

Abstract
Scope:
Cancer cachexia is characterized by the loss of skeletal muscle resulting in functional impairment, reduced quality of life and mortlity. Naringenin, a flavonoid found in citrus fruits, improves insulin sensitivity and reduces inflammation and tumor growth in preclinical models. Therefore, we hypothesized that dietary supplementation of naringenin would improve insulin sensitivity, decrease inflammation, slow body weight loss and delay tumor growth in a mouse model of cancer cachexia.
Methods & Results:
Mice were fed 2 wt% dietary naringenin before and during initiation of cancer cachexia using inoculated adenocarcinoma-26 cells (C-26). Food intake, body weight, body composition, muscle function, insulin tolerance, and inflammatory status were assessed. Although naringenin-fed tumor-bearing mice exhibited reductions in body weight and food intake earlier than control diet-fed tumor-bearing mice, dietary naringenin was protective against loss of muscle strength, and attenuated the onset of insulin resistance and markers of inflammation.
Conclusions:
Dietary supplementation of naringenin improved multiple aspects of metabolic disturbance and inflammation during cancer cachexia progression in [C-26 tumor-bearing] mice. However, the acceleration of anorexia and weight loss was also observed. These findings emphasize the link between inflammation and insulin resistance as a basis for understanding their roles in the pathogenesis of cancer cachexia.
Keywords: cancer cachexia, inflammation, insulin resistance, naringenin, skeletal muscle
Graphical Abstract
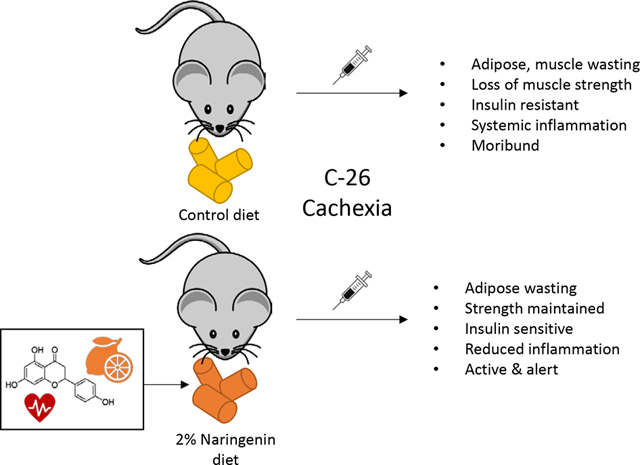
Lifestyle methods to reduce or prevent the progression of cancer cachexia are needed. Naringenin is a flavonoid that was able to maintain muscle strength and insulin sensitivity in a mouse model for cachexia. Future studies should interrogate the mechanism(s) of action and determine safety and efficacy of naringenin and other flavonoids, as delivered through the diet, to maintain muscle strength in humans.
1. Introduction
Cancer cachexia is a multifactorial disease characterized by the loss of skeletal muscle mass (with or without adipose loss), and leads to functional impairment [1]. Many factors contribute to wasting, including inflammation and alterations in systemic metabolic pathways [2]. Cancer cachexia leads to weakness, loss of volitional effort, resistance to antineoplastic therapies, and decreased quality of life and survival [3]. It is estimated that cancer cachexia contributes to 20% of all cancer-related deaths [4]. Wasting in cancer cachexia cannot be prevented or reversed by conventional nutritional therapies, due to the development of abnormal energy metabolism and anorexia, which leads to chronic negative nitrogen and energy balance. Currently, there are no effective clinical therapeutic treatments for cancer cachexia; however, metabolic modulators have successfully slowed cancer cachexia development in some clinical studies [5, 6], underscoring the importance of metabolic regulation in cancer cachexia treatment. There is a need to identify therapeutic treatments that can help improve quality of life by maintaining skeletal muscle mass and function in patients suffering from the disease. Potential dietary treatments that could be combined with adjuvant therapies are a desirable option, considering that cancer cachexia patients are often taking additional therapeutic drugs for cancer treatment.
Naringenin is a flavonoid most commonly found in citrus fruits and tomatoes that has a wide range of beneficial impacts on inflammation, metabolism, and anti-cancer properties by inhibiting pro-inflammatory pathways in a variety of cell types and tissues [7–11]. Due to the known link between inflammation and metabolism [12], it is not surprising that naringenin also exhibits bioactivity in vitro and in vivo which results in improved glucose clearance [13–17], insulin sensitivity [13, 14, 18, 19], lipid metabolism [16, 19, 20], and energy metabolism [18, 19, 21]. Furthermore, we and others have shown that dietary supplementation of naringenin preserves lean mass in other animal models of metabolic disturbances [19, 21]. To our knowledge, the impact of naringenin on muscle function has not been reported.
An extension of naringenin’s role in attenuating metabolic disturbances is its potential therapeutic means for the prevention of cancer development. Several epidemiological studies report that higher consumption of naringenin-containing foods is associated with lower cancer incidence [22–25] and a number of preclinical studies have identified anti-cancer properties of naringenin [18, 26–29]. The body of evidence highlighting the metabolically beneficial effects of naringenin underscores the important relationship between inflammation and metabolic health, which are both major contributors to the development of cancer cachexia. We therefore hypothesized that dietary naringenin supplementation would delay the pathogenesis of cancer cachexia by reducing inflammation, improving insulin sensitivity, and possibly delaying tumor growth, leading to maintenance of body weight, muscle mass, and strength.
In the current study we utilized the Colon-26 (C-26) mouse model of cancer cachexia to address this hypothesis. In this model, cachexia is driven by systemic inflammation as a result of persistently elevated interleukin-6 (IL-6), resulting in reduction in food intake, body weight, and lean and adipose tissue mass [30–32]. Therefore, we assessed the impact of consuming diets with or without 2% dietary naringenin on body composition, muscle strength, and insulin sensitivity over the course of cancer cachexia progression in the C-26 tumor-bearing mice.
2. Experimental Section
2.1. Colon-26 adenocarcinoma cell culture
Colon-26 adenocarcinoma cells (NCI, Bethesda Maryland; cell line referenced in [33]) were cultured in Roswell Park Memorial Institute media (RPMI 1640) + L-glutamine (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 5% fetal bovine serum and 1% Penicillin-Streptomycin at 37°C and 5% CO2.
2.2. Animals, diets, and experimental design
Twenty-to-28 day-old male CD2F1 mice (N=44) were obtained from Charles River (Wilmington, MA, USA). Mice were individually housed in a vivarium with room temperature of 22±0.5°C and on a 12-hour light/dark cycle, with free access to food and water and then allowed to acclimate to their new environment for 6 days. After the acclimation period, mice were matched by body weight into two diet groups. The control (CON) group (n=21) received a semi-purified AIN-93G diet (D12450J, Research Diets Inc. New Brunswick, NJ, USA), while the naringenin (NAR) group (n=23) received an AIN-93G diet supplemented with 2% naringenin (Sigma-Aldrich, St. Louis, MO, USA) prepared by Research Diets Inc (New Brunswick, NJ, USA; Supplemental Table S1). Due to the potential confounding effects of dietary naringenin on food intake [18, 21] in a disease and mouse model characterized by anorexia [1, 34], mice were acclimated to naringenin diets prior to tumor inoculation. After 13 days of diet consumption, mice in each group were matched by body weight into a non-tumor (−) group or tumor (+) group. The final experimental groups consisted of CON(+) (n=11), CON(−) (n=10), NAR(+) (n=12), and NAR(−) (n=11). To adequately control for variation in body weight, tumor or vehicle inoculation was carried out once mice reached a body weight of 18g (between 13 to 26 days after beginning experimental diets). The two tumor groups (+) were inoculated in the right flank with a 1×106 cell suspension of C-26 cells suspended in 100μl of phosphate-buffered saline (PBS). Mice in non-tumor groups (−) were inoculated with 100μl of PBS and served as non-tumor-bearing controls. At Day 0 (day of inoculation), NAR mice had an average body weight of 19.25 ± 0.27g while CON mice had an average body weight of 20.33g ± 0.25g (P = .005; data not shown). The experimental design is outlined in Fig. 1. Bodyweight and food intake of mice were measured daily.
Fig. 1. Details of experimental design.
