

Graphical abstract
COVID-19 is a pandemic disease caused by novel corona virus, SARS-CoV-2, initially originated from China. In response to this serious life-threatening disease, designing and developing more accurate and sensitive tests are crucial. The aim of this study is designing a multi-epitope of spike and nucleocapsid antigens of COVID-19 virus by bioinformatics methods. The sequences of nucleotides obtained from the NCBI Nucleotide Database. Transmembrane structures of proteins were predicted by TMHMM Server and the prediction of signal peptide of proteins was performed by Signal P Server. B-cell epitopes’ prediction was performed by the online prediction server of IEDB server. Beta turn structure of linear epitopes was also performed using the IEDB server. Conformational epitope prediction was performed using the CBTOPE and eventually, eight antigenic epitopes with high physicochemical properties were selected, and then, all eight epitopes were blasted using the NCBI website. The analyses revealed that α-helices, extended strands, β-turns, and random coils were 28.59%, 23.25%, 3.38%, and 44.78% for S protein, 21.24%, 16.71%, 6.92%, and 55.13% for N Protein, respectively. The S and N protein three-dimensional structure was predicted using the prediction I-TASSER server. In the current study, bioinformatics tools were used to design a multi-epitope peptide based on the type of antigen and its physiochemical properties and SVM method (Machine Learning) to design multi-epitopes that have a high avidity against SARS-CoV-2 antibodies to detect infections by COVID-19.

Keywords: SARS-CoV2, Serological tests, Multi-epitopes, Nucleocapsid phosphoprotein, Spike glycoprotein
Introduction
Coronavirus disease 2019 (COVID-19), that was first identified in Wuhan, China, in December 2019 and currently is an ongoing pandemic, is a contagious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Lai et al. 2020). On 3rd March 2020, the World Health Organization announced: Worldwide, 3.4% of reported cases died of COVID-19) Li, Xu et al. 2020). The symptoms of COVID-19 present most commonly as fever, dry cough, and fatigue. However, less commonly, it can present as diarrhea, headache, insomnia, and ageusia (Ho et al. 2020). Less frequently, more serious symptoms such as dyspnea and chest pain and acute respiratory distress syndrome (ARDS) also may be presented (Baj et al. 2020). Typically, 5–6 days after exposure to the virus the symptoms emerge, however, in some cases, the incubation period can be about 14 days (Lauer et al. 2020).The ARDS is not the only serious consequence of the infection (Acosta and Singer 2020). Patients can suffer from other life-threatening conditions due to strong and uncontrolled inflammatory responses such as cytokine storms, thrombosis, and coagulopathy and disseminated intravascular coagulation (DIC), multi-organ failure, and septic shock (Ferrer-Oliveras et al. 2021). Furthermore, damages to organs including the lungs and heart can become chronic and last even all over the life.
An effective suggested strategy for reducing the spread of the virus is to provide the facilities to track the virus spread and prevent it from circulating freely among the population, by testing as many individuals as possible, detecting infected people and whom they have contact with. Although this strategy is now implemented using an array of testing methods, but due to the large required resources, it can have a large number of limitations for health systems, especially in developing countries. Therefore, designing and developing more accurate and sensitive tests are crucial during the COVID-19 pandemic. There are different testing models that can detect the virus directly, e.g., by detecting the viral RNA, or indirectly for example through measuring antibodies against the virus in the bodies that are known as Immunoassays (Udugama et al. 2020; Liu and Rusling 2021). Sufficient sensitivity and accuracy are the requirements that make a diagnostic test method appropriate to be used during a pandemic. Real-time PCR that directly detects the viral genome is a most well-known laboratory test for the SARS-CoV-2 detection (Udugama et al. 2020). Recent studies have shown that the SARS-CoV-2 mainly infects the lower respiratory tract and that virus RNA can be detected through nasopharyngeal swabs and Bronchoalveolar lavage (BAL) specimens (Huang et al. 2020a, b; Liu et al. 2020a, b). However, sampling of the lower respiratory tract (especially BAL specimens) requires a proper suction device and a skilled operator. Serological methods are precise and efficient techniques for screening pathogenic organisms. Immunoassays are used to measure the specific antibodies against the virus (Chansaenroj et al. 2021). Another possibility is using point-of-care (POC) and rapid test methods that do not require laboratory instruments that dismiss the logistic and economic hurdles to a large extent and can be performed in a substantially shorter time (Chansaenroj et al. 2021).
There is a tough challenge between designing of both rapid and conventional immunoassays cross-reactivity with other viral diseases. SARS-CoV-2 has an RBD (receptor-binding domain) structure like that of the SARS-CoV. Functionally, major structural proteins, including the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins, are also well reported (Lu et al. 2020). According to previous studies, the M and E proteins are necessary for virus assembly. The spike glycoprotein is substantial for attachment to host cells, where the RBD (SARS-CoV-2 RNA-binding domain) of spike glycoprotein mediates the interaction with angiotensin-converting enzyme 2 (ACE2) (Zhou et al. 2020). The spike glycoprotein is located on the surface of the SARS-CoV-2 and the results of recent studies have shown that it is highly immunogenic (Woo et al. 2005). The nucleocapsid protein is one of the main structural proteins of the SARS-CoV-2, and plays an important role in transcription and replication of viral RNA and interference with cell cycle processes of the host cell (Liu et al. 2020a, b). Furthermore, in SARS-CoV and other coronaviruses, the nucleocapsid protein has high antigenic and immunogenic activity and is highly expressed during infection (Che et al. 2004; Liu et al. 2020a, b). Both nucleocapsid and spike proteins may be potential antigenic proteins for sero-diagnosis of the COVID-19, just as many diagnostic methods have been developed for diagnosing the SARS-CoV-2 based on nucleocapsid (N) and spike(S) proteins (Liu et al. 2020a, b). It seems that designing the most suitable immunogenic protein consisting of several immunogenic epitopes of two antigens S and N (proteins currently are being used in ELISA kits) is useful to increase the specificity and sensitivity of immunoassay-based tests. Having a multi-epitopic peptide that meets high specificity and sensitivity requirements that is a bottleneck in the immunoassay design processes facilitates development of rapid diagnostic tests with acceptable benefits (Aghamolaei et al. 2020; Habibi et al. 2020; Mamaghani et al. 2020). Therefore, the aim of the present study is to predict and design a novel synthetic (fusion) protein consisting of multiple immune dominant B-cell epitopes from N and S proteins of SARS-CoV-2.
