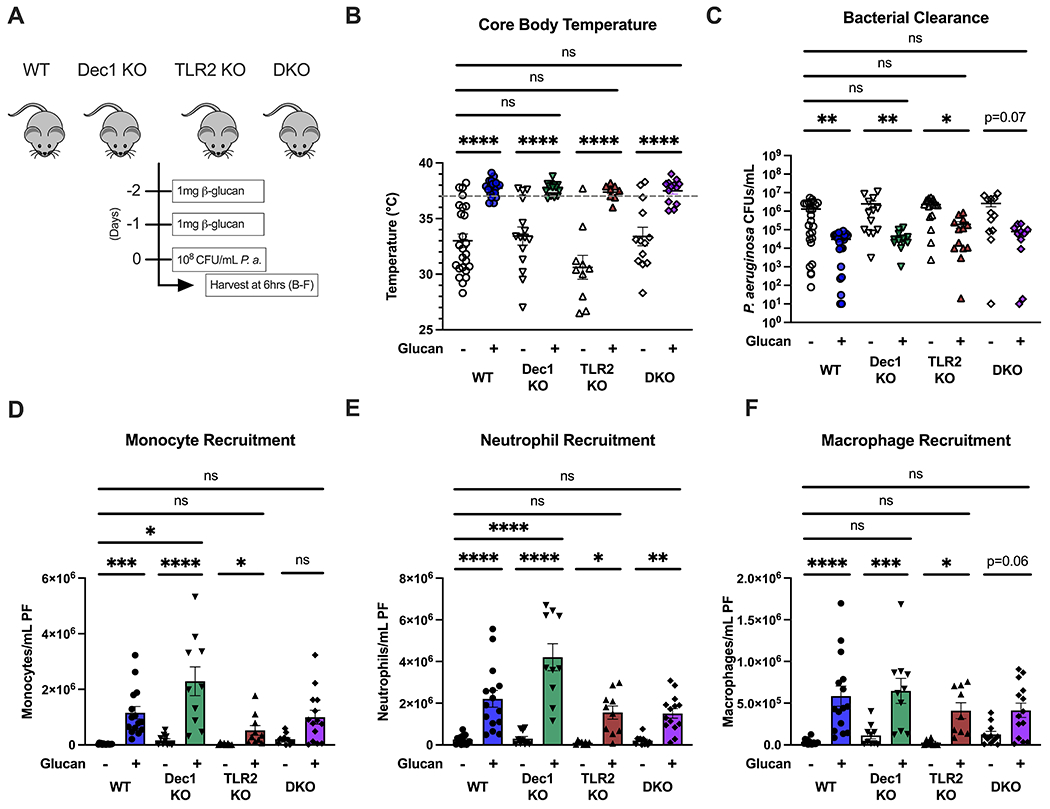
(A) Wildtype, Dectin-1 knockout, TLR-2, or Dectin-1/TLR2 double knockout C57Bl/6 mice were injected i.p. with β-glucan (1 mg) or vehicle 48 and 24 hours prior to i.p. inoculation with 108 CFU P. aeruginosa with subsequent harvest of plasma and peritoneal lavage fluid 6 hours later. (B) Core (rectal) body temperature in vehicle- or β-glucan treated mice 6 hours after i.p. P. aeruginosa. (C) CFUs of P. aeruginosa per mL of peritoneal fluid. (D-F) Number of monocytes (D), neutrophils (E), or macrophages (F) in infected vehicle- or β-glucan-treated mice. Body temperature and clearance data shown with median. All other data shown as mean ± SEM. N = 10-15 mice per group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test (B, C) or ANOVA with Tukey’s post-hoc multiple comparison test (D-F).
Additionally, we determined whether loss of Dectin-1 and TLR-2 impacted β-glucan-induced metabolic alterations in BMDM by performing extracellular flux analysis. Wild type and DKO BMDM were treated with β-glucan for 24 hours and assessed immediately or allowed to rest for 3 days to induce trained immunity (i.e., 3dp). DKO BMDM had reduced basal ECAR levels after both 24hr and 3dp β-glucan as compared to WT BMDM, however no differences were seen in max ECAR levels (Fig. 8A–C). WT and DKO BMDM showed no significant differences in basal and max OCR levels after 24h β-glucan and in the trained group (Fig. 8D–F).
Figure 8. Metabolic alterations in β-glucan-trained macrophages are independent of Dectin-1 and Toll-like receptor 2.
