

Intimal hyperplasia is widely studied because of its role in cardiovascular pathology. Pervasive proliferative vascular diseases such as atherosclerosis, bypass-graft failure, and coronary restenosis result from neointimal formation,1–3 in conjunction with comorbidities including diabetes, hypercholesterolemia, hypertension, and smoking. Each of these disease processes share a common pathway: narrowing of the arteries, reduced blood flow, and subsequent ischemia. The proliferation of vascular smooth muscle cells (vSMCs) and their migration to the intima have been shown to contribute to this pathway.4,5 Thus, elucidating the molecular mechanism underlying vSMC regulation may lead to the identification of pharmacologic targets for the treatment of these disorders.
The neointima is composed of vSMCs, although the origin of these cells remains unclear.1,3 It is well established that vSMCs of the ascending aorta have embryologic origins from the secondary heart field (SHF) and the cardiac neural crest (NC); however, their relative contribution to neointima formation is not well understood.2,6 vSMCs from the luminal side of the ascending aortic wall are derived from the NC (NC-SMCs), whereas vSMCs near the outer layer of the aortic wall originate from the SHF (SHF-SMCs).6 NC-SMCs have more potential to contribute to intimal hyperplasia in the absence of the elastin-induced inhibition of vSMC proliferation, because of their proximity to the intima.6 However, the contribution of SHF-SMCs to intimal hyperplasia is not known. Contractile vSMCs transform into a synthetic phenotype, which plays a key role in hyperplasia.5 In addition, cell studies using elastin-null murine models have shown that elastin can inhibit the proliferation of vSMCs and that it may act through direct G-protein–coupled signaling to induce a mature, contractile phenotype in vSMCs.2 This mechanism remains to be verified in animal models, and the effect of elastin integrity on SMC phenotypes and neointimal formation is not completely clear.7
In this issue of Arteriosclerosis, Thrombosis, and Vascular Biology, Lin et al.8 provide compelling evidence showing the contribution of SHF-SMCs to the neointima. The authors demonstrated that, in elastin-deficient mice, the neointima starts to build up in the ascending aorta shortly after birth. Using the lineage-tracing ROSA26mT/mG reporter with TagInCre for identifying all vSMCs, Wnt1Cre for NC-SMCs, and Isl1Cre for SHF-SMCs, they showed that both SHF- and NC-derived cells compose the neointima, with SHF-derived cells dominating the cell population.
Lin et al.8 also examined the effect of cell-specific elastin deficiency on neointima formation by using TaglnCre;Elnf/f (elastin-null in all vSMCs), Wnt1Cre;Elnf/f (elastin-null in NC-SMCs), and Isl1Cre;Elnf/f (elastin-null in SHF-SMCs). This series of investigations revealed that neointimal hyperplasia developed where there were breaches in the internal elastic lamina (IEL), supporting that elastic fragmentation facilitates neointimal hyperplasia. Furthermore, in a subgroup of Wnt1Cre;Elnf/f mice, they identified a fragmented IEL without neointima formation, implying that extracellular matrix (ECM) breakdown, while necessary, is not sufficient for inducing intimal hyperplasia (Figure). This finding is in concordance with a prior study showing that the neointima forms at the site of IEL disruption.9
