

Abstract
Untargeted liquid chromatography–mass spectrometry (LC–MS)-based metabolomics strategies are being increasingly applied in metabolite screening for a wide variety of medical conditions. The long-standing “grand challenge” in the utilization of this approach is metabolite identification—confidently determining the chemical structures of m/z-detected unknowns. Here, we use a novel workflow based on the detection of molecular features of interest by high-throughput untargeted LC–MS analysis of patient body fluids combined with targeted molecular identification of those features using infrared ion spectroscopy (IRIS), effectively providing diagnostic IR fingerprints for mass-isolated targets. A significant advantage of this approach is that in silico-predicted IR spectra of candidate chemical structures can be used to suggest the molecular structure of unknown features, thus mitigating the need for the synthesis of a broad range of physical reference standards. Pyridoxine-dependent epilepsy (PDE-ALDH7A1) is an inborn error of lysine metabolism, resulting from a mutation in the ALDH7A1 gene that leads to an accumulation of toxic levels of α-aminoadipic semialdehyde (α-AASA), piperideine-6-carboxylate (P6C), and pipecolic acid in body fluids. While α-AASA and P6C are known biomarkers for PDE in urine, their instability makes them poor candidates for diagnostic analysis from blood, which would be required for application in newborn screening protocols. Here, we use combined untargeted metabolomics–IRIS to identify several new biomarkers for PDE-ALDH7A1 that can be used for diagnostic analysis in urine, plasma, and cerebrospinal fluids and that are compatible with analysis in dried blood spots for newborn screening. The identification of these novel metabolites has directly provided novel insights into the pathophysiology of PDE-ALDH7A1.
Introduction
The metabolomics community increasingly relies on untargeted liquid chromatography–mass spectrometry (LC–MS)-based metabolic screening to detect novel metabolites that can be correlated to various conditions, treatments, and environments—so called biomarkers. What is widely acknowledged as the “grand challenge” or “bottleneck” of this approach is the identification of a metabolite’s molecular structure.2−4 While the detection of metabolites directly from body fluids or tissues and the identification of their elemental composition are now routinely performed down to low nanomolar (nM) abundances, these techniques cannot directly resolve molecular structures. The most common approach to molecular identification is tandem mass spectrometry (MS/MS), which maintains the high sensitivity of the LC–MS experiment. However, many isomeric structures show nearly identical MS/MS fragmentation spectra, and their interpretation, which is to date challenging to model in silico,3,5 requires physical reference standards. Conversely, NMR spectroscopy is a powerful technique to identify a molecular structure; however, it is limited in terms of sensitivity and cannot delve deeper into the metabolome than the micromolar (μM) level; moreover, tedious purification steps are usually required prior to analysis.
Very recently, several groups have suggested infrared ion spectroscopy (IRIS) as an alternative mass spectrometry-based technique for small-molecule identification.6−15 By combining the sensitivity and mass selectivity of MS(/MS) with infrared spectroscopy-based orthogonal structural characterization, IRIS is able to provide detailed structural information to annotate detected features arising from MS analyses. IRIS can provide a mass-selective fingerprint IR spectrum for essentially any ion detected in an MS experiment. These IR spectra can be matched to reference IR spectra measured from standards or predicted by quantum-chemical computations, which is now routinely done using commercially available software packages. Recently, several proof-of-principle studies have successfully demonstrated the potential for metabolite identification using this technology.6−9,16,17 Other studies have demonstrated the coupling of IRIS with online or offline LC and paved the way toward full integration of IRIS in untargeted metabolomics workflows.6,8,13,18
In this study, we use IRIS for metabolite identification in the search for novel biomarkers for the inborn error of metabolism pyridoxine-dependent epilepsy (OMIM 266100, PDE-ALDH7A1)19,20 (see Scheme 1). PDE is an error of lysine metabolism presenting intractable epileptic seizures in the neonatal period and known to result from pathogenic mutations in the ALDH7A1 gene that encodes the enzyme α-aminoadipic semialdehyde (α-AASA) dehydrogenase, also known as antiquitin (ATQ).21 ATQ deficiency leads to the accumulation of α-AASA and its imine form piperidine-6-carboxylic acid (P6C) in body fluids and tissues. P6C inhibits vitamin B6 (pyridoxine) function through deactivation in a spontaneous reaction.22,23
Scheme 1. Overview of the l-Lysine Metabolic Pathway Involved in Pyridoxine-Dependent Epilepsy.

(a) Inactivity of the enzyme antiquitin coded by the ALDH7A1 gene leads to (b) accumulation of α-aminoadipic semialdehyde (α-AASA) and its cyclic counterpart piperideine-6-carboxylic acid (P6C). (c) P6C can react spontaneously with several β-ketoacids or β-diacids, resulting in the formation of a range of P6C-derived biomarkers (1–4), several of which are identified here. Orange arrows indicate metabolites found at elevated levels in PDE.
The secondary pyridoxine deficiency that arises is thought to be pathophysiological for PDE-ALDH7A1,21 as pyridoxine is a crucial co-factor for many enzymatic processes. In most cases, treatment with high doses of pyridoxine prevents seizures in patients. However, mild to moderate intellectual disability is still highly prevalent, especially when therapy was not started early in life due to diagnostic delay. While PDE-ALDH7A1 is a relatively common metabolic disease (estimated to occur in 1:60,000 conceptions24), it likely remains underdiagnosed, as no reliable biomarkers are available for early detection of this disease in newborn screening. P6C and α-AASA are currently used as diagnostic PDE-ALDH7A1 biomarkers in urine.25 For plasma, an LC–MS/MS workflow has been demonstrated for simultaneous detection of P6C and α-AASA after derivatization.26−28 However, as a result of their high chemical instability, these biomarkers are not suited for application in newborn screening programs which are regularly based on direct infusion MS analysis of dried blood spots.29 Thus, there currently remains a clinical need for novel PDE-ALDH7A1 biomarkers that could be applied in newborn screening and that would enable early diagnosis and optimal clinical outcomes for patients.
