

Abstract
Both preclinical and clinical studies have demonstrated that exposures to acetaminophen (APAP) at levels that cause hepatic injury cause pulmonary injury as well. However, whether exposures that do not result in hepatic injury have acute pulmonary implications is unknown. Thus, we sought to determine how APAP exposures at levels that do not result in significant hepatic injury impact the mature lung. Adult male ICR mice (8–12 wk) were exposed to a dose of APAP known to cause hepatotoxicity in adult mice [280 mg/kg, intraperitoneal (ip)], as well as a lower dose previously reported to not cause hepatic injury (140 mg/kg, ip). We confirm that the lower dose exposures did not result in significant hepatic injury. However, like high dose, lower exposure resulted in increased cellular content of the bronchoalveolar lavage fluid and induced a proinflammatory pulmonary transcriptome. Both the lower and higher dose exposures resulted in measurable changes in lung morphometrics, with the lower dose exposure causing alveolar wall thinning. Using RNAScope, we were able to detect dose-dependent, APAP-induced pulmonary Cyp2e1 expression. Finally, using FLIM we determined that both APAP exposures resulted in acute pulmonary metabolic changes consistent with mitochondrial overload in lower doses and a shift to glycolysis at a high dose. Our findings demonstrate that APAP exposures that do not cause significant hepatic injury result in acute inflammatory, morphometric, and metabolic changes in the mature lung. These previously unreported findings may help explain the potential relationship between APAP exposures and pulmonary-related morbidity.
Keywords: acetaminophen, CYP2E1, FLIM, lung injury, paracetamol
INTRODUCTION
Acetaminophen (N-acetyl-p-aminophenol, APAP) is a widely used analgesic and generally perceived to be safe (1). However, APAP is the leading cause of acute liver failure in the United States and Europe (1). In addition, a growing body of literature has associated chronic APAP exposure and pulmonary morbidity, including asthma (2–14) and COPD (2). The mechanisms underlying this association are unknown, and the potential detrimental pulmonary effects of APAP exposure deserve further investigation.
Following exposure, the majority of ingested APAP (80%–90%) is metabolized in the liver through glucuronidation by glucuronyl transferase or sulfation by sulfotransferases to nontoxic metabolites (1, 15). Under normal conditions, a small percentage of the remaining APAP is metabolized by cytochrome p450 family 2 subfamily E member 1 (CYP2E1) to the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI). This toxic metabolite is inactivated by conjugation with glutathione (GSH). With toxic APAP exposure, glucuronyl transferase and sulfotransferase activity is saturated, and CYP2E1-generated NAPQI increases. Accumulation of this toxin leads to multiple deleterious effects, including glutathione (GSH) depletion, APAP-protein adducts, mitochondrial dysfunction, and hepatocyte death (1, 15).
Mitochondrial dysfunction plays a central role in the hepatocyte injury and death following toxic APAP exposures (16). The accumulation of mitochondrial APAP adducts and the resulting mitochondrial dysfunction occurs transiently at exposures that do not cause hepatotoxicity (17); however, with toxic exposures, they induce sustained metabolic changes that contribute to cellular injury (16). Studies of mitochondrial respiration following APAP exposure demonstrate disturbances in complex I and complex II function along with cellular ATP depletion (18–22). In fact, complex II is directly susceptible to inhibition by CYP2E1-generated NAPQI (23). The direct effect of NAPQI on complex II has important implications on cellular metabolism due to its key role in electron transport and the tricarboxylic acid (TCA, Krebs) cycle. Knowledge of this biochemical mechanism is important as bypassing complex II inhibition by directly providing TCA cycle substrate to restore cellular ATP and attenuates APAP-induced hepatocyte injury (24). Thus, our growing understanding of the metabolic implications of APAP toxicity in the liver has revealed potential therapeutic approaches to limit injury.
Importantly, CYP2E1 expression is not limited to the liver. Multiple cells in the lung express CYP2E1 (25). Consistent with the observation of pulmonary CYP2E1 expression, clinical studies have associated acute APAP overdose with pulmonary injury (2, 26–28). Supporting these clinical observations, preclinical studies have demonstrated that APAP is directly toxic to the lung, and is independent of hepatic injury (29–37). Importantly, APAP-protein adducts can be detected in the lung following exposure (29–31, 35, 38). Of note, APAP exposures known to cause hepatic toxicity are also reported to injure the bronchiolar epithelium (35–37, 39), and the distal pulmonary epithelium resulting in emphysematous-type histological changes (40). Despite the consistent and reliable observation that APAP exposures that result in hepatotoxicity also injure the mature lung, the mechanisms underlying this injury are unknown (41). In addition, whether the mature lung is susceptible to APAP toxicity at exposures that do not result in liver injury is unknown. Finally, whether APAP exposures induce metabolic changes in the lung, at what level of exposure these occur, and whether these effects are specific to a subset of CYP2E1 expressing cells within the lung is unknown. These fundamental gaps leave potential treatment options unexplored and unavailable to those with APAP-induced lung injury.
Therefore, we set out to determine if APAP exposures that do not cause significant hepatic toxicity resulted in transcriptional, morphometric, or metabolic changes in the mature murine lung. In this study, we exposed adult male mice to APAP [140 mg/kg or 280 mg/kg, intraperitoneal (ip)] and performed robust and blinded histopathological assessments of hepatic injury and assessed the pulmonary transcriptomic, morphometric, and metabolic responses. We found that APAP 140 mg/kg did not induce significant hepatic injury; however, this exposure resulted in a significant proinflammatory pulmonary transcriptional response and an influx of immune cells in the bronchoalveolar lavage fluid (BALF). These proinflammatory changes were associated with measurable morphometric changes suggesting parenchymal tissue remodeling and alveoli wall thinning at the low (140 mg/kg) APAP dose. Finally, using fluorescence lifetime imaging microscopy (FLIM) we were able to detect local pulmonary metabolic changes induced by APAP at both exposures. Exposures to low dose resulted in decreased glycolysis and increased Fluorescent Lifetime Redox Ratio (FLIRR) suggesting that mitochondrial overload with potential mitochondrial dysfunction. In contrast, exposure to high doses resulted in increased glycolysis with decreased mitochondrial function. These previously unrecognized transcriptomic, metamorphic, and metabolic alterations may help explain the association between APAP exposure and pulmonary morbidity and support further study of the use of APAP in settings where the effects on the lung are unknown.
METHODS
Murine Model of Acetaminophen Exposure
Adult (8- to 12-wk-old) male ICR mice were exposed to APAP [140 or 280 mg/kg, ip; dissolved in phosphate-buffered saline]. Mice were fasted overnight (∼16 h) before the day of exposure. Mice were euthanized 6 and 24 h following exposure, blood was collected by cardiac puncture after which normal saline was perfused through the right ventricle, and tissue samples were collected. All procedures were approved by the IACUC at the University of Colorado (Aurora, CO) and care and handling of the animals were in accord with the National Institutes of Health guidelines for ethical animal treatment.
Histological Evaluation of APAP-Induced Hepatic Injury
Liver tissue was fixed with 4% paraformaldehyde, paraffin-embedded, and sections were cut (5 µm) and stained with hematoxylin and eosin (H&E) at the University of Colorado Denver Morphology and Phenotyping Core. Histopathological scoring of the liver tissue was performed by a trained histologist blinded to the treatments or grouping of animals. Briefly, the acetaminophen-induced liver injury system relied on three semiquantitative and one quantitative criteria. These criteria included: 1) extent and locale of necrosis, 2) the extent of inflammatory cell infiltrate, 3) the extent of centrilobular sinusoidal dilatation, and 4) measurement of the serum alanine transaminase (ALT) all as indicated in the study by Martin–Murphy et al. (42) and previously used by our group (40).
Serum Alanine Aminotransferase Measurement
Serum ALT was quantitively determined by using the ALT (SGPT) reagent and colorimetric, end-point method according to the manufacturer’s instructions (Teco Diagnostics).
