

Summary
Vertebrate craniofacial morphogenesis is a highly orchestrated process that is directed by evolutionarily conserved developmental pathways1,2. Within species, canalized development typically produces modest morphological variation. However, as a result of millennia of artificial selection, the domestic pigeon displays radical craniofacial variation within a single species. One of the most striking cases of pigeon craniofacial variation is the short beak phenotype, which has been selected in numerous breeds. Classical genetic experiments suggest that pigeon beak length is regulated by a small number of genetic factors, one of which is sex-linked (Ku2 locus)3–5. However, the genetic underpinnings of pigeon craniofacial variation remain unknown. Using geometric morphometrics and quantitative trait loci (QTL) mapping on an F2 intercross between a short-beaked Old German Owl (OGO) and a medium-beaked Racing Homer (RH), we identified a single Z-chromosome locus that explains a majority of the variation in beak morphology in the F2 population. Complementary comparative genomic analyses revealed that the same locus is strongly differentiated between breeds with short and medium beaks. Within the Ku2 locus, we identified an amino acid substitution in the non-canonical Wnt receptor ROR2 as a putative regulator of pigeon beak length. The non-canonical Wnt pathway serves critical roles in vertebrate neural crest cell migration and craniofacial morphogenesis6,7. In humans, ROR2 mutations cause Robinow syndrome, a congenital disorder characterized by skeletal abnormalities, including a widened and shortened facial skeleton8,9. Our results illustrate how the extraordinary craniofacial variation among pigeons can reveal genetic regulators of vertebrate craniofacial diversity.
Keywords: Craniofacial, beak, pigeon, morphometrics, QTL mapping, comparative genomics, non-canonical Wnt signaling pathway, ROR2
Results and Discussion
The avian beak shows remarkable diversity among species. Variation in beak morphology within groups like Darwin’s finches and Hawaiian honeycreepers illustrates the diversifying potential of natural selection on beak skeletal structures and functions10,11. Although the underlying genetic basis of the extraordinary variation among birds remains relatively poorly understood, several genes associated with overall beak size or linear dimensions of beak shape are known in a modest number of species (e.g., COL4A5 in Great tits12; IGF1 in Black-bellied seedcrackers13; BMP4, CALM1, ALX1, and HMGA2 in Darwin’s finches14–18). Unlike in wild birds, the beak of the domestic pigeon is unconstrained by natural selection; the astounding level of morphological variation within this species is instead the product of intensive artificial selection. Some of the major axes of variation in craniofacial shape that distinguish distantly related avian species are recapitulated among breeds of domestic pigeon, despite different mechanisms of selection between wild and captive populations19,20. Therefore, pigeons provide a unique opportunity to uncover genetic variants associated with the types of beak variation that exist throughout the radiation of birds.
Pigeon beak length covaries with body size and braincase shape in an experimental cross
To determine the genetic architecture of beak length in pigeons, we established an F2 intercross between a male Racing Homer (RH) and a female Old German Owl (OGO). The RH, which “has been bred for one purpose – speed – almost to the exclusion of all other factors and traits”21, has a medium-length beak that resembles the ancestral condition in rock pigeons (Figure 1A,B). In contrast, the OGO beak “is one of the distinctive characteristics” of the breed and is “short in appearance, which is partly caused by the broad width of the beak in relation to its length”22 (Figure 1C,D).
Figure 1. Beak length variation in a pigeon F2 intercross.
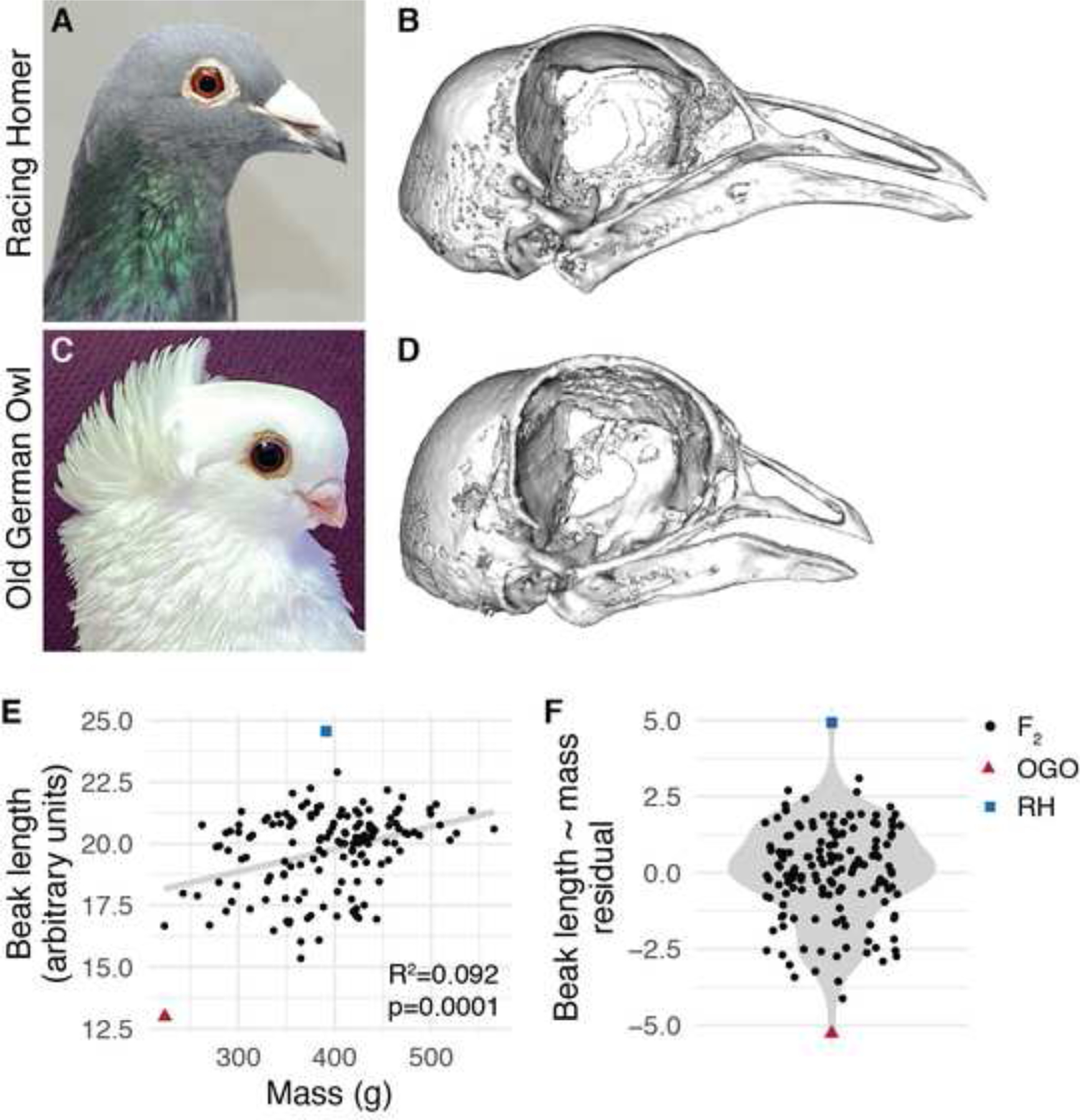
(A) Representative image of the medium beak Racing Homer (RH) breed. (B) 3D surface model of the craniofacial skeleton of the male RH founder. (C) Representative image of the short beak Old German Owl (OGO) breed. (D) Surface model of the craniofacial skeleton of the female OGO founder. (E) Raw beak length (measured in arbitrary units) vs. total body mass (measured in grams) in the RH × OGO cross. Gray line indicates linear model from beak length ~ mass regression. (F) Distribution of residuals from beak length ~ mass regression. For (E-F), black dots denote RH × OGO F2 individuals, red triangle is OGO founder, blue square is RH founder. Image credits: Sydney Stringham (A), Brian McCormick (B).
We scanned the RH × OGO cross founders and 145 F2 individuals using micro-CT, generated 3D surface models of the craniofacial skeleton, and applied a set of 49 landmarks to the beak and braincase (Figure S1, Table S1). By calculating the linear distance between the base and tip of the beak and using mass as a proxy for body size23, we found a significant positive association between beak length and body size in the F2 population (R2=0.092, p=0.0001, Figure 1E). We removed the effects of body size variation by fitting a beak length ~ body size linear regression model and found that beak length residuals remained highly variable in the F2 population, demonstrating that beak length varies independently of body size (Figure 1F).
We also measured three-dimensional (3D) variation in beak and braincase shape through geometric morphometric analysis. In the RH × OGO F2 population, cranium centroid size is negatively associated with body size (R2=0.03, p=0.02, Figure S2A). This result contrasts with broad patterns of cranium ~ body size allometry observed across diverse pigeon breeds and wild birds20, and is likely driven by the exceptionally large cranium and small body size selected in the OGO breed. In the F2 population, cranium centroid size is also negatively associated with curvature from the tip of the beak to the back of the braincase and, to a lesser extent, beak length (R2=0.073, p<0.001, Figure S2B). Taken together, the linear and 3D shape analyses reveal subtle but significant relationships between body and cranium size and craniofacial shape in the RH × OGO cross. By shuffling genetic programs for two generations in an experimental cross, we also find that body size, cranium size, and beak shape are modular and separable.
