

Abstract
Pathogenic mycobacteria pose a sustained threat to global human health. Recently, cytochrome bcc complexes have gained interest as targets for antibiotic drug development. However, there is currently no structural information for the cytochrome bcc complex from these pathogenic mycobacteria. Here, we report the structures of Mycobacterium tuberculosis cytochrome bcc alone (2.68 Å resolution) and in complex with clinical drug candidates Q203 (2.67 Å resolution) and TB47 (2.93 Å resolution) determined by single-particle cryo-electron microscopy. M. tuberculosis cytochrome bcc forms a dimeric assembly with endogenous menaquinone/menaquinol bound at the quinone/quinol-binding pockets. We observe Q203 and TB47 bound at the quinol-binding site and stabilized by hydrogen bonds with the side chains of QcrBThr313 and QcrBGlu314, residues that are conserved across pathogenic mycobacteria. These high-resolution images provide a basis for the design of new mycobacterial cytochrome bcc inhibitors that could be developed into broad-spectrum drugs to treat mycobacterial infections.
Research organism: Other
Introduction
Mycobacteria, which belong to the phylum Actinobacteria, have coevolved with humans over thousands of years (Chisholm et al., 2016). Approximately 200 species of mycobacteria have been identified that have diverse lifestyles, morphologies, and metabolic pathways (Tortoli et al., 2017). Mycobacteria can broadly be grouped into two categories: tuberculosis-causing mycobacteria and non-tuberculous mycobacteria (NTM). Mycobacterium leprae is often represented in a distinct genetic clade owing to its genetic and phenotypic differences compared to other mycobacterial species (Cole et al., 2001). Mycobacteria can be further classified into fast-growing and slow-growing species or species complexes; these assignments are according to the physiological, phenotypic and phylogenetic characteristics (Rastogi et al., 2001). Although nearly all mycobacteria are saprophytes or non-pathogenic to humans, a few species cause diseases, resulting in pulmonary and extra-pulmonary infections that can affect nearly all organs. Infections, which are caused by strict or opportunistic pathogenic mycobacteria (Figure 1), pose a sustained threat to human health. Of these, tuberculosis (TB), caused by Mycobacterium tuberculosis (M. tuberculosis), is the most serious, leading to ~1.2 million fatalities per year (World Health Organization, 2019). Infections involving other pathogenic mycobacteria, for example, Mycobacterium abscessus and Mycobacterium avium complex, are on the rise with some outnumbering those caused by M. tuberculosis in countries including the United States (Donohue, 2018; Johansen et al., 2020). These infections are notoriously difficult to treat due to intrinsic or emerging resistance to many common antibiotics, thus exacerbating the challenge to find suitable drug targets.
Figure 1. Sequence similarity comparison of M. tuberculosis QcrB with other pathogenic mycobacteria.

Oxidative phosphorylation (OXPHOS) has gained interest as a target space for antibiotic drug development (Cook et al., 2017; Hards et al., 2020). In OXPHOS, the protein complexes of the electron transport chain (ETC) establish a proton motive force (PMF) across a biomembrane that drives the synthesis of adenosine triphosphate (ATP) by ATP synthase (Mitchell, 1961). Maintenance of PMF and ATP homeostasis is required for the survival of both replicative and non-replicative (often referred to as dormant) mycobacteria, and its dissipation leads to a rapid loss of cell viability and cell death (Koul et al., 2008; Rao et al., 2008). The reliance on the PMF and ATP homeostasis thus highlights the importance of the mycobacterial proton-pumping cytochrome bcc-aa3 supercomplex, which consists of a bcc menaquinol reductase (complex III, CIII) and an aa3 oxidase (complex IV, CIV) that are tightly associated (Gong et al., 2018; Kim et al., 2015; Megehee et al., 2006). Several studies support the attractiveness of cytochrome bcc-aa3 for mycobacterial drug development (de Jager et al., 2020; Liu et al., 2019; Lu et al., 2019; Pethe et al., 2013; Scherr et al., 2018). Given the strict sequence conservation of this complex (Figure 1), broad-spectrum activity of a therapeutic within the pathogenic mycobacteria is likely (Lee et al., 2020b). Interestingly, all the cytochrome bcc-aa3 inhibitors published to date appear to target the QcrB subunit (Figure 1) of the cytochrome bcc complex and are likely bound to the menaquinol-binding (Qp) site of the QcrB subunit (Lee et al., 2020b). The most advanced of these are Q203 and TB47, which have been shown to clear infections due to M. tuberculosis (de Jager et al., 2020; Lu et al., 2019; Pethe et al., 2013) and Mycobacterium ulcerans (Liu et al., 2019; Scherr et al., 2018). Q203 has recently completed phase II clinical trials for TB treatment (ClinicalTrials.gov number, NCT03563599; de Jager et al., 2020). TB47 has also been evaluated in a preclinical study (http://www.newtbdrugs.org/pipeline/clinical).
To progress an understanding of cytochrome bcc and its interaction with new drug leads, here we have determined the atomic resolution cryo-electron microscopy (cryo-EM) structures of M. tuberculosis cytochrome bcc alone (2.68 Å resolution) and in complex with Q203 (2.67 Å resolution) and TB47 (2.93 Å resolution). These high-resolution structures will greatly accelerate efforts towards structure-guided drug discovery.
Results and discussion
Structure of M. tuberculosis cytochrome bcc
Considering that the hybrid supercomplex consisting of M. tuberculosis CIII and Mycobacterium smegmatis CIV can be stabilized as a functional assembly (Kim et al., 2015), we first chose to express and purify this complex to a high level of homogeneity (Figure 2—figure supplement 1). We confirmed this complex to be active with a turnover number of 23.3 ± 2.4 e-s–1 (mean ± SD, n = 4), in agreement with the previous study that shows M. tuberculosis CIII can functionally complement native M. smegmatis CIII and maintain the growth of M. smegmatis (Kim et al., 2015). The structure of this hybrid supercomplex was determined by cryo-EM to an overall resolution of 2.68 Å, allowing that the components to be clearly assigned in the density map (Figure 2—figure supplements 2 and 3, Table 1). The hybrid supercomplex including the M. tuberculosis CIII dimer forms a pseudo twofold symmetrical compact rod, but with a slight curvature in the membrane plane, as previously observed in the M. smegmatis CIII2CIV2 supercomplex (Gong et al., 2018; Wiseman et al., 2018; Figure 2A and B). The hybrid supercomplex is structurally similar to the supercomplex isolated from M. smegmatis (Gong et al., 2018; Wiseman et al., 2018; Figure 2—figure supplement 4), except that subunits LpqE and Unk (probably MSMEG_0987) (Wiseman et al., 2018) were not observed here. The absence of these two subunits may be due to their depletion during purification of the supercomplex or their map density signal was averaged to background noise during structural determination. As expected, the topology of M. tuberculosis cytochrome bcc and M. smegmatis CIV in the hybrid supercomplex is also similar to that of the equivalent subunits in the M. smegmatis CIII2CIV2 supercomplex (Figure 2C and D, Figure 2—figure supplements 4 and 5). As such, there is no notable non-native contacts that resulted from the hybrid assembly in the hybrid supercomplex. This is attributable to the high structural similarity between the M. tuberculosis cytochrome bcc and M. smegmatis cytochrome bcc. In M. tuberculosis cytochrome bcc, QcrA has three transmembrane helices (TMHs) and has a ‘U’-shaped structure (Figures 2D and 3A). The N-terminal region with TMH1/2 and the TMH3 make up the two arms of the ‘U’ structure. These arms are linked by the region located near the cytoplasmic side. Attached to QcrATMH3 is the C-terminal domain, which faces the periplasmic side of the membrane and holds the [2Fe-2S] cluster in place. QcrB has eight TMHs (Figures 2D and 3B). Four of these are responsible for burying two functionally important heme b cofactors (high potential heme bH and low potential heme bL). Heme bL and heme bH are located between TMH I/II and TMH III/IV, respectively. Heme bL is near the periplasmic side and heme bH near the cytoplasmic side. QcrC is a transmembrane protein with a C-terminal TMH located between QcrBTMH5 and QcrBTMH7 (Figures 2D and 3C). The N-terminal periplasmic portion of QcrC can be divided into two heme-containing cytochrome c domains designated D1 and D2. The D1 domain protrudes out of the core of CIII, whereas the D2 domain interacts extensively with QcrA and QcrB. In the M. smegmatis CIII2CIV2 supercomplex, the cytochrome cc head domain of QcrC adopts an ‘open’ or a ‘closed’ conformation (Gong et al., 2018; Wiseman et al., 2018). However, in this structure it is only the closed conformation (Figure 2—figure supplement 4). Considering the high topology similarity of the subunits and the arrangement of prosthetic groups compared to previous M. smegmatis CIII2CIV2 supercomplex, the mechanism of action of the hybrid supercomplex including M. tuberculosis cytochrome bcc is expected to be the same as that for the M. smegmatis CIII2CIV2 supercomplex (Gong et al., 2018).
Table 1. Cryo-electron microscopy data collection, refinement, and validation statistics of hybrid supercomplex.
| State | apo |
|---|---|
| Data collection | |
| Microscope | Titan Krios |
| Voltage (kV) | 300 |
| Magnification | 29,000× |
| Detector | Gatan K3 |
| Data collection software | SerialEM |
| Electron exposure (e–/Å2) | 60 |
| Defocus range (μm) | –1.2 to –1.8 |
| Pixel size (Å) | 0.82 |
| Data processing | |
| Number of micrographs | 4141 |
| Final particle images | 112,804 |
| Symmetry imposed | C1 |
| Map resolution (Å)Fourier shell correlation 0.143 threshold | |
| 2.68 | |
| Refinement | |
| Initial model used (PDB code) | 6ADQ |
| Map sharpening B factor (Å2)d FSC model (0.143) masked | –65.32.5 |
| Map correlation coefficient | 0.89 |
| Mean CC for ligands | 0.78 |
| Model composition | |
| Non-hydrogen atoms | 42,279 |
| Protein residues | 5122 |
| Ligands | |
| 9Y0: 2 | |
| CDL: 17 | |
| 9YF: 4 | |
| HEA: 4 | |
| HEC: 4 | |
| MQ9: 10 | |
| HEM: 4 | |
| PLM: 4 | |
| CU: 8 | |
| FES: 2 | |
| Root mean squared deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.057 |
| Validation | |
| MolProbity score | 1.86 |
| Clashscore | 7.26 |
| Poor rotamers (%) | 0.05 |
| Ramachandran plot | |
| Favored (%) | 92.76 |
| Allowed (%) | 6.97 |
| Outliers (%) | 0.28 |
| Cβ outliers (%) | 0.00 |
Figure 2. Overall architecture of the hybrid supercomplex.
(A) Front view and (B) top view of the cryo-electron microscopy (cryo-EM) map of hybrid supercomplex at 2.68 Å resolution. QcrA, QcrB, and QcrC of the M. tuberculosis cytochrome bcc are colored teal, purple, and salmon, respectively. Other subunits of the hybrid supercomplex are in gray. (C) Cartoon representation of cytochrome bcc, using the same color scheme as above. The twofold symmetry of the dimer is depicted by the gray axis. The heme groups (bH, bL, cD1, and cD2) and menaquinone/menaquinol (MKP/MKN) are shown as stick models. The [2Fe-2S] clusters are shown as spheres. (D) A cross-sectional view (top) of the helices in the cytochrome bcc dimer.