Isolation of mRNA, cDNA Synthesis, and Analysis of Relative mRNA Levels by RT-qPCR
Mice were euthanized with a fatal dose of pentobarbital sodium. Lungs were perfused with normal saline, removed, snap-frozen in liquid nitrogen, and stored at −80°C. Livers were removed and snap-frozen in liquid nitrogen, and stored at −80°C. Frozen tissue was placed in RLT buffer (QIAGEN) and tissue was homogenized using the Bullet Blender (NextAdvance). Pulmonary mRNA was collected from homogenized tissue using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Initially, tissue RNA was assessed for purity and concentration using the NanoDrop (Thermo Fisher Scientific), and cDNA was synthesized using the Verso cDNA synthesis kit (Thermo Fisher Scientific). Relative mRNA levels were evaluated by quantitative real-time PCR using exon spanning primers (Table 1), TaqMan gene expression, and StepOnePlus Real-Time PCR System (Applied Biosystems). Relative quantitation was determined via normalization to the endogenous control 18S using the cycle threshold (ΔΔCt) method. Primers used can be found in Table 1.
Table 1.
List of genes and primers used for qPCR analysis
| Target | Assay ID |
|---|---|
| Cyp2e1 | Mm00491127_m1 |
| Il6 | Mm00446190_m1 |
| Gclc | Mm00802655_m1 |
| Il1b | Mm01336189_m1 |
| Tnf | Mm00443258_m1 |
| Mcp1 | Mm00443258_m1 |
| Mmp9 | Mm00442991_m1 |
| 18s | Mm03928990_g1 |
Cyp2e1, cytochrome p450 family 2 subfamily E member 1; Il6, interleukin 6; Gclc, glutamate-cysteine ligase catalytic subunit; Il1b, interleukin 1 beta; Tnf, tumor necrosis factor alpha; Mcp1, monocyte chemoattractant protein-1; Mmp9, matrix metallopeptidase 9.
Glutathione Peroxidase Activity Level
The activity level of glutathione peroxidase was determined indirectly by a coupled reaction with glutathione reductase, as previously described (43).
Bronchoalveolar Lavage Fluid Analysis
After exposure to APAP, adult mice were euthanized, bronchoalveolar lavage fluid (BALF) was obtained, and BALF total cell count and cell differentials were determined as previously described (44).
Immunohistochemical Detection and Quantification of Pulmonary Macrophages
Control and acetaminophen-exposed adult murine lung tissues were collected and fixed with 4% PFA and stored in 70% ethanol until embedded with paraffin. Samples were then deparaffinized and rehydrated with xylene and ethanol. Antigen retrieval was performed with Tris/EDTA buffer (pH 9). For diaminobenzidine (DAB) immunohistochemistry, slides were quenched with Bloxall (SP-6000, Vector Labs, Burlingame, CA) and blocked with horse serum (MP-7401, Vector Labs, Burlingame, CA). They were then incubated with CD68 primary antibody (ab283654, 1:500 dilution; Abcam, Cambridge, MA), followed by an horseradish peroxidase (HRP) horse anti-rabbit IgG Polymer reagent (MP-7401, Vector Labs, Burlingame, CA), and DAB staining (SK-4100, Vector Labs, Burlingame, CA), according to the manufacturer’s protocol. Sections were counterstained with hematoxylin QS (H-3404, Vector Labs, Burlingame, CA), and scanned for total macrophage quantification with AperioCS2 slide scanner (Leica Biosystems Inc., Buffalo Grove, IL) with ×40 lens magnification. Total number of CD68-positive cells was quantified in 10 random fields of view (FOVs) per animal (n = 5, 8, animals for control, 140 mg/kg APAP, respectively, 5813 × 3245 pixels/image; image size was kept the same for all measurements). Average number of CD68-positive cells/FOV was calculated.
Morphometric Evaluation of APAP-Induced Pulmonary Injury
For morphometric assessments, mice were euthanized with a fatal dose of pentobarbital sodium. Following perfusion of the lungs with normal saline, the trachea was cannulated with a 24G angiocath and the left lungs were inflation-fixed at 25 cmH2O pressure for 10 min with 4% paraformaldehyde. Lungs were paraffin-embedded, and sections were cut (5 µm) and stained with H&E at the University of Colorado Denver Morphology and Phenotyping Core. Tissue sections were scanned using an AperioCS2 slide scanner (Leica Biosystems Inc., Buffalo Grove, IL) with ×40 lens magnification. Thirty random FOVs per animal (n = 8, 8, 5 animals for control, 140 mg/kg, and 280 mg/kg APAP, respectively, 1716 × 942 pixels/image; image size was kept the same for all measurements) excluding conducting airways and major blood vessels were taken and analyzed in ImageJ [National Institutes of Health (NIH), Bethesda, MD] as previously described (45, 46). FOVs acquisition and analysis were performed by different personnel (ED and SIA, respectively) with FOVs analyses performed in a blinded fashion. Images were opened in RGB format and converted to 8-bit grayscale. Airspaces were thresholded using the corresponding background histogram and analyzed with Analyze Particles plugin tool. Particles greater than 500 square pixels were segmented, circularity range was 0–1, and holes were included. Areas of each individual particle were recovered. Consequently, tissue areas were calculated by subtracting the sum of total airspace areas from image size. Number and size of airspaces, tissue-to-airspace ratios (TAR)s, and total tissue amount were calculated. TAR histograms were calculated and fitted to a least-square Gaussian fitting model using Igor Pro 8 (WaveMetrics, Inc., Lake Oswego, Oregon), and their means and standard deviations were extracted from the fits for statistical significance comparison purposes.
In Situ Hybridization for Cyp2e1 Expression
RNAScope detection mRNA was used to perform in situ hybridization according to the manufacturer’s protocol (Advanced Cell Diagnostics, Hayward, CA). Briefly, formalin-fixed paraffin-embedded murine lungs were cut into 5-µm thick tissue sections. Slides were deparaffinized in xylene, followed by rehydration in a series of ethanol washes. Following citrate buffer (Advanced Cell Diagnostics) antigen retrieval, slides were treated with protease plus (Advanced Cell Diagnostics) at 40°C for 30 min in a HybEZ hybridization oven (Advanced Cell Diagnostics). Probe directed against Cyp2e1 mRNA was applied at 40°C in the following order: target probes, preamplifier, amplifier, and label probe. After each hybridization step, slides were washed two times at room temperature. mRNA detection was followed by immunofluorescent staining for alveolar epithelial type II cells [proSPC (ab90716, Abcam; 1:500) positive] and secretory cells [uteroglobin/CCSP (ABS1673, Sigma; 1:500) positive]. Slides were blocked in 5% BSA buffer for 1 h at room temperature, incubated with primary antibodies overnight at +4°C, stained with appropriate fluorescently labeled secondary antibodies, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) at 1:20,000 (BioLegend, 422801). Staining was visualized using an Aperio Vectra 8 whole slide scanner using a ×40 lens (Leica, Buffalo Grove, IL). The number of total Cyp2e1 expressing cells, and Cyp2e1/proSCP or Cyp2e1/CCSP double-positive cells were quantified in lung parenchyma and terminal bronchioles using ImageJ (NIH).
Fluorescence Lifetime Imaging Microscopy Acquisition and Analysis
FLIM was performed to detect local metabolic changes in 20 different areas in fresh right lower lungs using a Zeiss 780 laser-scanning confocal/multiphoton-excitation fluorescence microscope with a 34-Channel GaAsP QUASAR Detection Unit and nondescanned detectors for 2-photon fluorescence (Zeiss, Thornwood, NY) equipped with an ISS A320 FastFLIM box and titanium:sapphire Chameleon Ultra II (Coherent, Santa Clara, CA) as previously described (46, 47).
For the acquisition of FLIM images, fluorescence for reduced form of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) was detected simultaneously by two photon-counting photon multiplying tube (PMT) detectors (H7422p-40; Hamamatsu). Images of the lung were obtained with Vista Vision software by ISS in 256 × 256 format with a pixel dwell time of 6.3 µs/pixel and averaging over 30 frames. The number of pixels covered with lifetimes for free and bound NADH and FAD was calculated in SimFCS (LFD) and the values were normalized to the total number of pixels detected as previously described (13).
The glycolytic index was calculated for all experimental groups using the following equation:
Glycolytic Index = free NADH fraction/bound to enzyme NADH fraction as described previously (46, 48).