Although allometry is an important correlate of shape23, we focused further analyses on non-allometric craniofacial shape variation. Principal components analysis (PCA) of geometric morphometric shape variables demonstrates that, in the RH × OGO F2 population, the principal axis of shape variation (PC1, 39.1% of shape variation) describes compound variation in beak length and braincase volume (Figure 2A, Movie S1) and is strongly correlated with linear measurements of beak length (R2=0.64, p<2.2e-16, Figure S3A). PC2 (10.9% of shape variation) is defined almost exclusively by changes in braincase shape (Figure S3D, Movie S2).
Figure 2. A major-effect QTL on the Z-chromosome is associated with principal component 1 (PC1) in the RH × OGO F2 population.
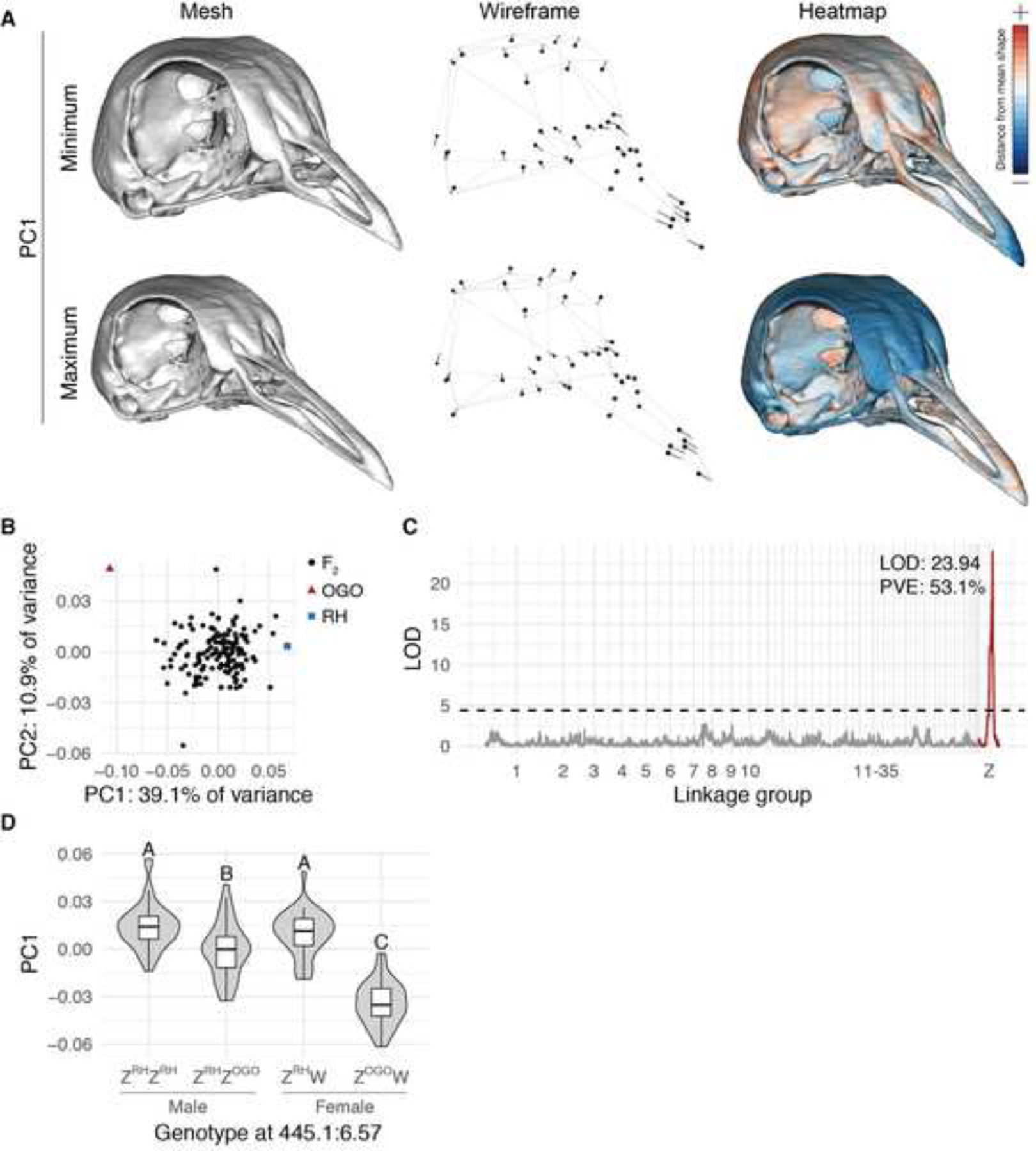
(A) Visualizations of geometric morphometric PC1 minimum and maximum shapes three ways: warped 3D surface meshes (left), wireframes showing displacement of landmarks from mean shape (center), and heatmaps displaying regional shape variation (right). For warped meshes and wireframes, shape changes are magnified 1.5x to aid visualization. (B) PCA plot of PC1 vs. PC2. (C) Genome-wide QTL scan for PC1 reveals significant QTL on Z linkage group. (D) PC1 effect plot estimated from QTL peak marker. Letters denote significance groups; RH = allele from RH founder, OGO = allele from OGO founder. See also Figure S3.
Similar to previous findings in domestic pigeons and wild birds20,24, 3D beak and braincase shape are strongly integrated in the RH × OGO F2 population (r-PLS=0.923, p<0.001, Figure S2B). Along the PC1 axis, all F2 individuals are confined to a morphospace defined by the cross founders, but cluster closer to the RH than the OGO (Figure 2B). This result is reminiscent of our analysis of pigeon beak curvature in a different study, in which F2 individuals derived from a straight-beaked Pomeranian Pouter and curved-beaked Scandaroon more closely resembled the Pomeranian Pouter and never achieved the extreme craniofacial curvature of the Scandaroon24. Therefore, two different genetic crosses using four different pigeon breeds suggest that the most exaggerated versions of craniofacial traits require coordination of multiple genetic factors.
Identification of a major-effect beak length QTL on the Z chromosome
Next, we used the PC1 scores, which primarily describe variation in beak length, to perform genome-wide QTL scans. A single major-effect QTL on the Z-chromosome linkage group was strongly associated with PC1 and explained more than half of phenotypic variance in the RH × OGO cross (log likelihood ratio (LOD)=23.72, percent variance explained (PVE)=53.2%, Figure 2C). Nearly identical results were obtained when beak length residuals were used for QTL mapping (Figure S3E–G). The identification of a major-effect QTL on the Z-chromosome is consistent with results from classical genetic studies that pointed to a sex-linked regulator of pigeon beak length3–5.
We next used the peak marker to estimate QTL effects. In the F2 population, male ZRH/ZRH homozygotes and female ZRH/W hemizygotes had the highest PC1 scores (longest beaks) and were not statistically different from one another (Figure 2D). Male ZRH/ZOGO heterozygotes had intermediate PC1 scores (Figure 2D), suggestive of an incompletely dominant pattern of inheritance. In contrast, female ZOGO/W hemizygotes had dramatically lower PC1 scores (shorter beaks) than all other F2 individuals carrying the RH allele (Figure 2D). Although the structure of our experimental cross did not generate homozygous ZOGO/ZOGO males in the F2 generation, previous classical genetic studies demonstrated that, in short-beaked pigeon breeds, body size is sex-associated but beak length is not5,25. Therefore, we predict that the beaks of ZOGO/ZOGO males would be indistinguishable from hemizygous ZOGO/W females, although we cannot rule out the possibility that an additional copy of the OGO allele could result in even shorter beaks in ZOGO/ZOGO males.
In summary, our results support the model that pigeon beak length is a polygenic trait controlled largely by one sex-linked factor. Additional minor-effect QTL are likely modifying beak length in the RH × OGO cross, some of which may be detectable in a larger F2 population or an F3 generation that includes ZOGO/ZOGO males.
A ROR2 coding variant is associated with beak length across diverse domestic pigeon breeds
The beak length QTL represents a relatively large (3.6-Mb) genomic region that includes several genes expressed during pigeon craniofacial development (Figure S3B–C, Table S2), thus limiting our ability to pinpoint the specific gene(s) and mutation(s) that regulate beak length in the RH × OGO cross. In addition, because the mapping population was derived from just two birds that represent a fraction of the morphological diversity across pigeon breeds, we have no way of knowing if the beak length QTL we identified is relevant beyond the RH × OGO cross. Short beaks are characteristic of numerous closely-related pigeon breeds that belong to the Owl family, but are also part of the breed standard, and thus under positive selection, in a variety of unrelated non-Owl breeds26–28. Pigeon breeders might have repeatedly selected the same standing variant in different breeds, independent variants of the same gene in different breeds, or different genes altogether in different breeds.
