

Abstract
Community efforts in the computational molecular sciences (CMS) are evolving toward modular, open, and interoperable interfaces that work with existing community codes to provide more functionality and composability than could be achieved with a single program. The Quantum Chemistry Common Driver and Databases (QCDB) project provides such capability through an application programming interface (API) that facilitates interoperability across multiple quantum chemistry software packages. In tandem with the Molecular Sciences Software Institute and their Quantum Chemistry Archive ecosystem, the unique functionalities of several CMS programs are integrated, including CFOUR, GAMESS, NWChem, OpenMM, Psi4, Qcore, TeraChem, and Turbomole, to provide common computational functions, i.e., energy, gradient, and Hessian computations as well as molecular properties such as atomic charges and vibrational frequency analysis. Both standard users and power users benefit from adopting these APIs as they lower the language barrier of input styles and enable a standard layout of variables and data. These designs allow end-to-end interoperable programming of complex computations and provide best practices options by default.
I. INTRODUCTION
The number of quantum chemistry (QC) programs is continuously increasing, building a rich spectrum of capabilities where varied levels of accuracy, performance, distributed computing, graphics processing units (GPU)-acceleration, or licensing can be obtained. While this is generally beneficial to the end user, the diversity of custom input and output makes it difficult to switch between programs without learning the vagaries of each. Even the simplest research tasks using QC programs require mastering layers of expertise. On the input side, users must know what model chemistry will treat the molecular system of interest with adequate physics in tractable time, as well as pertinent modifications like density-fitting (DF), convergence, and active space, which are all questions of scientific expertise (I-a). [Labels of non-scientific (i.e., beyond I-a) input I-x or output O-x problems enumerated here are referenced by solutions in Sec. II.] They must know the names given by a QC program to the knobs that dial up the model chemistry and modifications, a question of domain-specific-language (DSL) expertise (here, “domain” is the QC software) (I-b). They would benefit from knowing the insider best-practice knobs that select the most efficient algorithms, approximations, and implementations specialized to the model chemistry, a question of program expertise (I-c). They must know the structure of the input specification by which the QC program receives instruction, a question of formatting and DSL expertise (I-d). Last on the input side, they must know the dance of files, environment variables, and commands to launch the job, a question of program operational expertise (I-e).
On the output and analysis side, further skills are required to process the program-specific ASCII or structured data file. Users must know what strings in the output mark the desired result, a matter of DSL expertise (O-a). If the targeted quantity is not explicitly printed but is derivable, they must know the arithmetic or unit conversion, a question of QC expertise (O-b). If individual energies or derivatives are to be combined to create a more sophisticated model chemistry (e.g., basis set extrapolation,1,2 focal-point methods,3–5 G3 or HEAT procedures,6,7 or empirical correction8) or for molecular systems decomposition or perturbation [e.g., many-body expansion (MBE), counterpoise procedure,9 geometry optimization, or finite difference derivatives], users may be able to use routines built into QC programs (needing DSL expertise) but more generally must script the procedure themselves, requiring QC and programming expertise (O-c). More elaborately, they may want to combine the results with other programs—requiring recognizing and compensating for default knobs that render program results unmixable—a matter of QC and program expertise (I-f). Finally, users may hope that completed calculations can be stored and queried or even reused, matters of database expertise (O-d). Efforts to reduce non-scientific expertise burdens on the user have traditionally aggregated QC methods, geometry optimizers, and sundry procedures into vertically integrated “software silos” that, by increasing the DSL burden, risk locking users into one or a few programs. We pursue reducing the non-scientific expertise burdens on users by restructuring the QC software ecosystem while minimally disrupting longstanding, robust, and debugged computational molecular sciences (CMS) codes.
As a concrete example, in a high-accuracy spectroscopic application (see Sec. III), a user might want to include numerous small corrections, such as electron correlation effects beyond coupled-cluster (CC) through perturbative triples [CCSD(T)],10 basis set extrapolation, relativistic corrections,11 and Born–Oppenheimer (BO) diagonal corrections.12,13 The best implementation of each of these terms is not necessarily found in a single QC program. Careful users can evaluate different terms using different programs through a post-processing script to obtain a focal-point energy, but more complex procedures such as geometry optimizations14 are difficult due to tight coupling in QC programs that generally do not allow arbitrary gradients to be injected into the iterative optimizer.
Finally, in the emerging “data age” of computational chemistry, users increasingly want to treat QC results as a commodity, obtaining them on demand as part of complex workflows or generating datasets of millions of computations to use in force field (FF) parameterization, methodology assessment, machine learning (ML), or other data-driven pipelines. These users must be able to set up, execute, and extract computational results as easily as possible.
To address such challenges, the differing needs of workflows for uniform interaction with CMS codes have been separated into different layers of concern, resulting in the development of QCEngine and Quantum Chemistry Common Driver and Databases (QCDB).15
-
•
Consider a new QC practitioner learning which density functional theory (DFT) program best suits the local hardware or accessing the latest ML FF for many molecules. Such users would benefit from a uniform application programming interface (API) to evaluate these diverse capabilities without requiring knowledge of the specifics of each program’s DSL. QCEngine is designed to provide this uniform API and is an I/O runner around individual CMS codes’ core single-point capabilities. QCEngine communicates through a JavaScript Object Notation (JSON) Schema,16 denoted QCSchema, thus automatically generating program input files from a consistent and simple molecule and method specification.
-
•
Next, consider the systematic study of dipole moments at different levels of theory from different programs or a FF developer training on the many symmetry-adapted perturbation theory (SAPT) component results over thousands of molecules. These applications would benefit from output layout uniformity and programmatic access to detailed results. QCEngine covers these cases by harvesting binary, structured, or text output into standardized QCSchema fields.
-
•
Next, consider the maintainers of a CMS code whose users have been making the same formatting and incomplete input mistakes for the past decade and have been petitioning for quality-of-life features that would incur poor complexity-to-benefit ratio if implemented within the native framework and languages. These barriers to research would benefit from a shim layer in an easy and expressive language. QCDB provides a flexible input framework, helpful keyword validation, access to multijob procedures like MBE, and a place (besides documentation) to inject advice like context-dependent defaults.
-
•
Now, consider a spectroscopist modeling a molecule with a composite method or the QC beginner hoping to avoid learning multiple DSLs. These circumstances would benefit from uniformity of input and results across programs. QCDB compensates for variable defaults and conventions so that multi-program model chemistries can be safely defined and simple methods accessed interchangeably.
-
•
Finally, consider the experienced QC practitioner who writes inputs from memory and who turns keyword knobs as nimbly as organ stops but who would like to try another optimizer or an MBE procedure or not worry about capitalization and spaces today. This situation would benefit from a light hand in developing the QCSchema translation and common driver API so that existing expertise in direct interaction with CMS codes (DSL for keywords, for example) is applicable to these current projects.
In enabling uniformity at the input, output, and cross-program layers, both QCEngine and QCDB have striven to make their input predictable from customary input and to make customary output available.
Central to the ability of QCArchive17 and QCDB to provide generic I/O, driver, and database interfaces to CMS codes is a common standard QC data format. Of course, to develop such a standard information exchange format for all QC programs and to encourage its adoption by QC packages is a difficult approach for a single research group, or even a handful of research groups, to successfully prescribe to a broad developer community. However, here the Molecular Sciences Software Institute (MolSSI),18 funded by the U. S. National Science Foundation, provides a unique opportunity to sponsor community discussions and to advocate for standards. Members of our collaborative team and the codes represented have worked closely with MolSSI on their development of a QCSchema19 for quantum chemistry information exchange, and we have adopted it for QCEngine and QCDB.
There have been previous efforts to provide a unified interface to set up, drive, and analyze QC computations. For example, Newton-X20 and FMS9021–23 perform nonadiabatic dynamics computations using any of several QC programs. The Quantum Thermochemistry Calculator (QTC)24 interfaces to a handful of QC programs to provide unified thermochemistry analysis functions independent of the QC data source. Especially tailored to deal with excited state optimizations is Pysisyphus, an external optimizer that localizes stationary points on potential energy surfaces by means of intrinsic reaction coordinate (IRC) integration, chain-of-state optimization, and surface walking for several QC codes through a uniform interface.25 Among more general-purpose programs, Cuby26,27 is a uniform driver and workflow manager that works with multiple QC and force field tools. Cuby allows the combination of methods across its interfaced programs and provides mixed quantum mechanics/molecular mechanics (QM/MM) and molecular dynamics capabilities. The WebMO project is another that drives several QC programs as backends from a largely unified web portal frontend.28 Another popular tool is the Atomic Simulation Environment (ASE),29 which provides a Python interface to more than 40 QC or force field codes, along with drivers for geometry optimization and transition state searching with the nudged elastic band method and analysis and visualization functions. A recipes collection (ASR)30 supplies further spectroscopy and analysis tools. ASE and ASR are focused on solid-state computations; while molecular computations are also possible, they do not provide the level of detail required for the majority of quantum chemistry workflows. Compared to ASE, QCDB is more focused on high-accuracy quantum chemistry (providing, for example, built-in support for focal-point methods). Newer entrants to the field of computational chemistry workflow tools at the scope of QCArchive (rather than the narrower modular components QCEngine and QCDB discussed here) include AiiDA,31,32 which at present is materials focused, and ChemShell,33 which focuses on multiscale simulations. By interfacing with QCArchive, QCDB can also focus on high-throughput quantum chemistry and on creating large databases for force field parameterization and machine-learning purposes. Although not focused on running CMS codes, the cclib34,35 and HORTON36 projects also have extensive capabilities to regularize output and post-processing.
We describe the modular software built to facilitate interoperability, the community QC codes, and the technical challenges associated with an interoperability project in Sec. II. An example application demonstrating the use of multiple QC codes to perform very high accuracy computations of spectroscopic constants of some diatomic molecules is presented in Sec. III.
II. FEATURES AND DESIGN PHILOSOPHY
Discussed are the present software projects and their place in the CMS ecosystem in Sec. II A, interfaced software providing single-point energies and properties in Sec. II B, interfaced and built-in software providing more complex procedures in Sec. II C, how these are all linked by a common driver in Sec. II D, and further details about implementing interoperability in Sec. II E.
A. QCSchema and the quantum chemistry software ecosystem
The modular software components in our layered approach to QC interoperability and high-throughput computing are shown in Fig. 1. All are open-source projects, and community feedback and contributions through GitHub are welcome (links at Sec. V; QCEngine documentation includes the general process for adding a new QC program). The QCSchema19 definitions layer is foundational and encodes the community-developed data layouts and model descriptions useable in any language, from C++ to Rust to JavaScript to Fortran. Above that is the QCElemental37 data and models layer that implements QCSchema and imposes a Python language restriction to gain sophisticated validation and feature-rich models. Next is the QCEngine38 execution layer that adapts CMS codes for standardized QCSchema communication and imposes an execution environment restriction to gain easy access to many programs. Last is the QCFractal39 batch execution and database layer that imposes some calculation flexibility restrictions to gain multi-site distributed compute orchestration and provide structured-data storage and querying capabilities. [This layer, beyond the scope of the present work, addresses (O-d).] Together these compose the QCArchive Infrastructure, the Python software stack that backs the MolSSI QCArchive project.17,40 Enhancing QCEngine is the QCDB41 interoperability layer that imposes feature-registration and cross-program defaults restrictions to gain input uniformity and multi-program workflows.
FIG. 1.

Modular ecosystem around QCEngine and QCDB. QCEngine is the central, QCSchema-based QC program runner in the QCArchive Infrastructure software stack, while QCDB adds additional interoperability features atop it. User input routes to QC computations are shown as one or more turquoise boxes—“TXT” for a command-line interface, “PyAPI” for an interactive application programming interface in Python, or “JSON” for single-command QCSchema communication through command-line or Python.
QCElemental37 (see Fig. 1) provides data and utilities (like a QCSchema implementation) useable by all QC packages. For data, it exposes NIST Periodic Table and CODATA physical constants through a lightweight API and provides internally consistent unit conversion aided by the external module Pint.42 QCElemental supports multiple dataset versions for CODATA and for properties such as covalent and van der Waals radii. Additionally, QCElemental provides a Python reference implementation for the MolSSI QCSchema data layouts, including Molecule (example is given in Snippet 2), job input specification AtomicInput [examples at Figs. 2(b)–2(d)], and job output record AtomicResult. In addition to enforcing the basic key/value data layout inherent to a schema, QCElemental uses the external module Pydantic43 to collocate physics validation, serialization routines, extra helper functions (like Molecule parsing, alignment, and output formatting), and schema generation into a model for the QCSchema. Historically, many QCElemental capabilities were developed for QCDB in Psi4 and then refactored into QCElemental for broader community accessibility free from Psi4 and compiled-language dependence. QCEngine and QCDB use all the QCElemental capabilities mentioned, particularly for QCSchema communication and for uniform treatment of fragmented, ghosted, and mixed-basis molecules across differing QC program features.
SNIPPET 2.

QCSchema MOLECULE from Snippet 1. Translation described at Ref. 97.
FIG. 2.