To assess mitochondrial activity (OXPHOS), FLIM based optical redox ratios (Fluorescent Lifetime Redox Ratio, FLIRR) was calculated as follows:
FLIRR (OXPHOS) = bound to enzyme NADH fraction/bound to enzyme FAD fraction as previously described (48).
Statistical Analysis
For comparison between treatment groups, the null hypothesis that no difference existed between treatment means was tested by Student’s t test for two groups and two-way ANOVA for multiple groups with potentially interacting variables (time, APAP exposure), with statistical significance between and within groups determined by means of Bonferroni method of multiple comparisons (Prism, GraphPad Software, Inc.). Statistical significance was defined as P < 0.05. The two-sample t test (two-tailed unpaired Student’s t test) was performed for statistical analysis of histograms, using a 0.01 significance level and a two-sided P value. One-way ANOVA (significance threshold P < 0.05) was performed for FLIM results using Prism 9.0 Software (GraphPad, San Diego, CA).
RESULTS
A Single Exposure of APAP at 140 mg/kg ip Does Not Induce Hepatic Injury in ICR Mice
We have previously documented that a one-time, known hepatotoxic APAP exposure of 280 mg/kg given intraperitoneally induces both hepatic and pulmonary injury in adult ICR mice (40). Whether exposure to lower doses results in either liver or lung injury is unknown. Therefore, we exposed adult male ICR mice to a single dose of APAP at 140 mg/kg administered intraperitoneally. First, we evaluated exposed mice for evidence of hepatic histological injury. Compared with unexposed mice (Fig. 1A), liver sections from mice exposed to APAP 140 mg/kg for 6 h (Fig. 1B) or 24 h (Fig. 1C) revealed very little evidence of injury. All mice were assessed for APAP-induced hepatic injury using a previously validated histological scoring system (40). In mice exposed to APAP 140 mg/kg, we were unable to detect evidence of two key components of APAP-induced hepatic injury, namely, necrosis and inflammation, at either 6 or 24 h of exposure (data not shown). However, we did observe a transient and mild increase in sinusoidal dilation at 6 h of exposure, a finding that had resolved by 24 h (Fig. 1D). Consistent with a lack of histological hepatic injury, and in contrast to mice exposed to the 280 mg/kg dose, we could not detect an increase in serum ALT (Fig. 1E). Finally, we checked for APAP-induced hepatic expression of the proinflammatory mediators Tnf and Il1b, both of which have been associated with hepatic injury (49). Exposure to APAP at 140 mg/kg did not induce expression of these proinflammatory factors at 6 h, whereas there was a slight but significant increase by 24 h of exposure (Fig. 1, F and G). Induction of these proinflammatory mediators in the absence of histological or serological evidence of injury reliably demonstrates that while there is a hepatic response to APAP exposures of 140 mg/kg, there is a lack of significant hepatic injury, which is in contrast to the hepatotoxic exposure of 280 mg/kg.
Figure 1.
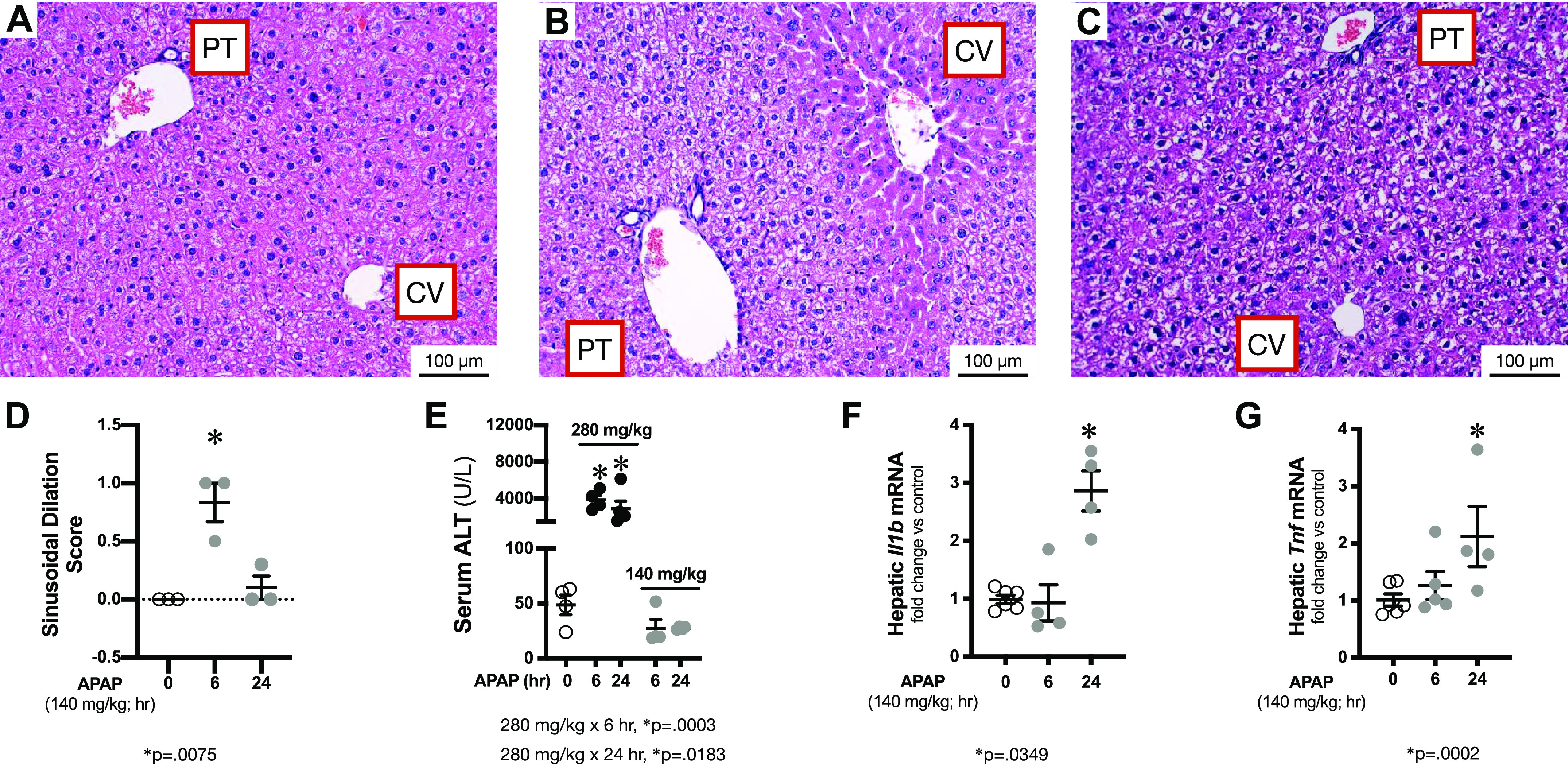
Time course of APAP-induced hepatic injury in ICR mice. A–C: representative H&E stained hepatic sections from control and APAP (140 mg/kg, ip) exposed (B: 6 h; C: 24 h) adult male ICR mice. Examples of portal triad (PT) and central vein (CV) have been added. Internal scale bar = 100 μm. D: blinded histopathological evaluation of H&E stained hepatic sections scored for sinusoidal dilatation. N = 3 per time point. Data are expressed as means ± SE; *P < 0.05 vs. unexposed control. E: serum ALT following APAP exposure (140 or 280 mg/kg ip; 6 or 24 h). N = 4 animals per time point. Data are expressed as means ± SE; *P < 0.05 vs. unexposed control. F and G: fold change in hepatic mRNA expression of (F) Tnf (tumor necrosis factor) and (G) Il1b (interleukin 1β) following APAP exposure (140 mg/kg, ip; 6 or 24 h) Data expressed as means ± SE (N = 4 animals per time point). *P < 0.05 vs. unexposed control. ALT, alanine transaminase; APAP, acetaminophen.