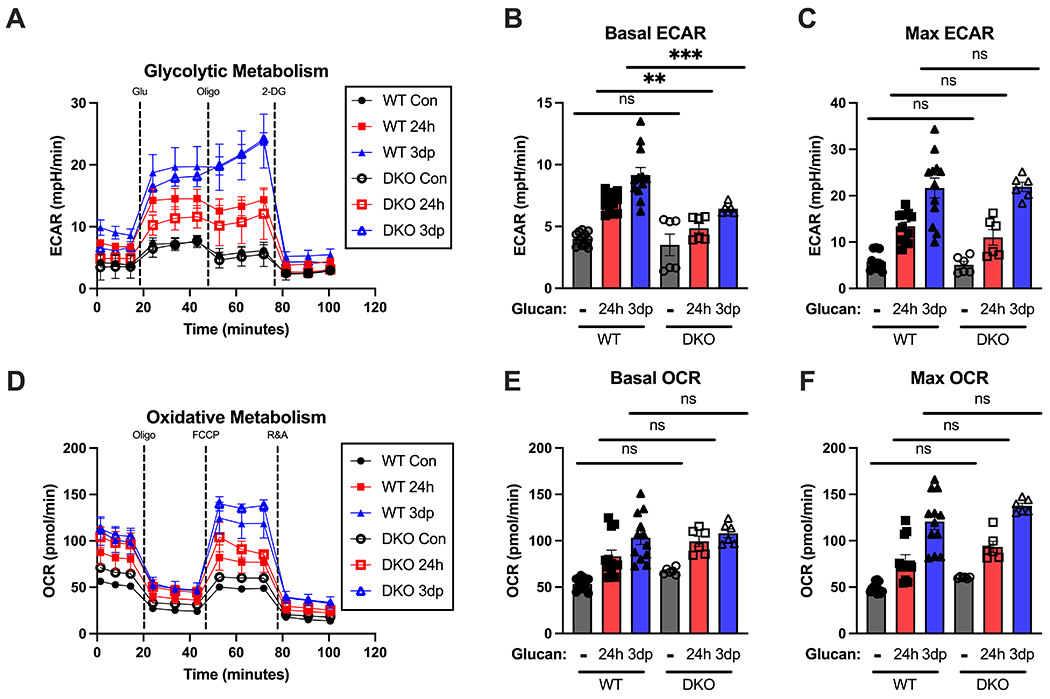
BMDM from WT and DKO mice were treated with β-glucan (5 μg) or vehicle for 24 hours (24h), washed and allowed to rest for 3 days (3dp) followed by assessment of macrophage metabolic phenotype. (A) Glycolysis stress test of control, 24h, and 3dp BMDM on the Seahorse XFe96 in wildtype and DKO mice. Extracellular acidification rate was measured over time at baseline and after glucose, oligomycin, and 2-deoxyglucose administration. (B) Basal ECAR in each group from multiple replicates. (C) Maximum ECAR in each group from multiple replicates. (D) Oxidative stress test of control, 24h, and trained BMDM on the Seahorse Xfe96. Oxygen consumption rate was measured over time at baseline and after oligomycin, FCCP, and R&A administration. (E) Basal OCR in each group from multiple replicates. (F) Maximum OCR in each group from multiple replicates. Data shown as mean ± SEM. Experiments were performed with 3 biological replicates. ** p<0.01, *** p<0.001 by ANOVA with Tukey’s post-hoc multiple comparison test.
To directly measure activation of the Dectin-1 and TLR-2 signaling pathways by the β-glucan training reagent, we treated BMDM with 5 mg/mL β-glucan (two separate batches) for 0.5, 1, and 2 hours and measured phosphorylation of inhibitor of kB kinase (IkK) and spleen tyrosine kinase (Syk) by Western blot. Concurrently, BMDM were treated with 5 mg/mL linear β-glucan for 0.5, 1, and 2 hours or with 100 ng/mL LPS for 1 hour as positive controls. LPS and linear glucan induced strong IkK phosphorylation and moderate Syk phosphorylation, while neither batch of β-glucan training reagent induced phosphorylation of either protein as compared to unstimulated BMDM (Fig. 9).
Figure 9. The β-glucan training reagent induces weak Dectin-1 and Toll-like receptor 2 activation.

Protein was isolated from BMDM treated with β-glucan for 0.5, 1.0, or 2.0 hours. BMDM treated with 100 ng/mL LPS for 1 hour or linear glucan for 0.5, 1.0, or 2.0 hours served as positive controls. Western blot of phosphorylated IkB kinase (pIkK), total IkK (IkK), phosphorylated spleen tyrosine kinase (pSyk), and total Syk (Syk). Blots are representative of three repeated experiments.
