

Abstract
Multi-drug-resistance (MDR) is a severe public health concern worldwide, and its containment is more challenging in developing countries due to poor antimicrobial resistance (AMR) surveillance and irrational use of antibiotics. The current study investigated 100 clinical E. coli isolates and revealed that 98% of them were MDR. PCR analysis using 25 selected isolates showed the predominance of metallo-β-lactamase gene blaNDM (80%) and ESBL genes blaOXA (48%) and blaCTX-M-15 (32%). The AmpC gene was detected in 68% of the isolates, while 32% was tetC positive. Notably, 34% of the isolates were resistant to carbapenem. Whole genome sequence (WGS) analysis of an extensively drug-resistant (XDR) isolate (L16) revealed the presence of the notorious sequence type 131 responsible for multi-drug-resistant infections, multiple antibiotic resistance genes (ARGs), virulence genes, and mobile genetic elements that pose risks to environmental transmission. Our results indicate that MDR is alarmingly increasing in Bangladesh that critically limits the treatment option against infections and contributes to further aggravation to the prevailing situation of MDR worldwide. The findings of this study will be valuable in designing sustainable strategies to contain MDR in the region.
Subject terms: Biochemistry, Biological techniques, Computational biology and bioinformatics, Genetics, Microbiology, Molecular biology, Pathogenesis
Introduction
Bacterial resistance to antibiotics is a global health crisis with far-reaching consequences. By 2050, Drug-resistant infections may cause 10 million deaths annually, with approximately 90% of the predicted deaths to happen in Asia and Africa1,2. Indiscriminate use of antibiotics, inappropriate dosing, and incomplete treatment in both humans and animals are significant factors leading to the development of resistance in bacteria3. The situation is deteriorated further by the rapid spread of resistance genes. Several intrinsic factors such as point mutation, gene amplification, and extrinsic factors like horizontal transfer of resistance genes between bacteria within and across species by transposons, integrins, or plasmids have been suggested for the development of resistance, that cannot be restricted once developed even by restricting the antibiotic usage4,5. Significant rise in the presence of extended-spectrum beta-lactamases (ESBLs) such as TEM, SHV, OXA, CMY, and CTX-M, and AmpC beta-lactamases in recent years has made the situation alarming, along with the presence of carbapenem resistance that is mediated chiefly by blaOXA and blaNDM variants3,6,7. Understanding the antimicrobial resistance determinants in bacteria at the genetic level plays a critical role in controlling and understanding the ever-changing dynamics of resistance8.
On the other hand, continuous accumulation of multiple mutations leads to possible emergence of genomes resistant to antimicrobials9. Hence, whole-genome sequencing (WGS) of microorganisms has become a powerful approach for screening antibiotic resistance and evaluating the number of mutations and functions of the mutated genes5,10. WGS also holds great promise in developing newer antibiotics, controlling antimicrobial resistance, and enhancing diagnostics and public health microbiology11,12.
Escherichia coli (E. coli) is one of the major causes of nosocomial and community-associated infections, including respiratory tract infections (RTIs), urinary tract infections (UTIs), and enteric infections in Bangladesh13,14. The clinical threat posed by E. coli is mainly attributed to its ability to rapidly acquire antibiotic resistance through multiple mechanisms. Moreover, the emergence of E. coli strains with ESBL and AmpC β-lactamase carriers also render them resistant to other antibiotics such as aminogylcosides, fluororquinolones, tetracycline, chloramphenicol, sulfonamides, and carbapenems—the last resort in the treatment of many life-threatening infections15. E. coli is also considered a good indicator of antibiotic resistance in bacterial communities as it has been known to be a significant reservoir of genes coding for antimicrobial drug resistance14. Robust antibiotic resistance surveillance strategies combined with high-throughput tools such as WGS are common in developed countries. However, in resource-limited countries such as Bangladesh, data to monitor trends and susceptibility patterns against antibiotics are infrequently produced2,16. During the last decade, several studies conducted in Bangladesh revealed the irrational use of antimicrobials, prevalence of self-treatment, and incomplete therapy that contributed to the emergence of resistant bacterial strains2,17,18. Lack of systematic and detailed studies on antibiotic resistance patterns from Bangladesh often makes physicians prescribe multiple antibiotics, including broad-spectrum antibiotics, which further aggravates the AMR situation in Bangladesh, where infectious diseases still hold the highest morbidity and mortality rate18,19.
In this study, we determined the antibiotic resistance pattern in E. coli isolates from clinical urine and sputum specimens collected from pathological laboratories of Dhaka, Bangladesh. The study also screened the ESBL genes (TEM, CTX-M-15, SHV, and OXA), AmpC, tetracycline, and carbapenem resistance genes such as OXA-47 and NDM in MDR isolates. We also report the whole genome characterization of an XDR E. coli strain isolated from a urine sample. In order to understand the phylogenomic changes, presence of genetic elements, and virulence, a whole-genome analysis of the isolate, including its plasmids and comparative multilocus sequence typing (MLST), was carried out. The concordance between WGS-based AMR prediction and the phenotype identified from conventional antimicrobial susceptibility tests was also evaluated.
Results
The current study has been conducted on 100 E. coli isolates obtained from clinical specimens collected from different diagnostic centers in Dhaka, Bangladesh. Isolates were subjected to antibiotic susceptibility testing after biochemical characterization. This was followed by PCR-based detection for ARGs and WGS analysis. A schematic illustration of the workflow of the entire procedure and analysis is shown in Fig. 1.
Figure 1.
Schematic illustration of workflow of the entire procedure and analysis.
Bacterial isolates and biochemical characterization
A total of 100 E. coli isolates were isolated from 100 clinical samples, which consisted of 90% urine and 10% sputum specimens. Based on gram-staining, morphological features, culture characteristics, and biochemical characterization, the isolates were confirmed as E. coli. All E. coli isolates appeared bright pink on MacConkey agar, and the colonies showed a characteristic metallic green sheen when cultured on EMB agar. All isolates were gram-negative and fermented dextrose, maltose, lactose, sucrose, and mannitol with acid and gas production. Catalase, Methyl red, and indole test of the E. coli isolates showed positive results, but the V-P test was negative.
Antimicrobial resistance profile
An antibiotic susceptibility test was carried out on all the 100 isolates against 12 different antibiotics. As shown in Table 1, the highest percentage of resistance was observed against amoxicillin (98%), followed by cefuroxime (75%) and cotrimoxazole (62%). Sixty percent of the E. coli isolates were resistant to piperacillin-tazobactam, a beta-lactam/beta-lactamase inhibitor combination. Resistance to gentamicin, ciprofloxacin, norfloxacin, chloramphenicol, and azithromycin varied between 34% and 49%. A significantly high degree of resistance was observed to carabapenems, 30% to meropenem and 38% to imipenem.
Table 1.
Antibiotic susceptibility patterns of E. coli isolates (n = 100).
| Antibiotic | Resistance% | Intermediate% | Sensitive% |
|---|---|---|---|
| Penicillins | |||
| Amoxicillin (10 μg) | 98 | 2 | |
| Aminoglycosides | |||
| Gentamicin (10 μg) | 39 | 13 | 48 |
| Fluoroquinolones | |||
| Ciprofloxacin (5 µg) | 34 | 8 | 58 |
| Norfloxacin (10 μg) | 39 | 5 | 56 |
| Cephalosporins | |||
| Cefuroxime (30 μg) | 75 | 5 | 20 |
| Carbapenems | |||
| Imipenem (10 μg) | 38 | 9 | 53 |
| Meropenem (10 μg) | 30 | 8 | 62 |
| Phenicols | |||
| Chloramphenicol (30 μg) | 40 | 12 | 48 |
| Macrolides | |||
| Azithromycin (15 μg) | 49 | 17 | 34 |
| Tetracyclines | |||
| Tetracycline (30 μg) | 55 | 12 | 33 |
| Sulfonamides | |||
| Cotrimoxazole (25 μg) | 62 | 11 | 27 |
| Penicillins with β-lactamase inhibitors (Beta-lactam/beta-lactamase inhibitor) | |||
| Piperacillin-tazobactam (100/10 μg) | 60 | 40 | |
n number of isolates.
