

Abstract
Chimeric antigen receptor (CAR)-modified T cell therapy is effective in treating lymphomas, leukemias, and multiple myeloma in which the tumor cells have high amounts of target antigen. However, achieving durable remission in patients and extending this therapy to patients with solid tumors will require CAR T cells that can eliminate tumors with a low density of target antigen. Although CARs were designed to mimic T cell receptor (TCR) signaling, TCRs are at least 100-fold more sensitive to antigen. To design a CAR with improved antigen sensitivity, we compared TCR and CAR signaling in primary human T cells in response to ROR1, a transmembrane neurotrophic tyrosine kinase that is associated with B-cell chronic lymphocytic leukemia. Phosphoproteomic analysis revealed that key T cell signaling proteins—such as CD3δ, CD3ε, and CD3γ, which comprise a T cell coreceptor, and the TCR adaptor protein LAT—were either not phosphorylated or were only weakly phosphorylated by CAR stimulation. Modifying the CAR sequence to better engage CD3ε and LAT (specifically, BB/ζ CARs containing CD3ε or GRB2 domains) resulted in enhanced T cell activation against tumor cells with a low density of antigen, in both culture and in vivo models of lymphoma, leukemia, and breast cancer. These CARs represent examples of alterations in receptor design that were guided by in-depth interrogation of T cell signaling.
Introduction
A single infusion of T cells engineered to express a synthetic CD19-targeted chimeric antigen receptor (CAR) can produce complete remissions in up to 90% of patients with acute lymphocytic leukemia and 40–50% of patients with non-Hodgkin lymphoma (1–5). Unfortunately, a substantial proportion of patients progress or relapse after therapy, with tumor cells expressing little to no detectable CD19. Efforts to rescue such patients by sequential CAR T cell targeting of a second molecule, CD22, similarly results in relapse of CD22low tumor cells (6–8). Clinical trials of B cell maturation antigen (BCMA)-targeted CAR T cells in patients with multiple myeloma have also demonstrated outgrowth of tumor cells expressing low levels of BCMA (9, 10). Thus, treatment failure due to reduced antigen expression is presently a barrier to effective CAR T cell therapy (11).
CARs have been designed empirically based on our understandings of T cell signaling. Traditional CAR designs link a tumor antigen binding domain, extracellular spacer, transmembrane domain, CD28 (28) or 4–1BB (BB) costimulatory domain, and CD3ζ (ζ) signaling endodomain into a single receptor (12). Unlike native TCRs, which can trigger T cell activation after recognition of as few as 1–10 agonist peptide-MHC (pMHC) complexes, both CAR formats require thousands of surface antigen molecules for robust signaling (13–17). CARs with 28/ζ signaling domains are more sensitive to antigen than their BB/ζ counterparts, but 28/ζ CAR T cells exhibit shorter persistence after adoptive transfer to patients compared to BB/ζ CAR T cells (18, 19). 28/ζ CARs also have an increased propensity to tonically signal, induce pro-inflammatory cytokine secretion that contributes to cytokine release syndrome, confer reduced metabolic fitness and promote T cell exhaustion compared to BB/ζ CARs (20–25).
There are many differences between CARs and TCRs that may account for the increased antigen density threshold necessary for CARs to evoke T cell effector functions. Unlike TCRs that bind agonist pMHC with micromolar affinity (26), CARs bind their MHC-independent ligands with nanomolar affinities (27). The increased binding affinity of CARs may alter receptor off-rate kinetics, serial triggering, and mechanoreceptor function, properties that are thought to contribute to the ability of TCRs to sense low levels of pMHC (28, 29). Upon antigen engagement, the TCR and its associated CD3δ, ε, γ, and ζ chains also assemble multi-component signaling complexes (30, 31). Although CARs do associate with some TCR signaling proteins, quantitative and qualitative alterations in complex assembly and synapse structure could alter antigen sensitivity (22, 32). Indeed, imaging of CAR and TCR synapses has shown that CAR synapses are less reliant on ICAM-1/LFA-1 interactions and exhibit disorganized patterns of LCK localization (33–35).
New CARs that either strengthen signaling to augment T cell recognition of tumor cells with lower antigen density (19, 21, 36, 37), or dampen signaling to prevent rapid effector differentiation and cytokine release syndrome (25, 38) have been identified by empirically altering known structural components. Designing CARs with novel structural features that more closely mimic TCR signaling could enable recognition of tumor cells with lower levels of target antigen and simultaneously promote long-term persistence. We surmised that a detailed comparison of TCR and CAR signaling events would provide insights to guide these design efforts. We therefore analyzed phosphoprotein signaling events after TCR or CAR activation in a population of human T cells that co-expressed a monospecific TCR and CAR. Phosphorylation of important upstream mediators of TCR signaling such as CD3δ, ε, and γ chains and LAT was markedly more prominent after TCR than 28/ζ or BB/ζ CAR stimulation. We then specifically modified the BB/ζ CAR backbone to engage CD3ε and LAT, yielding new receptors with a lower antigen threshold for activation and improved antitumor function in xenograft mouse models.
Results
Direct comparison of TCR and CAR antigen sensitivity in a single T cell population
The amount of antigen required for T cell activation is higher for CARs than TCRs, although the mechanisms underlying these differences remain speculative (13–15, 17, 39). To compare TCR and CAR antigen sensitivity in a single cell population, we derived human memory CD45RO+ CD8+ T cells specific for an epitope of Epstein Barr virus (EBV) presented by HLA-B8 (RAKFKQLL), and engineered these cells to express a 28/ζ CAR specific for ROR1, a tumor-associated antigen that is being targeted with CAR T cells in patients with advanced cancers (NCT02706392) (Fig. 1A). A 28/ζ CAR was utilized for the initial signaling studies to permit a direct comparison to TCR/CD28 signaling and because it is more sensitive than a BB/ζ CAR. T cells that both bound an HLA-B8/EBV tetramer and expressed the CAR transduction marker were purified by cell sorting and expanded for analysis (Fig. 1B).
Figure 1. Bi-specific T cells and magnetic beads enable analysis of TCR and CAR signaling in a single cell population.
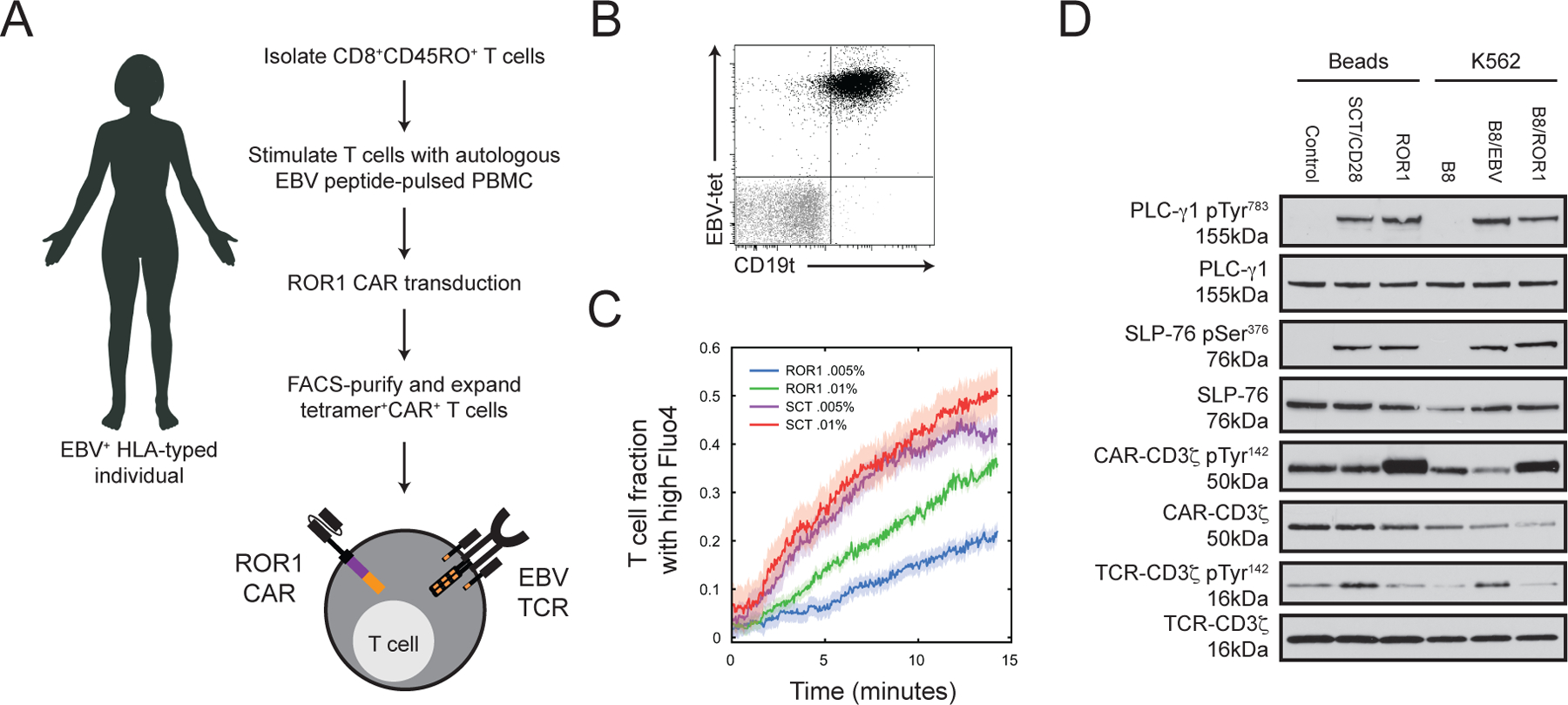
(A) Schematic describing formulation of bi-specific T cells possessing a EBV-specific TCR and ROR1-specific CAR. (B) Flow cytometry analysis of EBV-tetramer (EBV-tet) binding and CD19t transduction marker expression in expanded T cells. Plot shows stained (black) and isotype (grey) CD8+ singlet lymphocytes. Data are representative of three experiments using T cells from two unique donors. (C) Time plot shows mean ± SEM (shaded) Ca2+ mobilization in bi-specific T cells stimulated by ROR1 (green and blue) or SCT (red and purple) containing lipid bilayers as measured by Fluo-4 AM intensity of individual cell responses. Traces represent the fraction of cells above an activation threshold at any given time. Antigen density was modulated by altering the molar fraction of biotinylated lipids in the supported bilayer: 0.005% (magenta and blue) or 0.01% (red and green). (D) Western blot analysis for CD3ζ, CD3ζ pTyr142, SLP-76, SLP-76 pSer376, PLC-γ1 and PLC-γ1 pTyr783 in cell lysates after stimulation with indicated beads or K562 cells for 45 min. Data are representative of two experiments using T cells from two unique donors.
The “bi-specific” T cells expressed an endogenous antigen-specific TCR, which avoided altered levels of TCR expression as well as TCR α and β chain mispairing that can occur after transduction of an engineered TCR under non-physiologic regulatory control (40–42). Phenotyping of three independently derived bi-specific T cell populations immediately prior to stimulation showed that 80–93% bound the HLA B8/EBV tetramer and expressed the CD19t transduction marker, and a majority expressed CD62L and CD28 (fig. S1A–B). More than 83% of the cells were in the G0/G1 cell cycle phase, indicating the cells had returned to quiescence after transduction and expansion (fig. S1B). Notably, the functional TCR avidity of the EBV-specific T cells from each donor was similar (fig. S1C). CAR expression in bi-specific T cells, as measured by binding to ROR1-Fc, was also identical to polyclonal T cells transduced with the CAR (fig. S1D), demonstrating that the alternative T cell culture production method to obtain bi-specific T cells did not impact overall cell phenotype or function. Additionally, proximal signaling after CAR ligation in T cells generated from CD8+ naïve or memory precursors was nearly identical (fig. S1E), suggesting that studying early TCR and CAR events bi-specific T cells derived from memory precursors is relevant to other T cell subsets.
To provide an index of the relative sensitivity of TCR and CAR for antigen recognition, fluorescence microscopy was used to measure Ca2+ flux over time in individual bi-specific T cells after antigen engagement. We produced a recombinant single chain trimer (SCT) consisting of EBV-RAK peptide, HLA-B8, and β2 microglobulin, and a recombinant ROR1 ectodomain to stimulate the TCR or CAR, respectively (43). Varying quantities of biotinylated SCT or ROR1 were coated onto a supported lipid bilayer via streptavidin linkage. T cells were exposed to the bilayers and the fraction of T cells exhibiting high intracellular Ca2+ were compared. At two low antigen densities, a greater fraction of bi-specific T cells was triggered by TCR stimulation than by CAR stimulation (Fig. 1C). A similar frequency of T cells responded at both SCT antigen densities but the fraction of T cells responding to ROR1 stimulation was lower and directly proportional to antigen density. These data indicate that the TCR response was saturated over this 2-fold range in antigen density and confirm that TCRs possess an increased antigen sensitivity as compared to a 28/ζ CAR.