In our group, we have validated an untargeted metabolic profiling method based on ultrahigh pressure LC (UHPLC)–MS, coined “next-generation metabolic screening (NGMS),” for the diagnostic screening of inborn errors of metabolism.30 Apart from diagnosis based on known biomarkers, untargeted metabolomics offers the opportunity to detect new biomarkers. However, the chemical identification of these deviating features has been the limiting factor until now. As a major step forward, we have now combined NGMS with IRIS, which covers the complete route from biomarker discovery to chemical identification. In this study, we describe the successful application of this NGMS-IRIS strategy for the start-to-finish identification of several novel biomarkers for PDE-ALDH7A1.
Results and Discussion
LC–MS/MS Analysis of PDE Samples
We analyzed 11 plasma samples of 7 genetically confirmed PDE-ALDH7A1 patients with bi-allelic pathogenic ALDH7A1 mutations using NGMS. We detected elevated levels of α-AASA (m/z 146.0816, +ESI), P6C (m/z 128.0706, +ESI), and pipecolic acid (m/z 130.0816, +ESI), in line with what is reported in the literature (see Table 1).31 Apart from these metabolites, we observed several m/z features that were previously unreported. Two features, here labeled as A and B, both having an m/z value of 186.1123 (+ESI) and retention times of 2.33 and 2.55 min, respectively, were noted to be significantly increased in comparison to controls. This was also the case in urine and cerebrospinal fluid (CSF) of PDE-ALDH7A1 patients (Table 1). Figure 1a plots the summed intensity of features A and B in the plasma of 26 controls (gray) and 10 PDE patients (orange) measured in duplicate. Intensity plots of features A and B are shown in Figure S1. The average fold changes (PDE-ALDH7A1 patient/control) of features A and B are determined at 145 and 200, respectively, enabling the distinction of patients from the control group in all cases. A molecular formula of [C9H15NO3 + H]+ (Δm = −3.9 ppm) was assigned to these features; however, neither the HMDB,32−35 METLIN,36 nor ChemSpider37 libraries contained entries for this chemical formula that would be of biochemical relevance to PDE-ALDH7A1.
Table 1. Overview of Metabolites Identified by NGMS to Correlate with PDE.
| compound | m/z | RT (min) | CSF | plasma | urine |
|---|---|---|---|---|---|
| A | 186.1123 | 2.33 | + | + | + |
| B | 186.1123 | 2.55 | + | + | + |
| C | 188.0919 | 1.45 | + | + | + |
| D | 188.0919 | 1.49 | + | + | + |
| 6-oxoPIP | 144.0652 | 3.05 | + | + | + |
| α-ASAA | 146.0816 | 0.86 | + | + | + |
| P6C | 128.0706 | 0.86 | + | + | + |
Figure 1.

Results of LC–MS/MS analysis of PDE plasma samples. (a) Plot of the sum of feature A (m/z 186.1123, 2.33 min RT) and feature B (m/z 186.1123, 2.55 min RT) in the plasma of 26 controls (gray) and 11 PDE patients (orange). Each sample was analyzed as two technical replicates, shown as consecutive bars in the figure. All values are normalized on the value in a quality control sample. (b) CID MS/MS spectrum of feature A in one of the PDE patient plasmas. A scheme showing the hypothesized core structure of features A and B and their fragmentation to the m/z 128.0707 fragment is inlaid in the figure.
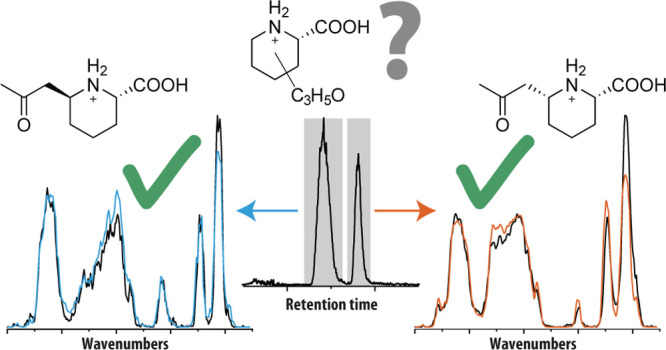
To resolve the molecular structures of features A and B, MS/MS spectra were recorded using collision-induced dissociation (CID); see Figure 1b for the MS/MS spectrum of feature A. Dissociation primarily leads to loss of CO + H2O (Δm = 1.7 ppm) and C3H6O (Δm = 4.6 ppm), giving fragments with m/z 140.1069 and m/z 128.0707, respectively. Moreover, the m/z 128.0707 fragment shows secondary fragmentation to m/z 100.0768, m/z 82.0652, and m/z 55.0542, which can be assigned as loss of CO (Δm = −36 ppm), CO + H2O (Δm = −0.5 ppm), and CO + H2O + HCN (Δm = −3.7 ppm), respectively. Of particular interest is the m/z 128.0707 fragment, as this corresponds to the mass of [P6C + H]+, a known biomarker of PDE-ALDH7A1 (see Scheme 1 and Table 1). Fragmentation of the unknowns A and B to P6C would suggest that their molecular structures are related to P6C. Specifically, this suggests a molecular structure consisting of P6C with an additional C3H5O-group, which is lost as a neutral fragment upon CID (see Figure 1b).
Identification of the m/z 128.0707 MS/MS Fragment Using IRIS and Quantum-Chemical Calculations
For further identification of features A and B, we relied on IRIS and quantum-chemically predicted IR spectra. LC fractions containing either feature A or B were obtained and used to obtain IR spectra of the LC-separated, mass-isolated features, as well as their CID-generated m/z 128.0707 fragment. Figure 2a shows the IR spectrum of the m/z 128.0707 fragment of feature A (black trace), which was found to be identical to the spectrum of the m/z 128.0707 fragment of feature B (see Figure S2). Qualitatively, one may correlate the peaks at ∼1800 and ∼1650 cm–1 with carbonyl and N–H functionalities, which are in agreement with the hypothesis that the fragment corresponds to protonated P6C, but other (positional) isomers cannot be directly excluded. A more confident structural assignment was derived from comparison against theoretical reference spectra of protonated P6C and a closely related isomer, protonated piperideine-2-carboxylic acid (P2C). Density functional theory (DFT) at the B3LYP/6–31++G(d,p) level was used to predict their vibrational spectra (orange traces in Figure 2a and Figure S3a). The IR spectrum computed for [P6C + H]+ was in excellent agreement with the experimental spectrum, whereas the computational [P2C + H]+ spectrum showed clear spectral discrepancies and shifts with respect to the experimental spectrum, especially in the 1200–1500 cm–1 region (see Figure S3a). The experimental peak around 1650 cm–1 was found to be red-shifted compared to both computational spectra. We assigned this peak as an N–H bending vibration, which are known to be inaccurately predicted by frequency calculations within the harmonic approximation due to its relatively strong anharmonic character.38,39 Overall, this led us to the conclusion that we have identified the m/z 128.1123 fragment ion of both features A and B as protonated P6C.