Figure 2—figure supplement 1. Purification and identification of the hybrid supercomplex consisting of M. tuberculosis CIII and M. smegmatis CIV.

Figure 2—figure supplement 2. Cryo-electron microscopy (cryo-EM) data processing of the apo hybrid supercomplex consisting of M. tuberculosis CIII and M. smegmatis CIV.

Figure 2—figure supplement 3. Cryo-electron microscopy (cryo-EM) map quality assessment of the hybrid supercomplex.

Figure 2—figure supplement 4. Structural comparison of the hybrid supercomplex III2IV2 and native CIII2CIV2 supercomplexes from M. smegmatis.

Figure 2—figure supplement 5. Structural alignment between M. tuberculosis CIII and equivalent CIIIs from other species.

Figure 3. Structure of the M. tuberculosis cytochrome bcc subunits.
Cartoon representation of the monomers of (A) QcrA, (B) QcrB, and (C) QcrC, with prosthetic groups. (D) The QP-binding site and (E) QN-binding site. The residues potentially involved in the binding of MK/MKH2 are shown with side chains in stick model representation. MK/MKH2 have their carbon atoms in green and are represented as stick models. The [2Fe-2S] and heme groups are shown as spheres and labeled.

Figure 3—figure supplement 1. Identification and comparison of MK/MKH2 in the hybrid supercomplex.

Figure 3—figure supplement 2. Comparison of M. tuberculosis CIII quinol-binding site (gray) with those from CIII from S. cerevisiae (PDB 3CX5) (blue) and (PDB 4PD4) (purple).

Figure 3—figure supplement 3. Structural superposition of native (left) and docked (right) MK9 with the Q203 in the Qp site.

