

Abstract
Targeting G protein-coupled receptors (GPCRs) through allosteric sites offers advantages over orthosteric sites in identifying drugs with increased selectivity and potentially reduced side effects. In this study, we developed a probe confined dynamic mapping protocol that allows the prediction of allosteric sites at both the GPCR extracellular and intracellular sides, as well as at the receptor–lipid interface. The applied harmonic wall potential enhanced sampling of probe molecules in a selected area of a GPCR while preventing membrane distortion in molecular dynamics simulations. The specific probes derived from GPCR allosteric ligand structures performed better in allosteric site mapping compared to commonly used cosolvents. The M2 muscarinic, β2 adrenergic, and P2Y1 purinergic receptors were selected for the protocol’s retrospective validation. The protocol was next validated prospectively to locate the binding site of [5-fluoro-4-(hydroxymethyl)-2-methoxyphenyl]-(4-fluoro-1H-indol-1-yl)methanone at the D2 dopamine receptor, and subsequent mutagenesis confirmed the prediction. The protocol provides fast and efficient prediction of key amino acid residues surrounding allosteric sites in membrane proteins and facilitates the structure-based design of allosteric modulators.
Short abstract
Using specific probes from G protein-coupled receptor allosteric ligands, our probe confined dynamic mapping outperforms standard cosolvents and detects allosteric sites at extra-/intracellular sides and the lipid interface.
Introduction
G Protein-coupled receptors (GPCRs) are the largest membrane protein family consisting of 800 members that transduce a signal inside cells from a variety of endogenous ligands including hormones, neurotransmitters, metabolites, pheromones, odorants, and light. As a result of their broad influence on human physiology, GPCRs are drug targets in many therapeutic areas such as inflammation, metabolic and neurological disorders, pain, addiction, infertility, viral infections, and cancer. A total of 475 GPCR drugs (34% of all drugs) are currently approved by the United States Food and Drug Administration (FDA), and ≥300 GPCR agents are currently in clinical trials.1 Although GPCR drugs have shown substantial therapeutic success, developing drugs for many GPCR subfamilies has proved challenging. A key challenge is to achieve selectivity when targeting highly conserved orthosteric sites where the endogenous ligands bind.
Most GPCRs can be modulated by small-molecule ligands binding to allosteric sites that are spatially and topologically distinct from the orthosteric sites. Both positive allosteric modulators (PAM), which enhance the binding and signaling of orthosteric agonists, and negative allosteric modulators (NAM), which reduce the activity of orthosteric agonists, have been described. Allosteric drugs have a better potential for receptor subtype selectivity due to greater sequence variability in allosteric sites. Furthermore, allosteric modulators confer agonist dependence and functional selectivity, causing selective receptor activation and thus different tolerance in chronic diseases.2−4 In addition, allosteric modulators have a diverse relationship between duration and intensity of the effect, which can prolong the therapeutic effect without drug overdose.5 Two recent FDA-approved allosteric drug examples are cinacalcet, a PAM of the calcium-sensing receptor against hyperparathyroidism, and maraviroc, a NAM of the chemokine CCR5 receptor to prevent the entry of HIV-1.1 Despite the clear potential benefit of GPCR allosteric modulation, the discovery of allosteric sites and lead compounds has been mostly serendipitous, often involving random screening of compound libraries.
Recent X-ray crystallography and cryoelectron microscopy (cryo-EM) structures of several GPCRs solved with bound allosteric modulators have revealed remarkably diverse locations of the allosteric binding sites.6,7 Allosteric drugs can reside inside the helical bundle from the extracellular (EC) and intracellular (IC) sides, as well as outside the helical bundle at the lipid interface (LI). For example, in the X-ray ternary complex of the M2 muscarinic receptor LY2119620, a PAM binds at an allosteric pocket facing the EC medium, including extracellular loop 2 and 3 (ECL2 and ECL3), which lies above the orthosteric site that is occupied by the agonist Iperoxo8 (Figure 1A). In the X-ray complex of the β2 adrenergic receptor with Cmp-15, this NAM occupies an allosteric site facing the IC side, involving intracellular loop 1 (ICL1) and the tips of helices 1, 2, 7, and 8, which is distant from the orthosteric site9 (Figure 1B). Allosteric modulators sitting at an IC allosteric site have also been found in the X-ray complexes of the chemokine CCR2, CCR7, and CCR9 receptors.10−12
Figure 1.

Locations of GPCR allosteric binding sites in the X-ray structures of the M2, β2, and P2Y1 receptors in complex with allosteric modulators. (A–C) The overall view of the receptors with a bound allosteric modulator. (A) The M2 receptor bound to LY2119620 (green), a PAM binding at the extracellular side, and the orthosteric ligand, Iperoxo (blue). (B) The β2 receptor bound to Cmp-15, a NAM, at the intracellular side. (C) The P2Y1 receptor in complex with BPTU, a NAM bound at the lipid interface. The receptors are in wild type with the rebuilt short intracellular loop 3 (ICL3) fragment. (D–F) The binding interactions between the allosteric modulator and the receptor obtained from MD simulations of the X-ray receptor–ligand complexes for the M2, β2, and P2Y1 receptors. The key residues forming strong interactions and the allosteric ligands are in gray and green sticks, respectively. Hydrogen bonds and π–π interactions are shown as pink and cyan dashed lines, respectively.
Of particular interest are the allosteric sites at the protein–lipid interface, identified by X-ray or cryo-EM and confirmed by site-directed mutagenesis. Figure 1C shows the X-ray complex of the purinergic P2Y1 receptor and BPTU, a NAM occupying an allosteric site facing the membrane environment and in contact with helices 1–3 and ECL1.13 Allosteric sites at different LI locations were found in the complexes with allosteric modulators of the cannabinoid CB1,14 complement C5a,15,16 corticotropin-releasing factor 1 CRF1,17 free fatty acid FFA1,18 glucagon GCG,19 glucagon-like peptide-1 GLP-120,21 and proteinase-activated PAR222 and β2 adrenergic23 receptors.
These structural data provide the first insights into GPCR allosteric regulation by small molecules and offer opportunities to develop computer-aided methodologies to search for allosteric sites. As the lipid bilayer plays a role in the formation of such sites, accounting for the receptor in a realistic environment and its dynamics is important for accurate mapping of the allosteric sites.
Molecular dynamics (MD) simulations have become an indispensable tool for studying the structure and dynamics of drug targets in the cellular environment and predicting ligand binding sites.24 Among MD-based computational techniques developed to identify binding sites in proteins, cosolvent mapping has recently garnered wide interest.25 In MD-based cosolvent mapping, small organic molecules such as isopropanol, acetamide, pyridine, and others are used as probes to map the binding sites. A molecular probe is a prototype molecule containing polar and/or nonpolar groups that can quickly diffuse into protein cavities during MD simulations, thus identifying such cavities as accessible and therefore as potential binding sites for allosteric modulators. In addition, MD-based cosolvent mapping can directly account for protein motion during the site identification process. This approach has been developed mainly for soluble proteins to map putative binding sites on the protein surface.25 There are only a few examples to date where cosolvent mapping has been used for membrane proteins.26−28 Although these studies have successfully mapped the binding sites of membrane-bound proteins, the proposed protocols could be challenging to sample all possible locations of allosteric sites in GPCRs as identified by X-ray crystallography and cryo-EM while avoiding probe nonspecific binding and membrane distortion.
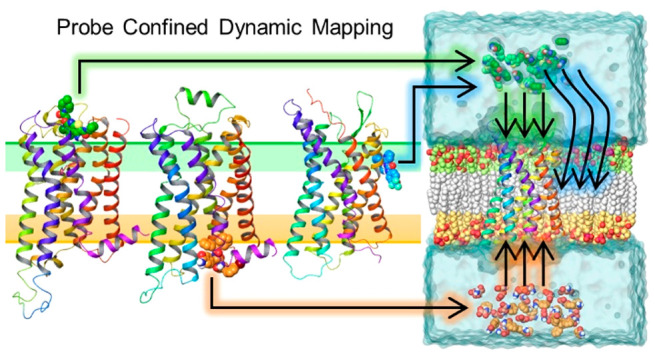
Here, we propose a novel and efficient MD-based probe mapping protocol that is capable of exploring all the possible scenarios of allosteric site locations known to date, including the most challenging case where the allosteric site resides at the LI. Our methodology overcomes the limitations of standard cosolvent mapping protocols through the application of a cylindrical harmonic wall potential that enhances probe sampling in a selected area of the receptor. In addition, we use probes derived from GPCR allosteric ligands that perform better in mapping allosteric sites compared to organic solvents. Our protocol represents a fully automatized pipeline including system setup and simulations for different scenarios. We used three exemplar receptors, i.e., the M2, β2, and P2Y1 receptors (Figure 1), for the protocol retrospective validation. We next applied the protocol in a prospective validation scenario by predicting the binding site of [5-fluoro-4-(hydroxymethyl)-2-methoxyphenyl]-(4-fluoro-1H-indol-1-yl)methanone (the UCB compound), a PAM at the dopamine D2 receptor and validating the prediction by site-directed mutagenesis. The outlined computational approach will facilitate structure-based allosteric drug design by predicting receptor binding sites of known allosteric modulators for further optimization and/or by identifying binding fragments that could be developed into new allosteric modulators.
Results
In line with previously published work on cosolvent simulations of soluble proteins25,29−31 and lipids,32−34 while the optimal cosolvent concentration range for soluble proteins is 5–20%, for membrane-embedded proteins the maximum tolerated concentration should be lower, in the 2.8–5.6% range. This is due to membrane distortion caused by a higher concentration of organic solvents. Cosolvents are also known to be technically challenging to use in combination with lipid bilayers as they tend to partition from the water layer and be adsorbed and redistribute into the membrane after just a few nanoseconds of simulation.32−34
To overcome the membrane distortion as the result of cosolvent diffusion into the membrane and to keep a suitable sampling of probe molecules in the receptor while using small concentrations, we applied a cylinder-shaped wall potential to allow the movement of the cosolvent molecules only within the GPCR EC and IC openings or within a defined area at the LI. With such a restraint, as a cosolvent molecule reaches the wall of the cylinder, a repulsive bias is applied to prevent it from visiting regions outside the cylinder. We considered the mapping of different allosteric site locations as separate simulation protocols. Thus, to explore allosteric sites at the EC side, probe molecules were placed at the top water layer (the M2 receptor case). In the case of an allosteric site at the IC side, the probe molecules were placed at the bottom water layer (the β2 receptor case). As the LI site in the P2Y1 receptor is located close to the EC side, the probe molecules were placed at the top water layer to accelerate probe diffusion to the membrane. This is also supported by the recent MD simulations of BPTU binding, which suggests its entrance to the binding site occurs from the EC side.35 The application of a wall potential allowed us to use a 10% probe concentration.