Figure 2.

Molecular identification of features A and B and their main fragment using computational modeling and IRIS and confirmation of the structural assignment from synthetic reference standards. (a) Comparison of the experimental IR spectra of the m/z 128.0707 fragment of feature A (black trace) and P6C reference compound (blue trace) and the theoretical IR spectrum of protonated P6C (orange trace). (b) Comparison of the experimental IR spectra of feature A (black trace) and feature B (dashed gray trace) and synthetic reference compounds (1 and 2, blue traces) and computational spectra (orange traces) of candidate structures 1 and 2. Candidate structures and conformations resulting from the quantum-chemical calculations are inlaid in each panel.
Preliminary Identification of Features A and B Using H/D Exchange and IRIS
Having identified the C3H6O-loss fragment as P6C, potential structures for features A and B could be hypothesized. MS/MS ion chemistry suggests that the precursor structure likely consists of a P6C-core with an additional C3H5O-functionalization on the ring at the location of the double bond (see the scheme in Figure 1b). Figure 2b shows the IR spectra of features A and B obtained using IRIS (black trace and dashed gray trace). The spectra show a high degree of similarity, which suggests that A and B possess highly similar molecular structures and that they could be, for example, diastereomers. The major difference between the two spectra is a peak shift near 1600 cm–1 and a difference in peak shape between 1200 and 1400 cm–1. Two carbonyl stretching vibrations above 1700 cm–1 are observed, suggesting that both features have two C=O groups in their structures. This suggests that, in addition to the carbonyl group of the carboxylic acid in the P6C core, the oxygen atom in the C3H5O-group is also present as a carbonyl.
A remaining question is whether the P6C-core is N- or C-functionalized. To determine this, we performed H/D-exchange experiments on features A and B (see the Methods section for details). We observed that both features undergo a 3 amu mass shift from m/z 186 to m/z 189, indicating the presence of three rapidly exchangeable protons. The C3H5O-group contains a carbonyl-group and thus no exchangeable protons and the carboxylic acid moiety contains one exchangeable proton. Taking into account the ionizing proton, this suggests that the ring nitrogen is a secondary amine, protonated on that nitrogen, and that our target is therefore C-functionalized.
Identification of Features A and B Using Quantum-Chemical Calculations
Based on the chemical characteristics outlined above, two plausible molecular structures can be proposed: 6-(2-oxopropyl)piperidine-2-carboxylic acid (1 and 2) and 6-propionylpiperidine-2-carboxylic acid (5 and 6, see Figure S3b). These molecules each have two stereocenters, which result in eight possible stereo-isomers. IR spectroscopy cannot distinguish between enantiomers (RR and SS), but diastereomers (SS and SR) may show differences in their IR spectra. As the unknowns are likely reaction products of P6C, a downstream metabolite of l-lysine, we only considered the stereoisomers that were S at the carboxylic acid position. We computed theoretical reference spectra of the remaining four structures using quantum-chemical calculations. Figure 2b compares the experimental and computationally predicted IR spectra of structures 1 and 2 (orange traces) to the experimental spectra of features A and B. A comparison to predicted IR spectra of unassigned alternative structures is shown in Figure S3b. All computed spectra show similarity to the experimental spectra, giving preliminary confirmation of the general chemical structure of features A and B. The frequencies of the two carbonyl stretching vibrations in the experimental spectra are very similar and correctly predicted only by the spectra of structures 1 and 2, suggesting that the C3H5O-group is a 2-oxopropyl-group. Additionally, in the spectral region below 1500 cm–1, the experimental spectrum of feature A is overall best predicted by the computed spectrum of structure 1 and that of feature B by the spectrum computed for structure 2. Note that the difference in peak shape in the 1200–1400 cm–1 region between the two features is relatively well explained by these two predicted spectra. In contrast, the experimentally observed bands around 1600 cm–1 of the two features are shifted relative to the predicted bands of structures 1 and 2. We attribute this discrepancy to the NH-bending character of the vibration which, as noted above for P6C as well, often involves a strongly anharmonic behavior, causing them to be shifted to higher wavenumbers in spectra predicted within the harmonic approximation. We note that the calculation does correctly predict the intensities and peak positions of this band in the two diastereomers relative to each other. Therefore, we tentatively identify feature A as (2S,6S)-6-(2-oxopropyl)piperidine-2-carboxylic acid and feature B as its (2S,6R)-diastereomer (structures 1 and 2 in Figure 2).
Confirmation Using IR Spectra of Synthetically Obtained Reference Standards
Having made this tentative assignment, we proceeded to synthesize the reference standards for candidate structures 1 and 2. To this end, two synthetic routes to a mixture of 1 and 2 were developed that allowed IRIS experiments on 1 and 2 and their m/z 128.0707 fragments and NMR characterization of the reference standards. To unambiguously assign the LC retention time to one diastereoisomer, a stereoselective synthesis route was developed, yielding only 2. Details of the synthesis and NMR characterization are described in the Supporting Information.
IR spectra of the protonated ions of the reference standards are inlaid and compared to the IR spectra of features A (left panel) and B (right panel) in Figure 2b. Figure 2a shows the IR spectrum of the m/z 128.0707 fragment ion generated from structure 1 in comparison to the IR spectrum of the m/z 128.0707 fragment generated from feature A. A similar comparison for the fragment of feature B and structure 2 is shown in Figure S4. All sets of spectra show an excellent match, and each of the small deviations between the spectra can be attributed to minor fluctuations of experimental conditions, such as laser power, between experiments.
Finally, the reference standard of structure 2 was used to unequivocally confirm the assignment of the relative stereochemistry of features A and B. The connectivity and relative stereochemistry of 2 was assigned on the basis of 1H, 13C, and NOESY NMR experiments, indicating the cis relationship between the substituents (see Supporting Information). With a pure standard of 2 in hand, its LC–MS retention time was measured, which matched the retention time of feature B.