Quinone and quinone-binding pockets of M. tuberculosis cytochrome bcc
Quinone-binding sites are essential to the function of respiratory chain complexes and thus are good targets for drug discovery (Harikishore et al., 2021; Lee et al., 2020b). Structurally diverse quinones such as ubiquinone and menaquinone (MK) bind in the mitochondrial ETC (Zhang et al., 1998) and mycobacterial ETC (Gong et al., 2018; Wiseman et al., 2018), respectively, indicating that there are structural differences in the quinone-binding sites of the different species. Structural differences in the ubiquinone-binding site are also observed when the mitochondrial and Escherichia coli respiratory chain complex IIs are compared (Huang et al., 2021). Therefore, it is feasible that species-specific quinone-binding-site inhibitors could be designed. In the present structure, two canonical quinone-binding sites are identified (Figure 3D and E): (i) the quinol oxidation site (QP site) and (ii) the quinone reduction site (QN site). In addition, other quinone-binding sites with quinone bound are also identified as observed in our previous study (Gong et al., 2018; Figure 3—figure supplement 1). These quinones are remote from the canonical quinone-binding sites and into the membrane space, suggesting that they have a structural rather than functional role. The QP site responsible for menaquinol (MKH2) oxidation is near heme bL, whereas the QN site responsible for MK reduction is close to heme bH (Figure 3D and E, Figure 3—figure supplement 1). The QP site is at the center of an inverted triangle structure surrounded by helices (Figure 3D). One MK molecule was identified at this site with its naphthoquinone ring surrounded by hydrophobic residues, QcrBPhe158, QcrBTyr161, QcrBLeu180, QcrBIle183, QcrBMet310, and QcrBMet342. Its hydrophobic tail, which contains multiple isoprenoid groups, wraps around QcrBTMH6 down to its cytoplasmic end and in so doing interacts with QcrBMet187, QcrBAla339, QcrBLeu344, and QcrBVal347. The edge-to-edge distance from MK to heme bL is 15 Å, exceeding the 14 Å limit for efficient physiological electron transfer (Page et al., 1999). Furthermore, there are no observed hydrogen bonds to the carbonyl groups of MK, which are needed to help deprotonate MKH2. In addition, structural superposition with the inhibitor-bound bc1 complex also shows that the MK binds deeper into the QP pocket in the bc1 complex (Birth et al., 2014; Solmaz and Hunte, 2008) than in the currently described complex (Figure 3—figure supplement 2). Hence, the endogenous electron donor MKH2 should bind deeper inside the pocket and close to polar residues such as QcrBTyr161, QcrBThr313, and QcrBAsp314 (Figure 3D) to facilitate electron transfer. In addition, MK could also be modeled to fit deeper inside the pocket (Figure 3—figure supplement 3). Thus, we speculate that what is observed here is the oxidized product as it leaves the QP site. It is worth noting that all the reported inhibitors including Q203 (Pethe et al., 2013) and TB47 (Lu et al., 2019) are suggested to interact with this QP site. In addition, the QN site is formed largely by QcrBTMH1, QcrBTMH4, QcrBTMH5 and one loop region of QcrB (Figure 3E). The head group of MK is bound in this pocket interacting with QcrBPhe39, QcrBGlu49, QcrBLeu225, QcrBLeu232, QcrBTrp236, and QcrBPhe262, and its long hydrophobic tail extends along QcrBTMH1 towards the periplasmic side. MK/MKH2 are part of the Q-cycle hypothesis and essential for electron transfer in the cytochrome bcc complex (Gong et al., 2018). Given the crossspecies activity of this complex (Lee et al., 2020b) and high homology of the QcrB subunits across mycobacterial pathogens (Figure 1), these data open the way for the discovery of broad-spectrum mycobacterial agents based on rational, structure-based inhibitor design principles.
Q203 interactions in M. tuberculosis cytochrome bcc binding pocket
Q203 has recently been subjected to a phase II clinical study for M. tuberculosis treatment (de Jager et al., 2020). This compound has also been shown to be strongly bactericidal against M. ulcerans (Scherr et al., 2018). It is suggested to be an inhibitor that competes with endogenous substrate binding (QP site) of the cytochrome bcc complex (Pethe et al., 2013), but this hypothesis is yet to be verified by direct experimental evidence. To obtain atomic information on the mode of binding of Q203 to cytochrome bcc, we have determined the structure of the hybrid supercomplex as described above in the presence of Q203 by cryo-EM to an overall resolution of 2.67 Å (Figure 4—figure supplements 1 and 2, Table 2). We observe that close to the Qp-binding pocket within the membrane of each QcrB of cytochrome bcc there is density for Q203 (Figure 4A and B). All of the Q203 molecules fill each QcrB subunit binding deeply into the QP pocket and with identical binding modes. The key interactions that anchor Q203 are (i) a hydrogen bond between the hydroxyl oxygen of the side chain of QcrBThr313 and the amine in the carboxamide linker of Q203 (3.0 Å), (ii) a halogen bond between the chlorine atom of the heterocyclic group and an ordered water molecule that simultaneously forms a hydrogen bond with the hydroxyl oxygen of the side chain of QcrBTyr164 (Figure 4—figure supplement 3), (iii) a hydrogen bond between the side chain of QcrBGlu314 and the nitrogen atom in the imidazopyridine ring (3.0 Å), and (iv) a hydrogen bond between the side chain of QcrAHis375 and the nitrogen atom in the imidazopyridine ring (2.98 Å) (Figure 4C). In addition, the carbon atoms of Q203 make hydrophobic interactions with QcrBGly175, QcrBAla179, QcrBLeu180, QcrBThr184, QcrBSer304, QcrBPro306, QcrBMet310, QcrBAla317, and QcrBMet342. These extensive interactions are in agreement with the fact that the activity of the supercomplex is inhibited by Q203 in vitro according to the DMNQH2/oxygen oxidoreductase activity assay. After addition of Q203, the turnover number of the hybrid supercomplex reduces to 5.8 ± 2.4 e-s–1 from 23.3 ± 2.4 e-s–1 (mean ± SD, n = 4; Figure 4—figure supplement 4). In addition, functional studies have shown that substitution of QcrBThr313 to alanine confers Q203 resistance (Pethe et al., 2013). The binding of Q203 involves residues from both QcrA and QcrB. Due to the need to form stabilizing interactions between subunits, resistance may be more difficult to achieve here than if the binding site is within only one subunit. Consistently, the mapping of reported mutations in Q203-resistant M. tuberculosis reveals that they are positioned directly where Q203 binds in this structure (Lupien et al., 2020; Figure 4—figure supplement 5).
Table 2. Cryo-electron microscopy data collection, refinement, and validation statistics for the Q203-bound hybrid supercomplex.
| State | Q203 |
|---|---|
| Data collection | |
| Microscope | Titan Krios |
| Voltage (kV) | 300 |
| Magnification | 29,000× |
| Detector | Gatan K3 |
| Data collection software | SerialEM |
| Electron exposure (e–/Å2) | 60 |
| Defocus range (μm) | –1.2 to –1.8 |
| Pixel size (Å) | 0.82 |
| Data processing | |
| Number of micrographs | 3763 |
| Final particle images | 106,770 |
| Symmetry imposed | C1 |
| Map resolution (Å)Fourier shell correlation (FSC) 0.143 threshold | |
| 2.67 | |
| Refinement | |
| Initial model used (PDB code) | 6ADQ |
| Map sharpening B factor (Å2)d FSC model (0.143) masked | –70.02.6 |
| Map correlation coefficient | 0.88 |
| Mean CC for ligands | 0.76 |
| Model composition | |
| Non-hydrogen atoms | 42,695 |
| Protein residues | 5132 |
| Ligands | |
| 9Y0: 2 | |
| CDL: 17 | |
| 9YF: 4 | |
| HEA: 4 | |
| HEC: 4 | |
| MQ9: 8 | |
| HEM: 4 | |
| PLM: 4 | |
| CU: 8 | |
| FES: 2 | |
| HUU (Q203): 2 | |
| Root mean squared deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.659 |
| Validation | |
| MolProbity score | 1.84 |
| Clashscore | 7.97 |
| Poor rotamers (%) | 6.23 |
| Ramachandran plot | |
| Favored (%) | 93.07 |
| Allowed (%) | 6.61 |
| Outliers (%) | 0.31 |
| Cβ outliers (%) | 0.00 |
Figure 4. Cryo-electron microscopy (cryo-EM) structure of the hybrid supercomplex in the presence of Q203.
(A) Side (left) and top (right) views of the cryo-EM structure of the M. tuberculosis cytochrome bcc complex presented as a cartoon representation. Q203 (orange) is bound to the Qp site. (B) Visualization of densities for Q203. Hydrogen bonds are shown as dotted lines. (C) Plot of distances of various parts of Q203 to residues in the Qp site as determined using LIGPLOT (https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/).

Figure 4—figure supplement 1. Cryo-electron microscopy (cryo-EM) data processing of the hybrid supercomplex consisting of M. tuberculosis CIII and M. smegmatis CIV in the presence of Q203.

Figure 4—figure supplement 2. Cryo-electron microscopy (cryo-EM) map quality assessment of M. tuberculosis cytochrome bcc complex.

Figure 4—figure supplement 3. The densities for Q203, H2O, and QcrBTyr164.

Figure 4—figure supplement 4. Rate of O2 reduction by the hybrid supercomplex before and after addition of Q203.

Figure 4—figure supplement 5. Reported mutations in Q203-resistant M. tuberculosis.

TB47-binding mode of M. tuberculosis cytochrome bcc
TB47, also currently being evaluated in preclinical studies, has been suggested to target the QcrB of cytochromes bcc from M. tuberculosis (Lu et al., 2019) and M. ulcerans (Liu et al., 2019). The 2.93 Å cryo-EM map shows density for TB47 and confirms that it binds in the same location as Q203 (Figure 5A and B, Figure 5—figure supplements 1 and 2, Table 3). Three hydrogen bond interactions are observed involving the side chains of QcrBThr313, QcrBGlu314, and QcrAHis375. Similar interactions are also observed when Q203 binds (Figure 5C). QcrBTyr161, QcrBLeu171, QcrBGly175, QcrBAla179, QcrBLeu180, QcrBThr184, QcrBMet187, QcrBLeu194, QcrBSer304, QcrBGly305, and QcrBMet342 also contribute to TB47 binding, largely through hydrophobic interactions (Figure 5C). Unlike when Q203 binds (Figure 4—figure supplement 3), there is no interaction between QcrBTyr164 and TB47 (Figure 5—figure supplement 3). However, after addition of TB47, the values of turnover number for the hybrid supercomplex reduced to 5.1 ± 2.9 e-s–1 (mean ± SD, n = 4; Figure 5—figure supplement 4), a similar value that was observed when Q203 binds. Thus, the absence of this does not greatly diminish inhibition. A mutation in TB47-resistant M. smegmatis (M. tuberculosis: H195Y) is close to the Qp-binding site (Lu et al., 2019; Figure 5—figure supplement 5). As a result of the change in shape, it would appear to contribute to steric interference with the binding of TB47, thus accounting for the observed resistance.
Figure 5. Cryo-electron microscopy (cryo-EM) structure of the hybrid supercomplex in the presence of TB47.
(A) Side (left) and top (right) views of the cryo-EM structure of the M. tuberculosis cytochrome bcc complex presented as a cartoon representation. TB47 (green) and Q203 (orange) are bound to the Qp site. (B) Visualization of the density for TB47. Hydrogen bonds are shown as dotted lines. (C) Plot of distances of various parts of TB47 to residues in the Qp site were determined using LIGPLOT (https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/).

Figure 5—figure supplement 1. Cryo-electron microscopy (cryo-EM) data processing of the hybrid supercomplex consisting of M. tuberculosis CIII and M. smegmatis CIV in the presence of TB47.

Figure 5—figure supplement 2. Cryo-electron microscopy (cryo-EM) map quality assessment for the hybrid M. tuberculosis cytochrome bcc complex.

Figure 5—figure supplement 3. The densities for TB47 and QcrBTyr164.
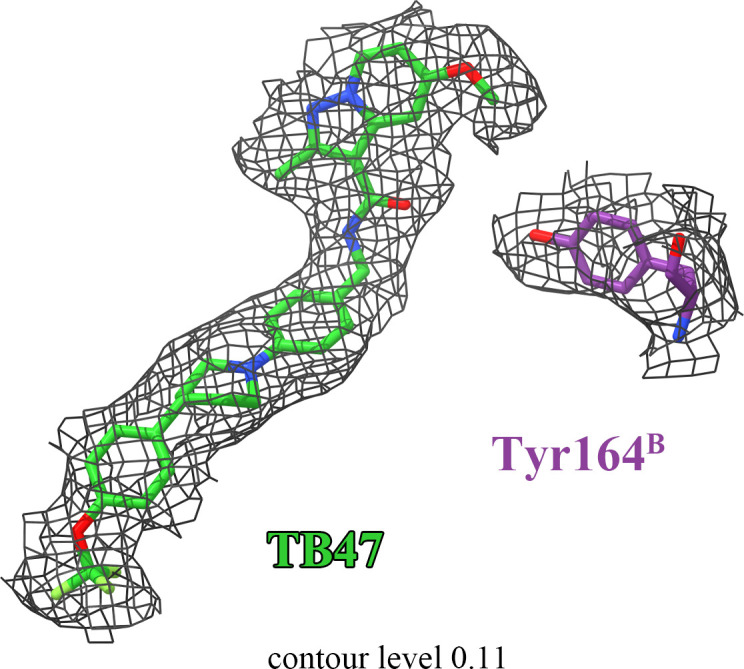
Figure 5—figure supplement 4. Rate of O2 reduction by the hybrid supercomplex before and after addition of TB47.