Figure.
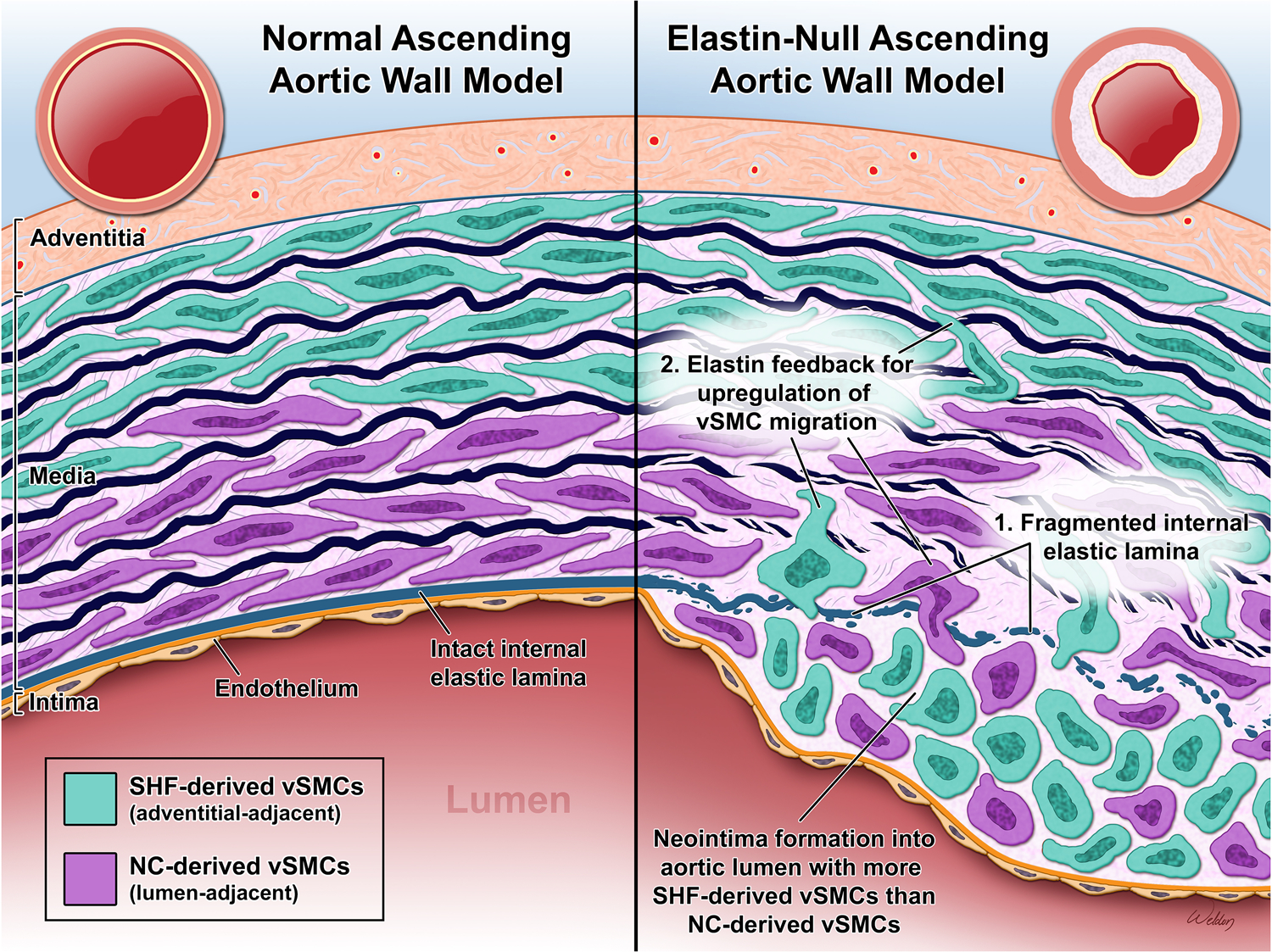
NC-SMCs are shown in the inner portion of the media with SHF-SMCs in the outer segment. Compared to normal, an elastin-null ascending aorta model has a fragmented IEL. The loss of a physical barrier as well as the absence of elastin feedback inhibiting vSMC migration allows vSMCs to migrate to the intima. There is an excess of SHF-derived vSMCs compared to NC-derived vSMCs in the neointima, despite their relative distances. The vSMCs lose their characteristic elongated shape and contractile properties during the phenotypic change that allows them to migrate and proliferate in the intima.
In a separate analysis of TagInCre;Elnf/f, Wnt1Cre;Elnf/f, and Isl1Cre;Elnf/f mice, the authors identified phenotypic differences in the formation of the IEL and neointima. Isl1Cre;Elnf/f mice developed medial hyperplasia, with neointimal cells of SHF origin. In contrast, Wnt1Cre;Elnf/f mice experienced premature death secondary to heart failure from preductal coarctation. Interestingly, the neointima was absent in two-thirds of these mice, implying that either the neointima requires both embryonic cell lines to develop completely (albeit, the SHF-derived cells may play a more important role), or there are other unknown secondary regulators of neointimal formation in the ECM.
To further elucidate the gene expression profiles and phenotypic features of vSMCs in the neointima, as well as the effects of elastin fragmentation on vSMC phenotypes, the authors performed single-cell RNA sequencing analyses on the ascending aortas of TagInCre;Elnf/f and control mice. Three distinct clusters of vSMC populations were identified in the aortic wall: contractile vSMCs (SMC1), myofibroblast vSMCs (SMC2), and proliferative vSMCs (SMC3). Compared with the contractile cluster, myofibroblast and proliferative vSMCs demonstrated less contractile phenotypes but increased ECM production. Whereas SMC1 cells were dispersed through the media, SMC2 and SMC3 subpopulations were exclusively seen in the neointima.
Additionally, the authors examined the effects of elastin deficiency on vSMC phenotypes. TaglnCre;Elnf/f mice showed upregulated ECM production and collagen formation in SMC1 and SMC2 compared with these clusters in Elnf/f and Elnf/+ mice, suggesting that elastin deletion in vSMCs promotes their transition from a contractile phenotype to an ECM-producing phenotype. The authors attributed this to a reactionary response to the loss of elastin, whereby the elastin loss initiates transcriptional and phenotypic changes in vSMCs, causing them to migrate to the intima and proliferate. Their study findings indicate that the compromise in elastin integrity plays a role in the initiation of vSMC migration and hyperplasia, but the exact mechanism and the role of other ECM components in the process are unclear.
