

Abstract
Background:
Checkpoint inhibitors and currently approved cellular products for metastatic castration-resistant prostate cancer have not resulted in the revolutionary changes in outcomes compared to other solid tumors. Much of this lack of progress is attributed to the unique tumor microenvironment of prostate cancer that is often immunologically cold and immunosuppressive. These unique conditions emphasize the need for novel therapeutic options. In this review, we will discuss progress made in design of T- and NK cell immune engagers in addition to chimeric antigen receptor products specifically designed for prostate cancer that are currently under investigation in clinical trials.
Methods:
We searched peer-reviewed literature on the PubMed and the ClinicalTrials.gov databases for active clinical trials using the terms “bispecific T-cell engager,” “bispecific killer engager,” “trispecific killer engager,” “chimeric antigen receptor,” “metastatic castration-resistant prostate cancer,” and “neuroendocrine prostate cancer.”
Results:
Ten bispecific T-cell engager studies and nine chimeric antigen receptor-based products were found. Published data was compiled and presented based on therapeutic class.
Conclusions:
Multiple immune engagers and cell therapies are in the development pipeline and demonstrate promise to address barriers to better outcomes for metastatic castration-resistant prostate cancer patients.
Introduction:
Cell-based therapies for prostate cancer have been under investigation and in use for a number of decades. Sipuleucel T, an autologous cellular product, was granted regulatory approval in 2010 after a placebo-controlled phase III study demonstrated a reduction in death in favor of its use(1). Despite its approval, and frequent use in mCRPC, questions remain about its mechanism of action(2). Further, remissions are rare and the main clinical effect of Sipuleucel T is improvement in overall survival(1). Unlike future cell therapies, prior development of agents like Sipulecuel T(2), Prostvac(3), and GVAX(4) focused on using the cellular product to present antigen, and not the direct application or stimulation of effector T or NK cells(5). A new generation of cell therapeutic strategies, in which immune effector cells are directly engaged, is demonstrating promise across a range of malignant diseases. This review will evaluate such strategies and their potential for integration into the prostate cancer treatment landscape.
While the therapeutic revolution that accompanied immune checkpoint inhibitor (ICI) therapy has vastly affected outcomes and survival of patients diagnosed with metastatic solid tumors such as melanoma(6), non-small cell lung cancer(7), and many other malignancies(8, 9), a similar effect has been observed only in rare situations in prostate cancer(10).
Prostate cancer tends to be immunologically “cold,” defined as a lack of pro-inflammatory cytokine production and T-cell infiltration (11) (12). “Cold” prostate tumors tend to respond poorly to single agent PD-L1 or CTLA-4 axis inhibition with some exceptions due to the lack of pro-inflammatory features(13). Rare prostate tumors considered “hot” typically are associated with microsatellite instability (10), increased neoantigens which are recognized by the immune system as foreign (14), and are more likely to respond to checkpoint inhibition (15)
PD-L1 expression has been proposed as a tumor-based predictive biomarker that is associated with response to immunotherapy in many solid tumors (15). Approximately 35% of prostate cancers express PD-L1 and higher expression is associated with higher Gleason score and androgen receptor positivity (16). There is additional evidence for higher PD-L1 expression in CRPC (10%) and neuroendocrine subtype (41%) compared to androgen sensitive (5%) (17).
The Immunological Microenvironment and Prognosis in Prostate Cancer:
Compared more responsive tumor types, regulatory T-cells (Treg)- the presence of which i to result in suppression of T-effector cells - are the predominant infiltrating lymphocyte and peripheral blood lymphocyte subtype in prostate cancer patients(18, 19)– a finding reproducible in murine prostate dysplasia models(20), supporting a generally T-cell suppressed microenvironment.
Other immune subsets may affect outcome in prostate cancer more than Tregs. In one study, the presence or absence of Tregs in patient biopsy tissue was not associated with distant metastasis-free survival (DMFS), yet an improved DMFS was observed in patients with increased ratios of activated to resting NK cells, hazard ratio (HR) of 0.72 (95% CI 0.56–0.93) (P=0.01), mast cells HR (0.67 (0.49–0.9) (P=0.009), and dendritic cells HR 0.66 (0.51–0.86) (P==0.002)(21). This study again noted similar patterns of worse distant metastasis free survival (DMFS) in patients with lower normalized ratios of classically-activated pro-inflammatory macrophage type 1 (M1) to alternatively-activated anti-inflammatory macrophage type 2 (M2) HR 1.67 (1.22–2.3) (P=0.002)(21). Higher levels of M2 macrophages, which are immunosuppressive, express CD163+, and produce anti-inflammatory transforming growth factor beta (TGF-beta) in the microenvironment were associated with a trend to higher metastatic rate at diagnosis HR 1.98 (1.17–3.33, P=0.11) and a higher Gleason score at diagnosis(22). M1 tumor-infiltrating macrophages are associated with increased production of nitric oxide synthase 2 (NOS2), associated with increased respiratory burst and subsequent inflammatory microenvironment characteristics. Additionally, there was a lower M1/M2 ratio in prostate cancer compared to colorectal cancer, demonstrating the unique differences and challenges in dealing with the tumor microenvironment of prostate cancer compared to malignancies that typically respond to ICI therapy(16). Such observations suggest that cellular immunity in the context of prostate cancer may be more dependent on non-T cell components than in other solid tumors.
While certain molecular subgroups of prostate cancer, such as microsatellite instability high (MSI-high), CDK12 biallelic inactivation, and BRCA1/2 and ATM mutated tumors demonstrate promising results using single agent ICI, these overall comprise a relatively small subset of patients with MCRPC(10). With these results in mind, the focus has subsequently shifted to different protocols utilizing sequencing of anti-androgen therapy or PARP inhibitors with ICIs(23). These studies have been subject to several recent, in-depth reviews and we will refer the reader to these excellent manuscripts for further review(24, 25).
Prostate cancers do express specific surface markers with relatively low physiologic expression in other normal tissues. Pursuit of immune therapies targeting these antigens specifically expressed on prostate malignancies has accelerated in recent years, particularly with focuses on targeting PSMA(26), PSA(27), EpCAM(28), STEAP1(29), and DLL-3 in the current drug-development pipeline. Products range from monoclonal antibodies targeting these surface markers to immune engager molecules such as bispecific- T (BiTE) and natural killer cell engagers (BiKE) and chimeric antigen receptor (CAR) T and NK cells. Monoclonal antibodies promoting passive immunotherapy for prostate malignancies or specific toxin/radiotherapy delivery have been extensively reviewed recently(30).