Prevalence of multiple drug resistance
In this study, 96% of isolates showed a MAR index greater than 0.3, whereas 64% showed a MAR index above 0.5. Figure 2A shows a high prevalence of multiple antibiotic resistance among the isolates where 98% of the isolates were MDR, and the highest frequency of resistance to multiple antibiotics classes observed was 38.78%, with resistance to six different classes of antibiotics. Moreover, 16% of the isolates showed extensive drug resistance (XDR), with one of the isolates being resistant to all the antibiotics used in this study.
Figure 2.
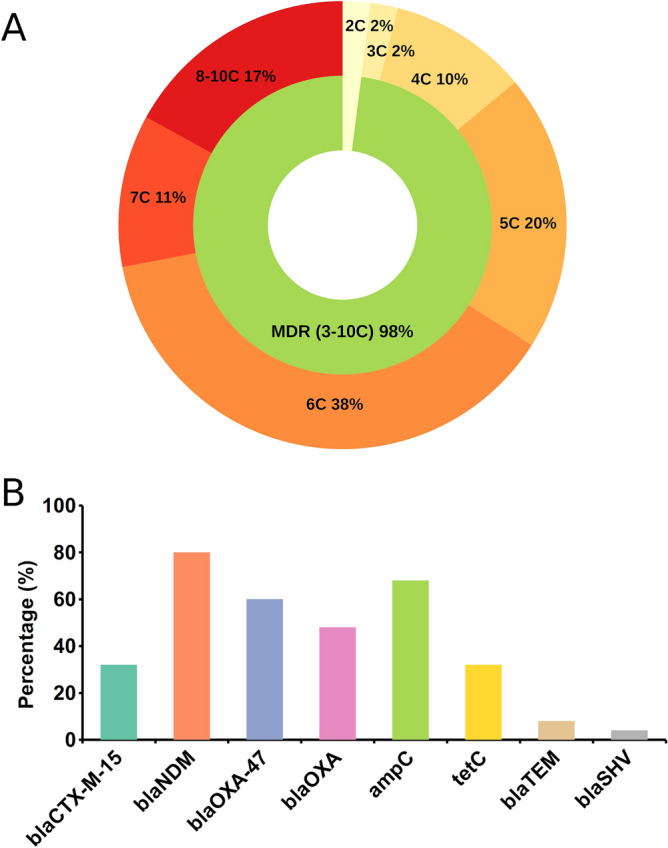
Prevalence of multiple antibiotic resistance among the isolates. (A) Distribution of resistance pattern against different classes of antibiotics. (B) Prevalence of ARGs among the tested E. coli isolates. C, classes.
Detection of antibiotic resistance genes
Based on the antibiogram data, 25 isolates were studied for the presence of antibiotic resistance genes. The overall prevalence of ARGs among the investigated E. coli isolates with their resistance phenotype is shown in Table 2. All the isolates were positive for at least one AMR gene. The most prevalent gene was metallo-β-lactamase blaNDM harbored by 80% of the isolates, which confers resistance to carbapenems. Another gene, blaOXA-47, which confers resistance to carbapenems, was present in 60% of the isolates. A total of 17 (68%) isolates were AmpC-positive, while tetC gene was detected in 8 (32%) isolates. Among the ESBL genes, blaCTX-M-15, blaTEM and blaOXA were detected in 32%, 8%, 48% of the isolates, respectively, while only one isolate (4%) exhibited the presence of blaSHV. The prevalence of different ARGs is shown in Fig. 2B.
Table 2.
Phenotypic antibiotic resistance and distribution of resistance genes among E. coli isolates.
| Isolate | Resistance phenotype | Resistance ESBL genes including carbapenemnase | AmpC and tetC genes |
|---|---|---|---|
| GSU-1 | Amx, Gtm, Cip, Nor, Cxm, Imp, Mem, Azm, Te, Ctm, Ptz | blaNDM, blaOXA-47 | ampC |
| GSU-2 | Amx, Cxm, Imp, C, Te, Ctm | blaOXA-47 | |
| GSU-3 | Amx, Cxm, Imp, Azm, Te, Ctm | blaOXA, blaCTX, blaOXA-47 | ampC |
| GSU-4 | Amx, Nor, Cxm, Imp, Ctm, Ptz | blaNDM | |
| GSU-5 (L16) | Amx, Gtm, Cip, Nor, Cxm, Imp, Mem, C, Azm, Te, Ctm, Ptz | blaTEM, blaCTX, blaOXA, blaOXA-47, blaNDM | ampC, tetC |
| GSU-6 | Amx, Gtm, Cip, Nor, Cxm, Imp, Azm, Te, Ctm | blaOXA-47, blaNDM | ampC, tetC |
| GSU-7 | Amx, Cxm, Imp, C, Azm, Te | blaCTX, blaOXA, blaNDM | ampC |
| GSU-8 | Amx, Nor, Cxm, Imp, Azm, Te, Ctm, Ptz | blaOXA, blaSHV, blaNDM, blaOXA-47 | ampC, tetC |
| GSU-9 | Amx, Cip, Cxm, Imp, Azm, Te, Ctm, Ptz | blaNDM | ampC |
| GSU-10 | Amx, Gtm, Nor, Cxm, Imp, Mem, Azm, Ctm, Te, Ptz | blaOXA, blaNDM, blaOXA-47 | |
| GSU-11 | Amx, Cip, Nor, Cxm, Imp, Mem, Ctm, Ptz | blaCTX, blaNDM, blaOXA-47 | ampC |
| GSU-12 | Amx, Cip, Nor, Cxm, Imp, Mem, Azm, Ptz | blaCTX, blaNDM | ampC, tetC |
| GSU-13 | Amx, Cip, Nor, Cxm, Imp, Azm, Te, Ctm, Ptz | blaOXA, blaTEM, blaNDM, blaOXA-47 | ampC, tetC |
| GSU-14 | Amx, Cip, Nor, Cxm, Imp, C, Azm,Te, Ctm, Ptz | blaOXA, blaNDM | tetC |
| GSU-15 | Amx, Nor, Cxm, Imp, Ptz | blaOXA | |
| GSU-16 | Amx, Gtm, Cip, Nor, Cxm, Imp, C, Azm,Ctm, Ptz | blaOXA-47, blaNDM | ampC |
| GSU-17 | Amx, Gtm, Nor, Cxm, Imp, C, Ptz | blaOXA, blaNDM | ampC |
| GSU-18 | Amx, Gtm, Nor, Cxm, Imp, Azm, Ctm, Ptz | blaOXA, blaNDM | ampC |
| GSU-19 | Amx, Gtm, Cxm, Imp, Azm, Ctm, | blaNDM | ampC |
| GSU-20 | Amx,Nor, Cxm, Imp, C, Azm,Te,Ctm,Ptz | blaOXA-47, blaNDM | ampC, tetC |
| GSU-21 | Amx, Nor, Cxm, Imp, Azm, | blaOXA-47 | |
| GSU-22 | Amx, Cip, Cxm, Imp,Ptz | blaOXA | |
| GSS-23 | Amx, Gtm, Nor, Cxm, Imp, Mem, C, Ctm, Ptz | blaCTX, blaOXA, blaOXA-47, blaNDM | ampC |
| GSS-24 | Amx, Gtm, Cip, Nor, Cxm, Imp, Mem,Te, Ctm, Ptz | blaCTX, blaNDM, blaOXA-47 | |
| GSS-25 | Amx, Gtm, Cip, Nor, Cxm, Imp, C, Te, Ctm, Ptz | blaCTX, blaOXA-47, blaNDM | ampC, tetC |
Amx amoxicillin, Cip ciprofloxacin, Gtm gentamycin, Imp imipenem, Mem meropenem, Nor norfloxacin, C Chloramphenicol, Te tetracycline, Ptz piperacillin & tazobactam, Cxm cefuroxime, Ctm cotrimoxazole, Azm azithromycin.