To distinguish between independent and shared genetic origins of short beaks, we scanned for genomic variants associated with beak length across diverse pigeon breeds by comparing resequenced genomes of 56 short-beaked individuals from 31 breeds (7 Owl and 24 non-Owl) to 121 genomes from 58 medium- or long-beaked breeds and feral pigeons (Figure 3A–B). We then searched for genomic regions that were differentiated between these groups using two related differentiation statistics (wcFST29 and pFST30). A ~293-kb segment on the Z-chromosome scaffold ScoHet5_445.1 stood out as significantly differentiated between the short- and medium/long-beaked groups (top 0.1% by wcFST; Figure 3C–D, Figure S4) and was located within the genomic interval identified in our QTL scan. In the peak differentiated region, short-beaked pigeons displayed elevated levels of haplotype homozygosity relative to medium/long-beaked individuals, providing further support for widespread positive selection on this locus in short-beaked breeds (Figure 3E). Thus, the short beak allele identified in our QTL mapping experiments is not specific to either the OGO cross founder breed or the Owl family. Instead, the short beak allele on the Z-chromosome likely arose once and was repeatedly selected in different unrelated breeds.
Figure 3. Comparison of short beak and medium/long beak pigeon genomes reveals ROR2 coding variant.
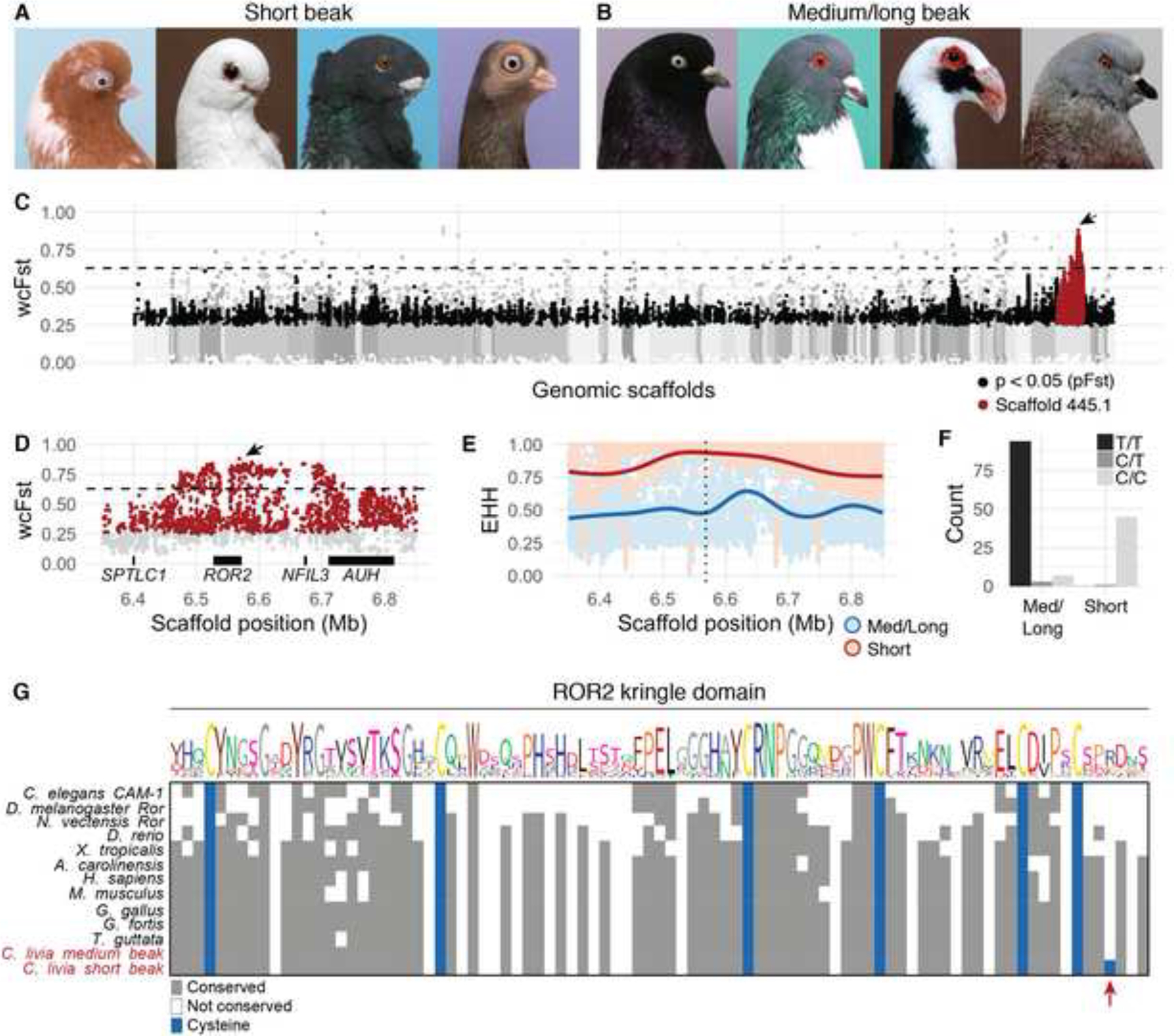
(A-B) Representative images of individuals representing short beak (A) and medium or long beak (B) pigeon breeds. (A) Short beak pigeons, from left to right: English Short Face Tumbler, African Owl, Oriental Frill, Budapest Tumbler. (B) Medium/long beak pigeons, from left to right: West of England, Cauchois, Scandaroon, Show King. (C) Genome-wide scan for allele frequency differentiation between short beak (n=56) and medium/long beak (n=121) pigeons. (D) Region of peak FST on ScoHet5_445.1; black horizontal bars represent four genes in the region. For (C-D), genomic scaffolds are colored in gray and ordered by genetic position in RH × OGO linkage map; black dots indicate SNPs that are significantly differentiated by pFST (Bonferroni-corrected p-value < 0.05); red dots are significant SNPs located on scaffold ScoHet5_445.1; dashed horizontal line represents threshold for genome-wide top 0.1% of differentiated SNPs by wcFST; arrow points to ScoHet5_445.1:6568443, the most differentiated SNP (FST=0.88) genome-wide. See also Figure S4. (E) Extended haplotype homozygosity in FST peak region; dotted vertical line indicates position of ScoHet5_445.1:6568443; smoothed lines represent local regression fitting53. See also Figure S4. (F) Histogram of genotypes at ScoHet5_445.1:6568443 in short beak and medium/long beak groups. (G) Amino acid alignment of kringle domain from vertebrate ROR2 and invertebrate Ror homologs. The Ku2 allele causes an arginine-to-cysteine substitution in short beak pigeon breeds.
The single most significantly-differentiated SNP genome wide (wcFST=0.88, pFST=0) is located at scaffold position ScoHet5_445.1:6568443. The non-reference allele causes a missense substitution in the seventh exon of ROR2 (ROR2C1087T, hereafter the Ku2 allele5) in short-beaked pigeons. ROR2 encodes a noncanonical Wnt receptor with well-established roles in cell polarity and motility in multiple embryonic tissues, including the neural crest31. This gene is required for normal craniofacial development: in humans, mutations in ROR2 cause autosomal recessive Robinow syndrome, a severe skeletal dysplasia characterized by extensive abnormalities, including a prominent forehead (frontal bossing), wideset eyes (hypertelorism), and a broad, short nose8,9. In mice, Ror2 knockout or knock-in of Robinow-associated mutations disrupts endochondral bone development and causes profound skeletal abnormalities, including craniofacial outgrowth defects32–34. Likewise, the OGO and morphologically similar pigeon breeds have reduced craniofacial outgrowths in the form of short beaks.
Within the short-beaked group, 98% of pigeons (45/46) with genotype data at ScoHet5_445.1:6568443 were homozygous or hemizygous for the Ku2 allele; only the Chinese Nasal Tuft, a breed that can have a short- or medium-length beak35, was heterogyzous. In contrast, 93% (97/104) of medium-beaked birds were homozygous, hemizygous, or heterozygous for the ancestral allele (Figure 3F). A genome-wide scan for putatively damaging coding variants predicted that the Ku2 allele is both highly deleterious and associated with short beaks (VAAST36 top-ranked feature, score=64.37, p=4e-8). At the amino acid level, the Ku2 allele causes an arginine-to-cysteine transition in the ROR2 extracellular kringle fold, a cysteine-rich, disulfide-bonded domain that is unlikely to tolerate mutations due to its small size and complex folding37. The precise number and spacing of cysteine residues in the kringle domain are deeply conserved in vertebrate ROR2 and invertebrate Ror homologs (Figure 3G), suggesting that the ectopic cysteine residue introduced by the Ku2 mutation may have a substantial impact on disulfide bond formation and kringle domain folding. Although the precise function of the ROR2 kringle domain remains unclear, it is thought to mediate protein-protein interactions and may modulate the affinity of the adjacent Frizzled-like ligand-binding domain for WNT5A38,39.