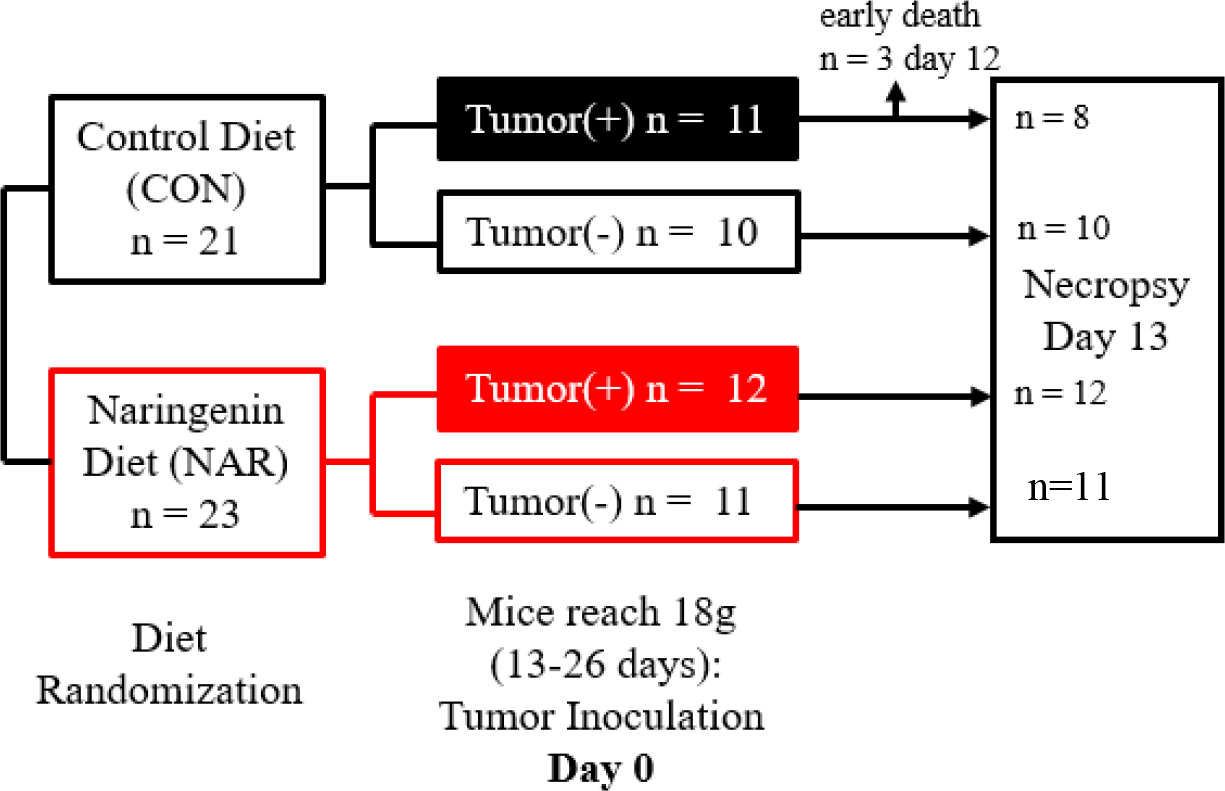
Mice were randomized to a control diet (CON; n=21) or a diet containing 2% naringenin (NAR; n=23). After 13 days of diet consumption, mice were matched by body weight into a no-tumor (−) or tumor (+) group. The final experimental groups consisted of CON(+) (n=11; black filled box), CON(−) (n=10; black outlined box), NAR(+) (n=12; red filled box), and NAR(−) (n=12; red outlined box). When mice reached a weight of 18g between 13–26 days after beginning diets, tumor(+) groups were inoculated with a 1×106 cell suspension of Colon-26 adenocarcinoma and tumor(−) groups with saline. At Day 12 post inoculation, 3 mice in the CON(+) group required early euthanasia. On Day 13, mice were sacrificed and necropsy was performed.
Mice were euthanized 13 days after inoculation under isoflurane anesthesia with cervical dislocation at 12pm after food was removed for 6 hours (mice had access to HydroGels- Cincinnati Lab Supply, Inc., Cincinnati, OH, USA- containing .62kcal/g during the 6 hours prior to euthanasia to prevent dehydration in moribund mice). Blood was collected by cardiac puncture, deposited into EDTA-coated blood collection tubes, and stored on ice until plasma was prepared. Tissues were collected, weighed, and immediately flash-frozen in liquid nitrogen. Plasma and frozen tissues were stored at −80°C until further analysis, as described below. All procedures involving mice were approved by The Ohio State University Institutional Animal Care and Use Committee (protocol # 2013A00000036-R2) and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
2.3. Dosage information / dosage regimen
For mice assigned to the NAR groups, 2% naringenin diets were consumed ad libitum 13–26 days prior to inoculation of C-26 cells or vehicle control for the remainder of the study (Day 13 post-inoculation). This experimental design addressed our hypothesis that consumption of naringenin in the diet would prevent the onset and progression of cancer cachexia. The dosage of naringenin chosen is based on our previous studies that determined that diets containing 1% and 3% naringenin were detectable in plasma, adipose and muscle tissues [18] and increased locomotor function [20]. Although difficult to achieve this amount of naringenin in the diet through food sources, it is possible through supplementation. However, this study is aimed to determine a proof of principle: whether naringenin supplementation has any beneficial effect against the detrimental effects of cancer cachexia development. For a mouse weighing 18g and consuming 2.5g of 2% naringenin diet per day, this equates to 2778mg of naringenin/kg body weight and a human equivalent dose of 226 mg/kg body weight [35]. For an individual with a body mass of 60kg, to achieve this dose they would need to consume 13.5g of naringenin per day.
2.4. EchoMRI for body composition
To assess time-course changes in body composition of live mice, EchoMRI (Houston, TX, USA) was used to measure lean body and adipose tissue masses at three timepoints: Days -1, 7, and 12 (1 day prior to necropsy).
2.5. Grip strength
Mice were acclimated to forelimb and hindlimb grip strength testing for 2 weeks prior to the first experimental measurement. Forelimb and hindlimb grip strength were measured on Day -1, 7, and 12 with the Columbus Instruments Grip Strength Meter (Columbus, OH, USA). Values reported represent the average of measurements taken in triplicate, with at least 5 minutes of rest between measurements for each mouse.
2.6. Fasting glucose and insulin-stimulated glucose clearance
Mice were moved to clean cages without food and to a quiet room for 6 hours prior to the start of the insulin tolerance test (ITT). The ITT was performed on Day 11 to determine the effect of naringenin diet and tumor inoculation on insulin-stimulated glucose clearance. Blood glucose was measured by pricking the tip of the tail with a 20-gauge needle. After measuring baseline blood glucose, mice were given a bolus of 0.75 U/kg insulin by intraperitoneal injection. Blood glucose was monitored using the OneTouch Ultra glucometer (Lifescan, Inc., Milpitas, CA, USA) at 0 (“fasting”), 15, 30, 45, 60, 90, and 120 minutes post-insulin injection.
2.7. IL-6, adiponectin and high molecular weight adiponectin
The concentration of plasma IL-6 (Invitrogen, Carlsbad, CA, USA), high molecular weight (HMW) and total adiponectin (Alpco, Salem, NH, USA) were measured by ELISA according to the manufacturer’s protocols.
2.8. Assessment of muscle fiber succinate dehydrogenase (SDH) activity
To determine the size and quantity of muscle fibers presenting with a more pronounced oxidative phenotype, freshly isolated quadriceps muscles were mounted in 7% gum tragacanth and snap-frozen in liquid nitrogen-cooled isopentane at necropsy. Ten μm transverse sections were acquired from frozen muscle tissue on a cryostat (Leica, Wetzlar, Germany). SDH activity of muscle fibers was evaluated using a colorimetric staining method. Briefly, 2–3 serial sections from each mouse were incubated in a working solution containing 1-methoxyphenzine methosulphate, nitroblue tetrazolium, disodium succinate, azide, EDTA, and PO4 for 15 minutes. To stop the reaction, slides were dipped in deionized water, air-dried in the dark at room temperature for 10 minutes, and mounted for imaging. Images of stained muscle sections were acquired using an Olympus IX71 microscope and cellSens Standard software (Center Valley, PA, USA). The cross-sectional area of each darkly stained SDH-positive muscle fiber was analyzed using ImageJ (National Institute of Health, Bethesda, MD, USA) by manually outlining each fiber section. A blinded evaluator grouped the results accordingly for data analysis and averaged each of the 2–3 sections per muscle prior to statistical analysis.
2.9. Real-time quantitative PCR
Approximately 50 mg of tissue from gastrocnemius muscle was cut on dry ice. Muscle RNA was isolated using qiazol reagent (Qiagen, Germantown, MD, USA) and reverse transcribed to cDNA (High Capacity DNA Archive Kit, Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cDNA was amplified by real-time quantitative PCR using pre-designed and validated primers under universal cycling conditions defined by Applied Biosystems (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The primers used were (4331182 Murf-1 Mm01185221_m1; Atrogin1 Mm00499523_m1; Bax Mm00432051_m1; Bcl-2 Mm00477631_m1; Bnip3 Mm01275600; Ppargc1a Mm01208835; Ppargc1b Mm00504730; Nrf1 Mm01135606 ). The thermocycling conditions were 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec, and 60°C for 1 min. Target gene expression was normalized to the endogenous control GAPDH (Product no.4352932E, Thermo Fisher Scientific, Inc., Waltham, MA, USA) in the same reaction and expressed as 2−ddct relative to the CON(−) group [36].