DISCUSSION
The major finding of this study is that β-glucan elicited a trained immune phenotype in differentiated macrophages that conferred protection from infection with P. aeruginosa. We found that β-glucan training augmented the host response to P. aeruginosa infection by facilitating recruitment of innate leukocytes and augmenting bacterial clearance at the site of infection. Adoptive transfer of macrophages treated ex vivo with β-glucan into naïve mice recapitulated the protective phenotype. Furthermore, β-glucan induced significant alterations in macrophage gene transcription and metabolism. These changes underpinned a robust enhancement of macrophage antimicrobial functions, including phagocytosis, ROS generation and cytokine production. This study advances our understanding of trained immunity by showing the contributions of differentiated macrophages and defining cellular and molecular mechanisms of training in these effector cells. Our findings suggest that differentiated macrophages, which survive for weeks to months in situ, contribute to the protective phenotype conferred by treatment with β-glucan. In addition, β-glucan-induced protection was preserved after loss of Dectin-1 and TLR-2, which suggests that alternative receptors drive the trained phenotype.
Beta-glucan has long been described as an immunomodulatory agent with the capacity to augment the host response against bacterial, viral, and fungal pathogens (46–49). More recent studies show that β-glucan-mediated innate immunomodulation lasts for several weeks (11, 50). Ciarlo and colleagues showed that training of mice with zymosan, of which β-glucan is the major biologically active component, induced training that conferred protection from E. coli peritonitis and listeriosis for 5 weeks (50). Furthermore, they demonstrated that zymosan treatment enhanced survival after P. aeruginosa pneumonia for 1 week. Our study shows sustained and robust protection from P. aeruginosa infection for at least 2 weeks after training with β-glucan. This raises the question of what mechanisms support this innate immune memory phenotype. One possibility is that β-glucan induces long-term changes to bone marrow progenitor cells that can serve to sustain the trained phenotype. Indeed, β-glucan promotes hematopoietic stem cell precursor (HSPC) expansion and myelopoiesis, which confers a survival benefit in mice repeatedly treated with the myeloablative drug 5-fluorouracil (51). Additionally, HSPCs treated with depleted zymosan, a β-glucan enriched for Dectin-1 signaling, and adoptively transferred into Dectin-1 deficient mice secreted higher levels of IL-6 and TNFα when isolated and challenged ex vivo with the synthetic triacylated lipopeptide Pam3CSK4 (52). Mice trained with β-glucan and subsequently infected with Mycobacterium tuberculosis (Mtb) undergo HSPC expansion after training and ultimately demonstrate improved survival after pulmonary infection (53). Ciarlo and colleagues also demonstrated myelopoiesis in mice trained with zymosan but did not observe alterations in protection from Listeria monocytogenes infection in mice depleted of neutrophils by treatment with anti-Ly6G (50). Their findings support the notion that enhanced local antimicrobial activity, rather than recruitment of β-glucan-trained myeloid cells, mediates protection against intracellular pathogens such as Mtb and L. monocytogenes. However, it is unclear whether β-glucan-trained HPSCs mediate augmented protection against an acute infection with common extracellular pathogens such as Pseudomonas aeruginosa. The present study indicates that augmented neutrophil recruitment is important for improved clearance of Pseudomonas aeruginosa in mice trained with β-glucan. These findings are consistent with our prior observations in β-glucan-trained mice infected with Escherichia coli and suggest that expansion and mobilization of neutrophil precursors from bone marrow may contribute to the trained phenotype in vivo (54). Nevertheless, further studies are needed to fully characterize the impact of immune training on HSPC function and the impact on neutrophil expansion and recruitment during infection.
Our study shows that macrophages trained with β-glucan confer resistance to infection after adoptive transfer, which supports the notion that β-glucan trains macrophages that are poised to immediately respond to infection. Very little is known about β-glucan-induced innate immune memory in differentiated macrophages, although there is some evidence to suggest that short-term treatment with β-glucan immediately before LPS influences BMDM activation (55). Bistoni and colleagues reported that systemic infection with a low-virulence variant of Candida albicans confers resistance to infection with a broad array of pathogens and macrophages were central mediators of the protective effect (56). Although they did not identify the C. albicans component causing the trained effect, their results are similar to the results of the present study in which we employed C. albicans-derived β-glucan and support the contention that macrophages have the capacity to facilitate the trained phenotype in vivo. Likewise, Ciarlo and colleagues showed that depletion of macrophages with clodronate-laden liposomes depleted the protective effects of zymosan against L. monocytogenes infection (50). Our findings extend those observations by showing that adoptive transfer of macrophages trained with β-glucan confers resistance to infection with P. aeruginosa. Macrophages represent a population of leukocytes that are well suited to sustain the trained phenotype due to their central role as regulators of the innate immune response to infection and a life span that is compatible with preserving the trained phenotype for weeks (57).