Whole-genome sequencing
We performed a paired-end WGS of the bacterial isolate showing the highest antimicrobial resistance. Sequencing analysis revealed that the quality of both the forward and the reverse reads was considerably high. There were approximately 2.8 million sequences, and no read had poor quality. Duplicate counts were 29% and 18.5%, and GC contents were 50% and 51% for the forward and reverse reads, respectively. For most reads, the mean quality values (Phred scores) across different base positions were greater than 30 (Fig. S1).
Genome assembly and annotation
The reads were subjected to the comprehensive genome analysis service at PATRIC20 where the strain was designated as Escherichia coli Strain-L16 (L16). This genome appears to be of good quality based on the annotation statistics and comparing other genomes in PATRIC within this same species. L16 reads were trimmed and assembled using unicycler, a de novo genome assembly tool21. There were 118 contigs, an estimated genome length of 5,137,608 bp, and an average GC content of 50.61%. The N50 length, defined as the shortest sequence length at 50% of the genome, is 225,781 bp. The L50 count, which is defined as the smallest number of contigs whose length sum produces N50, is 8. The lengths of the smallest and the largest contigs were 511 bp and 673,523 bp, respectively.
The L16 genome was annotated using the RAST tool kit (RASTtk)22 and assigned a unique genome identifier (562.73579) in PATRIC. This genome is in the superkingdom Bacteria and was annotated using genetic code 11. The taxonomy of this genome is: cellular organisms > Bacteria > Proteobacteria > Gammaproteobacteria > Enterobacterales > Enterobacteriaceae > Escherichia > Escherichia coli. This genome has 5222 protein-coding sequences (CDS), 79 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. The annotation included 593 hypothetical proteins and 4629 proteins with functional assignments. The functional assignments included 1319 proteins with Enzyme Commission (EC) numbers, 1098 with Gene Ontology (GO) assignments, and 925 proteins mapped to KEGG pathways. PATRIC annotation includes two types of protein families, and this genome has 5004 proteins belonging to the genus-specific protein families (PLFams), and 5088 proteins belong to the cross-genus protein families (PGFams). Figure S2 shows a circular representation of the L16 annotation features.
A subsystem is a set of proteins that implement a specific biological process or structural complex, and PATRIC annotation includes an analysis of the subsystems unique to each genome23. We compared the subsystems of L16 with two E. coli reference genomes, K-12 substr. MG1655 and O157:H7 str. Sakai (Fig. S3). K-12 substr. MG1655 is a nonpathogenic strain, whereas O157:H7 str. Sakai is a pathogenic strain. Overall, the subsystems of L16 are very similar to those of two reference genomes. The number of genes annotated in the subsystem superclasses of cellular processes, energy, membrane transport, and metabolism was slightly higher in the genome L16.
Many annotated genes in L16 have homology with known genes for antibiotic resistance, drug targets, transporters, and virulence factors. Table S1 provides a comparison of annotated gene numbers among L16 and two reference genomes.
In silico typing
Based on the allelic combination of the seven genes, isolate L16 was identified as having the sequence type (ST) 131. ST131 is a highly virulent pathogenic clone24 of the subgroup extraintestinal pathogenic E. coli (ExPEC)25.
Based on the wzx and wzy genes encoding cell surface O antigens and the fliC gene encoding the flagellar antigen H, L16 was identified as serotype O25:H4. In silico phylotyping predicted phylogroup B2 for the L16 genome.
Antimicrobial resistance genes
Annotated AMR genes in L16 are listed in Table 3, which shows that majority of them are involved in conferring resistance via efflux pumps and modified antibiotic targets.
Table 3.
AMR genes of L16 annotated in PATRIC.
| AMR mechanism | Genes |
|---|---|
| Antibiotic activation enzyme | KatG |
| Antibiotic inactivation enzyme | AAC(3)-II,III,IV,VI,VIII,IX,X, BlaEC family, CatB family, CTX-M family, OXA-1 family |
| Antibiotic resistance gene cluster, cassette, or operon | MarA, MarB, MarR |
| Antibiotic target in susceptible species | Alr, Ddl, dxr, EF-G, EF-Tu, folA, Dfr, folP, gyrA, gyrB, inhA, fabI, Iso-tRNA, kasA, MurA, rho, rpoB, rpoC, S10p, S12p |
| Antibiotic target protection protein | BcrC |
| Efflux pump conferring antibiotic resistance | AcrAB-TolC, AcrAD-TolC, AcrEF-TolC, AcrZ, EmrAB-TolC, EmrD, EmrE, EmrKY-TolC, MacA, MacB, MdfA/Cmr, MdtABC-TolC, MdtEF-TolC, MdtL, MdtM, SugE, TolC/OpmH |
| Gene conferring resistance via absence | gidB |
| Protein altering cell wall charge conferring antibiotic resistance | GdpD, PgsA |
| Regulator modulating expression of antibiotic resistance genes | AcrAB-TolC, EmrAB-TolC, GadE, H-NS, OxyR |
Analysis of L16 resistome using the RGI tool as mentioned in methods identified 8 perfect hits and 53 strict hits (Supplementary Dataset 1).
Notably, the presence of the CTX-M-15 allele renders the E. coli clone resistant to extended-spectrum-beta-lactamases (ESBLs). The L16 genome is positive for several genes and mutations, such as mdtH, emrA, emrB, emrR, mutated gyrA, and mutated parC, conferring fluoroquinolone resistance. Moreover, this clone also has several multidrug-resistant genes including acrB, AcrS, cpxA, evgA, hns, gadW, TolC, mdtM, mdtF, and marA. Together, the presence of multiple resistant genes is expected to make the strain resistant to aminoglycosides, carbapenem, cephalosporins, cephamycin, fluoroquinolones, macrolides, penicillin, and tetracyclines (Table 3, Supplementary Dataset 1).
Virulence factor genes
In PATRIC, L16 was annotated for 95 genes with homology to genes of the VFDB (Supplementary Dataset 2). There are 35 genes with iron uptake functions and 28 genes with adherence functions. Sixty of these genes were predicted to be of E. coli origin, whereas 35 genes originated in other species.
Mobile genetic elements
In the L16 genome, 14 mobile genetic elements (MGEs) were predicted using the MobileElementFinder web tool26. Three of the MGEs are miniature inverted repeats, and the remaining 11 are insertion sequences (Table 4). MITEEc1 is an IS630 family MGE (https://www-is.biotoul.fr/) and an Enterobacterial repetitive intergenic consensus (ERIC) sequence with possible functions in mRNA stability27. In the L16 genome, one copy of MITEEc1 is located in contig 1 between the genes YbhJ (encodes a conitase family protein) and YbhI (encodes an inner membrane protein). Contig 10 of L16 has a 2410 bp-log IS21 family transposase. The longest MGE predicted in L16 was the IS682, an IS66 family MGE consisting of three open reading frames (ORFs) including two accessory genes TnpA, TnpB, and a DDE transposase element (https://www-is.biotoul.fr/).
Table 4.