Bi- Specific T-cell Engagers (BiTE):
Several cellular therapies are currently in use against hematologic malignancies. The first of these, blinatumomab (Blincyto), an anti-CD3/anti-CD-19 BiTE (example in Figure 1A), received initial approval in December 2014 for relapsed/refractory acute lymphoblastic leukemia (ALL)(31) in adults and children. Catumaxomab (Revomab), the first BiTE approved for solid tumors, targeted EpCAM and CD3 and was approved in 2010 by the European Medicines Agency for EpCAM positive malignancies and received orphan drug approval from the FDA for EpCAM postive ovarian cancer in 2006 and gastric cancer in 2009(32, 33), but an application with the FDA for further approval was never submitted(34). Catumaxomab market authorization was voluntarily withdrawn from US and EU markets in 2013 and 2017(35), respectively at the request of the manufacturer, Neovii Biotech GmbH, for reasons related to the company’s insolvency(32, 36).
Figure 1:
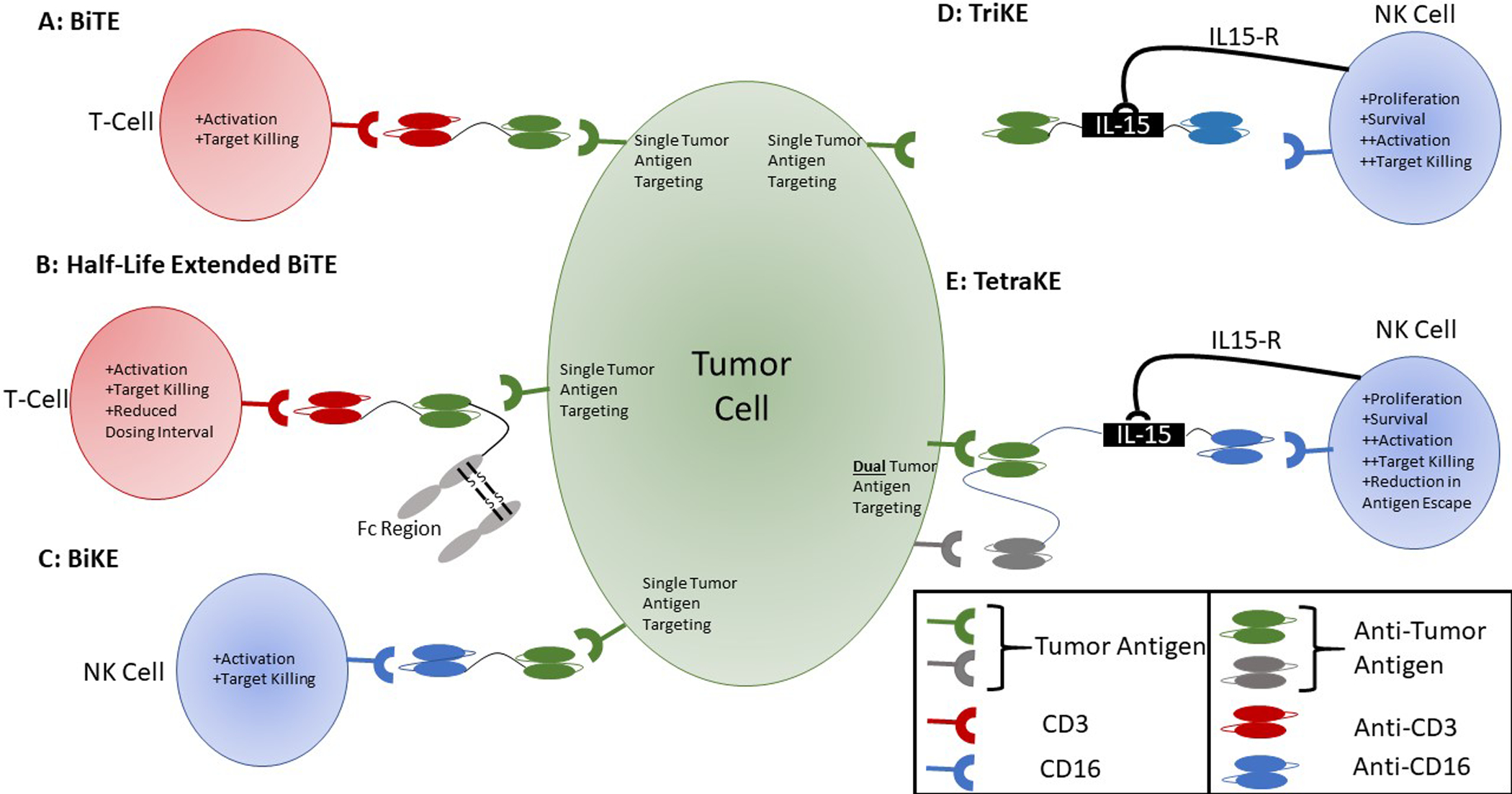
Structure of T- and NK cell immune engagers. A. Bi-specific T-cell engager consisting of an anti-CD3 ScFv to engage effector T-cells and anti-tumor antigen ScFv component bound by a flexible linker region. B. Extended half-life BiTE consisting of anti-CD3 ScFv to engage effector T-cells, anti-tumor antigen ScFv component and an Fc region bound by a flexible linker region. C. Bi-specific killer engager consisting of an anti-CD16 ScFv to engage effector NK cells and anti-tumor antigen ScFv bound by a flexible linker region. D. Tri-specific killer engager consisting of an anti-CD16 ScFv to engage effector NK cells, an IL-15 molecule to stimulate and activate NK cells via IL-15 receptor, and an anti-tumor ScFv. All components are joined by flexible linker regions to yield a single molecule. E. Tetra-specific killer engager consisting of an anti-CD16 ScFv to engage effector NK cells, an IL-15 molecule to stimulate and activate NK cells via IL-15 receptor, and two anti-tumor ScFv components. All components are joined by flexible linker regions to result a single molecule.
At the time of data compilation for this manuscript, 25 ongoing clinical trials studying BiTE technology for treatment of solid (10 studies, 4 specifically for prostate cancer) and hematologic malignancies were listed on ClinicalTrials.gov. Trials not discussed in the body of this manuscript are listed in Table 1. While effective for hematologic malignancies, there are significant drawbacks to BiTEs, particularly related to cytokine release syndrome (CRS) and neurologic adverse events mediated by particularly, but not exclusively by, IL-6 production, often leading to dose-limiting toxicities, drug discontinuation, or in rare cases even death(37, 38). Additionally, given the small size of the BiTE molecules and active filtration via the glomerulus, a continuous infusion over the course of weeks is required(39), making administration less convenient compared to other prospective therapies.