2.10. Statistical Analysis
Data are represented as the mean ± the standard error of the mean (SEM). Plasma adiponectin data were log-transformed for statistical analysis to reduce skewedness of the distribution. Differences between CON(−), NAR(−), CON(+) and NAR(+) groups were analyzed by 2-way analysis of variance (ANOVA) to determine the main effects of tumor, diet or their interaction. Group differences were distinguished using Sidak’s multiple comparisons test. A student’s t-test was utilized to determine the effect of diet on insulin-stimulated glucose clearance at each individual timepoint. All statistical tests were performed using STATA (StataCorp LLC, College Station, TX, USA) and graphed with Graphpad Prism (Graphpad Software, San Diego, CA, USA). All statistical tests were performed at the 5% significance level.
3. Results
3.1. Characteristics of the effects of tumor inoculation and naringenin-supplemented diet on mice.
Prior to inoculation, NAR mice began to exhibit reduced body weight compared to CON mice, which was apparent by Day 0 (day of inoculation) of the study (P=.006; Table 1). Post-inoculation (beginning at Day 0), while body weight increased over time for both control groups, NAR(−) mice had reduced body weight and developed lower cumulative food intake over the course of the study compared to CON(−) groups (Fig. 2a–b). On Day 13 (day of necropsy), there was no effect of diet on total body weight between CON(−) and NAR(−) groups (Fig. 2c); however, NAR(−) had reduced food intake compared to CON(−) mice (Fig. 2d; P<.05).
Table 1.
Characteristics of healthy and tumor-bearing mice fed either a control or naringenin-rich diet.
| Variable | No Tumor | Tumor | P-value | ||||
|---|---|---|---|---|---|---|---|
| Control | Naringenin | Control | Naringenin | Diet | Tumor | D x T | |
| Day 0 (day of inoculation) body weight (g) | 19.3 ± 0.3a | 20.3 ± 0.3b | - | - | .006 | - | - |
| Final tumor-free body weight (g) | 21.4 ± 0.4a | 20.2 ± 0.6a* | 18.0 ± 0.6b | 15.0 ± 0.3b* | .0001 | .0000 | .0791 |
| Cumulative food intake | 64.5 ± 1.9 a | 59.5 ± 1.4a* | 61.0 ± 1.5b | 52.1 ± 0.8b* | .0000 | .0008 | .2074 |
| Tumor weight (g) | - | - | 1.27 ± .06 | 1.05 ± .09 | .0685 | - | - |
| Tibia length (cm) | 1.65 ± .01 | 1.64 ± .01 | 1.66 ± .01 | 1.65 ± .01 | .3050 | .5612 | .9128 |
| Spleen weight (% tf-bw) | 0.24 ± .02a | 0.26 ± .01a | 0.82 ± .08b | 0.52 ± .04c | .0006 | .0000 | .0001 |
| Liver weight (%tf-bw) | 3.42 ± .12a | 3.66 ± .13a | 4.30 ± .27b | 3.93 ± .14b | .6953 | .0015 | .0823 |
| Quadriceps weight (% tf-bw) | 0.71 ± .01 | 0.71 ± .01 | 0.72 ±.01 | 0.71 ± .02 | .5453 | .5876 | .6063 |
| Tibialis anterior weight (% tf-bw) | 0.42 ± .01 | 0.44 ± .02 | 0.41 ± .04 | 0.44 ± .03 | .4569 | .9908 | .8078 |
| Gastrocnemius weight (%tf-bw) | 1.02 ±.01a | 1.03 ± .02b | 0.98 ± .02a | 1.09 ± .03b | .0226 | .4692 | .0668 |
| Inguinal adipose weight (%tf-bw) | 1.72 ±.11a | 1.25 ± .06a* | 0.82 ± .14b | 0.23 ± .05b* | .0000 | .0000 | .5461 |
| Epididymal adipose weight (%tf-bw) | 1.77 ± .18a | 1.29 ± .15a* | 1.09 ± .24b | 0.18 ± .11b* | .0012 | .0001 | .2672 |
| Brown adipose tissue weight (%tf-bw) | 1.63 ± .08a | 1.27 ± .08a* | 0.63 ± .14b | 0.36 ± .14b* | .0081 | .0000 | .6779 |
| Heart weight (%tf-bw) | 0.61 ± .02a | 0.59 ± .01a | 0.66 ± .04b | 0.65 ± .01b | .4387 | .0139 | .7415 |
| Plasma IL-6 (pg/mL) | - | - | 1059.4 ± 303.2a | 140.6 ± 40.19b | .0170 | ||
| LOG plasma total adiponectin (μg/mL) | 16.68 ± 1.0 a | 14.19 ± .44a* | 4.96 ± .24b | 4.14 ± .34b* | .0000 | .0046 | .6755 |
| LOG plasma HMW adiponectin (μg/mL) | 4.15 ± .36a | 3.55 ± .24ab | 3.51 ±.42b | 1.66 ±.16c | .0001 | .0000 | .0033 |
For each individual variable, a two-way ANOVA was conducted on the influence of the two independent variables (diet, tumor) on each outcome. P-values are bolded for significant effects (P<.05). Interaction effect were distinguished using Sidak’s multiple comparisons test. Significant differences between groups are highlighted with lowercase letters (a,b) and interaction effects are highlighted with an asterisk.
Fig. 2. Characteristics of the effects of tumor inoculation and naringenin-supplemented diet on male mice.
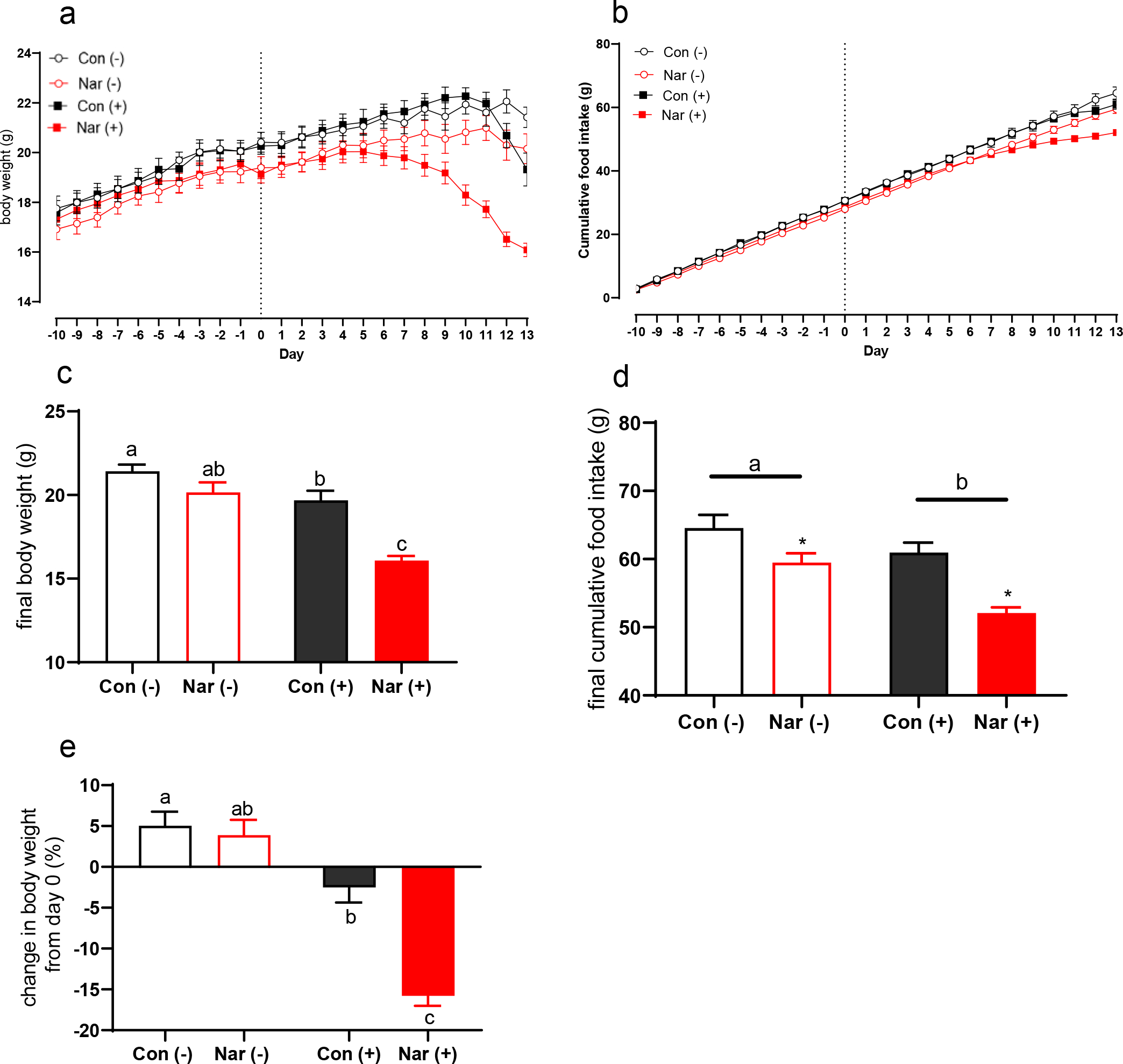
At study Day 0, mice were randomized to receive tumor or saline inoculation, resulting in four experimental groups: control diet saline CON(−), control diet tumor CON(+), naringenin diet saline NAR(−), and naringenin diet tumor NAR(+). (a) Daily body weights of all mice, beginning at diet randomization. Day 0 indicates the day of tumor or saline inoculation. (b) Cumulative food intake over time, beginning at day −10. (c) Final body weight in grams, including tumor mass. (d) Final cumulative food intake at Day 13 (day of necropsy). (e) Percent change in body weight from Day 0 (inoculation) to Day 13 at necropsy. Data are expressed as the mean ± SEM (n=10–12/group, unless otherwise indicated). (c-e) A two-way ANOVA was conducted on the influence of the two independent variables (diet, tumor) on each depicted outcome. Significant effects (P<.05) are shown in the graphs as follows: main effect of tumor is depicted with a bar above tumor and non-tumor groups; main effect of diet is depicted with an asterisk. Interaction effects were distinguished using Sidak’s multiple comparisons test; interaction effects between each individual group are depicted with differing letters (a,b) directly over bars.