Probe Confined Dynamic Mapping
Our ad hoc protocols (workflow of the M2 receptor as an example shown in Figure 2A) enable MD cosolvent and fragment system setup, equilibration, and production of membrane-bound receptors. In the system setup, a water-probe (cosolvents or fragments) box is generated and placed at a user-defined distance at either the receptor EC and IC side with (for PAMs) or without (for NAMs) a bound orthosteric ligand. The box height (Z dimension) is specified by the user, whereas the box width and depth (x and y sizes) are calculated based on minimum/maximum protein dimensions on the EC/IC sides. After the box is placed, the protein is embedded in the membrane, and the system is solvated and neutralized.
Figure 2.

The GPCR probe confined dynamic mapping workflow. (A) An example of the protocol for the M2 receptor extracellular allosteric site. System setup: A box filled with a mixture of water and probe molecules at a defined concentration is specified using a Z dimension by the user, whereas the box X and Y lengths are calculated based on protein size at the EC side. The protein is then embedded into the POPC membrane, and the system is solvated. System equilibration: A cylinder-shaped harmonic wall potential to hold probe molecules away from the protein and membrane is applied during the equilibration. System production: An extended cylinder-shaped harmonic wall potential is applied to allow the molecules to move toward and interact with the receptor avoiding partitioning to the membrane during the production step. (B) The cylinder-shaped harmonic wall potential with addition of two collective variables (CV1 and CV2) to confine the movement of the probes at the lipid interface of helices 1–3 (in red, orange, and yellow) in the P2Y1 receptor production simulations. The collective variables that define a cylinder were selected with lower and upper boundaries (10 and 35 Å, respectively).
During the equilibration, the probe molecules are confined in a closed cylinder and not allowed to interact with the protein–membrane system (Figure 2A). During the production, to map EC/IC allosteric sites, the cylinder boundary facing the protein is removed to allow the probes to diffuse toward and interact with the protein. During the production, the distanceZ collective variable facing the system boundary is maintained, thus defining a semiclosed cylinder, and its boundary lowered by ∼10 Å (Figure 2A). This enables the probes to be confined in the periodic cell and increases their probability to interact with the protein. To sample putative allosteric sites at the LI, during the system production, two additional distanceZ collective variables were added to confine the probe movement in the specific area of the cylinder defined by target receptor transmembrane helices to sample. Thus, in the case of the P2Y1 receptor, the area of the cylinder that is sampled by the probes is defined based on helices 1–3 (Figure 2B). In addition, to increase probe membrane penetration, the van der Waals radii of the carbon atoms of the bilayer lipid tails was decreased by 10%. This slight artifact enabled us to preserve membrane integrity during the simulation while allowing the probe to diffuse more easily into the lipid bilayer.
Probe Selection from Organic Solvents and Privileged Fragments
Organic solvents such as isopropanol, acetamide, and pyridine (Table 1) are often used as standard probes to sample donor and acceptor hydrogen bonds and hydrophobic interactions in the dynamic mapping of putative binding sites for soluble proteins.29−31 Allosteric modulators of various drug targets, however, are characterized by high aromaticity and rigidity in their structures.36,37 Thus, the probes derived from common/privileged substructures of GPCR allosteric modulators could be more suitable for mapping GPCR allosteric sites. The muscarinic PAMs including LY2119620 (Figure 1) and its analogues were subjected to a maximum common substructure search that yielded N-methylthieno[2,3-b]pyridine-2-carboxamide as a “core fragment” (Figure S1). From this structure, two substructures, thieno[2,3-b]pyridine (THP) and N-methylformamide (NMF) were identified by ring–chain fragmentation as probes (Table 1). For the β2 receptor, the fragmentation of Cmp-15, (Figures 1 and S1) resulted in the selection of benzamide (BZA) and butyramide (BTA) as probes. For the P2Y1 receptor, the BPTU compound (Figures 1 and S1) was fragmented by functional groups, and phenol (PHX) and 2-hydroxypyridine (P2O) were selected as probes. The choice of these fragments fits the recent docking structure–activity analysis of the P2Y1 allosteric antagonists.38 The three selected receptors were simulated in the presence of the standard cosolvents and the above-mentioned fragments (Table 1).
Table 1. Standard Cosolvent and Specific Probes Used for MD Mapping of GPCR Allosteric Sites.

MD Trajectory Analysis
We conducted probe confined dynamic mapping of two empty receptor structures taken from X-ray complexes of the receptors with and without an allosteric modulator. In the case of the M2 receptor to map the PAM binding site, we also simulated the receptor structure in the presence of Iperoxo, the orthosteric agonist. From the trajectory visual inspection, probe molecules reached and interacted with the amino acid residues of the EC, IC, or LI allosteric binding sites in all the receptors (Videos 1, 2, and 3 in Supporting Information). This is particularly true for the specific fragments that are retained in allosteric sites for a long time. The harmonic wall potential prevented the probes from penetrating the lipid bilayer and diffusing to distant water layers. To assess the performance of the protocol, we analyzed the trajectories in two different ways. First, we quantified the probe presence in the allosteric binding sites (retrospective analysis). We then evaluated the possibility of allosteric site detection from our probe simulations assuming the location of the allosteric sites is unknown (predictive analysis).
Retrospective Analysis: Probe Allosteric Site Occupancy
To validate the ability of the probes to occupy the allosteric site of the M2, β2, and P2Y1 receptors, we calculated the probe occupancy at allosteric interaction spots (Table 2), i.e., residues contributing most to the ligand–receptor interaction energy as obtained from conventional MD simulations of the X-ray structures of the receptors bound to the allosteric modulators (Figure 1D–F, Table S1). The probe occupancy was calculated from the 40 ns production runs and expressed as the percentage of the simulated time in which each spot was occupied by a probe averaged over (at least) three independent trajectories.
Table 2. Probe Occupancy at Allosteric Interaction Spotsa.
| receptor | occupancy (%) |
||||
|---|---|---|---|---|---|
| M2 | probe (no.) | SB | HB1 | HB2 | HYD |
| Agonist4MQT | ISO (31) | 32 ± 9 | 67 ± 19 | 64 ± 16 | 71 ± 13 |
| Empty4MQT | ISO (31) | 36 ± 5 | 80 ± 8 | 75 ± 12 | 76 ± 11 |
| Agonist4MQT | ACE (32) | 38 ± 9 | 81 ± 6 | 64 ± 18 | 76 ± 11 |
| Empty4MQT | ACE (32) | 41 ± 7 | 76 ± 15 | 52 ± 26 | 73 ± 15 |
| Agonist4MQT | PYR (24) | 30 ± 9 | 69 ± 11 | 45 ± 19 | 59 ± 19 |
| Empty4MQT | PYR (24) | 38 ± 10 | 73 ± 12 | 54 ± 1 | 79 ± 6 |
| Agonist4MQT | NMF (33) | 52 ± 1 | 94 ± 4 | 88 ± 11 | 91 ± 7 |
| Empty4MQT | NMF (33) | 48 ± 10 | 83 ± 13 | 66 ± 11 | 81 ± 14 |
| Agonist4MQT | THP (14) | 43 ± 13 | 93 ± 2 | 85 ± 7 | 92 ± 2 |
| Empty4MQT | THP (14) | 26 ± 9 | 59 ± 21 | 54 ± 7 | 47 ± 36 |
| Agonist4MQS | ISO (31) | 31 ± 5 | 49 ± 10 | 35 ± 3 | 66 ± 14 |
| Agonist4MQS | ACE (32) | 27 ± 6 | 29 ± 10 | 29 ± 6 | 27 ± 10 |
| Agonist4MQS | PYR (24) | 14 ± 3 | 80 ± 7 | 24 ± 15 | 73 ± 6 |
| Agonist4MQS | NMF (33) | 30 ± 1 | 50 ± 15 | 33 ± 2 | 42 ± 13 |
| Agonist4MQS | THP (14) | 23 ± 3 | 82 ± 3 | 63 ± 16 | 79 ± 5 |
| β2 | probe | HB1 | HB2 | HYD1 | HYD2 |
|---|---|---|---|---|---|
| Empty5X7D | ISO (42) | 63 ± 7 | 52 ± 9 | 46 ± 11 | 21 ± 26 |
| Empty 5X7D | ACE (43) | 77 ± 20 | 55 ± 16 | 58 ± 21 | 80 ± 7 |
| Empty5X7D | PYR (32) | 87 ± 9 | 71 ± 21 | 65 ± 25 | 51 ± 38 |
| Empty5X7D | BZA (21) | 87 ± 10 | 80 ± 10 | 77 ± 14 | 80 ± 12 |
| Empty5X7D | BTA (29) | 85 ± 12 | 42 ± 33 | 68 ± 8 | 62 ± 38 |
| Empty2RH1 | ISO (42) | 72 ± 8 | 0 | 6 ± 2 | 65 ± 31 |
| Empty2RH1 | ACE (43) | 15 ± 6 | 0 | 6 ± 2 | 46 ± 40 |
| Empty2RH1 | PYR (32) | 41 ± 34 | 0 | 6 ± 5 | 52 ± 10 |
| Empty2RH1 | BZA (21) | 66 ± 26 | 0 | 48 ± 27 | 75 ± 43 |
| Empty2RH1 | BTA (29) | 53 ± 22 | 0 | 55 ± 22 | 3 ± 4 |
| P2Y1 | probe | HB | HYD |
|---|---|---|---|
| Empty4XNV | ISO (30) | 4 ± 7 | 18 ± 11 |
| Empty4XNV | ACE (30) | 0 | 11 ± 4 |
| Empty4XNV | PYR (23) | 12 ± 19 | 19 ± 12 |
| Empty4XNV | PHX (19) | 22 ± 19 | 63 ± 11 |
| Empty4XNV | P2O (19) | 48 ± 12 | 79 ± 7 |
| Empty4XNW | ISO (30) | 6 ± 10 | 39 ± 39 |
| Empty4XNW | ACE (30) | 0 | 5 ± 4 |
| Empty4XNW | PYR (23) | 5 ± 4 | 45 ± 28 |
| Empty4XNW | PHX (19) | 40 ± 10 | 73 ± 13 |
| Empty4XNW | P2O (19) | 19 ± 11 | 68 ± 23 |
The occupancy is expressed as a percentage of the simulated time averaged over three independent trajectories. Allosteric interaction spots are identified based on ligand-residue interaction energy obtained from the MD simulations of X-ray receptor-ligand complexes (Table S1). The allosteric interaction spots include the following residues: SB: E172ECL2, HB1: N4106.58 and Y177ECL2, HB2: Y802.61 and N419ECL3, and HYD: W4227.35 and F181ECL2 for the M2 receptor; HB1: N692.40, D3318.49, and backbone of R63IL3, HB2: T2746.36, HYD1: V54, L64ILC1, and F3328.50 and HYD2: I722.43, L2756.37, and Y3267.53 for the β2 receptor; and HB: L1022.55 backbone and HYD: T1032.56 and M1233.24 for the P2Y1 receptor. “Agonist” and “Empty” are for the receptor agonist-bound and empty forms, respectively. X-ray structures used in the MD simulations with PDB code: 4MQT, 5X7D, and 4XNV contain an allosteric modulator and with PDB code: 4MQS, 2RH1, and 4XNW are without an allosteric modulator. The number of probes in the box/cylinder used in the simulations is indicated in parentheses. Occupancy with the highest values and mentioned in the text are in bold.