Figure 5—figure supplement 5. Reported mutations in TB47-resistant M. tuberculosis.
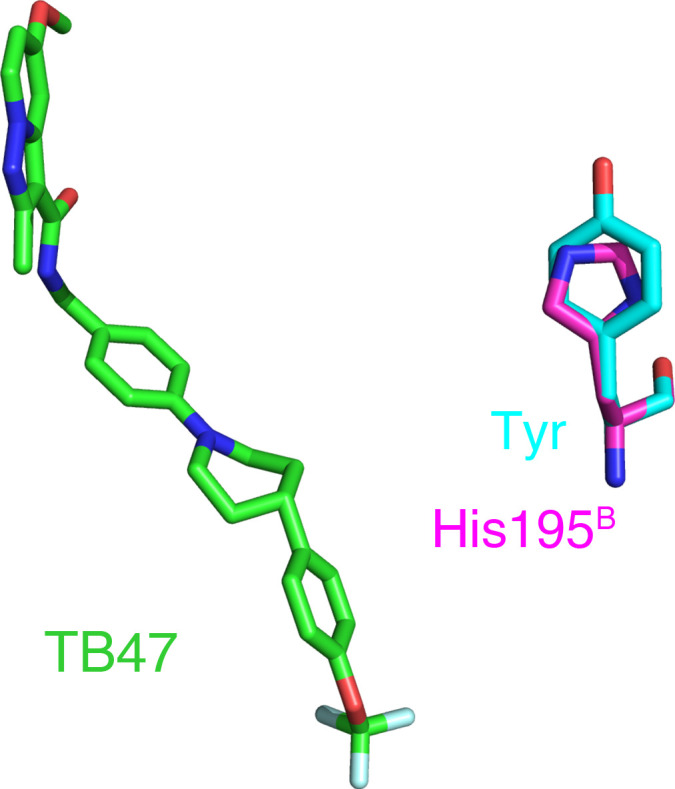
Table 3. Cryo-electron microscopy data collection, refinement, and validation statistics for the TB47-bound hybrid supercomplex.
| State | TB47 |
|---|---|
| Data collection | |
| Microscope | Titan Krios |
| Voltage (kV) | 300 |
| Magnification | 29,000× |
| Detector | Gatan K3 |
| Data collection software | SerialEM |
| Electron exposure (e–/Å2) | 60 |
| Defocus range (μm) | –1.2 to –1.8 |
| Pixel size (Å) | 0.82 |
| Data processing | |
| Number of micrographs | 2698 |
| Final particle images | 169,988 |
| Symmetry imposed | C1 |
| Map resolution (Å)Fourier shell correlation (FSC) 0.143 threshold | |
| 2.93 | |
| Refinement | |
| Initial model used (PDB code) | 6ADQ |
| Map sharpening B factor (Å2)d FSC model (0.143) masked | –97.52.9 |
| Map correlation coefficient | 0.90 |
| Mean CC for ligands | 0.79 |
| Model composition | |
| Non-hydrogen atoms | 42,679 |
| Protein residues | 5119 |
| Ligands | |
| 9Y0: 2 | |
| CDL: 17 | |
| 9YF: 4 | |
| HEA: 4 | |
| HEC: 4 | |
| MQ9: 8 | |
| HEM: 4 | |
| PLM: 4 | |
| CU: 8 | |
| FES: 2 | |
| HV0 (TB47): 2 | |
| Root mean squared deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.739 |
| Validation | |
| MolProbity score | 1.87 |
| Clashscore | 8.75 |
| Poor rotamers (%) | 6.23 |
| Ramachandran plot | |
| Favored (%) | 92.39 |
| Allowed (%) | 7.25 |
| Outliers (%) | 0.36 |
| Cβ outliers (%) | 0.00 |
Specificity of Q203 and TB47 for mycobacterial cytochrome bcc complex
The basis for the high specificity of Q203 and TB47 toward the Qp site of mycobacterial cytochromes bcc becomes apparent in the structural comparison between the QcrB subunit of M. tuberculosis and counterparts from other species (Figure 6). The highly conserved residues that are involved in the binding of these two molecules in this region (Figure 6—figure supplement 1) suggest a consistent overall fold and binding site exists in mycobacteria. This is also in agreement with the fact that Q203 and TB47 show antimycobacterial activity across many species (de Jager et al., 2020; Liu et al., 2019; Lu et al., 2019; Pethe et al., 2013; Scherr et al., 2018). It is worth noting that the structures of cytochrome bcc from M. tuberculosis and M. smegmatis have high similarity (Figure 2—figure supplement 5), and no steric hindrance is observed between the Q203 and M. smegmatis cytochrome bcc (Figure 6). This observation indicates that Q203 should have a similar binding mechanism and a similar effect on the activity of cytochrome bcc from M. smegmatis and M. tuberculosis. This is in good agreement with previous antimycobacterial activity data and inhibition data for the bcc complexes from M. smegmatis and M. tuberculosis (Gong et al., 2018; Lu et al., 2018). In contrast, in other prokaryotic, eukaryotic and human Qp-binding pockets, for example, from Saccharomyces cerevisiae (Lange and Hunte, 2002), Rhodobacter sphaeroides (Esser et al., 2008), and human (Guo et al., 2017), many of the observed interactions would not be possible (Figure 6). This suggests that Q203 and TB47 should have low-binding affinity toward its counterpart QcrB in non-mycobacterial bacteria and in eukaryotes. Even if there is some flexibility in the Qp-binding pocket that enables some level of binding, key residues that enable the binding of Q203 and TB47 in the mycobacteria are not present in other bacteria and eukaryotes (Figure 6—figure supplement 1). In addition, the binding free energies of Q203 between wild-type and mutant hybrid supercomplex were calculated and used to further elucidate the importance of some key residues that could play a role in determining the inhibitor specificity. According to a previous functional study (Pethe et al., 2013), structural comparison (Figure 6), and sequence alignment (Figure 6—figure supplement 1), QcrBThr313 and QcrBGlu314 of hybrid supercomplex were selected and mutated to their counterparts in the human supercomplex, namely residues Ala and Tyr, respectively. It is believed that Ala would decrease the affinity of Q203 (Figure 4; Pethe et al., 2013) and the Tyr could lead to steric hindrance of Q203 binding (Figure 6). 75 ns simulations were used for the calculation of binding free energy (Figure 6—figure supplement 2). The different values for the root mean squared deviation (RMSD) suggest that three mutants might have the different effects on the structural conformation of QcrB, and the dual mutant (Thr313Ala and Glu314Tyr) induced significantly structural changes compared to the wild-type complex. The 75 ns simulation was further equally divided into three phases, and the average values were used as the final relative binding free energy (Table 4), which was calculated as the binding free energy of Q203 in the mutant minus that of the wild-type systems. It is suggested that the wild-type system has stronger affinity than the mutant systems (Thr313Ala or Glu314Tyr). The relative free energy of binding calculation for Q203 in the dual mutant (Thr313Ala and Glu314Tyr) indicates that its binding affinity is significantly weaker compared to the wild-type or single-mutant systems. Therefore, the suggestions drawn from the calculation of binding free energy of Q203 provide further evidence for specificity of Q203 inhibition. In summary, these observations correlate with the observed low general antibacterial activity and low cytotoxicity of Q203 and TB47 (Liu et al., 2019; Lu et al., 2019; Pethe et al., 2013; Scherr et al., 2018).
Figure 6. Structural alignment between the M. tuberculosis Qp-binding pocket where Q203 or TB47 binds with homologous subunits from four other species.
These subunits are from (A) S. cerevisiae (pink, PDB: 1KYO), (B) R. sphaeroides (blue, PDB: 2QJP), (C) Homo sapiens (green; PDB: 5XTE), and (D) M. smegmatis (violet, PDB: 6ADQ). Residues causing steric clashes in the homologous subunits are labeled.

