

Summary
Cortical visual processing transforms features of the external world into increasingly complex and specialized neuronal representations. These transformations arise in part through target-specific routing of information; however, within-area computations may also contribute to area-specific function. Here, we sought to determine whether higher-order visual cortical areas LM, AL, PM, and AM have specialized anatomical and physiological properties by using a combination of whole-cell recordings and optogenetic stimulation of V1 axons in vitro. We discovered area-specific differences in the strength of recruitment of interneurons through feed-forward and recurrent pathways, as well as differences in cell-intrinsic properties and interneuron densities. These differences were most striking when comparing across medial and lateral areas, suggesting that these areas have distinct profiles for net excitability and integration of V1 inputs. Thus, cortical areas are not defined simply by the information they receive, but also by area-specific circuit properties that enable specialized filtering of these inputs.
Keywords: excitation, inhibition, parvalbumin, somatostatin, hierarchical processing, electrophysiology, synaptic transmission, circuit, connectivity
Graphical Abstract
eTOC
Visual processing engages multiple cortical areas in parallel to generate increasingly specialized representations. Li et al. demonstrate that specialization in higher-order visual areas could arise from distinct anatomical and physiological properties of feedforward inputs from primary visual cortex and recurrent connections within each area.
Introduction
The neocortex is involved in a diverse array of computations that are fundamental to sensory and motor processing. It encompasses areas that represent basic features like the contrast of a visual stimulus or the frequency of a tone, and also areas directly linked to decision-making, working memory, and motor planning1-3. Given the wide range of these specialized functions, the overall structure of the areas that perform them is remarkably similar2,4. Neocortical areas are characterized by a six-layer architecture broadly comprised of two neuronal cell types: excitatory, glutamatergic pyramidal neurons and inhibitory, GABAergic interneurons. Consideration of genetically defined subpopulations of these cell types and their connectivity profiles reveals consistent motifs of circuit wiring. For example, feed-forward inhibition involving co-activation of pyramidal cells and interneurons is thought to involve parvalbumin-expressing (PV) interneurons, whereas late-onset, feedback inhibition is mediated by somatostatin-expressing (SOM) interneurons5-8. These types of feed-forward and feedback inhibitory connections are observed throughout cortex and work in conjunction to combine hierarchical inputs across areas with local processing within areas9-12. Understanding how the repetition of basic cortical motifs can give rise to specialized representations is crucial for insight into how the brain transforms neuronal activity across areas to generate higher-order computations.
Historically, two hypotheses have emerged to explain how specialization arises across cortical areas. On one hand, the consistent patterning of cortical structure could translate to a similar uniformity in local processing. In this model, termed the protocortex hypothesis, higher order computations are performed by fundamental cortical units that acquire specialization from inputs routed through target-specific anatomical pathways13. Such functional organization is found in macaque primary visual cortex (V1), where segregated projections to thin stripe and thick stripe/inter-stripe regions in V2 enable the parallel transmission of color and orientation signals, respectively14,15. Similarly, motion information is selectively routed to MT through a subset of V1 neurons that are more likely than chance to be direction-selective16. In further support of the protocortex hypothesis, sensory cortical areas can remap across modalities to compensate the loss of one; this flexibility of representation could arise from a large degree of anatomical and functional similarity of structure across areas17. Yet, additional variation in individual components of these canonical cortical motifs could create specialized microcircuits that further tune responses within an area. This idea is central to the protomap hypothesis, which predicts that variations in the expression patterns of genes across cortical areas produce differences in response properties of a circuit18-21. For example, response timing and sensitivity have been shown to vary across different sensory cortical areas, even after controlling for sensory modality and subcortical activity22. These specializations could be related to a variety of features: differences in intrinsic excitability of neurons, recurrent connectivity, and relative recruitment of excitation and inhibition as a function of neocortical topography9,23-25. However, evidence of specialization of multiple features across specific, functionally defined areas remains relatively sparse9,20,26,27.
The visual system is an excellent model for understanding the relative contribution of different mechanisms to neocortical specialization. As information is transferred hierarchically between primary and higher visual areas (HVAs), there is divergence of visual representations from basic features such as orientation and contrast to objects and self-generated motion28-33. Thus, visual perception involves the coordinated evolution of stimulus representation across many areas of the brain. While studies of hierarchical transformation have largely been conducted in primates, recent studies have demonstrated similar functional specialization in areas of mouse cortex27,34-38. In the mouse, V1 projects to many HVAs immediately surrounding it, including areas LM (lateromedial), AL (anterolateral), PM (posteromedial), and AM (anteromedial)39. Similar to primate cortex, V1 projections to HVAs are selectively routed and are more likely to target areas with matched function40. Consequently, different areas can share similar circuit motifs but have divergent functional properties depending on the inputs they receive41-43. However, this divergence alone is insufficient to account for all differences in stimulus selectivity across areas27,35,44. The organization of HVAs around V1 separates many of these areas by millimeters of cortical space. Therefore, divergence could also arise from relatively localized specialization of cellular and circuit properties that imbue different HVAs with distinct computational capabilities24,25,45.
To investigate these properties of mouse HVAs, we performed whole-cell patch clamp recordings of pyramidal cells and PV- and SOM-expressing subpopulations of interneurons in retinotopically matched regions of LM, AL, PM, and AM. We made systematic measurements of intrinsic properties of these cells, as well as features of their feed-forward and local connections using paired recordings and optogenetic stimulation. Our data demonstrate significant variation in anatomical and physiological properties across cortical areas, specifically across areas separated on medial versus lateral sides of V1. Compared to lateral areas, pyramidal cells in medial areas had higher input resistance and lower relative feedforward excitatory input onto neighboring PV and SOM interneurons, indicating that the primary difference might be in the overall excitability between these areas. However, we found that this difference could be compensated for by stronger recurrent inhibition: in medial areas we find an enrichment in SOM cells and an increase in local connectivity between pyramidal cells and interneurons. Altogether, our results indicate that higher order visual areas are not uniform in their cellular and circuit properties and these distinctions may contribute to differences in response properties across areas.
Results
Functional identification of mouse HVAs in coronal slices
Typically, HVAs are functionally identified with in vivo imaging that reveals their retinotopic organization46. However, in order to study these areas with histology and in vitro physiology we needed to develop an alternative method to consistently identify each area in coronal sections ex vivo. To do this, we combined functional imaging and histology within mice to compare in vivo, retinotopically identified positions of HVAs to their relative location ex vivo in the coronal slice (Figure 1A). First, we identified the HVAs in vivo using intrinsic autofluorescence imaging. Then, using this retinotopic map we targeted these areas for injection with fluorescent dyes (Figure 1B) and visualized the injection sites ex vivo in coronal sections of the brain (Figure 1C). Across mice (n=3), we find anatomical markers in coronal sections that allow us to reliably identify four HVAs: LM (lateromedial), AL (anterolateral), PM (posteromedial) and AM (anteromedial). LM is the most posterior of these HVAs, and appears in sections with the superior colliculus and the most posterior portion of the hippocampus (before clear segregation of the dentate gyrus; Figure 1C, top); AL and PM are anterior to LM, typically in the same sections, at the level of the medial geniculate nucleus of the thalamus (Figure 1C, middle); AM is the most anterior of these HVAs, appearing at the level of the lateral geniculate nucleus of the thalamus (Figure 1C, bottom). For all other experiments, we visualized HVAs by anterograde labeling of V1 neurons with opsins fused to fluorescent reporters (see STAR Methods). Using the projection pattern of fluorescently labeled V1 axons and these landmarks as a reference, we could clearly distinguish HVAs along the anterior-posterior axis in coronal slices (Figure 1D).
Figure 1. In vivo retinotopic mapping enables identification of HVAs in coronal slices.

A. Schematic of procedure for identifying and labeling HVAs using both dye injections directly into the HVAs (top) and adeno-associated virus (AAV)-mediated fluorophore expression in V1 axons (bottom). B. Left: Retinotopic map of left visual cortex with stimuli presented at 3 positions (azimuth: −10°, red; +10°, green; +30°, blue). Right: Same field of view as on left, with blue and magenta dye injections in LM/AM and AL/PM, respectively. C. Coronal sections from the brain in B ordered from posterior (top) to anterior (bottom) with HVAs and other landmarks labeled (SC = superior colliculus; MGN = medial geniculate nucleus; LGN = lateral geniculate nucleus). Locations of coronal sections in anterior-posterior axis correspond to dotted lines in B. D. Coronal sections, at the same anterior-posterior locations as in C, from a different mouse with viral fluorophore expression in V1 neurons and their axons in the HVAs and other target regions (SC, LGN). Note the alignment of the V1 axon arborizations with the areas labeled via dye injection in C.
Density and laminar distribution of feedforward excitatory inputs are key determinants for how inputs are integrated in cortical areas, and these appeared to vary across HVAs (Figure 1D). We systematically compared V1 axons in retinotopically matched regions in each HVA, using fluorescence intensity to measure input density47 (Figure 2A; see STAR Methods). We find that V1 axons are densest in area LM and in layer 2/3, consistent with previous studies (two-way ANOVA, LM vs all other areas: p<0.001, AL vs AM: p=0.009; L2/3 vs all other layers: p<0.001; n=8 mice; Figure 2B-C)12,48. We also find significant differences in laminar distribution across areas when normalizing density to the maximum within an area. Normalization revealed that inputs to layer 5 of AM and layer 1 of PM are comparatively denser than projections to these layers in other areas (Figure 2D-E; Layer 5: AM vs LM p=0.002, AM vs PM p<0.001; Layer 1: PM vs LM p=0.002, PM vs AL p<0.001, PM vs AM p<0.001), and a higher fraction of inputs target layer 4 of lateral areas than medial areas (LM vs PM p<0.001, LM vs AM p=0.002. AL vs PM p<0.001, AL vs AM p=0.007). Differences across areas were generally larger than individual animal variability within areas (Figure S1A) and could not be explained by differences in the level of fluorophore expression in individual axons (Figure S1B-D). These differences in the density and distribution of V1 axons across areas suggests there is heterogeneity in the impact of V1 on its targets. Moreover, the match between these data and the literature indicate that this viral-expression/landmark-based method can accurately identify the HVAs in coronal sections.
Figure 2. V1 axon densities are distinct across layers and HVAs.

A. Confocal images of V1 and HVAs from an example EMX-Cre mouse injected with an AAV driving Cre-dependent oChIEF-tdTomato expression in V1 neurons. Dashed lines indicate layer boundaries as defined by DAPI staining. Scale bar = 100 μm. B. Fluorescence density by depth (left) or binned by layer (right), normalized to maximum fluorescence across all areas (typically in L2/3 of LM). Error bars are SEM across mice (n = 6 mice; LM- green, AL- orange, PM- blue, AM-pink). See also Figure S1. C. Heatmap summary of B (right). D-E. Same as B-C, normalized to maximum density within each area.