Mobile genetic elements (MGEs) in the L16 genome.
| Name | Type | Allele_length | E_value | Identity (%) | Coverage (%) |
|---|---|---|---|---|---|
| MITEEc1 | Miniature inverted repeat | 123 | 2.3E−49 | 96.7 | 100.0 |
| ISKpn8 | Insertion sequence | 1443 | 0 | 94.7 | 100.0 |
| MITEEc1 | Miniature inverted repeat | 123 | 2.0E−36 | 90.2 | 100.0 |
| ISEc10 | Insertion sequence | 2410 | 0 | 100.0 | 100.0 |
| MITEEc1 | Miniature inverted repeat | 122 | 8.0E−53 | 98.4 | 99.2 |
| ISEc1 | Insertion sequence | 1291 | 0 | 96.6 | 100.0 |
| ISEc53 | Insertion sequence | 1885 | 0 | 100.0 | 100.0 |
| ISEc38 | Insertion sequence | 1722 | 0 | 95.2 | 100.0 |
| IS30 | Insertion sequence | 1221 | 0 | 99.8 | 100.0 |
| IS629 | Insertion sequence | 1310 | 0 | 94.7 | 100.0 |
| ISKpn37 | Insertion sequence | 1262 | 0 | 97.1 | 99.3 |
| IS629 | Insertion sequence | 1310 | 0 | 94.1 | 100.0 |
| IS682 | Insertion sequence | 2532 | 0 | 90.9 | 99.6 |
| IS26 | Insertion sequence | 820 | 0 | 100.0 | 100.0 |
Pangenome analysis
E. coli harbors an open pangenome that is evolving continuously. Rasko et al. determined a reservoir of more than 13,000 E. coli genes in 200828. We performed pangenomic analysis of L16 and 46 genome assemblies of Bangladeshi origin from NCBI (https://www.ncbi.nlm.nih.gov/assembly). The pangenome consisted of 14,057 genes, of which 2937 are in the core (Fig. 3A). The pangenome matrix shows clusters of genes and dendrogram of the isolates (Fig. 3B). With 13 other isolates, L16 is in a cluster where more genes are in the core than two other clusters. The isolate ASM1676131 is the closest relative in the dendrogram. Notably, all but one genome (ASM1676075v1) are ST131 clones. Analyzing the gene presence-absence, we found 26 unique genes (Table S2) in L16. The L16 genome has at least 90 prophages, including four unique prophages listed in Table S2.
Figure 3.

Pangenome analysis of L16 and 46 other strains of Bangladeshi origin. (A) Number of genes in the pangenome. (B) Roary matrix shows clustering of genes among 47 strains.
Considering the global dissemination of the E. coli ST131, we next explored the relatedness of the L16 genome to all publicly available complete E. coli genomes. We downloaded 1253 assemblies from NCBI (https://www.ncbi.nlm.nih.gov/assembly, accessed on April 11, 2021) and mapped the L16 genome to each of the public E. coli genomes using lastz29. After filtering out the hits below 90% match, average percent identity and total mapped sequences were tabulated for each chromosome from the assemblies and plotted on a scatter chart (Fig. 4A). Except for one outlier, all public sequences matched with 3.5–5.1 Mbp of the L16 genome. The eight genomes showing the highest identity are shown in Fig. 4B. These isolates were collected between 2009 and 2018 from Australia, Germany, Italy, Japan, Sweden, and the USA (Table S3).
Figure 4.

Mapping of L16 genome to other public E. coli genomes. (A) Mapping of L16 genome to all complete public E. coli genomes. Inset shows eight genomes with longest mapped regions. (B) Mapping to eight closely related genomes (amplified view of the inset in (A). (C) Circos presentation of L16 mapping to three closest public genomes.
From lazt mapping, we identified three public E. coli genomes with the highest mapped sequences for L16 and visualized the mapping in a circos plot (Fig. 4C). Apparently, the L16 genome is almost identical to those public genomes (Fig. 4C). Next, we aligned the AMR protein features of L16 to the eight closely related genomes (Fig. S4). As expected, the features were almost identical among the genomes except that L16 carries the chloramphenicol O-acetyltransferase (Fig. S4).
Discussion
Among all the WHO regions, southeast Asian countries including Bangladesh are at the highest risk of AMR18, and ESBL-producing carbapenem-resistant E. coli are among the global priority pathogens list of WHO categorized as critical30. The strategies to contain AMR rely on a robust antibiotic surveillance system, assessing the threats imposed due to the emergence of MDR and implementation of the findings. Scarcity of data on AMR patterns and minimal information on the presence of ARGs and molecular determinants of resistance prompted this study in which we investigated resistance phenotypes among 100 E. coli isolates collected from clinical specimens obtained from diagnostic centers in Dhaka, Bangladesh.
In the current investigation, 98% of isolates were MDR, defined as resistant to three or more antibiotics classes, which is alarming (Fig. 2A). The highest resistance was observed against amoxicillin (98%) which is more or less similar to recent study reports from Bangladesh13,18,31. We observed higher resistance to cefuroxime (75%) compared to 53. 81% as reported by Nahar et al. in 201731, which could be attributed to the sampling area. Among the antimicrobials tested, imipenem, ciprofloxacin, norfloxacin, and meropenem were found as the most effective agents against E. coli in this study. However, the rate of resistance against carbapenem was much higher (> 30%) in comparison to recent reports involving E. coli isolates from clinical specimens by Nahar et al. 2017 (< 1.5%)31, Hosaain et al. 2021 (< 3%)32 and Nobel et al. 2021 (< 10%)13 from Bangladesh. A study from India reported the presence of 29.54% carbapenem resistance in E. coli isolates33, which is in line with the current study. However, some studies from India and Nepal34,35 reported much higher resistance to meropenem (60–75%). Carbapenems are considered the last shelter for gram-negative bacteria treatment, but carbapenem resistance is increasing in Southeast Asian countries, which is dangerous. Resistance observed against gentamicin (39%) and cotrimoxazole (62%) in this study is consistent with the findings of Nahar et al.31. However, some studies from Bangladesh showed lesser resistance to gentamicin (< 15%), cotrimoxazole (< 50%), and also to tetracycline (34%)32,36. These variations could be attributed to the prevailing usage and drug abuse in the study area, which warrants stringent and continuous surveillance on antibiotic resistance patterns. Resistance to other drugs are in agreement with some previously published reports31,36 with some minor variations. Sixty four percent of the isolates showed MAR index greater than 0.5, which indicates a very high prevalence of multiple drug resistance among clinical E. coli isolates in Bangladesh. Out of the 100 isolates, 38.78% showed resistance to 6 out of 10 classes of antibiotics tested, and 16% of isolates were XDR. Although the number of isolates from sputum samples (10) was smaller than urine samples (90), similar or higher resistance was observed against all the antibiotics except cefuroxime, meropenem, and tetracycline. The percentage of isolates from urine with a MAR index greater than 0.3 was higher (96.67%) than sputum isolates (90%). Reports emanating from developing countries like Bangladesh indicate a high prevalence of multidrug-resistant E. coli in clinical as well as in community settings such as drinking water and poultry2,3,14. However, limited information is available on the prevalence and the genotypic characteristics of antibiotic-resistant strains associated with different ecological niches in Bangladesh3,14. The widespread availability of antimicrobials without prescription in the country, self-medication, non-compliance of dosage, and irrational use across agriculture, poultry and health sectors have contributed significantly to the spread of resistant strains in Bangladesh18. The presence of antimicrobial residues such as tetracycline, ciprofloxacin, and amoxicillin have been observed in a high percentage of poultry meat and eggs18,37. Discharge of untreated medical wastes resulting in the presence of high levels of resistant E. coli in the water has also been reported38. This implies co-selection of resistance towards multiple classes of antibiotics leading to the rapid dissemination of MDR in same or distantly related species, which could be the greatest threat to public health in the twenty-first century. The prevalence of ARGs is the major contributor to the development of multidrug and pandrug-resistant bacteria. However, data on the prevalence of antibiotic resistance genes in clinical E. coli isolates and the molecular basis of resistance is very limited in Bangladesh2. In this study, blaNDM was predominant (80%) among the eight tested antibiotic resistance genes. Among metallo-β-lactamases (MBLs), NDM-1 has received wide attention because of its broad hydrolytic activity on β-lactam antibiotics, including carbapenems, and carriage of NDM-1 enables drug resistance to move between communities and hospitals. The prevalence of blaNDM-1 has been previously reported in several studies39,40. Safain et al. reported NDM-1 (55%) as the most prevalent resistance gene in Bangladesh2. In the current study, we found that 80% of the isolates harbored blaNDM-1, which is alarming as therapeutic options against NDM-1 producers are rare. NDM-producers have been reported to be on the rise in