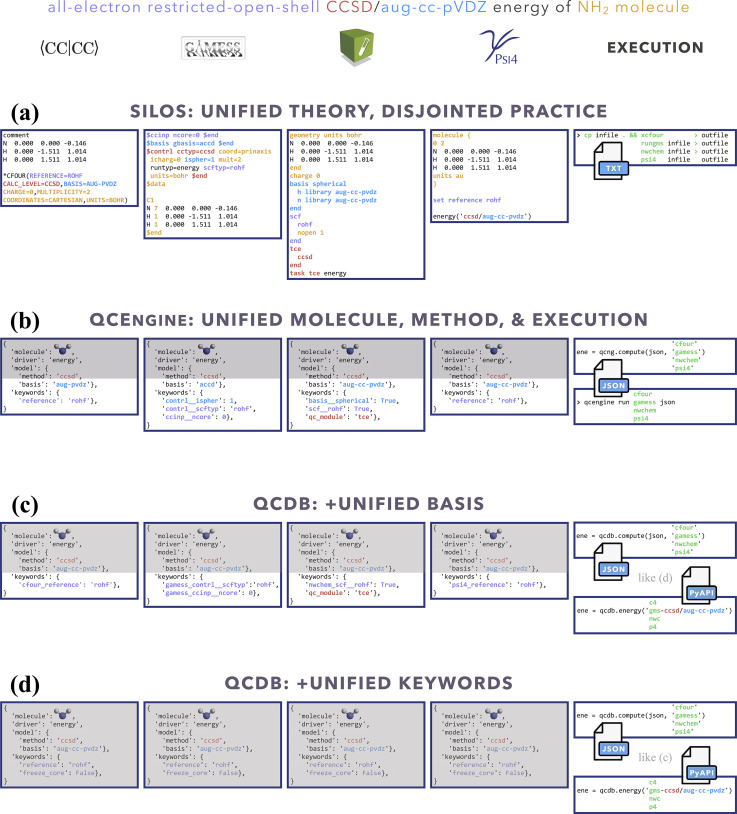
Degrees of unifying access to quantum chemical calculations illustrated through an open-shell CCSD energy computation. Black text and gray shading are aspects not requiring user knowledge of multiple DSLs. See penultimate paragraph of Sec. II A for discussion.
QCEngine38 provides a uniform execution interface whereby community CMS codes consume QCSchema AtomicInputs and emit AtomicResults via adaptors, called ProgramHarnesses. Depending on the degree of programmatic access a QC package provides, the ProgramHarness may be simple, as for a package that already provides a QCSchema interface; moderate, as for a package that supports a Python API or has serialized output, be it binary, Extensible Markup Language (XML), or JSON; or involved, as for an executable with ASCII I/O; further details may be found in Sec. II E 10. A typical ProgramHarness consists of taking an AtomicInput, translating it into input file(s) and execution conditions, executing it, collecting all useful output, parsing the results into an AtomicResult, and returning it to the user. A ProgramHarness is written to cover analytic single-point computations, namely, energies, gradients, Hessians, and properties, as discussed further in Sec. II B. Adaptors for more complicated actions are classified as ProcedureHarnesses and are discussed in Sec. II C. QCEngine additionally collects runtime data such as elapsed time, the hardware architecture of the host machine, memory consumption of the job, software environment details, and execution provenance (e.g., program, version, and module). As suggested by Fig. 1, adaptors written in QCDB have been migrated to QCEngine so that both projects access more QC codes and share the maintenance and development burden.
QCDB41 supplements QCEngine’s program and procedure capabilities with interoperability-enhanced ProgramHarnesses and multi-program procedures; furthermore, it links QCEngine calls into an interactive driver interface. From the user’s viewpoint, this layered approach to uniform QC computation is shown in Fig. 2 by an open-shell CCSD single-point energy. Running a QC code directly, as in Fig. 2(a), requires considerable DSL knowledge for method, basis, and keywords, not to mention details of layout and execution; essentially only the geometry (black text) is uniform. By molding the text inputs of Fig. 2(a) into the QCSchema data layout Fig. 2(b), QCEngine unifies the gray-shaded fields but still requires DSL from multiple codes. QCDB imposes more dependencies, like its own basis set library and utilities, to allow uniform basis specification and molecule symmetry as in Fig. 2(c). By imposing keyword registration and precedence logic, QCDB can provide the uniform and single-DSL input of Fig. 2(d). In practice, QCDB harnesses are minimal wrappers around QCEngine harnesses.
By choosing an entry point (software component in Fig. 1) and interface (CLI, Python API, JSON), external projects can satisfy a number of interoperability use cases: convention for data layout (stop after QCSchema), molecule string parsing (stop after QCElemental), uniform CMS execution (stop after QCEngine), tolerant Python interface to single venerable CMS code (QCDB), or multicode workflows (QCDB).
B. Program capabilities
For several community codes or programs [Fig. 3(i); not comprehensive] capable of computing analytic energies, gradients, or Hessians, the authors have written QCSchema adaptors for QCEngine known as ProgramHarnesses [Fig. 3(ii)]. The primary returns can be full scalars or arrays, as for most QC methods, or partial, as for dispersion corrections. So long as program communication fits into the AtomicResult data layout, semi-empirical and molecular mechanics programs can also formulate QCEngine adaptors. A summary of interfaced codes can be seen in Table I. QCDB asserts greater control over codes to assure consistent output values, so its capabilities are centered on CFOUR, GAMESS, NWChem, Psi4, and select partial calculators [Fig. 3(iii)]. Note that output harvesting capabilities (results available programmatically as opposed to text files) may lag behind those for input execution. A test suite that ensures matching values can be extracted from different programs has been established for both QCEngine and QCDB to document differing conventions (e.g., canonicalization for ROHF CC, all-electron vs frozen-core). Uncovered incorrect values or missing properties have been reported to the code developers for further investigation.
FIG. 3.

Layout and access pattern between selected existing and planned (marked by *) community quantum chemistry codes, QCEngine, and QCDB. Community codes (i) in a variety of languages are wrapped in QCSchema input/output by a QCEngine harness (ii) and (iv), which may be light (if the code has an API or structured output) or heavy (if only text output available). The QCDB harnesses (iii) and (v) add unifying and ease-of-use layers atop the QCEngine calls. Whereas analytic energies and derivatives are classified as programs (ii) and (iii) and call QC codes directly, multi-stage and post-processing jobs are written as procedures (iv) and (v) for composability and distributability and call programs in turn. The QCDB driver provides API access to both sets. Labels (a)–(d) correspond to the stages of unified input in Fig. 2.
TABLE I.
Interfaced programs in QCEngine and QCDB. For each, availability of one or
more methods for energy (E), gradient (G), and Hessian (H) is shown, as well as
collection of properties (e.g., one-electron energy or dipole) and wavefunction
quantities (e.g., number of basis functions and orbitals). Symbols are present
(), absent
( ), or
inapplicable (). Non-QC programs are not suitable for QCDB. Program I/O is handled
primarily through QCSchema API (Q), API (A), structured XML, JSON, binary
(S), or text (T).
), or
inapplicable (). Non-QC programs are not suitable for QCDB. Program I/O is handled
primarily through QCSchema API (Q), API (A), structured XML, JSON, binary
(S), or text (T).
| CMS program | QCEngine | QCDB | Cite | I/O | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E | G | H | Prop. | Wfn | E | G | H | |||
| Quantum chemistry | ||||||||||
| ADCC |
|

|
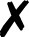
|

|

|

|

|

|
44 and 45 | A |
| CFOUR |

|

|

|

|

|

|

|

|
46 | TS |
| GAMESS |

|

|
|

|
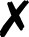
|

|
|
|
47 | T |
| Molpro |

|

|

|

|
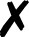
|
|
|
|
48 and 49 | S |
| MRChem |

|

|

|

|
|
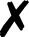
|
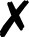
|
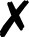
|
50 and 51 | S |
| NWChem |

|

|

|

|

|

|

|

|
52 | T |
| Psi4 |

|

|

|

|

|

|

|

|
53 | Q |
| Q-Chem |

|

|

|
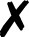
|
|
|
|
|
54 | TS |
| Qcore |

|

|

|

|

|

|

|

|
55 | S |
| TeraChem |

|

|

|

|
|

|
|
|
56 and 57 | Q,T |
| Turbomole |

|

|

|
|
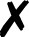
|
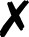
|
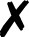
|
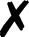
|
58 and 59 | T |
| Semi-empirical | ||||||||||
| MOPAC |

|

|

|

|
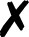
|
60 | T | |||
| xtb |

|

|

|

|

|
61 | Q | |||
| Molecular mechanics | ||||||||||
| OpenMM |

|

|

|

|
62 | A | ||||
| RDKit |

|

|

|

|
63 | A | ||||
| Analytical corrections | ||||||||||
| DFTD3 |

|

|

|

|

|

|

|
8 and 64 | T | |
| DFTD4 |

|

|
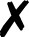
|
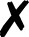
|