Like the pigeon Ku2 allele, the majority of known Robinow-associated missense mutations in human patients are clustered in the kringle and Frizzled-like extracellular domains. All of the characterized disease variants cause increased ROR2 protein retention in the endoplasmic reticulum, suggesting that the extracellular domain must be properly folded before transport to the plasma membrane37,40. Based on available evidence, we hypothesize that the Ku2 allele disrupts ROR2 protein folding in short-beaked pigeons, resulting in craniofacial outgrowth anomalies similar to Robinow syndrome in humans.
ROR2 and WNT5A are expressed during pigeon craniofacial morphogenesis
In chicken and mouse embryos, ROR2 expression is widespread with regions of strong expression in the facial prominences, dorsal root ganglia, and limb buds31,41,42. Using RNA-seq, we found that both ROR2 and WNT5A are also strongly expressed in short- and medium-beaked pigeon facial primordia (n=5 each), with higher expression in the frontonasal and maxillary prominences (upper beak) relative to the mandibular prominence (lower beak; Figure 4A–B). Neither ROR2 nor WNT5A is differentially expressed between short- and medium-beaked embryos at the pigeon equivalent of chicken stage HH29 (Ref. 43, Figure 4A–B), an early embryonic stage at which distinct craniofacial morphologies are evident among avian species43.
Figure 4. ROR2 and WNT5A are expressed in pigeon facial primordia.
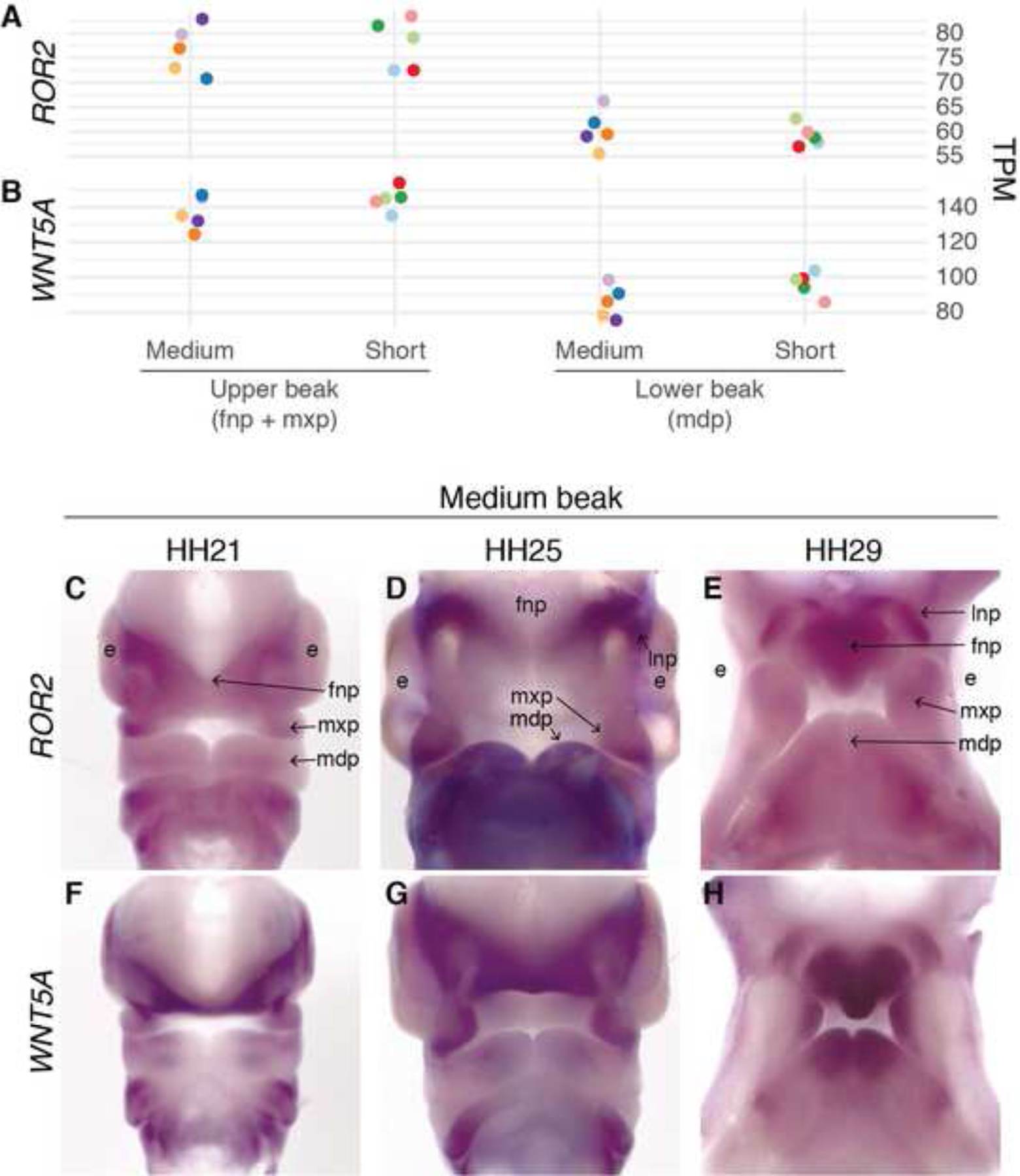
(A-B) ROR2 (A) and WNT5A (B) mRNA expression in facial primordia that will form the upper and lower beak from HH29 short and medium beak pigeon embryos. Each individual embryo is displayed in a different color. (C-H) Whole-mount in situ hybridization for ROR2 (C-E) and WNT5A (F-H) in medium beak pigeon embryos at HH21, HH25, and HH29. ROR2 is broadly expressed in facial primordia at all stages. WNT5A is strongly expressed in the FNP and at the lateral edges of the MXP and LNP, with increased expression at the edge of the MDP at HH29. Letters indicate embryonic tissues/structures: e=eye, fnp=frontonasal prominence, l=lateral nasal prominence, mdp=mandibular prominence, mxp=maxillary prominence.
Spatial expression of ROR2 is broad during early pigeon facial development (HH21–29; Figure 4C–E). WNT5A expression domains overlap with ROR2, but are more spatially restricted to the regions of the facial primordia that will grow out to form the beak (Figure 4F–H), similar to mouse and chicken44,45. Thus, ROR2 and WNT5A are expressed together in pigeons in a spatial and temporal manner that is consistent with their role as regulators of craniofacial morphogenesis. The lack of differential ROR2 expression in short- and medium-beaked pigeon embryos implicates the Ku2 coding mutation, rather than differences in the regulation of expression, in the development of the short beak phenotype.
Pigeons model vertebrate evolution and disease
Several developmental pathways have been implicated in the evolution of beak diversity in other birds, including Darwin’s finches, Great tits, and Black-bellied seedcrackers12–16. Although ROR2 has a well-established role in mammalian craniofacial development, to our knowledge, the noncanonical Wnt signaling pathway has not been implicated in regulation of craniofacial development and diversity in birds. Our results provide evidence that ROR2 is associated with beak size variation in domestic pigeons. Although additional studies are required to determine the functional consequences of the ROR2 coding variant, this finding suggests that genetic control of beak variation contrasts with other examples of recurrent evolution of derived traits via changes in the same genes in pigeons and other species, including head crests (EPHB2, also in ringneck doves27,46), feathered feet (PITX1 and TBX5, also in chickens30,47), and plumage color patterning (NDP, also in crows48–50). Considering the deep evolutionary conservation of developmental pathways that regulate craniofacial morphogenesis, our findings raise the possibility that noncanonical Wnt signaling is modulated in other cases of avian craniofacial variation. We did not identify noncanonical Wnt pathway genes in our recent study of the genetic basis of pigeon beak elaboration24, suggesting that distinct genetic programs underlie reduction and exaggeration of the same tissues and structures.
The identification of ROR2 as a putative regulator of beak length adds to a growing list of genes that underlie morphological variation in the domestic pigeon and are associated with human diseases, including congenital defects and cancer30,49,51,52. In addition, prior work in pigeons has predicted the molecular basis of diverse morphological traits in other wild and domestic species27,30,46,48–50. Thus, the pigeon is an exceptional model to interrogate the genetic underpinnings of vertebrate evolution, development, and disease.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Shapiro (mike.shapiro@utah.edu).
Materials Availability
Plasmids generated to synthesize RNA in situ hybridization probes against pigeon ROR2 and WNT5A are available upon request.
Data and Code Availability
Whole genome sequencing and RNA-sequencing datasets generated for this study have been deposited to the NCBI SRA database under BioProject PRJNA680754. Additional short-beaked genomes are available under BioProject PRJNA513877 (SRR8420387-SRR8420391, SRR8420393, SRR8420394, SRR8420397).