Layer-specific differences in density of inhibitory interneurons across HVAs
Using this approach to identify HVAs, we next addressed whether local inhibitory networks differed across the HVAs. Synaptic inhibition provided by local inhibitory interneurons is a critical factor in determining the time scale of integration in cortical neurons and their visual response properties49-53. PV and SOM interneurons are highly prevalent throughout cortex, but differences in their relative proportions, recruitment, or connectivity can influence integration and visual response properties in the HVAs45. First, we used an anatomical approach to measure the relative cell densities of PV and SOM interneurons in the HVAs.
To label PV or SOM cells, we crossed PV-Cre or SOM-Cre mice with Ai14 reporter mice to drive tdTomato expression. We virally expressed eGFP or Chronos-GFP in V1 neurons and used the axonal arborizations to define the boundaries of retinotopically matched regions of the HVAs. The number of labeled cells within each area was divided by the cortical volume used for counting to calculate the density of interneurons in each area, thus normalizing for any differences in HVA size (# labeled cells/mm3; Figure 3A, Figure S2). Comparing overall densities of the two cell types across areas, we find that SOM cells are significantly more abundant in AM and PM than in LM (Figure 3B).
Figure 3. Different relative densities of PV and SOM interneurons across layers and HVAs.

A. Confocal images from two example mice with either PV (top) or SOM (bottom) interneurons transgenically labeled with tdTomato (magenta) and V1 axons virally labeled with Chronos-GFP (blue). Layer boundaries are defined using a DAPI stain. B. Top: PV cell densities summed across all layers, normalized to the maximum across areas. Blue lines are individual mice (n=8), black circles are mean. Error bars are SEM across mice. Bottom: Same as top, for SOM cells (n=5 mice). *, **, and *** denote p < 0.05, 0.01, and 0.001.C. Left, blue: PV cell densities across HVAs for each layer z-scored within layer. Right, green: Same as left for SOM cells. Colored circles- individual mice; black circles- average across mice. Error bars are SEM across mice. See also Figure S2. D. Scatter plot of z-scored PV and SOM densities z-scored across layers and areas. Each marker represents the z-scored average density across all mice of each cell type. Shape indicates layer, color indicates area. Gray line measures example Euclidian distance. Note that distances within each layer (across areas) are shorter than distances within each area (across layers). E. Euclidian distance between points in D within the same layer, grouped by medial or lateral areas (LM-AL and PM-AM), anterior or posterior areas (LM-PM and AL-AM), or neither (LM-AM and AL-PM). Each point is a pairwise distance within layer (symbols corresponding to same layer shapes as D). Example distance in D is shown in dark gray. Error bars are SD across layers.
To determine whether this difference in SOM cell density exists across all layers, we used DAPI staining to define laminar boundaries (Figure 3A). Given the large variability in density across layers (Figure S2), we z-scored densities within layers to compare across areas. We find a higher density of SOM cells in L2/3 of AM and L6 of PM as compared to LM and AL (SOM: one-way ANOVA with post hoc Tukey tests; L2/3 p=0.002, LM vs AM p=0.002, AL vs AM p=0.008; L6 p=0.023, LM vs PM p=0.049, AL vs PM, p=0.047; all other within-layer area comparisons p > 0.05; n=5 mice; Figure 3C). In addition, we find that despite a lack of an overall difference in PV cell density across areas, there are some significant differences when separating by layers: PV cells in L6 are significantly more dense in AM compared to LM and AL, and PV cells in L5 are significantly denser in LM and AL versus AM (PV: one-way ANOVA with post hoc Tukey tests; L5 p=0.049, LM vs PM p=0.044; L6 p=0.008; LM vs PM p=0.011, AL vs PM p=0.037; n=8 mice). Thus, SOM cells tend to be more abundant in medial areas, while the relative density of PV cells varies depending on layer.
Notably, when comparing the relative ratio of PV and SOM densities divided by layer and area (z-scored across all conditions within cell type), we find that interneuron densities are more similar within a particular layer across areas than within a particular area across layers (Euclidian distance within layer, across areas [distances between matched shapes]: 0.56±0.31, Euclidian distance within areas, across layers [distances between matched colors]: 2.11±0.73, p<0.001 Figure 3D, Figure S2). This reveals some broader trends that hold across areas: relative enrichment of PV cells compared to SOM cells in L4, relative sparsity of PV and SOM cells in L2/3, and relative enrichment of PV and SOM cells in L5. Comparing Euclidian distances between areas within each layer, we find that areas that share the same medial-lateral axis (i.e. LM/AL and PM/AM) are more similar than those that share the same anterior-posterior axis (i.e. LM/PM and AL/AM) or share neither axis (i.e. LM/AM and PM/AL) (Figure 3E; one-way ANOVA with post hoc Tukey tests; shared M-L vs shared A-P, p=0.007; shared M-L vs neither axis, p=0.003). Together, these differences in density of both PV and SOM cells indicate that inhibition may shape excitability in an area- and layer-specific manner.
Intrinsic properties of pyramidal cells and interneurons are similar across HVAs
Activity within a region is also influenced by neurons’ intrinsic membrane properties, which determine how they integrate inputs and fire action potentials54-56. Neurons in layer 2/3 receive dense input from V1 (Figure 2C) and send major excitatory projections to further downstream cortical areas57. Thus, differences in intrinsic properties of layer 2/3 pyramidal cells and local interneurons could alter input-output functions in an area-specific manner. These properties differ significantly between cell types, such as between pyramidal cells and interneurons, but also within cell type as a function of layer, cortical region, and projection target4,25,56,58,59. To determine whether these features varied within cell types across HVAs, we made whole-cell current clamp recordings to measure intrinsic properties of pyramidal cells and interneurons in all four areas (Figure 4A).
Figure 4. Intrinsic properties of pyramidal cells, but not interneurons, are distinct across areas.

A. Example Pyr (black; left), PV (blue; center), and SOM (green; right) voltage traces (top) in response to current steps (bottom). All depolarizing steps until first spikes are shown. B. Spike rate adaptation (ISIlast/ISIfirst) for pyramidal cells (LM = 55 cells, AL = 31, PM = 50, AM = 62), PV interneurons (LM = 22 cells, AL = 20, PM = 19, AM = 19), and SOM interneurons (LM = 19 cells, AL = 20, PM = 25, AM = 19). Colored circles- individual cells; black circles-average across cells. Error is SEM across cells. *, **, and *** denote p < 0.05, 0.01, and 0.001. C. Same as B, for voltage sag amplitude from activation of hyperpolarization-activated current. D. Same as B, for input resistance (Rin). See also Figure S3. E. Firing rate (FR) vs current injection (Iinj) for pyramidal cells (left), PV interneurons (center), and SOM interneurons (right) for each area. Shaded error is SEM across cells. *, **, and *** denote p < 0.05, 0.01, and 0.001 for effect of area from two-way ANOVA with post hoc Tukey test. See also Table S1.
While we find significant differences across cell types, within a cell type most intrinsic properties are consistent across areas. Unlabeled neurons with clear apical dendrites tend to have broad action potentials, strong spike frequency adaptation, and a small Vm sag due to the activation of a hyperpolarization-activated depolarizing channel (Figure 4B-C; Table S1), consistent with these being excitatory pyramidal cells. In comparison, PV interneurons have narrow action potentials, marginal spike frequency adaptation, and less Vm sag, while SOM interneurons have slightly broader action potentials, moderate spike frequency adaptation, and large Vm sag. Furthermore, SOM cells have characteristically high input resistance and a slow membrane time constant, whereas pyramidal cells and PV cells have lower input resistance as well as faster membrane time constants (Figure 4D; Table S1). These intrinsic properties result in significant differences in the input-output functions of each cell type as measured by spiking in response to depolarizing current injections (Figure 4E). Our results are consistent with previous measurements in primary sensory areas and demonstrate that significant differences in characteristic features of each of these major cell types’ intrinsic properties are maintained in the HVAs58,59 (two-way ANOVA for spike frequency adaptation, half-width, Vm sag, Rin, and tau; main effect of cell type: p<0.05 for all features).
We find significant differences across areas for Vm sag and input resistance of pyramidal neurons in the medial versus lateral HVAs (Figure 4C, Vm sag: AL vs PM p=0.003, AL vs AM p=0.032; Figure 4D, Rin: AL vs PM p=0.009, AL vs AM p=0.019). Although differences in cortical thickness across cortex have been shown to create gradients in input resistance25, there was no significant effect of depth in our analysis (two-way ANOVA, main effect of area: p=0.011, main effect of depth: p=0.079). Furthermore, this mediolateral difference was still present when between-area comparisons were restricted to measurements recorded within the same depths (Figure S3; LM vs PM, p=0.001, LM vs AM p=0.009, AL vs PM p=0.0001, AL vs AM p=0.011). Consistent with these differences in input resistance, there is also a significant difference in the input-output functions of pyramidal cells across areas, revealing area-specific differences in excitability (two-way ANOVA, main effect of Iinj: p<0.001 main effect of area: p=0.019; LM vs PM p=0.035; AL vs PM p=0.005; AL vs AM p=0.036; Figure 4E). Intrinsic properties of PV and SOM interneurons are consistent across all HVAs—spike frequency adaptation and Vm sag are not significantly different, nor are input resistance and F-I relationships (Figure 4B-D; center and right). Although we sampled fewer PV and SOM interneurons than pyramidal cells, the differences in detectable differences were minimal when performing power analyses (see STAR Methods). Collectively, these data demonstrate that pyramidal cells across medial and lateral areas have subtle differences in cell-intrinsic excitability, but most intrinsic passive and active properties of both excitatory and inhibitory neurons within layer 2/3 are relatively consistent between HVAs, suggesting similar filtering functions of V1 inputs.
Area-specific differences in the excitation of interneurons and pyramidal cells by V1
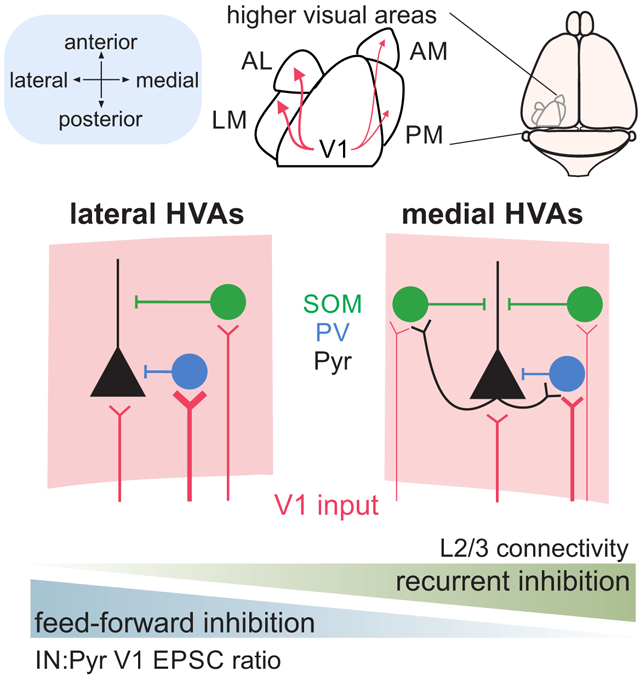
Parallel excitatory input onto pyramidal cells and interneurons ties the strength and timing of local inhibition to that of feed-forward excitation in a process called feed-forward inhibition4,60,61. Typically, in primary sensory areas, feed-forward excitation drives PV cells more strongly than neighboring pyramidal cells such that PV cells provide robust, short-latency feed-forward inhibition11. Notably, SOM cells generally receive weaker feed-forward excitation. As a result, SOM cells are not as robustly engaged in generating feed-forward inhibition5,62-64. Thus, the relative strength of feed-forward V1 excitation onto different inhibitory cell types is an important mechanism for regulating the balance and timing of excitation and inhibition within an area.