|

|

|
65 and 66 | Q | |
| gCP |
|

|
|

|

|

|

|
67 and 68 | T | |
| MP2D |

|

|
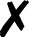
|
|

|

|
|
69 and 70 | T | |
| Machine learning inference | ||||||||||
| TorchANI |

|

|

|

|
71–73 | A | ||||
1. ADCC
The interface to ADCC allows for computations of excited states based on the algebraic-diagrammatic construction (ADC) scheme for the polarization propagator. Several methods are available, including ADC(2), ADC(2)-x, and ADC(3), together with the respective core-valence separation (CVS) and spin-flip variants. For all aforementioned methods, excitation energies and properties are accessible. The interface uses Psi4 to compute the SCF reference state first and then calls adcc via its Python API. A minimum adcc v0.15.1 is required.
2. CFOUR
Many CFOUR features are available to both QCEngine and QCDB, including most ground-state many-body perturbation theory and coupled-cluster energies, gradients, and Hessians: Hartree–Fock, MP2, MP3, MP4, CCSD, CCSD(T) with RHF, UHF, and ROHF references. Excited states are available for running but not parsing. Special features include CC with quadruple excitations through the NCC module, the ability to compute the diagonal Born–Oppenheimer correction using coupled-cluster theory, and, after revision, second-order vibrational perturbation theory (VPT2) (see Sec. II C 6). The interface generates text input and collects mixed text and binary output. A minimum CFOUR v2.0 is required.
3. GAMESS
The GAMESS interface for QCEngine and QCDB provides Hartree–Fock, DFT, MP2, and coupled-cluster methods. Special features include full configuration interaction. In the future, the GAMESS interface will also provide effective fragment potential (EFP) capability through potential file generation (see Sec. II C 7) and running pure EFP calculations on molecular clusters, energy ("gms-efp"). A particular complication for GAMESS is the controlled molecule and custom basis syntax, which led to QCDB feeding only symmetry-unique atoms and their full basis sets into the GAMESS input file. As QCEngine does not have symmetry capabilities, QCEngine-based GAMESS calculations are restricted to C1. The interface generates text input and collects text output. The harness has been tested with the GAMESS 2017 R1 version.
4. Molpro
Energies and gradients are available in QCEngine from Hartree–Fock, DFT, MP2, CCSD, and CCSD(T) levels of theory, including some local methods. The interface generates text input and collects XML output. A minimum Molpro v2018.1 is required.
5. MRChem
Thanks to a harness to the MRChem software package, quasi-exact energies and selected properties in the multiwavelet, multiresolution basis are available with QCEngine. MRChem provides an efficient implementation for Hartree–Fock and DFT. Electric dipoles, quadrupoles, static and frequency-dependent polarizabilities, magnetizabilities, and NMR shielding constants are available. At variance with GTO-based quantum chemical software packages, the basis used in MRChem is adaptively refined: thanks to the multiwavelet framework, these results are exact to within the user-requested precision.74 As a practical consequence, only the method keyword is required to define an input model to MRChem. JSON files are used to handle communication between QCEngine and MRChem. The harness can leverage the hybrid MPI/OpenMP parallelization of MRChem, provided suitable resources are available. A minimum MRChem v1.0.0 is required.
6. NWChem
The NWChem interface for QCEngine and QCDB provides a large selection of the quantum mechanical methods available, including Hartree–Fock, DFT, MP2, and coupled-cluster methods [both the code automatically derived and implemented with the Tensor Contraction Engine75 (TCE) and the hand-coded implementations, where available]. Additional calculations available in the TCE include configuration interaction through single, doubles, triples, and quadruples level of theory and MBPT methods through the fourth order. Special features include CCSDTQ energies, excited states through equation of motion (EOM) coupled-cluster energies, and relativistic approximations. The interface generates text input and collects text output. The harness has been tested with NWChem v6.6 and v7.0.
7. Psi4
Essentially, all Psi4 features are available to QCEngine and QCDB, as Psi4 communicates natively in QCSchema (psi4 – qcschema in.json) and QCDB began as the Psi4 driver. These include conventional and density-fitted Hartree–Fock, DFT, MP2, and coupled-cluster methods. Special features are symmetry-adapted perturbation theory, coupled-cluster response properties, density-fitted CCSD(T) gradients, and optimized-orbital MP2, MP2.5, and MP3 energies and gradients. Wavefunction information is returned in QCSchema format. The interface generates JSON (QCSchema) input and collects JSON output. A minimum Psi4 v1.3 is required for QCEngine and v1.4 for QCDB.
8. Q-Chem
Energies, gradients, Hessians, and some properties are available in QCEngine at the SCF (Hartree–Fock and tens of DFT functionals) and MP2 levels (both conventional and density-fitted). The interface generates text input and collects mixed text and binary output. A minimum Q-Chem v5.1 is required.
9. Qcore
Energies, gradients, and Hessians are available in QCEngine from Hartree–Fock, DFT, and extended tight-binding (xTB). Qcore along with Psi4 are the two programs that can return wavefunction information in QCSchema. The interface generates JSON input and collects JSON output. A minimum of Qcore v0.7.1 is required.
10. TeraChem
TeraChem features two modes for driving computations via QCEngine: a standard text interface and a typed Protocol Buffers76 interface. The former generates text input and collects text output to provide energies and gradients from Hartree–Fock and DFT levels of theory. A minimum TeraChem v1.5 is required.
TeraChem’s Protocol Buffers (TCPB) server57 interface offers a second way to drive computations using QCEngine. It provides energies and gradients from Hartree–Fock and DFT levels of theory, molecular properties including dipoles, charges, and spins, and limited wavefunction data including alpha- and beta-spin orbitals and orbital occupations. The TCPB interface also accelerates calculations by performing GPU initialization routines once at server startup. As a result, subsequent computations can begin instantaneously, thereby providing substantial speed-up for small systems (∼10 heavy atoms) and minor speed-up for medium systems (∼100 atoms).77 The TCPB interface requires the installation of an additional Python package tcpb78 minimum v0.7.0 to power the QCEngine integration. Subsequent updates to the tcpb package will expand the set of properties and wavefunction data available from TeraChem via QCEngine.
11. Turbomole
Energies, gradients, and Hessians are available in QCEngine for Hartree–Fock, many DFT functionals, and define-fitted MP2, MP3, MP4, and CC2. Turbomole’s interactive define function for processing input proved an extra challenge to integrate with QCSchema. The interface generates interactive text input and collects text output. The harness has been tested with Turbomole v7.3 and v7.4.
12. XTB
The interface uses the Python API of XTB, which provides QCSchema support, to generate JSON (QCSchema) input and collect JSON output. A minimum of XTB v6.3 is required.
13. dftd3 and dftd4
A Python API to Grimme’s dftd3 executable for computing variants of -D2 and -D3 for arbitrary QCSchema Molecule with automatic or custom parameter sets has been available in Psi4 for several years.8,79,80 This has been adapted as a ProgramHarness for QCEngine and QCDB. The interface generates text input and collects text output. A minimum of dftd3 v3.2.1 is required.
For the separate dftd4 software, the interface uses the Python API, which provides QCSchema support, to generate JSON (QCSchema) input and collect JSON output. A minimum of dftd4 v3.1 is required.
14. GCP
Energies and gradients are available for the geometrical counterpoise correction GCP program developed by Kruse and Grimme that corrects the inter- and intramolecular basis set superposition error (BSSE) in Hartree–Fock and DFT calculations.68 It also offers the GCP-part of the “3c” correction used in composite methods like HF-3c or PBEh-3c.81 The interface generates text input and collects text output. The harness was tested with gCP v2.02.
C. Procedure capabilities
Whenever a quantum chemistry work sequence takes in QC-program-agnostic energies, gradients, Hessians, or properties (i.e., AtomicResults) but requires multiple ones (e.g., a finite difference derivative) or needs additional software [e.g., EFP potentials or symmetry-adapted linear combination (SALC) coordinates] or needs to take action in multiple stages (e.g., a geometry optimizer) or could combine AtomicResults from different programs (e.g., a composite method), it is classified in QCEngine or QCDB as a procedure [see Fig. 3(iv-v)]. Procedures are implemented in a ProcedureHarness to facilitate modularity and address O-c. Because procedures act upon generalized quantities, any code interfaced with QCEngine or QCDB gets all of the applicable procedures “for free.” Together, programs and procedures are elements that can be composed into workflows both simple (e.g., opt + freq + vib) or complex as in Sec. III.
Presently available in QCEngine are the geomeTRIC, PyBerny, and (Python) OptKing geometry optimizers, the first of which has been used extensively (>380k optimizations) by the Open Force Field82 community. Presently available or anticipated (*) for QCDB are the Composite, FiniteDifference,* ManyBody, diatomic, and vib routines inherited from the Psi4 recursive driver.14 The Psi4 OptKing geometry optimizer, written in C++, has been redeveloped in Python as a more versatile tool for future development and with the independence suitable for QCDB, while resp* and CrystaLattE* have been expanded from Psi4 to work with QCDB. Procedures makefp* and vpt2* make use of specially extractable features from GAMESS and CFOUR, respectively, and require installation of the parent code. Similarly, findif retains for the short term a dependence on Psi4. Note that the full capabilities from proven software components that were once or are presently partially or fully interfaced are in the procedure descriptions below. Procedures in QCEngine and QCDB have passed through the proof-of-principle stage and are presently being reworked and expanded into the below forms; current availability is limited.
1. Geometry optimizers
To be used by QCEngine or QCDB, a geometry optimizer must be able to take an input geometry in Cartesian coordinates and to take an arbitrarily sourced gradient and produce a next-candidate geometry displacement rather than be in control of both gradient and geometry-step stages. Regrettably, this eliminates most optimizers embedded in QC programs. Some alternatives are Wang’s geomeTRIC project,83,84 which uses the TRIC coordinate system to specialize in interfragment and constrained optimizations, King’s OptKing,85 which is a conventional IRC- and TS-capable QC optimizer, and Hermann’s PyBerny,86 also a QC-focused optimizer. OptKing can apply flexible convergence criteria including those related to energy change and the maximum or root-mean-square of the gradient or displacement, and it has the most common settings for many embedded/native optimizers conveniently accessible as keywords. QCEngine presently has available geomeTRIC, PyBerny, and the Python OptKing, while QCDB only has the original C++ OptKing. After a planned driver update, all three Python optimizers will work with QCEngine and hence with QCDB. All optimizers communicate through schema, in particular, a QCSchema OptimizationInput that contains an ordinary AtomicInput as template for the gradient engine. Optimizations are called through QCEngine using qcng.compute_procedure({“input_molecule”: …, “keywords”: {“program”: “gamess”}, “input_specification”: {“model”: {“method”: “mp2”, “basis”: “6-31G”}}}, “geomeTRIC”) or qcdb.optking(“gms-mp2/6-31G”), where the latter can take as model chemistry any sensible combination of other procedures (i.e., qcdb.optking(“gms-mp2/[23]zapa-nr”, bsse_type=“cp”)).
2. vib: Harmonic vibrational analysis
The harmonic vibrational analysis routine is automatically run after any qcdb.frequency() computation.87 Taking in a Hessian matrix, the molecule, basis set information, and optional dipole derivatives, vib() performs the usual solution of whole or partial Hessians into normal modes and frequencies, reduced masses, turning points, and infrared intensities, all returned in schema. Other features include rotation-translation space projection, isotopic substitution analysis, Molden output, and a full thermochemical report incorporating the best features of several QC programs’ vibrational output.
3. FiniteDifference: Derivatives
As QCEngine and QCDB are focused on interfacing QC programs’ analytic quantum chemical methods or unique features, user calls for non-analytic derivatives in QCDB are by default routed through the finite difference procedure.87 This procedure (originally from Psi4) performs three- or five-point stencils for gradients and Hessians (full or partial), communicates through schema, and is parallelism-ready. The alternative of letting the internal finite difference of a QC program run and then parsing output files for multiple energies or gradients has been implemented in some cases, but this is not preferred (nor for internal geometry optimization).
4. Composite: Composite method and basis extrapolation treatments
Whenever an additive model chemistry is designated that involves differences of method (i.e., a focal point analysis or “delta” correction), basis [i.e., a complete basis set (CBS) extrapolation], keywords (e.g., all-electron minus frozen-core), or any combination thereof, the Composite procedure can encode it. Here, one can mix QC programs to perform conventional coupled cluster with CFOUR and DF-MP2 with Psi4, for example. Implementing new basis extrapolation formulas is simple, and it works on gradients and Hessians, as well as energies. If a subsidiary method energy can be obtained in the course of a target method, the procedure will recognize and avoid the unnecessary calculation (thus a TQ MP2 correlation energy extrapolation atop a DTQ HF energy will do 3, not 5, jobs). Input specification can be through API, schema, or strings (a user-friendly example is in the final paragraph of Sec. II E 5). All Composite communication is through schema, and the procedure is parallelism-ready.
5. ManyBody: Fragmentation and many-body approaches
All fragmentation and basis set superposition error (BSSE) treatments are collected into the ManyBody wrapper for many-body expansion (MBE) inherited from Psi4. The fragmentation pattern known from the QCSchema Molecule is applied to determine the degree of decomposition into monomers, dimers, etc., up to the full molecule, or the user can set the max_nbody level. Total quantities (energy, gradient, or Hessian) and interaction quantities are accessible through uncounterpoise (noCP), couterpoise (CP), and Valiron–Mayer functional counterpoise (VMFC) schemes.9,88,89 Geometry optimization with many-body-adapted quantities is also available. The wrapper can act on uniform single-method quantities or apply different model chemistries to each expansion level or interface with Composite or FiniteDifference results or both. All ManyBody communication is through schema, and the procedure is parallelism-ready.
6. vpt2: Anharmonic vibrational analysis