Dietary naringenin caused early onset of body weight loss in the tumor group that preceded the onset of control tumor mice (Fig. 2a). NAR(+) mice began to exhibit a decline in body weight compared to non-tumor control mice at Day 6 post-inoculation. Body weight loss in the NAR(+) group corresponded with reduced cumulative food intake observed beginning at study Day 6 (Fig. 2b). On Day 13 (day of necropsy), there was an interaction effect of naringenin and tumor on final body weight: both tumor groups exhibited reduced body weight compared to their respective non-tumor bearing controls, while the reduction was significantly larger in mice fed the naringenin diet (−4g) than those fed the control diet (−2g; P<.05) .The final body weight (including tumor mass) of the NAR(+) mice was also about 4g lower on average compared to the CON(+) group (Fig. 2c; P<.05). Analysis of final cumulative food intake revealed that both tumor inoculation and naringenin diet caused a reduction in cumulative food intake. NAR(−) mice cumulatively consumed about 5g less than CON(−) mice, while NAR(+) consumed about 8g less food than CON(+) mice; both tumor groups had lower cumulative food intake than CON(−) mice (Fig. 2d; all P<.05). Compared to the CON(−) group, CON(+) had reduced food intake beginning on Day 11. By Day 13, NAR(+) mice developed cachexia as defined by >5% body weight loss compared to their body weight at Day 0 (inoculation), while the CON(+) mice did not, indicating a main effect of dietary naringenin in the tumor groups that accelerated loss of body weight from Day 0 (inoculation) to study termination (Fig. 2e; P<.05). On Day 13, NAR(+) mice exhibited greater than 15% body weight loss from Day 0 (inoculation), while CON(+) mice exhibited negligible change in body weight (Fig. 2e; P<.05)
Although NAR(+) mice exhibited an accelerated decline in body mass and food intake compared to CON(+) mice, they appeared bright and active (Suppl. Video S1–2). This is supported by the observation that while 3/11 CON(+) mice required early sacrifice due to clear lethargy and moribundity (78% survival), 12/12 NAR(+) mice survived to the day of necropsy (Fig. 1). These findings suggest that although the naringenin-supplemented diet accelerated the progression of body weight loss typically associated with cancer cachexia, the severity of body weight loss did not predict for earlier decline or worse outcomes.
3.2. Effects of tumor and dietary naringenin on body composition and grip strength.
By Day 7 post-inoculation, NAR(+) mice exhibited reduced adipose mass when expressed as a percent change from their initial adipose mass (Fig 3a; P<.05). By Day 12, NAR(+) mice exhibited a nearly 100% reduction in adipose mass as a percent of total body weight (Fig 3a; P<.05). This reduction of adipose mass was significantly greater than the 25% reduction of adipose mass in the CON(+) group (Figure 3a, P<.05). These results remained statistically significant when controlling for body mass at Day 0, adipose mass at Day 0, and cumulative food intake at Day 0 (data not shown). At Day 13 (day of necropsy), NAR (+) mice had significantly reduced inguinal, epididymal, and brown adipose tissue depots analyzed as a percent of tumor-free body weight compared to CON(+) mice (Table 1; all P<.05).
Fig. 3. Effect of tumor inoculation and naringenin-supplemented diet on body composition and grip strength in male mice.
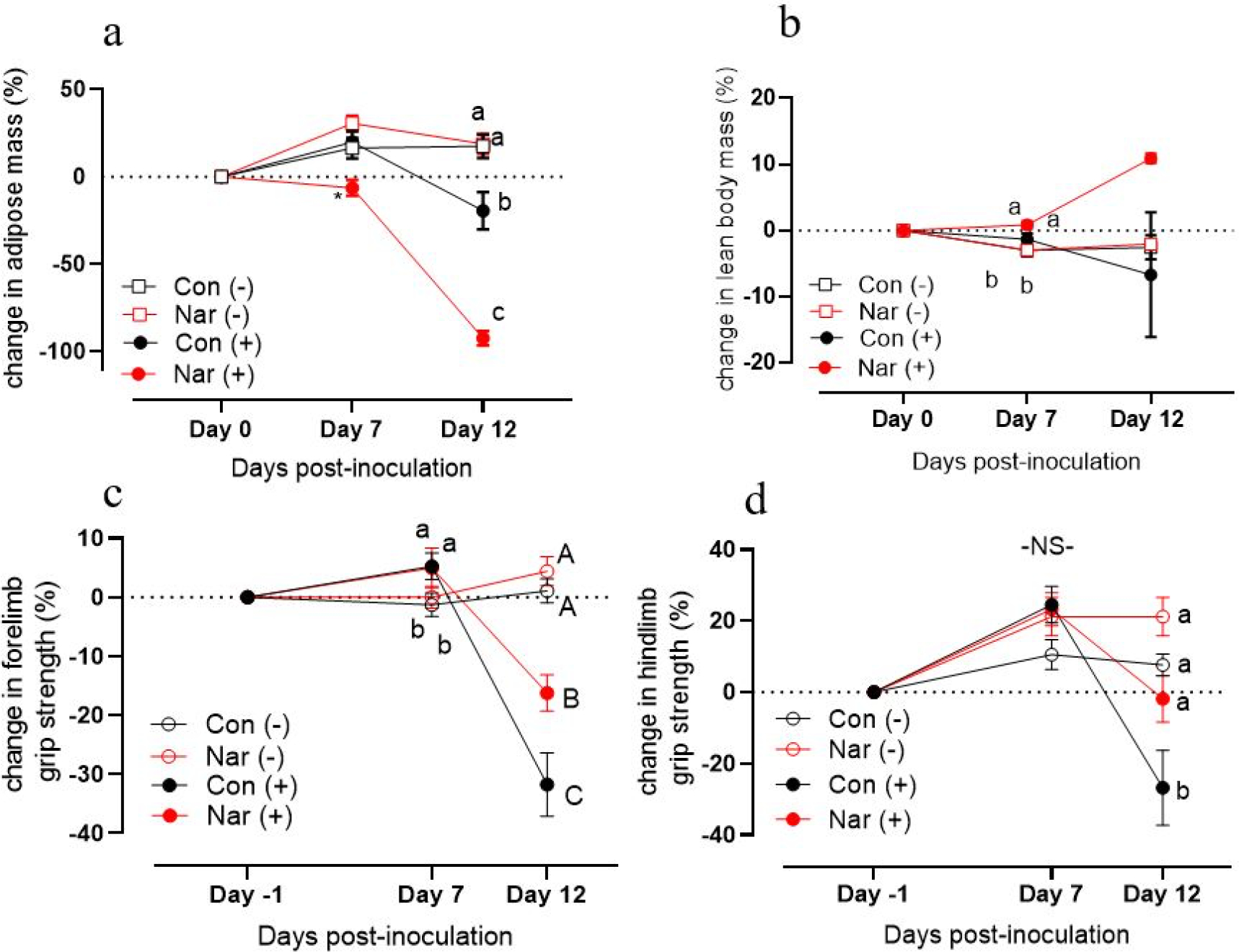
(a) Percent change from Day −1 measurements of adipose mass and (b) lean body mass over the experimental period, measured using EchoMRI. Percent change from Day −1 measurements of (c) forelimb and (d) hindlimb grip strength over the course of the experimental period. For each individual timepoint, a two-way ANOVA was conducted on the influence of the two independent variables (diet, tumor) on each outcome. Significant effects (p <.05) are shown in the graphs as follows: main effect of tumor is depicted with ‘a’ next to tumor(−) groups and ‘b’ next to tumor(+) groups. Interaction effects were distinguished using Sidak’s multiple comparisons test and are depicted with differing letters (a,b) over bars. (c) Capital letters (A,B) indicate interaction effects at Day 12.
Naringenin supplementation has previously been shown to exert positive metabolic effects on skeletal muscle [13, 20]. At Day 7, tumor-bearing mice had increased lean body mass compared to non-tumor groups (Fig. 3b), likely due to increased overall body mass from growth between Days 0–7 (Fig. 2a). At Day 7, tumor-bearing groups had higher grip strength than non-tumor-bearing groups (Fig. IIIc); however, CON(+) mice and NAR(+) mice were indistinguishable despite the decline in NAR(+) average body weight (Fig. 2a). At Day 13 (day of necropsy), NAR(+) mice exhibited increased gastrocnemius mass as a percent of tumor-free body weight when compared to CON(−) mice (Table 1). Raw tissue weights can be found in Supplementary Table S2.