Table 1:
Remaining immune engager and cellular therapies with active clinical trials. These trials were not discussed in detail in the manuscript due to limited information availability.
| Trial Phase | Target Antigen | Engager | Additional Information | Eligibilty | NCT | Publication |
|---|---|---|---|---|---|---|
| 2 | HER2-neu | CD3 | With pembrolizumab | Progression despite castrate testosterone level. | NCT03406858 | None |
| 1 | PSMA | CD3 | None | Must have progression on abiraterone or enzalutamide. | NCT02262910 | 91 |
| 1 | STEAP | CD3 | Dual anti-STEAP Fab | Must have progression on novel antiandrogen. | NCT04221542 | 92 |
| 1 and 2 | PSMA | CD28 | With cemiplimab (anti PD-1) | Progression on 2 or more approved therapies for mCRPC. | NCT03972657 | 93 |
| Phase 1 | PSMA | CD3 | With AMG-404 targeting PD-1 and either enzalutamide or aberaterone | Castrate levels of testosterone. Subject planning to receive enzalutamide for the first time |
NCT04631601 | None |
| 1 | PSMA | LIGHT technology | Progression on abiraterone or chemotherapy. >50% PSMA+ on repeat biopsy. |
NCT04053062 | None | |
| 1 and 2 | EpCAM | Second generation CD3zeta/CD28 | Relapsed/refractory EpCAM+ cancer. | NCT03013712 | None | |
| 1 | PSCA | 4–1BB/TCRzeta-CD19 | Progression on abiraterone or enzalutamide. PSCA+ tumor expression by central lab. |
NCT03873805 | 94 | |
| 1 | PSMA | Unknown | PSMA antigen on tumor tissue by IHC or flow cytometry. No limit on prior treatment regimens. |
NCT04429451 | None | |
| 1 | NKG2D-ligand | Gamma/Delta T Cell Rather than alpha/beta | Metastatic cancer receiving at least two prior regimens for recurrent or persistent disease. | NCT04107142 | 95 |
AMG-160 (Amgen) is an anti-PSMA BiTE under development by Amgen for use with and without concurrent PD-1 targeting antibodies. It is a fully-humanized, half-life extended BiTE consisting of two scFv fragments fused to an Fc domain (Figure 1B). Pre-clinical studies by Bailis et al and Tran et al demonstrated T-cell activation in non-human primate and human samples and an extended half-life of approximately 1 week (40, 41). Subsequent reports demonstrated AMG-160 paired with pembrolizumab targeted C4–2B cells engineered to overexpress PD-L1(42, 43). AMG-160 induced upregulation of PD-L1 on T-cells and co-treatment with pembrolizumab increased T-cell killing of targets 2–5 fold compared to AMG-160 alone(43). Murine tumor explant models induced autologous T-cell cytokine production and reduction in tumor size. Importantly, 68-Ga-PSMA-11 did not interfere with AMG-160 binding or activity in vitro and in vivo(42). This combination therapeutic strategy has subsequently progressed to a Phase 1 trial as reported at the annual meeting of the American Society of Clinical Oncology in 2020(41).
Updates presented at the annual meeting of the European Society for Medical Oncology (ESMO) in 2020 reported results from 43 patients enrolled in the Phase 1 study. In this cohort, 68.6% of patients experienced any level of PSA decline and 34.3% with greater than 50% reduction. Six patients were maintained on therapy for 6 or more months with no reports of grade 4 or 5 CRS or treatment discontinuations, although maximum tolerated dose had not been achieved at the time of publication. Twenty-six (60.5%) and 11 (25.6%) patients developed grade 2 or 3 CRS, respectively, at worst (44).
MT-110/AMG-110 (Solitomab) is an anti-EpCAM BiTE that was demonstrated to redirect cytotoxic T-cells to pancreatic cancer cells also expressing EpCAM in pre-clinical studies(45) is of particular interest due to EpCAM expression on solid tumor stem cells. Kebenko et al published preliminary results of a Phase 1 dose-escalation study enrolling refractory solid tumors known to express EpCAM(46). Of the 65 patients enrolled, three (5%) were men with mCRPC. Fifty-four of 65 patients were able to be assessed for response using RECIST criteria, with 17 patients showing stable disease, 1 unconfirmed partial response, and 28 progressive disease. Twenty percent of patients had Grade 3 or higher adverse events (AE), particularly diarrhea, LFT elevations, or elevated lipase. Significant AEs were limited to diarrhea, abdominal pain, and one diarrhea-related fatal event; however, there are no additional follow-up publications. This program was ultimately discontinued by the manufacturer (47).
AMG-212 (formerly BAY2010112) pasotuxizumab is another anti-PSMA BiTE under development and was the first BiTE demonstrated to be effective targeting solid tumors(48). Hummel et al presented data at ASCO 2019 from a Phase 1 dose-escalation and safety study (NCT01723475) on 16 patients, a dose-dependent decline in PSA compared to baseline levels at the highest tested dose (80ug/day)(49). One patient showed a 54.9% reduction in PSA at the highest dose and one patient at the 40ug dose responded for 14 months and one patient at 80ug responded for 19.4 months. Recruitment for this study was stopped before the MTD was reached to allow for recruitment to a new study. The patient with 80ug and a long-term response noted >90% reduction in PSA and alkaline phosphatase with resolution of soft-tissue metastases and decreased bone metastases. No grade 5 adverse events (AE) were reported, but all patients had at least one AE, including fever (94%), chills (69%) and fatigue (50%). Thirteen patients developed grade 3 AE or higher, 44% of each decreased lymphocytes and infections. Patients did not develop anti-drug antibodies. Notably, this was the first published study demonstrating effectiveness of BiTE therapy for solid tumors. Recruitment for this study was stopped before the MTD was reached to allow for recruitment to a new study and this was ultimately terminated in favor of AMG-160(50).
AMG-509, a STEAP1 targeting BiTE, consisting of two humanized anti-STEAP1 Fab domains and an anti-CD3 scFv. Data presented at AACR 2020 by Nolan-Stevaux showed primary prostate cancers expressed STEAP1 in 80%, 83%, and 77% of primary, metastatic, or bony metastases, respectively(51). AMG-509 was 50-fold more effective in lysis of STEAP1-expressing prostate cancer cells in vitro than a companion molecule composed of a single anti-STEAP1 domain and demonstrated activity in prostate cancer xenograft models. Addition of the second Fab region also extended serum-half-life compared to prototypical BiTE molecules. A Phase-1 trial (NCT04221542) is currently recruiting for this study.
AMG-577 a second half-life extended BiTE targeting DLL-3 under investigation for prostate cancer with neuroendocrine transformation (NET) (NCT04702737). DLL-3 is an inhibitory ligand in the notch signaling pathway commonly upregulated on NET prostate cancer and small-cell lung cancers, but minimally expressed in normal prostate tissue and non-NET prostate cancers(52). DLL-3 is upregulated despite the associated losses of PSA, STEAP1, and PSMA(53), offering an option for therapy in the tumors undergoing lineage plasticity and loss tumor antigen expression for more common antigens utilized for targeting mCRPC. While there is no data published for prostate cancers with NET, patient-derived xenograft small-cell lung cancer samples treated with AMG-577 demonstrated tumor regression and led to increased tumor infiltration by CD4+ and CD8+ T-cells(47). AMG-577 was well-tolerated by non-human primates with a half-life of approximately 9.8 days(47). A second DLL-3 targetging BiTE, BI 764532, is under development by Boehringer Inhgelheim and consists of an anti-CD3 scFv and an anti-DLL3 scFV linked by an engineered Fc region. This also demonstrated target specificity, T-cell redirection and a half-life of 10 days(54). A Phase 1 study (NCT04429087) is currently enrolling patients with neuroendocrine tumors including prostate to evaluate the maximum-tolerated dose.