Methods and methods
Protein sequences
The sequences of nucleotides were obtained from the National Centre for Biotechnology Information (NCBI) Nucleotide Database [N protein (GenBank: QIZ15545.1) and S protein (GenBank: P0DTC2.1)], and the protein sequences were acquired from UniProt (Universal Protein resource) database of proteins. The protein sequences were recovered in an FASTA format by their accession number.
Membrane protein topology and signal peptide prediction
The transmembrane structures of proteins were predicted by TMHMM Server v. 2.0 (https://www.cbs.dtu.dk/services/ TMHMM/). The sequences of proteins were presented to the server as input, and outside, transmembrane and inside regions, were analyzed (Krogh et al. 2001). The prediction of signal peptide of proteins was performed by Signal P 4.1 Server (https://www.cbs.dtu.dk/services/Signal P/) (Geourjon and Deleage 1995).
Predicting the secondary structure of S and N proteins
The secondary structures of S and N proteins were predicted by the “self-optimized prediction method with alignment” (SOPMA) server (https://www.npsaprabi.ibcp.fr/cgibin/npsa_automat.plpage = npsa_sopma.html). It can predict with a prediction accuracy of 69.5% the three-state description of secondary structure (random coil, β-sheet, and α-helix) in a collection of non-homologous proteins that have less than 25% identity (Geourjon and Deleage 1995).
Tertiary structure prediction and validation
Three-dimensional protein structures of S and N proteins were predicted using the I-TASSER online prediction server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). It is an online platform that implements the I-TASSER-based algorithms to predict protein 3D structure and function (He et al. 2002; Yang and Zhang 2015 https://doi.org/10.1038/s41586-020-2772-0). Consequently, the predicted structures were validated by Ramachandran plots by PROCHECK program (https://saves.mbi.ucla.edu/) (PROCHECK; https://servicesn.mbi.ucla.edu/PROCHECK). Furthermore, they were validated using the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php). Finally, the Verify 3D program (https://saves.mbi.ucla.edu/) (https://servicesn.mbi.ucla.edu/Verify3D/) was applied to determine the compatibility of the 3D model with its amino acid sequence (ID) by assigning the structural class (alpha, beta, loop, polar, nonpolar, etc.) based on its location and environment.
Prediction of S and N proteins’ B-cell epitopes
B-cell epitopes’ prediction was performed by the online prediction server Immune Epitope Database (IEDB; http://tools.immuneepitope.org/main/bcell/). In the IEDB server, linear epitopes’ prediction is implemented using the Bepipred-1.0 Linear Epitope Prediction method (http://tools.iedb.org/maim/bcell/). It uses combination of a hidden Markov model and propensity scale method. Residues with a score above 0.35 showed in yellow color and are considered as the predicted epitope candidates. Bepipred-2.0 Linear Epitope Prediction tool (https://services.healthtech.dtu.dk/) was also applied to confirm the predictions of linear epitopes.
Beta turn structure of linear epitopes was also performed using the IEDB server that is based on the Chou and Fasman beta-turn prediction method. For this prediction, threshold was set on about 1.162; the accuracy of this method is about 50–60%. The accessibility scale of linear B-cell epitopes was evaluated using the Emini Surface accessibility scale with the threshold score defined at 1.00. Numbers greater than 1.00 indicate an increased probability for being found on the surface. This method was also accessed through IEDB. The Karplus and Schulz flexibility scale method, also available on IEDB server, was used to predict the flexible area of linear B-cell epitopes. The prediction threshold for this method is defined at 1.054. On the same server, the semi-empirical Kolaskar and Tongaonkar antigenicity scale method was administered to predict the high antigenicity areas on linear B-cell epitopes. This method implements the physicochemical properties of amino acid residues and their frequencies of occurrence in experimentally known segmental epitopes for the prediction. It has been shown that the accuracy of the result is about 75% when the threshold is adjusted at about 0.991.
Parker Hydrophobicity Prediction (https://tools.iedb.org/bcell/) method was used to identify the regions with high hydrophilicity. In this method, the hydrophilic scale was calculated using peptic retention time during High-Performance Liquid Chromatography (HPLC) on a reversed-phase column. The threshold for this method was set at 3.769. Accordingly, the predictions of linear B-cell epitopes were performed using artificial neural network-based B-cell epitope prediction server (ABCpred; https://crdd.osdd.net/raghava/abcpred/). The accuracy of the predictions in this method is about 65.93%. The LBtope server (linear B-cell epitope prediction; https://crdd.osdd.net/raghava/lbtope/protein.php) discriminates between the linear B-cell epitopes and non-epitopes for a given sequence of protein with an overall accuracy of ~ 81%. In this server, a machine learning techniques or SVM (Support Vector Machine) score ranging from 20 to 100% (default threshold is set at 60%) is acquired during the prediction of each overlapping 20 mers of the sequence. The overall accuracy of this server is ~ 81%.
Conformational epitope prediction was performed using the CBTOPE (Conformational B-cell Epitope Prediction Server; https://crdd.osdd.net/raghava/cbtope/) to distinguish the residues composing antibody epitopes in the protein sequences. Similar to LBtope, this server also implements the SVM scoring method based on the amino acid composition of the query sequence. When the threshold is set at − 0.3, CBTOPE has accuracy of 85% approximately.
In the next step, ElliPro (derived from Ellipsoid and Protrusion) server was used for the prediction of B-cell epitopes. This model is based on solvent-accessibility and flexibility. ElliPro server is a structure-based web tool that predicts and visualizes antibody epitopes in protein sequences and structures (https://tools.iedb.org/ellipro/).