A major predictor of the decline of quality of life and mobility in patients with cancer cachexia is loss of muscle strength and function [1]. To track changes in muscle function corresponding with body composition, grip strength was measured the day prior to inoculation (Day −1), Day 7, and Day 12. At Day 7, both tumor-bearing groups had greater forelimb grip strength than non-tumor bearing mice (Fig. 3c; P<05). By Day 12, there was a significant decrease in forelimb grip strength in both tumor-bearing groups (Fig. 3c; P<.05). However, naringenin supplementation appeared to attenuate loss of strength at this timepoint, despite significant loss of body mass indicative of a more severe cachexia phenotype (Fig. 3c). Hindlimb grip strength showed a more pronounced pattern: while CON(+) mice experienced a ~30% loss in hindlimb grip strength (P<.05), NAR(+) mice did not experience any decrease in grip strength and had similar maintenance of grip strength compared to both non-tumor controls despite their lower body weight (Fig. 3c). At Day 12, although tumor-bearing mice had decreased grip strength compared to non-tumorbearig controls, NAR(+) mice had an attenuated decline in grip strength compared to CON(+) mice. Grip strength measurements, both raw and standardized to body weight on Day 12 further demonstrate that NAR(+) mice exhibit improved grip strength compared to CON(+) mice despite their lower body mass (Suppl. Table S2). Taken together, the naringenin-rich diet attenuated the decline in grip strength during the progression of cancer cachexia.
3.3. Effects of tumor inoculation and dietary naringenin on insulin-stimulated glucose clearance.
We previously showed in models of metabolic disturbance that dietary naringenin supplementation reduced plasma glucose [20] and insulin [21]. To determine the effects of dietary naringenin and tumor burden on insulin-stimulated glucose clearance, we conducted an ITT on Day 11. In non-tumor bearing mice, naringenin diet lowered glucose levels at 2 hours-post insulin injection (Fig. 4a; P<.05), indicating greater insulin sensitivity. Similar to its effect in non-tumor bearing mice, diet had an effect on measures of insulin-stimulated glucose disappearance in tumor-bearing mice (Fig. 4b). CON(+) mice exhibited an increase in blood glucose after insulin injection with most significant differences at 15 and 30 minutes post-injection (Fig. 4b). Comparatively, NAR(+) mice exhibited an insulin-stimulated reduction in blood glucose levels that was indistinguishable from NAR(−) control mice and a significantly greater area under the curve for the change in glucose over time compared to CON(−) mice (Fig. 4b–c; P<.05). These results indicate that naringenin increases insulin sensitivity in both healthy and tumor-bearing mice.
Fig. 4. Impact of tumor inoculation and naringenin diet on insulin-stumulated glucose uptake in male mice.

On Day 11, mice received an intraperitoneal injection of insulin (0.75 U/kg body weight) and blood glucose was measured over a two-hour period. (a,b) Change in glucose over time for tumor and no-tumor mice, respectively. (c) Area under the curve (AUC) of change in glucose over time for all four groups. For each individual timepoint of the ITT glucose curves (a-b) a t-test was used to determine whether there was a significant effect of diet on ITT outcomes for tumor(−) and tumor(+) groups, respectively. Significant effects (P<.05) are shown in the graphs with an asterisk at each significant time point. A two-way ANOVA was conducted of the influence of the two independent variables (diet, tumor) on glucose AUC. Interaction effects are depicted with letters above each individual bar.
3.4. Effects of tumor and dietary naringenin on skeletal muscle oxidative cell type and markers of proteolysis, apoptosis, and mitochondrial biogenesis.
It is well-established in the C-26 model of cancer cachexia that the progression of the disease results in muscle loss, explained by the reduction of individual muscle fiber size [30, 34, 37]. To address skeletal muscle fiber size and the contribution of skeletal muscle to insulin-stimulated glucose clearance, we assessed quadriceps muscle SDH activity using a colorimetric staining technique on cryosectioned quadriceps muscle. While there were no significant differences between the darkly-stained SDH-positive fiber size distribution of mice fed a naringenin diet and their respective control groups (Fig. 5a–b), CON(+) and NAR(+) groups showed an increase in darkly-stained SDH-positive fibers counted (Fig. 5c; P<.05). This finding was likely due to the decrease in SDH-positive fiber cross-sectional area seen in both of the tumor-bearing groups (Fig. 5d; P<.05). Representative images from each group illustrate the decrease in muscle fiber size as well as the decrease in the size of SDH-positive fibers in the tumor groups (Fig. 5e).
Fig. 5. Effect of tumor inoculation and naringenin-supplemented diet on skeletal muscle oxidative cell types and markers of proteolysis, apoptosis, and mitochondrial biogenesis.

(a,b) Frequency distribution of darkly-stained SDH-positive muscle fibers in quadriceps muscle of non-tumor-bearing mice and tumor-bearing mice, respectively. (c) Average number of darkly-stained SDH-positive muscle fibers in quadriceps of mice. (d) Average cross-sectional area of darkly-stained SDH-positive muscle fibers in quadriceps of mice. (e) Representative images of transverse sections of quadriceps muscle stained for SDH activity, used to quantify darkly-stained SDH-positive fiber size. (f) Relative gene expression markers related to muscle wasting and mitochondrial activity; relative fold change was normalized to control no-tumor mice. For each fiber cross-sectional area bin and other experimental outcomes depicted in these graphs, a two-way ANOVA was conducted on the influence of the two independent variables (diet, tumor) on each outcome. Significant effects (P<.05) are shown in the graphs as follows: main effect of tumor is shown with a bar with ‘a’ or ‘b’ denoting the differences between tumor(+) and tumor(−) groups; main effect of diet is shown with an asterisk above each red bar.
Due to the contribution of skeletal muscle to whole-body energy metabolism during the progression of cancer cachexia, we measured several markers of cachexia progression and mitochondrial biogenesis by quantitative real-time PCR (Fig. 5f). Both tumor-bearing groups exhibited significantly elevated levels of mRNA of proteolytic degradation markers Murf1 and Atrogin1, with no effect of diet in tumor bearing mice (NAR(+) vs. CON(+)). Similar patterns were observed for pro-apoptotic marker Bax and anti-apoptotic marker Bcl-2. While naringenin supplementation increased marker of mitochondrial biogenesis Pgc1a expression in both NAR(−) and NAR(+) groups compared to their respective CON groups, both tumor-bearing groups had significantly higher Pgc1a expression than the non-tumor groups (Figure 5; P<.05). However, other mitochondrial genes did not follow a similar pattern: there was no significant effect of diet or tumor on Pgc1b gene expression, and Nrf-1 gene expression was significantly increased in all tumor-bearing mice irrespective of diet treatment. Overall, the effect of tumor burden had a greater effect than the naringenin-rich diet on gene expression of cachexia-related genes in quadriceps skeletal muscle (Figure 5f).
3.5. Impact of tumor inoculation and dietary naringenin on inflammatory and adiponectin response.
Adiponectin, an adipokine secreted from adipose tissue, has well-established roles in regulating insulin sensitivity and in reducing plasma free fatty acids, glucose, triacylglycerols [38–40] and pro-inflammatory cytokines [41]. Adiponectin is present in circulation in several multimeric forms; of these forms, high molecular weight (HMW) adiponectin is thought to have the greatest bioactivity in its relation to its aforementioned regulatory functions [38, 42]. In other models of metabolic disturbances, naringenin supplementation has yielded mixed reports about its ability to increase levels of circulating adiponectin [21, 43, 44]. Although we saw significant improvements in insulin sensitivity in NAR(+) mice compared to CON(+) mice, NAR(+) and CON(+) mice exhibited reduced levels of circulating total adiponectin (Fig. 6a) and HMW adiponectin (Fig. 6b). Interestingly, dietary naringenin sustained the HMW/Total adiponectin ratio which was similar to HMW/total adiponectin ratios in both diet groups of non-tumor-bearing mice (Fig. 6c).
Figure 6. Impact of tumor inoculation and naringenin-supplemented diet on elements of inflammatory response in male mice.

(a) Plasma total adiponectin. (b) Plasma high molecular weight adiponectin. (c) Ratio of plasma high molecular weight to total adiponectin. (d) Plasma IL-6 in tumor groups. Because non-tumor-bearing controls had IL-6 levels that were too low to be detected in the standard curve, results are not reported here. (e) Relative gene expression markers related to inflammation in quadriceps muscle. (f) Spleen mass as a percent of total body mass. A two-way ANOVA was conducted on the influence of the two independent variables (diet, tumor) on each outcome. Plasma adiponectin was log-transformed for statistical analyses; raw values are reported in graphs. Significant effects (P<.05) are shown in the graphs as follows: a main effect of tumor is shown with a bar with ‘a’ or ‘b’ denoting the differences between tumor(+) and tumor(−) group; main effect of diet is shown with an asterisk above each red bar; interaction effects are depicted with individual letters (a,b) over each bar. Interaction effects were distinguished using Sidak’s multiple comparisons test.