Although ISO, PYR, and NMF probes occupy two interaction spots for over 70% of the simulated time, in the simulations of the M2 empty receptor form, the occupancy tends to be higher in the agonist-bound form of the M2 receptor (Table 2). The THP and NMF probes occupy three interaction spots (HB1, HB2, and HYD) for over 85% of the simulation time, whereas the ACE probe occupies the HB1 spot in the agonist-bound form of the M2 receptor for over 70% of the time. This supports the hypothesis that the Iperoxo agonist stabilizes the allosteric binding site of LY2119620, which is in line with the recent conventional MD simulations of both the M2 receptor forms.39 The probes derived from the maximum substructure search (THP and NMF) performed better by yielding a higher occupancy of several interaction spots than the standard probes. In most of the THP and NMF trajectories, we observed one probe molecule occupying the allosteric site and forming persistent interactions with W4227.35 (the Ballesteros-Weinstein numbering is given in superscripts40) or N419ECL3 and Y802.61 (Figure S2, Video 1 for the THP probe). Other probes were less persistent and only occupied the site intermittently. Up to two probe molecules were detected around the residue interaction spots at a 2.5 Å distance (Figure S2).
We next simulated the X-ray structure of the M2 receptor bound to Iperoxo in the absence of the allosteric modulator (PDB ID: 4MQS) to assess whether the probes were able to recognize and occupy the allosteric site. The THP and PYR occupancies were high, over 60%, for the HB1, HB2, and HYD interaction spots and over 70% for the HB1 and HYD spots, respectively. The occupancies of other probes were lower with ISO and NMF probes occupying the HB1 and HYD interaction spots by above 40% of the time.
In the β2 receptor simulations, three probes (PYR, BZA, and BTA) occupied for over 85% of the time the HB1 interaction spot (Table 2). The ACE probe was retained in HB1 and HYD2 spots for over 77% of the time, whereas BZA occupies all four interaction spots for over 77% of the time. The occupancy of the ISO probe ranged from 21 to 63%. The probes derived from ligand fragmentation, particularly BZA, had the highest occupancy in all the hotspots. In the trajectories, one or two BZA molecules interacted with N692.40, S3298.47, D3318.49, and R63ICL1 (Figure S3). In particular, one BTA molecule formed stable hydrogen bonds with N692.40 and T2746.36 (Figure S3, Video 2). Other probes formed less persistent interactions in the allosteric site. The simulations of the β2 X-ray structure (PDB ID: 2RH1) without an allosteric modulator showed that the BZA and ISO probes occupied the HB1 and HYD2 interaction spots for over 65% of the simulation time. The BTA probe occupied the HB1 and HYD1 spots for above 50% of the time, whereas the PYR molecules occupied the HYD2 spot for over 50% of the time. None of the ACE probe molecules occupied the receptor cavity persistently. The HB2 spot was not sampled in all the simulations.
In the P2Y1 receptor simulations, the probe occupancy was generally lower compared to the receptors with the EC and IC allosteric sites (Table 2). This is due to the need for a probe to pass through the lipid layer. However, the P2O and PHX probes derived from the allosteric ligand fragmentation yielded a higher occupancy of the HYD spot (79% and 63% for P2O and PHX, respectively) and the HB (48% and 22% for P2O and PHX, respectively) compared to ISO, ACE, and PYR probes. One to three molecules of P2O and PHX molecules occupied the allosteric site by forming hydrogen bonds to the backbone of M1233.24 and L1022.55 (Figure S4 and Video 3). The simulations of the P2Y1 X-ray structure in complex with an orthosteric antagonist (MRS2500) and in the absence of the allosteric modulator (PDB ID: 4XNW) demonstrated that the PHX and P2O yielded better performance than other probes and occupied the HB and HYD binding spots for 40% and 73%, and 19% and 68% of the time, respectively. We also performed probe simulations by sampling the LI area around helices 2, 3, and 4 (1); 3, 4, and 5 (2); 6 and 7 (3); and 1 and 7 (4) at the P2Y1 receptor (PDB ID: 4XNW) (Figure S5). The selection of these lipid–helix interfaces was based on the MDpocket cavity prediction41 from the P2Y1 receptor conventional MD simulations (see the section below) and the available receptor X-ray complexes bound to an allosteric modulator at the LI. For example, allosteric ligands sit at the LI of helices 2, 3, and 4 in the PAR2 and CB1 receptors and at the LI of helices 3, 4, and 5 in the C5a, β2, and FFA1 receptors.14−16,18,22,23 Both probes displayed either low occupancy or no occupancy at all in the selected LI areas (Table S2), thus demonstrating the specificity of P2O and PHX in binding at the LI of helices 1–3.
Overall, the three receptor examples demonstrate that specific probe molecules perform better in both receptor conformations. The probe occupancy is generally lower for the receptor conformation derived from the X-ray structure obtained in the absence of the allosteric modulator, although most of the allosteric interactions could still be mapped. The performance of standard probes is particularly low for such a conformation. In addition, the probe occupancy was higher in the presence of an orthosteric agonist when the binding site of the M2 PAM was mapped, thus indicating the importance of adding an agonist in probe simulations in search for PAM binding sites. Although more probe-MD simulation tests are required, the results obtained on the P2Y1 receptor show that the specific probes are capable of detecting a distinct allosteric site.
Predictive Analysis: Probe Density and Cavity Detection
We next analyzed the trajectories blind to the nature of the allosteric interaction spots as it would be the case for novel ligands or previously unexplored GPCRs to assess if allosteric sites can be predicted ab initio. Therefore, a probe density analysis for the MD simulation trajectories was carried out using the VolMap tool.42 In Figure 3A, the aggregated view of probe distribution (isovalue 0.5) calculated from the three replicates for each probe obtained for the two X-ray structures considered for each receptor is depicted. From this analysis, we accessed the coordinates of the points with isovalues higher than a threshold (>0.5) and determined the list of residues that interacted with the probe molecules. As shown in Table 3, the initial number of residues around a putative allosteric site was high.
Figure 3.

Probe density and cavity detection analyses for MD simulations of the receptor empty forms obtained from X-ray structures of the receptors with (PDB codes: 4MQT, 5X7D, and 4XNV) and without an allosteric modulator (PDB codes: 4MQS, 2RH1, and 4XNW). (A) The aggregated view of probe density from the probe-MD simulations. The probe density was calculated using the VMD VolMap tool (isovalue = 0.5). The probe density is in yellow, red, and pink for the standard probes and in cyan and green for specific probes. (B) Binding cavities detected by MDpocket from the three conventional MD simulation runs of the empty receptors. The cavities are shown in transparent surface representation and generated with Maestro 2019-3. (C) Druggable cavities predicted by Fpocket. (D) The overlap of probe density and druggable cavities for the M2 receptor and the overlap of probe density with detectable cavities for the β2 and P2Y1 receptors. (E) The X-ray structure of the receptors bound to the allosteric modulator is shown for comparison.
Table 3. Prediction of Allosteric Interaction Spot Residues from Probe Mapping, Cavity Detection, and Probe–Residue Interaction Energy.
| number
of residues |
||||||
|---|---|---|---|---|---|---|
| receptor | receptor structure used for MD simulations | from probe density | from cavity detection | from the intersection between probe density and cavity detection | from probe-residue interaction energy < −1 kcal/mol | identified allosteric interaction spot residues |
| M2 | 4MQT | 55 | 35 | 35 | 23 | 8 |
| 4MQS | 52 | 46 | 40 | 24 | 8 | |
| β2 | 5X7D | 83 | 45 | 45 | 23 | 9 |
| 2RH1 | 72 | 34 | 33 | 26 | 9 | |
| P2Y1 | 4XNV | 27 | 18 | 18 | 11 | 3 |
| 4XNW | 24 | 10 | 10 | 10 | 3 | |
To further narrow down the residue selection, we next linked the probe density analysis with cavity detection. The MDpocket tool41 was used to track putative ligand binding cavities and predict their druggability in conventional MD simulations of the receptor empty forms. On the basis of the assumption that not all locations where a probe molecule resides in the probe-MD simulations may represent ligand binding cavities, we intersected the residues obtained from the probe density analysis with the residues obtained from cavity detection to find allosteric site residues. Figure 3B,C shows all detectable and druggable cavities in the receptors from the three simulation replicates of the two receptor X-ray structures. MDpocket identified multiple putative ligand binding sites in different regions of the three receptors. The allosteric sites of the M2, β2 and P2Y1 receptors were all detected as ligand binding cavities by MDpocket. Among them, the allosteric site at the EC side in the M2 receptor has also been identified as a druggable cavity in all simulation replicates of the M2 empty receptor obtained from the X-ray complex bound to an allosteric modulator and in one replicate of the M2 empty receptor obtained from the X-ray complex not containing an allosteric modulator, according to the Fpocket druggability criteria.43−45 A small region of the allosteric site at the IC side has been only identified as druggable in one simulation of the empty β2 receptor obtained from the X-ray complex bound to an allosteric modulator. None of the conventional MD simulations of the P2Y1 receptor empty forms predicted the allosteric site at the LI side as a druggable cavity. The druggable cavity at the EC side of the M2 receptor and detectable cavities at the IC site of the β2 receptor and at the LI side of the P2Y1 receptor overlapping with probe density (Figure 3D) were used to determine residues lining these cavities (Table 3).