Experimental Model and Subject Details
Columba livia
Pigeons were utilized in accordance with protocols approved by the University of Utah Institutional Animal Care and Use Committee (protocols 10-05007, 13-04012, and 19-02011). Further information is provided throughout the Method Details section.
Method Details
RH × OGO F2 intercross and 3D imaging
A F2 intercross was established between a male Racing Homer (RH) and a female Old German Owl (OGO). F1 hybrids (n=15) were interbred to generate a F2 mapping population. F2 offspring that reached 6 months of age (n=145) were euthanized and basic biometrics (e.g. mass) were recorded. All F2 offspring and cross founders were submitted to the University of Utah Preclinical Imaging Core Facility for micro-CT imaging. For each bird, a whole-body scan was performed on a Siemens Inveon micro-CT using the following parameters: voxel size=94 μ, photon voltage=80 kV, source current=500 μA, exposure time=200 ms. Scans were reconstructed using a Feldkamp algorithm with Sheep-Logan filter and a calibrated beam hardening correction.
Surface model generation and landmarking
Surface model generation and landmarking were performed as described24. Briefly, a substack containing the cranium was extracted from the whole-body DICOM file stack in ImageJ v1.52q, exported as a NifTI file (*.nii), and imported into Amira v6.5.0 (ThermoFisher Scientific). Using the Segmentation Editor threshold feature, the cranial skeleton was segmented from soft tissue and exported as a HxSurface binary (*.surf) file. Surface meshes were converted to Polygon (Stanford) ASCII (*.ply) files using i3D Converter v3.80 and imported into IDAV Landmark Editor v3.0 (UC Davis) for landmarking. We applied a set of landmarks (Figure S1, Table S1) to the braincase (n=29 landmarks) and upper beak (n=20) of all F2 individuals and the cross founders. Landmark coordinates were exported as a NTsys landmark point dataset (*.dta) for geometric morphometric analysis.
Blood collection and genomic DNA extraction
Blood samples from adult pigeons used for whole-genome sequencing were collected at local pigeon shows, at breeder’s homes, or in the Shapiro lab loft. RH × OGO F2 offspring were bled at time of fledging (approximately 1 month of age). For each individual, a blood sample was collected from the brachial vein and stored in an EDTA-coated sample tube at −80°C. RNase-treated genomic DNA was extracted using a DNeasy Blood and Tissue Kit and eluted in Buffer EB (Qiagen).
Genotyping-by-sequencing (GBS) and linkage map assembly
Genomic DNA samples from RH × OGO cross founders and 171 F2 offspring were submitted to the University of Minnesota Genomics Core for GBS library prep and sequencing. Genomic DNA samples were digested with ApeKI (NEB), then ligated with T4 ligase (NEB) and phased adaptors with CWG overhangs. The ligated samples were purified with SPRI beads and amplified for 18 PCR cycles with 2X NEB Taq Master Mix to add barcodes. Libraries were purified, quantified, pooled, size selected for the 624–724 bp library region (480–580 DNA insert), and treated with ExoVII to remove any remaining single stranded material. The final pool was diluted to 1 nM for sequencing on the Illumina NovaSeq 6000 using single-end 1×100 reads. Target sequencing volume was ~4.75M reads/sample. Sequencing read quality was assessed with FastQC (Babraham Bioinformatics) and Illumina adapters were trimmed with Cutadapt54. Reads were mapped to the Cliv_2.1 reference assembly55 using Bowtie 256. Genotypes were called using the Stacks v2.52 ref_map.pl program57,58, which executes the Stacks pipeline programs gstacks and populations. The following options were passed to populations: -H -r 0.75 --map-type F2 --map-format rqtl.
The RH × OGO genetic map was constructed with the R package R/qtl v1.46-259 using genotype data from 171 F2 individuals. Because of differences in segregation patterns, autosomal and Z-linked scaffolds were assembled separately. For autosomal scaffolds, markers with identical genotypes or displaying segregation distortion (chi-square p < 0.005) were eliminated. Preliminary filtering was performed to remove markers missing in more than 20% (34/170) of F2 individuals. Pairwise recombination fractions were calculated and a preliminary genetic map was estimated using the est.rf and est.map functions, respectively. The droponemarker and calc.errorlod functions were used with the parameter (error.prob = 0.005) to identify problematic markers and likely genotyping errors, which were eliminated from the genetic map. Linkage groups were formed using the function formLinkageGroups with parameters (max.rf = 0.25, min.lod = 6). For the Z-chromosome, the same workflow was carried out, except that distorted markers were not removed. Preliminary marker ordering was done for all linkage groups using the orderMarkers function with the parameter (window size = 7). Final marker ordering was completed manually based on calculated recombination fractions and LOD scores. The compareorder function was used to test alternative marker orders; changes in marker ordering that resulted in an increased LOD score and decreased linkage group length were retained. The final RH × OGO genetic map is composed of 6128 markers (5553 autosomal, 575 Z-linked) on 35 linkage groups (34 autosomal, 1 Z-linked) with a genotyping rate of 90.1%.
Whole-genome resequencing
For the current study, we resequenced genomes for 33 pigeons from 24 short-beaked breeds: African Owl, Australian Tumbler, Berlin Short Face Tumbler, Budapest Tumbler, Canario Cropper, Chinese Nasal Tuft, Classic Old Frill, Damascene, Egyptian Swift, English Long Face Tumbler, English Short Face Tumbler, Granadino Pouter, Hamburg Sticken, Helmet, Italian Owl, Long Face Muff Tumbler, Nun, Old German Owl, Oriental Frill, Rafeno Pouter, Russian Tumbler, Taganrog Tumbler, Temeschburger Schecker, Uzbek Tumbler. We also resequenced 29 pigeons from 24 medium- or long-beaked breeds: Berlin Long Faced Tumbler, Dragoon, English Carrier, English Magpie, Scandaroon, Racing Homer, Danzig Highflier, Schalkaldener Mohrenkopf, Fairy Swallow, Hungarian Giant House Pigeon, Crested Saxon Field Color Pigeon, Saint, Franconian Trumpeter, American Highflier, Bokhara Trumpeter, Komorner Tumbler, Brunner Pouter, Mindian Fantail, Naked Neck, Turkish Tumbler, Norwich Cropper, Miniature American Crest, Rhine Ringbeater, Vienna MF Tumbler.
Genomic DNA samples were submitted to the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at the University of Utah for library preparation and sequencing. DNA libraries were prepared using the Illumina TruSeq DNA PCR-Free Sample Preparation Kit with an average insert size of 350 bp. 125-cycle paired-end sequencing was performed on an Illumina HiSeq 2500 instrument (3–4 libraries/lane).
Embryonic tissue isolation and RNA extraction
Pigeon eggs were collected from Racing Homer (medium beak) and Oriental Frill (short beak) breeding pairs and incubated to embryonic day 6 (Hamburger-Hamilton (HH) stage 28–29,60). Facial prominences that form the upper beak (frontonasal and maxillary, FNP+MXP) and lower beak (mandibular, MDP) were dissected and stored separately in RNAlater (ThermoFisher Scientific) at −80°C. Additional tissue was harvested from each embryo and used for DNA extraction and sex determination following a previously published PCR-based assay61. Total RNA was extracted from embryonic tissue samples using the RNeasy Mini Kit with RNase-Free DNAse Set and a TissueLyser LT (Qiagen).
RNA-sequencing
Total RNA from FNP+MXP and MDP samples from HH28–29 female Racing Homer (n=5) and Oriental Frill (n=5) embryos was submitted to the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at the University of Utah for library preparation and sequencing. RNA sample quality was assessed using the RNA ScreenTape Assay (Agilent). For each sample, a stranded sequencing library was prepared using the TruSeq Stranded mRNA Sample Prep Kit with oligo(dT) selection (Illumina). 125-cycle paired-end sequencing was performed on an Illumina HiSeq 2500 instrument (12 libraries/lane). An average of 23.4 million reads was generated for each sample.
ROR2 multiple sequence alignment
Amino acid sequences for vertebrate ROR2 and invertebrate ROR homologs were downloaded from Ensembl (ensembl.org) or NCBI (ncbi.nlm.nih.gov/gene). Clustal Omega multiple sequence alignments were performed and visualized with the R package msa v1.18.062.
Whole-mount RNA in situ hybridization (ISH)
ISH probe templates were generated by PCR amplification of a portion of pigeon ROR2 (692 bp amplicon) or WNT5A (783 bp amplicon) from a pooled cDNA library generated from HH21, HH25, and HH29 Racing Homer embryos using the following primer sets: ROR2-forward: 5’-GGAACCGACAGGTTCTACCA-3’, ROR2-reverse: 5’-TGCTTCGTCCATCTGAAGTG-3’, WNT5A-forward: 5’-CATAGTGGCTCTGGCCATTT-3’, WNT5A-reverse: 5’-CCCCGACTGTTGAGTTTCAT-3’. ROR2 and WNT5A amplicons were cloned into pGEM-T Easy (Promega) and confirmed by Sanger sequencing. Antisense and sense RNA probes were generated by in vitro transcription as previously described63. For ROR2, pGEM-ROR2 was digested with NcoI or SalI and transcribed with SP6 or T7 RNA polymerase, respectively. For WNT5A, pGEM-WNT5A was digested with Kpn1 or Nco1 and transcribed with T7 or SP6 RNA polymerase, respectively.