Figure 6—figure supplement 1. Sequence alignment of M. tuberculosis QcrB with their counterparts in other species including Homo sapiens.

Figure 6—figure supplement 2. Molecular dynamics simulation plot for the root mean squared deviation (RMSD) of the heavy atoms in the inhibitor (A) and main chain atoms of QcrB (B).

Table 4. Relative binding free energy (kcal/mol) for Q203 in three mutants of QcrB compared to the wild-type (WT).
| Mutant | 25–50 ns | 50–75 ns | 75–100 ns | Average | Standard deviation |
|---|---|---|---|---|---|
| T313A | 6.32 | 11.01 | 8.11 | 8.48 | 2.37 |
| T314Y | 4.54 | 6.00 | 8.07 | 6.20 | 1.77 |
| T313A + E314Y | 10.28 | 16.30 | 13.64 | 13.41 | 3.02 |
Implications of the Q203 and TB47 inhibitory mechanism
To gain further insights into the mechanism of action of Q203, we compared the structures of M. tuberculosis cytochrome bcc in the presence and absence of Q203 (Figure 7—figure supplement 1A). The structure of apo cytochrome bcc is almost identical with the Q203-bound structure (rmsd of 0.50 Å for all Ca atoms), which suggests that Q203 binding does not significantly affect the overall architecture of cytochrome bcc. A comparison of the Q203-bound and Q203-free cytochrome bcc structures shows residues involved in the binding pocket move outward, thus adapting to the shape of Q203 (Figure 7—figure supplement 1B). Specifically, the side chains of QcrBSer304, QcrBGlu313, QcrBGlu314, and QcrBMet342 undergo significant conformational changes to form hydrogen bonds with Q203. The binding of TB47 to M. tuberculosis cytochrome bcc also induces very similar conformational changes in the Qp-binding pocket to those seen for Q203 (rmsd of 0.454 Å for all Ca atoms) (Figure 7—figure supplement 1B). Differences in binding are due to the different ethyl group and methyl moieties in the head groups of Q203 and TB47. It is also important to note that one endogenous substrate molecule is also bound to the Qp site in the apo structure of cytochrome bcc, which potentially affects the evaluation of the conformational changes upon the binding of Q203 or TB47.
When analyzing the superimposed structures (Figure 7—figure supplement 1), it is apparent that Q203 and TB47 act competitively with the quinol binding as they almost completely occupy the Qp pocket. We therefore conclude that Q203 and TB47 are bona fide analogs of the substrate, and thus ultimately function by hindering the downstream synthesis of ATP (Figure 7). These two compounds are also highly bactericidal against M. ulcerans, almost certainly targeting the Qp-binding site (Liu et al., 2019; Scherr et al., 2018). In summary, the sequences of the QcrB subunits have high homology across pathogenic mycobacteria (Lee et al., 2020b) and the essential residues (QcrBGlu313 and QcrBGlu314) that are involved in hydrogen-bonding interactions with the inhibitors (Pethe et al., 2013; Scherr et al., 2018) are conserved across pathogenic mycobacteria (Figure 7—figure supplement 2).
Figure 7. Schematic of M. tuberculosis cytochrome bcc inhibition by Q203 and TB47.
The two monomers of M. tuberculosis cytochrome bcc are colored magenta and cyan, respectively. The binding of Q203 (orange spheres) or TB47 (green spheres) prevents substrate access (gray spheres).

Figure 7—figure supplement 1. Comparison of apo and Q203/TB47-bound structures of M. tuberculosis cytochromes bcc.

Figure 7—figure supplement 2. Sequence alignment for the QP site within pathogenic mycobacteria.