Bi- and Tri- and Tetra-Specific Killer Engagers (BiKE/TriKE/TetraKE):
Bispecific Killer engagers (BiKE) work in a similar fashion to BiTEs with the key difference being an anti-CD16 rather than anti-CD3 component (Figures 1C). CD16 is the strongest known inducer of NK cell activity and ligation of CD16 alone is sufficient to induce proliferation and activation of NK cells against a target(55). Ideally, BiKEs offer a lower risk of toxicity compared to BiTEs. Pre-clinical evidence by(56) demonstrated that BiKEs increased killing of a broad range of carcinoma cells, including PC3 prostate carcinoma cell lines. The choice of tumor targets for BiKEs in prostate cancer remains undefined, however targeting CD133 on prostate cancer cells with a CD16/CD133 targeting BiKE is under evaluation (57).
Tri-specific killer engagers (TriKE) may offer even further enhancement of NK approaches to solid tumors. In addition to the NK-cell engaging CD16 and target cell engaging moieties, an IL-15 linker has been added to promote NK cell proliferation and activation once engaged by the TriKE (Figure 1D). An EpCAM TriKE targeting prostate cancer cell lines published by Schmohl et al in 2016 led to increased antibody dependent cell-mediated cytotoxicity (ADCC), proliferation and activation of NK cells(58).
Phase 1/2 clinical development of the TriKE approach, sponsored by GT Biopharma, is currently underway in patients with hematologic malignancies (NCT03214666). This Trike, GTB-3350, a CD16/IL-15/CD33, engages with CD33 on malignant cells in acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) TriKE targeting high-risk hematologic malignancies (C). A similar technology, DF1001, developed by Dragonfly and known as a TriNKET (Tri-specific NK Engager Therapy), is currently enrolling patients with HER2+ malignancies (NCT04143711) although this structure has not been published(59). Cheng et al have also published pre-clinical data utilizing a TriKE targeting CD19(60). Taken together, these studies demonstrate the viability of NK cell engagers for clinical testing. Further approaches with TriKEs offer to retain the CD16/IL-15 component while utilizing targets that are specific for prostate cancer.
Due to observations of tumor antigen escape under the selective pressure of a single immune therapy as described for rituxumab targeted lymphomas (61), there is concern that a similar resistance will rapidly arise in prostate cancers treated with single antigen targeting therapies as well via lineage plasticity or NET. To address this from the perspective of prostate-specific immune engagers, Schmohl et al developed a CD16/IL-15/EpCAM/CD133 TetraKE to target cancer stem cells with two prostate cancer-specific domains (Figure 1E) (62). This TetraKE induced increased NK cell degranulation by CD107a release assay when co-incubated with PC3 prostate cancer cell lines compared to a BiKE targeting EpCAM alone. No prostate-cancer specific dual-antigen targeting CARs currently under clinical investigation; however, such approaches will likely be critical developments as immune therapies targeting solid tumors gain clinical approval.
Chimeric antigen receptor T-cells (CAR-T):
The CAR construct has evolved over the past 30 years from the initial development of a chimeric receptor expressing T-cell in 1989 by (63). CAR-T typically consist of three key components. First, an extracellular antigen-recognition domainthat recognize cell surface-expressed tumor associated antigens. A transmembrane domain (typically derived from CD3 or CD28) anchors the CAR to the cell membrane and facilitates signal transmission to the signal transduction domain(64). The signal transduction domain containing immune-receptor tyrosine-based activation motifs (ITAM) that are the final component in transmitting signals to the T-cell and promoting target cell lysis (65).
To receive CAR-T cell therapy, the patient undergoes leukapheresis to collect peripheral blood mononuclear cells (PBMC). T-cells are then selected from the bulk PBMCs, activated, and subsequently transduced with retroviral constructs expressing the CAR construct of interest. Transduced cells are then expanded to generate a therapeutic dose of CAR-T cells and then frozen prior to transport. Finally, approximately 23 days (range 21–37) after initial apheresis, patients are treated with lymphodepleting chemotherapy and then the thawed CAR-T product is reinfused into the patient (66, 67).
Chimeric antigen receptor T-cells (CAR-T) were first approved by the FDA for use against refractory DLBCL in adults and ALL in pediatric/young adult patients in 2017. Tisagenlecleucel was approved in August (68) and axicabtagene ciloluecel was approved in October of the same year(69). These products were indicated in patients with relapsed/refractory disease to at least two lines of therapy and were subsequently approved for adult patients with relapsed refractory diffuse large B-cell lymphoma. Since their initial FDA approval, CAR-T technology has rapidly developed in order to improve response to target antigens, persistence and proliferation. Despite the rapid development and approval of multiple CAR-T therapies for hematologic malignancies, there is yet to be an approved solid-tumor targeting CAR-T product.
While most of the elements so far discussed have remained relatively stable between CAR-T generations, the intracellular signaling domains have continuously evolved with each progressive generation. First generation CAR-T constructs consist of CD3-zeta as the lone intracellular activating domain and were initially described by Eshhar et al in 1993 (Figure 2A) (70). Second generation CAR-T constructs consist of two intracellular activating domains (Figure 2B), while third generation contain three intracellular activating domains with CD28 and 4–1BB the most-commonly used (Figure 2C). Fourth generation CAR-T contain additional modifications to the signaling domain to optimize response, survival, and proliferation of cell product and were designed to overcome additional deficiencies in earlier CAR-T constructs. Fourth generation CAR-T cells typically also contain 3 intracellular activating domains as in third generation products. These modifications address the need for more fine-tuned activation parameters or add suicide switches such as herpes simplex virus thymidine kinase, leading to CAR-T cell death in response to exogenously administered gancyclovir. Such mechanisms allow for termination of the CAR-T product in the case of cytokine release syndrome (71).