Designing multi-epitope
Finally, eight antigenic epitopes with high physicochemical properties were selected, and then, all eight epitopes were blasted using the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi.PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). A multi-epitope peptide was designed based on eight antigenic epitopes that were predicted by physicochemical methods from S and N Proteins predicted epitopes which were connected using linkers consisting of glycine and serine (Flexible linker). The three-dimensional structure of the multi-epitope peptide was predicted by Iterative Threading ASSEmbly Refinement (I-TASSER) server, and validated using Ramachandran plots, ProSA, and Verify3D servers. Consequently, the multi-epitope peptide properties were surveyed by ProtParam and SOL pro-tools. Physicochemical parameters including molecular weight, theoretical pI (isoelectric point), amino acid composition, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) were calculated. To obtain the highest foreigner gene expression level in the prokaryote host cell (Escherichia coli K12 strain) reverse translation, codon optimization and restriction enzyme cutting region removal were done using the Java codon adaptation tool (J-CAT) (Sandhu et al. 2008).
Results
The secondary structure, signal peptide, and transmembrane helices prediction of the S and N proteins
The analyses revealed that the α-helices, extended strands, β-turns, and random coils were 28.59%, 23.25%, 3.38%, and 44.78% for S protein, 21.24%, 16.71%, 6.92%, and 55.13% for N Protein, respectively (Fig. 1). The highest ratio of α-helices and random coils in the structure of antigens indicate the probability of their existence at antigenic epitopes. The amino acid positions 1–20 in the S protein sequence were coding the signal peptide region (Table 1). Furthermore, following transmembrane helices prediction analysis was found that the outside regions of S and N Proteins are located at positions 35–1287 and 1–419, respectively (Tables 1 and 2).
Fig. 1.

The secondary structure of retrieved amino acid sequences by SOPMA server. A Spike glycoprotein (S protein); B nucleocapsid phosphoprotein (N protein)
Table 1.
Prediction of linear and discontinuous B-cell epitopes using LBTope, ABCpred, IEDB, CBTope, and DiscoTope for nucleocapsid protein
| Type of prediction | Server name | Signal peptide and transmembrane topology prediction | Position of epitope |
|---|---|---|---|
| Linear epitope prediction | LBTope | Signal peptide: There is no signal peptides | 1–15, 8–22, 20–34, 153–155, 155–177 |
| ABCpred | Transmembrane topology prediction: out (extracellular: 1–419) | 91–107, 249–265, 136–152, 354–370, 24–40, 127–143, 327–343, 59–75,182–198, 376–392,12–28,77–93, 362–378, 389–305, 268–284,114–130, 243–259, 300–316, 206–222, 348–368, 334–350, 275–291, 237–253,162–178, 32–48, 317–333, 257–273, 169–185, 71–87, 49–65, 1–17 | |
| Linear epitope (Prediction by IEDB server) | BepiPred linear epitope | 4–15, 17–48, 59–105, 119–127, 137–163, 165–216, 226–267, 276–299, 343–348, 358–402, 404–416 | |
| Prediction | Antigenicity prediction Kolaskar and Tongaonkar | 52–59, 69–75, 83–89, 106–115, 119–124, 130–136,154–166, 217–227, 243–249, 267–273, 299–315, 333–339, 347–363, 379–385, 389–401, 403–411 | |
| Emini surface accessibility Prediction | 4–11, 36–42, 87–92, 185–197, 237–242, 254–264, 277–282, 295–300, 340–346, 365–377, 384–390 | ||
| Chou and FasmanBeta-Turn Prediction | 1–10, 19–40, 40–50, 65–87, 95–110, 115–130, 140–156, 165–215, 230–240, 275–290, 295–300, 340–350, 360–370, 410–420 | ||
| Parker hydrophilicity prediction | 5–11, 20–40, 59–105, 135–155, 175–220, 230–270, 275–290, 340–345, 365–390 | ||
| Karplusand Schulz Flexibility Prediction | 4–12, 18–50, 60–85, 90–105, 160–170, 175–215, 228–252, 255–265, 275–300, 325–330, 360–390 | ||
| ELLIPRO | 367–394, 1–39, 399–419, 4–58, 9–139, 27–282, 30–309, 31–335, 34–369, 6–65, 7–75, 21–214 | ||
| Conformational epitope | CBTope (Antigen regions were selected) | 1–30, 41–60, 70–90, 100–120, 140–150, 160–180, 230–240, 267–300, 310–330, 350–383, 390–418 |
Table 2.