To directly compare the relative strength of V1 inputs to the HVAs across cell types and areas, we made paired whole-cell voltage-clamp recordings from layer 2/3 pyramidal cells and interneurons while optogenetically stimulating V1 axons expressing the excitatory opsin Chronos (Figure 5A-B). Cells were voltage clamped at the reversal potential for inhibition to isolate excitatory post-synaptic currents (EPSCs). This approach evoked EPSCs with short latency and fast rise time in all three cell types, consistent with these EPSCs being due to monosynaptic V1 excitation (Figure S4A-B). However, stimulation of V1 axons can drive other postsynaptic neurons within the HVAs to fire, resulting in the recruitment of additional excitation from polysynaptic circuits with some temporal delay. To determine whether the early component of the multiphasic EPSCs we observed could be used to specifically study feedforward excitatory V1 input, we pharmacologically suppressed local population spiking to assay the origin of the input (Figure 5C). In a subset of experiments we applied the GABAA receptor agonist muscimol (50 μM) to hyperpolarize the local population and prevent recurrent excitation65 (Figure S5). Application of muscimol does not affect the EPSC onset latency (Pyr p=0.495, PV p=0.826, SOM p=0.436; Figure 5E). Furthermore, in an early time window (0-4 ms from laser onset), there was no significant change in the ratio of excitation onto the interneuron and a paired pyramidal cell (paired t-test, PV:Pyr p=0.935, SOM:Pyr p=0.803; Figure 5F). Therefore, we used this early window as a temporal boundary for measuring monosynaptic V1 inputs on interneurons and pyramidal cells in the HVAs.
Figure 5. Early-onset, feedforward excitation from V1 is stronger onto interneurons in lateral areas.

A. Example image from slice with recording pipettes positioned in L2/3 and laser site photobleached after recording. B. Left: Schematic depicting setup for paired interneuron (IN) and Pyr recording with L5 optogenetic stimulation of V1 axons expressing Chronos. Local inputs from neighboring pyramidal cells come from both within L2/3 and across layers (black lines). Right: Example EPSCs following V1 axon stimulation recorded in a SOM and Pyr pair (SOM green, Pyr black) in the presence of CGP54626 (1 μM). Blue square indicates time of laser. C. Same as B, after muscimol (50 μM) application. Greyed out connections indicate local inputs from cells suppressed by muscimol application. See also Figure S5. D. Grand average EPSCs across cells before (dark) and after (light) muscimol for Pyr (n = 23), PV (n = 5), and SOM (n = 18) cells. Shaded pink window indicates analysis window used to quantify feedforward V1 inputs. E. Change in latency between baseline and muscimol condition. Colored circles are individual cells, colored by cell type. Black circle is mean. Error is SEM across all cells. F. Percent change in IN:Pyr for PV:Pyr and SOM:Pyr pairs between baseline and muscimol conditions. Colored circles are pairs, black circles are mean across pairs. G. Example EPSCs from a PV:Pyr pair (left) and SOM:Pyr pair (right) following optogenetic stimulation. Shaded pink window indicates analysis window obtained from muscimol experiments used to quantify feedforward V1 inputs. Arrows reflect amplitude measurement in that window. H. Ratio of excitation onto each IN and pyramidal cell (IN:Pyr ratio) for pairs divided by HVA. Colored circles- individual pairs; black circles- average across pairs. Error is SEM across pairs. Left: PV:Pyr; LM = 17 pairs, AL = 11, PM = 12, AM = 13; right: SOM:Pyr; LM = 17 pairs, AL = 15, PM = 15, AM = 17. *, **, and *** denote p < 0.05, 0.01, and 0.001. See also Figure S4.
In order to compare the relative excitation of cell types, we normalized the monosynaptic EPSC onto each interneuron by its paired pyramidal cell (IN:Pyr ratio). Similar to what has been observed at thalamocortical inputs onto V1 neurons, V1 inputs onto PV cells are significantly larger than onto paired pyramidal cells (IN:Pyr>1; p<0.001; paired t-test; n=49 pairs; Figure 5G-H), while V1 inputs onto SOM cells are significantly weaker (IN:Pyr<1; p<0.001; paired t-test; n=56 pairs). Divided by area, this trend is similar but we do find significant differences in the strength of excitation onto interneurons and pyramidal cells across medial and lateral areas. Lateral areas tend to have larger IN:Pyr ratios than medial areas for both PV (effect of area p=0.002; one-way ANOVA; Figure 5H, left; LM vs AM p=0.014; AL vs PM p=0.039; AL vs AM p=0.008) and SOM interneurons (effect of area p<0.001; LM vs AM p<0.001; AL vs AM p=0.019 one-way ANOVA with post hoc Tukey test; Figure 5H, right). This effect is particularly salient for PV cells, which have EPSC ratios below 1 only in medial areas. Importantly, this effect is not dependent on the strength of activation: when varying the laser strength within recordings of a pair, we find the ratio to be independent of the laser intensity (p=0.070; n=13 PV:Pyr pairs; n=10 SOM:Pyr pairs; one-way ANOVA; Figure S4C). Thus, inhibitory interneurons are more strongly activated by V1 inputs in lateral areas as compared to medial areas.
Late-onset, local recruitment of excitation differs across HVAs
Our pharmacology experiments suggest that in recordings without muscimol, stimulation of V1 axons generates not only feedforward monosynaptic excitation, but also late-onset excitation that could originate from recruitment of recurrent circuits within the HVAs (Figure 6A)66. Multi-phasic events were often observed in SOM cells and were sometimes also seen in pyramidal cells (Figure 6B). To quantify the relative recruitment of feed-forward and local excitation, we measured the time to half-max for the cumulative excitatory charge (Figure 6B-C). Shorter times reflect fast decaying EPSCs in which the majority of the current is monosynaptic, while longer times reflect additional recruitment of polysynaptic inputs. EPSCs recorded in pyramidal cells have similar times to half-max across areas (Figure 6D, left), indicating a similar temporal distribution of excitatory inputs after stimulation. In comparison, EPSCs recorded in SOM cells have shifted temporal distributions of charge in medial compared to lateral areas. EPSCs onto SOM cells in PM and AM have significantly longer times to half-max than those in LM and AL (Figure 6D, right; LM vs PM: p<0.001; LM vs AM: p=0.033; AL vs PM: p=0.002). A similar, though non-significant, trend is seen in PV cells (Figure 6C-D, center). Importantly, this difference in the time course cannot be explained by differences in EPSC kinetics, since evoked EPSC rise times and spontaneous EPSC rise and decay times are not significantly different across areas (Figure S4; Figure S6).
Figure 6. Interneurons in medial areas receive more late-onset, polysynaptic excitation.

A. Schematic depicting setup for paired recordings in HVAs with local excitatory inputs within the HVA. Local inputs from neighboring pyramidal cells come from both within L2/3 and across layers (black lines). B. Top: Cumulative charge for a PV (blue) and Pyr (black) pair (left) and a SOM (green) and Pyr (black) pair (right) with time to 50% charge labeled (shaded region, arrowhead). Bottom: EPSCs used for analysis in top. Same example cells as in Figure 5G. C. Grand average of cumulative charges for Pyr (left), PV (center), and SOM (right) cell types. D. Summary of time to 50% charge for each cell type. Colored circles- individual cells; black circles- average across cells. Error is SEM across cells. Pyr: LM = 34 cells, AL = 26, PM = 27, AM = 30; PV: LM = 17 cells, AL = 11, PM = 12, AM = 13; SOM: LM = 17 cells, AL = 15, PM = 15, AM = 17. *, **, and *** denote p < 0.05, 0.01, and 0.001. See also Figure S6. E. Normalized EPSCs averaged across neurons, grouped by lateral (dark; LM and AL) and medial (light; PM and AM) areas. Shaded error is SEM across cells.
To more carefully assess the temporal properties of the optogenetically-evoked inputs, we deconvolved these evoked EPSCs with a template fit to spontaneous EPSCs in each cell type (see STAR Methods). This analysis confirms that only SOM cells receive significantly stronger inputs in medial areas during a late time window (7.5-15 ms; Figure S6). Indeed, inspection of the grand average EPSCs recorded across areas reveals that our measure reflects differences in the recruitment of secondary, longer latency EPSCs (Figure 6E). While the average time courses of EPSCs onto pyramidal cells are similar across medial and lateral areas, there is robust, late-onset excitation onto SOM cells selectively in medial HVAs where feed-forward excitation of interneurons is comparatively weaker. PV cells in medial areas also showed somewhat greater late-onset excitation, but this difference was not significant. Thus, stimulation of V1 inputs to the HVAs differentially recruits local inhibitory networks in medial versus lateral areas.
Different connectivity probabilities between L2/3 pyramidal cells and interneurons across HVAs
We reasoned there are two possible mechanisms that might explain the stronger local input onto interneurons in medial HVAs. One possibility is that stimulation of V1 inputs to medial areas drives stronger activation of local circuits. This mechanism would be consistent with the comparatively weaker feed-forward excitation onto interneurons that we observed (Figure 5H) enabling increased excitability in medial areas. If overall activity was higher in medial areas, we would also expect to see a similar increase in local inputs onto pyramidal cells. However, we observed no difference in the amplitude of either feed-forward EPSCs (Figure S4D) or late-onset EPSCs measured in pyramidal cells (Figure 6E). While we cannot rule out a contribution of differences in pyramidal cell spiking across areas, this observation suggests it is not the only factor driving the increased late excitation onto SOM cells in medial areas. Another possibility is that there is a difference in the strength or probability of local excitation onto interneurons within the HVAs.