Phosphoproteomic analysis of TCR and CAR signaling
We next studied phosphoprotein signaling within bi-specific T cells using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) to gain insights into the mechanisms of differential antigen sensitivity between the TCR and CAR. To facilitate LC-MS/MS analysis of phosphoproteomic changes in T cells, we formulated SCT- or ROR1-coated beads that would initiate signaling without contamination by proteins from antigen presenting cells (APC) (22, 44). T cell stimulation with SCT or with ROR1 beads resulted in phosphorylation of PLC-γ1 at Tyr783 and of the endogenous TCR-associated CD3ζ chain or CAR CD3ζ domain, respectively (fig. S2A–B). Consistent with prior studies of CD28/CD3ζ CARs, basal phosphorylation of CAR CD3ζ domain was observed and was augmented by stimulation with ROR1 beads (22, 45). Because CARs deliver both ‘signal 1’ (CD3ζ) and “signal 2” (CD28), we added CD28 monoclonal antibody to SCT beads to provide signal 2 for TCR stimulation. Stimulation with SCT/CD28 beads promoted a similar intensity of phosphorylation of CAR CD3ζ, ZAP-70, SLP-76, and PLC-γ1 as did the SCT beads but enhanced T cell proliferation after 72 hours (fig. S2C–D), consistent with the costimulatory function of CD28. As validation, we compared SCT/CD28 and ROR1 bead stimulation to that with K562 cells expressing the respective TCR and CAR target antigens. After 45 minutes of stimulation with pMHC or CAR ligand by APC or beads, phosphorylation of native CD3ζ or exogenous CAR-CD3ζ chains at Tyr142 appeared nearly identical, and that of SLP-76 Ser376 and PLC-γ1 Tyr783 were also highly similar after APC or bead stimulation (Fig. 1D).
Cellular lysates of bi-specific T cells incubated with SCT/CD28, ROR1, or uncoated magnetic beads for 10, 45, or 90 minutes were prepared for LC-MS/MS (Fig. 2A). Three independent experiments were performed with bi-specific T cells formulated from two donors. In total, LC-MS/MS identified 30,669 distinct phosphorylation sites corresponding to 4,997 gene products, with 64% of those sites observed in at least two experiments (fig. S3A). 715 phosphorylation sites (2.3%) were tyrosines, 5,056 (16.5%) were threonines, and 24,898 (81.2%) were serines, consistent with prior studies of unstimulated and stimulated T cells (22, 46, 47).
Figure 2. CAR stimulation promotes less intense phosphorylation of CD3 chains and proximal TCR signaling adaptors.
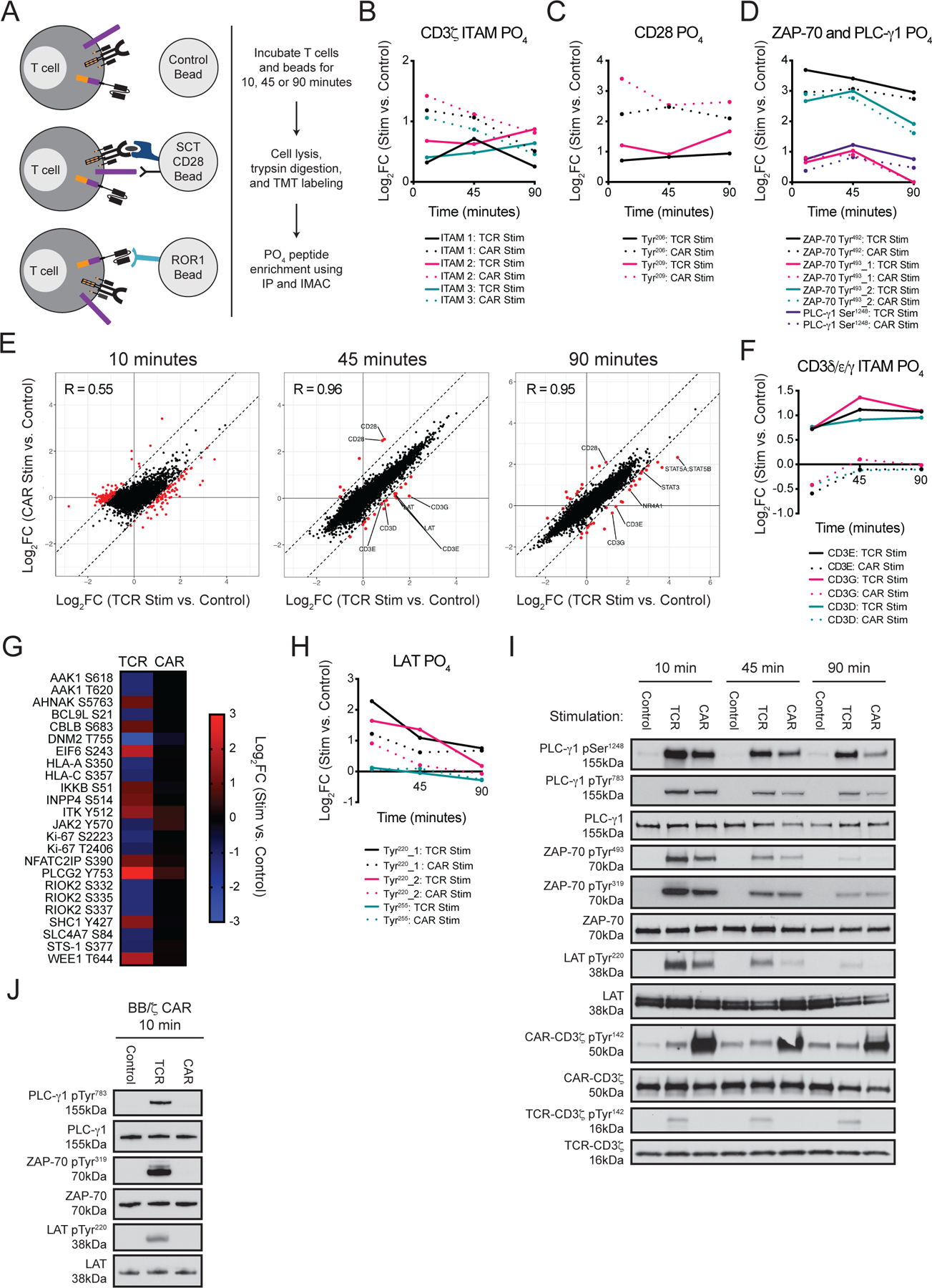
(A) Schematic of experimental design stimulating bi-specific T cells with beads coated with TCR (SCT/CD28) or CAR (ROR1) antigens. (B to D) Graphs show mean log2FC of the indicated phosphorylation (PO4) sites as depicted in (A) from two or three LC-MS/MS experiments with T cells from two unique donors. (E) Comparison of the mean log2FC of phosphorylation sites after TCR or CAR stimulation with SCT/CD28 or ROR1 beads. Red dots specify sites that possessed mean log2FC values differing by ≥ 1 between TCR and CAR stimulated samples at 10, 45, and 90 min relative to control bead stimulation. (F) Graph shows mean log2FC of phosphorylation of CD3δ, CD3ε, and CD3γ ITAMs. The log2FC in phosphorylation of both ITAM tyrosines was averaged. Data are means from two or three LC-MS/MS experiments with T cells from two unique donors. (G) Heat map shows mean log2FC of select phosphorylation sites at the 10-min time point. (H) Graph shows mean log2FC of select phosphorylation sites in LAT. (I) Western blot analysis for CD3ζ, CD3ζ pTyr142, LAT and LAT pTyr220, ZAP-70, ZAP-70 pTyr319, ZAP-70 pTyr493, PLC-γ1, and PLC-γ1 pTyr783, and PLC-γ1 pSer1248 (p = phosphorylation) in the bi-specific T cell lysates utilized for LC-MS/MS experiments. Blots are representative of three experiments using T cells prepared from two unique donors; compiled data is quantified in fig. S4A. (J) Western blot analysis for LAT, LAT pTyr220, ZAP-70, ZAP-70 pTyr319, PLC-γ1, and PLC-γ1 pTyr783 in lysates from bi-specific T cells expressing a BB/ζ CAR after 10 min of stimulation with control, SCT/CD28, or ROR1 beads. Blots are representative of three experiments using T cells prepared from two unique donors; compiled data is quantified in fig. S4B.
CAR stimulation promotes early phosphorylation of CD3ζ but less intense phosphorylation of of other CD3 chains and signaling adaptors
Differences between CAR and TCR stimulation at each time point were examined by analyzing the log2FC of phosphorylation sites in TCR and CAR stimulated samples relative to the control sample (table S1). Query of the dataset for phosphorylation sites on CD3ζ, CD28, ZAP-70, and PLC-γ1 confirmed that each canonical signaling protein was phosphorylated after TCR or CAR engagement (48). All three CD3ζ ITAMs as well as CD28 Tyr206 and Tyr209 were more intensely phosphorylated by CAR stimulation than by TCR stimulation at the 10- and 45-minute time points (Fig. 2B–C) (49). Although there is no established mechanism for recruitment of the CD8 coreceptor and its associated pool of LCK by CARs, we and others have demonstrated that LCK directly associates with the CAR CD28 endodomain (21, 22, 50), which may in part account both for basal phosphorylation of CD3ζ and the rapid phosphorylation of CD3ζ and CD28 observed with CAR stimulation. Despite stronger CD3ζ phosphorylation after CAR engagement, ZAP-70 Tyr493 displayed a similar log2FC in phosphorylation after TCR and CAR stimulation, and PLC-γ1 Ser1248 was more weakly phosphorylated by CAR than TCR stimulation (Fig. 2D–E). These findings were surprising since the more intense phosphorylation of CD3ζ and CD28 after CAR stimulation might be expected to result in increased phosphorylation of ZAP-70 and PLC-γ1.
Global analysis of the 10-minute time point revealed a weak, non-1-to-1 association (R = 0.55) between TCR and CAR stimulation-induced changes in protein phosphorylation, suggesting that early signaling events were different (Fig. 2E). Filtering for phosphorylation sites that differed by at least two-fold between TCR and CAR stimulated samples identified 282 PO phosphorylation sites (table S2). Of these, 267 (95%) were more intensely modulated by TCR than CAR stimulation and 15 (5%) were more intensely modulated by CAR than TCR stimulation. It was notable that at 10 minutes the ITAMs on CD3D, CD3E, and CD3G were phosphorylated by TCR stimulation and de-phosphorylated by CAR stimulation (Fig. 2F). Signaling intermediates (AHNAK, CBLB, IKKB, INPP4A and SHC1) as well as RNA binding proteins (EIF3A, EIF3C, EIF6, and RBM25) were also phosphorylated by TCR stimulation but not by CAR stimulation (Fig. 2G). log2FC values comparing TCR stimulation to control treatment or CAR stimulation to control treatment were similar across replicate experiments (fig. S3B). Individual phosphosite localizations for certain T cell signaling molecules were also confirmed (table S3).
At the 45-minute time point, TCR and CAR stimulated samples exhibited near 1-to-1 concordance (R = 0.96) and only 28 phosphorylation sites differed more than two-fold between TCR- and CAR-stimulated samples (Fig. 2E). Specifically, tyrosine residues on both positive (CD3D, CD3E, CD3G, and LAT) and negative (LAX1 and PAG1) regulators of T cell signaling were more intensely phosphorylated by TCR than CAR stimulation. LAT and PAG1 had displayed slight (<2-fold) differences in protein phosphorylation at the 10-minute time point that did not meet our cutoffs, indicating that certain differences in proximal signaling after CAR and TCR engagement were magnified over time. After 90 minutes, 37 sites were associated with fold change values that differed by more than 2-fold between TCR and CAR stimulated samples (Fig. 2E). Tyrosine residues in CD3E and CD3G ITAMs remained more intensely phosphorylated by TCR than CAR stimulation, and we also detected preferential phosphorylation of BTAF, TAGAP, SOS1, STAT3, STAT5A/B, and NR4A1 after TCR but not CAR stimulation.
The finding that ITAMs on CD3 chains other than CD3ζ were phosphorylated after TCR but not CAR stimulation was anticipated, because CARs only include CD3ζ and have no mechanism for recruiting other CD3 chains. However, our finding that LAT, which is essential for T cell development and a critical adaptor for coupling antigen receptor and downstream signaling pathways (51), was less intensely phosphorylated by CAR stimulation than TCR stimulation was not previously appreciated (Fig. 2H). To validate the difference in the phosphorylation of LATas well as the previously discussed differences in the phosphorylation of PLC-γ1 and ZAP-70, we analyzed the original whole cell lysates from bi-specific T cells by Western blot. Consistent with the LC-MS/MS data, CAR stimulation resulted in similar phosphorylation of ZAP-70 Tyr493, a transiently weaker phosphorylation of PLC-γ1 Ser1248, and a significantly weaker phosphorylation of LAT Tyr220 compared to each by TCR stimulation (Fig. 2I and fig. S4A).
A 28/ζ CAR was utilized for LC-MS/MS analyses; however, CARs containing a 4–1BB costimulatory domain are also highly effective in patients (12). We therefore asked whether differences in the phosphorylation of ZAP-70, PLC-γ1 and LAT identified between TCR and 28/ζ CAR signaling were also present with a BB/ζ CAR. Bi-specific T cells possessing an EBV-specific TCR and ROR1-specific BB/ζ CAR were derived and signaling was assessed by Western blot. BB/ζ CAR stimulation led to reduced phosphorylation of ZAP-70 at Tyr319, of PLC-γ1 at Tyr783 and of LAT at Tyr220 compared to each by TCR stimulation (Fig. 2J and fig. S4B). Notably, the relative differences in phosphorylation appeared to be of greater magnitude with the BB/ζ CAR than the 28/ζ CAR, as there was negligible phosphorylation of each after 10 minutes of CAR stimulation. We could not evaluate the phosphorylation of CD3δ, CD3ε, and CD3γ ITAM by Western blot due to a lack of commercially available antibodies, but prior LC-MS/MS analysis showed that the ITAMs of these CD3 chains are not phosphorylated by stimulation of BB/ζ CAR T cells (22). Together, our data suggest that phosphorylation of certain key downstream signaling intermediates are delayed and/or weaker after CAR stimulation than TCR stimulation, with potential consequences for the antigen threshold necessary to convey signals that elicit effector activation.