As negative controls, we obtained reference standards for two closely related compounds, 1-(2-oxopropyl)piperidine-2-carboxylic acid and 1-propanoylpiperidine-2-carboxylic acid containing a C3H5O-substituent attached to the nitrogen of the piperidine. The spectral comparison in Figure S5 shows a clear mismatch between their IR spectra and the spectra of features A and B, further confirming the assignments mentioned above.
Additional Biomarkers Identified by IRIS
In addition to features A and B, two additional features, C and D, were identified to have elevated levels in comparison to controls (though with a smaller fold change than features A and B). These m/z 188.0919 features have retention times of 1.45 and 1.49 min and were not baseline separated. We therefore obtained an IR spectrum of a mixture of the co-eluting features C and D (black trace in Figure 3). Interestingly, the recorded IR spectrum shows a high-degree of similarity with the IR spectra of features A and B (see Figure 2b). Additionally, features C and D were also found to fragment to an m/z 128.0707 (assigned as protonated P6C for features A and B above). Taking this together, it suggests that features C and D are the two diastereomers of 6-(carboxymethyl)piperidine-2-carboxylic acid (structures 3 and 4 in Figure 3a). Figure 3a compares the experimental IR spectrum of the metabolite mixture with the calculated IR spectra of structures 3 and 4 (orange traces). We see that both theoretical spectra generally match the experimental IR spectrum of the mixture well. Overall, the IR spectrum of structure 3 (left panel of Figure 3a) seems to have a more significant contribution to the experimental spectrum, suggesting that it is present at a higher concentration than structure 4. Three features in the experimental IR spectrum directly suggest a contribution from structure 4: being the shoulder of the ∼1100 cm–1 peak, explained by the calculated band indicated by the red star, the intensity around 1300 cm–1, explained by the band indicated by the blue star, and the shoulder of the band around 1550 cm–1, explained by the calculated band indicated with the green star. Note that the latter is again red-shifted compared to the calculation due to the anharmonic character of the NH-bending vibration.38,39
Figure 3.

Molecular identification of feature C and D using computational modeling and IRIS. (a) Experimental IR spectrum of the co-eluting features C and D (black trace in both panels) in comparison to the theoretical IR spectra of structures 3 and 4 (left and right panel, orange trace). Input structures for the computations and the conformations resulting from the quantum-chemical calculations are inlaid in each panel. Colored stars indicate bands that are particularly diagnostic for structure 4; see text. (b) Comparison of the experimental IR spectra of the co-eluting features C and D (black trace) and protonated 4 (blue trace). The molecular structure of the protonated reference standard is inlaid in the panel.
We confirmed this assignment by developing a synthesis route yielding a reference standard of structure 4 (see Supporting Information). We recorded an IR spectrum of structure 4, and Figure 3b shows a comparison of this IR spectrum with the IR spectrum of the feature C/D mixture. The three IR features predicted to originate from structure 4 are more prevalent in this IR spectrum (again indicated by the color-coded stars) and support its structural assignment.
Biochemical Origins of Biomarkers A–D
In a separate study, we focus on the implications of structures 1 and 2 on the pathophysiological understanding and diagnosis of PDE-ALDH7A1.1 In that study, we explore the (bio)chemical origins of structures 1 and 2 by conducting incubation experiments of P6C under physiological conditions. The synthesis of the reference standard described here showed that acetoacetic acid can react with P6C to afford 1 and 2. Acetoacetic acid is also known to be available in human body fluids. Indeed, incubation of P6C with acetoacetate under physiological conditions produced 1 and 2. We expect that the enol form of acetoacetic acid reacts with P6C and after decarboxylation affords features A and B.
Here, we hypothesize that malonic acid, which is similarly available in human body fluids, reacts with P6C to yield 3 and 4. We performed similar experiments by incubating malonic acid with P6C under physiological conditions in water, phosphate-buffered saline, human control plasma, and human control urine. The reaction of P6C with malonic acid occurred under all conditions tested and was confirmed by analysis with LC–MS and IRIS. Figure S6 shows the IR spectra of the observed features in all samples, identifying them as a mixture of structures 3 and 4. These observations point in a more general sense to the fact that P6C reacts as an imine with any carbonyl containing species that can form stable enol tautomers, such as the β-dicarbonyl compounds acetoacetate and malonate. In contrast, P6C reacts in its enamine tautomeric form in a cross-coupling with the aldehyde of PLP, leading to the secondary pyridoxine deficiency in PDE-ALDH7A1.
Conclusions
Using our NGMS/IRIS pipeline, we have now shown that the molecular structures of unknown m/z-features detected in untargeted metabolomics workflows can be annotated using IRIS, theoretically predicted IR spectra, and synthesized reference compounds. Features are preliminarily annotated by comparison of their experimentally measured IR spectra with theoretically predicted IR spectra generated for candidate molecular structures. Furthermore, the most relevant candidate structures necessary to consider in this approach can often be determined directly from structural information (functional group vibrational assignments) contained in the IR spectra of the unknowns. Importantly, this approach dramatically limits the scope of synthetic standards required for final identification. Here, we have applied this technique to determine the molecular structures of four previously unknown biomarkers for PDE-ALDH7A1. We have identified them as two sets of diastereomeric derivatives of P6C, a known metabolite in the degradation of l-lysine that accumulates in patients with PDE-ALDH7A1. This logically connects these metabolites to known biochemical routes of l-lysine metabolism, but also suggests a disease-specific expansion of this metabolic pathway in PDE-ALDH7A1. The identification of the molecular structures of the biomarkers allows us to deduce their likely biochemical origins, which may lead to a more complete understanding of the chemical basis for the pathophysiology of PDE-ALDH7A1 and to new treatment targets.1
Methods
Sample Preparation
Plasma, CSF, and urine samples were stored at −80 °C and thawed at 4 °C just before sample preparation. A total of 100 μL of the sample was combined with 400 μL of ice-cold methanol/ethanol 50:50 [v/v] and mixed for 15 s using a vortex mixer. For the samples used in metabolic profiling, the methanol/ethanol mixture contained five internal standards [caffeine-d3 0.88 μmol/L, hippuric-d5 acid 0.22 μmol/L, nicotinic-d4 acid 0.88 μmol/L, octanoyl-l-carnitine-d3 0.22 μmol/L, and l-phenyl-d5-alanine 0.44 μmol/L (all from C/D/N Isotopes, Pointe-Claire, Canada)]. The resulting mixture was incubated for 20 min at 4 °C and centrifuged for 15 min at 4 °C (18,600 g). The supernatant (350 μL) was dried in a centrifugal vacuum evaporator (Eppendorf). The dried sample was reconstituted in 100 mL of deionized water/methanol 90:10 [v/v] with 0.1% formic acid, mixed for 15 s with a vortex mixer at room temperature and centrifuged for 15 min at 18600 g. The supernatant (90 μL) was used for LC–MS analysis.