Figure 2:
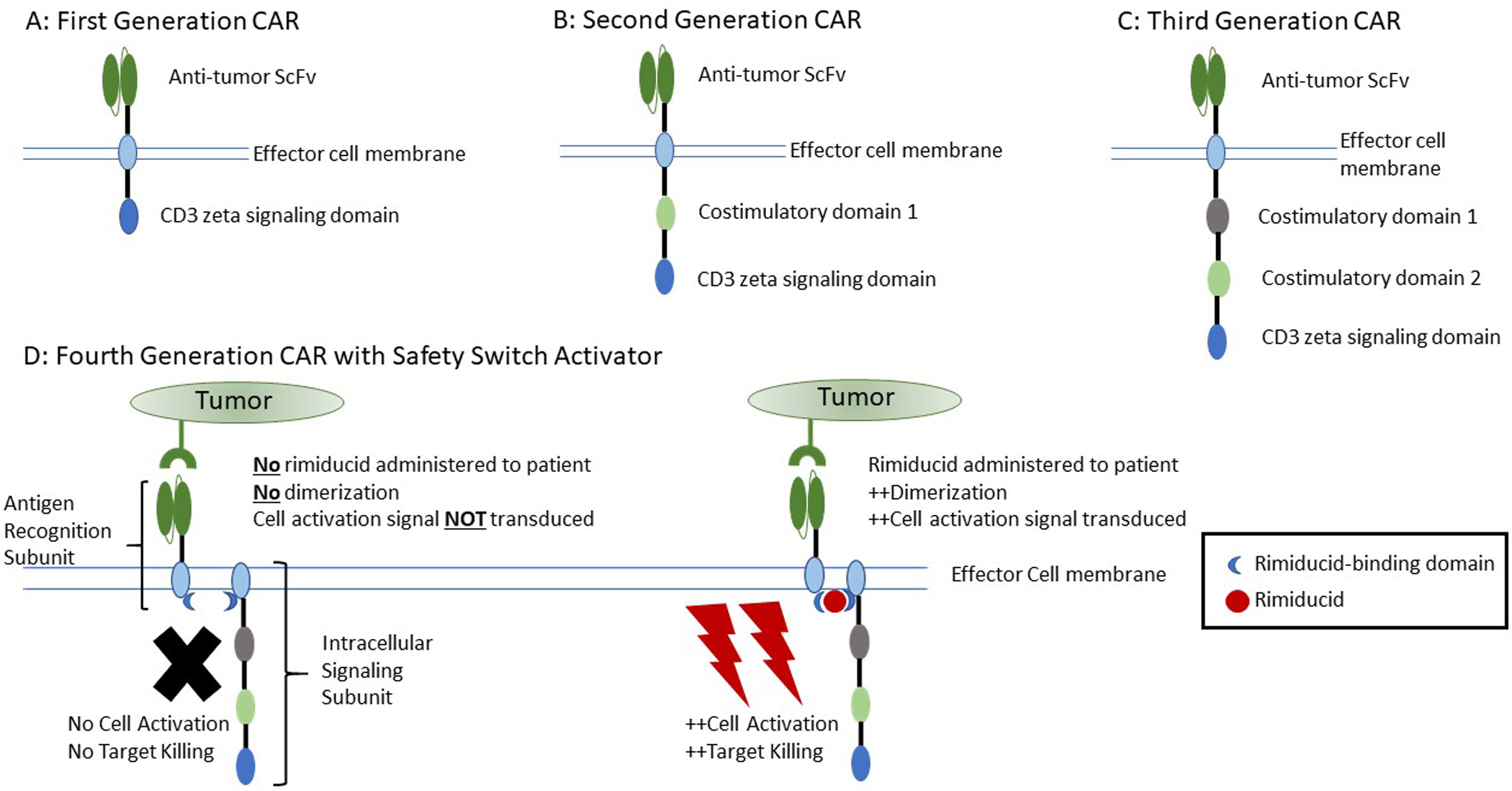
Structure of chimeric antigen receptors. A. First generation consisting of an extracellular antigen recognition domain and a single CD3 zeta region for intracellular signaling. B. Second generation composed of a conserved extracellular antigen recognition domain, a single costimulatory domain, and a single CD3 zeta signaling domain. C. Third generation consisting of an extracellular antigen recognition domain, two costimulatory domains, and a single CD3 zeta signaling domain. D. Example of a fourth generation, consisting of a third-generation chimeric antigen receptor divided into two separate components (antigen recognition and costimulatory/signaling domains) and a rimiducid safety switch. In the left figure, despite the presence of target antigen, there is no signaling or effector cell activation under conditions without rimiducid due to lack of dimerization of the antigen recognition subunit and intracellular signaling subunit. In the right portion of the figure, administration of rimiducid to the patient in the presence of target tumor antigen allows for dimerization of the two subunits of the chimeric antigen receptor and results in intracellular signaling and effector cell activation.
Nine CAR-T trials targeting prostate cancer are currently listed on clinicaltrials.gov (Table 1); and limited result data are available The first noted trial was published by Slovin et al at ASCO 2013 using a 2nd generation CAR targeting PSMA(72). This construct also conferred ganciclovir sensitivity as a safety mechanism allowing for rapid elimination of these CAR-T cells if necessary. CAR-T transduced with this construct were found to persist in recipients for two weeks based on cytokine analysis of peripheral blood samples. Of the four patients receiving this product, two had progressive disease, one had stable disease for more than 6 months and one had stable disease for more than 16 months(73)
Additional advances in CAR-T cell development have included a trial of a PSMA-targeting CAR transduced into transforming growth factor beta (TGF-beta) dominant-negative receptor. In vivo modelling demonstrated increased proliferation, increased cytokine secretion, reduced T-cell exhaustion and increased persistence of CAR-T cells (74). A first-in-human Phase 1 trial results were initially published at ASCO 2020 by Narayan et al. There was a note of increased CRS that was treated with tocilizumab. In the Phase 1 portion of the study (NCT03089203), there was no note of dose-limiting toxicity in the first two cohorts receiving 1–3×10^7 or 1–3×10^8 cells per meter squared.
Two CAR-T cells with inducible “ON” safety switches are currently under study. NCT04249947 is a Phase I study of PSMA 101–001 using rimiducid as a safety switch activator to enhance proliferation and activation while minimizing toxicity. The CAR-T receptor in such systems is composed of a heterodimer with one component containing the antigen receptor domain and the second the signaling domain. Heterodimerization of the two domains to allow for a functional CAR signal only occurs in the presence of both antigen and rimiducid (Figure 2D). Such “ON” switches allow for more targeted activation of the CAR-T product days to weeks after the initial product infusion and would terminate activity without CAR-T death if rimiducid dosing was held (75).
A second rimiducid induction strategy via an MyD88/CD40 co-stimulatory domain was published by Becerra et al at ASCO 2019 using BPX-601 from Bellicum Pharmaceuticals. Patients with advanced pancreatic, stomach, or prostate cancers were eligible for enrollment using this PSCA-targeting CAR-T construct(76). This trial, NCT02744287, demonstrated engraftment by Day+4 and reported no dose-limiting toxicities, neurotoxicity or CRS. Interestingly, patients in this Phase 1 trial did not undergo lymphodepleting chemotherapy prior to infusion of the CAR-T product, but still had expansion, cytokine release and persistence. Four patients had stable disease at 8 weeks or more, two had minor responses and two patients had progressive disease.
The remaining products are listed in Table 1 given the limited data available.