Rational modification of BB/ζ CAR structure enhances T cell antigen reactivity
We hypothesized that the lack of phosphorylation of CD3δ, CD3ε, and CD3γ observed by LC-MS/MS after CAR engagement might limit ZAP-70 recruitment and the bridging between ZAP-70 and LAT (52). In turn, reduced LAT phosphorylation could result in less efficient assembly of the LAT–SLP-76 signaling apparatus that is critical to activate Ca2+ flux, PKC, and Ras and amplify early signaling (53–55). This led us to design new CAR structures that might increase LAT and PLC-γ1 phosphorylation after stimulation. We focused on improving the antigen sensitivity of CARs with a 4–1BB costimulatory domain since 28/ζ CARs have a higher propensity to tonically signal (20, 21), generate larger quantities of pro-inflammatory cytokines that promote the development of cytokine release syndrome (22, 56), and appear more likely to promote T cell exhaustion and dysfunction than BB/ζ CAR T cells (25).
Because CD3ε is as plentiful as CD3ζ in the TCR complex and has been used in other immunotherapy receptor formats (37), we first asked whether incorporating all or part of the CD3ε endodomain into a BB/ζ CAR would provide a functional receptor and enhance antigen sensitivity. CD3ε has three known signaling motifs – a membrane-adjacent basic residue-rich sequence (BRS) that binds LCK (57), a proline-rich sequence (PRS) that may enhance TCR antigen sensitivity by interacting with NCK1 (58, 59), and an ITAM that activates ZAP-70 (48). We designed CD19- and ROR1-specific CARs in which the entire CD3ε endodomain was placed before or after the CD3ζ domain and an STII sequence was placed in the CAR extracellular hinge (Fig. 3A) (60). We also designed a CAR variant that contained only CD3ε PRS and ITAM sequences to determine if signaling could be augmented without increased LCK association that may occur by inclusion of BRS sequences. The addition of full-length CD3ε either before or after CD3ζ resulted in lower CAR surface expression and impaired IFN-γ production by T cells in response to target antigen; however, CARs containing only the CD3ε PRS and ITAM domains were expressed and functioned similarly to a conventional BB/ζ CAR (Fig. 3B and fig. S5A). After incubation with ROR1+ or CD19+ tumor cells, T cells engineered with ROR1 or CD19 BB/εPRS_ITAM/ζ CARs secreted similar amounts of IFN-γ and TNF-α, but more IL-2, than did conventional BB/ζ CAR T cells (Fig. 3C and fig. S5A). Notably, in both cases, T cell cytokine secretion was antigen-dependent, demonstrating that the BB/εPRS_ITAM/ζ CAR did not confer autonomous T cell activation. Together, our analyses indicated that the BB/εPRS_ITAM/ζ CAR was highly functional and did not markedly increase pro-inflammatory cytokine production, which is characteristic of 28/ζ CARs and can lead to cytokine release syndrome (61).
Figure 3. MS-guided CAR designs are expressed by primary T cells.
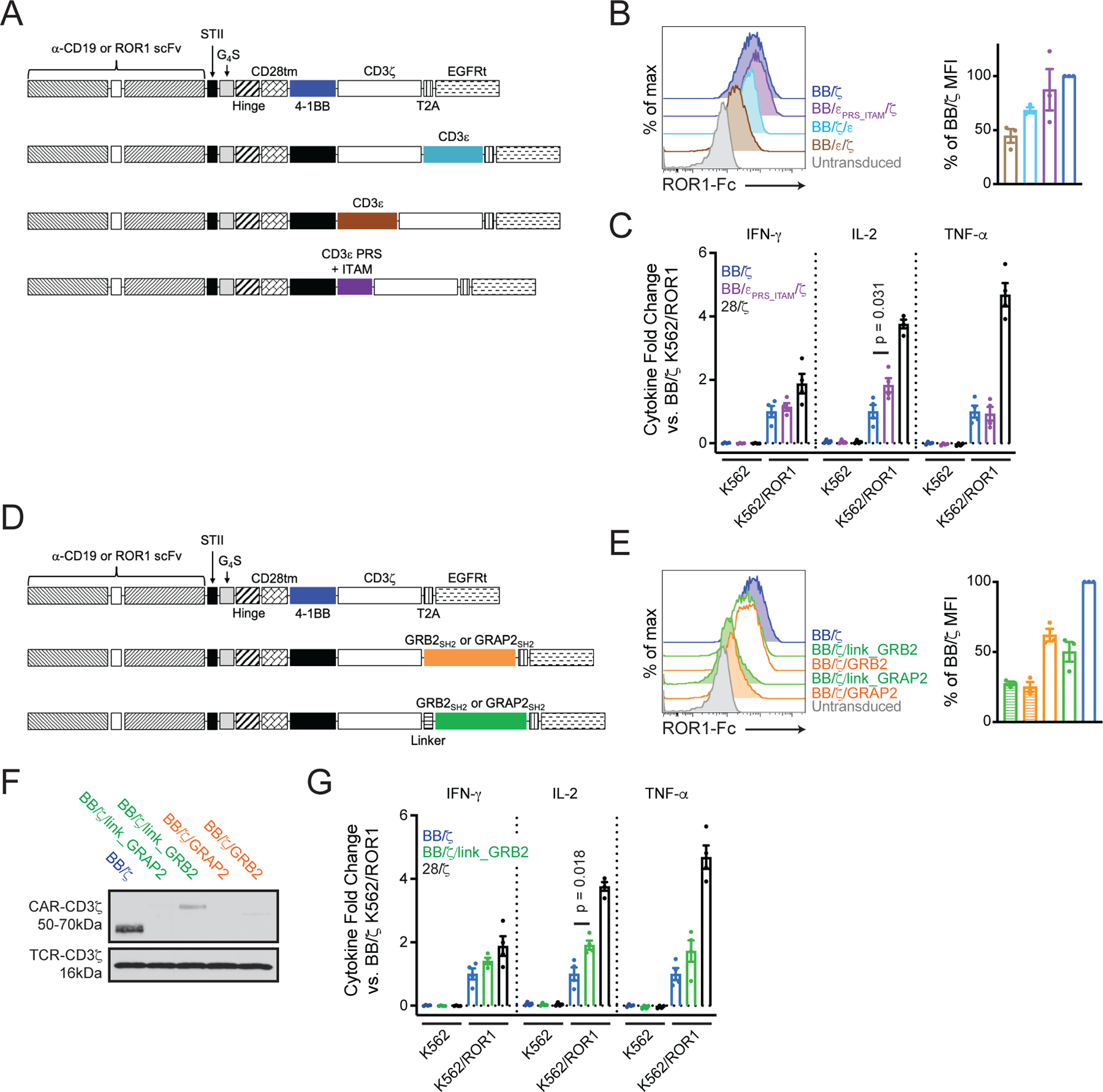
(A) Schematics of CARs with CD3ε sequences. (B) Flow cytometry analysis of ROR1-Fc binding to measure CAR expression on transduced T cells. Plots show untransduced (grey) or CD8+EGFRt+ (colors) singlet lymphocytes. Bar graph shows mean ± SEM of relative percent mean fluorescence intensity (MFI) compared to BB/ζ CAR T cells. N = three experiments using T cells from three unique donors. (C) Mean ± SEM fold change of IFN-γ, IL-2, or TNF-α concentration in cellular supernatant 24 hours after co-culture of indicated CAR T cells with K562 or K562/ROR1 tumor cells. N = four unique T cell donors. P value by an ordinary one-way ANOVA with Tukey’s post test. (D) Schematics of CARs with GRB2 or GRAP2 SH2 domains. (E) Flow cytometry analysis of ROR1-Fc binding, as in (B). Bar graph shows mean ± SEM of relative percent MFI compared to BB/ζ CAR T cells. N = three experiments using T cells from three unique donors. (F) Western blot analysis for CD3ζ in CAR T cell lysates. Blot is representative of three independent experiments using T cells from two unique donors. (G) Mean ± SEM fold change of IFN-γ, IL-2, or TNF-α concentration in cellular supernatant, as in (C). N = 4 unique T cell donors. Note, the BB/ζ and 28/ζ CAR data in (G) are the same as those in (C), altogether run in the same experiments. P value by an ordinary one-way ANOVA with Tukey’s post test.
CAR constructs that improved LAT activation were also designed. We first encoded the LAT transmembrane domain in a BB/ζ CAR hypothesizing that this domain would localize the CAR to lipid rafts and improve LAT association. However, CARs containing two unique LATTMD variants, which differed in the number of extracellular and intracellular residues flanking the transmembrane sequence, were poorly expressed on the surface and in cell lysates (fig. S6A–C) (62). Analysis of CAR-LATTMD-GFP fusion proteins revealed that a LATTMD-GFP fusion protein that lacked any CAR structural elements was highly expressed on the T cell surface, but GFP fusion proteins containing either the extracellular or intracellular CAR sequences were weakly expressed and existed in intracellular compartments (fig. S6D). These results indicated that incorporating a LATTMD into a conventional CAR structure was problematic, and led us to pursue an alternative strategy to improve LAT activation.
The adaptor molecules GRB2, GRAP2, and SOS1 aid formation of signal-amplifying microclusters through binding of their SH2 domains to phosphotyrosine residues on LAT and SLP-76 (62–64). We therefore constructed four CARs bearing a GRB2 or GRAP2 SH2 domain after ζ with or without a flexible linker separating the ζ and SH2 domains (Fig. 3D) (65). CARs containing GRAP2 SH2 sequences were poorly expressed, whereas CARs with GRB2 SH2 sequences were surface expressed at higher levels than CARs with GRAP2 SH2 sequences, but all were lower than that of a conventional BB/ζ CAR (Fig. 3E). Western blotting further showed that the CAR with a GRB2 SH2 domain connected by a flexible linker (BB/ζ/link_GRB2) was best expressed (Fig. 3F). Despite lower levels of BB/ζ/link_GRB2 CAR surface expression, co-culture of BB/ζ/link_GRB2 CAR T cells with K562 and K562/ROR1 tumor cells led to antigen-dependent production of similar quantities of IFN-γ and TNF-α and more IL-2 than BB/ζ CAR T cells (Fig. 3G).
To determine if the comparable cytokine responses elicited by BB/ζ, BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR activation reflected an equivalent sensitivity to antigen, we studied the kinetics and fraction of T cells exhibiting Ca2+ flux when exposed to a ROR1 containing lipid bilayer. We found that fewer than 20% of BB/CD3ζ CAR T cells were activated after 20 minutes of exposure to a ROR1-containing bilayer, even through a wide range of ROR1 ligand densities (Fig. 4A). In contrast, 40–80% of T cells expressing a structurally identical 28/ζ CAR were triggered, consistent with published findings (19). Among the new CAR designs, BB/εPRS_ITAM/ζ CAR T cells exhibited the largest fractional response, approaching that of 28/ζ CAR T cells at the highest ligand concentration; BB/ζ/link_GRB2 CAR T cells were intermediate between BB/εPRS_ITAM/ζ and BB/ζ. Further analysis of the kinetics of Ca2+ mobilization demonstrated that the novel CARs shortened the onset of Ca2+ flux responses and increased the percentage of T cells that mobilized Ca2+ stores compared to the BB/ζ CAR (Fig. 4B–C). Thus, the inclusion of a CD3ε or GRB2 domain onto a BB/ζ CAR enhanced T cell Ca2+ mobilization in response to low levels of antigen. However, no CAR, including 28/ζ, could approximate the robust Ca2+ flux generated by the natural TCR on bi-specific T cells (fig. S5B).
Figure 4. Novel CAR designs possess increased antigen sensitivity.