Our characterization of innate immune memory in macrophages reveals interesting distinctions when compared to the canonical monocyte profile. Trained monocytes activate hypoxia-inducible factor-1α to shift energy production primarily toward glycolysis and away from oxidative metabolism, even in oxygen-rich environments, a phenomenon known as aerobic glycolysis (58). In contrast, training of macrophages with β-glucan augments both glycolytic and oxidative metabolism. Here, we show that enhancement of oxidative metabolism seems to be achieved by expansion of the functional mitochondria pool and increased mitochondrial membrane potential. Furthermore, we show that inhibition of either glycolytic or oxidative metabolism with 2-deoxyglucose and oligomycin, respectively, blunted the ability of trained macrophages to constitutively secrete chemokines. This suggests that augmentation of broad metabolic pathways contributes to the antimicrobial phenotype in β-glucan trained macrophages.
A key tenet of β-glucan trained immunity is augmented cytokine production in response to infection or LPS challenge and its purported ability to reverse endotoxin tolerance in monocytes previously exposed to LPS (18). Results of the present study show that macrophages trained with β-glucan sustain the ability to secrete cytokines in response to LPS. Macrophages trained with β-glucan secrete comparable levels of IL-6 and TNFα after stimulation with LPS compared to untrained controls. Furthermore, transcriptomic profiling of macrophages after LPS challenge revealed similarity in cytokine and chemokine expression between β-glucan-trained and control macrophages. Interestingly, we observed decreased cytokine and chemokine concentrations in peritoneal lavage from β-glucan-trained mice after i.p. P. aeruginosa challenge compared to vehicle-treated controls. The disparity in cytokine production in vivo versus that observed in cultured macrophages could be explained by the augmented ability of β-glucan-trained mice to clear bacteria from the site of infection, thus decreasing local and systemic inflammation. We previously published data showing that a training agent’s ability to increase or decrease cytokine production in macrophages does not necessarily correlate with its ability to protect against infection (38, 59). This interpretation has been corroborated by work in human monocytes (60, 61). Instead, we propose that examination of direct antimicrobial functions, such as phagocytosis, ROS production, microbial killing, and leukocyte recruitment, rather than reliance on cytokine production alone, is a more accurate metric of the ability of an agent to confer trained immunity. Trained macrophages showed enhanced phagocytosis and ROS generation in the present study, indicating augmentation of direct antimicrobial functions. Taken together, our findings indicate that β-glucan induces a macrophage phenotype that is more effective at recruiting leukocytes to the site of infection and mediating direct microbial clearance.
Dectin-1 and TLR-2 are both well-characterized PRRs for β-glucan (62, 63). Dectin-1 deficiency diminishes cellular responses to β-glucan in vitro, but its role in response to fungal infection has been disputed (64, 65). Similarly, the contribution of TLR-2 to β-glucan-induced immune responses and the host response to infection has not been fully elucidated in vivo (66). Here, we show that neither Dectin-1 nor TLR-2 are required for β-glucan-induced protection against P. aeruginosa infection or β-glucan-induced metabolic reprogramming in macrophages. Given that different fungi containing β-glucan trigger different surface receptors on leukocytes, this phenomenon should be confirmed in infections with other pathogens. Interestingly, β-glucan has been shown to stimulate macrophages independently of Dectin-1 and TLR-2, adding further evidence to the possibility that β-glucan trained immunity occurs independently of either receptor (32, 67). Other receptor families that have been reported to mediate β-glucan recognition include complement receptor 3 (CR3, CD18/CD11b), LacCer and scavenger receptors (68–70). These varying results may be due to the fact that β-glucan of varying structural motifs can be derived from wide ranging species of fungi (71, 72). Our finding that a linear β-glucan derived from C. albicans induces Syk and IKK phosphorylation whereas the branched β-glucan training reagent does not, supports this contention. Clearly, further research is needed to further identify β-glucan structure, receptors and signaling pathways.
Our findings support the development of agents that induce trained immunity to fight infection in the clinical realm and show that training with β-glucan provides protection against P. aeruginosa infection. Our results are complemented by the work of Ciarlo and colleagues, who showed that training with zymosan confers protection from P. aeruginosa pneumonia (50). Among the critically ill, P. aeruginosa has emerged as a common pathogen leading to serious healthcare-associated infections (73). P. aeruginosa accounts for nearly twenty percent of infections in intensive care units, and the mortality rate of these infections approaches forty percent (74). Furthermore, the prevalence of antimicrobial-resistant P. aeruginosa is increasing dramatically, due in part to the spread of mobile genetic elements that convey antibiotic resistance and poor antibiotic stewardship among health care providers (75–77). Thus, there is an acute need for the development of alternative strategies to prevent and abate P. aeruginosa infections. Induction of trained immunity by agents such as β-glucan provides an opportunity to augment host resistance to common pathogens.