Challenges and Limitations of CAR-T for Prostate Cancer:
CAR-T cells, despite demonstrating potent anti-tumor effects in otherwise-refractory leukemias and lymphomas, may present safety challenges in a subset of patients with prostate cancer that are be older and have multiple comorbidities. Cytokine-release syndrome (CRS) is a hyperinflammatory state notable for excess production of IL-2Ralpha, IL-6, interferon gamma, and GM-CSF due to activation and proliferation of CAR-T cells to the targeted antigen. While due to the desired effects of the infused CAR-T cell, without treatment, CRS can rapidly be fatal and often requires ICU level care due to encephalopathy, hypotension, tachycardia, and hypoxia(77). In the initial studies of Axicabtagene Ciloleucel for diffuse large B-cell lymphoma, 93% of patients experienced some level of CRS within 2 weeks of infusion (78). 64% of patients on this trial developed neurological events such as encephalopathy, confusion, tremor, aphasia, or somnolence and 28% developed Grade 3 or higher neurological events.
Off-target effects in solid tumor targets have led to deaths in Phase 1 trials due to tissue cross-reactivity. A solid tumor-specific CAR targeting erythroblasosis oncogene 2 (ERBB2) led to development of CRS and death in one patient (79). This patient required intubation within an hour of CAR-T infusion due to rapid development of pulmonary edema due to proposed cross-reactivity with normal lung tissue. Death and organ dysfunction secondary to on-target effects of CAR-T therapy has been described by several other groups including liver and biliary tract injury (80)and neurologic damage (81).
Taken together, the emerging toxicity data on CAR-T suggest that its development must proceed with greater than usual attention to patient selection, pre-therapy evaluations and limited use in patients with comorbidities. In its early development stages, clinical development of CART therapies in prostate cancer will, as a result, focus on fitter, younger patients who have experienced disease progression on standard AR targeted therapies and taxane-based chemotherapy.
Antigen down-regulation and subsequent resistance to single antigen CAR-T cells is also documented in hematologic malignancies (82). Prostate cancer, whether via NET or lineage plasticity, would likely exhibit similar escape mechanisms and target downregulation. This limitation is being addressed in pre-clinical studies of CAR-T cells targeting two multiple myeloma-specific antigens to prevent antigen escape(83).
In addition to concerns of on-target activation and tissue damage, CAR-T cells require an average of 3 weeks to manufacture and there is risk that at the end of this time period, although low, there will be failure to generate a product CAR-T for clinical use. Although it is less likely to be a concern in the setting of prostate cancer, the time frame to manufacture CAR-T products is a key consideration given the refractory and often aggressive nature of disease for patients to be eligible for CAR-T therapy to start. Additionally, patients are often rendered leuko- and lymphopenic by chemotherapy regimens used just prior to the collection period. This requires patients to be off chemotherapy for at least two weeks prior to collection to minimize the risk of failed collection (78), but extends the time period for which patients must await delivery of the final CAR-T product.
Chimeric Antigen Receptor Natural killer (CAR-NK) Cells
In response to concerns regarding the potential toxicity of CAR-T therapies, significant interest in developing a clinical-grade CAR-NK cell product has arisen due to the inherent characteristics of these cells. Unlike T-cells, NK cells do not induce graft-versus host disease in the allogeneic, haploidentical setting (84). NK cells retain their innate ability to recognize target cells that have downregulated the CAR-specific antigen by using MHC-I downregulation as an additional method to recognize malignant or otherwise physiologically stressed cells by the “missing self” mechanism (85). There is additional hope for development of an “off-the shelf” CAR-NK product that is not HLA-specific to the recipient, allowing for universal patient use rather than requiring individualized manufacture Additional benefits of “off-the-shelf” products include the ability to be stored and thawed for use on demand, and reduced cost (86).
The difficulty of expanding and virally transducing NK cells in vitro are also significant limitations leading to use of NK cell lines or induced-pleuripotent derived NK cell lines instead of peripheral blood or cord blood-derived NK cells (87–89)
There is one CAR-NK product with a registered clinical trial (NCT03692663). PSMA CAR NKZGZYZL is being developed by Allife Medical Science and Technology Company using the NK-92 cell line rather than autologous NK cells. This study is not yet recruiting and no additional information is available.
No current CAR-NK product is approved; however, there are multiple products undergoing clinical trials for a variety of malignancies both hematologic and solid tumor(90). Despite being in early stage development, CAR-NK approaches promise to be a source of cellular therapy product that may suit prostate cancer patients well due to the potential for lower toxicity and an opportunity to engage with appropriate targets in the disease.
Conclusions:
The lack of neoantigens, relatively immunologically cold tumor microenvironment, in addition to tumor heterogeneity, have led to unique challenges in the development of immune based therapies for prostate cancer thus far. These challenges also provide opportunities to develop improved cellular therapy products that will be broadly applicable to other solid tumors once optimized. Numerous methodologies are being pursued to address limitations, including T- and NK-cell engagers, multiple CAR-T and a single CAR-NK product. The challenges of treating solid tumors with immune and cellular therapies may also be overcome by utilization of combination therapies with monoclonal antibodies and chimeric antigen-expressing effector cells, taking advantage of unique advantages of each treatment. Innovative modes of delivery, cellular products, and novel target development promises to bring cellular therapies to men with prostate cancer.
Acknowledgements:
Nicholas A. Zorko is supported by 2T32HL007062 Hematology Research Training Program T32, University of Minnesota (PI: Dr Gregory Vercellotti) and an Academic Investment Education Program grant from the University of Minnesota Medical School.
Charles J. Ryan is the B.J. Kennedy Chair of Clinical Medical Oncology at the University of Minnesota Medical School.
Charles J. Ryan receives the following support:
Receipt of grants/research supports
Clovis Oncology- Research Grant
Sanofi-Genzyme- Research Grant
Bayer - Research Grant and Consultation
Receipt of honoraria or consultation fees
Advisory Boards:
Advanced Accelerator Applications - Consultation
Roivant - Consultation
Pfizer - Consultation
Dendreon - Consultation
Myovant (Payment to U of M)
Clovis (Payment to U of M)
Bayer (Payment to U of M)
Footnotes
Nicholas A. Zorko does not have any competing financial interests to declare.