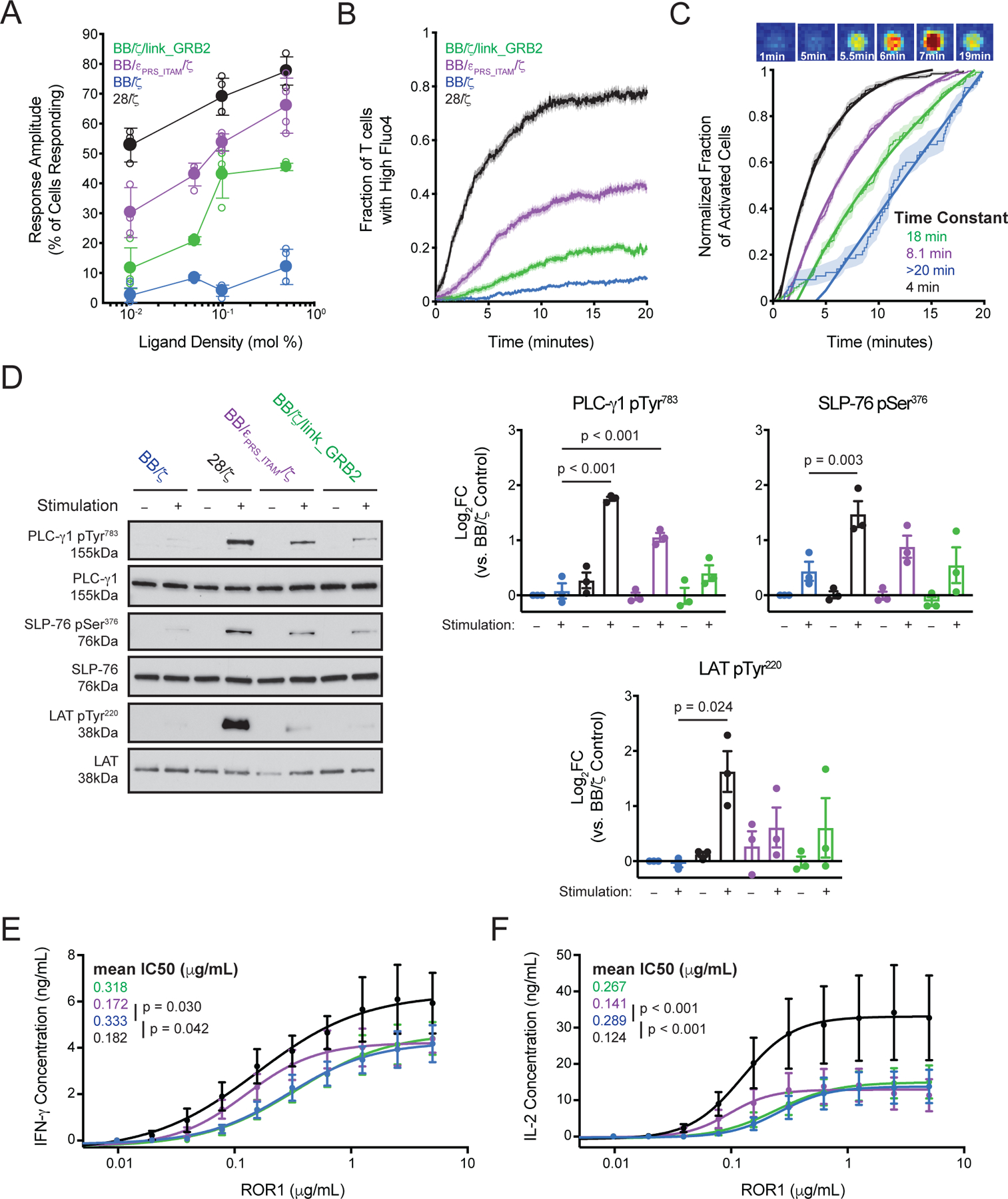
(A) X-Y plot shows the mean ± SEM fraction of cells exhibiting Ca2+ responses after 20 min of exposure to bilayers possessing a range of ROR1 densities as measured by Fluo-4 AM fluorescence intensity. Mol % denotes the molar percentage of ROR1-coated lipids in the bilayer. N = 3 or 4 experiments using T cells from two unique donors. (B) Fluo-4 Ca2+ mobilization measurements for cells stimulated on 0.1% ROR1-labeled bilayers. Time plot shows mean ± SEM (shaded) of the cumulative fraction of Ca2+ mobilization across time after exposure to bilayers. N = 4 independent experiments. (C) Above, image sequence shows a representative example of changing Fluo-4 AM fluorescence intensity as a cell lands on and is activated by a ROR1-containing bilayer. Below, data from (B) is represented as normalized cumulative curves to highlight differences in the kinetics of Ca2+ mobilization. Thick lines are exponential fits to the data with activation time constants for the whole population of cells extracted. (D) Western blot analysis for LAT, LAT pTyr220, SLP-76, SLP-76 pSer376, PLC-γ1 and PLC-γ1 pTyr783 in T cell lysates after 10 minutes of incubation with ROR1 (+) or control (–) beads. Bar graphs show mean ± SEM of log2FC of normalized band intensity from three experiments using T cells from three unique donors. P values were calculated by repeated-measures one-way ANOVA with Tukey’s post test. (E and F) Graphs show mean ± SEM of IFN-γ (E) or IL-2 (F) concentration in supernatant 24 hours after stimulation with the indicated amounts of plate-bound ROR1. A sigmoidal curve was fitted to each CAR construct and mean IC50 is indicated. N = 5 experiments using T cells from five unique donors. P values were calculated by repeated-measures one-way ANOVA with Tukey’s post test.
We then measured phosphorylation of signaling proteins by Western blot to determine whether increased Ca2+ mobilization correlated with downstream signaling. After 10 minutes of stimulation with ROR1 beads, BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CARs promoted modest increases in the phosphorylation of LAT at Tyr220, SLP-76 at Ser376 and PLC-γ1 at Tyr783 relative to that promoted by BB/ζ CAR T cells (Fig. 4D). The phosphorylation intensity matched the trends in Ca2+ mobilization, with progressively increased protein phosphorylation after stimulation of BB/ζ, BB/ζ/link_GRB2, BB/εPRS_ITAM/ζ, and 28/ζ CAR T cells, respectively. We attempted to immunoprecipitate potential binding partners of the novel CARs; however, we did not detect differences in the relative amounts of ZAP-70, NCK1 or LAT associated with each CAR. As previously shown, LCK was differentially associated with BB/ζ and 28/ζ CARs (22, 50).
Subsequently, we asked whether the increased Ca2+ mobilization and PLC-γ1 phosphorylation led to improved T cell effector responses at low ligand density. The various CAR T cells were incubated with titrated quantities of ROR1 for 24 hours prior to measurement of IFN-γ and IL-2 secretion in supernatant. 28/ζ and BB/εPRS_ITAM/ζ CAR T cells secreted more IFN-γ and IL-2 after exposure to limiting amounts of antigen than BB/ζ or BB/ζ/link_GRB2 CAR T cells (Fig. 4E–F). Importantly, mutation of the CD3ε PRS and ITAM abrogated the improved sensitivity (fig. S5D), indicating that the enhanced antigen sensitivity resulted from signaling properties of the PRS and ITAM domains. A mutated form of the BB/ζ/link_GRB2 CAR with an SH2 domain engineered for enhanced affinity for phosphotyrosine motifs was poorly expressed and less functional than the unmutated BB/ζ/link_GRB2 CAR (fig. S5E–F) (66).
BB/ζ CARs containing CD3ε or GRB2 domains maintain antitumor function in settings of high antigen expression
Intense signaling mediated by 28/ζ CARs is linked to the development of T cell exhaustion in settings of high antigen density (19, 22, 25, 67). We therefore sought to determine the sequelae of BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR signaling in xenograft models where tumor cells express high antigen levels. For this, we used models of disseminated CD19+ Raji lymphoma and CD19+ Nalm-6 leukemia, in which the tumor cells express a high number of CD19 molecules per cell (68).
Infusion of a low sub-curative dose of purified CD8+EGFRt+ CD19-specific 28/ζ CAR T cells into Raji-bearing recipient mice transiently reduced tumor burden and modestly improved survival compared to a saline injection (Fig. 5A–C). In contrast, an equivalent dose of purified CD8+EGFRt+ T cells expressing a BB/εPRS_ITAM/ζ, BB/ζ/link_GRB2, or conventional BB/ζ CAR promoted marked improvements in tumor control and survival. Analyses of T cells in the bone marrow 21 days after tumor injection, at a timepoint when the survival curves begin to diverge, showed that BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR T cells expressed lower amounts of the exhaustion-associated inhibitory molecules PD-1 and Lag-3 than 28/ζ CAR T cells, and were present at higher frequencies than 28/ζ CAR T cells (Fig. 5D–F). The graded patterns of PD-1 and Lag-3 expression in the Raji model are consistent with prior work showing that 28/ζ CARs promote higher levels of inhibitory receptor expression that correlate with less effective tumor control (22).
Figure 5. BB/ζ CARs containing CD3ζ or GRB2 domains maintain in vivo antitumor function in settings of high antigen expression.
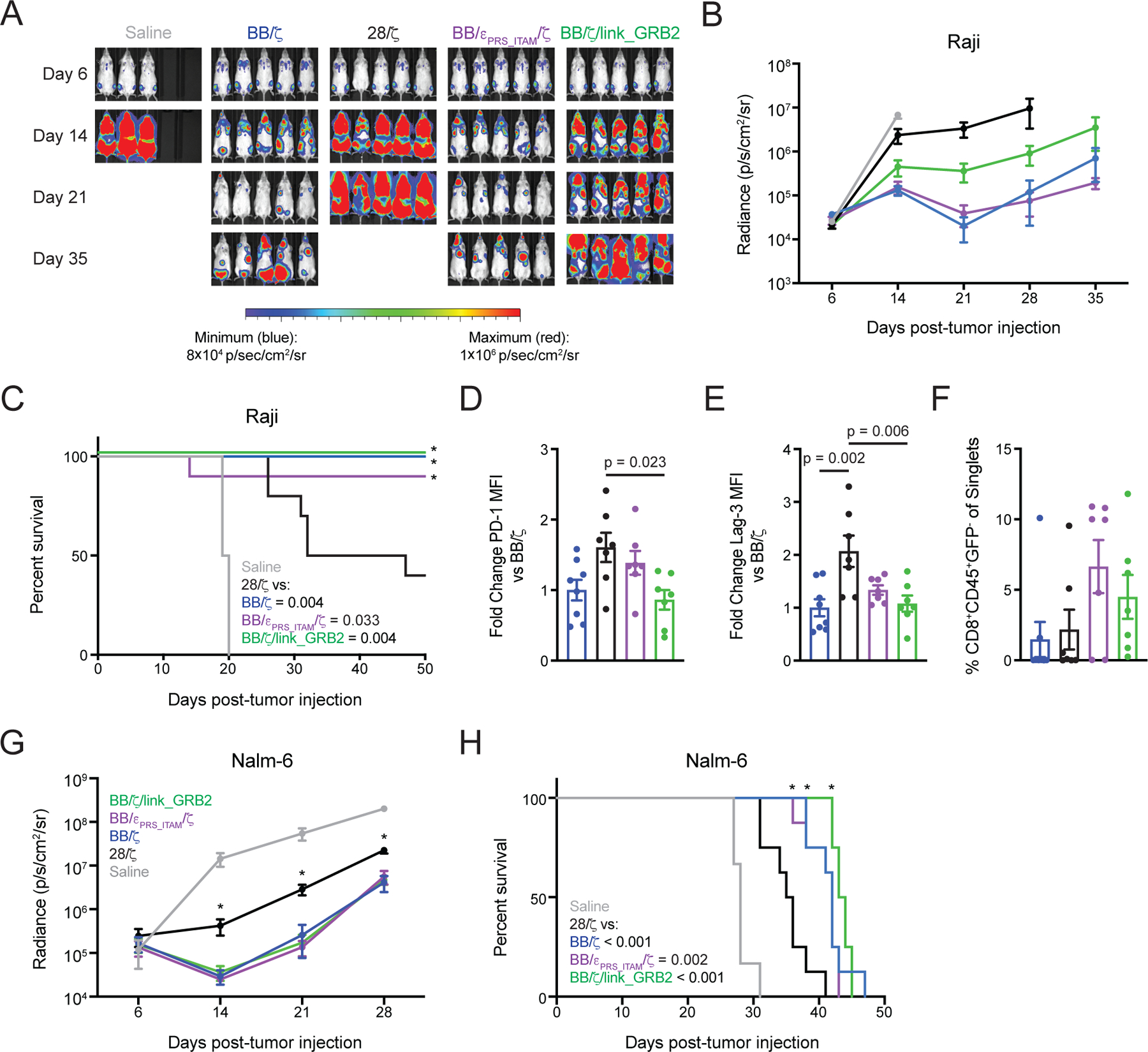
(A and B) Representative bioluminescence images (A) and mean ± SEM radiance (photons/second/cm2/sr) of (B) Raji/ffluc tumor burden in mice treated with CD19-specific CD8+ CAR T cells. N = 6 or 10 mice per group pooled from two independent experiments using T cells from unique donors. (C) Survival analysis of mice as in (B). Significance (*) and P values (inset legend) were calculated by log-rank test. (D to F) Graphs show mean ± SEM of normalized PD-1 (D) and Lag3 (E) mean fluorescence intensity (MFI) on CD8+CD45+GFP- CAR T cells as well as percent frequency of such cells (F) within the bone marrow 21 days after Raji cell tumor injection. N = 7 or 8 mice per group pooled from two independent experiments using T cells from unique donors. P values by ordinary one-way ANOVA with Tukey’s post test. (G) Graph shows mean ± SEM radiance (photons/second/cm2/sr) of Nalm-6/ffluc tumor burden in mice treated with CD19-specific CD8+ CAR T cells. N = 6 or 8 mice per group pooled from two independent experiments using T cells from unique donors. Significance (* P < 0.05) was assessed by one-way ANOVA with Tukey’s post test. (H) Survival analysis of mice as in (G). Significance (*) and P values (inset legend) assessed by log-rank test.
Separate experiments treating disseminated Nalm-6 leukemia with a sub-curative dose of purified CD8+EGFRt+ CAR T cells also showed that 28/ζ CAR T cells were less effective than BB/ζ, BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR T cells at reducing tumor burden or prolonging survival (Fig. 5G–H). In both Raji and Nalm-6 models, we found statistically significant differences in survival when comparing 28/ζ to BB/ζ, BB/εPRS_ITAM/ζ or BB/ζ/link_GRB2 CARs but statistical significance was not reached for pair-wise comparisons of the BB/ζ, BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CARs. Thus, whereas the most highly antigen-sensitive CAR possessing 28/ζ signaling domains promoted T cell dysfunction in the setting of disseminated CD19+ tumor cells, the new 4–1BB-based CARs with CD3ε and GRB2 sequences that exhibit enhanced antigen sensitivity retained antitumor activity.