India, Pakistan, and Nepal, indicating inappropriate and non-prescription antibiotic use as a probable cause of the development of resistance in this subcontinent2,6,35. Apart from NDM, other genes frequently associated with carbapenem resistance are blaOXA-47 and blaOXA-1, which were detected in 60% and 48% of the isolates. These findings are in accordance with the high phenotypic resistance observed against carbapenems (Tables 1, 2). The blaOXA-1 gene has frequently been associated with genes encoding extended-spectrum β-lactamases (ESBLs) and suggested imparting resistance to penicillin/inhibitor combinations41. Out of the 25 isolates tested for ARGs, 15 were resistant to piperacillin/tazobactam and ten harbored the blaOXA-1 gene (67%) (Tables 1, 2) which is approximately three times higher than that reported by Livermore et al. (12/59)41. Co-occurrences of blaNDM along with variants of blaOXA have been reported previously42. Similar results were observed in our study with the presence of blaNDM + blaOXA-47 and blaNDM + blaOXA-1 combinations in 12 and 9 isolates, respectively, while 5 isolates showed co-occurrence of blaNDM along with blaOXA-1 and blaOXA-47 genes. Frequent association of blaOXA-1 with the CTX-M-15, the most abundant ESBL determinant in human E. coli isolates, has been reported from diverse geographical origins, contributing to resistance towards beta-lactam lactamase inhibitor combinations42. Among the isolates tested for ARGs, 32% showed the presence of blaCTX-M-15. A recent study reported a higher prevalence of blaCTX-M-15 (52%)43 among ESBL-producing E. coli isolates, causing extraintestinal infections in Bangladesh. A study from Egypt reported dominance of CTX-M-15 (87%) among CTX-M genotypes44. Isolates producing β-lactamases such as CTX-M-15 are associated with clonal lineages such as ST131, which spread extensively via MGEs, leading to a rapid increase in the prevalence of urinary tract and bloodstream infections worldwide43,45. The other most frequently described enzymes in E. coli include TEM and SHV types in which point mutations are suggested to give rise to ESBLs44. Although the abundant presence of blaTEM has been described in several studies, including some from Bangladesh, the current study revealed the presence of blaTEM only in 8% of the isolates, which is lower than a previously published report from Bangladesh46. The presence of blaSHV was least in this study (4%) which is in line with the findings of Ahsan and Islam46. As the representatives of efflux mechanism of tetracycline, tetA and tetC genes are predominant in E. coli47. In this study, we determined the presence of tetC, which is higher (32%) than some previous studies48,49, indicating increased resistance to tetracycline. AmpC β-lactamases are a major clinical concern. They confer resistance to a wide range of β-lactam drugs, including penicillins, cephamycin, 1st-3rd generation cephalosporins, and classical β-lactamase inhibitors like clavulanic acid and tazobactam50,51. The expression level of the AmpC gene could be changed due to mutations in the promoter region which implies that phenotypic resistance detection against beta-lactam antibiotics52,53 is vital for clinical interventions. In this study, all the AmpC positive isolates showed 100% resistance to three (amoxicillin, imipenem, and cefuroxime) out of five beta-lactam antibiotics tested. In contrast, resistance against piperacillin-tazobactam and imipenem was observed in 76.47% and 29.41% of the isolates, respectively. Also, the AmpC gene was the second most prevalent among the ARGs, with 68% of the tested isolates being positive for AmpC.
The higher prevalence of AmpC beta-lactamases is also reported by Satter et al., 2020 from Bangladesh54. However, Ahsan and Islam reported a much lower prevalence (6%)46. This calls for continuous surveillance studies to fill the gaps in knowledge regarding the prevalence of ARGs. In Asia, the percentage of amp C positive clinical E. coli isolates varies over a wider range55, with the highest prevalence in India and Nepal35,56.
In the present study, we investigated the genome of an extensively drug-resistant isolate designated as L16 that showed resistance against all the antibiotics and presence of all the ARGs tested except SHV to determine the molecular mechanisms of resistance. L16 also showed the presence of multiple resistant genes (Table 3) to make the strain resistant to aminoglycosides, carbapenem, cephalosporins, cephamycin, fluoroquinolones, macrolides, penicillin, and tetracyclines (Table 3, Supplementary Dataset 1). These findings strongly support the AMR phenotype of L16 determined by antibiotic susceptibility test as well as PCR assay (Table 2). Many annotated genes in L16 showed homology with known genes for antibiotic resistance, drug targets, transporters, and virulence factors. MLST, one of the gold standards for determining epidemiological relatedness of organisms, showed that the isolate L16 belonged to the sequence type (ST) 131. ST131 has been notorious for causing worldwide pandemic multidrug-resistant infections crossing borders of countries, communities, and hospitals57. A serious concern for public health, the ST131 clones show broad resistance to extended-spectrum beta-lactams, fluoroquinolones, and also to carbapenems6,57. The isolate L16 belonged to phylogroup B2, which is predominant in blood and urinary tract infections that have a poor prognosis58 and also related to inflammatory bowel disease (IBD)59. L16 was annotated for 95 genes with homology to virulence genes of the VFDB (Supplementary Dataset 2), which shows the pathogenicity of the clone. The extreme pathogenicity of ST131 clones has been attributed to the presence of a large number of virulence factors, in addition to the appearance of multidrug-resistant genes24. The study of mobile genetic elements showed transposase ISEc10 apart from other MGEs, which might lead to overproduction of the AmpC-type beta-lactamase60. This also supports the findings from the PCR assay. Also, the longest MGE predicted in L16 was the IS682, which belongs to the IS66 family that is usually present in gram-negative rods and cocci61,62, and an association has been proposed between the presence of Helicobacter pylori (H. pylori) genes TnpA and TnpB and development of gastrointestinal cancer63. The pangenome analysis carried out in this study revealed 26 unique genes (Table S2) in L16 with four unique prophages. Although the functions of prophage genes are still enigmatic, they could contribute to the bacterial acquisition of novel genetic information64. Two sopB genes of L16 encode for the SopB proteins with suggested roles in E. coli F plasmid partitioning65,66.
In this study, AMR protein features of L16 were aligned to the eight closely related genomes (Fig. S3). As expected, the features were almost identical among the genomes except that L16 carries the chloramphenicol O-acetyltransferase. This enzyme is known to confer chloramphenicol resistance to pathogens via acetylation-mediated inactivation of the drug67,68. The findings from the analysis of the L16 genome demonstrate the great potential of WGS techniques in providing adequate and reliable data for surveillance and monitoring of antimicrobial resistance. However, the high cost and a lack of expertise in sequencing and bioinformatics data analysis are significant challenges in resource-limited countries. The phenotypic resistance pattern observed in the current study shows that conventional antimicrobial susceptibility testing complements the genotypically predicted resistance and molecular determinants. Hence, a combined approach will be the most appropriate for monitoring resistance. Such comprehensive investigations with an integrated approach are highly recommended to fully understand the AMR landscape and follow the dynamic virulence of the rapidly evolving pathogens in Bangladesh. The overall findings of the present study will provide helpful information to the local authorities towards the development of a robust antimicrobial stewardship program.
This study has several limitations. We investigated clinical samples from the Dhaka division of Bangladesh, which might not represent the overall situation in Bangladesh. A more extensive study including all the broader territories or divisions and a larger sample size is highly desirable to monitor the trends in resistance patterns over a time period. Determination of ESBL phenotypes could have been a valuable addition to this study from the epidemiological perspective as unexpressed ESBL genes in antibiotic-susceptible isolates may lead to horizontal gene transfer through such strains69. Also, screening more isolates for the presence of ARGs and WGS analysis and inclusion of ARGs for each class of antibiotics would provide better correlation between phenotypic resistance and molecular markers. The presence of mobile genetic elements and whether the resistance genes are plasmid-encoded may further be studied to ascertain the threats associated with the transmission of resistance.