To test this hypothesis, we made paired recordings from neighboring layer 2/3 pyramidal cells and interneurons within 50 μm of each other (Figure 7A). The amplitude and short-term dynamics of these connections are consistent with what was expected for these cell types (based on measurements in primary sensory areas4,6,60,66,67). We find that pyramidal cells receive significantly stronger inhibition from PV cells than SOM cells but provide equal excitation to both cell types (one-way ANOVA PV→Pyr vs SOM→Pyr: p=0.011; Pyr→PV vs Pyr→SOM: p=0.291; Table S2). When dividing by areas we find no significant differences between the amplitude of the unitary EPSCs recorded in PV and SOM interneurons (one-way ANOVA, all comparisons: p>0.05; Figure 7B-C). Among inputs from interneurons to pyramidal cells, one comparison emerged statistically significant: uIPSCs measured in PV→Pyr connections were significantly larger in LM than in PM (p=0.026), consistent with stronger recruitment of feedforward inhibition in this area by V1 inputs (Figure 5H). While short-term plasticity of unitary connections is different based on interneuron type (one-way ANOVA, PV vs SOM, Pyr→IN P10/P1: p<0.001; IN→Pyr P10/P1: p<0.001; Table S2), within cell types there are no differences in the paired-pulse ratios between areas (one-way ANOVA, P2/P1 and P10/P1: p>0.05 for all area comparisons). Thus, differences in strength and short-term plasticity of connectivity between pyramidal cells and interneurons cannot explain differences in local excitatory inputs onto interneurons across different HVAs.
Figure 7. Higher recurrent connectivity between pyramidal cells and interneurons in medial HVAs.

A. Example PV (blue) and Pyr (black) pair with both Pyr→IN connection (left) and IN→Pyr connection (right). Thin lines are individual trials, thick lines are mean. B. Amplitude of monosynaptic connections for Pyr→PV inputs (top) or Pyr→SOM inputs (bottom). Colored circles- individual pairs; black circles- average across pairs. Error is SEM across pairs. *, **, and *** denote p < 0.05, 0.01, and 0.001. C. Same as B, for IN→Pyr connections. D. Fraction of connections found out of total connections tested for PV/Pyr pairs, sorted by area and connection type. Shading of box indicates connection probability. See also Table S2. E. Same as D, for SOM/Pyr pairs.
When we quantified the probability of connection between pyramidal cells and interneurons, we found significant differences in the probability of these connections across areas. Moreover, these differences are consistent with interneurons receiving more excitatory inputs in medial areas. Both PV and SOM cells in medial areas are more likely than those in lateral areas to receive input from a neighboring pyramidal cell (Fisher’s exact test; Pyr→PV: AM vs LM p =0.014, AM vs AL p=0.007; Pyr→OM: PM vs LM p =0.037, PM vs AL p = 0.007; Figure 7D-E). SOM cells were also more likely to inhibit neighboring pyramidal cells in medial versus lateral areas (Fisher’s exact test; SOM→Pyr: PM vs LM p=0.048, AM vs LM p=0.009, AM vs AL p=0.033; Figure 7E) and showed greater connectivity overall in medial areas compared to lateral areas—only one pair tested within PM and AM had no connections (Fisher’s exact test; LM vs PM p=0.009, LM vs AM p<0.001, AL vs PM p=0.040, AL vs AM p=0.006). In V1, interneurons and pyramidal cells form segregated subnetworks where an interneuron forms inhibitory synapses with a bias towards pyramidal cells providing direct input to that interneuron68. We found a similar bias in the HVAs with a large proportion of Pyr→IN connections that were reciprocated (Figure 7D-E). Comparing the likelihood of these reciprocal connections across areas, we find PV/Pyr and SOM/Pyr pairs have even greater reciprocal connectivity in medial areas compared to lateral areas (Fisher’s exact test; PV: PM vs AL p=0.041, AM vs LM p=0.029, AM vs AL p=0.007, SOM: PM vs AL p=0.003, AM vs AL p=0.030). Thus, mediolateral differences in local inputs appear to arise through differences in both the number and selectivity of connections in these inhibitory networks.
Discussion
The neocortex is a highly conserved anatomical structure that performs a wide variety of computational tasks. Evidence suggests that specialization of function could arise through targeted routing of inputs to specific cortical areas (protocortex hypothesis)13,15,16,42,69. However, some features of cellular and circuit properties have been shown to vary as a function of neocortical topography, such that areas have demonstrable differences along spatial axes12,24,25,45. Indeed, we found significant variation in anatomical and physiological properties across visual cortical areas. These effects suggest a net difference in excitability across areas–in medial areas pyramidal cells had higher input resistance and neighboring interneurons received relatively weaker feed-forward excitatory input. However, these differences may be compensated by complementary increases in local excitation and density of interneurons within medial HVAs. Thus, in this study we provide support for the protomap hypothesis: cortical microcircuits share motifs but are not mirror images of each other21. Moreover, variation of these motifs could shape how inputs are filtered in an area and ultimately support specialization of cortical function.
This spatial divide of cellular and circuit properties across medial and lateral areas is reminiscent of specialization of dorsal/ventral streams of visual processing in primates33,70. Our findings are consistent with studies separating areas PM and AM from LM, but depart from previous studies that distinguish AL and LM as parts of the dorsal and ventral streams, respectively47,48,71. Instead, we find properties of AL to be more similar to LM than PM or AM such that the segregation of function is biased to process stimuli more similarly within medial and lateral areas than across them. Notably, medial and lateral HVAs are separated by larger distances than those along the anterior-posterior axis. If these differences arise in part due to molecular gradients24,25,45, then larger medial-lateral distances might explain some of the bias. Regardless of origin, in this study we have identified significant anatomical and physiological specializations that could contribute to functional specialization of responses within each area.
Significant differences in these cell-intrinsic, synaptic, and circuit features could converge to generate broader differences in how inputs are transformed through the canonical circuit motifs present across neocortex. Inhibitory circuits are particularly important for determining the spatial and temporal dynamics of cortical responses52,72,73. In order to dissect properties of inhibitory cells across cortical areas, we utilized transgenic lines that label PV and SOM cells. Previous studies find that the overlap between fluorophore expression and these cell-type specific markers is high74,75, and the significant differences in intrinsic properties that we find across cell types confirms their specificity. Furthermore, the larger relative V1 input to PV cells than neighboring pyramidal cells (PV:Pyr EPSC > 1) and smaller input onto SOM interneurons than pyramidal cells (SOM:Pyr EPSC < 1) also supports the appropriate classification of these cell types4,9,12,64. Interestingly, we also found an area-specific bias in these ratios, where both classes of interneurons in medial areas receive comparatively less excitation than neighboring pyramidal cells. This finding is different from previous studies measuring V1 inputs in HVAs using subcellular mapping, which found a larger PV:Pyr EPSC ratio in PM than LM9,12. Here we evoked transmitter release in layer 2/3 via action potentials generated with axon stimulation at a single site in layer 5, whereas subcellular mapping involves localized, direct depolarization of axon terminals in tetrodotoxin (TTX) and 4-AP to block action potential generation and optimize terminal depolarization. These differences in stimulation could be a source of variability—whereas vesicle release associated with action potential generation is more physiological, it can also trigger recurrent inputs. While we have made an effort to isolate the early EPSC component and found that this component has similar kinetics to EPSCs recorded in TTX and 4AP, it is possible that some of the early input we measured is not solely from V1 axons.
At a population level, network models show that decreasing the ratio of feedforward excitatory inputs onto interneurons versus pyramidal cells (in a manner similar to what we see in medial HVAs) leads to increases in activity levels that may impair network stability72,76-78. Our results suggest that in medial HVAs, stability is maintained by balancing weaker feedforward inhibitory pathways with stronger feedback inhibitory pathways. In particular, we find a robust increase in both the density and the degree of recurrent excitation of SOM cells, a cell type known for its role in recurrent inhibition49,51,79. Notably, modeling also demonstrates that increasing the weight of recurrent excitatory synapses onto interneurons is a robust mechanism for counteracting the effects of weak feed-forward inhibition on network firing rates76. While some of the increased recurrent excitation onto SOM cells might be explained by increased excitability in medial areas (due to the reduced feed-forward inhibition), it also likely reflects area-specific differences in recurrent connectivity. Indeed, while the range of pyramidal cell and interneuron connectivity rates is consistent with the literature6,67,68,80,81, when separating by area we found a striking difference in the probability of connectivity from pyramidal cells to interneurons—the likelihood of connection was nearly twice as high in medial compared to lateral areas. This difference is unlikely to be explained by different slicing angles between medial and lateral areas differentially disrupting connectivity, as V1 excitatory input to pyramidal cells is similar between areas and our optogenetic stimulation requires action potential propagation in intact V1 axons in L5 onto intact dendrites in L2/3.
How could differences in cell-intrinsic and circuit properties across areas ultimately lead to the differences in stimulus specificity observed across these areas? Differences in recruitment of local inhibition may serve to alter excitability across areas and modulate response thresholds and sensitivity51,82. While no significant differences in contrast response functions have been observed across areas35, contrast normalization is thought to engage both feed-forward and feedback inhibitory pathways. Thus, opposing effects may result in little net effect on contrast responses83,84. Conversely, surround suppression is a phenomenon thought to be generated specifically through feedback recruitment of SOM cells49,52. Given the stronger recruitment of SOM cells in medial areas, it is surprising that neurons in PM actually have larger receptive fields and less surround suppression than neurons in lateral areas35. However, spatial integration also relies on recurrent connections between pyramidal cells 10,85 as well as connections between pyramidal and SOM cells separated by hundreds of microns52,67,81. Here, we have only tested connectivity of immediately neighboring pyramidal cells and interneurons, which we have shown to exhibit increased connection probability in medial areas. Further study of the probabilities for both of these types of connections over larger scales will be important to understand how connectivity patterns might contribute to spatial integration within an area.
Stronger feed-forward inhibition could also influence representation of stimuli in the temporal domain. Engagement of feed-forward inhibition, which is primarily thought to be mediated by PV interneurons, could improve representation of the timing of sensory input by narrowing the integration time window in neurons61. Thus, stronger recruitment of feed-forward inhibition in lateral areas could improve encoding of higher frequency temporal features and amplify the anatomical divergence in temporal frequency preferences of V1 inputs to HVAs34,42. This could support the higher temporal frequency preferences in LM and AL compared to PM34,37. However, AM which shares many anatomical and physiological similarities with PM, has temporal frequency preferences more similar to LM. This highlights the important point that while most of the variation in circuit properties was found across the medio-lateral axis, there are significant functional differences that have been observed along the anterior-posterior axis. Instead, differences along the anterior-posterior axis may be explained by variation in other properties that we did not investigate. For example, the short-term plasticity of synaptic transmission at excitatory synapses is theorized to affect the temporal dynamics of a stimulus response86. Future experiments investigating the short-term plasticity of inputs to the HVAs may account for additional differences across areas. Additionally, while we have focused on feedforward excitation from V1 and recurrent excitation within the HVAs, the organization and strength of feedback and thalamic connections are other promising candidates for revealing mechanisms of functional specialization9,12,44,57. Indeed, a recent study suggested that the inputs from the lateral posterior nucleus of the thalamus are more important than inputs from V1 in determining HVA function44. Alternatively, major functional differences along the anterior-posterior axis may largely originate from functionally-specific routing of inputs to these areas.