Overall, this study serves to further knowledge regarding the origin of vSMCs, the role of elastin in the regulation of vSMCs, and the relation of vSMCs to intimal hyperplasia. The authors presented a novel isolation of elastin-null NC-SMCs and SHF-SMCs in a murine model to assess phenotypic changes. Their study demonstrates that the loss of elastin is not only essential to phenotypic change, but also to migration. This is a key distinction from prior studies that focus on NC-SMCs populating the neointima, illustrating that there is a variety of noncontractile vSMCs of different lineages that compose the neointima.
Caveats to consider when interpreting these findings include the use of a TagInCre driver, which may show other non-vSMCs or adventitial cells that have differentiated into noncontractile vSMCs. In addition, the lineage-tracing method for single-cell RNA sequencing analysis cannot be applied retroactively to back trace the cellular origins of neointimal cells as from either SHF-SMCs or NC-SMCs. Most importantly, it should be noted that the potential for neointimal formation cannot be directly compared between the Wnt1Cre;Elnf/f and Isl1Cre;Elnf/f murine models because of the relative complexities of the Isl1 and Wnt1 genes and their roles in IEL synthesis.
The finding of NC- and SHF-derived cells in the neointima limits this mechanism to the ascending aorta and arch, where these cells are predominantly found. This may limit the clinical utility of the derived mechanism to congenital cardiac disorders, such as supravalvular aortic stenosis, unless other neointimal vSMC origins are isolated. Studies focusing on vSMCs arising from pro-epicardial, somatic, splanchnic mesodermal, and nephrogenic stromal origins will be important for exploring the mechanisms of intimal hyperplasia in other aortic segments, the coronary arteries, and branch arteries distal to the left subclavian artery.10
Going forward, although this study demonstrates the complex mechanism underlying neointimal hyperplasia, as well as the interconnection between elastin and other ECM components, the exact mechanism remains to be uncovered. Success in dissecting the key points in the mechanism may reveal novel pharmacologic targets, enabling the development of new preventive and therapeutic approaches against a host of proliferative vascular diseases. Further studies are needed to analyze mechanisms concerning vSMC phenotypic changes that induce a proliferative and ECM-producing phenotype and how these vSMCs migrate to the intima. Additionally, the absence of a neointima in a subset of Wnt1Cre;Elnf/f mice suggests that elastin’s absence alone is not sufficient for neointimal formation. Clarifying whether elastin works through ECM-cell interaction or direct signaling to induce phenotypic changes would be ideal, given that, as Li et al.7 suggest, vSMC proliferation is uninhibited in the absence of elastin.
Acknowledgments
The authors thank Scott A. Weldon, MA, CMI for creating the associated figure and Nicole Stancel, PhD, ELS(D), of the Department of Scientific Publications at the Texas Heart Institute, for editorial support.
Sources of Funding
K.R. Rebello is supported by NIH T32-HL139430 Research Training Program in Cardiovascular Surgery at Baylor College of Medicine. S.A. LeMaire’s work is supported in part by the Jimmy and Roberta Howell Professorship in Cardiovascular Surgery at Baylor College of Medicine.
Footnotes
Disclosures
S.A. LeMaire serves as a consultant for Terumo Aortic and Cerus; serves as a principal investigator for clinical studies sponsored by Terumo Aortic and CytoSorbents; and serves as a co-investigator for clinical studies sponsored by W.L. Gore & Associates. The authors report no conflicts of interest.
REFERENCES
- 1.Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR, Jorgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016:119:1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003:130:411–423. [DOI] [PubMed] [Google Scholar]
- 3.Wu W, Zhang W, Choi M, Zhao J, Gao P, Xue M, Singer HA, Jourd’heuil D, Long X. Vascular smooth muscle-MAPK14 is required for neointimal hyperplasia by suppressing VSMC differentiation and inducing proliferation and inflammation. Redox Biol. 2019:22:101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA, Weiser-Evans MC. Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by Klf4. Circ Res. 2017:120:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees B, Schurgers L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2019:39:1351–1368. [DOI] [PubMed] [Google Scholar]
- 6.Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler Thromb Vasc Biol. 2017:37:1722–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998:393:276–280. [DOI] [PubMed] [Google Scholar]
- 8.Lin CJ, Hunkins B, Roth R, Lin CY, Wagenseil JE, Mecham RP. Vascular smooth muscle cell subpopulations and neointimal formation in mouse models of elastin insufficiency. Arterioscler Thromb Vasc Biol. 2021:##:###–###. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CJ, Staiculescu MC, Hawes JZ, Cocciolone AJ, Hunkins BM, Roth RA, Lin CY, Mecham RP, Wagenseil JE. Heterogeneous cellular contributions to elastic laminae formation in arterial wall development. Circ Res. 2019:125:1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G, Jacquet L, Karamariti E, Xu Q. Origin and differentiation of vascular smooth muscle cells. J Physiol. 2015:593:3013–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]