Prediction of linear and discontinuous B-cell epitopes using LBTope, ABCpred, IEDB, CBTope, and DiscoTope for spike glycoprotein
| Type of prediction | Server name | Signal peptide and transmembrane topology prediction | Position of epitope |
|---|---|---|---|
| ABCpred | LBTope | Signal peptide: 1–20 | 20–30, 67–83, 111–136, 246–260, 268–282, 297–322, 297–411, 436–450, 490–518, 527–545, 559–573, 607–627, 653–667, 696–710, 727–747, 760–774, 807–821, 840-856, 880–894, 926–940, 943–957, 980–994, 1131–1145, 1156–1170, 1161-1175, 1164–1175, 1179–1193, 1259–1273 |
| Linear epitope (Prediction by IEDB server) | ABCpred | Transmembrane topology prediction: out (extracellular:35–1287) | 879–895, 594–610, 257–273, 11112–1128, 245–261, 1180–1196, 476–492, 307–323, 648–664, 492–508, 1247–1263, 470–486, 236–252, 1206–1222, 1058–1074, 931–947, 898–914, 1064–1080, 786–802, 525–541, 464–480, 266–282, 151–167, 1240–1256, 732–748, 406–422, 391–407, 1050–1066, 97–113, 739–755, 630–646, 415–431, 200–216, 1234–150, 1084–1100, 847–863, 604–620, 280–296, 1195–1211, 1129–1145, 70–86, 689–705, 564–580, 329–345, 347–363, 1041–1057, 803–819, 674–690, 583–599, 49–65, 320–336, 288–304, 194–210, 124–140, 829–845, 360–376, 159–176, 107–123, 994–1010, 719–735, 657–673, 501–517, 1227–1243, 941–957, 838–854, 709–725, 695–711, 374–390, 301–317, 251–267, 1146–1162 |
| BepiPred Linear Epitope Prediction | 13–37, 59–81, 97–98, 138–154, 177–189, 206–221, 250–260, 293–296, 304–322, 329–363, 369–393, 404–426, 440–501, 516–536, 555–562, 580–583, 602–606, 616–632, 634–644, 656–666, 672–690, 695–710, 773–779, 786–800, 807–814, 828–842, 988–992, 1035–1043, 1107–1118, 1133–1172, 1203–1206, 1252–1267 | ||
| Kolaskar and Tongaonkar AntigenicityPrediction | 34–41, 44–51, 53–60, 65–70, 81–87, 115–121, 125–134, 136–146, 168–174, 210–216, 223–230, 239–248, 263–270, 272–278, 288–295, 333–339, 359–371, 376–385, 430–435, 488–495, 505–527, 592–599, 607–615, 617–627, 647–653, 667–674, 687–693, 723–730, 735–741, 750–763, 781–788, 803–808, 837–843, 847–853, 858–864, 873–880, 959–966, 973–979, 1003–1011, 1030–1037, 1057–1070, 1079–1085, 1123–1132, 1174–1179, 1221–1256 | ||
| Emini surface accessibility Prediction | 20–32, 35–43, 73–80, 110–115, 144–153, 179–185, 202–208, 250–255, 278–284, 314–323, 352–357, 419–428, 437–442, 455–468, 495–500, 569–581, 601–606, 627–636, 655–660, 674–685, 773–779, 786–794, 808–817, 914–920, 1068–1076, 1105–1111, 1139–1162, 1179–1186, 1202–1210, 1256–1261 | ||
| Chou and FasmanBeta–Turn Prediction | 20–47, 60–85, 95–120, 132–155, 178–191, 203–223, 245–263, 289–301, 310–325, 327–343, 364–376, 378–392, 408–433, 434–455, 472–510, 520–538, 540–551, 585–597, 596–609, 631–645, 652–669, 671–686, 697–717, 737–753, 772–781, 786–816, 832–848, 902–916, 923–934, 935–946, 1032–1051, 1079–1091, 1116–1131, 1133–1149, 1152–1176, 1234–1255 | ||
| Parker Hydrophilicity Prediction | 20–37, 38–47, 48–55, 66–83, 87–103, 106–118, 119–128, 131–140, 141–175, 177–191, 193–202, 203–212, 213–225, 234–240, 245–270, 276–305, 306–329, 351–377, 378–391, 401–433, 435–451, 456–489, 493–510, 520–542, 543–551, 552–560, 563–584, 586–597, 598–610, 611–623, 634–650, 652–665, 670–692, 697–715, 728–740, 742–752, 768–784, 789–801, 806–820, 833–848, 931–947, 950–961, 982–995, 1014–1049, 1051–1061, 1066–1080, 1081–1094, 1111–1131, 1133–1142, 1145–1176, 1180–1197, 1200–1211, 1245–1267 | ||
| Karplusand Schulz Flexibility Prediction | 20–37, 47–57, 68–85, 91–102, 105–119, 143–159, 177–192, 193–205, 211–222, 233–242, 246–261, 267–278, 278–290, 291–304, 306–320, 321–327, 352–362, 380–392, 400–419, 421–433, 434–451, 454–473, 474–487, 495–506, 523–540, 542–561, 562–584, 596–610, 625–635, 636–648, 653–663, 672–964, 698–708, 720–738, 742–753, 770–783, 787–801, 803–819, 832–843, 850–860, 861–869, 877–888, 906–918, 919–926, 929–943, 945–960, 962–973, 979–991, 992–1014, 1024–1031, 1033–1041, 1050–1060, 1067–1080, 1082–1096, 1102–1112, 1114–1128, 1133–1175, 1177–1189, 1190–1199, 1200–1210, 1253–1272 | ||
| ELLIPRO | 1113–1273, 390–528, 239–265, 329–382, 64–83, 117–190, 94–103, 784–799, 1079–1092, 207–215, 555–565, 883–897, 806–815, 477–758, 577–584, 1098–1103, 837–844, 981–987, 863–869 | ||
| Conformational epitope | CBTope (Antigen regions were selected) | 20–42, 66–76, 133–147, 161–182, 201–216, 257–274, 283–297, 301–325, 341–361, 371–390, 400–420, 421–443, 449–460, 471–491, 492–515, 519–515, 519–530, 551–561, 598–618, 621–643, 670–690, 736–753, 761–780, 781–790, 801–830, 900–920, 921–941, 961–980, 1020–1030, 1032–1051, 1061–1080, 1097–1110, 1120–1130, 1132–1170, 1220–1250 |
Prediction and validation of tertiary structure
The S and N protein three-dimensional structure was predicted using the prediction I-TASSER server (Fig. 2). The Ramachandran analysis was performed for S and N Proteins. For S protein structure, it revealed that 201aa, 92.2% were in favored regions, 17aa, 7.8%, were in allowed regions and there was no amino acid in outlier regions. In the case of N protein, the number of residues in favored regions was (198aa, 57.2%), residues in allowed regions were (115aa, 33.2%), generously allowed regions were the (22aa, 6.4%), and outlier regions were the (11aa, 3.2%) (Fig. 3a). The z-score that implies overall quality of protein was –5.67 and − 2.43 for S and N Proteins, respectively (Fig. 3b).
Fig. 2.

Three-dimensional structure prediction of SARS-CoV-2 antigens and selection of antigenic epitopes; A spike glycoprotein; B nucleocapsid protein using I-TASSER server
Fig. 3.