Anharmonic vibrational analysis has long been a feature of CFOUR. It requires a high-quality harmonic frequency procedure as input. It then performs further Hessian computations at geometry displacements along the normal coordinates. These are then combined into a third-order and partial fourth-order potential followed by vibrational analysis. Although many analytic Hessians are available in CFOUR itself, the qcdb.vpt2() procedure focuses on the formulation through analytic gradients, as being suited to distributed computing and generalization to program-generic gradients. Thus, CFOUR is a helper program that, with the QCDB procedure, can perform anharmonic analyses of, for example, CCSD (from CFOUR gradients called through QCDB), DFT (from another QC program’s gradients), or CBS (that produces a generalized gradient). All qcdb.vpt2() communication is through schema, and the procedure is parallelism-ready.
A complication is that the vpt2() procedure is essentially a series of invocations of CFOUR subcommands like xcubic, which expect files in native JOBARC form with energies, dipoles, and gradients. To accommodate this, QCDB uses Python modules to write imitations of the native files in string representations of binary form, which is lossless. Hence, a Psi4 DFT gradient is represented as a JOBARC to pass through the CFOUR mechanisms.
7. makefp: EFP library generation
The two engines for computing EFP interactions, LibEFP90,91 and GAMESS,47 use the same parameter file for storing the EFP potential at a given basis set and monomer geometry. Only GAMESS can generate that file, and the routine has been wrapped by QCDB for access through qcdb.makefp(). The resulting .efp file contents are returned in the QCSchema output and so are available for writing to a personal library or to feed to subsequent qcdb.energy(“gms-efp”) (or “lefp-efp” or “p4-efp”) calls to determine non-covalent interactions between EFP fragments. Certain EFP integrations await expansion of QCSchema Molecule.
8. Diatomic: Spectroscopic constants
The electronic potential analysis for diatomic molecules has long been encoded in Psi4 as a post-processing procedure from a list of electronic energies along the interatomic coordinate. This has been reworked as a procedure and is demonstrated in Sec. III.
9. RESP: Charge fitting
The restrained electrostatic potential (RESP) charge model92 is obtained by an iterative fitting of the electrostatic potential emerging from QC calculations on one or several conformers of a molecule to a classical point-charge potential. An existing RESP plugin93,94 drives the property calculations with Psi4, and this has been expanded to alternately draw from GAMESS using the QCDB API.
10. CrystaLattE: Crystal lattice energies
The process of estimating the lattice energy of a molecular monocrystal via the many-body expansion is encoded in the CrystaLattE software.95,96 Starting with extracting a subsample from a cif file, the program handles fragmentation into dimers, trimers, etc., identifies unique N-mers, prepares QC inputs, and keeps track of many-body results into final quantities. Although the thousands of component calculations mean that it will only become practical after QCDB upgrades to the distributed driver (see Sec. II D), CrystaLattE is ready to be integrated in serial mode in QCDB.
D. QCDB common driver
The driver component of QCDB [Fig. 3(vi)] is the fairly lightweight coordinator code that (1) facilitates the interactive API of set_molecule, set_keywords, energy ("nwc-b3lyp/6-31g*"), print(variable("b3lyp dipole")) rather than communicating through QCSchema; (2) imposes cross-QC-program suggestions like tightening convergence for higher derivatives or for finite difference; and (3) weaves together procedures and programs so that optimize("mp6") commences finite difference or energy("ccsd/cc-pv[tq]z", bsse_type="vmfc") runs ManyBody, Composite, and program harnesses in the right sequence. The driver is primarily concerned with processing user-friendly input [“User API” in Fig. 3(vi)] into QCSchema as directly as possible and then routing it into a program harness [Fig. 3(iii) for analytic single-points] or through procedures [Fig. 3(v)] on their way to program harnesses (e.g., for Composite, FiniteDifference) or through procedures after program harnesses [e.g., for resp(), vib()]. In order to make good use of the QCDB common driver, a QC program must register capabilities and information. These include the available analytic methods (for appropriate use of finite difference), insider best-practice options from the program’s developers (see Sec. II E 9), and all keywords and their defaults (for flexible and informative keyword validation through Python).
The common driver is based upon the Psi4 v1.0 recursive driver described in Ref. 14 that unifies many complex treatments (e.g., MBE and CBS) into a few user-facing functions that focus on what, not how. After polishing in Psi4 v1.5, a new distributed driver with the same interface but tuned to QCSchema communication and embarrassingly parallel execution through QCArchive Infrastructure will be substituted. See Sec. IV and Fig. 2 of Ref. 53 for details.
E. Technical aspects to interoperability
Details of specifying and running QC computations, particularly arbitrating the expression of QCSchema by QCEngine and QCDB, are collected below. Readers who prefer a software overview should proceed to Sec. III. Symbols like (I-b) mark strategies for overcoming or unifying the expertise barriers to using QC programs enumerated in the initial paragraphs of Sec. I.
1. Memory
User specification of memory resources is managed by QCEngine and is outside the QCSchema. By default, the job is given all of the compute node’s memory (less some buffer). If user-specified, input units are in GiB, e.g., qcdb or qcng.compute(…, local_options={"memory": 10}) (I-b). In either case, the memory quantity is translated into DSL keyword names such as memory_size and mem_unit for CFOUR. Because QCEngine exercises total control over memory, any specification misplaced as a keyword into QCSchema is ignored and overwritten in QCEngine or raises an error if conflicting in QCDB. An exception is cases like NWChem, where aggregated memory is managed by QCEngine but distribution between heap, stack, and global is editable through keywords (e.g., memory__total or memory__stack).
2. Disk
The working directory and execution environment are also governed by QCEngine, and user modifications are outside QCSchema. Each job is run in a quarantined scratch directory created for it and populated by input and any auxiliary files. Execution occurs through Python subprocess (or less often through Python API). Output files and any program-specific files in text or binary format (including the generated input) are collected and returned in QCSchema fields before scratch directory deletion (I-e).
3. Parallelism
The execution flags or environment variables that control CMS program parallelism and their single- or multi-node capabilities are built into their respective QCEngine harnesses. A job gets the full single-node resources (max cores and near-max memory) assigned to it by default; multinode execution (only for NWChem at present) requires explicit specification. Assigning instead an optimal portion of the full resources on the basis of method and memory could be implemented in a harness, but none presently do. User specification of parallelism is managed by QCEngine and is outside QCSchema [e.g., qcdb or qcng.compute(…, local_options={"ncores": 4})] (I-e).
4. Molecule specification
Molecule specification is the most important aspect that QCEngine and QCDB control via QCSchema to the exclusion of a program’s DSL. The QCSchema Molecule can store mass, isotope, charge/multiplicity, fragmentation, ghostedness, and connectivity information (and more), along with the basic element and Cartesian geometry data (I-d). All quantities are stored in amu or Bohr to avoid imprecision from multiple unit conversions through different revisions of physical constants.
Initializing a molecule can occur through a variety of string formats (of Cartesian coordinates) or directly by arrays. Extensive validation and application of physics-based defaults follows such that string Snippet 1 becomes (Ref. 97 for details) the schema Snippet 2. In the QCDB API, molecules can additionally be specified via Z-matrix, mixed Cartesian/Z-matrix, and with variable and deferred coordinates. QCSchema Molecule holds almost all data relevant to molecular system specification in QC, including EFP fragments, which are parseable without additional software and are stored in a secondary object. Items that appear in the molecule specification sections of some programs but do not fit in QCSchema Molecule, such as the stars signaling optimizable internal coordinates in CFOUR, reside in an extras section. (EFP and extras are future extensions.)
SNIPPET 1.
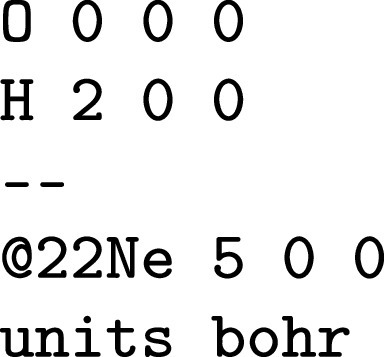
A string molecule input with complicating mass number, fragments, and implicit multiplicity.
Like memory or other aspects monopolized by QCSchema, user specification of the molecule in the DSL through keywords (e.g., scf__nopen in NWChem or contrl__icharg in GAMESS) is ignored and overwritten in QCEngine or raises an error if inconsistent in QCDB.
A requirement for combining vector data from multiple jobs is that the data be in a common frame of reference. Although each QC program has a standard internal orientation, these can be different between programs or between input specifications, and not all programs can return quantities in an arbitrary input frame and atom ordering. To smooth over inconsistent capabilities, the input geometry and the output geometry are both collected from output data, and an aligner computes the displacement, rotation matrix, and atom mapping needed to transform between them. Then, any vector results have the appropriate transformations applied so that all results in AtomicResult are in input orientation (O-a). This occurs for both QCEngine and QCDB when the Molecule fields fix_com and fix_orientation are True. (Here, “fix” is used in the “fasten” sense, not the “repair” sense.) When False, QCEngine returns in program native frame, while QCDB returns in Psi4 native frame.
5. Methods
Perhaps the most compelling element of QCSchema is the ability to request methods by a single string rather than piecemeal (e.g., "blyp-d3(bj)", "mp2", "cis" in place of {"method": "blyp","dft_d": "d3_bj"}, {"mplevl": 2}, {"calclevel": "hf","excite": "cis"}), thereby closely tying results to the model section (with subfields method and basis) of the data layout (barring algorithm, space, auxiliary basis set choices). As far as possible, all method specification and no extraneous information are consolidated into the atomicinput.model.method field. This is the primary translation effort of each QCEngine harness, as shown by the uniformity of the field in Fig. 2(b). In calling QCEngine, the user supplies the canonical method name (I-b). There is no compensation for program peculiarities; for example, "b3lyp" returns different answers if submitted to programs that have made a different choice of VWN3 vs VWN5, consistent with the principle that users can translate an input directly into QCSchema.
A complication to this principle is when programs conflate non-method information like algorithm (e.g., rimp2) or alternate code paths (e.g., task tce energy) into the primary method call. To maintain QCSchema integrity for model.method, the project invents top-level keywords like {"qc_module": "tce"} to allow deliberate choice of the TCE over hand-coded CC in NWChem and {"mp2_type": "df"} to instruct DF in GAMESS, NWChem, or Q-Chem. Keyword qc_module can also control choice of VCC/ECC/NCC in CFOUR and DFMP2/DFOCC/DETCI in Psi4, although these also have local knobs cfour_cc_program and psi4_qc_module.
Method specification in QCDB is similar to QCEngine except a compound program-method argument like optimize("nwc-mp2") is used. This difference is historical and endures for ease of specifying composite model chemistries like gradient("p4-mp2/cc-pv[56]Z + d: nwc-ccsd/cc-pv[tq]z + d: c4-ccsdtq/cc-pvdz")98 employing Psi4, NWChem, and CFOUR for different stages. Additionally, QCDB tests the major methods to ensure the same string yields the same result (I-f). It also maintains a list of capabilities, so, for example, ROHF CCSD in NWChem can be automatically routed to TCE [see Fig. 2(d)]. User specification of method information in keywords instead of through the model field is overwritten without warning in QCEngine, while in QCDB, contradictory information yields an error.
6. Basis sets
Notwithstanding the curation efforts of the Basis Set Exchange99 (BSE), every QC program maintains an internal library of basis sets with uneven upstream (from the basis set developer) updates applied, uneven downstream (by the program owner) specializations applied, and different spellings for accessing a given basis, not to mention different data formats. In QCEngine, only the internal library of a program is used, accessed from the atomicinput.model.basis field. Thus, due to DSL, the same string value directed toward different programs can lead to different results, and different strings can lead to the same results, as in Fig. 2(b). To allow consistency between programs and to reduce user DSL demands, QCDB pulls basis sets from a single library (Psi4’s in .gbs format, which is amply stocked with Pople, Dunning, Peterson, Karlsruhe, and other orbital and fitting basis sets) and performs the translation into the custom per-atom specification and format for each program, including setting spherical or Cartesian for d-shells and higher according to basis set design. In this way, a standard case-insensitive label and a consistent interface to custom and mixed basis sets is available (I-b). Alternatively, QCDB can act like QCEngine to access a program’s internal basis set library through program-specific keywords (e.g., set gamess_basis__gbasis accd vs set basis aug-cc-pvdz). While the Psi4 basis set library is used at present, future work will switch to the new MolSSI BSE.
7. Execution
Apart from CMS programs, QCEngine requires only QCElemental and some common Python packages. It is readily installed by conda install qcengine -c conda-forge or pip install qcengine. Execution occurs through CLI or one-call API with JSON-like input. For example, if AtomicInput specification {…, "model": {"method": "ccsd","basis": "aug-cc-pvdz"}} was in a file, qcengine spec run cfour would run CFOUR and return QCSchema AtomicResult (I-e). If the specification was a dictionary in a Python script, then qcengine.compute(spec, "cfour") produces the same results, as in the “execution” column of Fig. 2(b). QCEngine can be run through a queue manager, but for more than incidental jobs, users should consider the job orchestration capabilities of QCFractal.
QCDB requires only QCEngine and is installed similarly by conda install qcdb -c psi4. Execution modes CLI and one-call API are called analogously, only replacing qcng by qcdb (and ccsd by c4-ccsd) as shown in Figs. 2(c) and 2(d). Additionally, though, QCDB can function through an interactive driver API to reuse molecule and keyword sets and perform more complex sequences. This is shown in Snippet 3 that scans an energy potential and then performs a computation at the optimum distance at a better level of theory. This is analogous to the PsiAPI mode in Psi4. A simplified, plain-text input that gets processed into the API and is analogous to the PSIthon mode of Psi4 will be available after further integration with Psi4; an example is at Snippet 4.
SNIPPET 3.