Metabolic Profiling
A full description of the untargeted metabolic screening procedure and statistical analysis leading to the detection of the biomarkers discussed in this work are described separately.1 In short, LC–MS runs were preformed using an Agilent 1290 UHPLC system coupled to a 6545 QTOF mass spectrometer. Data acquisition and analysis was done using Agilent Mass Hunter (version B.08.00). Separations were performed using a flow rate of 0.4 mL/min on a Waters Acquity HSS T3 C18 column (100 × 2.1 mm i.d., 1.8 μm particles, 100 Å pore size) held at 40 °C. The mobile phase consisted of 0.1% (v/v) formic acid in water and 0.1% (v/v) formic acid in 99:1 (v/v) methanol: water (mobile phase B). After a hold at 99% A for 1 min, a gradient of 15 min was run to 100% B, followed by a hold of 4 min at 100% B and a return to 99% A of 1 min. An equilibration time of 4 min was used, leading to a total analysis time of 25 min. Injection volumes of 2 μL were used for all samples. Alignment and feature extraction were performed using the open access software package XCMS,40 and two-sided t-tests were performed to identify significantly altered features between patients and controls. See Coene et al.30 and Engelke et al.1 for details regarding this procedure.
Infrared Ion Spectroscopy
IRIS experiments were performed in a quadrupole ion trap mass spectrometer (Bruker, AmaZon Speed ETD) that has been modified to provide optical access to the stored ions for spectroscopy experiments. Details of these modifications and operation of the experiment are described elsewhere.41 Fractions containing metabolic features of interest were collected with a Bruker Elute SP HPLC system and a two-position six-port switching valve controlled by the quadrupole ion trap. The separations were performed using the same separation procedure as described above but with a mobile phase consisting of 10 mM acetic acid in water (mobile phase A) and 10 mM acetic acid in methanol (mobile phase B). For features A and B, the switching valve was used to divert the eluent containing the features to Eppendorf tubes, which were analyzed by IRIS in a separate experiment by infusing the fractions with a syringe pump (120–180 μL/h). For features C and D, the LC and IRIS experiments were combined by installing an 80 μL sample loop between the switching valve and the ion source of the ion trap. As soon as the LC peak of interest arrived at the mass spectrometer, the valve was switched, such that the remainder of the feature material was infused by a syringe pump (120 μL/h) for IRIS experiments. For reference IR measurements, 1-propanoylpiperidine-2-carboxylic acid [Enamine Ltd. (Kyiv, Ukraine)] and 1-(2-oxopropyl)piperidine-2-carboxylic acid (see Supporting Information for the synthesis procedure) and compound 2 and compound 4 were dissolved in 50:50 (v/v) acetonitrile:water (∼10–7 M) and infused directly with the syringe pump for IRIS experiments (180 μL/h). Compound 1 was isolated from a mixture of 1 and 2 by performing the chromatographic separation detailed above and collecting the fraction of the eluent containing 1 using a Foxy R2 fraction collector equipped with two 96-well plates. This fraction was infused with the syringe pump for IRIS experiments at 120–180 μL/h flow rates.
To obtain IRIS spectra, features of interest were mass-isolated and stored for ∼200 ms in the quadrupole ion trap. During this time, they were irradiated using the FELIX infrared free electron laser,42 which produced IR radiation in the form of ∼10 μs macropulses of 30–200 mJ at a 10 Hz repetition rate (bandwidth ∼0.4% of the center frequency). IR spectra were obtained by scanning the laser with 3 cm–1 steps and recording a mass spectrum after irradiation at each frequency step. MS spectra were recorded as an average of four to eight scans. When the laser frequency is resonant with a molecular vibration of the trapped ions, absorption takes place, which increases the internal energy of the stored ions, leading, after the absorption of multiple photons, to unimolecular dissociation. The amount of IR absorption is measured by the degree of fragmentation at each wavelength point by relating the precursor and fragment ion intensities [yield = Σ(fragment ions)/(Σ (parent + fragment ions)]. The laser frequency was calibrated using a grating spectrometer and the fragmentation yield was linearly corrected for frequency-dependent pulse energy variation. IR spectra of fragment ions were obtained by first using CID (50 ms, fragmentation amplitude optimized on the fragment ion signal) on the relevant precursor ion before mass isolation of the desired fragment ion and IR laser irradiation as described above.
H/D Exchange
In order to perform H/D exchange on features A and B, an LC fraction containing both unknown analytes was collected according to the procedure described above. The fraction was dried in a centrifugal vacuum evaporator (Eppendorf) and reconstituted in 100 μL of deuterated methanol. Subsequently, the H/D-exchanged fraction was infused at 180 μL/h to the quadrupole ion trap mass spectrometer. It was observed that the majority (>70%) of the ions corresponding to features A and B had undergone three exchanges. The remaining part of the ion population was observed at m/z 188, which corresponds to two exchanges. It is well-known that the trapping region of the ion trap used in these experiments is not completely water-free. Therefore, the observation of ions with m/z 188 is likely due to back-exchange inside the mass spectrometer.
Quantum-Chemical Calculations
To obtain predicted IR spectra, DFT calculations were performed using the Gaussian16 software package.43 For each ion of interest, several molecular structures were manually defined based on chemical intuition and known gas-phase ion chemistry. Structures were optimized at the B3LYP/6–31++G(d,p) level of theory, followed by a vibrational frequency calculation within the harmonic approximation. Calculated spectra were scaled using a linear scaling factor of 0.975 and broadened using a Gaussian line shape function (20 cm–1 full width at half maximum) to facilitate comparison with the experimental IR spectra.