A Single Exposure of APAP at 140 mg/kg ip Induces a Proinflammatory Pulmonary Response in ICR Mice
We have previously documented that a one-time APAP exposure of 280 mg/kg given intraperitoneally induces an increase in pulmonary transcription of Il1b, Il6, Mcp1, Mmp9, and Gclc in adult ICR mice (40). Analysis of bronchoalveolar lavage (BALF) showed that exposure to APAP 140 mg/kg leads to a statistically significant increase in the immune cells (Fig. 2A). Having noted an increase in the total cells in the BALF from mice exposed to APAP 140 mg/kg ip, we assessed BALF cell differentials. Of note, we have previously reported that within 4 h of exposure to APAP 280 mg/kg, murine BALF neutrophil content significantly increases and that this increase persists through 24 h of exposure (40). In addition, BALF macrophage content increases following APAP 280 mg/kg at 8 h of exposure and this persists through 24 h of exposure (40). At 24 h following exposure to APAP 140 mg/kg, macrophages significantly increase, and lymphocytes with neutrophils having a trend of being increased (Fig. 2B). Next, we assessed the lung parenchyma for the presence of macrophages by staining for CD68 (Fig. 2, C–D). We found that at 24 h following exposure to APAP 140 mg/kg, macrophages significantly increase in the lung parenchyma (Fig. 2D). Whole lung mRNA analysis revealed that a single dose of APAP at 140 mg/kg induced significant pulmonary upregulation of Il1b (Fig. 2E), Il6 (Fig. 2F), Ccl2 (Fig. 2G), and Gclc (Fig. 2I) but not Mmp9 (Fig. 2H). It is well known that APAP decreases hepatic glutathione peroxidase (GPx) activity (50), which is thought to result from APAP-adduct formation (51, 52). We found that pulmonary GPx activity decreased 24 h after APAP exposure of either 140 or 280 mg/kg (Fig. 2J). These results demonstrate a pulmonary proinflammatory response to APAP occurs following an exposure that does not cause hepatic toxicity.
Figure 2.

Pulmonary transcriptional and bronchoalveolar lavage response to APAP exposure (140 mg/kg, ip). A and B: total number of infiltrating cells and cells differentials quantification in control and following APAP exposure (140 mg/kg, ip, 24 h). Data are expressed as means ± SE (N = 5 animals per time point). *P < 0.05 and ****P < 0.0001 vs. unexposed control. C and D: assessment of pulmonary parenchymal macrophage content by CD68 DAB staining. C: representative histological images of control and APAP (140 mg/kg ip × 1, 24 h). Scale bar = 60 μm. Inset on the original image identifies a region of higher magnification image shown below the original image. D: objective quantification of the average number of CD68+ cells per field of view. N = 6–8 animals per condition. *P < 0.05 vs. unexposed control. E–I: fold change in pulmonary mRNA expression of (E) Il1b, (F) Il6 (interleukin 6), (G) Ccl2 (C-C motif chemokine ligand 2, monocyte chemotactic protein 1), (H) Mmp9 (matrix metallopeptidase 9), and (I) Gclc (glutamate-cysteine ligase catalytic subunit) following APAP exposure (140 mg/kg, ip; 6 or 24 h) Data are expressed as means ± SE (N = 4–6 animals per time point). *P < 0.05 vs. unexposed control. J: pulmonary glutathione peroxidase activity following APAP exposure (140 or 280 mg/kg, ip; 24 h). Data are expressed as means ± SE (N = 4–6 animals per time point). *P < 0.05 vs. unexposed control. APAP, acetaminophen; DAB, diaminobenzidine.
A Single Exposure of APAP at 140 mg/kg ip Induces Morphometric Changes
We have previously found emphysematous-type morphological changes in the lung parenchyma after APAP 280 mg/kg ip exposure (40). Here, we deployed tissue-to-airspace ratio (TAR), average airspaces size, and average airspaces number measurements and histogram analyses to assess parenchymal morphometric changes in adult ICR lungs after 24 h of 140 mg/kg and 280 mg/kg of APAP ip exposures in comparison with unexposed lungs. These morphometric variables were calculated from H&E stained lung sections (Fig. 3, A–C) for each experimental group, as described in the methods section. Their histograms were then generated and fitted to a least-squares Gaussian fitting model, from which means and standard deviations were recovered for each group for statistical comparisons purposes. Both 140 mg/kg and 280 mg/kg APAP ip exposed lung parenchyma had TAR relative frequency distributions shifted toward smaller TAR values (Fig. 3D), which were statistically different from the control counterpart, suggesting emphysematous parenchymal response (i.e., airspaces expansion). This response was confirmed for 280 mg/kg APAP ip exposure as evidenced by the accompanying average airspace size relative frequency distribution, which had a statistically significant shift toward greater values (Fig. 3E). However, average airspaces size relative frequency distributions for 140 mg/kg APAP ip exposure and unexposed were statistically indifferent (Fig. 3E). We also examined the average number of airspaces to check whether the reduction in 140 mg/kg APAP ip exposed TAR values relative to control (Fig. 3D) was due to an increase in the number of airspaces rather than their average size. In fact, the average number of airspaces relative frequency distributions for 140 mg/kg APAP ip and unexposed lungs were not statistically different either (Fig. 3F). Finally, we calculated the total tissue amount for the aforementioned groups. The total tissue amount histogram analyses revealed that while 140 mg/kg APAP ip and unexposed groups had similar average airspaces sizes and number of airspaces, the former had significantly less total tissue amount (Fig. 3G); indicating alveolar wall thinning. The exact mechanism underlying these observed changes change (for example, epithelial, endothelial, or mesenchymal cell death and/or extracellular matrix remodeling, etc.) deserves further investigation.
Figure 3.

Changes in pulmonary parenchymal morphometrics following APAP exposure (140 mg/kg or 280 mg/kg, ip; 24 h). A–C: representative H&E stained lung sections from (A) control, (B) APAP (140 mg/kg, ip), and (C) APAP (280 mg/kg, ip) exposed adult male ICR mice. Scale bar = 60 µm. D–G: statistical comparisons of the relative frequency distributions of (D) tissue-to-airspace ratios (TARs), (E) airspaces average size, (F) average number of airspaces, and (G) total tissue amount histograms for control and APAP 140 mg/kg and 280 mg/kg ip exposures. N = 240, 240, and 150 samples for control (n = 8 animals), APAP (140 mg/kg, ip; n = 8 animals), and APAP (280 mg/kg, ip; n = 5 animals), respectively. Histograms were fitted to a least-squares Gaussian fitting model and compared against each other using a two-sample t test (two-tailed unpaired Student’s t test). Statistical significance P = 0.02. Although both 140 mg/kg and 280 mg/kg APAP ip exposed lung parenchyma TAR (D) and total tissue amount (G) relative frequency distributions were not statistically different from each other, they were statistically different from control counterparts. On the other hand, 140 mg/kg APAP ip exposed lung parenchyma airspaces average size (E) and average number of airspaces (F) relative frequency distributions were not statistically different from controls but they both were statistically different from 280 mg/kg APAP counterparts. APAP, acetaminophen.
Importantly, exposure to 280 mg/kg APAP ip exposure resulted in airspaces enlargement (Fig. 3E). To examine the prevalence of these enlarged airspaces, we calculated their percentages in lung sections after 280 mg/kg APAP intraperitoneal dose (as well as 140 mg/kg APAP ip dose) relative to unexposed (control) (Fig. 4, A–C). To do that, we plotted airspaces’ average sizes as a function of their corresponding numbers for all groups. We then calculated the mean of the average airspaces size for control (indicated by a dotted line in Fig. 4). Percentages of the number of airspaces whose average size is greater than that of mean control counterpart were calculated. We found ∼ 67% (27% increase) of the 280 mg/kg APAP ip exposed lung parenchymal airspaces had sizes greater than that of mean unexposed (Fig. 4C) demonstrating the extent of the pulmonary emphysematous response to this dose. Whereas 140 mg/kg APAP ip exposed (Fig. 4B) and unexposed (Fig. 4A) lungs had similar percentages of the number of airspaces whose average size is greater than that of mean unexposed, supporting the alveolar wall thinning argument (rather than airspaces expansion—which would have resulted in an increase in the corresponding percentage of the enlarged airspaces, as seen with 280 mg/kg dose.)
Figure 4.