We next calculated the interaction energy between the residues selected from the overlap of the probe density and cavity detection results, and the probe molecules to identify the residues forming interactions with the probe below 1 kcal/mol (Table 4). Less than 10 residues were identified (Table 4), and among them there were residues of the allosteric interaction spots, which were scored at the top of the list for both receptor conformations of the M2 and P2Y1 receptors (Table 4). In the case of the β2 adrenergic receptor, the allosteric interaction spot residues were more scattered, particularly from the simulations obtained based on the X-ray structure of the receptor without the allosteric ligand. This is because of the partially closed IC cavity in this structure. The specific probes had a major contribution to the interaction energy.
Table 4. Probe–Residue Interaction Energya.
| Probe–Residue
Interaction Energy (kcal/mol) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M2_4MQT | M2_4MQS | β2_5X7D | β2_2RH1 | P2Y1_4XNV | P2Y1_4XNW | ||||||||||||
| E175 | –6.2 | NMF | D97 | –8.4 | NMF | R63 | –7.3 | BZA | R328 | –6.0 | ISO | F119 | –4.5 | P2O | F119 | –4.8 | PHX |
| N419 | –5.6 | THP | E175 | –6.5 | NMF | D331 | –6.6 | BZA | R63 | –5.7 | BZA | M123 | –4.3 | P2O | L102 | –3.9 | PHX |
| W422 | –5.1 | THP | N410 | –4.2 | THP | R131 | –6.2 | BTA | S329 | –5.1 | ISO | L102 | –2.9 | PHX | Q127 | –3.4 | ISO |
| N410 | –4.0 | NMF | E172 | –4.0 | NMF | A271 | –3.6 | BZA | R131 | –5.1 | BTA | T103 | –2.6 | P2O | T103 | –3.2 | P2O |
| Y177 | –3.9 | NMF | Y177 | –3.6 | ISO | S329 | –3.2 | BZA | D331 | –4.5 | BZA | A122 | –2.3 | PHX | M123 | –2.8 | PHX |
| E172 | –3.6 | NMF | W422 | –3.6 | THP | N69 | –3.2 | BZA | K140 | –4.3 | ACE | L126 | –2.1 | P2O | W117 | –1.9 | ISO |
| F181 | –2.6 | THP | A414 | –3.4 | THP | Q142 | –3.0 | BZA | K243 | –3.8 | BZA | G120 | –1.8 | ACE | A106 | –1.6 | P2O |
| Y80 | –2.4 | NMF | N419 | –3.3 | THP | T274 | –2.9 | BZA | E244 | –3.1 | BTA | Q127 | –1.5 | P2O | L126 | –1.1 | PHX |
| Y426 | –2.2 | NMF | Y80 | –2.7 | PYR | F264 | –2.3 | BTA | N69 | –2.8 | ACE | A106 | –1.5 | P2O | P105 | –1.0 | PHX |
| T170 | –2.2 | THP | Y83 | –2.5 | PYR | L275 | –2.2 | BZA | T274 | –2.8 | ACE | W117 | –1.4 | ACE | |||
| S182 | –2.1 | THP | W162 | –2.5 | ISO | K267 | –2.2 | BTA | Q142 | –2.5 | BZA | P105 | –1.0 | PHX | |||
| A414 | –2.1 | THP | F181 | –2.2 | THP | L145 | –1.7 | BZA | F332 | –2.5 | PYR | ||||||
| T84 | –2.0 | THP | P415 | –2.2 | THP | L272 | –1.5 | BZA | P306 | –2.5 | BZA | ||||||
| Y83 | –2.0 | THP | T423 | –2.2 | PYR | T68 | –1.5 | BZA | Y326 | –2.4 | ISO | ||||||
| T423 | –1.8 | THP | T170 | –2.1 | ACE | F332 | –1.4 | BZA | Y219 | –2.3 | ISO | ||||||
| Y88 | –1.6 | THP | I417 | –2.0 | THP | T66 | –1.3 | BZA | Y141 | –2.2 | ACE | ||||||
| Y403 | –1.5 | NMF | T84 | –1.6 | PYR | Y326 | –1.3 | BZA | A247 | –2.1 | PYR | ||||||
| T187 | –1.5 | NMF | T411 | –1.6 | THP | Y219 | –1.2 | ISO | T68 | –1.4 | ACE | ||||||
| I417 | –1.2 | ACE | Q179 | –1.5 | ISO | C327 | –1.1 | PYR | L275 | –1.4 | PYR | ||||||
| N183 | –1.1 | THP | Y88 | –1.5 | THP | I72 | –1.1 | BZA | L248 | –1.3 | PYR | ||||||
| G87 | –1.1 | NMF | S182 | –1.3 | THP | L143 | –1.0 | ACE | S143 | –1.2 | ACE | ||||||
| T411 | –1.0 | ACE | V171 | –1.2 | NMF | T146 | –1.0 | BTA | I72 | –1.2 | PYR | ||||||
| Y104 | –1.0 | NMF | G174 | –1.2 | PYR | L64 | –1.0 | BZA | V67 | –1.1 | BZA | ||||||
| P418 | –1.1 | THP | T66 | –1.1 | BZA | ||||||||||||
| L64 | –1.1 | BZA | |||||||||||||||
The residues selected from the overlapping region obtained from the probe density and cavity detection analyses were used to calculate the probe–residue interaction energy. The interaction energies < −1 kcal/mol are shown for the probe that forms the strongest interaction with a residue. Residues that contribute to the allosteric interaction spots (from Table 2) are indicated in bold.
Overall, the binding interactions of probe molecules are matched with the interactions of the allosteric ligands in the X-ray structures. Not all allosteric cavities meet an established druggability rule; this is especially true with cavities at the LI; therefore, all detectable cavities should be potentially considered in a predictive analysis. Our computational protocol was then validated in a prospective study, where the key receptor residues predicted by probe MD mapping were assessed by mutagenesis.
Experimental Validation of Probe Confined Dynamic Mapping
To test our methodology in an experimental setting, we applied our pipeline to the D2 receptor. As a proof of concept, we explored how the protocol performed in predicting where the previously published UCB PAM might bind (Figure 4A).46 Two probe molecules indole (IND) and benzyl alcohol (BAL) were generated from the UCB compound.46 Probe confined dynamic mapping on the D2 crystal structure47 in a dopamine-bound form was carried out to explore putative binding sites at the EC and IC sides and the LI. From probe density analysis, druggable cavities detection and probe–residue interaction energies, as described above, we have predicted a putative allosteric site for the UCB compound at the EC side of the D2 receptor consisting of helices 2 and 7, ECL1 and ECL2 (Figure 4B–C). From our approach, the final list of residues involving V912.61, L942.64, E952.65, W100ECL1, I184ECL2, and W4137.40 was selected for site-directed mutagenesis by taking into consideration a docking pose of the UCB compound in the predicted allosteric site (Figure 4D).
Figure 4.

Computational and experimental prediction of the allosteric site for the UCB compound in the D2 receptor. (A) The UCB compound and probe molecules used in allosteric site mapping. (B) The structure of the D2 receptor in cartoon representation with the results of MD simulation analysis. From left to right: the aggregated view of probe density from the probe simulations, IND and BAL are in cyan and purple, respectively; detectable cavities predicted by MDpocket from the MD trajectories are shown in transparent surface representation; a druggable cavity predicted by Fpocket; the overlap of probe density and a druggable cavity. (C) The probe–residue interaction energy for the residues selected from probe density and druggable cavity detection. The interaction energy < −1 kcal/mol is shown for the probe that forms the strongest interaction with a residue. (D) Docking pose of the UCB compound at the putative allosteric site. The UCB compound and dopamine are shown in green and orange sticks, respectively. Only residues selected from probe–residue interaction energy calculation are shown. The druggable cavity is shown in transparent surface representation. Labels of residues selected for mutagenesis are in blue, and labels of residues in contact with the orthosteric agonist are in red. (E) cAMP accumulation assays in the absence and presence of the UCB compound at the D2 receptor WT and mutants. Concentration–response curves of the endogenous agonist, dopamine, measuring Forskolin-induced (7.5 μM) cAMP accumulation at the D2 WT receptor (1) and the mutants V91A (2), L94A (3), L94W (4), W413A (5), and E95A (6) in the absence and presence of UCB compound 1 (10 μM). The absence of the PAM is indicated with a solid line, while the presence of 10 μM of the PAM is indicated by a dotted line. Each data point represents the mean ± SEM of triplicate wells of three independent experiments. Analysis of the pharmacological parameters for these curves can be found in Supplementary Table S3.
To test if these residues were involved in the function of the UCB compound, we first confirmed the compound modulated cAMP production in line with an allosteric modulator of a Gi-coupled receptor (Figure 4E.1). We then tested each mutant in transfected HEK293 cells to ensure that the mutation of each residue did not significantly impact the ability of the receptor to reduce Forskolin-stimulated production of cAMP after agonist addition (Figure 4E, Table S4) or the expression (Figure S7). We found either a tryptophan or alanine at position L942.64 was well tolerated (pEC50 = 9.7 and 8.7, respectively). At positions 2.61 and 2.65, we were able to mutate these residues to an alanine while conserving activity with minimal changes in pEC50 (Figure 4 and Table S3). Altering W100ECL1, however, led to a significant decrease in activity, in line with published results47 (pEC50 = 8.1) (Figure S6) and showed altered expression (Figure S7). A similar change in activity was seen with the I184AECL2 mutation (pEC50 = 8.3) (Figure S6). Thus, these two constructs were not explored further. For the W4137.40 position, we tested mutation to alanine which preserved receptor function (pEC50 = 8.8).