Validation of the quality of nucleocapsid protein and spike glycoprotein: A validation of 3D models by Ramachandran plot; B validation of 3D models by ProSA-web
Prediction of epitopes
The S and N Proteins epitopes were predicted by the IEDB server (Tables 1, 2). B-cell epitope prediction was based on physicochemical properties (linear epitopes, accessible surface, flexibility, antigenicity, hydrophilic regions, and β-turn). The conformational B-cell epitope prediction (CBTOPE) server was used to predict conformational B-cell epitopes in S and N Proteins (Tables 1, 2). A total of eight epitopes for the two antigens were predicted, consisting of five epitopes for S protein and three epitopes for N protein (Table 3).
Table 3.
Final selected amino acid sequence of each of the six epitopes predicted for nucleocapsid protein and spike glycoprotein
| Antigens | Epitope number | Sequence | Position |
|---|---|---|---|
| Spike protein | EP1 | VSVITPGTNTSNQVAVLYQDVNCTEVPVAIH | 595–625 |
| EP2 | SNVTWFHAIHVSGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGT | 60–108 | |
| EP3 | KQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGDCL | 786–841 | |
| EP4 | MSFPQSAPHGVVFLHVTYVPAQEKNFTTAPAICHD | 1050–1084 | |
| EP5 | NVYADSFVIRGDEVRQIAPGQTGKIADYNYKLPDDFTGCVI | 394–434 | |
| Nucleocapsid phosphoprotein | EP1 | SSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGA | 78–138 |
| EP2 | LPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGDAALAL | 167–221 | |
| EP3 | ALPQRQKKQQTVTLLPAADLDDFSKQLQQSMSSADSTQA | 381–419 |
Construction of a multi-epitope peptide of S and N proteins
Epitopes were blasted individually for each protein. Selected epitopes from each (N and S protein) had 100% homology with sequences recorded on the website, and had no homology with antigens from other viruses of the Cronavaridae family. The epitopes that were predicted for S and N Proteins were used along with glycine and serine sequences that provide flexible linkers to design and construct a multi-epitope peptide (MEP) (Fig. 4). The B-cell multi-epitope secondary structure analysis showed that α-helices, extended strands, β-turns, and random coils account for 8.93%, 26.8%, 11.41%, and 52.85%, respectively.
Fig. 4.

A Predicting and designing multi-epitope; B three-dimensional structure of a multi-epitope designed by I-TASSER sever and the position of the amino acid sequence of nucleocapsid protein and spike glycoprotein epitopes on the 3D structure of the multi-epitope using the Chimera Version 1.8
Calculation of different physical and chemical parameters of the B-cell multi-epitope
The aliphatic index for B-cell multi-epitope was 61.99 that implies the stability of multi-epitope peptide in a wide range of temperatures. Nevertheless, the instability index indicates moderate stability. Grand Average Hydropathicity (GRAVY) was negative (− 0.426) implying the high hydrophilicity nature of the B-cell multi-epitope (Table 4).
Table 4.
Physical and chemical parameters of the final multi-epitope
| Number of amino acids | 403 |
| Molecular weight (KD) | 42.24 |
| Theoretical pI | 8.99 |
| Instability index | 58.67 |
| Estimated half-life | 1.9 hours (mammalian reticulocytes, in vitro) |
| > 20 hours (yeast, in vivo) | |
| > 10 hours (Escherichia coli, in vivo) | |
| Aliphatic index | 61.99 |
| Grand average of hydropathicity (GRAVY) | – 0.426 |
| Predicted Solubility upon Overexpression: INSOLUBLE with probability | 0.611709 |
Modeling, validation, and evaluation of the quality of final construct and codon optimization
Final construct is modeled by I-TASSER server (Fig. 5) (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). The final construct was Validated by Ramachandran plot and the number of residues in favored regions was 195aa (61.7%), in allowed regions 101aa (32%) and generously allowed regions was 16aa (5.1%) (Fig. 6a). Analyzing of the model quality by ProSA-web server demonstrated that final construct is located within the normal range of X-ray 3D structures with a z-score of – 2.1 (Fig. 6b). Furthermore, plotting energies of the final construct showed local model quality (Fig. 6c). A negative score below the threshold was obtained for most of the residues that implies that the final construct has the lowest problematic or erroneous parts. Analysis by Verify3D showed a score of > = 0.2 in at least 80% of the amino acids in the 3D/1D profile that indicate the structure is valid (Fig. 6d). The multi-epitope sequence was translated into nucleotide sequence using the J-CAT server (http://www.jcat.de/) Condon optimization, G and C contents were J-CAT server (Fig. 7).
Fig. 5.

3D structure of final multi-epitope predicted using I-TASSER server. A Model 1: C-score = − 2.76; B model 2: C-score = − 1.95; C model 3: C-score = − 3.21; d model 4: C-score = − 3.29; e model 5: C-score = − 4.78
Fig. 6.

Validation of final construct of multi-epitope by A Ramachandran plot; B and C ProSA-web plot and local model quality for 3D structure of final construct, plotting energies of the final construct, respectively; D Verify3D method to evaluate a 3D model of final multi-epitope
Fig. 7.

The multi-epitope protein sequence was reverse translated into nucleotide sequence using the J-CAT
Discussion
The recent outbreak and rapid spread of the novel coronavirus SARS-CoV-2 is a great threat to the world (Arab-Mazar et al. 2020; Sharma et al. 2020). Diagnostic methods are crucial to control pandemic. Although SARS-CoV-2 can be detected using RT-PCR, but this technique has some limitations including inadequate access to reagents and equipment, restrictive biosafety level facilities, and technical sophistication. Furthermore, this method is associated with a high rate of false-negative results mainly because of unstandardized collection of respiratory specimens. Previous studies imply that virus-specific IgM and IgG levels are useful in serologic diagnosis of SARS (Hou et al. 2020). The development of a highly specific and sensitive immunoassay to detect the presence of anti- SARS-CoV-2 antibodies can improve the diagnosis (Liu et al. 2020a, b).