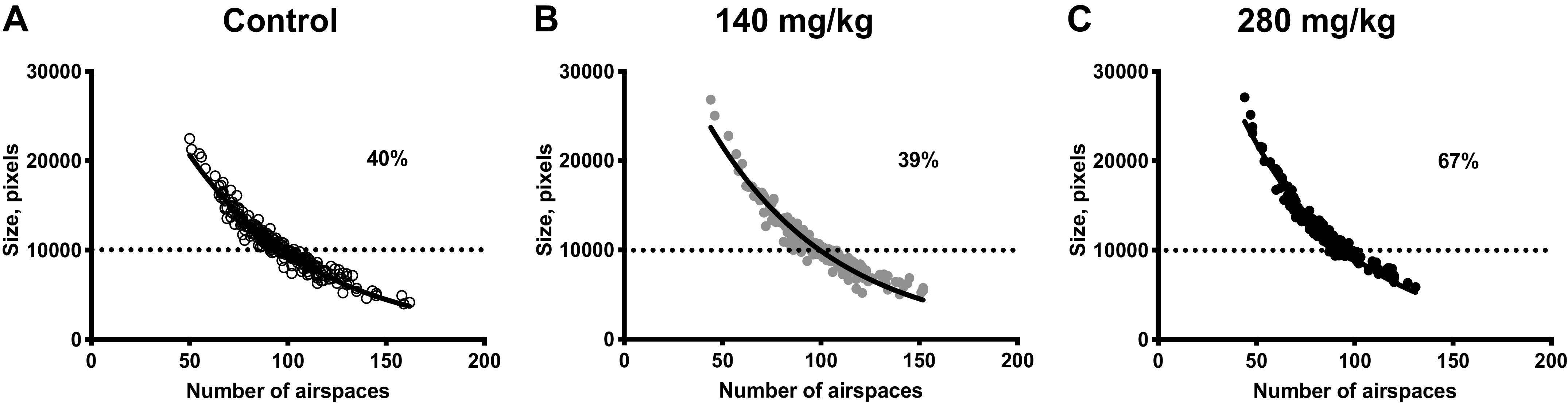
Prevalence of enlarged airspaces in pulmonary parenchyma following APAP exposure (140 mg/kg or 280 mg/kg, ip; 24 h). Correlations between airspaces average size and corresponding number per fields of view (FOVs) for (A) control, (B) APAP (140 mg/kg, ip), and (C) APAP-exposed (280 mg/kg, ip) adult male ICR mice. N = 240, 240, and 150 samples for control (n = 8 animals), APAP (140 mg/kg, ip; n = 8 animals), and APAP (280 mg/kg, ip; n = 5 animals), respectively. Data were fitted to a nonlinear single exponential fitting model (solid line). Dashed line represents the mean of airspaces average sizes for control. APAP, acetaminophen.
A Single Exposure of APAP at 140 mg/kg ip Stimulates Pulmonary Cyp2e1 Expression
Previously, we have shown that a single toxic APAP dose (280 mg/kg) leads to Cyp2e1 mRNA increase in a whole lung homogenate and CYP2E1 protein increase in the EpCAM-isolated pulmonary epithelial cells and macrophages (40). Here, when assessing the whole lung, we could not detect any significant change in pulmonary Cyp2e1 mRNA expression following exposure to APAP 140 mg/kg (Fig. 5A). However, we did assess peripheral lungs for Cyp2e1 expressing cell locality and distribution by performing RNAScope in the tissue sections from control animals and mice, treated with 140 mg/kg and 280 mg/kg APAP for 24 h. We found that control distal lungs predominantly had mesenchymal Cyp2e1 (magenta signal) expression, with some alveolar epithelial type II cells (cyan signal) having Cyp2e1 mRNA and almost absent secretory cells (yellow signal) containing Cyp2e1 (Fig. 5B). Single APAP exposure led to an increase in number of mesenchymal cells and secretory cells, expressing Cyp2e1 following 24 h of both, 140 mg/kg (Fig. 5C) and 280 mg/kg (Fig. 5D), APAP doses. Quantification of all cell types and two examined epithelial cell types revealed a dose-dependent increase of a total number of cells, expressing Cyp2e1 mRNA (Fig. 5E). Furthermore, we analyzed two epithelial cell types known to express Cyp2e1 and found a significant increase of both epithelial cell types in 280 mg/kg dose (Fig. 5F), compared with control and 140 mg/kg APAP exposure. Yet, when we stratified the result for each individual cell type, we found that number of alveolar epithelial type II cells expressing Cyp2e1 mRNA had no significant decrease after APAP exposure compared with controls (Fig. 5G). Oppositely, the number of secretory cells expressing Cyp2e1 significantly increased in a dose-dependent manner (Fig. 5H). These results demonstrate that APAP exposure leads to the Cyp2e1 mRNA increase, and this increase is dose-dependent.
Figure 5.
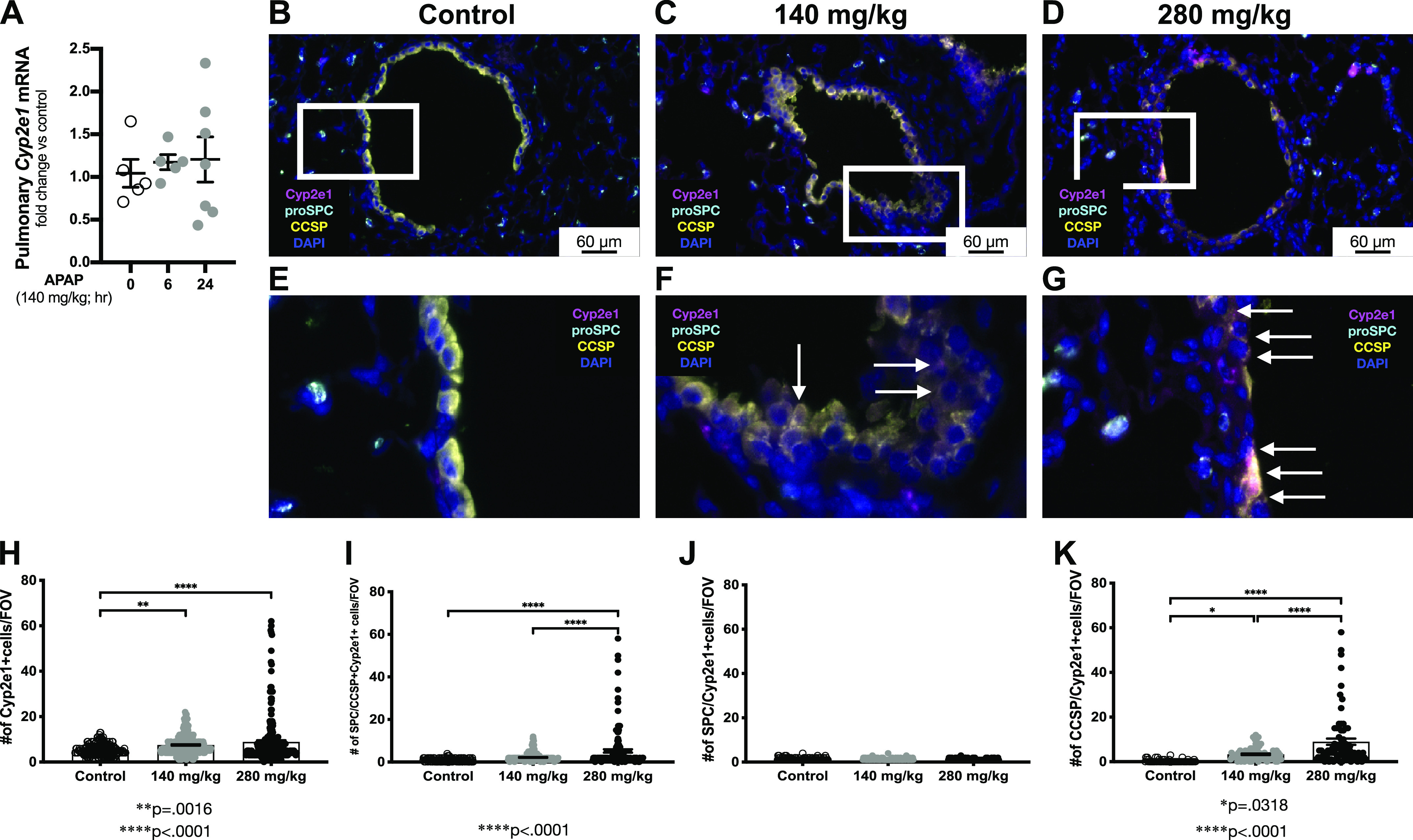
Pulmonary Cyp2e1 expression following exposure to APAP exposure (140 mg/kg, ip). A: fold change in pulmonary Cyp2e1 mRNA expression following APAP exposure (140 mg/kg, ip; 6 or 24 h). Data are expressed as means ± SE (N = 4–6 animals per time point). *P < 0.05 vs. unexposed control. B–G: in situ hybridization of lung specimens with Cyp2e1 mRNA (magenta), costained with proSPC (cyan, type II cell marker) and uteroglobin/CCSP [yellow, secretory Club (Clara) cell marker]. DAPI (blue) was used to stain the nuclei. Scale bar = 60 µm. Inset on the original image identifies region of higher magnification image shown below the original image. White arrows indicate secretory cells (CCSP positive, yellow) coexpressing Cyp2e1 mRNA (magenta). H–K: quantification of the number of total and epithelial Cyp2e1 expressing cells in different conditions. Data are expressed as means ± SE [N= 180 FOVs measured in control (n = 5 animals), APAP (140 mg/kg, ip, n = 5), and APAP (280 mg/kg, ip; n = 5 animals), respectively]. *P < 0.05, **P < 0.005, and ****P < 0.0001. APAP, acetaminophen; FOVs, fields of view.