In this study, we provide evidence for specialization of cell-intrinsic, synaptic, and circuit features across multiple areas of the neocortex that may complement area-specific routing of inputs. By performing systematic measurements of various anatomical and physiological features across a subset of mouse higher visual areas, we have characterized distinct but complementary mechanisms governing feedforward and feedback inhibition that allow for flexibility—and therefore specialization—of local processing in higher visual areas. We identified a medio-lateral division of local processing in microcircuits across higher-order visual areas that is similar to dorsal and ventral streams in other mammals. These findings are relevant not only for understanding how visual information is transformed across areas, but also highlight a set of potential mechanisms for larger-scale specialization of cortical computations across the brain.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to Lindsey Glickfeld (glickfeld@neuro.duke.edu).
Materials availability
No new reagents were generated as a result of this study.
Data availability
All raw and analyzed anatomical and electrophysiological data supporting this manuscript are available on Mendeley Data. DOIs are listed in the key resources table. Data were analyzed offline using custom MATLAB code. The code for data analysis is available on Github, linked in the key resources table. Any additional information required for reanalysis is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| rAAV2/1-Flex-rev-oChIEF-tdTomato | Addgene89 | Plasmid #: 30541 |
| AAV8-Syn-Chronos-tdTomato | Addgene87 | Plasmid #: 62726 |
| AAV9-Syn-Chronos-GFP-WPRE-bGH | Addgene87 | Plasmid #: 59170 |
| AAV1.CB7.CI.eGFP.WPRE.rBG | University of Pennsylvania Viral Vector Core | Lot #: CS0326 |
| Experimental models: Organisms/strains | ||
| EMX1-IRES-Cre | Jackson Labs | Stock #: 005628 |
| SST-IRES-Cre | Jackson Labs | Stock #: 013044 |
| PV-Cre | Jackson Labs | Stock #: 008069 |
| Ai14 | Jackson Labs | Stock #: 007914 |
| Ai3 | Jackson Labs | Stock #: 007903 |
| Ai148 | Jackson Labs | Stock #: 030328 |
| Ai162 | Jackson Labs | Stock #: 031562 |
| C57/B6J | Jackson Labs | Stock #: 000664 |
| Chemicals, peptides, and recombinant proteins | ||
| Dextran, Alexa Fluor 488, 10,000 MW | ThermoFisher | Cat #: D22910 |
| Dextran, Alexa Fluor 594, 10,000 MW | ThermoFisher | Cat #: D22913 |
| NBQX | Tocris Bioscience | Cat #: 1044; CAS: 479347-86-9 |
| D-APV | Tocris Bioscience | Cat #: 0106; CAS: 79055-68-8 |
| CGP 54626 hydrochloride | Tocris Bioscience | Cat #: 1088; 149184-21-4 |
| Muscimol | Tocris Bioscience | Cat #: 0289; CAS: 2763-96-4 |
| Deposited data | ||
| Raw and analyzed data | This paper | Mendeley Data DOI: 10.17632/rtnkp83yxs.1 |
| Software and algorithms | ||
| Micromanager | NIH93 | https://micro-manager.org |
| ImageJ | NIH95 | https://imagej.nih.gov/ij/ |
| MWorks | MWorks | https://mworks.github.io/ |
| pClamp 10 Software Suite | Molecular Devices | |
| MATLAB | Mathworks | https://www.mathworks.com/ |
| Custom MATLAB scripts | This paper | https://github.com/Glickfeld-And-Hull-Laboratories/Manuscripts/tree/master/HVA%20inhibition |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures conformed to standards set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Duke University's Animal Care and Use Committee. Mice were housed on a normal 12:12 light-dark cycle. 151 mice (85 female) were used in this study. The following driver lines were used to express Cre-dependent optogenetic or fluorescent proteins: EMX1-IRES-Cre (Jackson Labs #005628, n=63), SST-IRES-Cre (Jackson Labs #013044, n=49), PV-Cre (Jackson Labs #008069, n=48). To fluorescently label genetically identified interneurons, we crossed Cre-driver lines with Ai14 (Jackson Labs #007914), Ai3 (Jackson Labs #007903), Ai162 (Jackson Labs #031562), or Ai148 (Jackson Labs #030328) reporter mice. All transgenic mice were heterozygous and bred on a C57/B6J (Jackson Labs #000664) background. Mice were 28-60 days old at the time of injection and 32-74 days old at the time of sacrifice for histology or recording. Animals were not used for any other experimental procedures prior to sacrifice. Our anatomical and physiology data are consistent with veridical labeling in the animals that we have used, however it is possible that a small subset of labeled interneurons (~3-6% in previous literature75) do not express the peptide of interest.
METHOD DETAILS
Viral expression in V1 neurons
For slice electrophysiology experiments we expressed the light-gated cation channel Chronos87 or oChIEF88,89 in V1 neurons. For the majority of optogenetic activation experiments we used Chronos; in a small subset (n=5 mice), Cre-dependent oChIEF was injected into EMX-Cre mice and interneurons were identified by their spiking properties (oChIEF: rAAV1/2-Flex-rev-oChIEF-tdTomato, 2.2x1013 GC/ml; Chronos: pAAV-Syn-Chronos-tdTomato. 1.6x1013 GC/ml). For anatomical experiments, we expressed GFP or tdTomato in V1 neurons. To label projections to the HVAs to measure expression density we injected rAAV1/2-Flex-rev-oChIEF-tdTomato (2.2x1013 GC/ml) in V1 of EMX-Cre mice. To facilitate area identification for interneuron counts we injected AAV1.CB7.CI.eGFP.WPRE.rBG (2.04x1013 GC/ml) or AAV9-Syn-Chronos-GFP-WPRE-bGH (5.63x1013 GC/ml) in V1 of mice with either PV or SOM interneurons labeled with tdTomato.
Viruses were pressure injected into the brain via a burr-hole. Briefly, isoflurane anesthetized mice were positioned in a stereotax (Kopf Instruments) and a small hole was drilled though the skull −2.6 mm lateral from lambda and directly anterior to the lambdoid suture (targeting the posterior and medial aspect of the primary visual cortex, V1). A glass micropipette containing the virus was mounted on a Hamilton syringe, lowered 350 – 500 μm into the brain, and 100 nL of virus was pressure injected using an UltraMicroPump (World Precisions Instruments). We waited a minimum of 2 weeks for opsin expression. Some AAV serotypes have been shown to exhibit retrograde and transsynaptic labeling abilities90,91. We observed cell bodies in the HVAs in only a small subset of our experiments and excluded data from those mice after confirming with histology (Figure S7).
In vivo retinotopic mapping
To find the correspondence between areas identified in vivo and in coronal section, mice were implanted with a headpost and a 5 mm cranial window 92. After recovery from surgery, retinotopic maps were generated from intrinsic autofluorescence. The brain was illuminated with blue light 473 nm LED (Thorlabs), and emitted light was measured through a green and red filter (500 nm longpass). Images were collected using a CCD camera (Rolera EMC-2, Qimaging) at 2 Hz through a 5x air immersion objective (0.14 numerical aperture (NA), Mitutoyo), using Micromanager acquisition software93. Visual stimuli were presented on a 144-Hz (Asus) LCD monitor, calibrated with an i1 Display Pro (X-rite). The monitor was positioned 21 cm from the contralateral eye. Visual stimuli were controlled with MWorks (http://mworks-project.org). Circular gabor patches (30° diameter) containing static sine-wave gratings alternated with periods of uniform mean luminance (60 cd/m2). Images were analyzed in ImageJ95 to measure changes in fluorescence (dF/F; with F being the average of all frames) to identify primary visual cortex (V1) and the higher visual areas. Vascular landmarks were used to identify targeted sites for injection of Alexa-conjugated dextrans (488 or 594; 10,000 MW, Thermo Fisher). To inject the dye, the cranial window was transiently removed, and the same protocol used to inject virus was used to inject 50 nL dye (5% in water).
Histology and imaging
Three weeks following the viral injection (or the day after for dye injections), mice were transcardially perfused with 4% paraformaldehyde. Whole brains were removed and post-fixed <24hrs in 4% paraformaldehyde. The paraformaldehyde was washed out of the tissue with 3x1hr rinses with PBS buffer containing (in mM): 137 NaCl, 2.7 KCl, 10 Na2HPO4, 1.8 KH2PO4. Fixed tissue was then soaked in a 30% sucrose solution until the tissue sunk. Brains were then sectioned in coronally (70 μm) using a freezing microtome and mounted onto microscope slides using a DAPI mounting medium (DAPI Fluoromount G, Southern Biotech).
Coronal brain slices were first imaged at 2x magnification using an epifluoresence microscope to determine the locations of the HVAs using a combination of the axonal arborizations of the V1 projections and anatomical landmarks. Next, confocal z-stacks (10x objective, 0.3 NA, Zeiss Axiovert 200M) were collected from the four consecutive slices with the densest V1 projections in each HVA. For measurements of projection density, the PMT gain was optimized for area LM (since this area generally has the strongest expression) and kept constant for all other areas to remove any variability due to differences in image acquisition.
In vitro slice preparation and recordings
Mice were anesthetized with isoflurane, the brain was removed and then transferred to oxygenated (95% O2 and 5% CO2), ice-cold artificial cerebrospinal fluid (ACSF, in mM: NaCl 126, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, glucose 20, CaCl2 2, MgCl2 1.3). Coronal brain slices (350 μm) were prepared using a vibrating microtome (VT1200S, Leica) and transferred to a holding solution (at 34° C) for 12 minutes, and then transferred to storage solution for 30 min before being brought to room temperature. The holding solution contained (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2, 2 MgSO4. The storage solution contained (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, 10 MgSO4. Intracellular recordings of were obtained using the whole-cell patch-clamp technique. Micropipettes pulled from borosilicate glass were filled with an internal solution containing (in mM): 142 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 5 phosphocreatine-tris, 5 phosphocreatine-Na2, 3 Mg-ATP, 0.5 GTP. Recording pipets had resistances of 2-5 MOhms.