The S protein that is involved attachment to host cells is located on the surface of the viral particles and is highly immunogenic (Huang et al. 2020a, b; Dai and Gao 2021). The N protein is a major structural protein of the virus and medicates various functions such as viral assembly and RNA replication (Khan et al. 2021; Yadav et al. 2021). The N protein is highly immunogenic and is expressed in a high rate during infection (Khan et al. 2020; Zeng et al. 2020). These features make both S and N proteins as potential antigens for sero-diagnosis of COVID-19, explaining why they have been interesting for many authors. In a study by Liu et al., in a total of 214 proved COVID-19 patients, recombinant N-based IgM and IgG were detected in 146 (68.2%) and 150 (70.1%) patients, respectively, by ELISA method; Furthermore, recombinant S-based IgM and IgG were detected in 165 (77.1%) and 159 (74.3%) patients, respectively (Liu et al. 2020a, b).
The application of bioinformatics methods is very helpful in predicting protein structure, their functions, biological features, specific immune response, avidity of antigen–antibody binding in vaccine design, and serologic diagnosis (Ebrahimi et al. 2019; Mamaghani et al. 2019; Habibi et al. 2020; Karimi et al. 2020). The epitope prediction servers based on the artificial intelligence have higher accuracy and sensitivity than those that perform epitope prediction based on physicochemical properties. In the present study, we used a combination of two servers (artificial intelligence and physicochemical properties methods) to predict the best epitopes (both in terms of high antigenic properties and desirable physicochemical properties). In the other hand, due to the higher accuracy of artificial intelligence methods in predicting epitopes, we focused on the results obtained from this server.
The recombinant antigens can be used to detect the presence of antibodies in the serum. In addition, multi-epitopic antigens used in other studies on different pathogenic microorganisms have improved the sensitivity and specificity of the immunoassays. Furthermore, it has been shown that multi-epitopic recombinant antigens can be very useful in the serologic diagnosis of some diseases (Dai et al. 2012; Hajissa et al. 2017). Immunologic B-cell epitopes’ identification can improve serological diagnostic tests for detection of specific antibodies in patients with COVID-19 (Phan et al. 2021). The benefits of prediction of B-cell epitopes include the exact identification of antigenic sites, identifying more than one B-cell epitope, combining several epitopes of different antigens of COVID-19, and facilitating the standardization of the diagnostic methods. Another application of multi-epitope peptides can be the detection of previous history of the infection. Kar et al. designed a multi-epitope vaccine using bioinformatics methods in which the spike glycoprotein (S protein) of SARS-CoV-2 was used. They reported that the multi-epitope vaccine candidate is structurally stable and can successfully induce specific immune responses (Kar et al. 2020).
In the current study, immunogenic B-cell epitopes were predicted based on S and N proteins’ amino acid sequences. ABCpred, Bepipred Linear Epitope Prediction, and CBTOPE servers were used to predict antigenic epitopes. The immunogenic epitopes were predicted by the IEDB server that implements physicochemical properties and machine learning methods. Consequently, the three-dimensional structure of designed epitopes and the locations of β-turns and α-helices and their accessibility were evaluated. Second structure analysis of the protein showed that the designed epitope structure has a high antigenicity and avidity due to its hydrophilic properties. The β-turns and α-helices regions have high antigenicity due to their complex spatial structure. Due to this high antigenicity property, paratopes (the part of an antibody which recognizes and binds to an antigen) of antibodies interact with high avidity with these regions. As the predicted epitopes in this study have the β-turns and α-helices conformation, they potentially have high antigenicity that is an advantage in diagnostic kits. The hydrophilicity was selected as a property of the designed epitopes in this study, because it makes it possible for the peptide to be dissolved in hypotonic environments and can easily interact with the paratope region of the antibodies. Although theoretically the epitopes selected in this study are good candidates for designing of serological diagnosis tests, but to assess the sensitivity and specificity, the designed multi-epitopes should be evaluated by serum of patients to prove its potential to be used as a diagnostic immunoassay.
Conclusion
In the current study, bioinformatics tools were used to design a multi-epitope peptide based on the type of antigen and its physiochemical properties to design multi-epitopes that have a high avidity against SARS-CoV-2 antibodies to detect infections by COVID-19. The results of the current study indicated that the recombinant S and N proteins’ multi-epitope antigens of SARS-CoV-2 can be a potential option to develop diagnostic test for COVID-19 Fig 8. The results are displayed in Supplementary Fig. 8.
Fig. 8.