A Single Exposure of APAP at 140 mg/kg ip Results in Local Metabolic Changes in the Lung
It is known that the accumulation of CYP2E1-generated toxic APAP metabolites leads to mitochondrial dysfunction, which will lead to a metabolic change in the liver (53) and neonatal lung tissues (40). Here, we used FLIM as a tool to investigate local metabolic changes in the fresh right lungs from control and APAP-treated animals 24 h after the exposure. We found that like left lungs analyzed in Fig. 5, right lungs following nontoxic APAP dose (140 mg/kg) had similar to control airspace sizes (Fig. 6A, top row, left and middle columns, respectively), whereas airspaces in the lungs after toxic APAP dose (280 mg/kg) were enlarged (Fig. 6A, top row, right column). After mapping the lifetimes for free/bound NADH (Fig. 6A, middle row, free NADH is yellow, bound NADH is magenta) and free/bound FAD (Fig. 6A, bottom row, free FAD is magenta, bound FAD is yellow) we found that the toxic APAP dose (280 mg/kg) led to a significantly increased glycolysis (Fig. 6B, black circles), not significantly compared with control decreased OXPHOS (FLIRR) (Fig. 6C, black circles) and significantly decreased free FAD fraction (Fig. 6D, black circles), suggesting decreased mitochondrial load and in particular reduced complex II activity, which aligns with previously published data (18). Contrary, a single APAP exposure at 140 mg/kg dose led to a significantly decreased glycolysis (Fig. 6B, gray circles) and significantly increased OXPHOS (FLIRR) (Fig. 6C, gray circles) with significantly increased free FAD fraction (Fig. 6D, gray circles), suggesting increased mitochondrial and complex II load following the treatment with low APAP dose. These results demonstrate that APAP exposure leads to a tissue metabolic change: toxic dose leads to a decreased mitochondrial function, especially associated with the complex II activity, whereas nontoxic exposure leads to a decreased glycolysis and mitochondrial overload.
Figure 6.
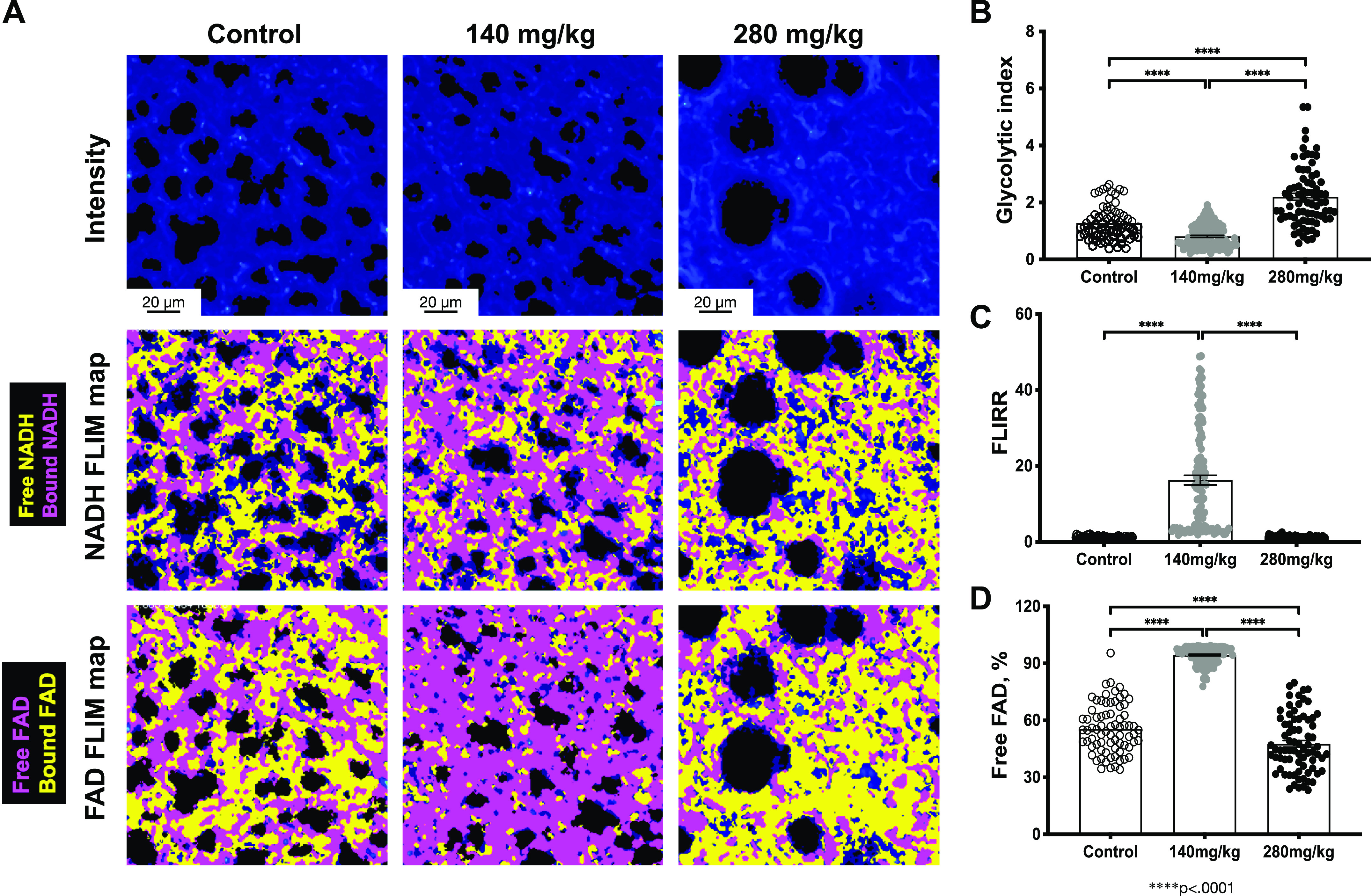
Pulmonary metabolic changes following exposure to APAP exposure (140 mg/kg or 280 mg/kg, ip; 24 h). A: representative intensity images (top), NADH (middle), and FAD (bottom) Fluorescent Lifetime Imaging Microscopy (FLIM) maps for control and APAP-treated tissues are shown. Quantified (B) Glycolytic index, (C) Fluorescent Lifetime Imaging Redox Ratio (FLIRR), and (D) free FAD fraction. Data are expressed as means ± SE (250 FOVs for each condition were measured in control (n = 5 animals), APAP (140 mg/kg, ip; n = 5 animals), and APAP (280 mg/kg, ip; n = 5 animals), respectively). ****P < 0.0001. Scale bar = 20 µm. APAP, acetaminophen; FAD, flavin adenine dinucleotide; FOVs, fields of view; NADH, nicotinamide adenine dinucleotide.