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance from the FELIX group and of Siebolt de Boer, Ed van der Heeft, and Joris Reintjes from the Translational Metabolic Laboratory. We thank the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. The authors are appreciative of the friendly and helpful support provided by Bruker related to hardware and software modifications of our mass spectrometers, in particular Dr. Christoph Gebhardt. Financial support for this project was provided by NWO Chemical Sciences under projects NWO-TTW nr. 15769 and TKI-LIFT nr. 731.017.419 awarded to J.O. This work was also supported by an ERC-Stg awarded to T.J.B. (GlycoEdit, 758913). We thank NWO Exact and Natural Sciences and the SURFsara Supercomputer Centre for providing the computational time and resources (grant 2019.062). Parts of this work were financially supported by an Interfacultary Collaboration Grant from Radboud University (awarded to K.L.M.C. and J.M.). This research made use of metabolomics infrastructure that is part of the NWO-funded Netherlands X-omics initiative, project 184.034.019.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c02896.
LC–MS, IRIS spectra, computed structures, IR spectra, and a full description of the synthetic methods (PDF)
Author Contributions
○ R.E.vO., U.F.H.E. and J.M. contributed equally to this work and share first authorship on this study. J.O., J.M., R.A.W., and L.A.J.K. conceptualized and supervised the study. R.E.vO. carried out IRIS measurements under guidance of G.B. and J.M. and performed quantum-chemical calculations. Analysis of IRIS data was performed by R.E.vO. with help from J.M., J.O., and G.B. U.F.H.E. performed untargeted LC–MS experiments under supervision of K.L.M.C. and R.A.W. Synthesis of reference standards at Nijmegen was designed by T.J.B. and F.P.J.T.R. and conducted by J.M. and J.M.; synthesis at Cologne was performed by M.P. and T.T. under supervision of A.B. Biochemical and clinical aspects of this study were contributed by M.C.D.H., K.L.M.C., R.A.W., L.A.J.K., and C.D.M.vK. The initial manuscript was written by R.E.vO and J.M., with contributions on synthesis from J.Mer. and on medical aspects from K.L.M.C. The manuscript was then edited by J.O., T.J.B., and R.A.W., with help from all authors. Acquisition of main grants upon which this study is based was by J.O., L.A.J.K., T.J.B., K.L.M.C., and J.M.
The authors declare no competing financial interest.
Notes
All data are included in the manuscript and the Supporting Information. Raw research data are available upon request from the corresponding authors.
Notes
The work described in this study has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. All patients (or their guardians) approved of the possible use of their anonymized left-over samples for method validation purposes, in agreement with institutional and national legislation.
Supplementary Material
References
- Dunn W. B.; Erban A.; Weber R. J. M.; Creek D. J.; Brown M.; Breitling R.; Hankemeier T.; Goodacre R.; Neumann S.; Kopka J.; Viant M. R. Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. 10.1007/s11306-012-0434-4. [DOI] [Google Scholar]
- Hufsky F.; Böcker S. Mining molecular structure databases: Identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom. Rev. 2017, 36, 624–633. 10.1002/mas.21489. [DOI] [PubMed] [Google Scholar]
- Cui L.; Lu H.; Lee Y. H. Challenges and emergent solutions for LC-MS/MS based untargeted metabolomics in diseases. Mass Spectrom. Rev. 2018, 37, 772–792. 10.1002/mas.21562. [DOI] [PubMed] [Google Scholar]
- Hufsky F.; Scheubert K.; Böcker S. Computational mass spectrometry for small-molecule fragmentation. TrAC, Trends Anal. Chem. 2014, 53, 41–48. 10.1016/j.trac.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Outersterp R. E.; Houthuijs K. J.; Berden G.; Engelke U. F.; Kluijtmans L. A.; Wevers R. A.; Coene K. L. M.; Oomens J.; Martens J. Reference-standard free metabolite identification using infrared ion spectroscopy. Int. J. Mass Spectrom. 2019, 443, 77–85. 10.1016/j.ijms.2019.05.015. [DOI] [Google Scholar]
- Martens J.; Berden G.; Bentlage H.; Coene K. L. M.; Engelke U. F.; Wishart D.; van Scherpenzeel M.; Kluijtmans L. A. J.; Wevers R. A.; Oomens J. Unraveling the unknown areas of the human metabolome: the role of infrared ion spectroscopy. J. Inherit. Metab. Dis. 2018, 41, 367–377. 10.1007/s10545-018-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Koppen V.; Berden G.; Cuyckens F.; Oomens J. Combined Liquid Chromatography-Infrared Ion Spectroscopy for Identification of Regioisomeric Drug Metabolites. Anal. Chem. 2017, 89, 4359–4362. 10.1021/acs.analchem.7b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; van Outersterp R. E.; Kluijtmans L. A. J.; Engelke U. F.; van Karnebeek C. D. M.; Wevers R. A.; Oomens J. Molecular identification in metabolomics using infrared ion spectroscopy. Sci. Rep. 2017, 7, 3363. 10.1038/s41598-017-03387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismesia A. P.; Bell M. R.; Tesler L. F.; Alves M.; Polfer N. C. Infrared ion spectroscopy: an analytical tool for the study of metabolites. Analyst 2018, 143, 1615–1623. 10.1039/C8AN00087E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlova O.; Colvin S. M.; Brathwaite A.; Menges F. S.; Craig S. M.; Miller S. J.; Johnson M. A. Identification and Partial Structural Characterization of Mass Isolated Valsartan and Its Metabolite with Messenger Tagging Vibrational Spectroscopy. J. Am. Soc. Mass Spectrom. 2017, 28, 2414–2422. 10.1007/s13361-017-1767-z. [DOI] [PubMed] [Google Scholar]