BB/ζ CARs containing CD3ε or GRB2 domains enhance in vivo antitumor function in settings of low antigen expression
To evaluate whether the BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR designs improved tumor control in the context of low antigen expression, we studied the antitumor activity and phenotype of the CAR T cells in additional xenograft mouse models. We first utilized a disseminated model of Jeko-1 mantle cell lymphoma with low levels of ROR1 expression (5,204 molecules per cell) (fig. S7A). Mice were intravenously engrafted with Jeko-1 cells and treated one week later with CD8+EGFRt+ ROR1-specific CAR T cells. All CAR formats yielded transient reductions in tumor burden (Fig. 6A–B), however mice treated with conventional 28/ζ CAR T cells showed more rapid tumor outgrowth and diminished survival (Fig. 6B–C). BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CAR T cells provided the most effective anti-tumor responses and survival in this model, although the differences in survival between the new CARs and the BB/ζ CAR group did not reach statistical significance. There were also no substantial differences in peak T cell frequency in the peripheral blood at day 13 or in the bone marrow at day 20, indicating that the various CAR T cells expanded equivalently at early time points after infusion (Fig. 6D). Thus, in three models with various antigen levels, BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CARs targeting CD19 or ROR1 were as effective in eliminating disseminated tumor cells as BB/ζ CAR T cells and superior to 28/ζ CAR T cells.
Figure 6. BB/ζ CARs containing CD3ζ or GRB2 domains enhance in vivo antitumor function in settings of low antigen expression.
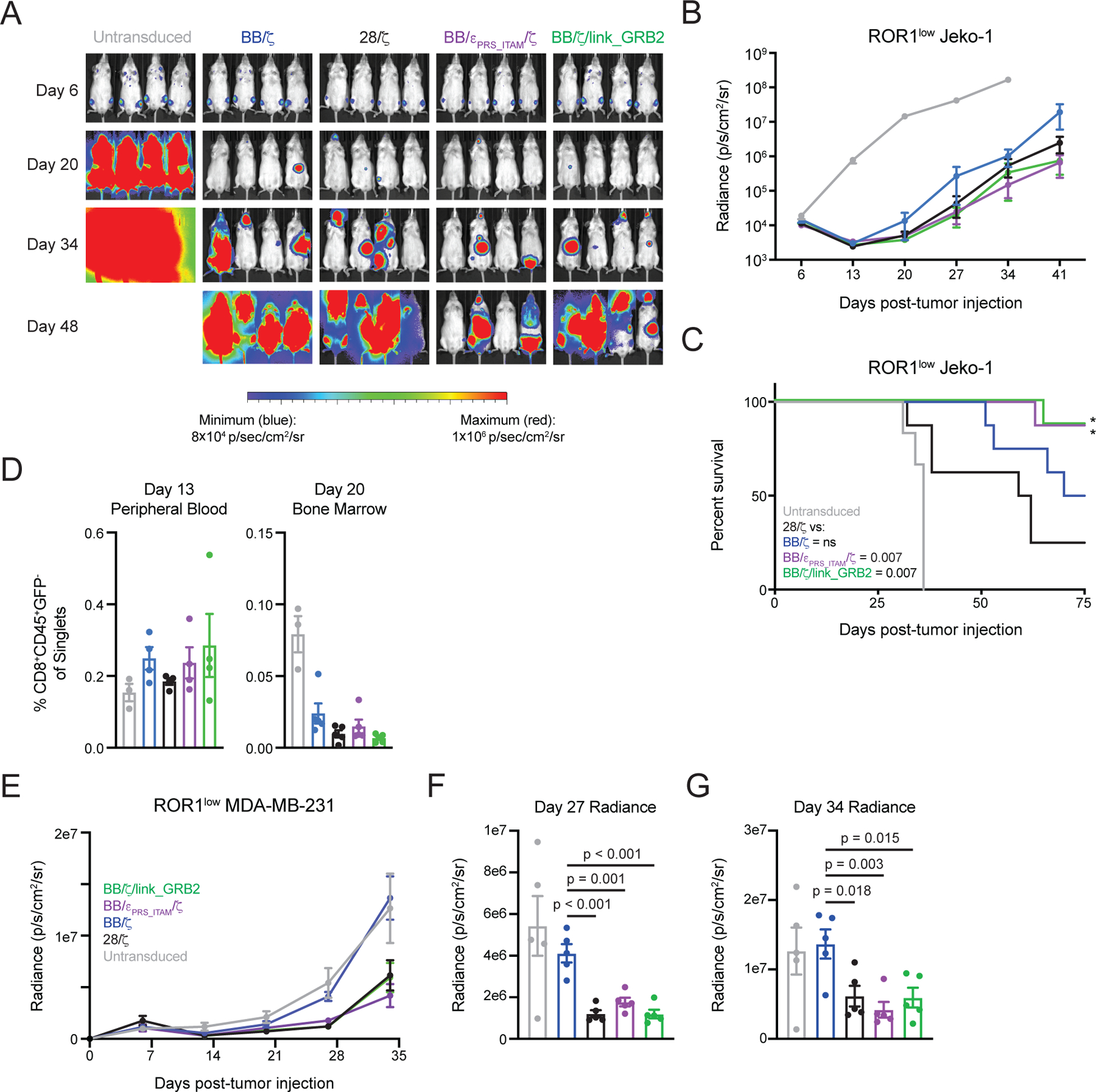
(A and B) Representative bioluminescence images (A) and mean ± SEM radiance (photons/second/cm2/sr) of (B) Jeko-1/ffluc tumor burden in mice treated with ROR1-specific CD8+ CAR T cells. N = 7 or 8 mice per group pooled from two independent experiments using T cells from unique donors. (C) Survival analysis of mice as in (B). Significance (*) and P values (inset legend) assessed by log-rank test. (D) Graphs show mean ± SEM of CAR T cell frequency within the blood (left) or bone marrow (right) at the indicated time points after Jeko-1 tumor cell injection. N = three or four mice per group. (E to G) Mean ± SEM of radiance of luciferase-expressing MDA-MB-231 tumors over time (E), or at days 27 (F) and 34 (G) after MDA-MB-231 tumor cell injection and treatment with ROR1-specific CD8+ CAR T cells on day 7. N = 5 mice per group. P values determined by an ordinary one-way ANOVA with Tukey’s post test.
Finally, we treated a solid tumor xenograft model of MDA-MB-231 breast adenocarcinoma that also has low ROR1 expression (~5,978 molecules per cell) with purified CD8+EGFRt+ T cells expressing the individual CARs (fig. S7A). In this model, MDA-MB-231 tumors grow slowly and are not rapidly fatal, making early tumor control the most appropriate measurement of T cell potency. Here, BB/ζ CAR T cells did not mediate an antitumor effect, despite recognizing MDA-MB-231 cells in vitro (Fig. 6E and fig. S7B). In contrast, BB/εPRS_ITAM/ζ, BB/ζ/link_GRB2, and 28/ζ CAR T cells demonstrated comparable antitumor effects. On days 27 and 34 after treatment, mice that received BB/εPRS_ITAM/ζ, BB/ζ/link_GRB2, and 28/ζ CAR T cells had significantly lower tumor burden than mice treated with BB/ζ or untransduced T cells (Fig. 6F–G). As observed in other xenograft solid tumor models, conventional 28/ζ CAR T cells displayed improved antitumor efficacy as compared to BB/ζ CAR T cells (21, 69). Nonetheless, together with the other three disseminated tumor models in this study, the new 4–1BB-based CARs with CD3ε and GRB2 sequences promoted the strongest and most durable antitumor functions.
Discussion
CARs are empirically designed synthetic receptors that require thousands of antigen molecules per cell to elicit potent antitumor functions (16, 17). This lack of sensitivity can result in ineffective elimination of tumors that express the target antigen at low levels (8, 10, 70). Thus, there is a need for new CAR designs with improved antigen sensitivity that simultaneously promote in vivo persistence and function, which are likely to be critical for achieving durable remissions in patients.
Prior studies tried to improve CAR function by modifying the receptor backbone to include cytokine signaling domains (36), a second CD3ζ endodomain or altered hinge and transmembrane domains (19), and, most recently, a CD3ε signaling domain (71). Entirely novel structures, like a TRUC or T cell antigen coupler, are also being evaluated (37, 72). Although some of these receptors improve antigen sensitivity, they were all designed empirically. Our approach was to study the TCR, which possesses exquisite antigen sensitivity (13, 14), and ask whether we could derive insights into the mechanisms for differential antigen sensitivity that might further guide CAR engineering. Global phosphoproteomic analyses of TCR and CAR signaling in identical T cells revealed differences in the phosphorylation of canonical T cell signaling molecules – CD3δ, ε, and γ chains, as well as LAT. These proteins were either not phosphorylated, or more weakly phosphorylated by CAR stimulation than by TCR stimulation. It is surprising that CARs are capable of initiating robust antitumor functions without fully engaging the LAT signaling hub that is believed to be critical for assembling T cell receptor signaling complexes and initiating effector functions (51, 73). Our signaling data suggest that CARs might compensate for weak LAT P phosphorylation O4 through supraphysiological phosphorylation of CD3ζ, CD28 or other proteins, and indicate that further research is necessary to elucidate the precise mechanisms of CAR T cell activation.
We utilized the phosphoproteomic data to design BB/εPRS_ITAM/ζ and BB/ζ/link_GRB2 CARs. When compared to BB/ζ CARs, these new CAR designs had superior antigen reactivity in vitro, but were not as sensitive as a 28/ζ CAR. The improved sensitivity of the BB/εPRS_ITAM/ζ CAR was abrogated by mutation of the CD3ε PRS and ITAM domains, implicating signaling as the mechanism for improved function. None of the CARs could approximate the sensitivity of a natural TCR illustrating the importance of attributes of the TCR like reduced affinity, serial triggering, mechanoreceptor function, and co-receptor binding that are difficult to engineer into CAR designs. Nevertheless, in four xenograft mouse models of hematologic malignancy and breast cancer, the new CARs possessed equivalent or improved antitumor activity compared to BB/ζ CARs. Furthermore, the new CARs were superior to 28/ζ CARs in three hematologic malignancy models, which coincided with less upregulation of inhibitory receptors and superior persistence. Therefore, our phosphoproteomics data enabled the design of new receptors with both improved in vitro antigen sensitivity and in vivo functionality.
A potential limitation of our approach using bi-specific T cells is that our work only examined CD8+ T cells since the comparison between CAR and TCR utilized a class I MHC restricted response to EBV. Additional studies are required to determine if our results can be extended to clinical CAR T cell products that are often derived from a bulk T cell population containing both CD8+ and CD4+ cells, as well as naïve and memory T cells. Basal CAR phosphorylation and tonic signaling appear to be common properties of CARs possessing a CD28 costimulatory domain (20, 22), however it is important to note that the 28/ζ CAR construct utilized in this study did not cause sufficient tonic signaling in the absence of antigen to upregulate PD-1 or induce constitutive growth.
In summary, our findings indicate that the functionality of “second-generation” BB/ζ CARs can be augmented through rational insertion of new signaling and protein-binding domains. It is important to note that insertion of additional domains may improve receptor signaling in a manner distinct from the addition or selection of alternative costimulatory domains, which have to this point been a major focus of optimization in the CAR field (12). Thus, new ways to modulate CAR structure remains an important area of investigation. Whereas CD3ε and GRB2 are two representative signaling domains derived from our phosphoproteomic dataset, the dataset provides a wealth of additional information to draw inferences about metabolic fitness and other aspects of T cell function. Another implication of our work is that future CARs may benefit from incorporation of “stripped down” functional domains that contain only those peptide sequences that are essential for driving signaling and/or protein recruitment. For CD3ε, partial removal the endodomain yielded a CAR that was expressed at higher levels and was more functional than a CAR containing the entire CD3ε endodomain. Before that possibility becomes a reality, we must gain a better understanding of how individual peptide sequences within T cell signaling endodomains promote T cell activation and effector responses. High-throughput screening strategies harnessing advances in synthetic biology and sequencing technology should improve the feasibility of these studies in future work.
Materials and Methods
Acquisition of peripheral blood T cells from healthy donors
Healthy adults (>18 years-old) were enrolled in Institutional Review Board-approved studies for peripheral blood collection. Informed consent was obtained from all donors. Researchers were provided donor age, nondescript donor ID number, human leukocyte antigen haplotype, Epstein-Barr virus serology results, and were blinded to all other personally identifiable information about study participants. PBMC were isolated by density gradient using Lymphocyte Separation Media (Corning). CD8+, naïve CD8+ and memory CD8+ T cells were further isolated using EasySep Human CD8+, Human Naïve CD8+, or Human Memory CD8+ T Cell Isolation/Enrichment Kits (StemCell Technologies) in accordance with manufacturer’s instructions. To obtain central memory (CD8+CD45RO+CD62L+) T cells, the resulting cell fraction after a Human Memory CD8+ T cell Enrichment Kit was stained with CD62L fluorochrome-conjugated monoclonal antibody and CD62L+ cells were enriched on a FACS Aria II (BD Biosciences).
Cell Culture
LentiX cells (Clontech) were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum, 1mM L-glutamine (Gibco), 25mM HEPES (Gibco), and 100U/mL penicillin/streptomycin (Gibco). Jeko-1 (CRL-3006), K562 (CCL-243), MDA-MB-231 (HTB-26), Nalm-6 (CRL-3273) and Raji (CCL-86) cells were obtained from American Type Culture Collection and cultured in RPMI-1640 (Gibco) supplemented with 5% fetal bovine serum, 1mM L-glutamine, 25mM HEPES, and 100U/mL penicillin/streptomycin. Primary human T cells were cultured in RPMI-1640 supplemented with 10% human serum, 2mM L-glutamine, 25mM HEPES, 100U/mL penicillin/streptomycin and 50μM β-mercaptoethanol (Sigma). All cells were cultured at 37°C and 5% CO2, and tested bi-monthly for the absence of mycoplasma using MycoAlert Mycoplasma Detection Kit (Lonza).