DISCUSSION
We found that the adult lung is susceptible to the toxic effects of APAP at exposure levels that cause minimal hepatic response without histological or serological evidence of significant toxicity. Specifically, following a one-time, intraperitoneal exposure of 140 mg/kg, the liver of adult ICR mice shows very little evidence of hepatic injury. In contrast, the lung demonstrates an increase in the expression of proinflammatory mediators, an increase in inflammatory cells in the BALF and lung parenchyma, and acute morphometric changes. This injury is associated with mesenchymal and club cell upregulation of the APAP-metabolizing enzyme Cyp2e1. Finally, this exposure causes metabolic changes in the lung that are consistent with increased mitochondrial workload and injury.
A large body of literature supports a dose-response relationship between APAP exposures and hepatic injury, this relationship has not been established for the lung. Of note, an APAP exposure of 300–500 mg/kg ip or 600 mg/kg orally (by mouth) reliably causes hepatic injury in adult CD-1 mice (54–60). In addition, we have recently reported that APAP exposures of 280 mg/kg ip × 1 injure the developing lung, in the absence of histological or serological evidence of hepatotoxicity (61). Our work here was guided by a body of literature showing a lack of hepatic injury in ICR mice exposed to lower doses of APAP. Other studies have shown that a single exposure of 150 mg/kg ip does not cause hepatic injury in adult male ICR mice (62–64). A temporal relationship between a single APAP exposure of 150 mg/kg, hepatic APAP protein adducts, and minor histological changes in the liver has been reported, but the effect was transient and not associated with an increase in serum ALT or AST (aspartate aminotransferase) (56). Importantly, we could find no reports interrogating the pulmonary findings after lower, “nontoxic” exposures. Thus, the acute pulmonary implications of these lower-dose exposures are not well understood.
Previous work has convincingly demonstrated that the lung is susceptible to injury at exposure that causes hepatic toxicity. Toxic APAP overdose has been reported to induce pneumonitis (27), alveolar injury (28), and acute respiratory distress syndrome (ARDS) (26) in humans. In the laboratory, APAP-induced lung injury occurs in rats, mice, and pigs with exposures known to cause hepatic injury (32, 35–37, 39, 65). Importantly, APAP-induced pulmonary injury in mice has been documented with exposures necessary to cause hepatic injury. An early study of APAP toxicity in adult male CD-1 mice demonstrated hepatic and pulmonary injury at an exposure of 600 mg/kg PO, whereas a lower dose (300 mg/kg) caused slight hepatic damage, pulmonary findings were not described (37). Furthermore, it has been reported that this higher dose exposure (600 mg/kg PO) results in APAP-protein adducts in the lung, specifically the club cells of the bronchioles and the ciliated epithelium (29, 31, 35). The developing murine lung is also susceptible to these higher dose exposures (61). However, studies that interrogate the effect of lower dose exposures on the lung are lacking. Importantly, one study showed that APAP adducts accumulated in the lung at exposures as low as 15 mg/kg ip and within 30 min of exposure (38). Furthermore, exposures as low as 15 mg/kg ip induced a significant increase in emigrated elastase-positive neutrophils in the tracheal epithelial and subepithelial space, and exposures of 60 mg/kg intragastrically increased pulmonary expression of the proinflammatory mediators keratinocyte chemoattractant and tumor necrosis factor (TNF) α (38). Importantly, these are exposures that result in no or minimal hepatic injury. Our report adds to the body of literature demonstrating that the lung is susceptible to the toxic effect of APAP at exposures that do not result in significant hepatic injury. We have demonstrated a proinflammatory, pulmonary transcriptional response, an increase in the immune cell content of the BALF, and morphometric changes in response to a single administration of 140 mg/kg APAP ip. These findings are important, as it has been argued that the pulmonary injury observed in rodents exposed to very large doses of APAP is not relevant to human disease (41). Our work suggests that more investigations must be done to understand how APAP exposures across a wide range of doses impact the lung.
Previously, we have used tissue-to-airspace ratio (TAR) histogram analyses, in addition to TAR-associated variables (namely, airspaces average size and number), in normal (46) and left congenital diaphragmatic hernia (CDH) model (45) rabbit fetal lungs after tracheal occlusion (TO), to investigate heterogeneity in the parenchymal morphometric response. In this study, similar morphometric analyses have been implemented to investigate the pulmonary morphometric changes after APAP ip exposure. A one-time 280 mg/kg APAP ip for 24 h resulted in the formation of enlarged airspaces (Fig. 3E), which were more prevalent in the lung parenchyma (Fig. 4C), confirming the emphysematous morphometric response, we previously reported with this dose (40). On the other hand, 140 mg/kg APAP ip exposure induced no significant changes in the average size of airspaces, nor their numbers compared with unexposed. However, this exposure did result in a significant reduction in the total tissue amount (Fig. 3G), leading to an overall decrease in the TARs (Fig. 3D) consistent with alveolar wall thinning. Further study is necessary to determine the mechanisms underlying these observations. Whether this pulmonary morphometric response will develop to an exacerbated more pronounced emphysematous one over time (>24 h) and/or with multiple repetitive hits of the same low dose (140 mg/kg) is yet to be explored.
Here, we present data demonstrating that APAP exposure that does not result in hepatic toxicity significantly impacts pulmonary mitochondrial function. It is well known that in the presence of CYP2E1 metabolism, APAP exposures adversely affect mitochondrial function. Multiple studies have implicated the formation of APAP-mitochondrial protein adducts and the subsequent mitochondrial dysfunction as the main contributors to APAP-induced cellular injury (16). Early studies of APAP-induced hepatotoxicity revealed both changes in mitochondrial ultrastructure (66) and function (18, 19, 21, 67). Importantly, pharmacological interventions to prevent mitochondrial dysfunction prevent APAP-induced cellular injury (23, 68). Although most of these studies interrogate hepatic mitochondrial function with toxic exposures, lower dose exposures also have metabolic implications. APAP exposures (150 mg/kg) consistent with those used in the current study cause reversible hepatic mitochondrial dysfunction with no associated increase in ALT or histological liver injury (17). Here, we demonstrate that the lung is also susceptible to APAP-induced mitochondrial overload and potential dysfunction coincides with alveoli wall thinning at the lower dose exposure, with an opposite metabolic shift to a glycolysis and alveoli enlargement following toxic APAP exposure. Congruently, we have previously demonstrated that airspace enlargement is associated with a shift to glycolysis (45, 46, 69).
There are multiple possible reasons why the lung would demonstrate susceptibility to APAP exposures that cause minimal if any hepatic injury. Constitutive pulmonary CYP2E1 expression is well described (25, 35, 70–74). Under homeostatic conditions in the lung of adult male ICR mice, Cyp2e1 is expressed in the club cells and minimally expressed in type II alveolar epithelial cells (75). Previously, we have reported that with the higher 280 mg/kg exposure, total pulmonary Cyp2e1 induction and pulmonary epithelium and the resident macrophages CYP2E1 protein increase (40). Here, we confirm our previous report that pulmonary Cyp2e1 expression increases in response to APAP toxic exposure (40). The stimulation of Cyp2e1 expression in the lung could contribute to the tissue-specific response to APAP exposure. In contrast to what is observed in the lung, hepatic Cyp2e1 expression significantly decreases following APAP exposure (76). The mechanisms underlying APAP-induced Cyp2e1 expression in the lung are unknown, and whether this tissue-specific response contributes to injury deserves further study. In addition, we demonstrate that pulmonary glutathione peroxidase activity is inhibited at these lower dose APAP exposures. Inhibition of glutathione peroxidase activity could potentially decrease cellular resistance to the toxic effects of APAP exposure. Interestingly, glutathione peroxidase has been identified as an APAP-adduct target in the liver (77). Our results demonstrate that these same mechanisms may be present in the APAP exposed lung.