Recordings occurred between 1.5 and 5 hours after the animal was sacrificed. Brain slices were transferred to a recording chamber and maintained at 34° C in oxygenated ACSF perfused at 2 mL/min. Higher visual areas were identified using the fluorescence of the infected V1 axons in conjunction with anatomical landmarks to identify location along the anterior-posterior axis (Figure 1D). Electrophysiological recordings were restricted to layer 2/3 and within 300 μm of the center of the labeled V1 axonal arborization. We recorded in multiple areas across medial and lateral areas from a single animal to minimize differences between recording days (median 3 HVAs/recording day). Online and post-hoc histological analysis ensured that we did not record from locations where cell bodies were infected with the optogenetic proteins (Figure S7). Thus, our measurements reflect the signals transmitted anterogradely from V1 to the higher areas. In all experiments, pyramidal cells were identified based on morphology and spiking responses. The majority of interneurons were identified with transgenic fluorescent labeling, but a small subset of unlabeled cells were assigned by somatic morphology and spiking properties (PV: 7/80; SOM: 2/83).
Neural signals were amplified using a MultiClamp 700B, low-pass filtered at 6 kHz, and digitized at 20 kHz using a Digidata 1550 (Axon Instruments). Data acquisition and stimulus presentation was controlled using the Clampex software package (pClamp 10.5, Axon Instruments). For characterization of intrinsic properties, we recorded in current clamp configuration and applied a series of current injections ranging from −680 pA to +680 pA. All neurons had <−55 mV resting membrane potential. For V1 axon stimulation experiments, optogenetically-evoked EPSCs were recorded in voltage clamp configuration while holding the membrane potential at the chloride reversal potential (−85 mV, uncorrected for liquid junction potential). Series resistance was monitored using −5 mV steps preceding each stimulus. We collected at least 10 sweeps for each recording. Only pairs where both neurons had <20 MOhms series resistance and stable holding current (<100 pA baseline variation) were included for analysis. Light pulses (350 μs) were generated using a 450 nm laser (Optoengine) coupled to the epifluorescence path (Olympus BX-RFA) and projected through a 40x water immersion lens (Olympus, 0.8 NA). The laser stimulation site had a diameter of 200 μm and was operated at a power of 20 mW/mm2 measured at the sample (Figure 5A). Light was targeted to layer 5 to avoid direct depolarization of the axon terminals.
We first determined the effects of muscimol in extracellular field recordings (Figure S5). A glass pipet (1-2 MOhms) filled with 3 M NaCl was placed in layer 2/3 to record the optogenetically evoked field potential. To isolate the presynaptic fiber volley, we applied glutamate receptor antagonists (10 μM NBQX and 30 μM D-APV) to block the fEPSP and the GABAB receptor antagonist CGP54626 (1 μM) to block off-target effects of muscimol. We then applied muscimol (50 μM) and waited a minimum of 3 minutes for the response to stabilize before measuring the effect on the fiber volley. We flowed in TTX (1 μM) to block action potentials and isolate the opsin potential. The fiber volley was obtained by subtracting the opsin potential from the potential measured after blocking glutamate receptors, either before or after muscimol. To determine the effect on intracellularly recorded EPSCs, we measured a baseline response in ACSF that contained CGP54626 before application of muscimol (Figure 5).
For IN:Pyr EPSC ratios and L2/3 connectivity analysis we performed paired recordings of neighboring interneurons and pyramidal cells separated by fewer than 50 μm. All pairs were recorded within 50-90 μm depth from the surface of the slice. For paired connectivity measurements between pyramidal cells and interneurons, we elicited a train of action potentials in one neuron while voltage clamping the other neuron to measure excitatory or inhibitory currents. The stimulus train consisted of ten 1 ms depolarizing steps at 12 Hz with a magnitude sufficient to reliably elicit an action potential on each step. EPSCs from pyramidal cells onto interneurons was measured by holding interneurons at −85 mV; IPSCs from interneurons onto pyramidal cells was measured by holding pyramidal cells at −40 mV.
QUANTIFICATION AND STATISTICAL ANALYSIS
Anatomical analysis
For all post-hoc analysis of tissue, we manually selected regions of interest within each confocal z-stack. First, we aligned the image to be perpendicular to the surface of the brain; then we cropped the image to include only the pia to the white matter, and to span 250 μm tangentially; finally, we only analyzed the subset of imaging frames from each confocal stack that exceeded half-maximal raw fluorescence to avoid including sections that were above or below the sample. Layer boundaries were assigned using size and density of cell bodies visualized with DAPI staining.
To quantify the density of axonal expression in the HVAs, we measured the profile of fluorescence intensity along the cortical depth. This was done by summing the raw fluorescence along the tangential dimension of the cortex. These depth-profiles were then aligned to the boundary between layer 1 and layer 2/3 and averaged across slices (4 per HVA per mouse). To compare axon density for all layers across HVAs, we first normalized the intensity to the maximum across areas, and then averaged across mice. This normalization serves to eliminate variability (e.g. injection amounts and incubation periods) across mice. To compare relative axon density distribution, we normalized to the maximum of the depth profile within each area, and then averaged across mice.
For quantification of single axon fluorescence intensity across areas, three regions of interest at the edge of each HVA were imaged at 63x. The distribution of pixel intensities was pooled across all HVAs and then fit with a single Gaussian. The signal threshold was set at 1 full-width at half-max above the peak of the distribution, below which pixel intensities were set to zero. For each area, pixel intensity values were summed and divided by the number of pixels above threshold in that area to obtain an area-specific average fluorescence intensity/pixel (Figure S1).
To quantify the density of interneurons in the HVAs, we developed a counting software written in MATLAB that allowed for the manual selection of fluorescent neurons. This allowed us to localize each neuron in the 3D volume of the coronal slice. Only interneurons with somas completely contained within the boundaries of an area were counted and cells were tracked across the z-plane to prevent over-counting of cells split between multiple slices for a particular area. Interneuron densities were calculated using the raw counts and a tissue volume determined by the laminar boundaries, imaging window, and slice thickness. Interneuron densities were z-scored within layer and cell type for comparing across areas. For calculating distances between areas and layers, PV and SOM densities were averaged across animals and grouped across all layers and then z-scored, and quantified as the Euclidean distance between each pair of areas within a layer or layers within an area.
Electrophysiology analysis
Spike width, input resistance (Rin), spike frequency adaptation, and frequency-current relationship (FI) were calculated from current injection steps. Spike width was calculated as the full-width at half-max of the spike amplitude for the first spike at minimal depolarizing step intensity. Input resistance was calculated as the slope of the subthreshold V-I curve around rest. Membrane time constant was calculated by fitting a single exponential to the first 100 ms of membrane voltage change during the smallest hyperpolarizing step (−100 pA). Vm sag was calculated as the amplitude of the change in minimum membrane potential between initial and steady state time windows at the beginning and end of the hyperpolarizing step, respectively, that produced an initial membrane potential closest to −90 mV. Spike frequency adaptation was determined using the smallest depolarizing current injection sweep that elicited at least a 5 Hz initial firing rate, calculated as ISIlast/ISIfirst. FI curves were generated by dividing total spikes during the current injection by the duration of the injection.
For analysis of optogenetically-evoked EPSCs in simultaneously recorded interneurons and pyramidal cells, we averaged over all sweeps and smoothed the trial-averaged trace over 0.25 ms bins. IN:Pyr EPSC ratio, latency, rise time, and cumulative charge distributions were all calculated using this trial-averaged, smoothed trace. For calculating IN:Pyr EPSC ratios, we wanted to measure the maximum EPSC amplitude of V1 excitation and not polysynaptic connections from other pyramidal cells. Therefore, we used only the “early-onset” EPSC amplitude, which was defined as the maximum amplitude in the time window 4 ms after laser onset. This time window was determined based on comparison of EPSCs measured in the baseline condition and with muscimol to inactivate local, polysynaptic inputs (Figure 5). We chose a time window that minimized the contribution of these late, presumably polysynaptic inputs. EPSC maximum amplitude in each interneuron was divided by the paired pyramidal cell EPSC maximum amplitude to generate the IN:Pyr EPSC ratio. Latency was calculated as time from laser onset to 20% of the EPSC maximum amplitude. Rise time was calculated as time from 20 to 80% of EPSC maximum amplitude.
To determine the relative distribution of excitatory charge in time, we created cumulative distributions of charge for all of the EPSCs recorded. Using each cell’s calculated latency, we generated a cumulative distribution of charge over a 100 ms window beginning from response onset. The half-point was defined as the time to 50% of the total charge. Normalized traces used to compare currents across medial and lateral areas (Figure 6E) were generated by dividing the EPSC in each cell by its respective EPSC maximum amplitude that was used to calculate IN:Pyr ratios.
Spontaneous EPSCs were isolated using Fourier transform-based deconvolution performed on the baseline period of traces recorded for IN:Pyr EPSC ratios. Events were identified by first deconvolving the baseline trace with an initial spontaneous EPSC template (Templateinitial; F −1[F(baseline EPSC)/F(Templateinitial)], where F is the discrete Fourier transform and F −1 is its inverse)94. The initial template used to isolate events had a τrise = 0.3 ms and τdecay = 1.2 ms. The deconvolution was band-pass filtered between 1-200 Hz and events that passed a threshold of 5 standard deviations above the peak of the noise distribution and were averaged to generate a new spontaneous event template for each cell type (Templatefitted). Fitted templates were generated from spontaneous events pooled across areas because area-specific template fits were highly correlated (r>0.98 for all area comparisons within cell type). These templates were then used to deconvolve optogenetically-evoked EPSCs and determine the time course of events in each cell type and area (F −1[F(optogenetically-evoked EPSC)/F(Templatefitted)]). Traces were averaged in 2.5 ms time bins and normalized to the peak of the response before being averaged across cells. Late maximum of the deconvolution analysis was calculated as the peak normalized response in each cell in the time window 7.5-15 ms after laser onset.
For testing connectivity in each direction in our paired recordings, we averaged across all sweeps in the voltage clamped neuron. Current amplitudes were determined using the response to the first stimulus in the train. We categorized neurons as connected if there was an excitatory or inhibitory response to any of the stimuli in the train. Responses above baseline were determined with visual inspection but blinded to the area the pair was recorded in.
Statistics
Unless otherwise stated in the results all data are reported as mean ± SEM. Ratio data and unitary currents were log-transformed prior to statistical analysis. In other cases data were assumed to be normal. Unless otherwise stated in the results, we used standard parametric tests (i.e. t-test and ANOVA with post-hoc Tukey test) and adjusted for multiple comparisons when necessary. In all figures, if the data passed an ANOVA, we made all possible post-hoc comparisons, but only illustrated significant results. Sample numbers are indicated in the figure legends.
In our measurements of intrinsic properties, we obtained recordings from many more pyramidal cells than from either interneuron type. To assess how this affected sensitivity of detecting significant differences across areas, we performed power analyses on the Rin data set for each cell type using the following equation:
Where ∣μ1 – μ2∣ is the difference between two groups, σ is the standard deviation of the data, ntotal is the total number of cells for a cell type, and nareas was 4. α and β are the probabilities of type I and type II errors, respectively. is the value from the standard normal distribution holding below it, Z1–β is the value from the standard normal distribution holding 1- β below it. With a 5% level of significance and 80% power to detect a difference, values of and Z1–β were 1.96 and 0.84, respectively. With this analysis, we found a slight difference in sensitivity: for pyramidal cells we could detect a difference of 12 MΩ between groups, whereas in PV and SOM cells the threshold was 18 MΩ.