Generation of transduced tumor cell lines
K562 cells expressing HLA-B8 were derived by transduction with lentivirus supernatant prepared from LentiX cells that were transiently transfected with psPAX2, pMD2.G, and a lentiviral vector encoding HLA-B8. psPAX2 and pMD2.G were gifts from Didier Trono (Addgene plasmid #’s 12259 and 12260). K562/B8 cells expressing EBV antigen were derived by transduction with retrovirus supernatant prepared from LentiX cells that were transiently transfected with MLV g/p, 10A1, and a retroviral vector encoding GFP and a minigene for the EBV peptide sequence RAKFKQLL. K562 and K562/B8 cells expressing human ROR1 were derived by transduction with lentivirus supernatant prepared from LentiX cells that were transiently transfected with MLV g/p, 10A1, and a retroviral vector encoding human ROR1 (UniProt Q01973, aa1–973). K562 cells expressing human CD19 were derived by transduction with lentivirus supernatant prepared from LentiX cells that were transiently transfected with psPAX2, pMD2.G, and a lentiviral vector encoding human CD19 (UniProt P15391, aa1–326). Jeko-1/ffluc, MDA-MB-231/ffluc, Nalm-6/ffluc and Raji/ffluc cells were derived by transduction with lentivirus supernatant prepared from LentiX cells that were transiently transfected with psPAX2, pMD2.G, and a lentiviral vector encoding GFP and firefly luciferase. For all transductions, viral supernatant was harvested 48 hours after transfection of LentiX cells, filtered using a 0.45μm PES syringe filter (Millipore), and added to tumor cells in the presence of Polybrene (Millipore) at a final concentration of 4.4μg/mL. Five to seven days later, transduced tumor cells were enriched on a FACSAria II to greater than 97% purity.
Generation of CAR constructs and CAR-encoding lentivirus
ROR1-specific 4–1BB/CD3ζ and CD28/CD3ζ CARs with a R12 scFv, modified IgG4 hinge and CD28 transmembrane domain were previously described (27). Optimizations to CAR design were performed using CD19- and ROR1-specific CD28/CD3ζ and 4–1BB/CD3ζ CARs containing a StrepTag II (STII) sequence and two G4S linkers inserted between the scFv and immunoglobulin G4 (IgG4) hinge (22). The entire CD3ε endodomain (UniProt P07766, aa153–207) or a truncated endodomain (aa179–207) containing PRS and ITAM components was inserted either immediately before or after the CD3ζ endodomain. A CAR with mutated CD3ε PRS and ITAM components was constructed by mutating each proline residue in the PRS to an alanine residue, as well as both tyrosine residues in the ITAM to phenylalanine residues. LATTMD CARs were developed by swapping in two versions of the LAT transmembrane domain in place of the CD28 transmembrane domain. The HCH transmembrane domain utilized amino acids 5–30 of human LAT (UniProt O43561); the EXT transmembrane domain utilized amino acids 1–35 of human LAT. CARs containing GRAP2 and GRB2 sequences were constructed by adding an 18-amino acid Whitlow linker (linker 218) followed by the GRAP2 SH2 domain (UniProt O75791, aa58–149) or GRB2 SH2 domain (UniProt P62993, aa60–152) to the C-terminus of CD3ζ (65). A CAR with an engineered GRB2 SH2 domain was also constructed with the following mutations in the SH2 domain: A91V, S96A and K109L (66). CAR constructs were codon-optimized, linked by T2A sequence to truncated forms of human CD19 (CD19t) or epidermal growth factor receptor (EGFRt) and cloned into a HIV7 lentiviral vector. All cloning was performed by PCR, enzyme digest and/or Gibson assembly. Plasmids were verified by capillary sequencing prior to use. Replication-deficient lentivirus was generated by transient transfection of LentiX cells using psPAX2, pMD2.G, and the CAR-encoding lentiviral vector. After 48 hours, lentiviral supernatant was harvested and filtered using a 0.45mm PES syringe filter.
T cell transduction and culture
To prepare bi-specific T cells, CD45RO+CD8+ T memory cells were isolated and stimulated using irradiated autologous PBMC that had been pulsed with 5μg/mL EBV peptide (RAKFKQLL, Elim Biopharmaceuticals) in AIM V media (Gibco). T cells and PBMC were cultured at a 1:1 ratio in CTL medium supplemented with 50 IU/mL human IL-2 (Prometheus). Two days later, CAR-encoding lentiviral supernatant was added to the T cell and PBMC co-culture. Polybrene (Millipore) was added at a final concentration of 4.4μg/mL and T cells were spinoculated at 800g and 32°C for 90 minutes. Viral supernatant was replaced 8 hours later with fresh CTL supplemented with 50 IU/mL IL-2. Half-volume media changes were performed every 48 hours using CTL supplemented with 50 IU/mL IL-2. Transduced CD8+ tetramer+ CD19t+ T cells were sorted on a FACSAria II on day 11.
To prepare conventional CAR T cells, polyclonal CD8+ T cells were activated using Dynabeads Human T-Activator CD3/CD28 (ThermoFisher) at a 3:1 bead to T cell ratio and cultured in CTL medium supplemented with 50 U/mL IL-2. The next day, CAR-encoding lentiviral supernatant was added to the activated T cells. Polybrene was added at a final concentration of 4.4μg/mL and the T cells were spinoculated at 800g and 32°C for 90 minutes. Viral supernatant was replaced 8 hours later with fresh CTL medium supplemented with 50 IU/mL IL-2. Half-volume media changes were performed every 48 hours using CTL supplemented with 50 IU/mL IL-2. Dynabeads were removed on day 5 and CD8+EGFRt+ transduced T cells were FACS-sorted on day 8. FACS-purified CD8+ tetramer+ CD19t+ or CD8+ EGFRt+ cells were either cultured in CTL supplemented with 50 IU/mL IL-2 until days 12–14 or immediately expanded if large numbers were necessary for experimentation.
T cell expansion for signaling, CAR expression, and Ca2+ flux analyses
If necessary, T cells were expanded using 30ng/mL purified OKT3, γ-irradiated LCL (8,000 rad), and γ-irradiated (3,500 rad) allogeneic PBMC at a LCL to T cell ratio of 100:1 and a PBMC to T cell ratio of 600:1. 50 IU/mL IL-2 was added on day 1, OKT3 was washed out on day 4, cultures were fed with fresh CTL medium supplemented with 50 IU/mL IL-2 every 2–3 days and resting T cells were used for assays 11–12 days after stimulation.
Flow cytometry and cell phenotyping
T cells and tumor cells were stained with a 1:100 dilution of fluorophore-conjugated monoclonal antibodies specific for human CD8 (SK1), CD19 (HIB19), CD28 (CD28.2), CD45 (HI30), CD45RO (UCHL1), CD62L (DREG56), CD223 (3DS223H), CD279 (eBioJ105), EGFR (AY13), HLA-B8 (REA145), human IgG Fc (HP6017), or ROR1 (2A2) purchased from Biolegend, BD Biosciences, Miltenyi Biotec or ThermoFisher. T cells were also stained with isotype control fluorophore-conjugated antibodies when appropriate. Fluorescein isothiocyanate (FITC)-conjugated Strep-tag II monoclonal antibody was purchased from GenScript. Phycoerythrin (PE)-conjugated HLA-B8/EBV tetramer was generated by the Immune Monitoring Core Facility at the Fred Hutchinson Cancer Research Center (FHCRC). ROR1-Fc recombinant protein was generated by the FHCRC Molecular Design and Therapeutics Program. Biotinylated Cetuximab (anti-EGFR, Bristol Myers Squibb) was prepared and used in conjunction with Streptavidin-APC (ThermoFisher) as previously described (22). DNA content staining was performed by fixing T cells with 70% ice-cold ethanol, permeabilizing cells with 1% Triton-X (Sigma), degrading RNA with 100μg/mL RNAse A (ThermoFisher), and staining DNA with 20μg/mL Propidium Iodide (ThermoFisher). Antigen density was calculated using Quantibrite beads (BD Biosciences). All data was collected on a FACSCanto II, FACSCelesta, or FACSAria II (BD Biosciences). FlowJo version 9 (Treestar) was used to analyze flow cytometry files.
SCT, SCT/CD28, ROR1 and control bead preparation
Recombinant single chain trimer (SCT) and human ROR1 proteins containing a C-terminal Avi tag were produced by the FHCRC Molecular Design and Therapeutics Program. SCT and ROR1 were biotinylated using BirA Ligase (Avidity) and desalted using PD-10 columns (GE Healthcare). 1mL Streptavidin Coated Magnetic Particles (Spherotech) was washed once in excess 1× PBS supplemented with 100 U/mL penicillin/streptomycin (PBS+P/S) using a benchtop magnet. SCT and ROR1 beads were prepared by resuspending beads in 1mL PBS+P/S and then slowly adding biotinylated recombinant proteins while vortexing the solution. Protein was added to fill half of the particles’ predetermined molar binding capacity. SCT/CD28 beads were prepared by resuspending beads in 1mL PBS+P/S and then slowly adding recombinant SCT protein and biotinylated CD28 mAb (CD28.2, ThermoFisher) at a 3:1 molar ratio. Beads were incubated overnight at 4°C on a 3D orbital shaker, washed three times with excess PBS+P/S using a benchtop magnet, and resuspended in 1mL PBS+P/S. To generate control beads, 1mL Streptavidin Coated Magnetic Particles was washed once using a benchtop magnet and the bead pellet was resuspended in 1mL PBS+P/S. All beads were stored at 4°C.
In vitro functional assays
CAR T cells were co-cultured with γ-irradiated (10,000 rad) K562, K562/B8, K562/CD19, K562/ROR1, or MDA-MB-231 cells at a T cell to tumor cell ratio of 2:1. In some experiments, CAR T cells were also incubated with control, SCT or SCT/CD28 beads at a ratio of 7.5μL beads per million cells. For antigen titration experiments, CAR T cells were incubated in 96-well plates that had previously been coated with varying quantities of ROR1 recombinant protein. Human IFN-γ, IL-2, and TNF-α concentrations in cellular supernatant were quantified by ELISA (ThermoFisher) 24 hours after stimulation. T cell proliferation was quantified by staining T cells with a 0.2μM solution of carboxyfluorescein succinimidyl ester (CFSE) dye (ThermoFisher) prior to incubation with beads for 72 hours.
Fluorescence microscopy
CD8+ T cells were transduced as previously described. Instead of FACS purification on day 9, cells were imaged on a DeltaVision Elite microscope (GE Healthcare). At least ten cells were visualized per condition. Raw images were subjected to a linear adjustment of brightness and contrast using ImageJ (NIH) and these modifications were uniformly applied to the entire image.
TCR or CAR stimulation and protein lysate generation
T cells were washed and resuspended in warm CTL medium. T cells were incubated with control, SCT, SCT/CD28 or ROR1 beads in a 37°C water bath at a final cell concentration of 2×107 cells per mL and a bead to cell ratio of 7.5μL per million cells. In some experiments, bi-specific T cells were incubated with K562/B8, K562/B8/EBV, and K562/B8/ROR1 tumor cells at a T cell to tumor cell ratio of 4:1. After the allotted time, cells were quickly washed twice using ice-cold PBS, then lysed in either a 6M Urea, 25mM Tris (pH 8.0), 1mM EDTA, 1mM EGTA solution for mass spectrometry assays or NP40 Cell Lysis Buffer (Thermo Fisher) for immunoprecipitations. Both lysis buffer solutions were supplemented with protease (Sigma) and phosphatase inhibitors (Sigma) at a 1:100 dilution prior to use. When using the urea-based lysis buffer, lysates were sonicated for 15 seconds immediately. When using the NP40 Cell Lysis Buffer, lysates were incubated on ice for 15 minutes with gently mixing every 5 minutes. After sonication or incubation, lysates were cleared by centrifugation at 10,000g and 4°C for 10 minutes. Beads were removed during lysate clearing and protein concentration was quantified by BCA Assay or Micro BCA Assay (ThermoFisher).
Western blotting
Equal masses of protein lysate were loaded into 4–12% Bis-Tris NuPAGE Gels (ThermoFisher). After protein transfer onto nitrocellulose membranes (ThermoFisher), membranes were blocked with Western Blocking Reagent (Sigma) diluted 1:10 in 1× Tris-buffered saline (TBS). Membranes were stained with primary and secondary antibodies diluted 1:5,000–1:10,000 in SuperBlock TBS (ThermoFisher) supplemented with 0.1% Tween. The following antibodies were used: anti-CD247 (8D3, BD Biosciences), anti-CD247 pTyr142 (K25–407.69, BD Biosciences), anti-LAT (polyclonal, Cell Signaling), anti-LAT pTyr220 (polyclonal, Cell Signaling), anti-LCK (D88, Cell Signaling), anti-NCK1 (15B9, Cell Signaling), anti-PLC-γ1 (D9H10, Cell Signaling), anti-PLC-γ1 pTyr783 (D6M9S, Cell Signaling), anti-PLC-γ1 pSer1248 (D25A9, Cell Signaling), anti-SLP-76 (polyclonal, Cell Signaling), anti-SLP-76 pSer376 (D9D6E, Cell Signaling), anti-ZAP-70 (D1C10E, Cell Signaling), anti-ZAP-70 pTyr319 (65E4, Cell Signaling), anti-ZAP-70 pTyr493 (polyclonal, Cell Signaling), anti-mouse horseradish peroxidase (HRP) (polyclonal, Cell Signaling), and anti-rabbit HRP (polyclonal, Cell Signaling). Band intensities were quantified using ImageJ [National Institutes of Health (NIH)]; normalized to total protein or loading control, and then renormalized to a control sample.