Naringenin has been previously reported to reduce plasma inflammatory cytokines in a model of metabolic disturbance [45]. In our study, Dietary naringenin suppressed the increase in plasma IL-6 levels in tumor bearing mice: NAR(+) mice exhibited a 5-fold reduction in plasma IL-6 (Fig. 6d; P<.05) compared to CON(+) mice. We also found that although tumor presence increased gene expression of pro-inflammatory markers TNFα and IL-6 in skeletal muscle, dietary naringenin did not alleviate these changes (Fig. 6e). Corresponding with our finding that plasma IL-6 was reduced in NAR(+) mice compared to CON(+) mice, while tumor presence elevated spleen mass, NAR(+) mice display a reduction in spleen mass compared to CON(+) mice (Figure 6f; P<.05). These data suggest the possibility that dietary naringenin alleviated the inflammatory burden on the spleen, which may have decreased plasma inflammation in response to tumor-induced physiological changes, as described by Bronte and Pittet [46].
4. Discussion
The aim of this study was to determine whether dietary naringenin supplementation could ameliorate the pathogenesis of cancer cachexia, through its characterized anti-inflammatory, insulin-sensitizing, and anti-cancer activities. Early-onset of anorexia and weight loss have long been considered predictors of poor prognosis in cancer cachexia in both experimental models and patients. In the current study, while NAR(+) mice exhibited accelerated anorexia (Fig. 2b,d) and loss of adipose mass (Fig. 3a), naringenin supplementation significantly reduced plasma IL-6 inflammation (Fig. 6d) and improved insulin sensitivity (Fig. 4b–c) compared to CON(+) mice. Even in non tumor-bearing mice, we observed an effect of naringenin diet on food intake (Fig. 2b,d). It is possible that mice perceived the diets as bitter, which may have contributed to reduced acceptability and lower food consumption. Furthermore, increased sensitivity to bitterness increases plasma TNF-α in tumor-bearing mice [47], and these factors may have contributed to the reduced food intake and body weight loss in NAR(+) group. This possibility suggests that naringenin may exert some of its effects on inflammation through neuromodulatory responses in addition to its direct activity in target tissues. The changes observed in body weight were not entirely explained by tumor burden or difference in growth: there was no significant difference in tumor mass or tibia length between diet groups (Table 1; P=.0685 and P=.3050, respectively). These changes may have protected mice from skeletal muscle wasting associated with advanced development of cancer cachexia symptoms. Further, the partial maintenance of muscle mass and strength (Fig. 3b–d) appeared to occur coincident with maintenance of mobility and alertness (Suppl. Videos S1–2), which may be indicative of improvements in end-stage quality of life.
The C-26 model is a well-characterized model of cancer cachexia driven by IL-6 inflammation [48, 49]. Transcriptional control of IL-6 is regulated by NFK κB-mediated pro-inflammatory pathways in activated macrophages, and these pro-inflammatory cytokines can exert their effects in an endocrine fashion by producing chronic systemic inflammation. Importantly, naringenin has been described as an inhibitor of pro-inflammatory pathways, as it reduces the levels of pro-inflammatory cytokines [50] . Many groups have identified that in a wide range of different cell types, including cancer cells, immune cells, adipocytes, and skeletal muscle naringenin regulates transcription, ultimately leading to a decrease in pro-inflammatory cytokine production [13, 29, 51–55]. In addition, others have shown that naringenin operates in a post-transcriptional fashion by promoting lysosome-mediated cytokine degradation [56]. In accordance to these prior findings, our results suggest that naringenin supplementation may cause reductions in systemic inflammation driven by the tumor in the C-26 model and prevent characteristic symptoms of cachexia that lead to poor prognosis. Indeed, in the current study, we identified that a 2% naringenin diet reduced plasma IL-6 levels and spleen mass in tumor-bearing mice (Figure 6), indicating a less robust systemic pro-inflammatory response when compared to tumor-bearing control mice. However, persistent IL-6 inflammation is known to reduce food intake and body weight [57, 58]: In the current study, naringenin reduced systemic IL-6 (Fig. 6d), but also reduced food intake and body weight (Fig. 2a–b). This suggests two possibilities: that IL-6 inflammation has a certain threshold at which it causes these changes, and/or that dietary naringenin reduces food intake and body weight through a mechanism independent of IL-6 inflammation. The latter is supported by our findings in previous studies [18, 21] and here (Fig. 2c–d) where naringenin reduced food intake and body weight in healthy [non-tumor bearing] mice, which had undetectable levels of circulating IL-6.
Another intriguing possibility is that a significant portion of IL-6 is produced by adipose tissue during the progression of the disease in this model. In the current study, NAR(+) mice had reduced adiposity (Fig. 3a) and plasma IL-6 levels (Fig. 6d). in patients with cancer cachexia, plasma IL-6 is positively correlated to free fatty acids in the blood, suggesting increased adipose tissue lipolysis in both early and late-stage cachexia [59]. Furthermore, treatment of IL-6 antibody in mice with cachexia prevents the reduction of adipocyte size and beiging, indicating that increases in IL-6 contribute to these pathological changes to adipose tissue [59]. Indeed, a recent study highlights the contributions of inflammation from adipose tissue to cachexia progression: in a in vitro model, palmitate and IL-6 added to media to replicate inflammation and lipolysis from adipose tissue induced wasting in C2C12 myotubes [60].
In our study, we observed that NAR(+) mice had an accelerated rate of adipose tissue depletion, compared with CON(+) mice (Fig. 3a). These findings paired with the evidence cited above suggest the alternative possibility that elevated inflammation during cancer cachexia may have slowed the rate of adipose wasting, despite the detrimental effects inflammation has on other disease outcomes in this model. Conversely, dietary naringenin, through its ability to reduce inflammation, may have accelerated adipose tissue wasting. Therefore, although unfavorable to maintenance of adiposity in cancer cachexia, naringenin likely exerted effects on muscle independent of effects on adipose tissue. Although NAR(+) mice exhibited more rapid weight loss attributed to accelerated wasting of adipose tissues (Fig. 3a), these mice were bright, alert, active (Suppl. Video S1–2), and maintained muscle strength (Fig. 3c–d). In contrast, at comparable timepoints CON(+) mice were lethargic and appeared very ill despite being larger in body weight than NAR(+) mice.
There is growing evidence that insulin resistance is an early and contributing factor to cancer cachexia progression [61, 62]. Additionally, our findings highlight the important relationship between inflammatory responses and regulation of insulin sensitivity. Through several pro-inflammatory pathways, macrophages can release inflammatory mediators, including IL-6, that inhibit insulin signaling [12] and are thought to contribute to the development of insulin resistance. Naringenin may increase glucose uptake into skeletal muscle by reducing IL-6 via AMPK-dependent signal transduction events downstream of the insulin receptor, simultaneously improving insulin sensitivity and whole-body energy metabolism in peripheral tissues [13]. The beneficial effects of dietary naringenin supplementation in models of dysregulated energy metabolism have been well-described and corroborated by studies in our lab and others [16, 18–21]. We previously identified that naringenin supplementation reduced adipose mass and inflammation in a mouse model of postmenopausal breast cancer [18]. Our current findings demonstrate that while CON(+) mice exhibited insulin resistance previously well-characterized in C-26 mice, NAR(+) mice had increased insulin sensitivity that was comparable to non-tumor-bearing CON(−) and NAR(−) mice (Fig. 5). In fact, even healthy NAR(−) mice exhibited improved insulin response when compared to healthy mice fed the control diet, suggesting that the effect of dietary naringenin on insulin-stimulated glucose uptake is greater than that of the tumor burden (Fig. 5). Measurement of muscle-specific insulin sensitivity is worth pursuing in a future study using a multistage hyperinsulinemic-euglycemic clamp.
The loss of skeletal muscle mass is an important characteristic in the definition of cancer cachexia and its progression, and leads to faster decline, decreased mobility, and lethargy in patients [1]. Importantly, in skeletal muscle, insulin signaling regulates energy substrate utilization as well as skeletal muscle degradation in the presence of a catabolic energy state [63]. Furthermore, in prior work from our lab, treatment of C-26 tumor-bearing mice with the insulin-sensitizing drug thiazolidinedione attenuated the loss of body weight and skeletal muscle mass [61]. In the current study, NAR(+) mice exhibited rapid adipose depletion compared to CON(+) mice (Fig. 3), but surprisingly did not undergo any loss of skeletal muscle. In fact, while CON(+) mice displayed loss of strength previously characterized in the C-26 model [34, 37], NAR(+) mice maintained grip strength that was indistinguishable from non-tumor bearing controls (Fig 3c–d). Interestingly, dietary naringenin was not protective against the reduction of skeletal muscle fiber size (Fig. 5e) and markers of proteolytic degradation caused by the presence of the tumor (Fig. 5f). However, mRNA of PGC-1α, a marker of mitochondrial biogenesis, was significantly increased in the NAR(+) group (Fig. 5f), indicating the possibility that naringenin stimulated the maintenance of skeletal muscle mass by replenishing mitochondria. Together with our findings that NAR(+) mice have reduced IL-6 inflammation and improved insulin sensitivity, this suggests that inflammation is strongly linked to insulin resistance in cancer cachexia, and that restoring insulin sensitivity and muscle mitochondrial function are key factors that preserve skeletal muscle function in the disease.