An interfragment potential energy scan followed by composite energy in QCDB.
SNIPPET 4.

Contradictory input opportunities.
8. Modes
QCDB operates in two modes, which treat keywords, particularly keyword defaults, differently. QCDB supports distinct modes of operation to tailor its capabilities toward driver integration of multiple programs (when unified results are needed) or toward interfacing a single program (when user familiarity is preferred). Most controlling is the driver or unified mode, which endeavors to elicit from different QC programs identical results out of identical input conditions (roughly the combination of method, basis, reference, active space, and integrals treatment) (I-f). Here, the driver imposes QCDB-level defaults such as non-DF algorithms, all-electron spaces, and graduated convergence criteria for energy vs analytic derivative vs finite difference derivative. This mode is required for multi-program procedure runs [e.g., energy("p4-mp2/cc-pv[tq]z + d:c4-ccsd/cc-pvtz")] and is active by default.
Another mode, denoted sandwich since the QCDB pre- and post-processing is less intrusive, is for users focusing on a single QC program who want the driver routines, method mapping [e.g., energy("gms-ccsd(t)",bsse_type="vmfc")], and I/O-wrapping advantages of QCDB but do not want surprise resets of their accustomed defaults. Driver-suggested QCDB-level (e.g., frozen-core), driver-level (e.g., graduated derivative convergence), and best-practices (e.g., module selection) defaults are all turned off. This mode is effectively how QCEngine runs.
Some background facts to illustrate the modes:
-
•
For the default MP2 algorithm, Psi4 uses DF, while CFOUR, GAMESS, NWChem, and QCDB use CONV.
-
•
The CFOUR, GAMESS, NWChem, Psi4, and QCDB default HF density convergences are 10−7, 10−5, 10−4, 10−8, and 10−8, respectively.
-
•
For the CCSD energy from CFOUR, the default CC module is VCC, while QCDB best-practice is ECC.
-
•
The NWChem default task ccsd energy does not run for open-shell, while QCDB uses the CCSD module for RHF and TCE module for ROHF.
-
•
GAMESS freezes core by default, while CFOUR, NWChem, Psi4, and QCDB correlate all electrons.
In the unified mode, energy("gms-mp2") and energy("p4-mp2") both run all-electron MP2 without DF and with 10−8 convergence. After setting ROHF, energy("c4-ccsd") runs through ECC, and energy("nwc-ccsd") runs through TCE, again both HF to 10−8 and all-electron. In contrast, sandwich mode energy("gms-mp2") produces a conventional frozen-core MP2 energy converged to 10−5, while energy("p4-mp2") produces a DF all-electron value converged to 10−8. In the ROHF CCSD case, the CFOUR job runs as all-electron through VCC with HF converged to 10−7, while the NWChem submission declines to run.
9. Keywords
QC programs have hundreds of keywords controlling their operation on matters of substance (e.g., RAS3), strategy (e.g., DIIS), computer science (e.g., INTS_TOLERANCE), and research convenience (e.g., DFT_NEW). The variety in spelling and text arrangement by which the same ideas are communicated to different QC programs is staggering (and a considerable barrier to trying new codes). The necessity to represent any (single-stage, single-program) input file as QCSchema requires mapping rules so that a user familiar with the native DSL can readily translate into the key/value representation of an AtomicInput’s keywords field. The primary guideline is that the right-hand side value must be a simple data quantity in natural Python syntax (e.g., CFOUR’s 3-1-1-0/3-0-1-0 becomes [[3, 1, 1, 0], [3, 0, 1, 0]]), and the left-hand side key is a string that encodes any level of nesting with double-underscore (e.g., GAMESS’s contrl__scftyp or NWChem’s dft__convergence__density). A present/absent keyword (as opposed to a key/value pair) becomes a boolean, such as NWChem scf__rohf. The ProgramHarness handles formatting the keywords field (back) into the input grammar (I-d), including quashing unnecessary case-sensitivity (e.g., Qz2p converts to lowercase for CFOUR, while a filename option passes unchanged). For QCDB, prefixing a keyword by program name targets it toward a particular program; hence, reference becomes cfour_reference or psi4_reference.
The greatest challenge to mapping rules is that some programs have an input structure that blurs module nesting vs keyword name vs keyword value. An extra mapping rule not strictly required by QCEngine is for keywords to be independent and granular such that they are one-to-one with other programs, not overworked like dft__grid={"lebedev": (99, 11), "treutler": True} (insufficiently granular) nor underworked like scf__rhf=False plus scf__uhf=True (insufficiently independent). QCDB uses internal aliasing and mutually exclusive groups to help keyword specification be intuitive for native users.
Making a QCSchema fed to multiple programs produce uniform output is not within the scope of QCEngine. Barriers to accessing multiple QC backends through a single DSL or, more intricately, to compatibly mixing backends include (a) heterogeneous control knobs across QC programs each with its own keyword set and (b) incompatible results due to different defaults yielding slightly different answers. QCDB takes up the task of uniting keywords into a single DSL for a further layer of interoperability. Unlike QCEngine, QCDB registers valid keywords for each QC program and can apply custom validation functions to each. Additionally registered are unified keywords so that, for example, setting REFERENCE is translated into CFOUR_REFERENCE or GAMESS_CONTRL__SCFTYP, as shown in Figs. 2(c) and 2(d) (I-b, I-f). As mentioned above, insisting on granular keywords for the QCSchema representation allows cleaner mapping between QC programs. As mentioned below, QCDB also encodes best-practice keywords to allow shorter inputs, context-dependent defaults, and bridging the developer-user knowledge gap. QCSchema or QCDB API offer ample opportunities for users to submit contradictory input specification, several of which are shown in Snippet 4.
QCDB resolves competing keyword suggestions and requirements by the user, driver, schema, and best practices into a final keyword set that is passed to QCEngine for final formatting. Because of this step, incompatible keywords pass without warning in QCEngine, while in QCDB, contradictory information yields an error.
Codebase authors know best how to run a computation, but they may have conveyed that knowledge only through documentation and forum posts. Due to the unwieldiness of large legacy codebases and the circuity of research (and the burden of backward compatibility), it can happen that a method needs several keywords to express it or that valuable approximations or code-routing do not get turned on by default. Due to its layered Python/C++ structure, Psi4 naturally has a place to express such “best-practice” defaults based on method, basis, system size, etc. The advantage is that simple method + basis inputs yield production-grade results. Thus, QCDB takes advantage of working with codebase authors and the intermediate Python layer to implement best-practice keywords based on available calculation data (I-c). These take the form of routing to the best (or only capable) module for a given method, reference, derivative level, and active space; of supplying sensible defaults such as the number of electrons or roots; of tuning convergence to the derivative and needed precision (analytic vs finite difference) at hand; or of specifying C1 or highest-Abelian symmetry to modules with symmetry restrictions. Such options can be overridden by the user and can be disabled in sandwich mode (Sec. II E 8). These defaults are themselves subject to change as recommendations evolve, but their state is readily viewed in program inputs.
10. QCVariables
The QC output stream, whether ASCII, binary, or structured, is read immediately after program execution. Scalar and array result quantities, such as PBE TOTAL ENERGY, MP4 CORRELATION ENERGY and PBE TOTAL GRADIENT, CCSD DIPOLE, are extracted and held as significant-figure-preserving floats or NumPy arrays, respectively, and are known collectively as QCVariables (O-a). Extraction uses the most precise available source, whether the standard output stream or available auxiliary files (e.g., CFOUR GRD). The internal geometry is always collected, and any vector results are manipulated in concert with it, as described in Sec. II E 4. For QCEngine, many of the same harvested quantities are directed into QCSchema AtomicResultProperties lists. Results are available programmatically through qcdb.variable("mp2 total energy") or atomicresult.properties.mp2_total_energy in QCDB and QCEngine, respectively.
A mild vexation in QC output files is that they contain different quantities such as total vs correlation energy or opposite-spin vs triplet energy that are interconvertible but not directly comparable. QCVariables enforce the consistency of common QC definitions and encode common combining rules (O-b). They are applied in post-processing to ensure that a maximum of data gets harvested from each run, that exactly the same quantities are collected from each QC program, and that trivially defined methods such as SCS(N)-MP2 and B3LYP-D3(BJ) need not clutter either the QC code or its parsing.
Using binary representations of floats rather than truncated strings from output files is a powerful argument for API integration rather than parsing. Binary representation is essential when dealing with many numbers with slight differences, such as finite differences or MBE sums. Programs with Python APIs (and that use APIs for internal inter-language transfer like between C++ and Python in Psi4) can transfer data with full precision; for QCEngine, these are, for example, adcc, OpenMM, RDKit, TorchANI, dftd4, Psi4, TCPB TeraChem, and xtb. Of these, the last four have implemented QCSchema directly for API access. An intermediate step is to use structured output like XML or JSON from Molpro, MRChem, and Qcore. For certain programs, a combination of reading available binary files (e.g., 99.0 for return energy in Q-Chem and JOBARX/JAINDX for certain QC results and organizational data in CFOUR) and text parsing is employed. Results from other programs are collected solely through text parsing: e.g., dftd3, GAMESS, gCP, MOPAC, mp2D, NWChem, the classic interface to TeraChem, and Turbomole. Although results are collected into QCSchema from QC programs at the greatest accessible precision, in order to maintain that precision among the data transfers and storage of the QCDB and QCArchive Infrastructure ecosystem, the QCElemental implementation of QCSchema (nominally a JSON Schema,16 which does not handle binary or numpy.ndarray) includes MessagePack100 serialization.
III. EXAMPLE: DIATOMIC SPECTROSCOPIC CONSTANT FITTING
With contemporary QC software, it is entirely possible to approach the ab initio limit in the description of diatomic molecules.101 Such spectroscopically accurate calculations require extrapolating to the full configuration interaction and complete basis set limits under the non-relativistic Born–Oppenheimer (BO) approximation, followed by usually negligible corrections to account for both relativistic effects and the BO approximation itself. Not only does this type of calculation present a remarkable computational challenge [as it is significantly more expensive than CCSD(T), the usually sufficient target of quantum chemistry], it can also be practically difficult to incorporate multiple corrections and extrapolations into a workflow. While all of the necessary features are present across various QC software packages, no single package implements everything (let alone has the best implementation). Furthermore, enforcing consistent geometries, basis sets, convergence criteria, frozen orbitals, etc. between programs is a cumbersome, often error-prone task. The QCDB driver remedies this problem by providing an easy-to-use Python interface to multiple QC programs.
To showcase this capability of the QCDB driver, the ground states of a few diatomic molecules (BH, HF, and C2) are optimized at essentially the ab initio limit, and spectroscopic constants are computed and compared to experiment. Previous studies estimating the ab initio limit for the full set of standard spectroscopic constants of these molecules have been reported (see, e.g., Refs. 102–104). The present study provides improved treatments for some of the small corrections and/or includes more correction terms. Here, we include corrections for electron correlation beyond CCSD(T), basis set effects beyond an already high-quality core-valence quadruple/quintuple-ζ extrapolation, relativistic effects, and the Born–Oppenheimer diagonal correction using four different QC programs through the unified QCDB interface. The effect of each correction is examined separately, as well as the cumulative effect of all corrections. Understanding the cost and importance of each correction is helpful for designing reasonable extrapolations for larger systems.
A spectroscopically accurate model chemistry energy (ETotal) is defined as a base energy (EBase) with five separate corrections,
| (1) |
Each energy and the QC program(s) used to obtain it is defined in Table II.
TABLE II.
Composite level of theory for spectroscopic constants and associated QC programs.
| Name | Method | Program |
|---|---|---|
| E Base | CCSD(T)/cc-pCV[Q5]Z | NWChem |
| ΔEBasis | MP2/(aug-cc-pCV[56]Z − cc-pCV[Q5]Z) | Psi4 |
| ΔEDBOC | CCSD/cc-pCVDZ | CFOUR |
| ΔERel | X2C-CCSD(T)/cc-pCVTZ | Psi4 |
| ΔECCSDTQ | [CCSDTQ − CCSD(T)]/cc-pVTZ | CFOUR |
| ΔEFCI | (FCI − CCSDTQ)/cc-pVDZ | GAMESS/ |
| CFOUR |
The rovibrational spectrum of a diatomic molecule is often expressed with Dunham’s expansion,
| (2) |
The first few Dunham coefficients correspond to well-studied spectroscopic constants,
| (3) |
The following truncation of the expansion is used to describe a diatomic:
| (4) |
The spectroscopic constants are then describable in terms of the electronic PES U(r) and its derivatives,
| (5) |
| (6) |
| (7) |
Note that these are all “equilibrium” constants, i.e., they are with respect to the bottom of the potential well (but with inclusion of the Born–Oppenheimer diagonal correction).
Accessed through the QCDB interface, the Psi4 procedure fits a set of points [r, E(r)] to this truncation, solving for the spectroscopic constants via a least-squares optimization.105 This procedure was used in the following way for each diatomic:
-
1.
Through the QCDB driver, ETotal was calculated at seven values of r, spaced 0.005 Å apart and centered approximately at the minimum of the PES. The spectroscopic constants were calculated with Psi4, including an approximate re.
-
2.
This seven-point calculation was repeated using the approximate re from the first step as the central point. The spectroscopic constants calculated from these PES points are those tabulated here.