Immunoprecipitation
Protein G Dynabeads (ThermoFisher) were incubated with anti-STII antibody (GenScript) for 60 minutes, cross-linked for 30 minutes using 20mM dimethyl pimelimidate (ThermoFisher) diluted in 200 mM triethanolamine (Fisher Scientific), quenched with 150mM monoethanolamine (Fisher Scientific), and washed three times with 1× PBS. CAR T cells that had previously been stimulated with ROR1 beads for 45 minutes were lysed as previously described. Immunoprecipitations were performed according to manufacturer’s instructions where Dynabeads were incubated with equal masses of cleared lysates for 90 minutes at room temperature.
Ca2+ mobilization measurements
Ca2+ mobilization was measured using fluorecence microscopy of T cells loaded with the Ca2+-sensitive dye Fluo-4 AM. Ca2+ flux was monitored in hundreds of individual cells simultaneously as they came into contact with supported lipid bilayers functionalized with ROR1 via biotin/streptavidin linkage. Ligand-functionalized bilayers were prepared within imaging flow chambers as described (74). Briefly, bilayers were formed via deposition of large unilamellar vesicles (LUVs) onto NoChromix (Godax laboratories)-cleaned glass within imaging flow cells. Lipid mixtures contained mole fractions ranging from .005% to .5% of 16:0 biotinyl cap PE in Egg PC (Avanti). Bilayers were fluorescently labeled and functionalized with ligand through successive incubation with Atto655-streptavidin (Sigma Aldrich) followed by biotinylated SCT or ROR1.
For each imaging experiment, 2–5×105 CAR T cells were loaded with the calcium-sensitive dye Fluo-4 AM (Thermo Fisher). Cells were loaded for 2 min at 37°C with 2.5μg/mL Fluo-4 AM in HEPES-buffered saline (HBS) containing 1 mg/mL BSA and 0.25 mM sulfinpyrazone. The cell suspension was then diluted to a final volume of 8 mL with HBS/BSA/sulfinpyrazone and incubated at 37°C for 30 min. Cells were pelleted and resuspended twice to remove excess dye and resuspended a final time in 100μL HBS/BSA/sulfinpyrazone. Imaging experiments were conducted on an Olympus IX81-XDC inverted microscope using an Andor iXon-987 EMCCD and epi-fluorescence illumination at 488nm with a CoolLED pE light source. Cells were imaged at room temperature and 10× magnification at 2 frames per second for 20 min, beginning immediately after dye-loaded cells were added to the flow chamber. Imaging data was processed in MATLAB (Mathworks) using custom data analysis routines (74). Cells were localized using a watershed segmentation algorithm and pixel intensities within cell boundaries were quantified. Cell positions were tracked to monitor intensity signatures of individual cells over the course of the experiment. Cellular activation times were defined as the first time point where Fluo-4 intensity surpasses three times the baseline fluorescence level of each cell before Ca2+ mobilization. Cumulative plots of single cell activation times were fit to exponential functions to extract time constants and total fraction of activated cells for the population response.
Protein digestion, TMT labeling, and phosphotyrosine (pTyr) peptide immunoprecipitation
Lysates were diluted to 2mg/mL using lysis buffer. Lysates were reduced in 24mM TCEP (ThermoFisher) for 30 minutes at 37°C with shaking, followed by alkylation with 48mM iodoacetamide (Sigma) in the dark at room temperature for 30 minutes. Lysates were then diluted with 200mM Tris (pH 8.0), to a urea concentration of 2M. Lys-C (Wako) was dissolved in 25mM Tris (pH 8.0) at 200ug/mL and added to lysates at 1:100 (enzyme:protein) ratio by mass and incubated for 2 hours at 37°C with shaking. Samples were further diluted with 200mM Tris (pH 8.0) to a urea concentration of 1M before adding trypsin at a 1:50 trypsin:protein ratio. After 2 hours, a second trypsin aliquot was added at a 1:100 trypsin:protein ratio. Digestion was carried out overnight at 37°C with shaking. After 16 hours, the reaction was quenched with formic acid (FA) to a final concentration 1% by volume. Samples were desalted using Oasis HLB 96-well plates (Waters) and a positive pressure manifold (Waters). The plate wells were washed with 3 x 400μL of 50% MeCN/0.1% FA, and then equilibrated with 4 x 400μL of 0.1% FA. The digests were applied to the wells, then washed with 4 x 400μL 0.1% FA before being eluted drop by drop with 3 x 400μL of 50% acetonitrile (MeCN)/0.1% FA. The eluates were lyophilized, followed by storage at −80°C until use. For TMT labeling (ThermoFisher), desalted peptides were resuspended in 50 mM HEPES at 1mg/mL based on starting protein mass. TMT reagents were resuspended in 257μL MeCN and transferred to the peptide sample. Samples were incubated at room temperature for 1 hour with mixing. Labeling reactions were quenched by the addition of 50μL of 5% hydroxyl amine (Sigma) and incubated for 15 minutes at room temperature with mixing. The independent labeling reactions were then pooled together and lyophilized. The labeled peptides were desalted as above and then lyophilized and stored at −80°C. Immunoprecipitation of pTyr peptides was performed using the PTMScan P-Tyr-1000 Kit (Cell Signaling). The enriched pTyr peptide fraction was purified using a C18 Spin Tip (ThermoFisher), lyophilized, and stored at −80°C until analysis. The flow-through fraction was desalted, lyophilized, and stored at −80°C.
Basic (high pH) reverse phase liquid chromatography
The desalted and pTyr peptide-depleted flow-through was fractionated by high-pH reverse phase (RP) liquid chromatography. 4mg of the protein digest was loaded onto a LC system consisting of an Agilent 1200 HPLC with mobile phases of 5mM NH4HCO3 (pH 10) (A) and 5mM NH4HCO3 in 90% MeCN (pH 10) (B). The peptides were separated by a 4.6mm x 250mm Zorbax Extend-C18, 3.5μm, column (Agilent) over 96 minutes at a flow rate of 1.0mL/min by the following timetable: hold 0% B for 9 minutes, gradient from 0 to 10% B for 4 minutes, 10 to 28.5% B for 50 minutes, 28.5 to 34% B for 5.5 minutes, 34 to 60% B for 13 minutes, hold at 60% B for 8.5 minutes, 60 to 0% B for 1 minute, re-equilibrate at 0% B for 5 minutes. 1 minute fractions were collected from 0–96 minutes by the shortest path by row in a 1 mL deep well plate (ThermoFisher). The high pH RP fractions were concatenated into 24 samples by every other plate column starting at minute 15 (e.g. sample 1 contained fractions from wells B10, D10, F10, etc.). The remaining fractions were combined such that fractions from 12 to 14 minutes were added to sample 1, all fractions after 86 minutes were added to sample 24, and all fractions from 0 to 11 minutes were combined into sample ‘A’. 95% of every 12th fraction of the 24 samples was combined (1,13; 2,14; …) to generate 12 more samples, which were dried down and stored at −80°C prior to phosphopeptide enrichment by immobilized metal affinity chromatography.
Immobilized metal affinity chromatography (IMAC)
IMAC enrichment was performed using Ni-NTA-agarose beads (Qiagen) stripped with EDTA and incubated in a 10mM FeCl3 solution to prepare Fe3+-NTA-agarose beads. Fractionated lysate was reconstituted in 200μL of 0.1% TFA in 80% MeCN and incubated for 30 minutes with 100μL of the 5% bead suspension while mixing at room temperature. After incubation, beads were washed 3 times with 300μL of 0.1% TFA in 80% MeCN. Phosphorylated peptides were eluted from the beads using 200μL of 70% ACN, 1% ammonium hydroxide for 1 minute with agitation at room temperature. Samples were transferred into a fresh tube containing 60μL of 10% FA, dried down and re-suspended in 0.1% FA, 3% MeCN. Samples were frozen at −80°C until analysis.
Nano-liquid chromatography-tandem mass spectrometry
Phosphopeptide-enriched samples were analyzed by LC-MS/MS on an Easy-nLC 1000 (ThermoFisher) coupled to an LTQ-Orbitrap Fusion mass spectrometer (ThermoFisher) operated in positive ion mode. The LC system, configured in a vented format consisted of a fused-silica nanospray needle (PicoTip emitter, 50μm ID x 20cm, New Objective) packed in-house with ReproSil-Pur C18-AQ, 3μm and a trap (IntegraFrit Capillary, 100μm ID x 2 cm, New Objective) containing the same resin as in the analytical column with mobile phases of 0.1% FA in water (A) and 0.1% FA in MeCN (B). The peptide sample was diluted in 20 µL of 0.1% FA, 3% MeCN, and 8.5μL was loaded onto the column and separated over 210 minutes at a flow rate of 300 nL/min with a gradient from 5 to 7% B for 2 minutes, 7 to 35% B for 150 minutes, 35 to 50% B for 1 minute, hold 50% B for 9 minutes, 50 to 95% B for 2 minutes, hold 95% B for 7 minutes, 95 to 5% B for 1 minute, re-equilibrate at 5% B for 38 minutes. A spray voltage of 2000 V was applied to the nanospray tip. MS/MS analysis occurred over a 3 second cycle time consisting of 1 full scan MS from 350–1500 m/z at resolution 120,000 followed by data dependent MS/MS scans using HCD activation with 27% normalized collision energy of the most abundant ions. Selected ions were dynamically excluded for 45 seconds after a repeat count of 1.
Shotgun mass spectrometry data analysis
Raw MS/MS spectra from each experiment (n = 3) were searched together against the reviewed Human Universal Protein Resource (UniProt) sequence database (release 2016_01) with common laboratory contaminants using the MaxQuant/Andromeda search engine version 1.6.0.1 (75). The search was performed with a tryptic enzyme constraint for up to two missed cleavages. Variable modifications were oxidized methionine, phosphorylated serine, phosphorylated threonine, and phosphorylated tyrosine. Carbamidomethylated cysteine was set as a static modification. Peptide MH+ mass tolerances were set at 20 ppm. The overall FDR was set at ≤ 1% using a reverse database target decoy approach.
Phosphopeptide site localization was determined by MaxQuant and converted to phosphorylation sites using Perseus version 1.6.0.7 (76). The site table was expanded, reverse hits and potential contaminants were excluded, and any phosphorylation site with fewer than 6 values across the nine TMT channels was excluded. Data normalization was performed by scaling each TMT channel to the channel median. Data was then log2 transformed (table S1, sheet A). Stimulation vs. control ratios were calculated by subtracting the control channel from the TCR or CAR stimulated channels at each time point. Due to incomplete MS sampling, some phosphorylation sites were only found in one or two replicate experiments. We chose to further analyze only those phosphorylation sites that had values in at least two experiments and all TMT channels and a phosphorylation localization score > 0.75, leaving us with 18,148 phosphorylation sites (table S1, sheet B). Select sites are shown in Figure 2. If two phosphopeptides with different multiplicities were identified for a given site in the expanded Perseus table, both values were graphed in Figure 2.
Transfer of T cells in NOD/SCID/γc-/- (NSG) mice
The Fred Hutchinson Cancer Research Center Institutional Animal Care and Use Committee approved all experimental procedures. Six- to eight-week-old male or female NSG mice were obtained from the Jackson Laboratory or bred in-house. Mice were engrafted with 5×105 MDA-MB-231/ffluc subcutaneously in the right flank, or intravenously by tail vein injection with Raji/ffluc, Nalm-6/ffluc or Jeko-1/ffluc cells. Seven days later, mice were injected intravenously with purified CD8+EGFRt+ CAR T cells, mock transduced CD8+ T cells, or saline. Mice were followed for survival or sacrificed at indicated time points for collection of bone marrow. Single cell suspensions from peripheral blood were prepared lysing red blood cells using ACK Lysing Buffer (Gibco) and filtering the resulting cell fraction with a 0.7μm filter. Single cell suspensions from bone marrow were prepared by crushing hindlimbs with mortar and pestle, filtering using a 0.7μm filter, and lysing red blood cells using ACK Lysing Buffer (Gibco). Single cell suspensions from blood or bone marrow were stained with Live-Dead stain (ThermoFisher) and fluorochrome-conjugated monoclonal antibodies for flow cytometric analysis. Mice handlers were blinded to group allocation.
Bioluminescence imaging of tumor growth
Mice received intraperitoneal injections of luciferin substrate (Caliper Life Sciences) resuspended in 1× PBS (15mg per gram body weight). Mice were anesthetized with isoflurane and imaged using an Xenogen IVIS Imaging System (Caliper Life Sciences) 10, 12 and 13 min after luciferin injection in small binning mode at an acquisition time of 15 sec to 1 min to obtain unsaturated images. Luciferase activity was analyzed using Living Image Software 4.7.2 (Caliper Life Sciences) and the photon flux analyzed within regions of interest that encompassed the entire body of each individual mouse.
Analysis of T cell phenotype, function, and in vivo experiments
Prism version 8 (GraphPad Software) was used to plot data and calculate statistics. Only those P values meeting an α = 0.05 level of significance are indicated in the figures. The precise statistical tests used are indicated in the figure legends.
Supplementary Material
Acknowledgments:
We thank the M. J. Murdock Charitable Trust and members of the FHCRC Proteomics Shared Resource for help in data acquisition. We would also like to thank Don Evan S Parrilla and LaTrice King for performing mouse husbandry and tumor xenograft experiments.