Conclusions
We have determined the apo- and Q203 and Tb47-bound structures of a hybrid pathogenic M. tuberculosis/M. smegmatis cytochrome bcc complex. The study shows the structural features of M. tuberculosis cytochrome bcc and how it is specifically inhibited by Q203 and TB47. The extensive interactions between Q203 or TB47 and the Qp-binding pocket account for the highly specific binding of these two inhibitors to pathogenic M. tuberculosis cytochrome bcc compared to eukaryotic counterparts. Two conservative residues involved with the formation of hydrogen bonds are observed across the pathogenic mycobacteria. These structures provide a long-sought basis for rational, structure-based inhibitor design to accelerate the development of Q203 and TB47 analogs as drug leads for mycobacterial infections.
Materials and methods
Key resources table.
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Mycobacterium tuberculosis) | qcrC | Mycobrowser | Rv2194 | https://mycobrowser.epfl.ch/genes/Rv2194 |
| Gene (M. tuberculosis) | qcrA | Mycobrowser | Rv2195 | https://mycobrowser.epfl.ch/genes/Rv2195 |
| Gene (M. tuberculosis) | qcrB | Mycobrowser | Rv2196 | https://mycobrowser.epfl.ch/genes/Rv2196 |
| Strain, strain background (Mycobacterium smegmatis) | mc2 51 | Li et al., 2014 | ||
| Genetic reagent (include species here) | pVV16-QcrCAB-His10 | This paper | Construct contains the qcrCAB operon encoding three subunits | |
| Commercial assay or kit | ClonExpress II One Step Cloning Kit | Vazyme | C112-01 | |
| Chemical compound, drug | Q203 | MCE | HY-101040 | Prepare stock solution in DMSO |
| Chemical compound, drug | TB47 | Lu et al., 2019 | Prepare stock solution in DMSO | |
| Chemical compound, drug | LMNG | Anatrace | NG310 | |
| Chemical compound, drug | Digitonin | BIOSYNTH | D-3200 | |
| Software, algorithm | SerialEM | Mastronarde, 2003 | Version 3.6 | |
| Software, algorithm | MotionCor2 | Zheng et al., 2017 | Version 1.2.1 | |
| Software, algorithm | RELION | Zivanov et al., 2019 | Version 3.03 | |
| Software, algorithm | cryoSPARC | Punjani et al., 2017 | Version 3.2.0 | |
| Software, algorithm | Phyre2 | Kelley et al., 2015 | Version 2.0 | |
| Software, algorithm | UCSF Chimera | Pettersen et al., 2004 | Version 1.12 | |
| Software, algorithm | COOT | Emsley et al., 2010 | Version 0.8.9 | |
| Software, algorithm | PHENIX | Adams et al., 2010 | Version 1.16 | |
| Software, algorithm | PyMOL | Schrödinger LLC, 2017 | Version 2.0 | |
| Software, algorithm | ChemDraw | Li et al., 2004 | Version 19.0 |
Expression of hybrid supercomplex consisting of M. tuberculosis CIII and M. smegmatis CIV
The hybrid supercomplex was obtained according to a previous study (Kim et al., 2015) but with some modifications. The M. tuberculosis cytochrome bcc complex is encoded by three putative genes (Rv2194-2196). Genes were amplified from H37Rv genomic DNA by PCR using Phanta Max DNA polymerase (Vazyme), and two-step PCR was used to inset a 10× His tag at the C-terminus of the qcrB (Rv2196). Genes encoding the entire cytochrome bcc complex operon were then cloned into the pVV16 expression vector. The resultant plasmid was transformed into M. smegmatis mc2 51 (Li et al., 2014) cells whose qcrCAB operon encoding M. smegmatis cytochrome bcc had already been knocked out. The cells were cultivated in Luria-Bertani broth (LB) liquid media supplemented with 50 μg/mL hygromycin, 25 μg/mL streptomycin, and 0.1% Tween 80. Cell pellets were harvested by centrifugation when the cells were grown to an optical density (OD600 of 1.0–1.2) at 37°C (220 rpm). Harvested cells were frozen at –80°C until use.
Purification of the hybrid supercomplex
Cell pellets were thawed and resuspended in buffer A containing 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.4, 100 mM NaCl, and then lysed by passing through a French Press at 1200 bar three times. Cell debris and non-lysed cells were removed by centrifugation at 14,000 rpm for 10 min at 4°C. The supernatant was collected and ultra-centrifuged at 36,900 rpm and 4°C for 2 hr. The membrane fraction was solubilized by addition of 1% (w/v) lauryl maltose neopentyl glycol (LMNG) in buffer A and incubated for 2 hr at 4°C with slow stirring. The suspension was ultra-centrifuged, and the supernatant was applied to Ni-NTA agarose beads (GE Healthcare) at 4°C. The beads were further washed in buffer A with 50 mM imidazole and 0.004% (w/v) LMNG. The buffer was exchanged to buffer B (20 mM MOPS, pH 7.4, 100 mM NaCl, and 0.1% [w/v] digitonin) and then washed in resin in batch mode. The protein was eluted from the beads with buffer B containing 500 mM imidazole. Protein was then concentrated and loaded onto a Superdex 6 increase (10/300GL, GE Healthcare) column equilibrated in buffer B. Peak fractions were pooled and concentrated to ~8 mg/mL for electron microscopy studies. The protein sample was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the bands were then identified through mass spectrometry.
Activity assays
2,3-Dimethyl-1,4-naphthoquinone (DMNQ, CAS 2197-57-1) was synthesized by WuXi AppTec. In order to obtained reduced DMNQH2, 20 mM DMNQ was ultrasonically dissolved in 1 mL ethanol with 6 mM HCl and reduced with sodium borohydride (NaBH4) in the ice bath. An appropriate amount of 12 N HCl was added into quench unreacted NaBH4 under the protection of argon. Oxidase activity was determined by a method described previously (Bonner et al., 1986; Kusumoto et al., 2000; Safarian et al., 2019). The oxygen consumption was monitored using the Clark-type oxygen electrode (Oxytherm+, Hansatech) in 1 mL reaction buffer (20 mM MOPS, pH 7.4, 100 mM NaCl, 0.004% LMNG) at 25℃, containing 62.5 nM hybrid supercomplex. The reaction was started by addition of 100 μM DMNQH2. The oxygen consumption curve was plotted using GraphPad Prime 8.0 software, from which an estimate of the oxygen-reduction rate (v0) was obtained (corrected for autoxidation). In the inhibition assay, 500 nM Q203 or TB47, which is approximately the value of the median inhibitory concentration (IC50) according to our previous study (Gong et al., 2018), was chosen and incubated with 62.5 nM hybrid supercomplex for 20 min at 25℃. Inhibition curves and oxygen-reduction rates (vi) with the different inhibitors were recorded. The inhibition rate (1 v0/vi) of Q203 and TB47 at 500 nM is reported. This assay was conducted using four groups of parallel experiments.
Cryo sample preparation and data collection
300-mesh Quantifoil R0.6/1.0 grids (Quantifoil, Micro Tools GmbH, Germany) were glow-discharged at H2/O2 atmosphere for 25 s. 3 μL aliquots of protein complex at a concentration of 10 mg/mL were applied to the grid and then blotted for 3 s with force 0 at 8°C and 100% humidity using a Vitrobot IV (Thermo). Images were collected using a Titan Krios 300 keV electron microscope (Thermo), equipped with K3 Summit direct electron detector camera (Gatan). Data were recorded at 29,000× magnification with a calibrated super-resolution pixel size 0.82 Å/pixel. The exposure time was set to 2.4 s with 40 subframes and a total dose of 60 electrons per Å2. All images were automatically recorded using SerialEM with a defocus range from 1.2 μm to 1.8 μm (Mastronarde, 2003). For the datasets of apo, Q203-bound and TB47-bound M. tuberculosis cytochrome bcc, a total of 4141, 3763, and 2968 images were collected, respectively.
Image processing
All dose-fractioned stacks were motion-corrected and dose-weighted using MotionCorr2 (Zheng et al., 2017) in RELION 3.03 (Zivanov et al., 2019). CTF estimation was conducted using cryoSPARC patch CTF estimation (Punjani et al., 2017). For the dataset of apo hybrid M. tuberculosis cytochrome bcc, 1,208,054 particles were picked automatically using EMD-9610 map as the template and extracted with a box size of rescaled 256 pixels (binned 2). 327,188 particles were selected after two rounds of 2D classification. 100,000 particles were used to perform ab initio reconstruction in four classes, and these four classes were used as 3D volume templates for heterogeneous refinement with all selected particles. 112,804 particles converged into one class with clear signals and then re-extracted with 512 pixels (binned 1). Next, this particle set was used to do homogeneous refinement and local refinement, yielding the final resolution 2.68 Å. For the dataset of Q203-bound and TB47-bound M. tuberculosis cytochrome bcc, the data processing was performed in a similar pipeline, resulting in the final reconstruction resolution at 2.67 Å and 2.93 Å, respectively (detailed parameters shown in supplementary figures).
Model building and validation
The M. smegmatis respiratory complex CIII2CIV2 (PDB code: 6ADQ) model (Gong et al., 2018) as rigid body was fitted into EM density maps using UCSF Chimera 1.12 (Pettersen et al., 2004). Next, the resultant atomic model was manually modified according to the subunit sequences of M. tuberculosis cytochrome bcc and refined in COOT 0.8.9.1 (Emsley et al., 2010), followed by real-space refinement in PHENIX (Adams et al., 2010). The smile strings of Q203 and TB47 were generated and copied from ChemDraw (Li et al., 2004) and defined in PHENIX elBOW. Q203 and TB47 were manually built into the corresponding EM densities. The local resolution map was calculated in cryoSPARC (Punjani et al., 2017). All reported resolutions were based on the gold-standard FSC 0.143 criteria (Rosenthal and Henderson, 2003).
MK9 docking study
The binding pose of MK9 with QcrB was generated by molecular docking using the Glide (Schrödinger, LLC). The spatially neighboring subunits QcrA/B were extracted from the hybrid supercomplex and used as the model. The protein structure was processed using the Protein Preparation Wizard module. In this process, hydrogens were added to heavy atoms and bond orders were assigned to each bond. The protonation state of each amino acid was predicted at pH 7.0 using the Epik algorithm (Shelley et al., 2007). All resolved waters were removed from the structure. At the same time, the structure of MK9 was prepared using the LigPrep module, and a low-energy conformation of MK9 was generated. The binding pocket of Q203 in QcrB was defined as the docking site using the Receptor Grid Generation module. The docking site was confined to an enclosed cube with side length of 20 Å, which was centered on the centroid of Q203. MK9 was then docked using the Ligand Docking module. Finally, 10 docking poses were generated for MK9 after post-docking minimization based on the force field of OPLS3 (Harder et al., 2016). These poses were ranked by the scoring function of GlideScore. The pose with the lowest GlideScore was chosen for binding mode analysis.
Binding free energies of Q203 between wild-type and mutant hybrid supercomplex