Next, we tested the effect of these mutations on their ability to influence the effects of the UCB compound. In line with the predicted computational analysis, mutations of V912.61A and L942.64A did appear to reduce the effects of the PAM on dopamine stimulation as measured by forskolin-stimulated cAMP accumulation (Figure 4E.2 and E.3). However, the dose responses were nearly flat in the presence of the UCB compound, with a measured ΔEmax of ∼57% and ∼34%, respectively, which is not easy to interpret. Enlarging this pocket by either mutation may provide too much flexibility for the compound. This varied with the L942.64W mutant, where the PAM effect appeared to be completely lost (Figure 4E.4). The W4137.40A mutation increased the PAM effect by improving dopamine affinity (ΔpEC50 = 3) with no impact on Emax (Figure 4E.5). The E95A2.65 mutation too showed a similar but milder effect (ΔpEC50 = 3) with a measured ΔEmax of ∼35%, suggesting the potential importance of these amino acids in maintaining the orientation of the PAM (Figure 4E.6). To quantify effects of the UCB compound, we performed cross-titration curves and calculated a Kb value (Figure S8A–C). These showed dramatic changes from WT vs L942.64W and W4137.40A mutants (Figure S8D). Indeed, for the L942.64W, no Kb could be obtained as all signs of allosterism were lost, consistent with the data obtained above. Together, these data support the predicted binding site of the UCB compound and serve to validate our prediction pipeline.
Discussion
We have developed a probe confined dynamic mapping protocol for fast and efficient detection of allosteric sites in GPCRs. The application of the cylinder-shaped harmonic wall potential and the specific probes derived from GPCR allosteric ligand structures helped address the limitations of currently available cosolvent mapping protocols such as limited probe sampling, membrane distortion, probe nonspecific binding, and protein denaturation.
The cylinder-shaped harmonic wall potential allows the probes to explore thoroughly either the EC or IC sides of the receptor while preventing them from partitioning into the lipid bilayer or distant water layers. In the case of the allosteric site mapping at the LI, the probes are only allowed to move from a water layer to the lipid bilayer at the interface of the selected helices. To explore the entire protein–lipid interface in a blind allosteric site search, we propose to run probe simulations sampling separately two to three helices.
The default simulation length for the production run was set at 40 ns, although the protocol was able to identify an allosteric site in simulation times as short as 20 ns. On the basis of the inspection of recent X-ray and cryo-EM structures of GPCR LI allosteric sites located deeper in the membrane, we envision that the simulation time for the search of such sites could be extended to enhance the sampling at the interface. In blind studies, we suggest mapping allosteric sites at the EC, IC, and LI separately. Incorporation of all the scenarios in a single protocol will require too many restraints that would result in a reduced simulation speed.
Here, we extended the dynamic mapping approach with cosolvents to fragments derived from GPCR allosteric ligand structures to improve sampling of specific binding in allosteric sites. From the probe occupancy and probe-residue interaction energy analysis, the specific probes performed substantially better in mapping key allosteric interaction spots compared to the standard organic solvents. In the case of the allosteric sites at the LI, the standard probes yielded poor results. In the probe-MD simulations of the P2Y1 receptor, where we allowed probes to sample the LI for various helices, we show that specific probes were also selective in mapping a specific cavity at the LI. In a prospective search of allosteric sites for a receptor with unknown allosteric ligands, we suggest using a set of fragments derived from “privileged” or common substructures of allosteric modulators of a related receptor subtype or receptor subfamilies as probes. Our choice of 10% probe concentration in the starting box of the water–probe mixture was enough to sample the allosteric sites. In the case of PAMs, the probe mapping simulations should be performed in the presence of an orthosteric agonist.
We also investigated the performance of receptor X-ray structures without an allosteric ligand. Although the probe occupancy was generally low, the specific probes were able to sample the allosteric interaction spots in the M2 and P2Y1 receptors. The probes were not able to sample one of the interaction spots in the β2 receptor because the binding cavity was partially occluded. However, in the proposed predictive setting of MD trajectory analysis, involving a combination of probe density analysis, binding cavity detection, and probe–residue interaction energy calculations, these receptor conformations allowed identification of the key residues.
We have applied the developed protocol to identify the binding site of the UCB PAM at the D2 receptor. Currently, the experimental structure of the D2 receptor bound to an allosteric ligand is not available. Recently, the cryo-EM structure of the D1 receptor has been published bound to LY3154207, a PAM at the LI of helices 3–5.48 The known D2 NAM, SB269652,49 and compounds based on thieno[2,3-d]pyrimidine scaffold50,51 are believed to bind to the EC side.50,52 In our study, we explored the possible allosteric site of the UCB compound at the EC and IC sides and the LI. We found that the probe density was particularly high at the EC side around helices 2 and 7, ECL1 and ECL2. Given that this binding cavity is also predicted as druggable, we chose this cavity as a preferable one for residue selection. Six residues were selected for site-directed mutagenesis, and four residues were confirmed to have an impact on the UCB compound of dopamine-induced activation of the receptor, thus validating our computational protocol. Mutating the residues in the EC loops beneath the dopamine binding site in direct contact with PAM’s F-indole functional group affects receptor function, as expected. In particular, I184 is believed to play a role in ligands Kon and Koff as well as β-arrestin signaling in the D2 receptor and other aminergic GPCRs.53,54 In the X-ray D2 receptor bound to risperidone, an inverse agonist forms a hydrophobic patch involving W100, I194, and L942.64 that changes the ECL1/2 orientation with respect to the one previously observed in D2-like receptors.47 Mutation of these residues reduces the residence time of risperidone. Hence, the effects observed in our experiments are in line with acquired knowledge on the D2 receptor. Mutating the residues surrounding the PAM Ph-CH2OH does not affect receptor function but impacts the PAM activity. This evidence supports the role suggested for the W4137.40 and E952.65 side-chains in keeping the PAM in an orientation that allows it to occlude the orthosteric pocket and/or interfere with the agonist binding.
The mapping of the UCB compound in the D2 receptor has turned out to be straightforward as the allosteric site of the compound was found at the EC side, which was predicted as a detectable and druggable cavity. The probe simulations were complementary with MDpocket and Fpocket results and, in addition, guided the selection of a ligand docking pose by identifying the residues establishing specific interactions with the probes. In many cases, as we see here for the P2Y1 and β2 receptors, especially for the receptor X-ray structure obtained without an allosteric modulator, the detectable cavities might not be druggable. This could be often observed for the LI sites. In this case, all detectable cavities identified by MDpocket-like programs should be considered. Probe-based MD simulations would be of particular use here to prioritize a putative allosteric cavity and evaluate its accessibility, polarity, and dynamics among other detectable cavities in an efficient manner. The P2Y1 receptor simulations where specific probes were capable to recognize a particular cavity could be an example to demonstrate how various cavities could be evaluated by probe simulations.
FTMap and FTSite tools were used to map allosteric sites in GPCR X-ray structures55 and MD-derived receptor conformations of the M2 and A2A adenosine receptors.56,57 These tools were successful in mapping EC and IC allosteric sites but failed to map LI sites. The site identification by a ligand competitive saturation (SILCS) MD-based approach has been also recently applied to map EC allosteric sites of the M2 and GPR40 receptors.28 Here, we have developed a methodology for mapping allosteric sites at the receptor EC and IC sides and, particularly, in the most challenging case involving allosteric sites at the interface between and the receptor and membrane. The cylinder-shaped harmonic wall potential applied to probe molecules in MD simulations allows efficient mapping of allosteric sites at various locations. Our methodology is able to identify allosteric sites in a short simulation time, and the results of the simulations could be inspected in less than a day, which makes it suitable for industry settings. Implementation of a fragment-based drug design approach demonstrated that confined dynamic probe mapping can successfully be performed on membrane proteins. The specific probes based on allosteric modulators have shown a potential in detecting a distinct allosteric site, suggesting that the method could be sensitive to identify false positives from allosteric modulator screens. Our methodology is a computationally feasible solution to initiate rational search of allosteric sites and design of allosteric ligands for GPCRs and other membrane-bound drug targets.
Materials and Methods
System Preparation
The X-ray structures of the M2 (4MQT and 4MQS), β2 (5X7D and 2RH1), P2Y1 (4XNV and 4XNW), and D2 (6CM4) receptors were used to revert to the wild type receptors and subsequent MD simulations. The wild types were built based on the sequences with UniProt ID: P08172, P07550, P47900, and P14416 with a partial reconstruction of ICL3 using the Prime module of Schrödinger software.58
Probe Confined Dynamic Mapping Protocols
The automatic procedure of probe confined dynamic mapping are provided as jupyter notebooks (MIDAS_EC.ipynb, MIDAS_IC.ipynb, and MIDAS_LI.ipynb) available in the Supporting Information and at GitHub (https://github.com/irinat12/Probe-Confined-Dynamic-Mapping-Protocols-GPCRs_membrane_proteins). The required input files are (i) pdb of a receptor (preoriented with OPM59), membrane, and one copy of cosolvent/fragment structures and (ii) an input file containing the following information: a receptor, an orthosteric ligand (if applicable), cosolvent and membrane file names (1); an orthosteric ligand (if applicable), cosolvent, and lipid residue names (2); cosolvent/fragment molecular weight and desired water/probe m/m % (in the closed box) (3); and height of the water buffer region between the protein and the water/probe mixture box (4).
System Setup
A box of water/probe (cosolvents or fragments) mixture of user-defined m/m % concentration is generated with Packmol60 and placed at a user-defined distance (zoff) from the protein atom with the maximum/minimum z coordinate on the EC/IC side. The user can specify the box z dimension (zheight), whereas the box x and y sizes are automatically calculated based on minimum/maximum protein dimensions on the EC/IC sides (based on protein atoms with z > 0 and z < 0, respectively). After the box is placed, the protein is embedded in the membrane, and the system is solvated with water molecules and neutralized with 0.15 NaCl. Final system files are created for subsequent MD simulations. All the above-mentioned steps are carried out using HTMD 1.13.6.61 NAMD program62 input, constraints, and collective variable files are automatically generated.