Conclusion
High resistance rate to multiple antibiotics including carbapenems combined with multiple ARGs and sequence type 131 in clinical isolates, as revealed in this study, suggests that the situation is alarming in Bangladesh where irrational use of antibiotics combined with inadequate AMR surveillance and facilities to detect MDR and ESBL genes are common. The findings of this study strongly support the urgent need for a robust, comprehensive, and regular antibiotic surveillance system and the development of rapid diagnostic services to guide antibiotic treatment to check further emergence of dreadful isolates like L16. The inclusion of modern techniques such as continuous WGS level molecular surveillance is highly desirable to track the spread of MDR pathogens in Bangladesh and fill the current gaps in the knowledge of the molecular mechanisms of resistance.
Materials and methods
Sampling and isolates
A total of 100 clinical samples including urine and sputum were collected from different diagnostic centers (Popular Diagnostic Center, Dhanmondi; Popular Diagnostic Center, Mirpur; and the diagnostic lab of Bangabandhu Sheikh Mujib Medical University) located in Dhaka city, Bangladesh during January 2019 to April 2019. The samples were diluted serially and plated on MacConkey agar and Eosin Methylene Blue (EMB) agar media. Single colonies with the typical characteristic appearance and green metallic sheen on EMB agar media were picked and subcultured for obtaining pure isolates. The isolates were subjected to biochemical characterization as per standard microbiological procedures3. E. coli colonies were tested for growth on triple sugar iron agar (TSI) and lysine iron agar (LIA) and citrate utilization, urease production, indol fermentation, glucose degradation (methyl red test), and motility. One isolate per sample was selected and a total of 100 isolates were stored in nutrient broth with 15% glycerol at − 80 °C.
Antibiotic resistance profiling of the isolates
The isolates were subjected to antibiotic susceptibility tests against 12 antibiotics belonging to 10 different classes using Kirby-Bauer disk diffusion method in Mueller–Hinton agar media according to the guidelines of the Clinical and Laboratory Standard Institute70. The antimicrobial discs included were as follows: amoxycillin (10 µg), gentamycin (10 μg), ciprofloxacin (5 µg), norfloxacin (10 μg), cefuroxime (30 μg), imipenem (10 μg), meropenem (10 μg), chloramphenicol (30 μg), azithromycin (15 μg), tetracycline (30 μg), co-trimoxazole (25 μg) and piperacillin-tazobactam (100/10 μg). Results obtained were used to classify isolates as being resistant, intermediate resistant, or susceptible to a particular antibiotic using standard reference values according to Clinical and Laboratory Standards Institute70. E. coli strain ATCC 25922 was used as a reference. For each isolate, the multiple antibiotic resistance (MAR) index was calculated using the formula; MAR index = a/b, where a = number of isolates resistant to antibiotics and b = total number of antibiotics used. MDR (Multidrug-resistant) isolate was defined as the isolate showing resistance to three or more classes of antibiotics tested. Non-susceptibility to at least one agent in all but two or fewer antimicrobial categories were identified as XDR.
Genomic DNA extraction and detection of antibiotic resistance genes
Genomic DNA was extracted from overnight grown bacterial cultures using a Genomic DNA purification kit (Promega, Wizard DNA Purification Kit) according to the manufacturer’s instructions. The integrity of DNA samples was checked by electrophoresis on 0.8% (w/v) agarose gel. The isolates were tested for the presence of antibiotic resistance genes by PCR using primer sets listed in Table 5. The assays were carried out in 20 µL reaction mix containing 1 µL of forward and reverse primer each (20 pmol/µL), 2 µL of template DNA, and 10 µL of Taq master mix (Favorgen, USA). The following conditions were used: initial denaturation at 94 °C for 5 min, 30 cycles of denaturation at 94 °C for 30 s, annealing at corresponding temperature (Table 5) for 45 s, extension at 72 °C for 1 min, and final extension at 72 °C for 7 min. The presence of a target gene was confirmed by the presence of a band on the agarose gel.
Table 5.
Primers and PCR conditions for amplification of AMR genes.
| Target gene | Primer sequence (5’–3’) | Annealing temperature (°C) | Amplicon size (bp) | References |
|---|---|---|---|---|
| blaOXA-1 group |
F: ACACAATACATATCAACTTCGC R: AGTGTGTTTAGAATGGTGATC |
56 | 814 | 14 |
| blaOXA-47 |
F: TCAACTTTCAAGATCGCA R: GTGTGTTTAGAATGGTGA |
47 | 609 | 14 |
| blaTEM |
F: TCGGGGAAATGTGCGCG R: TGCTTAATCAGTGAGGACCC |
58 | 850 | 14 |
| blaCTX-M-15 |
F: CACACGTGGAATTTAGGGACT R: GCCGTCTAAGGCGATAAACA |
56 | 996 | 14 |
| blaSHV |
F: CACTCAAGGATGTATTGTG R: TTAGCGTTGCCAGTGCTCG |
56 | 861 | 14 |
| tetC |
F: CTTGAGAGCCTTCAACCCAG R: ATGGTCGTCATCTACCTGCC |
58 | 418 | 71 |
| blaNDM |
F: GGTGCATGCCCGGTGAAATC R: ATGCTGGCCTTGGGGAACG |
56 | 660 | 72 |
| ampC |
F: TGAGTTAGGTTCGGTCAGCA R: AGTATTTTGTTGCGGGATCG |
56 | 98 | 72 |
Genome sequencing, assembly and annotation
Whole genome sequencing (WGS) was carried out for the isolate (L16) that showed resistance to all the antibiotics used in the study. Genomic DNA from the cultured isolate was prepared using a Qiagen Genomic-tip kit, and the sequencing was conducted on an Illumina MiSeq platform at the facilities of Genome Research Institute, NSU, followed by Fastqc and MultiQC analysis. The reads were then subjected to the comprehensive genome analysis service at PATRIC20, where the strain was designated. The genome was annotated using the RAST tool kit (RASTtk)22 and assigned a unique genome identifier. Subsystems of the genome were compared with two Escherichia coli reference genomes using PATRIC.
In silico typing, AMR profile, virulence genes, and mobile genetic elements analysis
Sequence type (ST) was analyzed using the multilocus sequence typing (MLST) 2.0 tool73 from the Center for Genomic Epidemiology (CGE) webserver (https://cge.cbs.dtu.dk/). E. coli scheme 1 was selected, which employs seven housekeeping E. coli genes (adk, fumC, gyrB, icd, mdh, purA, recA) as proposed by Wirth et al. in 200674. To determine the serotype of L16, we used SerotypeFinder 2.075 from the CGE webserver. In silico phylotyping was performed by ClermonTyping76 which separates the isolates into 8 phylogroups (A, B1, B2, D, C, E, F, and cryptic clades) based on the presence or absence of 5 genes: chuA, yjaA, tspE4.C2, arpA and trpA. A k-mer-based AMR genes detection method was utilized in PATRIC’s genome annotation service, which assigned functional annotation to broad antibiotic resistance mechanisms. Isolate resistome was analyzed using the Resistance Gene Identifier (RGI) tool of the Comprehensive Antibiotic Resistance Database (CARD), where partial genes were excluded, and the predictions were made with contigs > 20,000 bp. Virulence genes were screened by VFDB, a reference database for bacterial virulence factors (http://cge.cbs.dtu.dk/services/VirulenceFinder/) that contains 139 virulence genes, including 44 ExPEC related genes77,78. Mobile genetic elements (MGEs) were predicted using the MobileElementFinder web tool26.
Pangenome analysis
For pangenomic analysis, we downloaded 46 genome assemblies from NCBI (https://www.ncbi.nlm.nih.gov/assembly, accessed on May 02, 2021) with the search term “(562[Taxonomy ID]) AND Bangladesh”. The test isolate and the 46 assemblies were subjected to Prokka79 annotation and Roary pangenome pipeline80.
Supplementary Information
Acknowledgements
Authors are thankful to Razmin Bari for English editing of the manuscript.
Author contributions
P.J. performed microbiological experiments, analyzed data, prepared figures, and wrote the manuscript. A.K.B. analyzed genomic data, prepared figures, and wrote the manuscript. P.K.S., T.R. and R.I. performed microbiological experiments and analyzed data. M.H. performed genomic sequencing experiments. H.M.R. conceptualized and supervised the study, analyzed the data, drafted the manuscript. All authors approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-02251-w.