Basis sets with spherical harmonics were used in all calculations, and basis set coefficients were standardized across all programs via QCDB. Electrons in core orbitals were frozen for computations using the cc-pVXZ basis set family, which lack core correlation functions. Energies were converged to at least 10−10 Hartrees in all programs. Even tighter convergence would be beneficial for the numerical differentiation performed in the fitting. Numerical tests suggest that this precision in energy can lead to uncertainties in αe [proportional to U‴(re)] and ωexe [proportional to Uiv(re)] as large as 0.0001 and 0.2 cm−1, respectively.
The calculations of all diatomics and spectroscopic constants are presented in Table III, and the results for re and ωe are shown in Fig. 4 for easier analysis. Prior to discussing the chemical and computational implications of these results, it is worthwhile to first note that the corrections for BH closely match those of a previous study103 by Temelso et al. (which used a similar but less exact extrapolation). This validates these results from a software perspective: each program must be using correct geometries, basis sets, convergence criteria, etc. The finite-difference nature of the fitting procedure makes close agreement between programs particularly important.
TABLE III.
Comparison between theory and experiment for bond lengths (Å) and spectroscopic constants (cm−1) of three diatomic molecules. All Δ terms correspond to the difference between a value and the base CCSD(T)/cc-pCV[Q5]Z calculation. Experimental values from Ref. 106 (BH), 107 (HF), and 108 (C2). All published experimental uncertainties are smaller than the displayed precision of the spectroscopic values presented here.
| Molecule and method | r e | ω e | ω e x e | B e | D e | α e |
|---|---|---|---|---|---|---|
| BH | ||||||
| Base | 1.228 90 | 2371.24 | 49.4 | 12.088 | 0.001 257 | 0.423 |
| ΔBasis | +0.000 18 | −0.44 | −0.4 | −0.004 | −0.000 001 | −0.001 |
| ΔDBOC | +0.000 65 | −2.33 | −0.2 | −0.013 | −0.000 002 | +0.000 |
| ΔRel | −0.000 01 | −0.57 | +0.1 | +0.000 | +0.000 001 | +0.000 |
| ΔCCSDTQ | +0.000 19 | −2.07 | +0.1 | −0.004 | +0.000 001 | +0.001 |
| ΔFCI | +0.000 00 | +0.00 | −0.2 | +0.000 | +0.000 000 | +0.000 |
| ΔTotal | +0.001 01 | −5.41 | −0.5 | −0.020 | +0.000 000 | +0.000 |
| Total | 1.230 00 | 2365.83 | 49.0 | 12.068 | 0.001 256 | 0.423 |
| Experiment | 1.232 16 | 2366.72 | 49.3 | 12.026 | 0.001 235 | 0.422 |
| HF | ||||||
| Base | 0.916 54 | 4147.01 | 90.5 | 20.968 | 0.002 144 | 0.793 |
| ΔBasis | +0.000 17 | −1.79 | −0.7 | −0.008 | −0.000 001 | −0.002 |
| ΔDBOC | +0.000 01 | +0.32 | −0.2 | −0.001 | −0.000 001 | +0.000 |
| ΔRel | +0.000 06 | −3.54 | −1.3 | −0.003 | +0.000 003 | +0.000 |
| ΔCCSDTQ | +0.000 21 | −4.49 | +0.1 | −0.009 | +0.000 002 | +0.002 |
| ΔFCI | +0.000 01 | −0.19 | +0.0 | +0.000 | +0.000 000 | +0.000 |
| ΔTotal | +0.000 47 | −9.70 | −2.2 | −0.021 | +0.000 004 | +0.000 |
| Total | 0.917 00 | 4137.31 | 88.3 | 20.947 | 0.002 148 | 0.792 |
| Experiment | 0.916 808 | 4138.32 | 89.0 | 20.956 | 0.002 151 | 0.798 |
| C2 | ||||||
| Base | 1.240 39 | 1873.63 | 12.6 | 1.826 | 0.000 007 | 0.017 |
| ΔBasis | +0.000 16 | −1.01 | +0.0 | +0.000 | +0.000 000 | +0.000 |
| ΔDBOC | +0.000 01 | +0.09 | +0.0 | +0.000 | +0.000 000 | +0.000 |
| ΔRel | −0.000 16 | −0.41 | +0.1 | +0.000 | +0.000 000 | +0.000 |
| ΔCCSDTQ | +0.001 46 | −11.76 | +0.8 | −0.004 | +0.000 000 | +0.001 |
| ΔFCI | +0.001 00 | −4.58 | +0.0 | −0.003 | +0.000 000 | +0.000 |
| ΔTotal | +0.002 48 | −17.81 | +0.8 | −0.007 | +0.000 000 | +0.001 |
| Total | 1.242 87 | 1855.82 | 13.4 | 1.819 | 0.000 007 | 0.018 |
| Experiment | 1.242 44 | 1855.01 | 13.6 | 1.820 | 0.000 007 | 0.018 |
FIG. 4.
Influence of post-CCSD(T)/CBS corrections on two spectroscopic constants, (a) re and (b) ωe, and three diatomic systems, BH, HF, and C2. For each system, the right gray bar is the difference between the experimental constant and the constant calculated at the EBase [CCSD(T)/CBS] level of theory. The left gray bar is the difference in constants calculated at the ETotal and EBase levels of theories. Within the left gray bar, contributions from each correction are shown as colored bars. Data are from Table III.
The total extrapolation procedure shows remarkable agreement with experiment for bond lengths re (within 0.0005 Å) except for BH, off by 0.0022 Å. However, this extrapolation lacks nonadiabatic BO effects, which were found by Martin102 to be unusually high for BH, ∼0.0025 Å. This is rather close to the overall difference of 0.0022 Å between experiment and our best estimate. Theoretical harmonic frequencies ωe are in excellent agreement with experiment, off by only 1 cm−1. The rotational constant Be is also well predicted, within 0.01 cm−1 for HF and C2 and off by a somewhat larger 0.04 cm−1 for BH. The latter error may be largely due to already-noted non-BO effects, which cause a larger discrepancy in re for BH. ωexe is in good agreement with experiment, matching within 0.2–0.4 cm−1 for BH and C2 but is off by a larger 1.6 cm−1 for HF. It is not clear that the corrections employed here actually improve this constant, and the remaining discrepancy could be due to the numerical precision limitations discussed earlier. is very well predicted already by the base method, and the various corrections are extremely small. Similarly, αe appears to not require corrections on top of the base method, each of which changes it by only ±0.002 cm−1 or less. Final values are within 0.005 cm−1 of experiment.
Figure 4 shows that the sum of the small corrections matches experiment very well for re and ωe, except for the bond length of BH, where non-BO effects are important as noted above. All of the small corrections considered can be important for re and ωe, although there is no consistency about their relative importance from one molecule to another. For example, the DBOC is rather important for BH (which has the lightest nuclei), but not for HF and even less so for C2. Similarly, the FCI correction (beyond CCSDTQ) is negligible for BH and HF but is important for C2 (worth 0.001 Å and 4.6 cm−1) In total, the corrections for C2 lower the value of ωe by a surprisingly large 17.81 cm−1 from the base CCSD(T) value, which is very close to the experimental ωe (18.61 cm−1 lower than the base). A large majority of this change is due to missing electron correlation: the CCSDTQ correction is responsible for about 12 cm−1 and the FCI correction by about another 5 cm−1. This is presumably due to the much larger degree of electron correlation in C2, arising from the close near-degeneracy of the [core] and [core] configurations.
IV. SUMMARY AND CONCLUSIONS
Users increasingly desire programmatic (i.e., API: application programming interface) access to QC results, either for their convenience or for incorporation into automated workflows. The interface, volume, and intricacy requirements of that access vary widely across applications and increasingly involve uniform results across QC programs. The QCElemental, QCEngine, and QCDB software modules [the former two being part of the Molecular Sciences Software Institute18 (MolSSI) QCArchive17 project] provide a framework to facilitate interoperability among community computational molecular sciences (CMS) programs.
QCArchive and QCDB have been designed to work with emerging tools and standards developed by MolSSI, particularly the QCSchema JSON format for information passing. QCElemental provides implementations and validators around QCSchema objects, while QCEngine provides QCSchema I/O adaptors for CMS codes. In addition to wrapping nearly a dozen QC programs for uniform execution and programmatic access to results, QCEngine interfaces with geomeTRIC and other geometry optimizers that can, in turn, call QCEngine for QC gradients. QCEngine easily expands to additional CMS codes, has parallel execution capabilities through QCFractal, and by definition allows uniform execution, yet it is not in itself a coherent QC driver due to the differing implementations, conventions, defaults, and DSL of QC codes.
The Quantum Chemistry Common Driver and Databases (QCDB) project provides a simple and powerful driver front-end to multiple QC programs, allowing users automatic access to several features formerly requiring specialized scripts or laborious post-processing. These include built-in composite methods, many-body expansion procedures, vibrational analysis, and combinations thereof for not only energies but also gradients, Hessians, and geometry optimizations. By adding the basis set, keywords, and result tools for uniformity and interoperability, QCDB also allows mixing and matching capabilities of multiple quantum chemistry programs within a single computation. These features have been demonstrated with an application computing spectroscopic constants of diatomic molecules at the ab initio limit, including corrections for post-CCSD(T) electron correlation, beyond-cc-pCV[Q5]Z basis set effects, relativistic effects, and the Born–Oppenheimer diagonal correction, combining total energies computed by CFOUR, GAMESS, NWChem, and Psi4.
V. EXTERNAL MATERIAL
Software repositories and documentation are available for QCElemental at https://github.com/MolSSI/QCElemental/ and https://molssi.github.io/QCElemental/, for QCEngine at https://github.com/MolSSI/QCEngine/ and https://molssi.github.io/QCEngine/, for QCDB at https://github.com/qcdb/qcdb/ and https://qcdb.github.io/qcdb/, and for general QCArchive INFRASTRUCTURE at http://docs.qcarchive.molssi.org/. These programs remain in active development. Production computations are under way using many features of the software, and test suites are expected to pass. However, users are encouraged to contact the developers as they venture afield of the verified tests. Many snippets from this work, including an abbreviated diatomic fitting, are demonstrated in the test suite: https://github.com/qcdb/qcdb/blob/master/qcdb/tests/test_manuscript.py.
ACKNOWLEDGMENTS
Several of the co-authors have been supported in their development of QCDB and QCEngine and affiliated projects by the U.S. National Science Foundation through Grant Nos. ACI-1449723, CHE-1566192, ACI-1609842, ACI-1547580, ACI-1047772, ACI-1450217, ACI-2003931, CHE-1664325, and CHE-2134792, by the Office of Basic Energy Sciences Computational Chemical Sciences (CCS) Research Program (Grant Nos. AL-18-380-057 and DE-SC0018412), and by the Exascale Computing Project (Grant No. 17-SC-20-SC), a collaborative effort of the U.S. Department of Energy (DOE) Office of Science and the National Nuclear Security Administration.
The Molecular Sciences Software Institute acknowledges the Advanced Research Computing at Virginia Tech for providing computational resources and technical support.
D.G.A.S. also acknowledges the Open Force Field Consortium and Initiative for financial and scientific support.
A.G.H. was supported, in part, by the National Science Foundation under Grant No. OAC-1931387 at Stony Brook University and made use of the high-performance SeaWulf computing system, which was made possible by the National Science Foundation (Grant No. 1531492).
L.W. was additionally supported by DOE’s Advanced Scientific Research Office (ASCR) under Contract No. DE-AC02-06CH11357.
H.J.K. and F.L. were partially supported by Grant No. DE-SC001896.
L.-P.W. acknowledges Grant No. ACS-PRF 58158-DNI6 and National Institutes of Health (Grant No. R01GM132386).
J.D.C. acknowledges support from NIH Grant No. P30 CA008748, NIH Grant No. R01 GM132386, and the Sloan Kettering Institute.
R.D.R. acknowledges support from the European High-Performance Computing Joint Undertaking under Grant Agreement No. 951732 and partial support from the Research Council of Norway through its Centres of Excellence scheme (Project No. 262695) and through its Mobility Grant scheme (Project No. 261873).
H. K. and J.Š. acknowledge funding from the Praemium academiae (CAS).
M.F.H. has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No 810367).
T.J.M. and C.B.H. acknowledge support from the Office of Naval Research (Grant Nos. N00014-18-1-2659 and N00014-17-1-2875). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1656518 for C.B.H. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
The contents of this paper are solely the responsibility of the authors and do not necessarily represent the views of the commercial partners of the Open Force Field Consortium.
AUTHOR DECLARATIONS
Conflict of Interest
J.D.C. is a current member of the Scientific Advisory Board of OpenEye Scientific Software, Redesign Science, and Interline Therapeutics and has equity interests in Redesign Science and Interline Therapeutics.
DATA AVAILABILITY
The data that support the findings of this study are available within the article.
REFERENCES
- 1.Truhlar D. G., “Basis-set extrapolation,” Chem. Phys. Lett. 294, 45–48 (1998). 10.1016/s0009-2614(98)00866-5 [DOI] [Google Scholar]
- 2.Halkier A., Helgaker T., Jørgensen P., Klopper W., Koch H., Olsen J., and Wilson A. K., “Basis-set convergence in correlated calculations on Ne, N2, and H2O,” Chem. Phys. Lett. 286, 243–252 (1998). 10.1016/s0009-2614(98)00111-0 [DOI] [Google Scholar]
- 3.East A. L. L. and Allen W. D., “The heat of formation of NCO,” J. Chem. Phys. 99, 4638–4650 (1993). 10.1063/1.466062 [DOI] [Google Scholar]
- 4.Császár A. G., Allen W. D., and Schaefer H. F., “In pursuit of the ab initio limit for conformational energy prototypes,” J. Chem. Phys. 108, 9751–9764 (1998). 10.1063/1.476449 [DOI] [Google Scholar]
- 5.Schuurman M. S., Muir S. R., Allen W. D., and Schaefer H. F., “Toward subchemical accuracy in computational thermochemistry: Focal point analysis of the heat of formation of NCO and [H, N, C, O] isomers,” J. Chem. Phys. 120, 11586–11599 (2004). 10.1063/1.1707013 [DOI] [PubMed] [Google Scholar]
- 6.Curtiss L. A., Raghavachari K., Redfern P. C., Rassolov V., and Pople J. A., “Gaussian-3 (G3) theory for molecules containing first and second-row atoms,” J. Chem. Phys. 109, 7764–7776 (1998). 10.1063/1.477422 [DOI] [Google Scholar]
- 7.Tajti A., Szalay P. G., Császár A. G., Kállay M., Gauss J., Valeev E. F., Flowers B. A., Vázquez J., and Stanton J. F., “HEAT: High accuracy extrapolated ab initio thermochemistry,” J. Chem. Phys. 121, 11599–11613 (2004). 10.1063/1.1811608 [DOI] [PubMed] [Google Scholar]
- 8.Grimme S., Antony J., Ehrlich S., and Krieg H., “A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu,” J. Chem. Phys. 132, 154104 (2010). 10.1063/1.3382344 [DOI] [PubMed] [Google Scholar]
- 9.Boys S. F. and Bernardi F., “The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors,” Mol. Phys. 19, 553–566 (1970). 10.1080/00268977000101561 [DOI] [Google Scholar]
- 10.Raghavachari K., Trucks G. W., Pople J. A., and Head-Gordon M., “A fifth-order perturbation comparison of electron correlation theories,” Chem. Phys. Lett. 157, 479–483 (1989). 10.1016/s0009-2614(89)87395-6 [DOI] [Google Scholar]
- 11.Thaller B., Grant I. P., Quiney H. M., Andrae D., Desclaux J., Saue T., Labzowsky L., Goidenko I., Engel E., Dolg M., Sapirstein J., Christensen N., Wolf A., Reiher M., Hess B. A., Kutzelnigg W., Sundholm D., Fægri K., Dyall K. G., Schwerdtfeger P., and Visscher L., in Relativistic Electronic Structure Theory—Part 1: Fundamentals, edited by Schwerdtfeger P. (Elsevier, Amsterdam, 2002). [Google Scholar]
- 12.Born M. and Oppenheimer R., “Zur quantentheorie der molekeln,” Ann. Phys. 389, 457–484 (1927). 10.1002/andp.19273892002 [DOI] [Google Scholar]