Equilibration
The cosolvent molecules are confined in a closed cylinder, which is set up by distanceXY and distanceZ collective variables available in the NAMD program and using the hypotenuse of the rectangle defined by the Packmol box x and y sizes as radius. A small constraint on the protein center of mass is applied during the third equilibration phase to avoid protein drifting and maintain cylinder absolute coordinate reference.
Production
During the production phase, the cylinder boundary (distanceZ collective variable) facing the protein is removed, and the cylinder radius decreased to the half of the Packmol x or y box size, whichever is smaller, to allow the probes to diffuse toward and interact with the protein. During the production, the distanceZ collective variable facing the system boundary is maintained, thus defining a semiclosed cylinder, and its boundary lowered by ∼10 Å. To enable probe adaptation to the new cylinder size, the system is subjected to 240 steps of minimization, before 40 ns of NPAT (constant normal pressure and lateral surface area of membranes and constant temperature) production is ran (at least in triplicate). Protein Cα atoms with z coordinates ±5 Å from the cell origin are restrained to avoid protein drifting.
To sample allosteric sites at the membrane/protein interface two additional distanceZ collective variables (one on the x and one on the y axis) were added to confine the probes in the specific area of the cylinder defined by the user based on target receptor transmembrane helices to sample. To enable probe adaptation to the new area size, at least 1800 minimization steps were required before running the actual production (40 ns, NPAT condition, at least five replicas). The collective variables that define a cylinder were selected with lower and upper boundaries (10 and 35 Å). The 10 Å was set to prevent the probes from sampling MRS2500 orthosteric site, which is readily accessible from the EC side. The van der Waal radii of POPC CLT2 and CTL3 atom types were decreased by 10%, while the parameters of all the other POPC atom types remained unaltered.
Molecular Dynamics Simulations
CHARMM36 force field was used for proteins, lipids, and water.63,64 The parameters for ligands and probes were derived from the CHARMM General Force Field (CgenFF), v1.0.0.65 The receptors were placed in a 90 × 90 Å 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane patch. The receptor–lipid system was solvated with a 30 Å buffer from both sides of the lipid bilayer. The systems were neutralized by 0.15 M Na+ and Cl– ions.
All MD simulations were performed using NAMD Git-2017-12-19, Linux-x86_64-multicore-CUDA.66 The first equilibration step included 1000 steps of minimization followed by 0.5 ns of NVT simulations with the protein, lipid headgroups, ions, cosolvent, and water atoms fixed. The second equilibration step comprised 500 minimization steps followed by 2.0 ns of NPT simulations, where harmonic restraints on all protein atoms were applied, and a small force was applied to water molecules to prevent them from entering the membrane. In the case of probe simulations, the probes were confined in a closed cylinder. The third equilibration step involved 10 ns of NPT ensemble with the receptor free to relax with translation on the center of mass removed. Probe molecules were kept in a closed cylinder. The production step included 240 steps of minimization and 40 ns of simulations, where the whole system was free to relax. In the case of probe simulations, harmonic restraints on the protein Cα atoms with z ± 5 Å from the origin (0,0,0) were applied, and translation on the protein center of mass was removed. The probe molecules were confined in a smaller semiclosed cylinder, with the boundary facing the receptor opened. The temperature of all simulations was 310 K.
Cheminformatics Analysis
Maximum common substructure search and ligand fragmentation based on a ring–chain method or functional groups were performed using the cheminformatics toolkit (frags2img.py, getcore.py, and enumfras2pdf.py of OpenEye (OEChem TK 2.2.0).67
Trajectory Analysis
The probe occupancy at the distance of 4 Å from the allosteric spots was calculated using an in-house tcl-script using VMD 1.9.3.42 The probe density was calculated using the Volmap tool of VMD 1.9.3 with a cell side of 1 Å, and the density was averaged over all frames of the top molecule. The Volmap probe density was analyzed at isovalues of 0.3, 0.5, and 0.8. Probe density at an isovalue of 0.5 was selected for the selection of residues in the interaction with probe molecules at a 5 Å distance. MDpocket41 with Fpocket 3.043,44 was used to predict detectable and druggable pockets in MD trajectories. The residue–probe or residue–ligand interaction energy was calculated using the “namdenergy.tcl” script v 1.6 of NAMD.62 The residues at 5 Å distance from a ligand/probe were selected for the interaction energy analysis. Modeling pictures were created with Maestro 2019-3,58 and MD videos were generated with VMD 1.9.3.
Molecular Docking
The induced fit docking program of Schrodinger software 2019-368,69 was used for docking of the UCB compound and dopamine. Prior to docking, ligands were prepared using the “Ligand Prep” module, and the D2 receptor was preprocessed according to the protein preparation procedure of the Schrodinger software. All docking calculations were run in the “Standard Precision” (SP) mode70 with default values for all parameters. The docking box was set based on the residues predicted for the putative allosteric site from the probe-MD simulations. The best-docked structure was chosen using the Glide Score.71 Dopamine was maintained in the orthosteric site during the docking of the UCB compound.
Site-Directed Mutagenesis
All cDNA templates and primer sequences are listed in Table S4. Per mutation, a reaction mix (25 μL) was prepared containing 1× PhusionTM HF buffer, 200 μM of dNTP mix, 0.5 μM of forward primer, 0.5 μM of reverse primer, 25 ng of template DNA, and 0.02 U/μL of PhusionTM high-fidelity DNA polymerase. PCR cycles were carried out using Veriti thermocycler (Applied Biosystems). A three-step protocol was set such that initial denaturation occurred at 98 °C for 30 s, followed by 35 amplification cycles. Each amplification cycle consisted of denaturation for 10 min, an annealing gradient of 2–3 temperatures depending on the Tm of the primers for 60 s followed by an extension at 72 °C (22.5 s/kb). To terminate the PCR amplification cycles, a final extension step at 72 °C for 10 min was applied. For the mutation W413A, a two-step protocol was used. Agarose gel electrophoresis (1% w:v) was then used to analyze PCR products. A total of 5 U of DpnI restriction enzyme (2% of total volume in PCR tube) was added and incubated for 37 °C overnight. PCR products were transformed using Esherichia coli DH5α competent cells, amplified, and purified. DNA Sanger sequencing (Eurofins) was then used to confirm if PCR had been successful.
Transient Transfection
Reverse transfection was used to transiently transfect HEK293 cells using Lipofectamine 3000 (Thermo Fisher), using the method provided by the manufacturer. Transfections were performed such that each well contained 150 ng of the D2 WT receptor and 50 ng of the pGlo-SensorTM-22F cAMP protein sensor with lipofectamine added in a 1:1.5 w:v ratio, respectively. A total of 50 μL of this mix was added to a poly-d-lysine (Sigma-Aldrich) coated F white clear-bottom plate (Greiner Bio-One). To this, 100 μL of HEK293 cells at a viable cell density of 75 000 cells was then added. Plates were then incubated in a 5% CO2 atmosphere at 37 °C for 24 h prior to performing intracellular cAMP accumulation assays.
Intracellular cAMP Accumulation Assays
Twenty-four hours post-transfection, the cell culture media were removed slowly, minimizing disruption to attached cells adhered to the bottom of the wells. Cells were initially washed using HBSS-based cAMP assay buffer (pH 7.4). Thereafter, cAMP buffer supplemented with firefly d-luciferin (0.45 mg/mL; NanoLight Technologies) was added (90 μL for functionality assays or 80 μL for testing the PAM assays). The plate was then left to pre-equilibrate in the dark at 28 °C for 1 h. During this time, the CLARIOstar PLUS (BMG Labtech) was set to 28 °C. Remaining cAMP buffer was used to prepare ascorbic acid (0.01% w:v) and then further supplemented with forskolin (7.5 μM). This was used to prepare dopamine dilutions. The allosteric modulator, (1) was custom synthesized by Enamine and resuspended in DMSO to a stock concentration of 10 mM and then aliquoted. Prior to assays, dilutions were prepared in filtered cAMP buffer absent of ascorbic acid or forskolin. Postequilibration, 10 μL of the allosteric modulator was added to the wells and left to equilibrate for a further 15 min. Bioluminescence readings were then conducted to measure basal luminescence signal (∼6–10 cycles) prior to agonist addition. Upon agonist addition (10 μL), luminescence readings were taken for ∼1 h. For functionality assays, postequilibration, basal luminescence reads were performed, followed by agonist addition (10 μL) only. Variability in the luciferase signal was taken into consideration by using the average of the last three stable basal luminescence reads to normalize the response of each well. GraphPad Prism 9.0 was used to plot data.
Cross-titration curves of the UCB compound (30 μM, 10 μM, 3 μM, 1 μM, 0.3 μM, and 0 μM) were performed to calculate the Kb value at the WT, L94W, and W413A. Luminescence readings were performed for ∼30 min. A nonlinear regression analysis fit GraphPad Prism 9.0 was used to calculate the Kb value by using a nonlinear regression analysis fit.
Immunofluorescence Assays
HEK293 cells were grown on coverslips and transiently transfected using polyethylenimine (PEI). Prior to fixation they were rinsed 3× with PBS and fixed at room temperature for 10 min with 4% v/v paraformaldehyde. The cells were then permeabilized using Triton X-100 (0.2%) in PBS buffer 1 min followed by 1× wash with PBS. Then a quenching of aldehyde step was performed to reduce background with NH4Cl (50 mM, 15 min). The coverslips were then washed 1× for 5 min in PBS and blocked for 1 h at room temperature in 5% w/v BSA. The coverslips were subsequently washed in PBS 1× and primary (rabbit D2, dopamine receptor 2 antibody) diluted in 5% w/v BSA added at 1:200 overnight at 4 °C. The next morning the coverslips were washed again in PBS 3×, and the secondary (goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 568) was added at 1:1000 in 5% w/v BSA for 1 h at room temperature. The cells were washed again 3× in PBS and then mounted using ProLong Gold antifade reagent with DAPI and imaged using a Zeiss LSM 880 laser scanning confocal microscope.