In addition to the preservation of insulin sensitivity, muscle strength, and improved inflammatory status, naringenin improved survival in the C-26 model, suggested by the 100% (12/12) survival of NAR(+) mice on Day 13 in comparison to 73% (8/11) of CON(+) mice (Figure 1). We visually observed that the locomotor activity of NAR(+) mice was greater than that of CON(+) mice; the CON(+) group appeared lethargic and immobile (Suppl. Videos S1–2). The maintained activity and alertness in NAR(+) mice [vs. CON(+) mice] aligns with past quantitative measures of increased locomotor activity in naringenin-fed mice conducted in our laboratory [20]. Our observations suggest the possibility that naringenin may have effects on the brain in models of metabolic disturbances. Similar to its effect in other tissues, naringenin acts to resolve inflammation in the brain by preventing pro-inflammatory cytokine release, as well as by blocking other markers of pro-inflammatory response [64]. Taken together, the data presented here highlight the need for future studies investigating the effect of naringenin on metabolic regulation in the brain during the development of cancer cachexia.
Despite the current study’s findings, there are several limitations to consider. Although the current dose of naringenin used has been demonstrated in the literature to have beneficial metabolic, anti-inflammatory, and anti-cancer effects, it is important to note that this dose is utilized to overcome the low bioavailability of naringenin. We determined in a prior study that diets containing 1–3% naringenin resulted in its detection in plasma, liver, adipose tissues and muscle [20]. Importantly, many of these tissues serve important metabolic regulatory roles during the progression of cancer cachexia. Therefore, this study served as a proof of principle to determine whether naringenin might attenuate cancer cachexia progression, and future studies must be conducted to explore means of improving naringenin bioavailability. Additionally, it is difficult to differentiate the effects of naringenin consumption versus decreased food intake based on the current study design. However, when controlling for food intake, the effects of naringenin are still significant for many experimental outcomes. Therefore, a future study regimen that includes pair-feeding would help to clarify the effects of naringenin consumption on cancer cachexia development. Finally, in the current study we utilized grip strength to quantify muscle strength. It is important to acknowledge that grip strength is regarded as a behavioral test that emcompasses both strength and willpower. This measurement was chosen for the current study because in addition to strength, fatigue is an important characteristic of cancer cachexia to consider in both animal models and patients. However, this does not directly address the question of whether naringenin supplementation improves muscle strength. Therefore, future studies should include more quantitative measurements of muscle strength and fatigue, such as single fiber force measurements and treadmill exhaustion tests to more adequately describe the effect of naringenin supplementation on muscle function in cancer cachexia.
In conclusion, in the C-26 model of cancer cachexia, an AIN-93G diet containing 2% dietary naringenin improves multiple aspects of metabolic disturbances commonly observed during cancer cachexia development. Most notably, dietary naringenin improved muscle strength, attenuated the decline in insulin sensitivity, and improved inflammatory status. These findings establish a strong link between inflammation and insulin resistance as drivers of the progression of cancer cachexia and provide us with further understanding of how dysregulated metabolism impacts the loss of muscle strength and mass. Importantly, increased rate of adipose tissue loss was not predictive of skeletal muscle wasting or overall survivorship in tumor-bearing mice fed a diet containing 2% naringenin. These observations suggest the possibility that the improvement of metabolic status may be a more valuable measure of prognosis than retention of adipose mass. Therefore, we conclude that through its ability to improve metabolic and inflammatory status and voluntary strength, naringenin may be a useful phytotherapeutic tool for preventing the progression of cancer cachexia. Based on our findings, future studies are warranted to determine the mechanism by which naringenin action improves disease outcomes durig the progression of cancer cachexia.
Supplementary Material
Acknowledgements
The authors thank Dr. Erin Talbert and Dr. Taylor Banh for technical support and scholarly discussions about the data and Dr. Denis Guttridge for supplying the C-26 cell line for our experiments. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Molecular Nutrition & Food Research.
DBS assised with study design and execution, and was responsible for data acquisition and analyses, interpretation of results, and manuscript preparation. YN led study execution and contributed to data acquisition, produced preliminary results and literature review that lead to further analyses and interpretation of data, and contributed to manuscript writing and editing. RMC assisted with study execution, data acquisition, analyses and interpretation of results, manuscript writing and editing. AN contributed to statistical interpretation of study design, oversaw and mentored DBS to ensure that the appropriate statistical methods were used and described for data analyses, contributed to manuscript writing and editing. ARA sssisted with study execution, and contributed to interpretation of data, manuscript writing and editing. YV was co-investigator, contributed to contributed to acquisition of funding and experimental hypotheses and design. MAB was co-investigator, contributed to acquisition of funding, experimental hypotheses and design, managed experimental execution, and contributed to manuscript preparation, discussion and interpretation of results. The authors have no conflicts to disclose.
This research was supported by NIHR21CA185140. Additional support was provided by the Ohio Agricutlure Research and Development Center, OSU’s Center for Advancement of Functional Foods Research and Entrepreneurship. Support for DS was provided by the American Oil Chemists’ Society Thomas H. Smouse Memorial Fellowship and The Ohio State University Center for Muscle Disease and Neuromuscular Disorders Predoctoral Fellowship.
Abbreviations:
- C-26
colon-26 mouse model
- CON
mice fed a control diet
- NAR
mice fed a naringenin diet
- (−)
non-tumor-bearing mice
- (+)
tumor-bearing mice
- ITT
insulin tolerance test
- SDH
succinate dehydrogenase
- HMW
high molecular weight
- ANOVA
analysis of variance
5. References
- [1].Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, and Baracos VE, Lanclet Oncol, 2011, 12, 489–95. [DOI] [PubMed] [Google Scholar]
- [2].Argiles JM, Busquets S, Stemmler B, and Lopez-Soriano FJ, Nat Rev Cancer, 2014, 14, 754–62. [DOI] [PubMed] [Google Scholar]
- [3].Williams A, Sun X, Fischer J, and Hasselgren P, Surgery, 1999, 126, 744–750. [PubMed] [Google Scholar]
- [4].Tisdale MJ, Physiol Rev, 2009, 89, 381–410. [DOI] [PubMed] [Google Scholar]
- [5].Dodson S, Baracos VE, Jatoi A, Evans WJ, Cella D, Dalton JT, and Steiner MS, Annu Rev Med, 2011, 62, 265–79. [DOI] [PubMed] [Google Scholar]
- [6].Du L, Yang YH, Wang YM, Xue CH, Kurihara H, and Takahashi K, Food Funct, 2015, 6, 3652–62. [DOI] [PubMed] [Google Scholar]
- [7].Yilma AN, Singh SR, Morici L, and Dennis VA, Mediators Inflamm, 2013, 2013, 102457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang P, Han J, and Hui L, Protein Cell, 2010, 1, 218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Manchope MF, Calixto-Campos C, Coelho-Silva L, Zarpelon AC, Pinho-Ribeiro FA, Georgetti SR, Baracat MM, Casagrande R, and Verri WA Jr., PLoS One, 2016, 11, e0153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ruiz-Miyazawa KW, Borghi SM, Pinho-Ribeiro FA, Staurengo-Ferrari L, Fattori V, Fernandes GSA, Casella AM, Alves-Filho JC, Cunha TM, Cunha FQ, Casagrande R, and Verri WA, Journal of Functional Foods, 2018, 48, 106–116. [Google Scholar]