Given the importance of differentiated macrophages for the protective phenotype induced by β-glucan, future studies should explore the efficacy of newly developed agents for inducing trained immunity in these cells. Because β-glucan is one of the most extensively characterized immune training agents, it is a logical candidate to inform future drug-discovery efforts. However, our work and other studies indicate that noncanonical signaling pathways may be responsible for the trained phenotype. Thus, β-glucan is a promising candidate for development for use against increasingly common and lethal nosocomial pathogens such as P. aeruginosa.
Supplementary Material
KEY POINTS.
Beta-glucan protects mice from intraperitoneal Pseudomonas aeruginosa infection
Macrophages contribute to β-glucan-induced protection in vivo
Beta-glucan rewires macrophage function and metabolism independently of Dectin-1
ACKNOWLEDGEMENTS
We thank the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core for performing RNASeq. We also thank Yaomin Xu, Yu Wang, and Caley Stothers for help with RNASeq analysis.
This work was supported by the National Institutes of Health (NIH) Grants R01 GM119197 (E.R.S. and D.L.W.), R01 AI151210 (E.R.S.), R01 GM121711 (J.K.B), R35 GM141927 (J.K.B.), R01 GM083016 (D.L.W.), T32 GM108554 (N.K.P) and T32 GM007347 (Vanderbilt MSTP: C.L.S. and M.A.M.), the American Heart Association Grant 19PRE34430054 (C.L.S.), and Vanderbilt Faculty Research Scholars Award (N.K.P). The Agilent Seahorse Extracellular Flux Analyzer is housed and managed within the Vanderbilt High-Throughput Screening Core Facility, an institutionally supported core, and was funded by NIH Shared Instrumentation Grant 1S10OD018015.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Mayr FB, Yende S, and Angus DC. 2014. Epidemiology of severe sepsis. Virulence 5: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K, and E. I. G. o. Investigators. 2009. International study of the prevalence and outcomes of infection in intensive care units. JAMA 302: 2323–2329. [DOI] [PubMed] [Google Scholar]
- 3.Manners C, Larios Bautista E, Sidoti H, and Lopez OJ. 2020. Protective Adaptive Immunity Against Severe Acute Respiratory Syndrome Coronaviruses 2 (SARS-CoV-2) and Implications for Vaccines. Cureus 12: e8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nogales A, and DeDiego ML. 2020. Influenza Virus and Vaccination. Pathogens 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gourbal B, Pinaud S, Beckers GJM, Van Der Meer JWM, Conrath U, and Netea MG. 2018. Innate immune memory: An evolutionary perspective. Immunol Rev 283: 21–40. [DOI] [PubMed] [Google Scholar]
- 6.Butcher SK, O’Carroll CE, Wells CA, and Carmody RJ. 2018. Toll-Like Receptors Drive Specific Patterns of Tolerance and Training on Restimulation of Macrophages. Front Immunol 9: 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu ZQ, and Dong X. 2013. Systemic acquired resistance: turning local infection into global defense. Annu Rev Plant Biol 64: 839–863. [DOI] [PubMed] [Google Scholar]
- 8.Kurtz J, and Franz K. 2003. Innate defence: evidence for memory in invertebrate immunity. Nature 425: 37–38. [DOI] [PubMed] [Google Scholar]
- 9.Kurtz J 2005. Specific memory within innate immune systems. Trends Immunol 26: 186–192. [DOI] [PubMed] [Google Scholar]
- 10.Irvine DJ, Aung A, and Silva M. 2020. Controlling timing and location in vaccines. Adv Drug Deliv Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Valtanen P, Guzman-Genuino RM, Williams DL, Hayball JD, and Diener KR. 2017. Evaluation of trained immunity by beta-1, 3 (d)-glucan on murine monocytes in vitro and duration of response in vivo. Immunology and cell biology 95: 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyuksutova OI, Murphey ED, Toliver-Kinsky TE, Lin CY, Cui W, Williams DL, and Sherwood ER. 2005. Glucan phosphate treatment attenuates burn-induced inflammation and improves resistance to Pseudomonas aeruginosa burn wound infection. Shock (Augusta, Ga.) 23: 224–232. [PubMed] [Google Scholar]
- 13.Murphey ED, and Sherwood ER. 2008. Pretreatment with the Gram-positive bacterial cell wall molecule peptidoglycan improves bacterial clearance and decreases inflammation and mortality in mice challenged with Pseudomonas aeruginosa. Microbes Infect 10: 1244–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Netea MG, Giamarellos-Bourboulis EJ, Dominguez-Andres J, Curtis N, van Crevel R, van de Veerdonk FL, and Bonten M. 2020. Trained Immunity: a Tool for Reducing Susceptibility to and the Severity of SARS-CoV-2 Infection. Cell 181: 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez A, Patil NK, Stothers CL, Luan L, McBride MA, Owen AM, Burelbach KR, Williams DL, Sherwood ER, and Bohannon JK. 2019. Immunobiology and application of toll-like receptor 4 agonists to augment host resistance to infection. Pharmacological research 150: 104502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quintin J 2019. Fungal mediated innate immune memory, what have we learned? Semin Cell Dev Biol 89: 71–77. [DOI] [PubMed] [Google Scholar]