Acknowledgments
This work was supported by the European Union’s Horizon 2020 Research and Innovation Programme under the Marie-Skłodowska-Curie Grant Agreement No. 748830 (awarded to A.C.) and the Biotechnology and Biosciences Research Council (BBSRC) Responsive Mode Award BB/R007101/1 to I.G.T., and BB/R006946/1, Medical Research Council (MRC) Response Mode Award MR/S008608/1, and Barts Charity Award MRC0227 to P.J.M. This project made use of computational time on Kelvin-2 (Grant No. EP/T022175/1) and ARCHER2 granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (hecbiosim.ac.uk), supported by EPSRC (Grant No. EP/R029407/1). I.G.T. and P.J.M. participate in the European COST Action CA18133 (ERNEST). We thank Prof Graeme Milligan for several helpful discussions and suggestions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00802.
Author Present Address
§ Department of Chemical, Pharmaceutical and Agricultural Sciences, University of Ferrara, 44121, Ferrara, Italy
Author Contributions
(A.C.): Led computational chemistry effort, data curation, software, formal analysis, validation, visualization, methodology, writing - review and editing. (A.K.G.): Led molecular pharmacology effort, data curation, formal analysis, and visualization. (T.D.) Data curation, formal analysis, and visualization. (D.S.K.): Validation and formal analysis. (G.C.): Data curation and formal analysis. (P.J.M.): Resources, supervision, funding acquisition, writing - review and editing. (I.G.T.): Conceptualization, resources, data curation, software, supervision, funding acquisition, validation, methodology, writing - original draft, project administration, writing - review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discovery 2017, 16 (12), 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Marti-Solano M.; Babu M. M.; Sexton P. M. Mechanisms of Signalling and Biased Agonism in G Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol. 2018, 19 (10), 638–653. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- Kenakin T. G Protein Coupled Receptors as Allosteric Proteins and the Role of Allosteric Modulators. J. Recept. Signal Transduction Res. 2010, 30 (5), 313–321. 10.3109/10799893.2010.503964. [DOI] [PubMed] [Google Scholar]
- Gentry P. R.; Sexton P. M.; Christopoulos A. Novel Allosteric Modulators of G Protein-Coupled Receptors. J. Biol. Chem. 2015, 290 (32), 19478–19488. 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P.; Sexton P. M.; Christopoulos A. Allosteric Modulation of G Protein-Coupled Receptors: A Pharmacological Perspective. Neuropharmacology 2011, 60 (1), 24–35. 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Thal D. M.; Glukhova A.; Sexton P. M.; Christopoulos A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018, 559 (7712), 45–53. 10.1038/s41586-018-0259-z. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Oswald C.; Marshall F. H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38 (9), 837–847. 10.1016/j.tips.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Kruse A. C.; Ring A. M.; Manglik A.; Hu J.; Hu K.; Eitel K.; Hubner H.; Pardon E.; Valant C.; Sexton P. M.; Christopoulos A.; Felder C. C.; Gmeiner P.; Steyaert J.; Weis W. I.; Garcia K. C.; Wess J.; Kobilka B. K. Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504 (7478), 101–106. 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Masoudi A.; Kahsai A. W.; Huang L.-Y.; Pani B.; Staus D. P.; Shim P. J.; Hirata K.; Simhal R. K.; Schwalb A. M.; Rambarat P. K.; Ahn S.; Lefkowitz R. J.; Kobilka B. Mechanism of B2AR Regulation by an Intracellular Positive Allosteric Modulator. Science 2019, 364 (6447), 1283–1287. 10.1126/science.aaw8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Qin L.; Zacarias N. V. O.; de Vries H.; Han G. W.; Gustavsson M.; Dabros M.; Zhao C.; Cherney R. J.; Carter P.; Stamos D.; Abagyan R.; Cherezov V.; Stevens R. C.; IJzerman A. P.; Heitman L. H.; Tebben A.; Kufareva I.; Handel T. M. Structure of CC Chemokine Receptor 2 with Orthosteric and Allosteric Antagonists. Nature 2016, 540 (7633), 458–461. 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K.; Bruenle S.; Weinert T.; Guba W.; Muehle J.; Miyazaki T.; Weber M.; Furrer A.; Haenggi N.; Tetaz T.; Huang C.-Y.; Mattle D.; Vonach J.-M.; Gast A.; Kuglstatter A.; Rudolph M. G.; Nogly P.; Benz J.; Dawson R. J.P.; Standfuss J. Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 2019, 178 (5), 1222–1230. 10.1016/j.cell.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C.; Rappas M.; Kean J.; Dore A. S.; Errey J. C.; Bennett K.; Deflorian F.; Christopher J. A.; Jazayeri A.; Mason J. S.; Congreve M.; Cooke R. M.; Marshall F. H. Intracellular Allosteric Antagonism of the CCR9 Receptor. Nature 2016, 540 (7633), 462–465. 10.1038/nature20606. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Gao Z.-G.; Zhang K.; Kiselev E.; Crane S.; Wang J.; Paoletta S.; Yi C.; Ma L.; Zhang W.; Han G. W.; Liu H.; Cherezov V.; Katritch V.; Jiang H.; Stevens R. C.; Jacobson K. A.; Zhao Q.; Wu B. Two Disparate Ligand-Binding Sites in the Human P2Y 1 Receptor. Nature 2015, 520 (7547), 317–321. 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z.; Yan W.; Chapman K.; Ramesh K.; Ferrell A. J.; Yin J.; Wang X.; Xu Q.; Rosenbaum D. M. Structure of an Allosteric Modulator Bound to the CB1 Cannabinoid Receptor. Nat. Chem. Biol. 2019, 15 (12), 1199–1205. 10.1038/s41589-019-0387-2. [DOI] [PubMed] [Google Scholar]
- Robertson N.; Rappas M.; Doré A. S.; Brown J.; Bottegoni G.; Koglin M.; Cansfield J.; Jazayeri A.; Cooke R. M.; Marshall F. H. Structure of the Complement C5a Receptor Bound to the Extra-Helical Antagonist NDT9513727. Nature 2018, 553 (7686), 111–114. 10.1038/nature25025. [DOI] [PubMed] [Google Scholar]
- Liu H.; Kim H. R.; Deepak R. K.; Wang L.; Chung K. Y.; Fan H.; Wei Z.; Zhang C. Orthosteric and Allosteric Action of the C5a Receptor Antagonists. Nat. Struct. Mol. Biol. 2018, 25 (6), 472–481. 10.1038/s41594-018-0067-z. [DOI] [PubMed] [Google Scholar]
- Hollenstein K.; Kean J.; Bortolato A.; Cheng R. K.; Doré A. S.; Jazayeri A.; Cooke R. M.; Weir M.; Marshall F. H. Structure of Class B GPCR Corticotropin-Releasing Factor Receptor 1. Nature 2013, 499 (7459), 438–443. 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Lu J.; Byrne N.; Wang J.; Bricogne G.; Brown F. K.; Chobanian H. R.; Colletti S. L.; Di Salvo J.; Thomas-Fowlkes B.; Guo Y. Structural Basis for the Cooperative Allosteric Activation of the Free Fatty Acid Receptor GPR40. Nature Structural & Molecular Biology 2017, 24 (7), 570–577. 10.1038/nsmb.3417. [DOI] [PubMed] [Google Scholar]
- Jazayeri A.; Dore A. S.; Lamb D.; Krishnamurthy H.; Southall S. M.; Baig A. H.; Bortolato A.; Koglin M.; Robertson N. J.; Errey J. C.; Andrews S. P.; Teobald I.; Brown A. J. H.; Cooke R. M.; Weir M.; Marshall F. H. Extra-Helical Binding Site of a Glucagon Receptor Antagonist. Nature 2016, 533 (7602), 274–277. 10.1038/nature17414. [DOI] [PubMed] [Google Scholar]
- Song G.; Yang D.; Wang Y.; de Graaf C.; Zhou Q.; Jiang S.; Liu K.; Cai X.; Dai A.; Lin G.; Liu D.; Wu F.; Wu Y.; Zhao S.; Ye L.; Han G. W.; Lau J.; Wu B.; Hanson M. A.; Liu Z.-J.; Wang M.-W.; Stevens R. C. Human GLP-1 Receptor Transmembrane Domain Structure in Complex with Allosteric Modulators. Nature 2017, 546 (7657), 312–315. 10.1038/nature22378. [DOI] [PubMed] [Google Scholar]
- Bueno A. B.; Sun B.; Willard F. S.; Feng D.; Ho J. D.; Wainscott D. B.; Showalter A. D.; Vieth M.; Chen Q.; Stutsman C.; Chau B.; Ficorilli J.; Agejas F. J.; Cumming G. R.; Jiménez A.; Rojo I.; Kobilka T. S.; Kobilka B. K.; Sloop K. W. Structural Insights into Probe-Dependent Positive Allosterism of the GLP-1 Receptor. Nat. Chem. Biol. 2020, 16 (10), 1105–1110. 10.1038/s41589-020-0589-7. [DOI] [PubMed] [Google Scholar]
- Cheng R. K.; Fiez-Vandal C.; Schlenker O.; Edman K.; Aggeler B.; Brown D. G.; Brown G. A.; Cooke R. M.; Dumelin C. E.; Doré A. S. Structural Insight into Allosteric Modulation of Protease-Activated Receptor 2. Nature 2017, 545 (7652), 112–115. 10.1038/nature2230. [DOI] [PubMed] [Google Scholar]
- Liu X.; Kaindl J.; Korczynska M.; Stoßel A.; Dengler D.; Stanek M.; Hubner H.; Clark M. J.; Mahoney J.; Matt R. A.; Xu X.; Hirata K.; Shoichet B. K.; Sunahara R. K.; Kobilka B. K.; Gmeiner P. An Allosteric Modulator Binds to a Conformational Hub in the β 2 Adrenergic Receptor. Nat. Chem. Biol. 2020, 16, 749–755. 10.1038/s41589-020-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. R.; Lee C. T.; Durrant J. D.; Malmstrom R. D.; Feher V. A.; Amaro R. E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116 (11), 6370–6390. 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanakota P.; Carlson H. A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics: Miniperspective. J. Med. Chem. 2016, 59 (23), 10383–10399. 10.1021/acs.jmedchem.6b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P.; Hancock J. F.; Gorfe A. A. Binding Hotspots on K-ras: Consensus Ligand Binding Sites and Other Reactive Regions from Probe-based Molecular Dynamics Analysis. Proteins: Struct., Funct., Genet. 2015, 83 (5), 898–909. 10.1002/prot.24786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkaraju S. K.; Yu W.; Raman E. P.; Hershfeld A. V.; Fang L.; Deshpande D. A.; MacKerell A. D. Jr Mapping Functional Group Free Energy Patterns at Protein Occluded Sites: Nuclear Receptors and G-Protein Coupled Receptors. J. Chem. Inf. Model. 2015, 55 (3), 700–708. 10.1021/ci500729k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Jo S.; Lakkaraju S. K.; Lind C.; Yu W. Identification and Characterization of Fragment Binding Sites for Allosteric Ligand Design Using the Site Identification by Ligand Competitive Saturation Hotspots Approach (SILCS-Hotspots). Biochim. Biophys. Acta, Gen. Subj. 2020, 1864 (4), 129519. 10.1016/j.bbagen.2020.129519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Garcia D.; Barril X. Molecular Simulations with Solvent Competition Quantify Water Displaceability and Provide Accurate Interaction Maps of Protein Binding Sites. J. Med. Chem. 2014, 57 (20), 8530–8539. 10.1021/jm5010418. [DOI] [PubMed] [Google Scholar]