Racing Homer embryos used for ISH were dissected from eggs at the desired embryonic stage and fixed overnight in 4% paraformaldehyde at 4°C on a shaking table. Embryos were subsequently dehydrated into 100% MeOH and stored at −20°C. Whole-mount ISH was performed following a protocol optimized for avian embryos (geisha.arizona.edu/geisha/protocols.jsp). For each experiment, antisense or sense probes were applied to stage-matched embryos.
Quantification and Statistical Analysis
Linear measurement analysis
For each F2 individual and the cross founders, beak and braincase length were determined by calculating the linear distance between landmark pairs (beak: landmarks 1 and 2; braincase: landmarks 1 and 3,24) using the interlmkdist function from the R package geomorph v3.3.164–66. Raw beak length measurements were fit to a linear regression model (beak length ~ body mass) and residuals were calculated in R v3.6.367.
Geometric morphometrics
Geometric morphometric analyses were performed in geomorph as described24. The NTsys landmark point dataset was imported with the readland.nts function. Missing landmarks were estimated using the function estimate.missing(method = “TPS”). Bilateral symmetry analysis was performed via the bilat.symmetry(iter = 1) function and the symmetrical component of shape variation was extracted. A Generalized Procrustes Analysis was performed using the gpagen function. To analyze allometry, a linear model (shape ~ centroid size) was fit using the procD.lm function and residuals were used for analysis of allometry-free shape. Principal components analysis was performed using the gm.prcomp function. Integration of beak and braincase shape was analyzed using the two.b.pls function.
Shape changes were visualized with geomorph and the R package Morpho v2.8 (https://github.com/zarquon42b/Morpho). The geomorph function plotRefToTarget was used to generate wireframes. Surface mesh deformations, heatmaps, and movies were generated in Morpho with the tps3d, shade3d, meshDist, and warpmovie3d functions. For all mesh-based visualizations, deformations were applied to a reference mesh, which was generated by warping a RH × OGO F2 mesh to the mean shape.
QTL mapping
QTL mapping was performed using the R package R/qtl v1.46-259. Single-QTL genome scans were performed using the scanone function with Haley-Knott regression and sex as a covariate. The 5% genome-wide significance threshold was calculated by running scanone with 1000 permutation replicates. For each QTL, the 1.5-LOD support interval was calculated with the lodint function, percent variance explained (PVE) was calculated with the fitqtl function, and QTL effects were estimated via the plotPXG function. We compared phenotypic means in RH × OGO F2 genotypic groups at peak markers via one-way ANOVA and Tukey Test for pairwise comparisons in R. Genes within QTL intervals were identified using a custom R script and visualized using the R packages ggplot2 v3.3.053 and gggenes v0.4.0 (https://github.com/wilkox/gggenes).
Variant calling and comparative genomic analyses
Variant calling was performed with FastQForward68, which wraps the BWA short read aligner69 and Sentieon (sentieon.com) variant calling tools to generate aligned BAM files (fastq2bam) and variant calls in VCF format (bam2gvcf). Sentieon is a commercialized GATK equivalent pipeline that allows users to follow GATK best practices using the Sentieon version of each tool (broadinstitute.org/gatk/guide/best-practices and support.sentieon.com/manual/DNAseq_usage/dnaseq/). FastQForward manages distribution of the workload to these tools on a compute cluster to allow for faster data-processing than when calling these tools directly, resulting in runtimes as low as a few minutes per sample. Raw sequencing reads from 54 newly resequenced individuals (described in Whole-genome resequencing section) were aligned to the Cliv_2.1 reference assembly55 using fastq2bam. Variant calling was performed for each newly resequenced individual, as well as 132 previously resequenced individuals27,30,49,70, using bam2gvcf and individual genome variant call format (gVCF) files were created. Joint variant calling was performed on a total of 186 individuals using the Sentieon GVCFtyper algorithm. The resulting VCF file was used for all subsequent genomic analyses.
Genome-wide Weir and Cockerham’s FST (wcFST) and probabilistic FST (pFST) were calculated using the GPAT++ toolkit within the VCFLIB software library (github.com/vcflib) as previously described27,30,49,70. Extended haplotype homozygosity (EHH) was calculated for genomic scaffold ScoHet5_445.1 using the GPAT++ sequenceDiversity tool. Putatively deleterious variants were identified using the Variant Annotation, Analysis, and Search Tool (VAAST2,36), which was implemented as previously described27. For all comparative genomic analyses, pigeons were binned into short and medium beak groups based on breed standards71,72 and qualitative assessment of photos for each individual.
Short beak (56 individuals, 31 breeds): African Owl, Australian Tumbler, Bacska Tumbler, Berlin Short Face Tumbler, Budapest Tumbler, Canario Cropper, Catalonian Tumbler, Chinese Nasal Tuft, Chinese Owl, Classic Old Frill, Damascene, Egyptian Swift, English Long Face Tumbler, English Short Face Tumbler, Granadino Pouter, Hamburg Sticken, Helmet, Italian Owl, Komorner Tumbler, Long Face Tumbler, Nun, Old German Owl, Oriental Frill, Portuguese Tumbler, Rafeno Pouter, Russian Tumbler, Spanish Barb, Syrian Dewlap, Taganrog Tumbler, Temeschburger Schecker, Uzbek Tumbler.
Medium or long beak (121 individuals, 58 breeds and feral): American Highflier, American Show Racer, Archangel, Armenian Tumbler, Berlin Long Face Tumbler, Birmingham Roller, Bokhara Trumpeter, Brunner Pouter, Carneau, Crested Saxon Field Color Pigeon, Cumulet, Danish Tumbler, Danzig Highflier, Dragoon, English Carrier, English Magpie, English Pouter, English Trumpeter, Fairy Swallow, Fantail, Feral, Franconian Trumpeter, Frillback, German Beauty, Hungarian Giant House Pigeon, Ice Pigeon, Indian Fantail, Iranian Tumbler, Jacobin, King, Lahore, Laugher, Lebanon, Marchenero Pouter, Mindian Fantail, Miniature American Crest, Modena, Mookee, Naked Neck, Norwich Cropper, Old Dutch Capuchin, Oriental Roller, Parlor Roller, Polish Lynx, Pomeranian Pouter, Pygmy Pouter, Racing Homer, Rhine Ringbeater, Runt, Saint, Saxon Monk, Saxon Pouter, Scandaroon, Schalkaldener Mohrenkopf, Shakhsharli, Starling, Turkish Tumbler, Vienna Medium Face Tumbler, West of England.
RNA-seq analysis
Analysis of RNA-seq data was performed as previously described73. Briefly, sequencing read quality was assessed with FastQC (Babraham Bioinformatics). Illumina adapters were trimmed and reads were aligned to the pigeon Cliv_2.1 reference assembly (Holt et al., 2018) using STAR v2.5.0a74 using the 2-pass mode. GTF annotation files were used to guide spliced read alignments. Mapped reads were assigned to genes using featureCounts from the Subread package version 1.5.175. Transcript abundance (TPM) was quantified using Salmon v1.3.076. Differential expression analyses were performed with the R package DESeq2 version 1.12.477.
Supplementary Material
Movie S1. PC1 shape variation. Minimum to maximum, magnified 1.5x.