- 17.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van der Ent MA, Sharifi N, Janssen-Megens EM, Ter Huurne M, Mandoli A, van Schaik T, Ng A, Burden F, Downes K, Frontini M, Kumar V, Giamarellos-Bourboulis EJ, Ouwehand WH, van der Meer JW, Joosten LA, Wijmenga C, Martens JH, Xavier RJ, Logie C, Netea MG, and Stunnenberg HG. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (New York, N.Y.) 345: 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, Kim B, Kuznetsova T, Kox M, Zwaag J, Matarese F, van Heeringen SJ, Janssen-Megens EM, Sharifi N, Wang C, Keramati F, Schoonenberg V, Flicek P, Clarke L, Pickkers P, Heath S, Gut I, Netea MG, Martens JHA, Logie C, and Stunnenberg HG. 2016. beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167: 1354–1368 e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJ, van der Veer BM, Deen PM, Logie C, O’Neill LA, Willems P, van de Veerdonk FL, van der Meer JW, Ng A, Joosten LA, Wijmenga C, Stunnenberg HG, Xavier RJ, and Netea MG. 2014. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (New York, N.Y.) 345: 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, and Xavier RJ. 2016. Trained immunity: A program of innate immune memory in health and disease. Science (New York, N.Y.) 352: aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, Bigley V, Flavell RA, Gilroy DW, Asquith B, Macallan D, and Yona S. 2017. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. The Journal of experimental medicine 214: 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden W, Bremmers MEJ, van Crevel R, Handler K, Picelli S, Schulte-Schrepping J, Klee K, Oosting M, Koeken V, van Ingen J, Li Y, Benn CS, Schultze JL, Joosten LAB, Curtis N, Netea MG, and Schlitzer A. 2020. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franken L, Schiwon M, and Kurts C. 2016. Macrophages: sentinels and regulators of the immune system. Cell Microbiol 18: 475–487. [DOI] [PubMed] [Google Scholar]
- 24.Ginhoux F, Schultze JL, Murray PJ, Ochando J, and Biswas SK. 2016. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nature immunology 17: 34–40. [DOI] [PubMed] [Google Scholar]
- 25.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, and Angus DC. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaw AC, Goldstein DR, and Montgomery RR. 2013. Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13: 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotchkiss RS, Monneret G, and Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, and Gordon S. 2002. Dectin-1 is a major beta-glucan receptor on macrophages. The Journal of experimental medicine 196: 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown GD, and Gordon S. 2003. Fungal beta-glucans and mammalian immunity. Immunity 19: 311–315. [DOI] [PubMed] [Google Scholar]
- 30.Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, Kasprowicz DJ, Kellar K, Pare J, van Dyke T, Ziegler S, Unutmaz D, and Pulendran B. 2006. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. The Journal of clinical investigation 116: 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, Jacobs L, Jansen T, Verheijen K, Masthoff L, Morre SA, Vriend G, Williams DL, Perfect JR, Joosten LA, Wijmenga C, van der Meer JW, Adema GJ, Kullberg BJ, Brown GD, and Netea MG. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361: 1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smeekens SP, Gresnigt MS, Becker KL, Cheng SC, Netea SA, Jacobs L, Jansen T, van de Veerdonk FL, Williams DL, Joosten LA, Dinarello CA, and Netea MG. 2015. An anti-inflammatory property of Candida albicans beta-glucan: Induction of high levels of interleukin-1 receptor antagonist via a Dectin-1/CR3 independent mechanism. Cytokine 71: 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bubonja-Sonje M, Matovina M, Skrobonja I, Bedenic B, and Abram M. 2015. Mechanisms of Carbapenem Resistance in Multidrug-Resistant Clinical Isolates of Pseudomonas aeruginosa from a Croatian Hospital. Microb Drug Resist 21: 261–269. [DOI] [PubMed] [Google Scholar]