- Bakan A.; Nevins N.; Lakdawala A. S.; Bahar I. Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8 (7), 2435–2447. 10.1021/ct300117j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ung P. M. U.; Ghanakota P.; Graham S. E.; Lexa K. W.; Carlson H. A. Identifying Binding Hot Spots on Protein Surfaces by Mixed-Solvent Molecular Dynamics: HIV-1 Protease as a Test Case. Biopolymers 2016, 105 (1), 21–34. 10.1002/bip.22742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posokhov Y. O.; Kyrychenko A. Effect of Acetone Accumulation on Structure and Dynamics of Lipid Membranes Studied by Molecular Dynamics Simulations. Comput. Biol. Chem. 2013, 46, 23–31. 10.1016/j.compbiolchem.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Odinokov A.; Ostroumov D. Structural Degradation and Swelling of Lipid Bilayer under the Action of Benzene. J. Phys. Chem. B 2015, 119 (48), 15006–15013. 10.1021/acs.jpcb.5b09420. [DOI] [PubMed] [Google Scholar]
- Reigada R. Atomistic Study of Lipid Membranes Containing Chloroform: Looking for a Lipid-Mediated Mechanism of Anesthesia. PLoS One 2013, 8 (1), e52631. 10.1371/journal.pone.0052631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X.; Raniolo S.; Limongelli V.; Xu Y. The Molecular Mechanism Underlying Ligand Binding to the Membrane-Embedded Site of a G-Protein-Coupled Receptor. J. Chem. Theory Comput. 2018, 14 (5), 2761–2770. 10.1021/acs.jctc.8b00046. [DOI] [PubMed] [Google Scholar]
- Smith R. D.; Lu J.; Carlson H. A. Are There Physicochemical Differences between Allosteric and Competitive Ligands?. PLoS Comput. Biol. 2017, 13 (11), e1005813. 10.1371/journal.pcbi.1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zheng M.; Huang Z.; Liu X.; Zhou H.; Chen Y.; Shi T.; Zhang J. Toward Understanding the Molecular Basis for Chemical Allosteric Modulator Design. J. Mol. Graphics Modell. 2012, 38, 324–333. 10.1016/j.jmgm.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Ciancetta A.; O’Connor R. D.; Paoletta S.; Jacobson K. A. Demystifying P2Y1 Receptor Ligand Recognition through Docking and Molecular Dynamics Analyses. J. Chem. Inf. Model. 2017, 57 (12), 3104–3123. 10.1021/acs.jcim.7b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Rosés M.; Matsoukas M.-T.; Caltabiano G.; Cordomí A. Ligand-Triggered Structural Changes in the M2 Muscarinic Acetylcholine Receptor. J. Chem. Inf. Model. 2018, 58 (5), 1074–1082. 10.1021/acs.jcim.8b00108. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Sealfon S. C., Ed.; Receptor Molecular Biology; Academic Press, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Schmidtke P.; Bidon-Chanal A.; Luque F. J.; Barril X. MDpocket: Open-Source Cavity Detection and Characterization on Molecular Dynamics Trajectories. Bioinformatics 2011, 27 (23), 3276–3285. 10.1093/bioinformatics/btr550. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinf. 2009, 10, 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke P.; Le Guilloux V.; Maupetit J.; Tufféry P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–W589. 10.1093/nar/gkq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke P.; Barril X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53 (15), 5858–5867. 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- Wood M.; Ates A.; Andre V. M.; Michel A.; Barnaby R.; Gillard M. In Vitro and In Vivo Identification of Novel Positive Allosteric Modulators of the Human Dopamine D2 and D3 Receptor. Mol. Pharmacol. 2016, 89 (2), 303–312. 10.1124/mol.115.100172. [DOI] [PubMed] [Google Scholar]
- Wang S.; Che T.; Levit A.; Shoichet B. K.; Wacker D.; Roth B. L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug resperidone. Nature 2018, 555 (7695), 269–273. 10.1038/nature25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y.; Krumm B.; Zhang H.; Zhou X. E.; Wang Y.; Huang X.-P.; Liu Y.; Cheng X.; Jiang Y.; Jiang H.; Zhang C.; Yi W.; Roth B. L.; Zhang Y.; Xu H. E. Mechanism of Dopamine Binding and Allosteric Modulation of the Human D1 Dopamine Receptor. Cell Res. 2021, 31, 593. 10.1038/s41422-021-00482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M.; Fasciani I.; Marampon F.; Maggio R.; Scarselli M. The First Negative Allosteric Modulator for Dopamine D2 and D3 Receptors, SB269652 May Lead to a New Generation of Antipsychotic Drugs. Mol. Pharmacol. 2017, 91 (6), 586–594. 10.1124/mol.116.107607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J. R.; Chubukov P.; Liu W.; Canals M.; Cherezov V.; Abagyan R.; Stevens R. C.; Katritch V. Structure-Based Ligand Discovery Targeting Orthosteric and Allosteric Pockets of Dopamine Receptors. Mol. Pharmacol. 2013, 84 (6), 794–807. 10.1124/mol.113.088054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe T. J.; Zarzycka B.; Lim H. D.; Kellam B.; Mistry S. N.; Katrich V.; Scammells P. J.; Lane J. R.; Capuano B. A Thieno[2,3- d]Pyrimidine Scaffold Is a Novel Negative Allosteric Modulator of the Dopamine D2 Receptor. J. Med. Chem. 2019, 62 (1), 174–206. 10.1021/acs.jmedchem.7b01565. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Michino M.; Verma R. K.; Herenbrink C. K.; Shonberg J.; Kopinathan A.; Scammells P. J.; Capuano B.; Thal D. M.; Javitch J. A.; Christopoulos A.; Shi L.; Lane J. R. The Structural Determinants of the Bitopic Binding Mode of a Negative Allosteric Modulator of the Dopamine D2 Receptor. Biochem. Pharmacol. 2018, 148, 315–328. 10.1016/j.bcp.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free R. B.; Chun L. S.; Moritz A. E.; Miller B. N.; Doyle T. B.; Conroy J. L.; Padron A.; Meade J. A.; Xiao J.; Hu X.; Dulcey A. E.; Han Y.; Duan L.; Titus S.; Bryant-Genevier M.; Barnaeva E.; Ferrer M.; Javitch J. A.; Beuming T.; Shi L.; Southall N. T.; Marugan J. J.; Sibley D. R. Discovery and Characterization of a G Protein-Biased Agonist That Inhibits β-Arrestin Recruitment to the D2 Dopamine Receptor. Mol. Pharmacol. 2014, 86 (1), 96–105. 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Butler K. V.; Kelly B.; Rechsteiner K.; Karpiak J.; Betz R. M.; Kormos B. L.; Shoichet B. K.; Dror R. O.; Jin J.; Roth B. L. Structure-Inspired Design of β-Arrestin-Biased Ligands for Aminergic GPCRs. Nat. Chem. Biol. 2018, 14 (2), 126–134. 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield A. E.; Mason J. S.; Vajda S.; Keserű G. M. Analysis of Tractable Allosteric Sites in G Protein-Coupled Receptors. Sci. Rep. 2019, 9 (1), 1–14. 10.1038/s41598-019-42618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Nichols S. E.; McCammon J. A. Mapping of Allosteric Druggable Sites in Activation-Associated Conformers of the M2Muscarinic Receptor. Chem. Biol. Drug Des. 2014, 83 (2), 237–246. 10.1111/cbdd.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliman A. D.; Miao Y.; McCammon J. A. Mapping the Allosteric Sites of the A2A Adenosine Receptor. Chem. Biol. Drug Des. 2018, 91 (1), 5–16. 10.1111/cbdd.13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2018-4: Maestro; Schrödinger, LLC: New York, NY, 2018.
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22 (5), 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Martínez L.; Andrade R.; Birgin E. G.; Martínez J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30 (13), 2157–2164. 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- Doerr S.; Harvey M. J.; Noé F.; De Fabritiis G. HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12 (4), 1845–1852. 10.1021/acs.jctc.6b00049. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone φ, ψ and Side-Chain X1 and X2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114 (23), 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2009, 31 (4), 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Hardy D. J.; Maia J. D.; Stone J. E.; Ribeiro J. V.; Bernardi R. C.; Buch R.; Fiorin G.; Hénin J.; Jiang W. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153 (4), 044130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OpenEye Toolkits 2020.1.0; OpenEye Scientific Software: Santa Fe, NM, http://www.eyesopen.com. [Google Scholar]
- Farid R.; Day T.; Friesner R. A.; Pearlstein R. A. New Insights about HERG Blockade Obtained from Protein Modeling, Potential Energy Mapping, and Docking Studies. Bioorg. Med. Chem. 2006, 14 (9), 3160–3173. 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49 (2), 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118 (45), 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.