- 13.Kutzelnigg W., “The adiabatic approximation I. The physical background of the Born–Handy ansatz,” Mol. Phys. 90, 909–916 (1997). 10.1080/00268979709482675 [DOI] [Google Scholar]
- 14.Warden C. E., Smith D. G. A., Burns L. A., Bozkaya U., and Sherrill C. D., “Efficient and automated computation of accurate molecular geometries using focal-point approximations to large-basis coupled-cluster theory,” J. Chem. Phys. 152, 124109 (2020). 10.1063/5.0004863 [DOI] [PubMed] [Google Scholar]
- 15.The QCDB project name, Quantum Chemistry Common Driver and Databases, describes its early scope. The database aspect has since departed and been properly developed in the QCArchive project, particularly QCFractal. See Sec. II A and Ref. 17 for details.
- 16.JSON SCHEMA: A vocabulary that allows you to annotate and validate JSON documents. For the current version, see https://json-schema.org/; accessed January 2020.
- 17.Smith D. G. A., Altarawy D., Burns L. A., Welborn M., Naden L. N., Ward L., Ellis S., Pritchard B. P., and Crawford T. D., “The MolSSI QCARCHIVE project: An open-source platform to compute, organize, and share quantum chemistry data,” Wiley Interdiscip. Rev.: Comput. Mol. Sci. 11, e1491 (2021). 10.1002/wcms.1491 [DOI] [Google Scholar]
- 18.Krylov A., Windus T. L., Barnes T., Marin-Rimoldi E., Nash J. A., Pritchard B., Smith D. G. A., Altarawy D., Saxe P., Clementi C., Crawford T. D., Harrison R. J., Jha S., Pande V. S., and Head-Gordon T., “Perspective: Computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science,” J. Chem. Phys. 149, 180901 (2018). 10.1063/1.5052551 [DOI] [PubMed] [Google Scholar]
- 19.Smith D. G. A., de Jong B., Burns L. A., Hutchison G., and Hanwell M. D., QCSCHEMA: A schema for quantum chemistry. For the current version, see https://github.com/MolSSI/QCSchema; accessed January 2020.
- 20.Barbatti M., Granucci G., Persico M., Ruckenbauer M., Vazdar M., Eckert-Maksić M., and Lischka H., “The on-the-fly surface-hopping program system NEWTON-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems,” J. Photochem. Photobiol., A 190, 228–240 (2007). 10.1016/j.jphotochem.2006.12.008 [DOI] [Google Scholar]
- 21.Toniolo A., Thompson A. L., and Martínez T. J., “Excited state direct dynamics of benzene with reparameterized multi-reference semiempirical configuration interaction methods,” Chem. Phys. 304, 133–145 (2004). 10.1016/j.chemphys.2004.04.018 [DOI] [Google Scholar]
- 22.Levine B. G., Coe J. D., Virshup A. M., and Martínez T. J., “Implementation of ab initio multiple spawning in the Molpro quantum chemistry package,” Chem. Phys. 347, 3–16 (2008). 10.1016/j.chemphys.2008.01.014 [DOI] [Google Scholar]
- 23.Gaenko A., DeFusco A., Varganov S. A., Martínez T. J., and Gordon M. S., “Interfacing the ab initio multiple spawning method with electronic structure methods in GAMESS: Photodecay of trans-azomethane,” J. Phys. Chem. A 118, 10902–10908 (2014). 10.1021/jp508242j [DOI] [PubMed] [Google Scholar]
- 24.Keceli M. and Elliott S., Quantum Thermochemistry Calculator; https://github.com/PACChem/QTC; accessed 25 November 2019.
- 25.Steinmetzer J., Kupfer S., and Gräfe S., “pysisyphus: Exploring potential energy surfaces in ground and excited states,” Int. J. Quantum Chem. 121, e26390 (2021). 10.1002/qua.26390 [DOI] [Google Scholar]
- 26.Řezáč J., Cuby—Ruby framework for computational chemistry, version 4, http://cuby4.molecular.cz; accessed 22 November 2019.
- 27.Řezáč J., “Cuby: An integrative framework for computational chemistry,” J. Comput. Chem. 37, 1230–1237 (2016). 10.1002/jcc.24312 [DOI] [PubMed] [Google Scholar]
- 28.Polik W. F. and Schmidt J. R., “WebMO: Web-based computational chemistry calculations in education and research,” Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2021, e1554. 10.1002/wcms.1554 [DOI] [Google Scholar]
- 29.Larsen A. H., Mortensen J. J., Blomqvist J., Castelli I. E., Christensen R., Dułak M., Friis J., Groves M. N., Hammer B., Hargus C., Hermes E. D., Jennings P. C., Jensen P. B., Kermode J., Kitchin J. R., Kolsbjerg E. L., Kubal J., Kaasbjerg K., Lysgaard S., Maronsson J. B., Maxson T., Olsen T., Pastewka L., Peterson A., Rostgaard C., Schiøtz J., Schütt O., Strange M., Thygesen K. S., Vegge T., Vilhelmsen L., Walter M., Zeng Z., and Jacobsen K. W., “The atomic simulation environment—A Python library for working with atoms,” J. Phys.: Condens. Matter 29, 273002 (2017). 10.1088/1361-648X/aa680e [DOI] [PubMed] [Google Scholar]
- 30.Gjerding M. and Larsen A. H., ASR: Atomic simulation recipes: Recipes for calculating material properties. For the current version, see https://gitlab.com/asr-dev/asr; accessed September 2021. Documentation at https://asr.readthedocs.io/en/latest/index.html.
- 31.Huber S. P., Zoupanos S., Uhrin M., Talirz L., Kahle L., Häuselmann R., Gresch D., Müller T., Yakutovich A. V., Andersen C. W., Ramirez F. F., Adorf C. S., Gargiulo F., Kumbhar S., Passaro E., Johnston C., Merkys A., Cepellotti A., Mounet N., Marzari N., Kozinsky B., and Pizzi G., “AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance,” Sci. Data 7, 300 (2020). 10.1038/s41597-020-00638-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhrin M., Huber S. P., Yu J., Marzari N., and Pizzi G., “Workflows in AiiDA: Engineering a high-throughput, event-based engine for robust and modular computational workflows,” Comput. Mater. Sci. 187, 110086 (2021). 10.1016/j.commatsci.2020.110086 [DOI] [Google Scholar]
- 33.Lu Y., Farrow M. R., Fayon P., Logsdail A. J., Sokol A. A., Catlow C. R. A., Sherwood P., and Keal T. W., “Open-source, Python-based redevelopment of the ChemShell multiscale QM/MM environment,” J. Chem. Theory Comput. 15, 1317–1328 (2019). 10.1021/acs.jctc.8b01036 [DOI] [PubMed] [Google Scholar]
- 34.O’Boyle N. M., Tenderholt A. L., and Langner K. M., “cclib: A library for package-independent computational chemistry algorithms,” J. Comput. Chem. 29, 839–845 (2008). 10.1002/jcc.20823 [DOI] [PubMed] [Google Scholar]
- 35.Langner K. M. and Berquist E., CCLIB: Parsers and algorithms for computational chemistry logfiles. For the current version, see https://github.com/cclib/cclib; accessed May 2021, 10.5281/zenodo.1407790. [DOI]
- 36.Verstraelen T., Tecmer P., Heidar-Zadeh F., González-Espinoza C. E., Chan M., Kim T. D., Boguslawski K., Fias S., Vandenbrande S., Berrocal D., and Ayers P. W., HORTON, version 2.1.1, a helpful open-source research tool for n-fermion systems, see http://theochem.github.com/horton/.
- 37.Smith D. G. A., Burns L. A., Naden L., and Welborn M., QCELEMENTAL: Periodic table, physical constants, and molecule parsing for quantum chemistry. For the current version, see https://github.com/MolSSI/QCElemental; accessed January 2020.
- 38.Smith D. G. A., Lee S., Burns L. A., and Welborn M., QCENGINE: Quantum chemistry program executor and IO standardizer (QCSchema). For the current version, see https://github.com/MolSSI/QCEngine; accessed January 2020.
- 39.Smith D. G. A., Welborn M., Altarawy D., and Naden L., QCFRACTAL: A distributed compute and database platform for quantum chemistry. For the current version, see https://github.com/MolSSI/QCFractal; accessed January 2020.
- 40.Smith D. G. A., Burns L. A., Altarawy D., Naden L., and Welborn M., QCARCHIVE: A central source to compile, aggregate, query, and share quantum chemistry data, https://qcarchive.molssi.org; accessed January 2020.
- 41.Burns L. A., Lolinco A. T., Glick Z. L., Lee J., and Silva N. D., QCDB: Quantum chemistry common driver and databases. For the current version, see https://github.com/qcdb/qcdb; accessed January 2020.
- 42.Grecco H., PINT: Operate and manipulate physical quantities in Python. For the current version, see https://github.com/hgrecco/pint; accessed April 2020.
- 43.Colvin S., PYDANTIC: Data parsing and validation using Python type hints. For the current version, see https://github.com/samuelcolvin/pydantic; accessed April 2020.
- 44.Herbst M. F., Scheurer M., Fransson T., Rehn D. R., and Dreuw A., “adcc: A versatile toolkit for rapid development of algebraic-diagrammatic construction methods,” Wiley Interdiscip. Rev.: Comput. Mol. Sci. 10, e1462 (2020). 10.1002/wcms.1462 [DOI] [Google Scholar]
- 45.Herbst M. F. and Scheurer M., ADCC: Seamlessly connect your program to ADC. For the current version, see https://github.com/adc-connect/adcc; accessed January 2020. For the originating project, see https://adc-connect.org.
- 46.Matthews D. A., Cheng L., Harding M. E., Lipparini F., Stopkowicz S., Jagau T.-C., Szalay P. G., Gauss J., and Stanton J. F., “Coupled-cluster techniques for computational chemistry: The CFOUR program package,” J. Chem. Phys. 152, 214108 (2020). 10.1063/5.0004837 [DOI] [PubMed] [Google Scholar]
- 47.Barca G. M. J., Bertoni C., Carrington L., Datta D., De Silva N., Deustua J. E., Fedorov D. G., Gour J. R., Gunina A. O., Guidez E., Harville T., Irle S., Ivanic J., Kowalski K., Leang S. S., Li H., Li W., Lutz J. J., Magoulas I., Mato J., Mironov V., Nakata H., Pham B. Q., Piecuch P., Poole D., Pruitt S. R., Rendell A. P., Roskop L. B., Ruedenberg K., Sattasathuchana T., Schmidt M. W., Shen J., Slipchenko L., Sosonkina M., Sundriyal V., Tiwari A., Galvez Vallejo J. L., Westheimer B., Włoch M., Xu P., Zahariev F., and Gordon M. S., “Recent developments in the general atomic and molecular electronic structure system,” J. Chem. Phys. 152, 154102 (2020). 10.1063/5.0005188 [DOI] [PubMed] [Google Scholar]
- 48.Werner H.-J., Knowles P. J., Knizia G., Manby F. R., Schütz M., Celani P., Györffy W., Kats D., Korona T., Lindh R., Mitrushenkov A., Rauhut G., Shamasundar K. R., Adler T. B., Amos R. D., Bennie S. J., Bernhardsson A., Berning A., Cooper D. L., Deegan M. J. O., Dobbyn A. J., Eckert F., Goll E., Hampel C., Hesselmann A., Hetzer G., Hrenar T., Jansen G., Köppl C., Lee S. J. R., Liu Y., Lloyd A. W., Ma Q., Mata R. A., May A. J., McNicholas S. J., Meyer W., Miller T. F. III, Mura M. E., Nicklass A., O’Neill D. P., Palmieri P., Peng D., Pflüger K., Pitzer R., Reiher M., Shiozaki T., Stoll H., Stone A. J., Tarroni R., Thorsteinsson T., Wang M., and Welborn M., molpro, version 2019.2, a package of ab initio programs, 2019, see https://www.molpro.net.
- 49.Werner H.-J., Knowles P. J., Manby F. R., Black J. A., Doll K., Heßelmann A., Kats D., Köhn A., Korona T., Kreplin D. A., Ma Q., Miller T. F., Mitrushchenkov A., Peterson K. A., Polyak I., Rauhut G., and Sibaev M., “The Molpro quantum chemistry package,” J. Chem. Phys. 152, 144107 (2020). 10.1063/5.0005081 [DOI] [PubMed] [Google Scholar]
- 50.Bast R., Bjørgve M., Di Remigio R., Durdek A., Frediani L., Gerez G., Jensen S. R., Juselius J., Monstad R., and Wind P., MRChem: MultiResolution Chemistry, 2020.
- 51.Jensen S. R., Saha S., Flores-Livas J. A., Huhn W., Blum V., Goedecker S., and Frediani L., “The elephant in the room of density functional theory calculations,” J. Phys. Chem. Lett. 8, 1449–1457 (2017). 10.1021/acs.jpclett.7b00255 [DOI] [PubMed] [Google Scholar]
- 52.Aprà E., Bylaska E. J., de Jong W. A., Govind N., Kowalski K., Straatsma T. P., Valiev M., van Dam H. J. J., Alexeev Y., Anchell J., Anisimov V., Aquino F. W., Atta-Fynn R., Autschbach J., Bauman N. P., Becca J. C., Bernholdt D. E., Bhaskaran-Nair K., Bogatko S., Borowski P., Boschen J., Brabec J., Bruner A., Cauët E., Chen Y., Chuev G. N., Cramer C. J., Daily J., Deegan M. J. O., Dunning T. H., Dupuis M., Dyall K. G., Fann G. I., Fischer S. A., Fonari A., Früchtl H., Gagliardi L., Garza J., Gawande N., Ghosh S., Glaesemann K., Götz A. W., Hammond J., Helms V., Hermes E. D., Hirao K., Hirata S., Jacquelin M., Jensen L., Johnson B. G., Jónsson H., Kendall R. A., Klemm M., Kobayashi R., Konkov V., Krishnamoorthy S., Krishnan M., Lin Z., Lins R. D., Littlefield R. J., Logsdail A. J., Lopata K., Ma W., Marenich A. V., Martin del Campo J., Mejia-Rodriguez D., Moore J. E., Mullin J. M., Nakajima T., Nascimento D. R., Nichols J. A., Nichols P. J., Nieplocha J., Otero-de-la Roza A., Palmer B., Panyala A., Pirojsirikul T., Peng B., Peverati R., Pittner J., Pollack L., Richard R. M., Sadayappan P., Schatz G. C., Shelton W. A., Silverstein D. W., Smith D. M. A., Soares T. A., Song D., Swart M., Taylor H. L., Thomas G. S., Tipparaju V., Truhlar D. G., Tsemekhman K., Van Voorhis T., Vázquez-Mayagoitia Á., Verma P., Villa O., Vishnu A., Vogiatzis K. D., Wang D., Weare J. H., Williamson M. J., Windus T. L., Woliński K., Wong A. T., Wu Q., Yang C., Yu Q., Zacharias M., Zhang Z., Zhao Y., and Harrison R. J., “NWChem: Past, present, and future,” J. Chem. Phys. 152, 184102 (2020). 10.1063/5.0004997 [DOI] [PubMed] [Google Scholar]
- 53.Smith D. G. A., Burns L. A., Simmonett A. C., Parrish R. M., Schieber M. C., Galvelis R., Kraus P., Kruse H., Remigio R. D., Alenaizan A., James A. M., Lehtola S., Misiewicz J. P., Scheurer M., Shaw R. A., Schriber J. B., Xie Y., Glick Z. L., Sirianni D. A., O’Brien J. S., Waldrop J. M., Kumar A., Hohenstein E. G., Pritchard B. P., Brooks B. R., Schaefer H. F. III, Sokolov A. Y., Patkowski K., DePrince A. E. III, Bozkaya U., King R. A., Evangelista F. A., Turney J. M., Crawford T. D., and Sherrill C. D., “PSI4 1.4: Open-source software for high-throughput quantum chemistry,” J. Chem. Phys. 152, 184108 (2020). 10.1063/5.0006002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao Y., Gan Z., Epifanovsky E., Gilbert A. T., Wormit M., Kussmann J., Lange A. W., Behn A., Deng J., Feng X., Ghosh D., Goldey M., Horn P. R., Jacobson L. D., Kaliman I., Khaliullin R. Z., Kuś T., Landau A., Liu J., Proynov E. I., Rhee Y. M., Richard R. M., Rohrdanz M. A., Steele R. P., Sundstrom E. J., Woodcock H. L. III, Zimmerman P. M., Zuev D., Albrecht B., Alguire E., Austin B., Beran G. J. O., Bernard Y. A., Berquist E., Brandhorst K., Bravaya K. B., Brown S. T., Casanova D., Chang C.-M., Chen Y., Chien S. H., Closser K. D., Crittenden D. L., Diedenhofen M., R. A. DiStasio, Jr., Do H., Dutoi A. D., Edgar R. G., Fatehi S., Fusti-Molnar L., Ghysels A., Golubeva-Zadorozhnaya A., Gomes J., Hanson-Heine M. W., Harbach P. H., Hauser A. W., Hohenstein E. G., Holden Z. C., Jagau T.-C., Ji H., Kaduk B., Khistyaev K., Kim J., Kim J., King R. A., Klunzinger P., Kosenkov D., Kowalczyk T., Krauter C. M., Lao K. U., Laurent A. D., Lawler K. V., Levchenko S. V., Lin C. Y., Liu F., Livshits E., Lochan R. C., Luenser A., Manohar P., Manzer S. F., Mao S.-P., Mardirossian N., Marenich A. V., Maurer S. A., Mayhall N. J., Neuscamman E., Oana C. M., Olivares-Amaya R., O’Neill D. P., Parkhill J. A., Perrine T. M., Peverati R., Prociuk A., Rehn D. R., Rosta E., Russ N. J., Sharada S. M., Sharma S., Small D. W., Sodt A., Stein T., Stück D., Su Y.-C., Thom A. J., Tsuchimochi T., Vanovschi V., Vogt L., Vydrov O., Wang T., Watson M. A., Wenzel J., White A., Williams C. F., Yang J., Yeganeh S., Yost S. R., You Z.-Q., Zhang I. Y., Zhang X., Zhao Y., Brooks B. R., Chan G. K., Chipman D. M., Cramer C. J., Goddard W. A. III, Gordon M. S., Hehre W. J., Klamt A., Schaefer H. F. III, Schmidt M. W., Sherrill C. D., Truhlar D. G., Warshel A., Xu X., Aspuru-Guzik A., Baer R., Bell A. T., Besley N. A., Chai J.-D., Dreuw A., Dunietz B. D., Furlani T. R., Gwaltney S. R., Hsu C.-P., Jung Y., Kong J., Lambrecht D. S., Liang W., Ochsenfeld C., Rassolov V. A., Slipchenko L. V., Subotnik J. E., Voorhis T. V., Herbert J. M., Krylov A. I., Gill P. M., and Head-Gordon M., “Advances in molecular quantum chemistry contained in the Q-Chem 4 program package,” Mol. Phys. 113, 184–215 (2015). 10.1080/00268976.2014.952696 [DOI] [Google Scholar]