- Cismesia A. P.; Bailey L. S.; Bell M. R.; Tesler L. F.; Polfer N. C. Making Mass Spectrometry See the Light: The Promises and Challenges of Cryogenic Infrared Ion Spectroscopy as a Bioanalytical Technique. J. Am. Soc. Mass Spectrom. 2016, 27, 757–766. 10.1007/s13361-016-1366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler B.; Laloy-Borgna G.; Barnes L.; Allouche A.-R.; Bouju E.; Dugas V.; Demesmay C.; Compagnon I. Online Separation and Identification of Isomers Using Infrared Multiple Photon Dissociation Ion Spectroscopy Coupled to Liquid Chromatography: Application to the Analysis of Disaccharides Regio-Isomers and Monosaccharide Anomers. Anal. Chem. 2018, 90, 11741–11745. 10.1021/acs.analchem.8b02801. [DOI] [PubMed] [Google Scholar]
- Corinti D.; Maccelli A.; Crestoni M. E.; Cesa S.; Quaglio D.; Botta B.; Ingallina C.; Mannina L.; Tintaru A.; Chiavarino B.; Fornarini S. IR ion spectroscopy in a combined approach with MS/MS and IM-MS to discriminate epimeric anthocyanin glycosides (cyanidin 3-O-glucoside and -galactoside). Int. J. Mass Spectrom. 2019, 444, 116179 10.1016/j.ijms.2019.116179. [DOI] [Google Scholar]
- Walhout E. Q.; Dorn S. E.; Martens J.; Berden G.; Oomens J.; Cheong P. H. Y.; Kroll J. H.; O’Brien R. E. Infrared Ion Spectroscopy of Environmental Organic Mixtures: Probing the Composition of α-Pinene Secondary Organic Aerosol. Environ. Sci. Technol. 2019, 53, 7604–7612. 10.1021/acs.est.9b02077. [DOI] [PubMed] [Google Scholar]
- Lagatie O.; Verheyen A.; Van Asten S.; Odiere M. R.; Djuardi Y.; Levecke B.; Vlaminck J.; Mekonnen Z.; Dana D.; T’Kindt R.; Sandra K.; van Outersterp R.; Oomens J.; Lin R.; Dillen L.; Vreeken R.; Cuyckens F.; Stuyver L. J. 2-Methyl-pentanoyl-carnitine (2-MPC): a urine biomarker for patent Ascaris lumbricoides infection. Sci. Rep. 2020, 10, 15780. 10.1038/s41598-020-72804-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Outersterp R. E.; Moons S. J.; Engelke U. F. H.; Bentlage H.; Peters T. M. A.; van Rooij A.; Huigen M. C. D. G.; Boer S.; van der Heeft E.; Kluijtmans L. A. J.; van Karnebeek C. D. M.; Wevers R. A.; Berden G.; Oomens J.; Boltje T. J.; Coene K. L. M.; Martens J. Amadori rearrangement products as potential biomarkers for inborn errors of amino-acid metabolism. Commun. biol. 2021, 4, 367. 10.1038/s42003-021-01909-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; van Outersterp R. E.; Vreeken R. J.; Cuyckens F.; Coene K. L. M.; Engelke U. F.; Kluijtmans L. A. J.; Wevers R. A.; Buydens L. M. C.; Redlich B.; Berden G.; Oomens J. Infrared ion spectroscopy: New opportunities for small-molecule identification in mass spectrometry - A tutorial perspective. Anal. Chim. Acta 2020, 1093, 1–15. 10.1016/j.aca.2019.10.043. [DOI] [PubMed] [Google Scholar]
- Struys E. A.; Jakobs C. Metabolism of lysine in α-aminoadipic semialdehyde dehydrogenase-deficient fibroblasts: Evidence for an alternative pathway of pipecolic acid formation. FEBS Lett. 2010, 584, 181–186. 10.1016/j.febslet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- Crowther L. M.; Mathis D.; Poms M.; Plecko B. New insights into human lysine degradation pathways with relevance to pyridoxine-dependent epilepsy due to antiquitin deficiency. J. Inherit. Metab. Dis. 2019, 42, 620–628. 10.1002/jimd.12076. [DOI] [PubMed] [Google Scholar]
- Mills P. B.; Struys E.; Jakobs C.; Plecko B.; Baxter P.; Baumgartner M.; Willemsen M. A. A. P.; Omran H.; Tacke U.; Uhlenberg B.; Weschke B.; Clayton P. T. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat. Med. 2006, 12, 307–309. 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- Takazawa O.; Kogami K.; Hayashi K. New Crossed Aldol Reaction. Reaction of Enamines with Aldehydes Activated by Lewis Acids. Bull. Chem. Soc. Jpn. 1985, 58, 2427–2428. 10.1246/bcsj.58.2427. [DOI] [Google Scholar]
- Babler J. H.; Atwood M. C.; Freaney J. E.; Viszlay A. R. The role of imine–enamine tautomerism in effecting cross-aldol condensations. Tetrahedron Lett. 2007, 48, 7665–7667. 10.1016/j.tetlet.2007.08.096. [DOI] [Google Scholar]
- Coughlin C. R.; Swanson M. A.; Spector E.; Meeks N. J. L.; Kronquist K. E.; Aslamy M.; Wempe M. F.; van Karnebeek C. D. M.; Gospe S. M.; Aziz V. G.; Tsai B. P.; Gao H.; Nagy P. L.; Hyland K.; van Dooren S. J. M.; Salomons G. S.; Van Hove J. L. K. The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: a common epileptic encephalopathy. J. Inherit. Metab. Dis. 2018, 10.1007/s10545-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys E. A.; Bok L. A.; Emal D.; Houterman S.; Willemsen M. A.; Jakobs C. The measurement of urinary Δ1-piperideine-6-carboxylate, the alter ego of α-aminoadipic semialdehyde, in Antiquitin deficiency. J. Inherit. Metab. Dis. 2012, 35, 909–916. 10.1007/s10545-011-9443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzyuk T.; Liu A.; Thomas A.; Wilson J. E.; De Biase I.; Longo N.; Pasquali M. A novel method for simultaneous quantification of alpha-aminoadipic semialdehyde/piperideine-6-carboxylate and pipecolic acid in plasma and urine. J. Chromatogr., B 2016, 1017-1018, 145–152. 10.1016/j.jchromb.2016.02.043. [DOI] [PubMed] [Google Scholar]