References
- 1.Islam S, Aldstadt J, Aga D. Global antimicrobial resistance: A complex and dire threat with few definite answers. Trop. Med. Int. Health. 2019;24:658–662. doi: 10.1111/tmi.13230. [DOI] [PubMed] [Google Scholar]
- 2.Safain KS, et al. Situation of antibiotic resistance in Bangladesh and its association with resistance genes for horizontal transfer. BioRxiv. 2020 doi: 10.1101/2020.04.06.027391. [DOI] [Google Scholar]
- 3.Rahman MDM, et al. Isolation and molecular characterization of multidrug-resistant Escherichia coli from chicken meat. Sci. Rep. 2020;10:21999. doi: 10.1038/s41598-020-78367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kpoda DS, et al. Distribution of resistance genes encoding ESBLs in Enterobacteriaceae isolated from biological samples in health centers in Ouagadougou, Burkina Faso. BMC Res. Notes. 2018;11:471. doi: 10.1186/s13104-018-3581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng X, et al. Comparative genome analysis of an extensively drug-resistant isolate of avian sequence type 167 Escherichia coli strain sanji with novel in silico serotype O89b:H9. mSystems. 2019;4:e00242. doi: 10.1128/mSystems.00242-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ragupathi NKD, et al. First Indian report on genome-wide comparison of multidrug-resistant Escherichia coli from blood stream infections. PLoS ONE. 2020;15:e0220428. doi: 10.1371/journal.pone.0220428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Algammal AM, et al. Virulence-determinants and antibiotic-resistance genes of MDR-E. coli isolated from secondary infections following FMD-outbreak in cattle. Sci. Rep. 2020;10:19779. doi: 10.1038/s41598-020-75914-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deter HS, Hossain T, Butzin NC. Antibiotic tolerance is associated with a broad and complex transcriptional response in E. coli. Sci. Rep. 2021;11:6112. doi: 10.1038/s41598-021-85509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herring CD, et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- 10.Ben Zakour NL, et al. Sequential acquisition of virulence and fluoroquinolone resistance has shaped the evolution of Escherichia coli ST131. MBio. 2016;7:e00347. doi: 10.1128/mBio.00347-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasman H, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niemann S, et al. Genomic diversity among drug sensitive and multidrug resistant isolates of Mycobacterium tuberculosis with identical DNA fingerprints. PLoS ONE. 2009;4:e7407. doi: 10.1371/journal.pone.0007407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nobel F, et al. Prevalence of multidrug resistance patterns of Escherichia coli from suspected urinary tract infection in Mymensingh city, Bangladesh. J. Adv. Biotechnol. Exp. Ther. 2021;4:256. [Google Scholar]
- 14.Talukdar PK, et al. Antimicrobial resistance, virulence factors and genetic diversity of Escherichia coli isolates from household water supply in Dhaka, Bangladesh. PLoS ONE. 2013;8:e61090. doi: 10.1371/journal.pone.0061090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassan MA, et al. Insight into multidrug-resistant microorganisms from microbial infected diabetic foot ulcers. Diabetes Metab. Syndr. 2019;13:1261–1270. doi: 10.1016/j.dsx.2019.01.044. [DOI] [PubMed] [Google Scholar]
- 16.Nji E, et al. High prevalence of antibiotic resistance in commensal Escherichia coli from healthy human sources in community settings. Sci. Rep. 2021;11:3372. doi: 10.1038/s41598-021-82693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faiz A, Basher A. Antimicrobial resistance: Bangladesh experience. Reg. Health Forum. 2011;15:9. [Google Scholar]
- 18.Hoque R, et al. Tackling antimicrobial resistance in Bangladesh: A scoping review of policy and practice in human, animal and environment sectors. PLoS ONE. 2020;15:e0227947. doi: 10.1371/journal.pone.0227947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman MS, Huda S. Antimicrobial resistance and related issues: An overview of Bangladesh situation. Bangl. J. Pharmacol. 2014;9:218–224. [Google Scholar]
- 20.Wattam AR, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and Analysis Resource Center. Nucleic Acids Res. 2017;45:D535–D542. doi: 10.1093/nar/gkw1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brettin T, et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Overbeek R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolas-Chanoine M-H, et al. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J. Antimicrob. Chemother. 2008;61:273–281. doi: 10.1093/jac/dkm464. [DOI] [PubMed] [Google Scholar]
- 25.Russo TA, Johnson JR. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J. Infect. Dis. 2000;181:1753–1754. doi: 10.1086/315418. [DOI] [PubMed] [Google Scholar]
- 26.Johansson MHK, et al. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021;76:101–109. doi: 10.1093/jac/dkaa390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson LA, Sharp PM. Enterobacterial repetitive intergenic consensus (ERIC) sequences in Escherichia coli: Evolution and implications for ERIC-PCR. Mol. Biol. Evol. 2006;23:1156–1168. doi: 10.1093/molbev/msj125. [DOI] [PubMed] [Google Scholar]
- 28.Rasko DA, et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris RS. Improved Pairwise Alignment of Genomic DNA. The Pennsylvania State University; 2007. [Google Scholar]
- 30.WHO . Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis. WHO; 2017. [Google Scholar]
- 31.Nahar A, Hasnat S, Akhter H, Begum N. Evaluation of antimicrobial resistance pattern of uropathogens in a tertiary care hospital in Dhaka city, Bangladesh. South East Asia J. Public Health. 2017;7:12–18. [Google Scholar]
- 32.Hossain A, et al. Age and gender-specific antibiotic resistance patterns among Bangladeshi patients with urinary tract infection caused by Escherichia coli. Heliyon. 2020;6:e04161. doi: 10.1016/j.heliyon.2020.e04161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaggi N, et al. Carbapenem resistance in Escherichia coli and Klebsiella pneumoniae among Indian and international patients in North India. Acta Microbiol. Immunol. Hung. 2019;66:367–376. doi: 10.1556/030.66.2019.020. [DOI] [PubMed] [Google Scholar]
- 34.Ansari S, et al. Community acquired multi-drug resistant clinical isolates of Escherichia coli in a tertiary care center of Nepal. Antimicrob. Resist. Infect. Control. 2015;4:15. doi: 10.1186/s13756-015-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Govindaswamy A, et al. Prevalence and characterization of beta-lactamase-producing Escherichia coli isolates from a tertiary care hospital in India. J. Lab. Phys. 2019;11:123–127. doi: 10.4103/JLP.JLP_122_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haroon RM, et al. Antibacterial resistance patterns of bacteria isolated from clinical specimens at Uttara IbnSina Diagnostic Centre, Dhaka. Afr. J. Microbiol. Res. 2020;14:175–181. [Google Scholar]
- 37.Rashid M, Rakib MM, Hasan B. Antimicrobial-resistant and ESBL-producing Escherichia coli in different ecological niches in Bangladesh. Infect. Ecol. Epidemiol. 2015;5:26712. doi: 10.3402/iee.v5.26712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islam MS. Irrational use of drugs, healthcare level and healthcare expenditure in Bangladesh. Int. J. Health Econ. Policy. 2017;2:152. [Google Scholar]
- 39.Begum N, Shamsuzzaman SM. Emergence of carbapenemase-producing urinary isolates at a Tertiary Care Hospital in Dhaka, Bangladesh. Tzu Chi Med. J. 2016;28:94–98. doi: 10.1016/j.tcmj.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rakhi NN, Alam ASMRU, Sultana M, Rahaman MM, Hossain MA. Diversity of carbapenemases in clinical isolates: The emergence of blaVIM-5 in Bangladesh. J. Infect. Chemother. 2019;25:444–451. doi: 10.1016/j.jiac.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Livermore DM, et al. OXA-1 β-lactamase and non-susceptibility to penicillin/β-lactamase inhibitor combinations among ESBL-producing Escherichia coli. J. Antimicrob. Chemother. 2019;74:326–333. doi: 10.1093/jac/dky453. [DOI] [PubMed] [Google Scholar]