- [11].Umukoro S, Kalejaye HA, Ben-Azu B, and Ajayi AM, Biomed Pharmacother, 2018, 105, 714–723. [DOI] [PubMed] [Google Scholar]
- [12].Lackey DE and Olefsky JM, Nat Rev Endocrinol, 2016, 12, 15–28. [DOI] [PubMed] [Google Scholar]
- [13].Zygmunt K, Faubert B, MacNeil J, and Tsiani E, Biochem Biophys Res Commun, 2010, 398, 178–83. [DOI] [PubMed] [Google Scholar]
- [14].Ortiz-Andrade RR, Sanchez-Salgado JC, Navarrete-Vazquez G, Webster SP, Binnie M, Garcia-Jimenez S, Leon-Rivera I, Cigarroa-Vazquez P, Villalobos-Molina R, and Estrada-Soto S, Diabetes Obes Metab, 2008, 10, 1097–104. [DOI] [PubMed] [Google Scholar]
- [15].Priscilla DH, Roy D, Suresh A, Kumar V, and Thirumurugan K, Chem Biol Interact, 2014, 210, 77–85. [DOI] [PubMed] [Google Scholar]
- [16].Pu P, Gao DM, Mohamed S, Chen J, Zhang J, Zhou XY, Zhou NJ, Xie J, and Jiang H, Arch Biochem Biophys, 2012, 518, 61–70. [DOI] [PubMed] [Google Scholar]
- [17].Purushotham A, Tian M, and Belury MA, Mol Nutr Food Res, 2009, 53, 300–307. [DOI] [PubMed] [Google Scholar]
- [18].Ke JY, Banh T, Hsiao YH, Cole RM, Straka SR, Yee LD, and Belury MA, Mol Nutr Food Res, 2017, 61, [DOI] [PubMed] [Google Scholar]
- [19].Mulvihill EE, Allister EM, Sutherland BG, Telford DE, Sawyez CG, Edwards JY, Markle JM, Hegele RA, and Huff MW, Diabetes, 2009, 58, 2198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ke JY, Cole RM, Hamad EM, Hsiao YH, Cotten BM, Powell KA, and Belury MA, Mol Nutr Food Res, 2016, 60, 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ke JY, Kliewer KL, Hamad EM, Cole RM, Powell KA, Andridge R, Straka SR, Yee LD, and Belury MA, Nutr Metab, 2015, 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].De Stefani E, Boffetta P, Oreggia F, Brennan P, Ronco A, Deneo-Pellegrini H, and Mendilaharsu M, Int J Cancer, 2000, 87, 129–132. [DOI] [PubMed] [Google Scholar]
- [23].Sun L, Subar AF, Bosire C, Dawsey SM, Kahle LL, Zimmerman TP, Abnet CC, Heller R, Graubard BI, Cook MB, and Petrick JL, J Nutr, 2017, 147, 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Freedman ND, Park Y, Subar AF, Hollenbeck AR, Leitzmann MF, Schatzkin A, and Abnet CC, Int J Cancer, 2007, 121, 2753–60. [DOI] [PubMed] [Google Scholar]
- [25].Li WQ, Kuriyama S, Li Q, Nagai M, Hozawa A, Nishino Y, and Tsuji I, Int J Cancer, 2010, 127, 1913–22. [DOI] [PubMed] [Google Scholar]
- [26].Kanno S, Tomizawa A, Hiura T, Osanai Y, Shouji A, Unbe M, Ohtake T, Kimura K, and Ishikawa M, Biol Pharm Bull, 2005, 28, 527–530. [DOI] [PubMed] [Google Scholar]
- [27].Harmon AW and Patel YM, Breast Cancer Res Treat, 2004, 85, 103–110. [DOI] [PubMed] [Google Scholar]
- [28].Manthey JA and Guthrie N, J. Agric. Food Chem, 2002, 50, 5837–5843 [DOI] [PubMed] [Google Scholar]
- [29].Lim W, Park S, Bazer FW, and Song G, J. Cell. Biochem. 2017, 118, 1118–1131. [DOI] [PubMed] [Google Scholar]
- [30].Talbert EE, Metzger GA, He WA, and Guttridge DC, J Cachexia Sarcopenia Muscle, 2014, 5, 321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tanaka Y, Eda H, Tanaka T, Udagawa T, Ishikawa T, Horii I, Ishitsuka H, Kataoka T, and Taguchi T, Cancer Res, 1990, 50, 2290–2295. [PubMed] [Google Scholar]
- [32].Bonetto A, Kays JK, Parker VA, Matthews RR, Barreto R, Puppa MJ, Kang KS, Carson JA, Guise TA, Mohammad KS, Robling AG, Couch ME, Koniaris LG, and Zimmers TA, Front. Physiol, 2016, 11, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Talbert EE, Yang J, Mace TA, Farren MR, Farris AB, Young GS, Elnaggar O, Che Z, Timmers CD, Rajasekera P, Maskarinec JM, Bloomston M, Bekaii-Saab T, Guttridge DC, and Lesinski GB, Mol Cancer Ther, 2017, 16, 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Banh T, Snoke D, Cole RM, Angelotti A, Schnell PM, and Belury MA, Oncol Rep, 2019, 41, 2909–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nair AB and Jacob S, J. Basic Clin. Pharm, 2016, 7, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Livak KJ and Schmittgen TD, Methods, 2001, 25, 402–8. [DOI] [PubMed] [Google Scholar]
- [37].Murphy KT, Chee A, Trieu J, Naim T, and Lynch GS, Dis Model Mech, 2012, 5, 533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, and Tobe K, J Clin Invest, 2006, 116, 1784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kubota N, Terauchi Y, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, and Noda T, J Biol Chem, 2002, 277, 25863–6. [DOI] [PubMed] [Google Scholar]
- [40].Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, and Kadowaki T, Nat Med, 2001, 8, 941–946. [DOI] [PubMed] [Google Scholar]
- [41].Wolf AM, Wolf D, Rumpold H, Enrich B, and Tilg H, Biochem Biophys Res Commun, 2004. 323, 630–635. [DOI] [PubMed] [Google Scholar]
- [42].Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, and Palin MF, Endocrine, 2010, 37, 11–32. [DOI] [PubMed] [Google Scholar]
- [43].Liu L, Shan S, Zhang K, Ning ZQ, Lu XP, and Cheng YY, Phytother Res, 2008, 22, 1400–3. [DOI] [PubMed] [Google Scholar]
- [44].Den Hartogh DJ and Tsiani E, Biomolecules, 2019, 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Al-Rejaie SS, Aleisa AM, Abuohashish HM, Parmar MY, Ola MS, Al-Hosaini AA, and Ahmed MM, Neurol Res, 2015, 37, 924–33. [DOI] [PubMed] [Google Scholar]
- [46].Bronte V and Pittet MJ, Immunity 2013, 14, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Feng P, Jyotaki M, Kim A, Chai J, Simon N, Zhou M, Bachmanov AA, Huang L, and Wang H, Brain Behav Immun, 2015, 49, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Strassmann G, Fong M, Kenney JS, and Jacob CO, J Clin Invest, 1992, 89, 1681–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fujita J, Tsijinaka T, Yano M, Ebisui C, Saito H, Katsume A, Akamatsu K, Ohsugi Y, Shiozaki H, and Monden M, Int J Cancer, 1996, 68, 637–43. [DOI] [PubMed] [Google Scholar]
- [50].Zeng W, Jin L, Zhang F, Zhang C, and Liang W, Pharmacol Res, 2018, 135, 122–126. [DOI] [PubMed] [Google Scholar]
- [51].Yoshida H, Watanabe W, Oomagari H, Tsuruta E, Shida M, and Kurokawa M, J Nutr Biochem, 2013, 24, 1276–1284. [DOI] [PubMed] [Google Scholar]
- [52].Liu X, Wang N, Fan S, Zheng X, Yang Y, Zhu Y, Lu Y, Chen Q, Zhou H, and Zheng J, Sci Rep, 2016, 6, 39735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Eanes L and Patel YM, Biochim Open, 2016, 3, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hatkevich T, Ramos J, Santos-Sanchez I, and Patel YM, Exp Cell Res, 2014, 327, 331–9. [DOI] [PubMed] [Google Scholar]
- [55].Nie YC, Wu H, Li PB, Xie LM, Luo YL, Shen JG, and Su WW, Eur J Pharmacol, 2012, 690, 207–13. [DOI] [PubMed] [Google Scholar]
- [56].Jin L, Zeng W, Zhang F, Zhang C, and Liang W, J Immunol, 2017, 199, 3466–3477. [DOI] [PubMed] [Google Scholar]
- [57].Franckhauser S, Elias I, Rotter Sopasakis V, Ferré T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F, and Smith U, Diabetologia, 2008, 51, 1306–16. [DOI] [PubMed] [Google Scholar]
- [58].Mishra D, Richard JE, Maric I, Porteiro B, Häring M, Kooijman S, Musovic S, Eerola K, López-Ferreras L, Peris E, Grycel K, Shevchouk OT, Micallef P, Olofsson CS, Wernstedt Asterholm I, Grill HJ, Nogueiras R, and Skibicka KP, Cell Reports, 2019, 26, 3011–3026.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Han J, Meng Q, Shen L, and Wu G, Lipids Health Dis, 2018, 17, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rupert JE, Narasimhan A, Jengelley DHA, Jiang Y, Liu J, Au E, Silverman LM, Sandusky G, Bonetto A, Cao S, Lu X, O’Connell TM, Liu Y, Koniaris LG, and Zimmers TA, J Exp Med, 2021, 218, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Asp ML, Tian M, Kliewer KL, and Belury MA, Cancer Biol Ther, 2011, 12, 957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Asp ML, Tian M, Wendel AA, and Belury MA, Int J Cancer, 2010, 126, 756–63. [DOI] [PubMed] [Google Scholar]
- [63].Honors MA and Kinzig KP, J Cachexia Sarcopenia Muscle, 2012, 3, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wu LH, Lin C, Lin HY, Liu YS, Wu CY, Tsai CF, Chang PC, Yeh WL, and Lu DY, Mol Neurobiol, 2016, 53, 1080–1091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.