Binding free energies of Q203 between wild-type and mutant hybrid supercomplex were computed using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method based on all-atom molecular dynamic simulations (Greene et al., 2019; Homeyer and Gohlke, 2012; Homeyer and Gohlke, 2015). This task can be divided into three phases: (i) simulation system construction, (ii) molecular dynamic simulation, and (iii) binding free energy computation.
Considering that QcrB is the core subunit interacting with Q203 in the hybrid supercomplex, the QcrB with Q203 bound was the only region extracted from the Q203-bound hybrid supercomplex for simulation system construction. Membrane Builder in CHARMM-GUI (Jo et al., 2007) was used to build the protein/membrane system solvated in water. PDB file containing the coordination information of Q203-bound QcrB was read while only protein residues and Q203 were retained. The CHARMM General Force Field (CGenFF) was applied to parameterize the Q203 (Vanommeslaeghe and MacKerell, 2012). DPPC lipid molecules were used to explicitly build the lipid bilayers and bulky water layers were added to the top and bottom sides of the membrane. In addition, 0.15 M KCl was added to neutralize the charge of system using the distance-based ion placing method. Next, the protein was inserted in the lipid bilayers and covered by the water layers despite exposure of some residues to the solvents. In order to generate topology and parameter files for subsequent AMBER dynamic simulations, the force fields of ff19SB (Tian et al., 2020) and Lipids17 (Lee et al., 2020a) were respectively applied to the parameterization of protein and lipid molecules. Four Q203-bound QcrB simulation systems were built, including wild-type QcrB, T313A mutant, E314Y mutant, and T313A/E314Y mutant.
Molecular dynamic simulation was carried out using AMBER 2020 (Case et al., 2005). Before dynamic simulation, 5000 steps of energy minimization were performed on waters to remove potential steric clashes between the solute and solvents. Harmonic restraints with a force constant of 10 kcal/mol Å2 and 2.5 kcal/mol Å2 were placed on protein and lipids in the minimization, respectively. Then, two stages of equilibrium simulations were carried out to relax the protein and lipid molecules by gradually decreasing the restraint force. The first stage was further divided into three short simulations each taking 375 ps with the time step set to 1 fs. The constant volume and temperature (NVT) ensemble was applied to this stage of simulation. The second one was a much longer simulation with the constant pressure and temperature (NPT) ensemble, which took 1.5 ns to completely relax the whole system with extremely low force. At this stage, the time step was increased to 2 fs while the SHAKE algorithm was used to fix the bonds with hydrogens (Bailey and Lowe, 2009). Subsequently, a 10 ns NPT simulation without any restraints was performed to further relax the system. Langevin dynamics was used to control the temperature at 303.15 K in NVT and NPT simulations (Grønbech-Jensen and Farago, 2014). In the NPT simulations, additional semi-isotropic pressure coupling and constant surface tension were applied to the simulation of lipid bilayers (Bennun et al., 2007). Once the equilibrium simulation was finished, a 100 ns NPT simulation free of restraints was carried out for each molecular system to produce the final trajectory used for energy calculation. Coordinates were printed every 100 ps so that in total 1000 frames were contained in the final trajectory. The CPPTRAJ module in AMBER was applied to analyze the trajectory (Roe and Cheatham, 2013).
MM/PBSA methods were employed to calculate the binding free energy of Q203 in wild-type and mutant QcrB (Homeyer and Gohlke, 2012). As shown in the equation (ΔG= EMM + Epol + Enp - TS), free energy can be calculated by combining the MM part (EMM) that represents the gas phase energy contribution as well as the solvation free energy components including polar (Epol) and non-polar (Enp) contributions. The gas phase free energy was calculated according to the force field. As for the solvation free energy, the polar part representing electrostatic contribution was calculated using the Poisson–Boltzmann (PB) equation based on implicit solvent model (Honig and Nicholls, 1995). In AMBER, the equation also provides an implicit membrane model to calculate the membrane-mediated electrostatic interactions. Here, geometric multigrid based on iterative solver was selected for PB equation calculation (Harris et al., 2013). On the other hand, the LCPO method was applied to calculate the non-polar part that represents the hydrophobic contribution (Weiser et al., 1999). The Python script MMPBSA.py (Miller et al., 2012) in AMBER was used for the computation based on previously produced simulation trajectories, from which one frame was extracted every 1 ns for the energy calculations. Ions, water, and lipid molecules were all striped from the molecular systems before calculation. For implicit membrane model, the membrane dielectric constant was set to 7.0, and membrane thickness was set to 42 Å. Entropy contributions (TS) could be neglected as the four molecular systems were assumed to have similar entropy changes due to the same protein-ligand structures except for the mutated residues.
Creation of figures
All the figures were created using UCSF Chimera (Pettersen et al., 2004) or PyMOL (Schrödinger LLC, 2017).
Acknowledgements
We thank Dr. Chao Peng of the Mass Spectrometry System at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, SARI, China, for data collection and analysis and Prof. Lei Li (College of Life Sciences, Nankai University) for their technical support on Clark-type oxygen electrode and oxygen consumption assay. We thank Prof. Kaixia Mi (CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, CAS) for sharing the strain M. smegmatis mc2 51. We would like to thank Prof. Gregory M Cook (School of Biomedical Sciences, University of Otago, New Zealand) and Prof. Xiaoyun Lu (School of Pharmacy, Ji′nan University, China) for kindly providing TB47 for this study. We would also like to thank the Bio-Electron Microscopy Facility of ShanghaiTech University.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Yan Gao, Email: gaoyan@shanghaitech.edu.cn.
Zihe Rao, Email: raozh@mail.tsinghua.edu.cn.
Hongri Gong, Email: gonghr@nankai.edu.cn.
David Drew, Stockholm University, Sweden.
Volker Dötsch, Goethe University, Germany.
Funding Information
This paper was supported by the following grants:
National Key Research and Development Program of China 2017YFC0840300 to Zihe Rao.
National Key Research and Development Program 2020YFA0707500 to Zihe Rao.
National Natural Science Foundation of China 81520108019 to Zihe Rao.
National Natural Science Foundation of China 813300237 to Zihe Rao.
National Natural Science Foundation of China 32100976 to Hongri Gong.
Natural Science Foundation of Tianjin City 20JCQNJC01430 to Hongri Gong.
Additional information
Competing interests
No competing interests declared.
No competing interests declared.
Author contributions
Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review and editing.
Data curation, Formal analysis, Methodology, Visualization, Writing – review and editing.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Formal analysis, Methodology.
Data curation, Formal analysis, Methodology.
Data curation, Formal analysis, Methodology.
Formal analysis, Methodology, Writing – review and editing.
Formal analysis, Methodology.
Data curation, Formal analysis, Methodology, Writing – review and editing.
Funding acquisition, Resources, Supervision, Writing – review and editing.
Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review and editing.
Additional files
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Source data files have been provided. The accession numbers for the 3D cryo-EM density map of apo, Q203-bound and TB47-bound hybrid supercomplex in present study are EMD-30943, EMD-30944 and EMD-30945, respectively. The accession numbers for the coordinates for the apo, Q203-bound and TB47-bound hybrid supercomplex in this study are PDB: 7E1V, PDB: 7E1W and PDB: 7E1X, respectively.
The following dataset was generated:
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of apo hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV. Electron Microscopy Data Bank. EMD-30943
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV in the presence of Q203. Electron Microscopy Data Bank. EMD-30944
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV in presence of TB47. Electron Microscopy Data Bank. EMD-30945
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of apo hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV. RCSB Protein Data Bank. 7E1V
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV in the presence of Q203. RCSB Protein Data Bank. 7E1W
Zhou S, Wang W, Gao Y, Gong H, Rao Z. 2021. Cryo-EM structure of hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis complexIII and Mycobacterium smegmatis complexIV in presence of TB47. RCSB Protein Data Bank. 7E1X
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica. Section D, Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AG, Lowe CP. MILCH SHAKE: An efficient method for constraint dynamics applied to alkanes. Journal of Computational Chemistry. 2009;30:2485–2493. doi: 10.1002/jcc.21237. [DOI] [PubMed] [Google Scholar]
- Bennun SV, Dickey AN, Xing C, Faller R. Simulations of biomembranes and water: Important technical aspects. Fluid Phase Equilibria. 2007;261:18–25. doi: 10.1016/j.fluid.2007.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birth D, Kao WC, Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nature Communications. 2014;5:1–11. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- Bonner WD, Clarke SD, Rich PR. Partial purification and characterization of the quinol oxidase activity of Arum maculatum mitochondria. Plant Physiology. 1986;80:838–842. doi: 10.1104/pp.80.4.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. Journal of Computational Chemistry. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm RH, Trauer JM, Curnoe D, Tanaka MM. Controlled fire use in early humans might have triggered the evolutionary emergence of tuberculosis. PNAS. 2016;113:9051–9056. doi: 10.1073/pnas.1603224113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honoré N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Cook GM, Hards K, Dunn E, Heikal A, Nakatani Y, Greening C, Crick DC, Fontes FL, Pethe K, Hasenoehrl E, Berney M. Oxidative phosphorylation as a target space for tuberculosis: success, caution, and future directions. Microbiology Spectrum. 2017;5:2016. doi: 10.1128/microbiolspec.TBTB2-0014-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager VR, Dawson R, van Niekerk C, Hutchings J, Kim J, Vanker N, van der Merwe L, Choi J, Nam K, Diacon AH. Telacebec (Q203), a new antituberculosis agent. The New England Journal of Medicine. 2020;382:1280–1281. doi: 10.1056/NEJMc1913327. [DOI] [PubMed] [Google Scholar]