- 34.Pena C, Suarez C, Gozalo M, Murillas J, Almirante B, Pomar V, Aguilar M, Granados A, Calbo E, Rodriguez-Bano J, Rodriguez F, Tubau F, Martinez-Martinez L, and Oliver A. 2012. Prospective multicenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob Agents Chemother 56: 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowman DW, Greene RR, Bearden DW, Kruppa MD, Pottier M, Monteiro MA, Soldatov DV, Ensley HE, Cheng SC, Netea MG, and Williams DL. 2014. Novel structural features in Candida albicans hyphal glucan provide a basis for differential innate immune recognition of hyphae versus yeast. The Journal of biological chemistry 289: 3432–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ifrim DC, Joosten LA, Kullberg BJ, Jacobs L, Jansen T, Williams DL, Gow NA, van der Meer JW, Netea MG, and Quintin J. 2013. Candida albicans primes TLR cytokine responses through a Dectin-1/Raf-1-mediated pathway. Journal of immunology (Baltimore, Md. : 1950) 190: 4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez A, Luan L, Stothers CL, Patil NK, Fults JB, Fensterheim BA, Guo Y, Wang J, Sherwood ER, and Bohannon JK. 2019. Phosphorylated Hexa-Acyl Disaccharides Augment Host Resistance Against Common Nosocomial Pathogens. Critical care medicine 47: e930–e938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, McAtee-Pereira AG, Guo Y, Trenary I, Hernandez A, Fults JB, Williams DL, Sherwood ER, and Bohannon JK. 2018. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. Journal of immunology (Baltimore, Md. : 1950) 200: 3777–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng Q, Vickers K, Zhao S, Wang J, Samuels DC, Koues O, Shyr Y, and Guo Y. 2017. Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief Funct Genomics 16: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao Y, Smyth GK, and Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 42.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, and Sherlock G. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gene Ontology C 2021. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49: D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kokoshis PL, Williams DL, Cook JA, and Di Luzio NR. 1978. Increased resistance to Staphylococcus aureus infection and enhancement in serum lysozyme activity by glucan. Science (New York, N.Y.) 199: 1340–1342. [DOI] [PubMed] [Google Scholar]
- 47.Rice PJ, Adams EL, Ozment-Skelton T, Gonzalez AJ, Goldman MP, Lockhart BE, Barker LA, Breuel KF, Deponti WK, Kalbfleisch JH, Ensley HE, Brown GD, Gordon S, and Williams DL. 2005. Oral Delivery and Gastrointestinal Absorption of Soluble Glucans Stimulate Increased Resistance to Infectious Challenge. Journal of Pharmacology and Experimental Therapeutics 314: 1079–1086. [DOI] [PubMed] [Google Scholar]
- 48.Quintin J, Saeed S, Joost, Evangelos, Daniela, Logie C, Jacobs L, Jansen T, Kullberg B-J, Wijmenga C, Leo, Ramnik, Jos, Hendrik, and Mihai. 2012. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell host & microbe 12: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams DL, Mueller A, and Browder W. 2012. Glucan-Based Macrophage Stimulators. Clinical Immunotherapeutics 5: 392–399. [Google Scholar]
- 50.Ciarlo E, Heinonen T, Theroude C, Asgari F, Le Roy D, Netea MG, and Roger T. 2020. Trained Immunity Confers Broad-Spectrum Protection Against Bacterial Infections. The Journal of infectious diseases 222: 1869–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, Chatzigeorgiou A, Schlitzer A, Beyer M, Joosten LAB, Isermann B, Lesche M, Petzold A, Simons K, Henry I, Dahl A, Schultze JL, Wielockx B, Zamboni N, Mirtschink P, Coskun U, Hajishengallis G, Netea MG, and Chavakis T. 2018. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172: 147–161 e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bono C, Martinez A, Megias J, Gozalbo D, Yanez A, and Gil ML. 2020. Dectin-1 Stimulation of Hematopoietic Stem and Progenitor Cells Occurs In Vivo and Promotes Differentiation Toward Trained Macrophages via an Indirect Cell-Autonomous Mechanism. mBio 11: e00781–00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, Divangahi M, and Netea MG. 2020. beta-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell reports 31: 107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Rakinic J, and Di Luzio NR. 1988. Effect of glucan on neutrophil dynamics and immune function in Escherichia coli peritonitis. J Surg Res 44: 54–61. [DOI] [PubMed] [Google Scholar]
- 55.Walachowski S, Tabouret G, Fabre M, and Foucras G. 2017. Molecular Analysis of a Short-term Model of beta-Glucans-Trained Immunity Highlights the Accessory Contribution of GM-CSF in Priming Mouse Macrophages Response. Front Immunol 8: 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bistoni F, Verducci G, Perito S, Vecchiarelli A, Puccetti P, Marconi P, and Cassone A. 1988. Immunomodulation by a low-virulence, agerminative variant of Candida albicans. Further evidence for macrophage activation as one of the effector mechanisms of nonspecific anti-infectious protection. J Med Vet Mycol 26: 285–299. [DOI] [PubMed] [Google Scholar]