- 42.Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob. Agents Chemother. 2010;54:24–38. doi: 10.1128/AAC.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazumder R, Abdullah A, Ahmed D, Hussain A. High prevalence of blaCTX-M-15 gene among extended-spectrum β-lactamase-producing Escherichia coli isolates causing extraintestinal infections in Bangladesh. Antibiotics. 2020;9:796. doi: 10.3390/antibiotics9110796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramadan AA, Abdelaziz NA, Amin MA, Aziz RK. Novel bla CTX-M variants and genotype-phenotype correlations among clinical isolates of extended spectrum beta lactamase-producing Escherichia coli. Sci. Rep. 2019;9:4224. doi: 10.1038/s41598-019-39730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jadhav S, et al. Virulence characteristics and genetic affinities of multiple drug resistant uropathogenic Escherichia coli from a semi urban locality in India. PLoS ONE. 2011;6:e18063. doi: 10.1371/journal.pone.0018063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahsan S, Islam R. Beta-lactamase-producing Escherichia coli in Bangladesh: Their phenotypic and molecular characteristics. Dhaka Univ. J. Biol. Sci. 2019;28:71–81. [Google Scholar]
- 47.Zhang X-X, Zhang T, Fang HHP. Antibiotic resistance genes in water environment. Appl. Microbiol. Biotechnol. 2009;82:397–414. doi: 10.1007/s00253-008-1829-z. [DOI] [PubMed] [Google Scholar]
- 48.Huang J, Lan F, Lu Y, Li B. Characterization of integrons and antimicrobial resistance in Escherichia coli sequence type 131 isolates. Can. J. Infect. Dis. Med. Microbiol. 2020;2020:e3826186. doi: 10.1155/2020/3826186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh T, et al. Changing paradigm of antibiotic resistance amongst Escherichia coli isolates in Indian pediatric population. PLoS ONE. 2019;14:e0213850. doi: 10.1371/journal.pone.0213850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Estaleva CEL, et al. High prevalence of multidrug resistant ESBL- and plasmid mediated AmpC-producing clinical isolates of Escherichia coli at Maputo Central Hospital, Mozambique. BMC Infect. Dis. 2021;21:16. doi: 10.1186/s12879-020-05696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacoby GA. AmpC beta-lactamases. Clin. Microbiol. Rev. 2009;22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khari FIM, Karunakaran R, Rosli R, Tay ST. Genotypic and phenotypic detection of AmpC β-lactamases in Enterobacter spp. isolated from a Teaching Hospital in Malaysia. PLoS ONE. 2016;11:e0150643. doi: 10.1371/journal.pone.0150643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satter S, Mahbub H, Shamsuzzaman SM. Phenotypic and molecular characterization of AmpC beta-lactamase enzyme producing Escherichia coli and Klebsiella species isolated from A Tertiary Care Hospital in Bangladesh. Mymensingh Med. J. 2020;29:895–900. [PubMed] [Google Scholar]
- 55.Rensing KL, et al. Prevalence of plasmid-mediated AmpC in Enterobacteriaceae isolated from humans and from retail meat in Zagazig, Egypt. Antimicrob. Resist. Infect. Control. 2019;8:45. doi: 10.1186/s13756-019-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aryal SC, et al. Plasmid-mediated AmpC β-lactamase CITM and DHAM genes among gram-negative clinical isolates. Infect. Drug Resist. 2020;13:4249–4261. doi: 10.2147/IDR.S284751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rogers BA, Sidjabat HE, Paterson DL. Escherichia coli O25b-ST131: A pandemic, multiresistant, community-associated strain. J. Antimicrob. Chemother. 2011;66:1–14. doi: 10.1093/jac/dkq415. [DOI] [PubMed] [Google Scholar]
- 58.Chakraborty A, et al. Characterization of Escherichia coli phylogenetic groups associated with extraintestinal infections in South Indian population. Ann. Med. Health Sci. Res. 2015;5:241–246. doi: 10.4103/2141-9248.160192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fang X, et al. Escherichia coli B2 strains prevalent in inflammatory bowel disease patients have distinct metabolic capabilities that enable colonization of intestinal mucosa. BMC Syst. Biol. 2018;12:66. doi: 10.1186/s12918-018-0587-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haldorsen B, et al. The AmpC phenotype in Norwegian clinical isolates of Escherichia coli is associated with an acquired ISEcp1-like ampC element or hyperproduction of the endogenous AmpC. J. Antimicrob. Chemother. 2008;62:694–702. doi: 10.1093/jac/dkn257. [DOI] [PubMed] [Google Scholar]
- 61.Han C-G, Shiga Y, Tobe T, Sasakawa C, Ohtsubo E. Structural and functional characterization of IS679 and IS66-family elements. J. Bacteriol. 2001;183:4296–4304. doi: 10.1128/JB.183.14.4296-4304.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahillon J, Chandler M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abadi ATB, Mobarez AM, Bonten MJ, Wagenaar JA, Kusters JG. Clinical relevance of the cagA, tnpA and tnpB genes in Helicobacter pylori. BMC Gastroenterol. 2014;14:33. doi: 10.1186/1471-230X-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramisetty BCM, Sudhakari PA. Bacterial ‘grounded’ prophages: Hotspots for genetic renovation and innovation. Front. Genet. 2019 doi: 10.3389/fgene.2019.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanai R, et al. Molecular dissection of a protein SopB essential for Escherichia coli F plasmid partition. J. Biol. Chem. 1996;271:17469–17475. doi: 10.1074/jbc.271.29.17469. [DOI] [PubMed] [Google Scholar]
- 66.Hanai R, Arai Y. New roles of DNA and SopB in polymerization of SopA of Escherichia coli F plasmid. J. Biochem. (Tokyo) 2015;157:459–466. doi: 10.1093/jb/mvv003. [DOI] [PubMed] [Google Scholar]
- 67.Biswas T, Houghton JL, Garneau-Tsodikova S, Tsodikov OV. The structural basis for substrate versatility of chloramphenicol acetyltransferase CATI. Protein Sci. Publ. Protein Soc. 2012;21:520–530. doi: 10.1002/pro.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shaw WV. The Enzymatic acetylation of chloramphenicol by extracts of R factor-resistant Escherichia coli. J. Biol. Chem. 1967;242:687–693. [PubMed] [Google Scholar]
- 69.Zhang Z, et al. Characterization of unexpressed extended-spectrum beta-lactamase genes in antibiotic-sensitive Klebsiella pneumoniae isolates. Microb. Drug Resist. 2018;24:799–806. doi: 10.1089/mdr.2017.0018. [DOI] [PubMed] [Google Scholar]
- 70.CLSI . Performance Standards for Antimicrobial Disk Susceptibility Tests. Clinical and Laboratory Standards Institute; 2012. [Google Scholar]
- 71.Ng L-K, Martin I, Alfa M, Mulvey M. Multiplex PCR for the detection of tetracycline resistant genes. Mol. Cell. Probes. 2001;15:209–215. doi: 10.1006/mcpr.2001.0363. [DOI] [PubMed] [Google Scholar]
- 72.Fernando DM, et al. Detection of antibiotic resistance genes in source and drinking water samples from a first nations Community in Canada. Appl. Environ. Microbiol. 2016;82:4767–4775. doi: 10.1128/AEM.00798-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Larsen MV, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wirth T, et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Joensen KG, Tetzschner AMM, Iguchi A, Aarestrup FM, Scheutz F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 2015;53:2410–2426. doi: 10.1128/JCM.00008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beghain J, Bridier-Nahmias A, Le Nagard H, Denamur E, Clermont O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genomics. 2018;4:e000192. doi: 10.1099/mgen.0.000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joensen KG, et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014;52:1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tetzschner AMM, Johnson JR, Johnston BD, Lund O, Scheutz F. In silico genotyping of Escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J. Clin. Microbiol. 2020 doi: 10.1128/JCM.01269-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 80.Page AJ, et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.