Funding:
This research was supported by NIH R01 CA136551 (S.R.R), R01 CA114536 (S.R.R), U01 CA214114 (A.G.P), R50 CA211499 (J.R.W.), R21 CA226962 (S.L.V), and FHCRC Bezos Immunotherapy Pilot Award. A.I.S. is supported by the FHCRC Interdisciplinary Training in Cancer Research Training Grant and Medical Scientist Training Program. S.A.S. is supported by an American Cancer Society Postdoctoral Fellowship Award.
Footnotes
Competing interests: A.I.S. is a shareholder of and scientific consultant for Lyell Immunopharma. S.R.R. is a founder and shareholder of Lyell Immunopharma, and was a founder and scientific advisor of Juno Therapeutics, a Celgene company. R.G. has received consulting income from Juno Therapeutics, Celgene, Illumina, Takeda, Infotech Soft, and Merck, research support from Janssen Pharmaceuticals and Juno Therapeutics, and has an ownership interest in CellSpace Biosciences, Ozette Technologies, and Modulus Therapeutics. A.I.S., A.R., and S.R.R. have filed provisional patents relating to the CAR designs described in this manuscript. The other authors declare that they have no competing interests.
Data and materials availability: All LC-MS/MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD013734. All other data needed to evaluate the conclusions in the paper are present in the main text or supplementary materials.
References and Notes
- 1.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG, CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients, J. Clin. Invest 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, Soma L, Wood B, Li D, Heimfeld S, Riddell SR, Maloney DG, Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells, Sci Transl Med 8, 355ra116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY, Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma, N Engl J Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, Hoglund V, Lindgren C, Oron AP, Li D, Riddell SR, Park JR, Jensen MC, Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults, Blood 129, 3322–3331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, Klebanoff CA, Kammula US, Sherman M, Perez A, Yuan CM, Feldman T, Friedberg JW, Roschewski MJ, Feldman SA, McIntyre L, Toomey MA, Rosenberg SA, Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels, J. Clin. Oncol, JCO2016713024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JH, Riviere I, Gonen M, Wang X, Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, Roshal M, Maslak P, Davila M, Brentjens RJ, Sadelain M, Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia, N Engl J Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA, Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia, N Engl J Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, Shalabi H, Fountaine TJ, Shern JF, Majzner RG, Stroncek DF, Sabatino M, Feng Y, Dimitrov DS, Zhang L, Nguyen S, Qin H, Dropulic B, Lee DW, Mackall CL, CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy, Nature Medicine 24, 20–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, Kanakry JA, Ali SA, Mikkilineni L, Feldman SA, Stroncek DF, Hansen BG, Lawrence J, Patel R, Hakim F, Gress RE, Kochenderfer JN, T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma, J. Clin. Oncol 36, 2267–2280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, Plesa G, Chen F, Davis MM, Hwang W-T, Young RM, Brogdon JL, Isaacs R, Pruteanu-Malinici I, Siegel DL, Levine BL, June CH, Milone MC, B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma, J. Clin. Invest 130, 25764 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majzner RG, Mackall CL, Tumor Antigen Escape from CAR T-cell Therapy, Cancer Discov 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Sadelain M, Riviere I, Riddell S, Therapeutic T cell engineering, Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN, Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response, Immunity 4, 565–571 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Huang J, Brameshuber M, Zeng X, Xie J, Li Q-J, Chien Y-H, Valitutti S, Davis MM, A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells, Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, Sakemura R, Goto T, Hanajiri R, Imahashi N, Shimada K, Tomita A, Kiyoi H, Nishida T, Naoe T, Murata M, Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells, The Journal of Immunology 194, 911–920 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, Lopomo P, Vigny M, Fry TJ, Orentas RJ, Mackall CL, Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase, Mol Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman KM, Garrett TE, Evans JW, Horton HM, Latimer HJ, Seidel SL, Horvath CJ, Morgan RA, Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells, Hum. Gene Ther 29, 585–601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, Li Z, Cunanan KM, Huse M, Hendrickson RC, Wang X, Riviere I, Sadelain M, CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape, Nature 545, 423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, Richards RM, Heitzeneder S, Nguyen SM, Wiebking V, Theruvath J, Lynn RC, Xu P, Dunn AR, Vale RD, Mackall CL, Tuning the Antigen Density Requirement for CAR T-cell Activity, Cancer Discov 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors, Nature Medicine 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, Ahn S, Xu Y, Suzuki K, Li G, Tsahouridis O, Su L, Savoldo B, Dotti G, THEMIS-SHP1 Recruitment by 4–1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells, Cancer Cell 37, 216–225.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, Whiteaker JR, Gottardo R, Paulovich AG, Riddell SR, Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function, Sci Signal 11, eaat6753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, Duan P, Lin Y, Zheng W, Wang X, Lin N, Tu M, Xie Y, Zhang C, Liu W, Deng L, Gao S, Ping L, Wang X, Zhou N, Zhang J, Wang Y, Lin S, Mamuti M, Yu X, Fang L, Wang S, Song H, Wang G, Jones L, Zhu J, Chen S-Y, A safe and potent anti-CD19 CAR T cell therapy, Nature Medicine 18, 676 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Patel PR, Guedan S, Scholler J, Keith B, Snyder N, Blair I, Milone MC, June CH, Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells, Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M, Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency, Nature Medicine 545, 423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogquist KA, Jameson SC, The self-obsession of T cells: how TCR signaling thresholds affect fate “decisions” and effector function, Nat. Immunol 15, 815–823 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR, Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells, Clin. Cancer Res 19, 3153–3164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A, Serial triggering of many T-cell receptors by a few peptide-MHC complexes, Nature 375, 148–151 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Feng Y, Brazin KN, Kobayashi E, Mallis RJ, Reinherz EL, Lang MJ, Mechanosensing drives acuity of αβ T-cell recognition, Proc. Natl. Acad. Sci. U.S.A 114, E8204–E8213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakraborty AK, Weiss A, Insights into the initiation of TCR signaling, Nat. Immunol 15, 798–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voisinne G, Kersse K, Chaoui K, Lu L, Chaix J, Zhang L, Goncalves Menoita M, Girard L, Ounoughene Y, Wang H, Burlet-Schiltz O, Luche H, Fiore F, Malissen M, Gonzalez de Peredo A, Liang Y, Roncagalli R, Malissen B, Quantitative interactomics in primary T cells unveils TCR signal diversification extent and dynamics, Nat Immunol 15, 798 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, Pabón-Saldaña M, Darville L, Fang B, Rix U, Yoder S, Berglund A, Koomen JM, Haura EB, Abate-Daga D, An immunoproteomic approach to characterize the CAR interactome and signalosome, Sci Signal 12, eaap9777 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James JR, Vale RD, Biophysical mechanism of T-cell receptor triggering in a reconstituted system, Nature 487, 64–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, Beavis PA, Trapani JA, Kershaw MH, Ritchie DS, Darcy PK, Neeson PJ, Jenkins MR, Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity, Proc. Natl. Acad. Sci. U.S.A 115, E2068–E2076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gudipati V, Rydzek J, Doel-Perez I, Gonçalves VDR, Scharf L, Königsberger S, Lobner E, Kunert R, Einsele H, Stockinger H, Hudecek M, Huppa JB, Inefficient CAR-proximal signaling blunts antigen sensitivity, Nat Immunol 21, 848–856 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang C-H, Saso K, Butler MO, Minden MD, Hirano N, A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects, Nature Medicine 5, 177ra38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, Scarfo I, Choudhary R, Kiner O, Krishnamurthy J, Le B, Morath A, Baldeviano GC, Quinn J, Tavares P, Wei Q, Weiler S, Maus MV, Getts D, Schamel WW, Hofmeister R, Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response, Nat Commun 10, 2087–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, Young RM, June CH, Posey AD, Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability, J. Clin. Invest 130, 3087–3097 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, Lever M, Dushek O, Schmitt TM, Greenberg PD, Kranz DM, Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains, The Journal of Immunology 200, 1088–1100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser ADM, Pouw N, Debets R, Kieback E, Uckert W, Song J-Y, Haanen JBAG, Schumacher TNM, Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy, Nature Medicine 16, 565–70– 1p following 570 (2010). [DOI] [PubMed] [Google Scholar]
- 41.van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JHF, Heemskerk MHM, Mixed T cell receptor dimers harbor potentially harmful neoreactivity, Proc. Natl. Acad. Sci. U.S.A 107, 10972–10977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schober K, Müller TR, Gökmen F, Grassmann S, Effenberger M, Poltorak M, Stemberger C, Schumann K, Roth TL, Marson A, Busch DH, Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function, Nat Biomed Eng 7, 280ps7 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Yu YYL, Netuschil N, Lybarger L, Connolly JM, Hansen TH, Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells, J Immunol 168, 3145–3149 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Philipson BI, O’Connor RS, May MJ, June CH, Albelda SM, Milone MC, 4–1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling, Sci Signal 13, eaay8248 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M, Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection, Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navarro MN, Goebel J, Feijoo-Carnero C, Morrice N, Cantrell DA, Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes, Nat. Immunol 12, 352–361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salek M, McGowan S, Trudgian DC, Dushek O, de Wet B, Efstathiou G, Acuto O, Quantitative phosphoproteome analysis unveils LAT as a modulator of CD3ζ and ZAP-70 tyrosine phosphorylation, PLoS ONE 8, e77423 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brownlie RJ, Zamoyska R, T cell receptor signalling networks: branched, diversified and bounded, Nature Reviews Immunology 13, 257–269 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK, CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery, Proc. Natl. Acad. Sci. U.S.A 107, 16916–16921 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohrs JA, Zheng D, Graham NA, Wang P, Finley SD, Computational Model of Chimeric Antigen Receptors Explains Site-Specific Phosphorylation Kinetics, Biophys. J 115, 1116–1129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A, LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway, Immunity 9, 617–626 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Lo W-L, Shah NH, Ahsan N, Horkova V, Stepanek O, Salomon AR, Kuriyan J, Weiss A, Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT, Nat Immunol 11, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Letourneur F, Klausner RD, Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3 epsilon, 255, 79–82 (1992). [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE, LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation, Cell 92, 83–92 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Balagopalan L, Kortum RL, Coussens NP, Barr VA, Samelson LE, The linker for activation of T cells (LAT) signaling hub: from signaling complexes to microclusters, J. Biol. Chem 290, 26422–26429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, Xie Y, Ping L, Zhang C, Liu W, Deng L, Qi F, Ding Y, Lu X-A, Song Y, Zhu J, Parallel Comparison of 4–1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma, Mol Ther Oncolytics 15, 60–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L, Guo X, Shi X, Li C, Wu W, Yan C, Wang H, Li H, Xu C, Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling, Proc. Natl. Acad. Sci. U.S.A 114, E5891–E5899 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tailor P, Tsai S, Shameli A, Serra P, Wang J, Robbins S, Nagata M, Szymczak-Workman AL, Vignali DAA, Santamaria P, The proline-rich sequence of CD3epsilon as an amplifier of low-avidity TCR signaling, J Immunol 181, 243–255 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mingueneau M, Sansoni A, Grégoire C, Roncagalli R, Aguado E, Weiss A, Malissen M, Malissen B, The proline-rich sequence of CD3epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes, Nat Immunol 9, 522–532 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Liu L, Sommermeyer D, Cabanov A, Kosasih P, Hill T, Riddell SR, Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy, Nat. Biotechnol 34, 430–434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, Westin J, Gulbis AM, Loghin ME, de Groot JF, Adkins S, Davis SE, Rezvani K, Hwu P, Shpall EJ, Chimeric antigen receptor T-cell therapy - assessment and management of toxicities, Nature Reviews Clinical Oncology 2018 15:4 15, 47–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Trible RP, Samelson LE, LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation, Immunity 9, 239–246 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Bilal MY, Houtman JCD, GRB2 Nucleates T Cell Receptor-Mediated LAT Clusters That Control PLC-γ1 Activation and Cytokine Production, Front Immunol 6, 141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD, Phase separation of signaling molecules promotes T cell receptor signal transduction, 352, 595–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, Rollence ML, Wood JF, Schott ME, Milenic DE, An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability, Protein Eng 6, 989–995 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Kaneko T, Huang H, Cao X, Li X, Li C, Voss C, Sidhu SS, Li SSC, Superbinder SH2 domains act as antagonists of cell signaling, Sci Signal 5, ra68–ra68 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS, Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition, J. Clin. Invest 126, 3130–3144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo, Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, Nagaraja S, Granja J, de Bourcy CFA, Majzner R, Satpathy AT, Quake SR, Monje M, Chang HY, Mackall CL, c-Jun overexpression in CAR T cells induces exhaustion resistance, Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot J-M, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV, A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma, Sci Transl Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hartl FA, Beck-García E, Woessner NM, Flachsmann LJ, Cárdenas RMHV, Brandl SM, Taromi S, Fiala GJ, Morath A, Mishra P, Yousefi OS, Zimmermann J, Hoefflin N, Köhn M, Wöhrl BM, Zeiser R, Schweimer K, Günther S, Schamel WW, Minguet S, Noncanonical binding of Lck to CD3ε promotes TCR signaling and CAR function, Nat Immunol 21, 902–913 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, Newhook L, Hayes DL, Aarts C, Bojovic B, Denisova GF, Kwiecien JM, Brain I, Derocher H, Milne K, Nelson BH, Bramson JL, The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity, Nat Commun 9, 3049 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE, Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line, Int. Immunol 11, 943–950 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Núñez MF, Wisser K, Veatch SL, Parton R, Ed. Synergistic factors control kinase-phosphatase organization in B-cells engaged with supported bilayers, Mol. Biol. Cell 31, 667–682 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification, Nat. Biotechnol 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data, Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.