- Donohue MJ. Increasing nontuberculous mycobacteria reporting rates and species diversity identified in clinical laboratory reports. BMC Infectious Diseases. 2018;18:163. doi: 10.1186/s12879-018-3043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica. Section D, Biological Crystallography. 2010;66:486–501. doi: 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Elberry M, Zhou F, Yu C-A, Yu L, Xia D. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. The Journal of Biological Chemistry. 2008;283:2846–2857. doi: 10.1074/jbc.M708608200. [DOI] [PubMed] [Google Scholar]
- Gong H, Li J, Xu A, Tang Y, Ji W, Gao R, Wang S, Yu L, Tian C, Li J, Yen HY, Man Lam S, Shui G, Yang X, Sun Y, Li X, Jia M, Yang C, Jiang B, Lou Z, Robinson CV, Wong LL, Guddat LW, Sun F, Wang Q, Rao Z. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science. 2018;362:eaat8923. doi: 10.1126/science.aat8923. [DOI] [PubMed] [Google Scholar]
- Greene DA, Qi R, Nguyen R, Qiu T, Luo R. Heterogeneous dielectric implicit membrane model for the calculation of MMPBSA binding free energies. Journal of Chemical Information and Modeling. 2019;59:3041–3056. doi: 10.1021/acs.jcim.9b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønbech-Jensen N, Farago O. Constant pressure and temperature discrete-time Langevin molecular dynamics. The Journal of Chemical Physics. 2014;141:194108. doi: 10.1063/1.4901303. [DOI] [PubMed] [Google Scholar]
- Guo R, Zong S, Wu M, Gu J, Yang M. Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell. 2017;170:1247–1257. doi: 10.1016/j.cell.2017.07.050. [DOI] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. Journal of Chemical Theory and Computation. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Hards K, Adolph C, Harold LK, McNeil MB, Cheung CY, Jinich A, Rhee KY, Cook GM. Two for the price of one: Attacking the energetic-metabolic hub of mycobacteria to produce new chemotherapeutic agents. Progress in Biophysics and Molecular Biology. 2020;152:35–44. doi: 10.1016/j.pbiomolbio.2019.11.003. [DOI] [PubMed] [Google Scholar]
- Harikishore A, Chong SSM, Ragunathan P, Bates RW, Grüber G. Targeting the menaquinol binding loop of mycobacterial cytochrome bd oxidase. Molecular Diversity. 2021;25:517–524. doi: 10.1007/s11030-020-10034-0. [DOI] [PubMed] [Google Scholar]
- Harris RC, Boschitsch AH, Fenley MO. Influence of grid spacing in Poisson-Boltzmann equation binding energy estimation. Journal of Chemical Theory and Computation. 2013;9:3677–3685. doi: 10.1021/ct300765w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Molecular Informatics. 2012;31:114–122. doi: 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Homeyer N, Gohlke H. Extension of the free energy workflow FEW towards implicit solvent/implicit membrane MM–PBSA calculations. Biochimica et Biophysica Acta (BBA) - General Subjects. 2015;1850:972–982. doi: 10.1016/j.bbagen.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Huang L-S, Lümmen P, Berry EA. Crystallographic investigation of the ubiquinone binding site of respiratory Complex II and its inhibitors. Biochimica et Biophysica Acta. Proteins and Proteomics. 2021;1869:140679. doi: 10.1016/j.bbapap.2021.140679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLOS ONE. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen MD, Herrmann JL, Kremer L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nature Reviews. Microbiology. 2020;18:392–407. doi: 10.1038/s41579-020-0331-1. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M-S, Jang J, Ab Rahman NB, Pethe K, Berry EA, Huang L-S. Isolation and characterization of a hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis cytochrome bcc and Mycobacterium smegmatis cytochrome aa3. The Journal of Biological Chemistry. 2015;290:14350–14360. doi: 10.1074/jbc.M114.624312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A, Vranckx L, Dendouga N, Balemans W, Van den Wyngaert I, Vergauwen K, Göhlmann HWH, Willebrords R, Poncelet A, Guillemont J, Bald D, Andries K. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. The Journal of Biological Chemistry. 2008;283:25273–25280. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- Kusumoto K, Sakiyama M, Sakamoto J, Noguchi S, Sone N. Menaquinol oxidase activity and primary structure of cytochrome bd from the amino-acid fermenting bacterium Corynebacterium glutamicum. Archives of Microbiology. 2000;173:390–397. doi: 10.1007/s002030000161. [DOI] [PubMed] [Google Scholar]
- Lange C, Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. PNAS. 2002;99:2800–2805. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hitzenberger M, Rieger M, Zacharias M, Im W. CHARMM-GUI supports the Amber force fields. The Journal of Chemical Physics. 2020a;153:035103. doi: 10.1063/5.0012280. [DOI] [PubMed] [Google Scholar]
- Lee BS, Sviriaeva E, Pethe K. Targeting the cytochrome oxidases for drug development in mycobacteria. Progress in Biophysics and Molecular Biology. 2020b;152:45–54. doi: 10.1016/j.pbiomolbio.2020.02.001. [DOI] [PubMed] [Google Scholar]
- Li Z, Wan H, Shi Y, Ouyang P. Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. Journal of Chemical Information and Computer Sciences. 2004;44:1886–1890. doi: 10.1021/ci049794h. [DOI] [PubMed] [Google Scholar]
- Li X, Liu F, Hu Y, Mi K. Draft Genome Sequence of mc251, a Highly Hydrogen Peroxide-Resistant Mycobacterium smegmatis Mutant Strain. Genome Announcements. 2014;2:e00092-14. doi: 10.1128/genomeA.00092-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gao Y, Liu J, Tan Y, Liu Z, Chhotaray C, Jiang H, Lu Z, Chiwala G, Wang S, Makafe G, Islam MM, Hameed HMA, Cai X, Wang C, Li X, Tan S, Zhang T. The compound TB47 is highly bactericidal against Mycobacterium ulcerans in a Buruli ulcer mouse model. Nature Communications. 2019;10:1–9. doi: 10.1038/s41467-019-08464-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Asseri AH, Kremer M, Maaskant J, Ummels R, Lill H, Bald D. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Scientific Reports. 2018;8:1–7. doi: 10.1038/s41598-018-20989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Williams Z, Hards K, Tang J, Cheung C-Y, Aung HL, Wang B, Liu Z, Hu X, Lenaerts A, Woolhiser L, Hastings C, Zhang X, Wang Z, Rhee K, Ding K, Zhang T, Cook GM. Pyrazolo[1,5- a]pyridine Inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infectious Diseases. 2019;5:239–249. doi: 10.1021/acsinfecdis.8b00225. [DOI] [PubMed] [Google Scholar]
- Lupien A, Foo CS-Y, Savina S, Vocat A, Piton J, Monakhova N, Benjak A, Lamprecht DA, Steyn AJC, Pethe K, Makarov VA, Cole ST. New 2-Ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLOS Pathogens. 2020;16:e1008270. doi: 10.1371/journal.ppat.1008270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. SerialEM: a program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microscopy and Microanalysis. 2003;9:1182–1183. doi: 10.1017/S1431927603445911. [DOI] [Google Scholar]
- Megehee JA, Hosler JP, Lundrigan MD. Evidence for a cytochrome bcc–aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology. 2006;152:823–829. doi: 10.1099/mic.0.28723-0. [DOI] [PubMed] [Google Scholar]
- Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim S-A, Nam J, Kang H, Kwon H, Oh C-T, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han S-J, No Z, Lee J, Brodin P, Cho S-N, Nam K, Kim J. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nature Medicine. 2013;19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Rao SPS, Alonso S, Rand L, Dick T, Pethe K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. PNAS. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi N, Legrand E, Sola C. The mycobacteria: an introduction to nomenclature and pathogenesis. Revue Scientifique et Technique. 2001;20:21–54. doi: 10.20506/rst.20.1.1265. [DOI] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. Journal of Molecular Biology. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Safarian S, Hahn A, Mills DJ, Radloff M, Eisinger ML, Nikolaev A, Meier-Credo J, Melin F, Miyoshi H, Gennis RB, Sakamoto J, Langer JD, Hellwig P, Kühlbrandt W, Michel H. Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science. 2019;366:100–104. doi: 10.1126/science.aay0967. [DOI] [PubMed] [Google Scholar]
- Scherr N, Bieri R, Thomas SS, Chauffour A, Kalia NP, Schneide P, Ruf M-T, Lamelas A, Manimekalai MSS, Grüber G, Ishii N, Suzuki K, Tanner M, Moraski GC, Miller MJ, Witschel M, Jarlier V, Pluschke G, Pethe K. Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nature Communications. 2018;9:5370. doi: 10.1038/s41467-018-07804-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC . Schrödinger LLC; 2017. [Google Scholar]
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. Journal of Computer-Aided Molecular Design. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Solmaz SRN, Hunte C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. The Journal of Biological Chemistry. 2008;283:17542–17549. doi: 10.1074/jbc.M710126200. [DOI] [PubMed] [Google Scholar]
- Tian C, Kasavajhala K, Belfon KA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q, Simmerling C. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. Journal of Chemical Theory and Computation. 2020;16:528–552. doi: 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- Tortoli E, Fedrizzi T, Meehan CJ, Trovato A, Grottola A, Giacobazzi E, Serpini GF, Tagliazucchi S, Fabio A, Bettua C, Bertorelli R, Frascaro F, De Sanctis V, Pecorari M, Jousson O, Segata N, Cirillo DM. The new phylogeny of the genus Mycobacterium: The old and the news. Fection, Genetics and Evolution. 2017;56:19–25. doi: 10.1016/j.meegid.2017.10.013. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. Journal of Chemical Information and Modeling. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO) Journal of Computational Chemistry. 1999;20:217–230. doi: 10.1002/(SICI)1096-987X(19990130)20:2<217::AID-JCC4>3.0.CO;2-A. [DOI] [Google Scholar]
- Wiseman B, Nitharwal RG, Fedotovskaya O, Schäfer J, Guo H, Kuang Q, Benlekbir S, Sjöstrand D, Ädelroth P, Rubinstein JL, Brzezinski P, Högbom M. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. Nature Structural & Molecular Biology. 2018;25:1128–1136. doi: 10.1038/s41594-018-0160-3. [DOI] [PubMed] [Google Scholar]
- World Health Organization Global Tuberculosis Report. WHO 2019
- Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Scheres SHW. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ. 2019;6:5–17. doi: 10.1107/S205225251801463X. [DOI] [PMC free article] [PubMed] [Google Scholar]