- Xue J.; Wang J.; Gong P.; Wu M.; Yang W.; Jiang S.; Wu Y.; Jiang Y.; Zhang Y.; Yuzyuk T.; Li H.; Yang Z. Simultaneous quantification of alpha-aminoadipic semialdehyde, piperideine-6-carboxylate, pipecolic acid and alpha-aminoadipic acid in pyridoxine-dependent epilepsy. Sci. Rep. 2019, 9, 11371. 10.1038/s41598-019-47882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E. M.; Moorkoth S.; Lewis L.; Rao P. Biomarker Profiling for Pyridoxine Dependent Epilepsy in Dried Blood Spots by HILIC-ESI-MS. Int. J. Analyt. Chem. 2018, 2018, 1. 10.1155/2018/2583215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S.; Tran N.-T. B.; Gospe S. M.; Hahn S. H. Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS. Mol. Genet. Metab. 2013, 110, 237–240. 10.1016/j.ymgme.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Coene K. L. M.; Kluijtmans L. A. J.; van der Heeft E.; Engelke U. F. H.; de Boer S.; Hoegen B.; Kwast H. J. T.; van de Vorst M.; Huigen M. C. D. G.; Keularts I. M. L. W.; Schreuder M. F.; van Karnebeek C. D. M.; Wortmann S. B.; de Vries M. C.; Janssen M. C. H.; Gilissen C.; Engel J.; Wevers R. A. Next-generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. 10.1007/s10545-017-0131-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Karnebeek C. D. M.; Tiebout S. A.; Niermeijer J.; Poll-The B. T.; Ghani A.; Coughlin Ii C. R.; Van Hove J. L. K.; Richter J. W.; Christen H. J.; Gallagher R.; Hartmann H.; Stockler-Ipsiroglu S. Pyridoxine-Dependent Epilepsy: An Expanding Clinical Spectrum. Pediatric Neurology 2016, 59, 6–12. 10.1016/j.pediatrneurol.2015.12.013. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Tzur D.; Knox C.; Eisner R.; Guo A. C.; Young N.; Cheng D.; Jewell K.; Arndt D.; Sawhney S.; Fung C.; Nikolai L.; Lewis M.; Coutouly M.-A.; Forsythe I.; Tang P.; Shrivastava S.; Jeroncic K.; Stothard P.; Amegbey G.; Block D.; Hau D. D.; Wagner J.; Miniaci J.; Clements M.; Gebremedhin M.; Guo N.; Zhang Y.; Duggan G. E.; MacInnis G. D.; Weljie A. M.; Dowlatabadi R.; Bamforth F.; Clive D.; Greiner R.; Li L.; Marrie T.; Sykes B. D.; Vogel H. J.; Querengesser L. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Eisner R.; Young N.; Gautam B.; Hau D. D.; Psychogios N.; Dong E.; Bouatra S.; Mandal R.; Sinelnikov I.; Xia J.; Jia L.; Cruz J. A.; Lim E.; Sobsey C. A.; Shrivastava S.; Huang P.; Liu P.; Fang L.; Peng J.; Fradette R.; Cheng D.; Tzur D.; Clements M.; Lewis A.; De Souza A.; Zuniga A.; Dawe M.; Xiong Y.; Clive D.; Greiner R.; Nazyrova A.; Shaykhutdinov R.; Li L.; Vogel H. J.; Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Jewison T.; Guo A. C.; Wilson M.; Knox C.; Liu Y.; Djoumbou Y.; Mandal R.; Aziat F.; Dong E.; Bouatra S.; Sinelnikov I.; Arndt D.; Xia J.; Liu P.; Yallou F.; Bjorndahl T.; Perez-Pineiro R.; Eisner R.; Allen F.; Neveu V.; Greiner R.; Scalbert A. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Marcu A.; Guo A. C.; Liang K.; Vázquez-Fresno R.; Sajed T.; Johnson D.; Li C.; Karu N.; Sayeeda Z.; Lo E.; Assempour N.; Berjanskii M.; Singhal S.; Arndt D.; Liang Y.; Badran H.; Grant J.; Serra-Cayuela A.; Liu Y.; Mandal R.; Neveu V.; Pon A.; Knox C.; Wilson M.; Manach C.; Scalbert A. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Maille G. O.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. METLIN: A Metabolite Mass Spectral Database. Therap. Drug Monitor. 2005, 27, 747. 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Pence H. E.; Williams A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. 10.1021/ed100697w. [DOI] [Google Scholar]
- Boles G. C.; Hightower R. L.; Berden G.; Oomens J.; Armentrout P. B. Zinc and Cadmium Complexation of l-Threonine: An Infrared Multiple Photon Dissociation Spectroscopy and Theoretical Study. J. Phys. Chem. B 2019, 123, 9343–9354. 10.1021/acs.jpcb.9b08184. [DOI] [PubMed] [Google Scholar]
- Coates R. A.; McNary C. P.; Boles G. C.; Berden G.; Oomens J.; Armentrout P. B. Structural characterization of gas-phase cysteine and cysteine methyl ester complexes with zinc and cadmium dications by infrared multiple photon dissociation spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 25799–25808. 10.1039/C5CP01500F. [DOI] [PubMed] [Google Scholar]
- Engelke U. F. H.; van Outersterp R.; Merx J.; van Geenen F. A. M. G.; van Rooij A.; Berden G.; Huigen M. C. D. G.; Kluijtmans L. A. J.; Peters T. M. A.; Al-Shekaili H. H.; Leavitt B. R.; de Vrieze E.; Broekman S.; van Wijk E.; Tseng L. A.; Kulkarni P.; Rutjes F. P. J. T.; Mecinović J.; Struys E. A.; Jansen L. A.; Gospe S. M.; Mercimek-Andrews S.; Hyland K.; Willemsen M. A. A. P.; Bok L. A.; van Karnebeek C. D. M.; Wevers R. A.; Boltje T. J.; Oomens J.; Martens J.; Coene K. L. M. Untargeted metabolomics and infrared ion spectroscopy identify biomarkers for pyridoxine-dependent epilepsy. J. Clin. Invest. 2021, 131 (15), e148272. 10.1172/JCI148272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R.; Patti G. J.; Rinehart D.; Siuzdak G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Infrared ion spectroscopy in a modified quadrupole ion trap mass spectrometer at the FELIX free electron laser laboratory. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Oepts D.; van der Meer A. F. G.; van Amersfoort P. W. The Free-Electron-Laser user facility FELIX. Infrared Phys. Technol. 1995, 36, 297–308. 10.1016/1350-4495(94)00074-U. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision A.03. Gaussian, Inc., Wallingford CT: 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.