- 57.Sherwood ER, and Toliver-Kinsky T. 2004. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol 18: 385–405. [DOI] [PubMed] [Google Scholar]
- 58.Stothers CL, Luan L, Fensterheim BA, and Bohannon JK. 2018. Hypoxia-inducible factor-1alpha regulation of myeloid cells. J Mol Med (Berl) 96: 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fensterheim BA, Guo Y, Sherwood ER, and Bohannon JK. 2017. The Cytokine Response to Lipopolysaccharide Does Not Predict the Host Response to Infection. Journal of immunology (Baltimore, Md. : 1950) 198: 3264–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez-Jimenez E, Toledano V, Cubillos-Zapata C, Rapisarda A, Chen J, Duan K, Yang H, Poidinger M, Melillo G, Nizet V, Arnalich F, Lopez-Collazo E, and Biswas SK. 2015. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity 42: 484–498. [DOI] [PubMed] [Google Scholar]
- 61.Fernandes ML, Mendes ME, Brunialti MK, and Salomao R. 2010. Human monocytes tolerant to LPS retain the ability to phagocytose bacteria and generate reactive oxygen species. Braz J Med Biol Res 43: 860–868. [DOI] [PubMed] [Google Scholar]
- 62.Brown GD, and Gordon S. 2001. Immune recognition. A new receptor for beta-glucans. Nature 413: 36–37. [DOI] [PubMed] [Google Scholar]
- 63.Kanjan P, Sahasrabudhe NM, de Haan BJ, and de Vos P. 2017. Immune effects of β-glucan are determined by combined effects on Dectin-1, TLR2, 4 and 5. Journal of Functional Foods 37: 433–440. [Google Scholar]
- 64.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, and Iwakura Y. 2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nature immunology 8: 39–46. [DOI] [PubMed] [Google Scholar]
- 65.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, and Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nature immunology 8: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Viriyakosol S, Fierer J, Brown GD, and Kirkland TN. 2005. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1. Infection and immunity 73: 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly MM, McNagny K, Williams DL, van Rooijen N, Maxwell L, Gwozd C, Mody CH, and Kubes P. 2008. The lung responds to zymosan in a unique manner independent of toll-like receptors, complement, and dectin-1. American journal of respiratory cell and molecular biology 38: 227–238. [DOI] [PubMed] [Google Scholar]
- 68.Desamero MJM, Chung SH, and Kakuta S. 2021. Insights on the Functional Role of Beta-Glucans in Fungal Immunity Using Receptor-Deficient Mouse Models. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Legentil L, Paris F, Ballet C, Trouvelot S, Daire X, Vetvicka V, and Ferrieres V. 2015. Molecular Interactions of beta-(1-->3)-Glucans with Their Receptors. Molecules 20: 9745–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takematsu H, Yamamoto H, Naito-Matsui Y, Fujinawa R, Tanaka K, Okuno Y, Tanaka Y, Kyogashima M, Kannagi R, and Kozutsumi Y. 2011. Quantitative transcriptomic profiling of branching in a glycosphingolipid biosynthetic pathway. J Biol Chem 286: 27214–27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muller A, Ensley H, Pretus H, McNamee R, Jones E, McLaughlin E, Chandley W, Browder W, Lowman D, and Williams D. 1997. The application of various protic acids in the extraction of (1-->3)-beta-D-glucan from Saccharomyces cerevisiae. Carbohydr Res 299: 203–208. [DOI] [PubMed] [Google Scholar]
- 72.Mueller A, Raptis J, Rice PJ, Kalbfleisch JH, Stout RD, Ensley HE, Browder W, and Williams DL. 2000. The influence of glucan polymer structure and solution conformation on binding to (1-->3)-beta-D-glucan receptors in a human monocyte-like cell line. Glycobiology 10: 339–346. [DOI] [PubMed] [Google Scholar]
- 73.Nathwani D, Raman G, Sulham K, Gavaghan M, and Menon V. 2014. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: a systematic review and meta-analysis. Antimicrob Resist Infect Control 3: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, and Pinsky MR. 2001. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Critical care medicine 29: 1303–1310. [DOI] [PubMed] [Google Scholar]
- 75.Poole K 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pang Z, Raudonis R, Glick BR, Lin TJ, and Cheng Z. 2019. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 37: 177–192. [DOI] [PubMed] [Google Scholar]
- 77.Jean SS, Gould IM, Lee WS, Hsueh PR, and C. International Society of Antimicrobial. 2019. New Drugs for Multidrug-Resistant Gram-Negative Organisms: Time for Stewardship. Drugs 79: 705–714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.