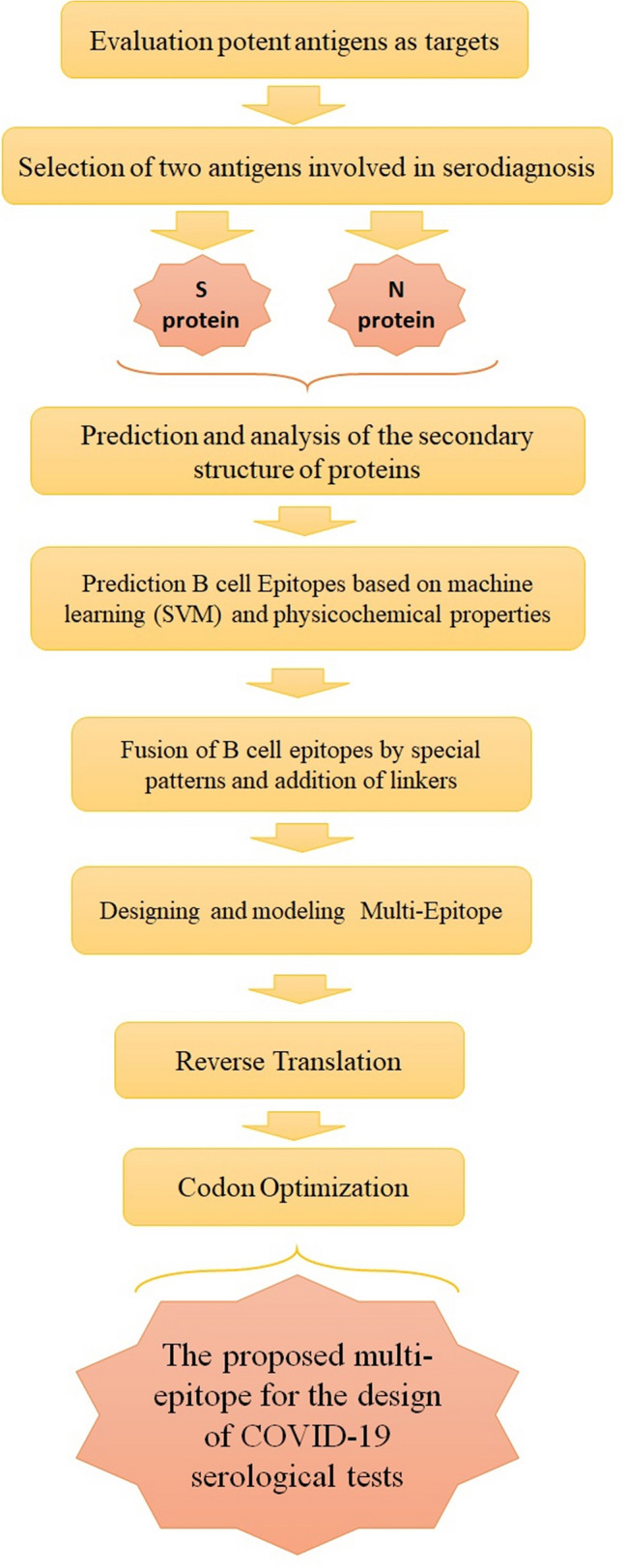
Workflow summary of SARS-CoV-2 multi-epitope design steps for designing a proposed serological diagnostic test for COVID-19 diagnosis
Acknowledgements
Hereby, the authors appreciate the cooperation of Shahid Beheshti University of Medical Sciences.
Declarations
Conflict of interest
All authors declared there is no conflict of interest.
Ethics approval
Informed consent was taken from all patients before taking fecal samples. The local committee ethics approved the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Acosta MAT, Singer BD. Pathogenesis of COVID-19-induced ARDS: implications for an ageing population. Eur Respir J. 2020;56(3):2002–2049. doi: 10.1183/13993003.02049-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghamolaei S, Kazemi B, Bandehpour M, Ranjbar M, Rouhani S, Mamaghani AJ, Tabaei S. Design and expression of polytopic construct of cathepsin-L1, SAP-2 and FhTP16 5 proteins of Fasciola hepatica. J Helminthol. 2020 doi: 10.1017/S0022149X20000140. [DOI] [PubMed] [Google Scholar]
- Arab-Mazar Z, Sah R, Rabaan AA, Dhama K, Rodriguez-Morales AJ. Mapping the incidence of the COVID-19 hotspot in Iran-implications for travellers. Travel Med Infect Dis. 2020;34:101630. doi: 10.1016/j.tmaid.2020.101630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baj J, Karakuła-Juchnowicz H, Teresiński G, Buszewicz G, Ciesielka M, Sitarz E, Forma A, Karakuła K, Flieger W, Portincasa P, Maciejewski R. COVID-19: specific and non-specific clinical manifestations and symptoms: the current state of knowledge. J Clin Med. 2020 doi: 10.3390/jcm9061753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chansaenroj J, Yorsaeng R, Posuwan N, Puenpa J, Sudhinaraset N, Chirathaworn C, Poovorawan Y. Detection of SARS-CoV-2-specific antibodies via rapid diagnostic immunoassays in COVID-19 patients. Virol J. 2021;18(1):52. doi: 10.1186/s12985-021-01530-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che X-Y, Qiu L-W, Pan Y-X, Wen K, Hao W, Zhang L-Y, Wang Y-D, Liao Z-Y, Hua X, Cheng VC. Sensitive and specific monoclonal antibody-based capture enzyme immunoassay for detection of nucleocapsid antigen in sera from patients with severe acute respiratory syndrome. J Clin Microbiol. 2004;42(6):2629–2635. doi: 10.1128/JCM.42.6.2629-2635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Gao GF. Viral targets for vaccines against COVID-19. Nat Rev Immunol. 2021;21(2):73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Jiang M, Wang Y, Qu L, Gong R, Si J. Evaluation of a recombinant multiepitope peptide for serodiagnosis of Toxoplasma gondii infection. Clin Vaccine Immunol. 2012;19(3):338–342. doi: 10.1128/CVI.05553-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi M, Seyyedtabaei SJ, Ranjbar MM, Tahvildar-biderouni F, Mamaghani AJ. Designing and modeling of multi-epitope proteins for diagnosis of Toxocara canis infection. Int J Pept Res Ther. 2019;26(3):1371–1380. doi: 10.1007/s10989-019-09940-1. [DOI] [Google Scholar]
- Ferrer-Oliveras R, Mendoza M, Capote S, Pratcorona L, Esteve-Valverde E, Cabero-Roura L, Alijotas-Reig J. Immunological and physiopathological approach of COVID-19 in pregnancy. Archf Gynecol Obstet. 2021;304:39–57. doi: 10.1007/s00404-021-06061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi A, Azizan A, Ehteshaminia Y, Jadidi-Niaragh F, Enderami E, Akbari E, Abediankenari S, Hassannia H. Design of a multi-epitope peptide vaccine against SARS-CoV-2 based on immunoinformatics data. J Mazandaran Univ Med Sci. 2020;30(190):126–132. [Google Scholar]
- Hajissa K, Zakaria R, Suppian R, Mohamed Z. An evaluation of a recombinant multiepitope based antigen for detection of Toxoplasma gondii specific antibodies. BMC Infect Dis. 2017;17(1):807. doi: 10.1186/s12879-017-2920-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BE, Ho AP, Ho MA, Ho EC. Case report of familial COVID-19 cluster associated with High prevalence of anosmia, ageusia, and gastrointestinal symptoms. IDCases. 2020;22:e00975. doi: 10.1016/j.idcr.2020.e00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Wang T, Zhang B, Luo Y, Mao L, Wang F, Wu S, Sun Z. Detection of IgM and IgG antibodies in patients with coronavirus disease 2019. Clin Transl Immunol. 2020;9(5):e01136. doi: 10.1002/cti2.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yang C, Xu X-F, Xu W, Liu S-W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41(9):1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM, Srivastava AP. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep. 2020;10(1):10895. doi: 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M, Tabaei SJS, Ranjbar MM, Fathi F, Jalili A, Zamini G, Mamaghani AJ, Nazari J, Roshani D, Bagherani N. Construction of a synthetic gene encoding the Multi-Epitope of Toxoplasma gondii and demonstration of the relevant recombinant protein production: A vaccine candidate. Galen Med J. 2020;9:1708. doi: 10.31661/gmj.v9i0.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MT, Irfan M, Ahsan H, Ahmed A, Kaushik AC, Khan AS, Chinnasamy S, Ali A, Wei D-Q. Structures of SARS-CoV-2 RNA-binding proteins and therapeutic targets. Intervirology. 2021;64(2):55–68. doi: 10.1159/000513686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Khan MT, Saleem S, Junaid M, Ali A, Ali SS, Khan M, Wei D-Q. Structural Insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput Str Biotechnol J. 2020;18:2174–2184. doi: 10.1016/j.csbj.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. 2020;55(3):105924. doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, Azman AS, Reich NG, Lessler J. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Int Med. 2020;172(9):577–582. doi: 10.7326/M20-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu L, Kou G, Zheng Y, Ding Y, Ni W, Wang Q, Tan L, Wu W, Tang S. Evaluation of nucleocapsid and spike protein-based enzyme-linked immunosorbent assays for detecting antibodies against SARS-CoV-2. J Clin Microbiol. 2020;58(6):e00461-00420. doi: 10.1128/JCM.00461-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu L, Kou G, Zheng Y, Ding Y, Ni W, Wang Q, Tan L, Wu W, Tang S, Xiong Z, Zheng S. Evaluation of nucleocapsid and spike protein-based enzyme-linked immunosorbent assays for detecting antibodies against SARS-CoV-2. J Clin Microbiol. 2020;58(6):e00461-00420. doi: 10.1128/JCM.00461-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Rusling JF. COVID-19 antibody tests and their limitations. ACS Sens. 2021;6(3):593–612. doi: 10.1021/acssensors.0c02621. [DOI] [PubMed] [Google Scholar]
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaghani AJ, Fathollahi A, Spotin A, Ranjbar mehdi M, Barati M, Aghamolaie S, Karimi M, Taghipour N, Ashrafi M, Tabaei SJS. Candidate antigenic epitopes for vaccination and diagnosis strategies of Toxoplasma gondii infection: a review. Microb Pathog. 2019;137:103788. doi: 10.1016/j.micpath.2019.103788. [DOI] [PubMed] [Google Scholar]
- Mamaghani AJ, Tabaei SJS, Ranjbar MM, Haghighi A, Spotin A, Dizaji PA, Rezaee H. Designing diagnostic kit for Toxoplasma gondii based on GRA7, SAG1, and ROP1 Antigens: An in silico strategy. Int J Pept Res Ther. 2020;26:2269–2283. doi: 10.1007/s10989-020-10021-x. [DOI] [Google Scholar]
- Phan IQ, Subramanian S, Kim D, Murphy M, Pettie D, Carter L, Anishchenko I, Barrett LK, Craig J, Tillery L, Shek R, Harrington WE, Koelle DM, Wald A, Veesler D, King N, Boonyaratanakornkit J, Isoherranen N, Greninger AL, Jerome KR, Chu H, Staker B, Stewart L, Myler PJ, Van Voorhis WC. In silico detection of SARS-CoV-2 specific B-cell epitopes and validation in ELISA for serological diagnosis of COVID-19. Sci Rep. 2021;11(1):4290. doi: 10.1038/s41598-021-83730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu KS, Pandey S, Maiti S, Pillai B. GASCO: genetic algorithm simulation for codon optimization. Silico Biol. 2008;8(2):187–192. [PubMed] [Google Scholar]
- Sharma A, Tiwari S, Deb MK, Marty JL. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int J Antimicrob Agents. 2020;56(2):106054. doi: 10.1016/j.ijantimicag.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugama B, Kadhiresan P, Kozlowski HN, Malekjahani A, Osborne M, Li VYC, Chen H, Mubareka S, Gubbay JB, Chan WCW. Diagnosing COVID-19: The disease and tools for detection. ACS Nano. 2020;14(4):3822–3835. doi: 10.1021/acsnano.0c02624. [DOI] [PubMed] [Google Scholar]
- Woo PC, Lau SK, Wong BH, Tsoi H-W, Fung AM, Kao RY, Chan K-H, Peiris JM, Yuen K-Y. Differential sensitivities of severe acute respiratory syndrome (SARS) coronavirus spike polypeptide enzyme-linked immunosorbent assay (ELISA) and SARS coronavirus nucleocapsid protein ELISA for serodiagnosis of SARS coronavirus pneumonia. J Clin Microbiol. 2005;43(7):3054–3058. doi: 10.1128/JCM.43.7.3054-3058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Chaudhary JK, Jain N, Chaudhary PK, Khanra S, Dhamija P, Sharma A, Kumar A, Handu S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10(4):821. doi: 10.3390/cells10040821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Liu G, Ma H, Zhao D, Yang Y, Liu M, Mohammed A, Zhao C, Yang Y, Xie J, Ding C, Ma X, Weng J, Gao Y, He H, Jin T. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem Biophys Res Commun. 2020;527(3):618–623. doi: 10.1016/j.bbrc.2020.04.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]