Movie S2. PC2 shape variation. Minimum to maximum, magnified 1.5x.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| DNeasy Blood and Tissue Kit | Qiagen | 69504 |
| RNAlater | ThermoFisher Scientific | AM7021 |
| RNeasy Mini Kit | Qiagen | 74004 |
| RNase-Free DNAse Set | Qiagen | 79254 |
| pGEM-T Easy Vector System | Promega | A1360 |
| Deposited Data | ||
| Pigeon whole genome sequencing and pigeon embryonic craniofacial RNA-sequencing datasets | NCBI SRA database | PRJNA680754, PRJNA513877 |
| Vertebrate ROR2 and invertebrate ROR homolog amino acid sequences | Ensembl (ensembl.org) and NCBI (ncbi.nlm.nih.gov/gene) | |
| Experimental Models: Organisms/Strains | ||
| Columba livia | Shapiro Lab loft, Utah Pigeon Club, various pigeon breeders | |
| Oligonucleotides | ||
| ROR2-forward | GGAACCGACAGGTTCTACCA | |
| ROR2-reverse | TGCTTCGTCCATCTGAAGTG | |
| WNT5A-forward | CATAGTGGCTCTGGCCATTT | |
| WNT5A-reverse | CCCCGACTGTTGAGTTTCAT | |
| Software and Algorithms | ||
| ImageJ v1.52q | https://imagej.nih.gov/ij/ | |
| Amira v6.5.0 | ThermoFisher Scientific | |
| i3D Converter v3.80 | ||
| IDAV Landmark Editor v3.0 | UC Davis | |
| FastQC | Babraham Bioinformatics | |
| Cutadapt | [54] | |
| Bowtie2 | [56] | |
| Stacks v2.52 | [57][58] | |
| R/qtl v1.46-2 | [67] | |
| msa v1.18.0 | [61] | |
| geomorph v3.3.1 | [63][64][65] | |
| R v3.6.3 | [66] | |
| Morpho v2.8 | https://github.com/zarquon42b/Morpho | |
| ggplot2 v3.3.0 | [53] | |
| gggenes v0.4.0 | https://github.com/wilkox/gggenes | |
| FastQForward | [68] | |
| VCFLIB GPAT++ toolkit | github.com/vcflib | |
| VAAST2 | [36] | |
| STAR v2.5.0a | [74] | |
| Subread featureCounts v1.5.1 | [75] | |
| Salmon v1.5.1 | [76] | |
| DESeq2 v1.12.4 | [77] | |
Acknowledgments
We are grateful to Nathan Young and Rich Schneider for generously sharing their time and expertise related to geometric morphometrics, craniofacial biology, and avian embryology. We thank all past and present members of the Shapiro Lab, including Rebecca Bruders, Alexa Davis, Anna Vickrey, and Ryan Wauer for help with animal husbandry, technical assistance, and advice. We thank members of the Utah Pigeon Club and National Pigeon Association for sample contributions. We acknowledge the University of Utah Preclinical Imaging Core Facility, especially Tyler Thompson, for micro-CT imaging; the Center for High Performance Computing at the University of Utah for computing resources; the University of Utah High-Throughput Genomics Shared Resource for RNA library preparation and sequencing; and the University of Minnesota Genomics Core for GBS library preparation and sequencing. This work was funded by the National Institutes of Health (F32DE028179 to E.F.B.; R35GM131787 to M.D.S.) and the National Science Foundation (DEB1149160 to M.D.S.). E.T.M. was supported by a fellowship from the Jane Coffin Childs Memorial Fund for Medical Research.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Twigg SRF, and Wilkie AOM (2015). New insights into craniofacial malformations. Hum. Mol. Genet 24, R50–R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fish JL (2016). Developmental mechanisms underlying variation in craniofacial disease and evolution. Developmental Biology 415, 188–197. [DOI] [PubMed] [Google Scholar]
- 3.Christie W, and Wriedt C (1924). Die Vererbung von Zeichnungen, Farben und anderen Charakteren bei Tauben. Z.Ver-erbungslehre 32, 233–298. [Google Scholar]
- 4.Hollander WF (1983). Origins and Excursions in Pigeon Genetics: A Compilation (The Ink Spot).
- 5.Sell A (2012). Pigeon Genetics: Applied Genetics in the Domestic Pigeon (Sell Publishing).
- 6.Topczewski J, Dale RM, and Sisson BE (2011). Planar cell polarity signaling in craniofacial development. Organogenesis 7, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayor R, and Theveneau E (2014). The role of the non-canonical Wnt-planar cell polarity pathway in neural crest migration. Biochem J 457, 19–26. [DOI] [PubMed] [Google Scholar]
- 8.Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tüysüz B, Murday VA, Patton MA, Wilkie AOM, et al. (2000). Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet 25, 419–422. [DOI] [PubMed] [Google Scholar]
- 9.van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, et al. (2000). Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat Genet 25, 423–426. [DOI] [PubMed] [Google Scholar]
- 10.Tokita M, Yano W, James HF, and Abzhanov A (2017). Cranial shape evolution in adaptive radiations of birds: comparative morphometrics of Darwin’s finches and Hawaiian honeycreepers. Phil. Trans. R. Soc. B 372, 20150481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navalón G, Marugán-Lobón J, Bright JA, Cooney CR, and Rayfield EJ (2020). The consequences of craniofacial integration for the adaptive radiations of Darwin’s finches and Hawaiian honeycreepers. Nat Ecol Evol 4, 270–278. [DOI] [PubMed] [Google Scholar]
- 12.Bosse M, Spurgin LG, Laine VN, Cole EF, Firth JA, Gienapp P, Gosler AG, McMahon K, Poissant J, Verhagen I, et al. (2017). Recent natural selection causes adaptive evolution of an avian polygenic trait. Science 358, 365–368. [DOI] [PubMed] [Google Scholar]
- 13.vonHoldt BM, Kartzinel RY, Huber CD, Le Underwood V, Zhen Y, Ruegg K, Lohmueller KE, and Smith TB (2018). Growth factor gene IGF1 is associated with bill size in the black-bellied seedcracker Pyrenestes ostrinus. Nat Commun 9, 4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abzhanov A (2004). Bmp4 and Morphological Variation of Beaks in Darwin’s Finches. Science 305, 1462–1465. [DOI] [PubMed] [Google Scholar]
- 15.Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR, and Tabin CJ (2006). The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. 442, 5. [DOI] [PubMed] [Google Scholar]
- 16.Mallarino R, Grant PR, Grant BR, Herrel A, Kuo WP, and Abzhanov A (2011). Two developmental modules establish 3D beak-shape variation in Darwin’s finches. Proceedings of the National Academy of Sciences 108, 4057–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, Promerová M, Rubin C-J, Wang C, Zamani N, et al. (2015). Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375. [DOI] [PubMed] [Google Scholar]
- 18.Lamichhaney S, Han F, Berglund J, Wang C, Almen MS, Webster MT, Grant BR, Grant PR, and Andersson L (2016). A beak size locus in Darwins finches facilitated character displacement during a drought. Science 352, 470–474. [DOI] [PubMed] [Google Scholar]
- 19.Baptista LF, Gómez JEM, and Horblit HM (2009). DARWIŃS PIGEONS AND THE EVOLUTION OF THE COLUMBIFORMS: RECAPITULATION OF ANCIENT GENES. 23. [Google Scholar]
- 20.Young NM, Linde-Medina M, Fondon JW, Hallgrímsson B, and Marcucio RS (2017). Craniofacial diversification in the domestic pigeon and the evolution of the avian skull. Nat Ecol Evol 1, 0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore Des (2018). Judging Racing Pigeons. Des Moore’s Pigeon Domain. http://desmoore.tripod.com/id56.html. [Google Scholar]
- 22.Old German Owl Club Standard (2019). Old German Owl Club. http://ogoc.org/standard.htm. [Google Scholar]
- 23.Hallgrímsson B, Katz DC, Aponte JD, Larson JR, Devine J, Gonzalez PN, Young NM, Roseman CC, and Marcucio RS (2019). Integration and the Developmental Genetics of Allometry. Integrative and Comparative Biology 59, 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boer EF, Maclary ET, and Shapiro MD (2021). Complex genetic architecture of three-dimensional craniofacial shape variation in domestic pigeons. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wexelsen H (1937). Size inheritance in pigeons. Journal of Experimental Zoology 76, 161–186. [Google Scholar]
- 26.Stringham SA, Mulroy EE, Xing J, Record D, Guernsey MW, Aldenhoven JT, Osborne EJ, and Shapiro MD (2012). Divergence, convergence, and the ancestry of feral populations in the domestic rock pigeon. Curr Biol 22, 302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapiro MD, Kronenberg Z, Li C, Domyan ET, Pan H, Campbell M, Tan H, Huff CD, Hu H, Vickrey AI, et al. (2013). Genomic diversity and evolution of the head crest in the rock pigeon. Science 339, 1063–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacheco G, van Grouw H, Shapiro MD, Gilbert MTP, and Vieira FG (2020). Darwin’s Fancy Revised: An Updated Understanding of the Genomic Constitution of Pigeon Breeds. Genome Biology and Evolution 12, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weir BS, and Cockerham CC (1984). Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358. [DOI] [PubMed] [Google Scholar]
- 30.Domyan ET, Kronenberg Z, Infante CR, Vickrey AI, Stringham SA, Bruders R, Guernsey MW, Park S, Payne J, Beckstead RB, et al. (2016). Molecular shifts in limb identity underlie development of feathered feet in two domestic avian species. Elife 5, e12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stricker S, Rauschenberger V, and Schambony A (2017). ROR-Family Receptor Tyrosine Kinases. In Current Topics in Developmental Biology (Elsevier), pp. 105–142. [DOI] [PubMed] [Google Scholar]
- 32.DeChiara TM, Kimble RB, Poueymirou WT, Rojas J, Masiakowski P, Valenzuela DM, and Yancopoulos GD (2000). Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat Genet 24, 271–274. [DOI] [PubMed] [Google Scholar]
- 33.Schwabe GC, Trepczik B, Süring K, Brieske N, Tucker AS, Sharpe PT, Minami Y, and Mundlos S (2004). Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome: Developmental Model for Robinow Syndrome. Dev. Dyn 229, 400–410. [DOI] [PubMed] [Google Scholar]
- 34.Raz R, Stricker S, Gazzerro E, Clor JL, Witte F, Nistala H, Zabski S, Pereira RC, Stadmeyer L, Wang X, et al. (2008). The mutation ROR2W749X, linked to human BDB, is a recessive mutation in the mouse, causing brachydactyly, mediating patterning of joints and modeling recessive Robinow syndrome. Development 135, 1713–1723. [DOI] [PubMed] [Google Scholar]
- 35.Mosca F, and Ipsom JP (2005). Standard of the Chinese Nasal Tuft. https://www.angelfire.com/ga3/pigeongenetics/nasaltuftstandard.html/.