Bibliography:
- 1.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. [DOI] [PubMed] [Google Scholar]
- 2.Madan RA, Antonarakis ES, Drake CG, Fong L, Yu EY, McNeel DG, et al. Putting the Pieces Together: Completing the Mechanism of Action Jigsaw for Sipuleucel-T. J Natl Cancer Inst. 2020;112(6):562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiPaola RS, Plante M, Kaufman H, Petrylak DP, Israeli R, Lattime E, et al. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7–1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Transl Med. 2006;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simons JW, Carducci MA, Mikhak B, Lim M, Biedrzycki B, Borellini F, et al. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naive prostate cancer. Clin Cancer Res. 2006;12(11 Pt 1):3394–401. [DOI] [PubMed] [Google Scholar]
- 5.Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotechnol. 2009;27(2):129–39. [DOI] [PubMed] [Google Scholar]
- 6.Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front Immunol. 2019;10:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis PM, Vella ET, Ung YC. Immune Checkpoint Inhibitors for Patients With Advanced Non-Small-Cell Lung Cancer: A Systematic Review. Clin Lung Cancer. 2017;18(5):444–59 e1. [DOI] [PubMed] [Google Scholar]
- 8.Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 2017;10(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonarakis ES. A New Molecular Taxonomy to Predict Immune Checkpoint Inhibitor Sensitivity in Prostate Cancer. Oncologist. 2019;24(4):430–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer. 2020;6(7):605–18. [DOI] [PubMed] [Google Scholar]
- 12.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol. 2019;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilusic M, Madan RA, Gulley JL. Immunotherapy of Prostate Cancer: Facts and Hopes. Clin Cancer Res. 2017;23(22):6764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. [DOI] [PubMed] [Google Scholar]
- 16.Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T, et al. The Immune Checkpoint Regulator PD-L1 Is Highly Expressed in Aggressive Primary Prostate Cancer. Clin Cancer Res. 2016;22(8):1969–77. [DOI] [PubMed] [Google Scholar]
- 17.Nappi L, Kesch C, Vahid S, Fazli L, Eigl BJ, Kollmannsberger CK, et al. Immunogenomic landscape of neuroendocrine small cell prostate cancer. Journal of Clinical Oncology. 2019;37(7_suppl):217-. [Google Scholar]
- 18.Miller AM, Lundberg K, Ozenci V, Banham AH, Hellstrom M, Egevad L, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol. 2006;177(10):7398–405. [DOI] [PubMed] [Google Scholar]
- 19.Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947–58. [DOI] [PubMed] [Google Scholar]
- 20.Tien AH, Xu L, Helgason CD. Altered immunity accompanies disease progression in a mouse model of prostate dysplasia. Cancer Res. 2005;65(7):2947–55. [DOI] [PubMed] [Google Scholar]
- 21.Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, et al. The Immune Landscape of Prostate Cancer and Nomination of PD-L2 as a Potential Therapeutic Target. J Natl Cancer Inst. 2019;111(3):301–10. [DOI] [PubMed] [Google Scholar]
- 22.Lundholm M, Hagglof C, Wikberg ML, Stattin P, Egevad L, Bergh A, et al. Secreted Factors from Colorectal and Prostate Cancer Cells Skew the Immune Response in Opposite Directions. Sci Rep. 2015;5:15651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall CH, Antonarakis ES. Emerging treatments for metastatic castration-resistant prostate cancer: Immunotherapy, PARP inhibitors, and PSMA-targeted approaches. Cancer Treat Res Commun. 2020;23:100164. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson LT, Fong L. Immune Checkpoint Inhibition in Prostate Cancer. Trends Cancer. 2020;6(3):174–7. [DOI] [PubMed] [Google Scholar]
- 25.Cha HR, Lee JH, Ponnazhagan S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020;80(8):1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang SS. Overview of prostate-specific membrane antigen. Rev Urol. 2004;6 Suppl 10:S13–8. [PMC free article] [PubMed] [Google Scholar]
- 27.Karan D. Prostate immunotherapy: should all guns be aimed at the prostate-specific antigen? Immunotherapy. 2013;5(9):907–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni J, Cozzi PJ, Duan W, Shigdar S, Graham PH, John KH, et al. Role of the EpCAM (CD326) in prostate cancer metastasis and progression. Cancer Metastasis Rev. 2012;31(3–4):779–91. [DOI] [PubMed] [Google Scholar]
- 29.Barroca-Ferreira J, Pais JP, Santos MM, Goncalves AM, Gomes IM, Sousa I, et al. Targeting STEAP1 Protein in Human Cancer: Current Trends and Future Challenges. Curr Cancer Drug Targets. 2018;18(3):222–30. [DOI] [PubMed] [Google Scholar]
- 30.Khalili N, Keshavarz-Fathi M, Shahkarami S, Hirbod-Mobarakeh A, Rezaei N. Passive-specific immunotherapy with monoclonal antibodies for prostate cancer: A systematic review. J Oncol Pharm Pract. 2019;25(4):903–17. [DOI] [PubMed] [Google Scholar]
- 31.Administration UFaD. Drug Approval Package-Blincyto (blinatumomab) Injection Company: Amgen, Inc. Application No.: 125557. 2017. [Google Scholar]
- 32.Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: A review. Pharmacol Ther. 2018;185:122–34. [DOI] [PubMed] [Google Scholar]
- 33.Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36(6):458–67. [DOI] [PubMed] [Google Scholar]
- 34.Larochelle M, Downing NS, Ross JS, David FS. Assessing the potential clinical impact of reciprocal drug approval legislation on access to novel therapeutics in the USA: a cohort study. BMJ Open. 2017;7(2):e014582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agency EM. Removab: Withdrawal of the marketing authorisation in the European Union.