Similarly, since pyramidal cell to interneuron connections were sparse in lateral areas, we obtained fewer samples for connection amplitude than in medial areas. With our samples size of connected pairs, the smallest difference we could resolve is 14 and 19 pA for PV and SOM uEPSCs, respectively. Thus, in these experiments we cannot rule out the possibility that there is some small difference that was not statistically significant due to relatively small sample size.
Supplementary Material
Highlights.
Higher visual areas (HVAs) in the mouse have distinct anatomy and physiology
V1 inputs to lateral areas excite interneurons more strongly than in medial areas
This different input is balanced by stronger recurrent connectivity in medial areas
Acknowledgements
We thank Emily Burke, Courtney Dobrott, Kyra Leonard, and Wenjuan Kong for technical assistance with viral injections and post hoc histology; Kevin Murgas for technical assistance with cranial window surgeries and dye injections, and intrinsic imaging; Greg Horwitz and Yasmine El-Shamayleh for the gift of a virus encoding oChIEF; Court Hull and members of the Hull and Glickfeld laboratories for suggestions on experimental design and analysis; and Greg Field and Stephen Lisberger for comments on the manuscript. This work was funded by grants DP2EY025439 and R01EY031328 to L.L.G. and F31EY031941 to J.Y.L.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gnadt JW, and Andersen RA (1988). Memory related motor planning activity in posterior parietal cortex of macaque. Exp. brain Res 70, 216–220. [DOI] [PubMed] [Google Scholar]
- 2.Douglas RJ, and Martin KAC (2004). Neuronal Circuits of the Neocortex. Annu. Rev. Neurosci 27, 419–451. [DOI] [PubMed] [Google Scholar]
- 3.Rushworth MFS, Noonan MAP, Boorman ED, Walton ME, and Behrens TE (2011). Frontal Cortex and Reward-Guided Learning and Decision-Making. Neuron 70, 1054–1069. [DOI] [PubMed] [Google Scholar]
- 4.Tremblay R, Lee S, and Rudy B (2016). GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma W.pei, Liu B. hua, Li Y. tang, Josh Huang Z, Zhang LI, and Tao HW (2010). Visual Representations by Cortical Somatostatin Inhibitory Neurons—Selective But with Weak and Delayed Responses. J. Neurosci 30, 14371 LP–14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapfer C, Glickfeld LL, Atallah BV, and Scanziani M (2007). Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat. Neurosci 10, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li LY, Xiong XR, Ibrahim LA, Yuan W, Tao HW, and Zhang LI (2015). Differential Receptive Field Properties of Parvalbumin and Somatostatin Inhibitory Neurons in Mouse Auditory Cortex. Cereb. Cortex 25, 1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang HJ, Chung H, Rowland JM, Richards BA, Kohl MM, and Kwag J (2020). Distinct roles of parvalbumin and somatostatin interneurons in gating the synchronization of spike times in the neocortex. Sci. Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Souza RD, Meier AM, Bista P, Wang Q, and Burkhalter A (2016). Recruitment of inhibition and excitation across mouse visual cortex depends on the hierarchy of interconnecting areas. Elife 5, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas RJ, Koch C, Mahowald M, Martin KAC, and Suarez HH (1995). Recurrent excitation in neocortical circuits. Science (80-.). 269, 981–985. [DOI] [PubMed] [Google Scholar]
- 11.Gabernet L, Jadhav SP, Feldman DE, Carandini M, and Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron 48, 315–327. [DOI] [PubMed] [Google Scholar]
- 12.Yang W, Carrasquillo Y, Hooks BM, Nerbonne JM, and Burkhalter A (2013). Distinct Balance of Excitation and Inhibition in an Interareal Feedforward and Feedback Circuit of Mouse Visual Cortex. J. Neurosci 33, 17373–17384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Leary DDM (1989). Do cortical areas emerge from a protocortex? Trends Neurosci. 12, 400–406. [DOI] [PubMed] [Google Scholar]
- 14.Livingstone MS, and Hubel DH (1984). Anatomy and physiology of a color system in the primate visual cortex. J. Neurosci 4, 309–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livingstone M, and Hubel D (1988). Segregation of form, color, movement, and depth: anatomy, physiology, and perception. Science (80-.). 240, 740 LP – 749. [DOI] [PubMed] [Google Scholar]
- 16.Movshon JA, and Newsome WT (1996). Visual response properties of striate cortical neurons projecting to area MT in macaque monkeys. J. Neurosci 16, 7733–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rauschecker JP (1995). Compensatory plasticity and sensory substitution in the cerebral cortex. Trends Neurosci. 18, 36–43. [DOI] [PubMed] [Google Scholar]
- 18.Takahata T, Komatsu Y, Watakabe A, Hashikawa T, Tochitani S, and Yamamori T (2009). Differential expression patterns of occ1-related genes in adult monkey visual cortex. Cereb. Cortex 19, 1937–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muñoz A, DeFelipe J, and Jones EG (2001). Patterns of GABABR1a,b receptor gene expression in monkey and human visual cortex. Cereb. Cortex 11, 104–113. [DOI] [PubMed] [Google Scholar]
- 20.Anderson KM, Krienen FM, Choi EY, Reinen JM, Yeo BTT, and Holmes AJ (2018). Gene expression links functional networks across cortex and striatum. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rakic P (1988). Specification of cerebral cortical areas. Science (80-. ). 241, 170–176. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, and Zador AM (2012). Differences in sensitivity to neural timing among cortical areas. J. Neurosci 32, 15142–15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luna VM, and Pettit DL (2010). Asymmetric rostro-caudal inhibition in the primary olfactory cortex. Nat. Neurosci 13, 533–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XJ (2020). Macroscopic gradients of synaptic excitation and inhibition in the neocortex. Nat. Rev. Neurosci 21, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher LN, and Williams SR (2019). Neocortical Topology Governs the Dendritic Integrative Capacity of Layer 5 Pyramidal Neurons. Neuron 101, 76–90.e4. [DOI] [PubMed] [Google Scholar]
- 26.Fulcher BD, Murray JD, Zerbi V, and Wang XJ (2019). Multimodal gradients across mouse cortex. Proc. Natl. Acad. Sci. U. S. A 116, 4689–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glickfeld LL, and Olsen SR (2017). Higher-Order Areas of the Mouse Visual Cortex. Annu. Rev. Vis. Sci 3, annurev-vision-102016-061331. [DOI] [PubMed] [Google Scholar]
- 28.Niell C, and Stryker M (2008). Highly Selective Receptive Fields in Mouse Visual Cortex. J. Neurosci 28, 7520–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubel DH, and Wiesel TN (1962). Receptive fields, binocular interaction and functional architecture in the cat’s visual cortex. J. Physiol 160, 106–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serre T, Kouh M, Cadieu C, Knoblich U, Kreiman G, and Poggio T (2005). A theory of object recognition : computations and circuits in the feedforward path of the ventral stream in primate visual cortex This version replaces the preliminary “ Halloween ” CBCL paper from November. 2005. Artif. Intell [Google Scholar]
- 31.Zemel RS, and Sejnowski TJ (1998). A model for encoding multiple object motions and self-motion in area MST of primate visual cortex. J. Neurosci 18, 531–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka K (1996). Inferotemporal Cortex and Object Vision. Annu. Rev. Neurosci 19, 109–139. [DOI] [PubMed] [Google Scholar]
- 33.Ungerleider LG, and Haxby JV (1994). ‘What’ and ‘where’ in the human brain. Curr. Opin. Neurobiol 4, 157–165. [DOI] [PubMed] [Google Scholar]
- 34.Andermann ML, Kerlin AM, Roumis DK, Glickfeld LL, and Reid RC (2011). Functional specialization of mouse higher visual cortical areas. Neuron 72, 1025–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murgas KA, Wilson AM, Michael V, and Glickfeld LL (2020). Unique spatial integration in mouse primary visual cortex and higher visual areas. J. Neurosci, 1919–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sit KK, and Goard MJ (2020). Distributed and retinotopically asymmetric processing of coherent motion in mouse visual cortex. Nat. Commun 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshel JH, Garrett ME, Nauhaus I, and Callaway EM (2011). Functional specialization of seven mouse visual cortical areas. Neuron 72, 1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siegle JH, Jia X, Durand S, Gale S, Bennett C, Graddis N, Heller G, Ramirez TK, Choi H, Luviano JA, et al. (2021). Survey of spiking in the mouse visual system reveals functional hierarchy. Nature 592, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, and Burkhalter A (2007). Area Map of Mouse Visual Cortex. J Comp Neurol 502, 339–357. [DOI] [PubMed] [Google Scholar]
- 40.Kim M-H, Znamenskiy P, Iacaruso MF, and Mrsic-Flogel TD (2018). Segregated Subnetworks of Intracortical Projection Neurons in Primary Visual Cortex. Neuron, 1–9. [DOI] [PubMed] [Google Scholar]
- 41.Han Y, Kebschull JM, Campbell RAA, Cowan D, Imhof F, Zador AM, and Mrsic-Flogel TD (2018). The logic of single-cell projections from visual cortex. Nature 556, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glickfeld LL, Andermann ML, Bonin V, and Reid RC (2013). Cortico-cortical projections in mouse visual cortex are functionally target specific. Nat. Neurosci 16, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsui T, and Ohki K (2013). Target dependence of orientation and direction selectivity of corticocortical projection neurons in the mouse V1. Front. Neural Circuits 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blot A, Roth MM, Gasler I, Javadzadeh M, Imhof F, and Hofer SB (2021). Visual intracortical and transthalamic pathways carry distinct information to cortical areas. Neuron 109, 1996–2008.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y, Yang GR, Pradhan K, Venkataraju KU, Bota M, García del Molino LC, Fitzgerald G, Ram K, He M, Levine JM, et al. (2017). Brain-wide Maps Reveal Stereotyped Cell-Type-Based Cortical Architecture and Subcortical Sexual Dimorphism. Cell 171, 456–469.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garrett ME, Nauhaus I, Marshel JH, and Callaway EM (2014). Topography and areal organization of mouse visual cortex. J. Neurosci 34, 12587–12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Gao E, and Burkhalter A (2011). Gateways of ventral and dorsal streams in mouse visual cortex. J. Neurosci 31, 1905–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q, Sporns O, and Burkhalter A (2012). Network analysis of corticocortical connections reveals ventral and dorsal processing streams in mouse visual cortex. J. Neurosci 32, 4386–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adesnik H (2017). Synaptic Mechanisms of Feature Coding in the Visual Cortex of Awake Mice. Neuron 95, 1147–1159.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson DE, Scholl B, and Fitzpatrick D (2018). Differential tuning of excitation and inhibition shapes direction selectivity in ferret visual cortex. Nature 560, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Priebe NJ, and Ferster D (2008). Inhibition, Spike Threshold, and Stimulus Selectivity in Primary Visual Cortex. Neuron 57, 482–497. [DOI] [PubMed] [Google Scholar]