- 55.Manby F., Miller T., Bygrave P., Ding F., Dresselhaus T., Batista-Romero F., Buccheri A., Bungey C., Lee S., Meli R. et al. , “Entos: A quantum molecular simulation package,” in ChemRxiv (Cambridge Open Engage, 2019). [Google Scholar]
- 56.Ufimtsev I. S. and Martínez T. J., “Quantum chemistry on graphical processing units. 3. Analytical energy gradients, geometry optimization, and first principles molecular dynamics,” J. Chem. Theory Comput. 5, 2619–2628 (2009). 10.1021/ct9003004 [DOI] [PubMed] [Google Scholar]
- 57.Seritan S., Bannwarth C., Fales B. S., Hohenstein E. G., Kokkila-Schumacher S. I. L., Luehr N., Snyder J. W., Song C., Titov A. V., Ufimtsev I. S., and Martínez T. J., “TeraChem: Accelerating electronic structure and ab initio molecular dynamics with graphical processing units,” J. Chem. Phys. 152, 224110 (2020). 10.1063/5.0007615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Furche F., Ahlrichs R., Hättig C., Klopper W., Sierka M., and Weigend F., “Turbomole,” Wiley Interdiscip. Rev.: Comput. Mol. Sci. 4, 91–100 (2014). 10.1002/wcms.1162 [DOI] [Google Scholar]
- 59.Balasubramani S. G., Chen G. P., Coriani S., Diedenhofen M., Frank M. S., Franzke Y. J., Furche F., Grotjahn R., Harding M. E., Hättig C., Hellweg A., Helmich-Paris B., Holzer C., Huniar U., Kaupp M., Marefat Khah A., Karbalaei Khani S., Müller T., Mack F., Nguyen B. D., Parker S. M., Perlt E., Rappoport D., Reiter K., Roy S., Rückert M., Schmitz G., Sierka M., Tapavicza E., Tew D. P., van Wüllen C., Voora V. K., Weigend F., Wodyński A., and Yu J. M., “TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations,” J. Chem. Phys. 152, 184107 (2020). 10.1063/5.0004635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stewart J. J. P., MOPAC: Semiempirical quantum chemistry. For the current version, see http://OpenMOPAC.net/; accessed January 2020, for Stewart Computational Chemistry, Colorado Springs, CO.
- 61.Bannwarth C., Caldeweyher E., Ehlert S., Hansen A., Pracht P., Seibert J., Spicher S., and Grimme S., “Extended tight-binding quantum chemistry methods,” Wiley Interdiscip. Rev.: Comput. Mol. Sci. 11, e1493 (2021). 10.1002/wcms.1493 [DOI] [Google Scholar]
- 62.Eastman P., Swails J., Chodera J. D., McGibbon R. T., Zhao Y., Beauchamp K. A., Wang L.-P., Simmonett A. C., Harrigan M. P., Stern C. D., Wiewiora R. P., Brooks B. R., and Pande V. S., “OpenMM 7: Rapid development of high performance algorithms for molecular dynamics,” PLoS Comput. Biol. 13, e1005659 (2017). 10.1371/journal.pcbi.1005659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landrum G., RDKIT: Cheminformatics and machine-learning software in C++ and Python. For the current version, see 10.5281/zenodo.591637; accessed January 2020. For the originating project, see https://www.rdkit.org/.
- 64.Grimme S., Antony J., Ehrlich S., and Krieg H., DFTD3: Dispersion correction for DFT, Hartree–Fock, and semi-empirical quantum chemical methods. For the current version, see https://github.com/loriab/dftd3; accessed January 2020. For the originating project, see https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/dft-d3.
- 65.Ehrlich S. and Caldewayher E., DFTD4: Generally applicable atomic-charge dependent London dispersion correction. For the current version, see https://github.com/dftd4/dftd4; accessed April 2021.
- 66.Caldeweyher E., Bannwarth C., and Grimme S., “Extension of the D3 dispersion coefficient model,” J. Chem. Phys. 147, 034112 (2017). 10.1063/1.4993215 [DOI] [PubMed] [Google Scholar]
- 67.Kruse H. and Grimme S., GCP: Geometrical counterpoise correction for DFT and Hartree–Fock quantum chemical methods. For the current version, see https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/gcp/gcp; accessed January 2020.
- 68.Kruse H. and Grimme S., “A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree–Fock and density functional theory calculations for large systems,” J. Chem. Phys. 136, 154101 (2012). 10.1063/1.3700154 [DOI] [PubMed] [Google Scholar]
- 69.Greenwell C., MP2D: A program for calculating the MP2D dispersion energy. For the current version, see https://github.com/Chandemonium/MP2D; accessed January 2020.
- 70.Řezáč J., Greenwell C., and Beran G. J. O., “Accurate noncovalent interactions via dispersion-corrected second-order Møller–Plesset perturbation theory,” J. Chem. Theory Comput. 14, 4711–4721 (2018). 10.1021/acs.jctc.8b00548 [DOI] [PubMed] [Google Scholar]
- 71.Gao X., TORCHANI: Accurate neural network potential on PyTorch. For the current version, see https://github.com/aiqm/torchani; accessed January 2020.
- 72.Smith J. S., Isayev O., and Roitberg A. E., “ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost,” Chem. Sci. 8, 3192–3203 (2017). 10.1039/c6sc05720a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao X., Ramezanghorbani F., Isayev O., Smith J. S., and Roitberg A. E., “TorchANI: A free and open source PyTorch-based deep learning implementation of the ANI neural network potentials,” J. Chem. Inf. Model. 60, 3408–3415 (2020). 10.1021/acs.jcim.0c00451 [DOI] [PubMed] [Google Scholar]
- 74.Frediani L., Fossgaard E., Flå T., and Ruud K., “Fully adaptive algorithms for multivariate integral equations using the non-standard form and multiwavelets with applications to the Poisson and bound-state Helmholtz kernels in three dimensions,” Mol. Phys. 111, 1143–1160 (2013). 10.1080/00268976.2013.810793 [DOI] [Google Scholar]
- 75.Hirata S., “Tensor contraction engine: Abstraction and automated parallel implementation of configuration-interaction, coupled-cluster, and many-body perturbation theories,” J. Phys. Chem. A 107, 9887–9897 (2003). 10.1021/jp034596z [DOI] [Google Scholar]
- 76.PROTOCOL BUFFERS: A language-neutral, platform-neutral extensible mechanism for serializing structured data. For the current version, see https://developers.google.com/protocol-buffers/docs/reference/proto3-spec; accessed May 2021.
- 77.Seritan S., Thompson K., and Martínez T. J., “TeraChem cloud: A high-performance computing service for scalable distributed GPU-accelerated electronic structure calculations,” J. Chem. Inf. Model. 60, 2126–2137 (2020). 10.1021/acs.jcim.9b01152 [DOI] [PubMed] [Google Scholar]
- 78.Seritan S., Hicks C. B., and Ford J. E., TCPB: Python client for TeraChem’s protobuf server mode. For the current version, see https://github.com/mtzgroup/tcpb-client; accessed May 2021.
- 79.Grimme S., “Semiempirical GGA-type density functional constructed with a long-range dispersion correction,” J. Comput. Chem. 27, 1787–1799 (2006). 10.1002/jcc.20495 [DOI] [PubMed] [Google Scholar]
- 80.Smith D. G. A., Burns L. A., Patkowski K., and Sherrill C. D., “Revised damping parameters for the D3 dispersion correction to density functional theory,” J. Phys. Chem. Lett. 7, 2197–2203 (2016). 10.1021/acs.jpclett.6b00780 [DOI] [PubMed] [Google Scholar]
- 81.Caldeweyher E. and Brandenburg J. G., “Simplified DFT methods for consistent structures and energies of large systems,” J. Phys.: Condens. Matter 30, 213001 (2018). 10.1088/1361-648x/aabcfb [DOI] [PubMed] [Google Scholar]
- 82.See https://openforcefield.org for OpenForceField.
- 83.Wang L.-P., Smith D. G. A., and Qiu Y., GEOMETRIC: A geometry optimization code that includes the TRIC coordinate system. For the current version, see https://github.com/leeping/geomeTRIC; accessed January 2020.
- 84.Wang L.-P. and Song C., “Geometry optimization made simple with translation and rotation coordinates,” J. Chem. Phys. 144, 214108 (2016). 10.1063/1.4952956 [DOI] [PubMed] [Google Scholar]
- 85.Heide A. and King R. A., OptKing: A Python version of the Psi4 geometry optimizer. For the current version, see https://github.com/psi-rking/optking; accessed January 2020.
- 86.Hermann J., PYBERNY: Molecular structure optimizer. For the current version, see https://github.com/jhrmnn/pyberny; accessed January 2020. Also, for Version 0.6.2 10.5281/zenodo.3695038. [DOI]
- 87.The finite difference and vibrational analysis procedures have been extracted from PSI4 as an independent module in QCDB. For the moment, there is a lingering library dependence on PSI4 for SALCs, so it, too, must be installed (as indicated in Fig. 3).
- 88.Wells B. H. and Wilson S., “van der Waals interaction potentials: Many-body basis set superposition effects,” Chem. Phys. Lett. 101, 429–434 (1983). 10.1016/0009-2614(83)87508-3 [DOI] [Google Scholar]
- 89.Valiron P. and Mayer I., “Hierarchy of counterpoise corrections for N-body clusters: Generalization of the Boys-Bernardi scheme,” Chem. Phys. Lett. 275, 46–55 (1997). 10.1016/s0009-2614(97)00689-1 [DOI] [Google Scholar]
- 90.Kaliman I., LIBEFP: Parallel implementation of the effective fragment potential method. For the current version, see https://github.com/ilyak/libefp; accessed January 2020. [DOI] [PubMed]
- 91.Kaliman I. A. and Slipchenko L. V., “LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library,” J. Comput. Chem. 34, 2284–2292 (2013). 10.1002/jcc.23375 [DOI] [PubMed] [Google Scholar]
- 92.Bayly C. I., Cieplak P., Cornell W., and Kollman P. A., “A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model,” J. Phys. Chem. 97, 10269–10280 (1993). 10.1021/j100142a004 [DOI] [Google Scholar]
- 93.Alenaizan A., RESP: A restrained electrostatic potential (RESP) plugin to PSI4. For the current version, see https://github.com/cdsgroup/resp; accessed January 2020.
- 94.Alenaizan A., Burns L. A., and Sherrill C. D., “Python implementation of the restrained electrostatic potential charge model,” Int. J. Quantum Chem. 120, e26035 (2020). 10.1002/qua.26035 [DOI] [Google Scholar]
- 95.Borca C. H., CRYSTALATTE: Automating the calculation of crystal lattice energies. For the current version, see https://github.com/carlosborca/CrystaLattE; accessed January 2020.
- 96.Borca C. H., Bakr B. W., Burns L. A., and Sherrill C. D., “CrystaLattE: Automated computation of lattice energies of organic crystals exploiting the many-body expansion to achieve dual-level parallelism,” J. Chem. Phys. 151, 144103 (2019). 10.1063/1.5120520 [DOI] [PubMed] [Google Scholar]
- 97.The QCElemental molecule parsing machinery is also used for PSI4, so its documentation and http://docs.qcarchive.molssi.org/projects/QCElemental/en/latest/model_molecule.html can be helpful. The following describes the particular case in the text. In Snippet 1, the units bohr string indicates that the Cartesian coordinates are already in QCSchema’s required atomic units, so these are unchanged in Snippet 2’s geometry field. The Snippet 1 strings O, H, and Ne specify the elements and are processed into Snippet 2 fields atomic_numbers and symbols. The prefix character @ to neon in Snippet 1 indicates it’s a ghost atom, so the Snippet 2 field real shows a T, T, F pattern. Gh(22Ne) would have been equivalent to the given @22Ne. The prefix string 22 to neon in Snippet 1 specifies the mass number, much like a nuclide symbol. Thus the Snippet 2 fields mass_numbers and masses use default values for the oxygen and hydrogen but 22Ne values for neon. @Ne@21.99138511 to specify the mass value would have been equivalent. The strings no_com and no_reorient were not given in Snippet 1, so the fields fix_com and fix_orientation in Snippet 2 are F, meaning that the origin and frame of geometry are incidental to the Molecule specification. A user label like O1 or O_bigbasis is parsed, but since Snippet 1 doesn’t include any, the atom_labels field of Snippet 2 are empty strings. The--line of Snippet 1 indicates there are two fragments in the molecule, the first with two atoms and the second with one. This is encoded in the fragments field of Snippet 2. No charge/multiplicity lines are present in Snippet 1, either overall or per-fragment, so defaults are assigned. The second fragment is all ghosts and so is a neutral singlet. Electrons are never added or removed to the specification, so the first fragment is assigned neutral doublet, and the overall molecule is a neutral doublet. These defaults are reflected in the Snippet 2 fields molecular_charge, molecular_multiplicity, fragment_charges, and fragment_multiplicities. The string parser also stamps the schema name and provenance information in Snippet 2.
- 98.In full, the command requests a Dunning 5ζ to 6ζ Helgaker-formula extrapolation of the MP2 correlation gradient performed by PSI4 with a coupled-cluster singles and doubles excitations correction (CCSD−MP2) at the Dunning triple-ζ to quadruple-ζ Helgaker-formula extrapolation gradient performed by NWCHEM with a CC up to quadruples excitations at cc-pVDZ performed by CFOUR, all atop an implicit 6-ζ Hartree–Fock.
- 99.Pritchard B. P., Altarawy D., Didier B., Gibson T. D., and Windus T. L., “New basis set exchange: An open, up-to-date resource for the molecular sciences community,” J. Chem. Inf. Model. 59, 4814–4820 (2019). 10.1021/acs.jcim.9b00725 [DOI] [PubMed] [Google Scholar]
- 100.Naoki I., MESSAGEPACK-PYTHON: MessagePack serializer implementation for Python. For the current version, see https://github.com/msgpack/msgpack-python; accessed January 2020. For the originating project, see https://msgpack.org/.
- 101.Bytautas L., Matsunaga N., Nagata T., Gordon M. S., and Ruedenberg K., “Accurate ab initio potential energy curve of F2. III. The vibration rotation spectrum,” J. Chem. Phys. 127, 204313 (2007). 10.1063/1.2805392 [DOI] [PubMed] [Google Scholar]
- 102.Martin J. M. L., “Benchmark ab initio potential curves for the light diatomic hydrides. Unusually large nonadiabatic effects in BeH and BH,” Chem. Phys. Lett. 283, 283–293 (1998). 10.1016/s0009-2614(97)01400-0 [DOI] [Google Scholar]
- 103.Temelso B., Valeev E. F., and Sherrill C. D., “A comparison of one-particle basis set completeness, higher-order electron correlation, relativistic effects, and adiabatic corrections for spectroscopic constants of BH, CH+, and NH,” J. Phys. Chem. A 108, 3068–3075 (2004). 10.1021/jp036933+ [DOI] [Google Scholar]
- 104.Boschen J. S., Theis D., Ruedenberg K., and Windus T. L., “Accurate ab initio potential energy curves and spectroscopic properties of the four lowest singlet states of C2,” Theor. Chem. Acc. 133, 1425 (2013). 10.1007/s00214-013-1425-x [DOI] [Google Scholar]
- 105.Bender J. D., Doraiswamy S., Truhlar D. G., and Candler G. V., “Potential energy surface fitting by a statistically localized, permutationally invariant, local interpolating moving least squares method for the many-body potential: Method and application to N4,” J. Chem. Phys. 140, 054302 (2014). 10.1063/1.4862157 [DOI] [PubMed] [Google Scholar]
- 106.Fernando W. T. M. L. and Bernath P. F., “Fourier transform spectroscopy of the A1Π-X1Σ+ transition of BH and BD,” J. Mol. Spectrosc. 145, 392–402 (1991). 10.1016/0022-2852(91)90126-u [DOI] [Google Scholar]
- 107.Huber K. P. and Herzberg G., Constants of Diatomic Molecules (Van Nostrand Reinhold, New York, 1979). [Google Scholar]
- 108.Douay M., Nietmann R., and Bernath P. F., “New observations of the transition (Phillips system) of C2,” J. Mol. Spectrosc. 131, 250–260 (1988). 10.1016/0022-2852(88)90236-6 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available within the article.