- 36.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stein AS, Schiller G, Benjamin R, Jia C, Zhang A, Zhu M, et al. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: management and mitigating factors. Ann Hematol. 2019;98(1):159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portell CA, Wenzell CM, Advani AS. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin Pharmacol. 2013;5(Suppl 1):5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailis J, Deegen P, Thomas O, Bogner P, Wahl J, Liao M, et al. Preclinical evaluation of AMG 160, a next-generation bispecific T cell engager (BiTE) targeting the prostate-specific membrane antigen PSMA for metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2019;37(7_suppl):301-. [Google Scholar]
- 41.Tran B, Horvath L, Dorff TB, Greil R, Machiels J-PH, Roncolato F, et al. Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE) immune therapy targeting prostate-specific membrane antigen (PSMA), in patients with metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2020;38(6_suppl):TPS261–TPS. [Google Scholar]
- 42.Deegen P, Thomas O, Nolan-Stevaux O, Li S, Wahl J, Bogner P, et al. The PSMA Targeting Half-Life Extended BiTE((R)) Therapy AMG 160 Has Potent Antitumor Activity in Preclinical Models of Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2021. [DOI] [PubMed] [Google Scholar]
- 43.Paweletz KL, Li S, Bailis JM, Juan G. Combination of AMG 160, a PSMA x CD3 half-life extended bispecific T-cell engager (HLE BiTE) immune therapy, with an anti-PD-1 antibody in prostate cancer (PCa). Journal of Clinical Oncology. 2020;38(6_suppl):155-.31693429 [Google Scholar]
- 44.Tran B, Horvath L, Dorff T, Retting T, Lolkema MP, Machiels J, Rottey S, Autio K, Greil R, Adra N, Lemech C, Minocha M, Cheng F, Kouros-Mehr H, Fizazi K 6090-Results from a phase I study of AMG 160, a half-life extended (HLE), PSMA-targeted, bispecific T-cell engager (BiTE) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). Annals of Oncology. 2020;31(suppl_4):S507–S49. [Google Scholar]
- 45.Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Res. 2012;18(2):465–74. [DOI] [PubMed] [Google Scholar]
- 46.Kebenko M, Goebeler ME, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8):e1450710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giffin MJ, Cooke K, Lobenhofer EK, Estrada J, Zhan J, Deegen P, et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small-Cell Lung Cancer. Clin Cancer Res. 2021;27(5):1526–37. [DOI] [PubMed] [Google Scholar]
- 48.Friedrich M, Raum T, Lutterbuese R, Voelkel M, Deegen P, Rau D, et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther. 2012;11(12):2664–73. [DOI] [PubMed] [Google Scholar]
- 49.Hummel H-D, Kufer P, Grüllich C, Deschler-Baier B, Chatterjee M, Goebeler M-E, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting Bispecific T cell Engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2019;37(15_suppl):5034-. [Google Scholar]
- 50.Hummel H-D, Kufer P, Grüllich C, Deschler-Baier B, Chatterjee M, Goebeler M-E, et al. Phase I study of pasotuxizumab (AMG 212/BAY 2010112), a PSMA-targeting BiTE (Bispecific T-cell Engager) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2020;38(6_suppl):124-.31411950 [Google Scholar]
- 51.Nolan-Stevaux O. Abstract DDT02–03: AMG 509: A novel, humanized, half-Life extended, bispecific STEAP1 × CD3 T cell recruiting XmAb® 2+1 antibody. Cancer Research. 2020;80(16 Supplement):DDT02–3–DDT-3. [Google Scholar]
- 52.Puca L, Gavyert K, Sailer V, Conteduca V, Dardenne E, Sigouros M, et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci Transl Med. 2019;11(484). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai HK, Lehrer J, Alshalalfa M, Erho N, Davicioni E, Lotan TL. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer. 2017;17(1):759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hipp S, Voynov V, Drobits-Handl B, Giragossian C, Trapani F, Nixon AE, et al. A Bispecific DLL3/CD3 IgG-Like T-Cell Engaging Antibody Induces Antitumor Responses in Small Cell Lung Cancer. Clin Cancer Res. 2020;26(19):5258–68. [DOI] [PubMed] [Google Scholar]
- 55.Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallera DA, Zhang B, Gleason MK, Oh S, Weiner LM, Kaufman DS, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm. 2013;28(4):274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target Oncol. 2016;11(3):353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK Cell Activation of an Anticarcinoma Bispecific Antibody by Genetic Insertion of a Modified IL-15 Cross-linker. Mol Ther. 2016;24(7):1312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Our Platform: TriNKET.
- 60.Cheng Y, Zheng X, Wang X, Chen Y, Wei H, Sun R, et al. Trispecific killer engager 161519 enhances natural killer cell function and provides anti-tumor activity against CD19-positive cancers. Cancer Biol Med. 2020;17(4):1026–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24(2):203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget. 2016;7(45):73830–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A. 2019;116(16):7624–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front Immunol. 2018;9:1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T Cell Manufacturing Processes during Pivotal Clinical Trials. Mol Ther Methods Clin Dev. 2020;16:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discovery. 2017;7(10):OF1–OF. [DOI] [PubMed] [Google Scholar]
- 69.Roberts ZJ, Better M, Bot A, Roberts MR, Ribas A. Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk Lymphoma. 2018;59(8):1785–96. [DOI] [PubMed] [Google Scholar]
- 70.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). Journal of Clinical Oncology. 2013;31(6_suppl):72-. [Google Scholar]
- 73.Slovin SF. Chimeric Antigen Receptor T-cell Therapy in Prostate Cancer: Reality or Folly? Eur Urol. 2020;77(3):309–10. [DOI] [PubMed] [Google Scholar]
- 74.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther. 2018;26(7):1855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Becerra CR, Hoof P, Paulson AS, Manji GA, Gardner O, Malankar A, et al. Ligand-inducible, prostate stem cell antigen (PSCA)-directed GoCAR-T cells in advanced solid tumors: Preliminary results from a dose escalation. Journal of Clinical Oncology. 2019;37(4_suppl):283-. [Google Scholar]
- 77.Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van den Berg JH, Gomez-Eerland R, van de Wiel B, Hulshoff L, van den Broek D, Bins A, et al. Case Report of a Fatal Serious Adverse Event Upon Administration of T Cells Transduced With a MART-1-specific T-cell Receptor. Mol Ther. 2015;23(9):1541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5(12):1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Larrea CF, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020;1(2):146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. [DOI] [PubMed] [Google Scholar]
- 85.Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol Res. 2018;6(4):467–80. [DOI] [PubMed] [Google Scholar]
- 86.Oberschmidt O, Kloess S, Koehl U. Redirected Primary Human Chimeric Antigen Receptor Natural Killer Cells As an “Off-the-Shelf Immunotherapy” for Improvement in Cancer Treatment. Front Immunol. 2017;8:654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mehta RS, Rezvani K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front Immunol. 2018;9:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front Immunol. 2017;8:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pfefferle A, Huntington ND. You Have Got a Fast CAR: Chimeric Antigen Receptor NK Cells in Cancer Therapy. Cancers. 2020;12(3):706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hernandez-Hoyos G, Sewell T, Bader R, Bannink J, Chenault RA, Daugherty M, et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Molecular Cancer Therapeutics. 2016;15(9):2155–65. [DOI] [PubMed] [Google Scholar]
- 92.Kelly WK, Danila DC, Edenfield WJ, Aggarwal RR, Petrylak DP, Sartor AO, et al. Phase I study of AMG 509, a STEAP1 x CD3 T cell-recruiting XmAb 2+1 immune therapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2020;38(15_suppl):TPS5589–TPS. [Google Scholar]
- 93.Drake CG, Zhang J, Stein MN, Xu Y, Seebach FA, Lowy I, et al. A phase I/II study of REGN5678 (Anti-PSMAxCD28, a costimulatory bispecific antibody) with cemiplimab (anti-PD-1) in patients with metastatic castration-resistant prostate cancer. Journal of Clinical Oncology. 2020;38(15_suppl):TPS5592–TPS. [Google Scholar]
- 94.Dorff TB, Blanchard S, Carruth P, Wagner J, Kuhn P, Chaudhry A, et al. A phase I study to evaluate PSCA-targeting chimeric antigen receptor (CAR)-T cells for patients with PSCA+ metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2020;38(6_suppl):TPS250–TPS. [Google Scholar]
- 95.Tao K, He M, Tao F, Xu G, Ye M, Zheng Y, et al. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemotherapy and Pharmacology. 2018;82(5):815–27. [DOI] [PubMed] [Google Scholar]