- 36.Hu H, Huff CD, Moore B, Flygare S, Reese MG, and Yandell M (2013). VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol 37, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Bellamy WP, Seabra MC, Field MC, and Ali BR (2005). ER-associated protein degradation is a common mechanism underpinning numerous monogenic diseases including Robinow syndrome. Human Molecular Genetics 14, 2559–2569. [DOI] [PubMed] [Google Scholar]
- 38.Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, et al. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway: Role of Ror2 in Wnt5a signalling pathway. Genes to Cells 8, 645–654. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Han X, Xu W, Rao Z, and Li X (2019). Purification and characterization of the extracellular region of human receptor tyrosine kinase like orphan receptor 2 (ROR2). Protein Expression and Purification 158, 74–80. [DOI] [PubMed] [Google Scholar]
- 40.Ali BR, Jeffery S, Patel N, Tinworth LE, Meguid N, Patton MA, and Afzal AR (2007). Novel Robinow syndrome causing mutations in the proximal region of the frizzled-like domain of ROR2 are retained in the endoplasmic reticulum. Hum Genet 122, 389–395. [DOI] [PubMed] [Google Scholar]
- 41.Matsuda T, Nomi M, Ikeya M, Kani S, Oishi I, Terashima T, Takada S, and Minami Y (2001). Expression of the receptor tyrosine kinase genes, Ror1 and Ror2, during mouse development. Mechanisms of Development 105, 153–156. [DOI] [PubMed] [Google Scholar]
- 42.Stricker S, Verhey Van Wijk N, Witte F, Brieske N, Seidel K, and Mundlos S (2006). Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in Brachydactyly type B and Robinow syndrome. Dev. Dyn 235, 3456–3465. [DOI] [PubMed] [Google Scholar]
- 43.Smith FJ, Percival CJ, Young NM, Hu D, Schneider RA, Marcucio RS, and Hallgrimsson B (2015). Divergence of craniofacial developmental trajectories among avian embryos: Craniofacial Trajectories Among Avian Embryos. Dev. Dyn 244, 1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geetha-Loganathan P, Nimmagadda S, Antoni L, Fu K, Whiting CJ, Francis-West P, and Richman JM (2009). Expression of WNT signalling pathway genes during chicken craniofacial development. Dev. Dyn 238, 1150–1165. [DOI] [PubMed] [Google Scholar]
- 45.Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE, and Mouse Genome Database Group (2019). Mouse Genome Database (MGD) 2019. Nucleic Acids Res 47, D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vickrey AI, Domyan ET, Horvath MP, and Shapiro MD (2015). Convergent Evolution of Head Crests in Two Domesticated Columbids Is Associated with Different Missense Mutations in EphB2. Mol Biol Evol 32, 2657–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Lee M, Davis BW, Lamichhaney S, Dorshorst BJ, Siegel PB, and Andersson L (2020). Mutations Upstream of the TBX5 and PITX1 Transcription Factor Genes Are Associated with Feathered Legs in the Domestic Chicken. Molecular Biology and Evolution 37, 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poelstra JW, Vijay N, Hoeppner MP, and Wolf JBW (2015). Transcriptomics of colour patterning and coloration shifts in crows. Mol Ecol 24, 4617–4628. [DOI] [PubMed] [Google Scholar]
- 49.Vickrey AI, Bruders R, Kronenberg Z, Mackey E, Bohlender RJ, Maclary ET, Maynez R, Osborne EJ, Johnson KP, Huff CD, et al. (2018). Introgression of regulatory alleles and a missense coding mutation drive plumage pattern diversity in the rock pigeon. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knief U, Bossu CM, Saino N, Hansson B, Poelstra J, Vijay N, Weissensteiner M, and Wolf JBW (2019). Epistatic mutations under divergent selection govern phenotypic variation in the crow hybrid zone. Nat Ecol Evol 3, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guernsey MW, Ritscher L, Miller MA, Smith DA, Schöneberg T, and Shapiro MD (2013). A Val85Met mutation in melanocortin-1 receptor is associated with reductions in eumelanic pigmentation and cell surface expression in domestic rock pigeons (Columba livia). PLoS One 8, e74475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Domyan ET, Guernsey MW, Kronenberg Z, Krishnan S, Boissy RE, Vickrey AI, Rodgers C, Cassidy P, Leachman SA, Fondon JW 3rd, et al. (2014). Epistatic and combinatorial effects of pigmentary gene mutations in the domestic pigeon. Curr Biol 24, 459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wickham H (2016). ggplot2: elegant graphics for data analysis Second edition. (Springer; ). [Google Scholar]
- 54.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10. [Google Scholar]
- 55.Holt C, Campbell M, Keays DA, Edelman N, Kapusta A, Maclary E, E TD, Suh A, Warren WC, Yandell M, et al. (2018). Improved Genome Assembly and Annotation for the Rock Pigeon (Columba livia). G3; (Bethesda: ) 8, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Catchen J, Hohenlohe PA, Bassham S, Amores A, and Cresko WA (2013). Stacks: an analysis tool set for population genomics. Mol Ecol 22, 3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Catchen JM, Amores A, Hohenlohe P, Cresko W, and Postlethwait JH (2011). Stacks: building and genotyping Loci de novo from short-read sequences. G3; (Bethesda: ) 1, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Broman KW, Wu H, Sen S, and Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. [DOI] [PubMed] [Google Scholar]
- 60.Hamburger V, and Hamilton HL (1992). A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 195, 231–72. [DOI] [PubMed] [Google Scholar]
- 61.Fridolfsson A-K, and Ellegren H (1999). A Simple and Universal Method for Molecular Sexing of Non-Ratite Birds. Journal of Avian Biology 30, 116. [Google Scholar]
- 62.Bodenhofer U, Bonatesta E, Horejš-Kainrath C, and Hochreiter S (2015). msa: an R package for multiple sequence alignment. Bioinformatics, btv494. [DOI] [PubMed] [Google Scholar]
- 63.Boer EF, Howell ED, Schilling TF, Jette CA, and Stewart RA (2015). Fascin1-Dependent Filopodia are Required for Directional Migration of a Subset of Neural Crest Cells. PLoS Genet 11, e1004946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams DC, Collyer ML, and Kaliontzopoulou A (2020). Geomorph: Software for geometric morphometric analyses. R package version 3.2.1 [Google Scholar]
- 65.Collyer ML, and Adams DC (2018). RRPP: An R package for fitting linear models to high-dimensional data using residual randomization. Methods Ecol Evol 9, 1772–1779. [Google Scholar]
- 66.Collyer ML, and Adams DC (2020). RRPP: Linear Model Evaluation with Randomized Residuals in a Permutation Procedure, R package version 0.5.2
- 67.R Core Team (2020). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- 68.Carson Holt FastQForward.
- 69.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bruders R, Van Hollebeke H, Osborne EJ, Kronenberg Z, Maclary E, Yandell M, and Shapiro MD (2020). A copy number variant is associated with a spectrum of pigmentation patterns in the rock pigeon (Columba livia). PLoS Genet 16, e1008274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levi WM (2013). Encyclopedia of Pigeon Breeds (Wendell Levi Publishing Company; ). [Google Scholar]
- 72.National Pigeon Association (2010). National Pigeon Association Book of Standards (Purebred Pigeon Publishing; ). [Google Scholar]
- 73.Boer EF, Van Hollebeke HF, Park S, Infante CR, Menke DB, and Shapiro MD (2019). Pigeon foot feathering reveals conserved limb identity networks. Dev Biol 454, 128–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–30. [DOI] [PubMed] [Google Scholar]
- 76.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. PC1 shape variation. Minimum to maximum, magnified 1.5x.
Movie S2. PC2 shape variation. Minimum to maximum, magnified 1.5x.
Data Availability Statement
Whole genome sequencing and RNA-sequencing datasets generated for this study have been deposited to the NCBI SRA database under BioProject PRJNA680754. Additional short-beaked genomes are available under BioProject PRJNA513877 (SRR8420387-SRR8420391, SRR8420393, SRR8420394, SRR8420397).