- 52.Adesnik H, Bruns W, Taniguchi H, Huang ZJ, and Scanziani M (2012). A neural circuit for spatial summation in visual cortex. Nature 490, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reinhold K, Lien AD, and Scanziani M (2015). Distinct recurrent versus afferent dynamics in cortical visual processing. Nat. Neurosci 18, 1789–1797. [DOI] [PubMed] [Google Scholar]
- 54.Bean BP (2007). The action potential in mammalian central neurons. Nat. Rev. Neurosci 8, 451–465. [DOI] [PubMed] [Google Scholar]
- 55.Connors BW, Gutnick MJ, and Prince DA (1982). Electrophysiological properties of neocortical neurons in vitro. J. Neurophysiol 48, 1302–1320. [DOI] [PubMed] [Google Scholar]
- 56.Yamashita T, Pala A, Pedrido L, Kremer Y, Welker E, and Petersen CCH (2013). Membrane potential dynamics of neocortical projection neurons driving target-specific signals. Neuron 80, 1477–1490. [DOI] [PubMed] [Google Scholar]
- 57.D’Souza RD, and Burkhalter A (2017). A laminar organization for selective cortico-cortical communication. Front. Neuroanat 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu H, Gan J, and Jonas P (2014). Fast-spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science (80-. ). 345, 1255263. [DOI] [PubMed] [Google Scholar]
- 59.Riedemann T (2019). Diversity and Function of Somatostatin-Expressing Interneurons in the Cerebral Cortex. Int. J. Mol. Sci 20, 2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beierlein M, Gibson JR, and Connors BW (2003). Two Dynamically Distinct Inhibitory Networks in Layer 4 of the Neocortex. J. Neurophysiol 90, 2987–3000. [DOI] [PubMed] [Google Scholar]
- 61.Pouille F, and Scanziani M (2004). Routing of spike series by dynamic circuits in the hippocampus. Nature 429, 717–723. [DOI] [PubMed] [Google Scholar]
- 62.Yavorska I, and Wehr M (2016). Somatostatin-expressing inhibitory interneurons in cortical circuits. Front. Neural Circuits 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silberberg G, and Markram H (2007). Disynaptic Inhibition between Neocortical Pyramidal Cells Mediated by Martinotti Cells. Neuron 53, 735–746. [DOI] [PubMed] [Google Scholar]
- 64.Hu H, and Agmon A (2016). Differential excitation of distally versus proximally targeting cortical interneurons by unitary thalamocortical bursts. J. Neurosci 36, 6906–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khibnik LA, Cho KKA, and Bear MF (2010). Relative contribution of feedforward excitatory connections to expression of ocular dominance plasticity in layer 4 of visual cortex. Neuron 66, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seeman SC, Campagnola L, Davoudian PA, Hoggarth A, Hage TA, Bosma-Moody A, Baker CA, Lee JH, Mihalas S, Teeter C, et al. (2018). Sparse recurrent excitatory connectivity in the microcircuit of the adult mouse and human cortex. Elife 7, e37349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avermann M, Tomm C, Mateo C, Gerstner W, and Petersen CCH (2012). Microcircuits of excitatory and inhibitory neurons in layer 2/3 of mouse barrel cortex. J. Neurophysiol 107, 3116–3134. [DOI] [PubMed] [Google Scholar]
- 68.Yoshimura Y, and Callaway EM (2005). Fine-scale specificity of cortical networks depends on inhibitory cell type and connectivity. Nat. Neurosci 8, 1552–1559. [DOI] [PubMed] [Google Scholar]
- 69.Churchland AK, and Lisberger SG (2005). Discharge properties of MST neurons that project to the frontal pursuit area in macaque monkeys. J. Neurophysiol 94, 1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mishkin M, Ungerleider LG, and Macko KA (1983). Object vision and spatial vision: two cortical pathways. Trends Neurosci. 6, 414–417. [Google Scholar]
- 71.Wang Q, and Burkhalter A (2013). Stream-related preferences of inputs to the superior colliculus from areas of dorsal and ventral streams of mouse visual cortex. J. Neurosci 33, 1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Isaacson JS, and Scanziani M (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cardin JA (2018). Inhibitory Interneurons Regulate Temporal Precision and Correlations in Cortical Circuits. Trends Neurosci. 40, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu H, Cavendish JZ, and Agmon A (2013). Not all that glitters is gold: off-target recombination in the somatostatin-IRES-Cre mouse line labels a subset of fast-spiking interneurons. Front. Neural Circuits 7, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vasuta C, Artinian J, Laplante I, Hebert-Seropian S, Elayoubi K, and Lacaille JC (2015). Metaplastic regulation of CA1 schaffer collateral pathway plasticity by hebbian MGluR1a-mediated plasticity at excitatory synapses onto somatostatin-expressing interneurons. eNeuro 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miska NJ, Richter LMA, Cary BA, Gjorgjieva J, and Turrigiano GG (2018). Sensory experience inversely regulates feedforward and feedback excitation-inhibition ratio in rodent visual cortex. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanzeni A, Akitake B, Goldbach HC, Leedy CE, Brunel N, and Histed MH (2020). Inhibition stabilization is a widespread property of cortical networks. Elife 9, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ozeki H, Finn IM, Schaffer ES, Miller KD, and Ferster D (2009). Inhibitory Stabilization of the Cortical Network Underlies Visual Surround Suppression. Neuron 62, 578–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kato HK, Asinof SK, and Isaacson JS (2017). Network-Level Control of Frequency Tuning in Auditory Cortex. Neuron 95, 412–423.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hofer SB, Ko H, Pichler B, Vogelstein J, Ros H, Zeng H, Lein E, Lesica NA, and Mrsic-Flogel TD (2011). Differential connectivity and response dynamics of excitatory and inhibitory neurons in visual cortex. Nat. Neurosci 14, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fino E, and Yuste R (2011). Dense Inhibitory Connectivity in Neocortex. Neuron 69, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu Y, Qiao W, Liu K, Zhong H, and Yao H (2015). Control of response reliability by parvalbumin-expressing interneurons in visual cortex. Nat. Commun 6, 6802. [DOI] [PubMed] [Google Scholar]
- 83.Carandini M, and Heeger DJ (2012). Normalization as a canonical neural computation. Nat. Rev. Neurosci 13, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nienborg H, Hasenstaub A, Nauhaus I, Taniguchi H, Josh Huang Z, and Callaway EM (2013). Contrast dependence and differential contributions from somatostatin- and parvalbumin-expressing neurons to spatial integration in mouse V1. J. Neurosci 33, 11145–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cossell L, Iacaruso MF, Muir DR, Houlton R, Sader EN, Ko H, Hofer SB, and Mrsic-Flogel TD (2015). Functional organization of excitatory synaptic strength in primary visual cortex. Nature 518, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chance FS, Nelson SB, and Abbott LF (1998). Synaptic depression and the temporal response characteristics of V1 cells. J. Neurosci 18, 4785–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin JY, Lin MZ, Steinbach P, and Tsien RY (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J 96, 1803–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bell KA, Shim H, Chen CK, and McQuiston AR (2011). Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain α4 and β2 subunits. Neuropharmacology 61, 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zingg B, Chou X. lin, Zhang Z. gang, Mesik L, Liang F, Tao HW, and Zhang LI (2017). AAV-Mediated Anterograde Transsynaptic Tagging: Mapping Corticocollicular Input-Defined Neural Pathways for Defense Behaviors. Neuron 93, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Castle MJ, Gershenson ZT, Giles AR, Holzbaur ELF, and Wolfe JH (2014). Adeno-associated virus serotypes 1, 8, and 9 share conserved mechanisms for anterograde and retrograde axonal transport. Hum. Gene Ther 25, 705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nat. Protoc 9, 2515–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Diamond JS, and Jahr CE (1995). Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15, 1097–1107. [DOI] [PubMed] [Google Scholar]
- 95.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and analyzed anatomical and electrophysiological data supporting this manuscript are available on Mendeley Data. DOIs are listed in the key resources table. Data were analyzed offline using custom MATLAB code. The code for data analysis is available on Github, linked in the key resources table. Any additional information required for reanalysis is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| rAAV2/1-Flex-rev-oChIEF-tdTomato | Addgene89 | Plasmid #: 30541 |
| AAV8-Syn-Chronos-tdTomato | Addgene87 | Plasmid #: 62726 |
| AAV9-Syn-Chronos-GFP-WPRE-bGH | Addgene87 | Plasmid #: 59170 |
| AAV1.CB7.CI.eGFP.WPRE.rBG | University of Pennsylvania Viral Vector Core | Lot #: CS0326 |
| Experimental models: Organisms/strains | ||
| EMX1-IRES-Cre | Jackson Labs | Stock #: 005628 |
| SST-IRES-Cre | Jackson Labs | Stock #: 013044 |
| PV-Cre | Jackson Labs | Stock #: 008069 |
| Ai14 | Jackson Labs | Stock #: 007914 |
| Ai3 | Jackson Labs | Stock #: 007903 |
| Ai148 | Jackson Labs | Stock #: 030328 |
| Ai162 | Jackson Labs | Stock #: 031562 |
| C57/B6J | Jackson Labs | Stock #: 000664 |
| Chemicals, peptides, and recombinant proteins | ||
| Dextran, Alexa Fluor 488, 10,000 MW | ThermoFisher | Cat #: D22910 |
| Dextran, Alexa Fluor 594, 10,000 MW | ThermoFisher | Cat #: D22913 |
| NBQX | Tocris Bioscience | Cat #: 1044; CAS: 479347-86-9 |
| D-APV | Tocris Bioscience | Cat #: 0106; CAS: 79055-68-8 |
| CGP 54626 hydrochloride | Tocris Bioscience | Cat #: 1088; 149184-21-4 |
| Muscimol | Tocris Bioscience | Cat #: 0289; CAS: 2763-96-4 |
| Deposited data | ||
| Raw and analyzed data | This paper | Mendeley Data DOI: 10.17632/rtnkp83yxs.1 |
| Software and algorithms | ||
| Micromanager | NIH93 | https://micro-manager.org |
| ImageJ | NIH95 | https://imagej.nih.gov/ij/ |
| MWorks | MWorks | https://mworks.github.io/ |
| pClamp 10 Software Suite | Molecular Devices | |
| MATLAB | Mathworks | https://www.mathworks.com/ |
| Custom MATLAB scripts | This paper | https://github.com/Glickfeld-And-Hull-Laboratories/Manuscripts/tree/master/HVA%20inhibition |