There are multiple limitations to the current study. We report a proinflammatory pulmonary response and metabolic changes to APAP exposures that do not injure the liver. These findings are consistent with previous reports demonstrating that APAP-induced hepatic and pulmonary injury occur independently (29, 30, 32). However, in the current study of the effect of lower dose APAP exposures we did not test whether the pulmonary changes were independent of any hepatic effect. We limited our investigation to 24 h after APAP exposure, and we did not test whether these pulmonary effects are reversible. Although we did not detect histological or serological evidence of hepatic injury following APAP 140 mg/kg ip, whether there are functional deficits or injury with repeated dosing was not evaluated. Furthermore, there is great interest in the long-term implications of these exposures or repeated low-dose exposures, and we have yet to investigate those outcomes. The assessments necessary to evaluate are both physiological measures of pulmonary health, including respiratory rate, oxygen saturation, as well as pulmonary mechanics. It is important to note that the findings reported here may not result in measurable changes in acute respiratory physiology, but whether these changes increase susceptibility to a second exposure or whether these changes are reversible deserve further study. These assessments would evaluate whether the findings presented here are permanent, or whether the lung can recover from these exposures. With this in mind, the effect of repeated exposures must be investigated in the future.
In conclusion, the murine lung is susceptible to the toxic effects of APAP at exposures that do not cause significant hepatic toxicity. Following this lower dose APAP exposure, there are measurable changes in the proinflammatory transcriptome, inflammatory cell BALF content, morphometrics, and metabolic function. It is possible that the acute changes described here may help explain the association between chronic use in adults and both asthma (2–14) and COPD (2). Furthermore, it could be speculated that the changes observed here following APAP exposure could increase susceptibility to a second exposure, including tobacco smoke, pollution/toxins, or infection. Thus, our description of acute pulmonary changes after a single-dose exposure supports the study of the effect of repeated lower-dose exposures, or the effect of a second pulmonary insult (oxygen, pollution, and smoking).
DATA AVAILABILITY
The data used to support the findings of this study are included within the article.
GRANTS
This work was supported by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grant R01HL132941 (to C.J.W.).
DISCLAIMERS
Contents are the authors’ sole responsibility and do not necessarily represent official National Institutes of Health views.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.D., S.I.A.-J., and C.J.W. conceived and designed research; E.D., S.I.A.-J., M.A.Z., L.Z., R.D.D., D.B., L.G.S., D.J.O., and C.J.W. performed experiments; E.D., S.I.A.-J., M.A.Z., R.D.D., L.G.S., D.J.O., and C.J.W. analyzed data; E.D., S.I.A.-J., and C.J.W. interpreted results of experiments; E.D., S.I.A.-J., and C.J.W. prepared figures; E.D., S.I.A.-J., and C.J.W. drafted manuscript; E.D., S.I.A.-J., M.A.Z., R.D.D., D.J.O., and C.J.W. edited and revised manuscript; E.D., S.I.A.-J., M.A.Z., L.Z., R.D.D., D.B., L.G.S., D.J.O., and C.J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The FLIM imaging experiments were performed in the Advanced Light Microscopy Core part of NeuroTechnology Center at University of Colorado Anschutz Medical Campus supported in part by Rocky Mountain Neurological Disorders Core Grant Number P30 NS048154 and by Diabetes Research Center Grant Number P30 DK116073.
REFERENCES
- 1.Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 4: 131–142, 2016. doi: 10.14218/JCTH.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKeever TM, Lewis SA, Smit HA, Burney P, Britton JR, Cassano PA. The association of acetaminophen, aspirin, and ibuprofen with respiratory disease and lung function. Am J Respir Crit Care Med 171: 966–971, 2005. doi: 10.1164/rccm.200409-1269OC. [DOI] [PubMed] [Google Scholar]
- 3.Shaheen SO, Sterne JA, Songhurst CE, Burney PG. Frequent paracetamol use and asthma in adults. Thorax 55: 266–270, 2000. doi: 10.1136/thorax.55.4.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newson RB, Shaheen SO, Chinn S, Burney PG. Paracetamol sales and atopic disease in children and adults: an ecological analysis. Eur Respir J 16: 817–823, 2000. doi: 10.1183/09031936.00.16581700. [DOI] [PubMed] [Google Scholar]
- 5.Kelkar M, Cleves MA, Foster HR, Hogan WR, James LP, Martin BC. Prescription-acquired acetaminophen use and the risk of asthma in adults: a case-control study. Ann Pharmacother 46: 1598–1608, 2012. doi: 10.1345/aph.1R430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Etminan M, Sadatsafavi M, Jafari S, Doyle-Waters M, Aminzadeh K, FitzGerald JM. Acetaminophen use and the risk of asthma in children and adults: a systematic review and metaanalysis. Chest 136: 1316–1323, 2009. doi: 10.1378/chest.09-0865. [DOI] [PubMed] [Google Scholar]
- 7.Thomsen SF, Kyvik KO, Skadhauge L, Steffensen I, Backer V. Intake of paracetamol and risk of asthma in adults. J Asthma 45: 675–676, 2008. doi: 10.1080/02770900802165998. [DOI] [PubMed] [Google Scholar]
- 8.Moral L, Marco N, Fuentes MJ, Toral T, Caño R, Pena MA. Asthma and paracetamol: could we really know what happens between them? Allergol Immunopathol (Madr) 41: 261–264, 2013. doi: 10.1016/j.aller.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Farquhar H, Stewart A, Mitchell E, Crane J, Eyers S, Weatherall M, Beasley R. The role of paracetamol in the pathogenesis of asthma. Clin Exp Allergy 40: 32–41, 2010. doi: 10.1111/j.1365-2222.2009.03378.x. [DOI] [PubMed] [Google Scholar]
- 10.Barr RG, Wentowski CC, Curhan GC, Somers SC, Stampfer MJ, Schwartz J, Speizer FE, Camargo CA Jr.. Prospective study of acetaminophen use and newly diagnosed asthma among women. Am J Respir Crit Care Med 169: 836–841, 2004. doi: 10.1164/rccm.200304-596OC. [DOI] [PubMed] [Google Scholar]
- 11.Henderson AJ, Shaheen SO. Acetaminophen and asthma. Paediatr Respir Rev 14: 9–15, 2013. doi: 10.1016/j.prrv.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Allmers H, Skudlik C, John SM. Acetaminophen use: a risk for asthma? Curr Allergy Asthma Rep 9: 164–167, 2009. doi: 10.1007/s11882-009-0024-3. [DOI] [PubMed] [Google Scholar]
- 13.Eneli I, Sadri K, Camargo C Jr, Barr RG. Acetaminophen and the risk of asthma: the epidemiologic and pathophysiologic evidence. Chest 127: 604–612, 2005. doi: 10.1378/chest.127.2.604. [DOI] [PubMed] [Google Scholar]
- 14.Nuttall SL, Williams J, Kendall MJ. Does paracetamol cause asthma? J Clin Pharm Ther 28: 251–257, 2003. doi: 10.1046/j.1365-2710.2003.00492.x. [DOI] [PubMed] [Google Scholar]
- 15.Ramachandran A, Jaeschke H. Acetaminophen toxicity: novel insights into mechanisms and future perspectives. Gene Expr 18: 19–30, 2018. doi: 10.3727/105221617X15084371374138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramachandran A, Jaeschke H. Acetaminophen hepatotoxicity: a mitochondrial perspective. Adv Pharmacol 85: 195–219, 2019. doi: 10.1016/bs.apha.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Ramshesh VK, McGill MR, Jaeschke H, Lemasters JJ. Low dose acetaminophen induces reversible mitochondrial dysfunction associated with transient c-Jun N-terminal kinase activation in mouse liver. Toxicol Sci 150: 204–215, 2016. doi: 10.1093/toxsci/kfv319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burcham PC, Harman AW. Acetaminophen toxicity results in site-specific mitochondrial damage in isolated mouse hepatocytes. J Biol Chem 266: 5049–5054, 1991. [PubMed] [Google Scholar]
- 19.Donnelly PJ, Walker RM, Racz WJ. Inhibition of mitochondrial respiration in vivo is an early event in acetaminophen-induced hepatotoxicity. Arch Toxicol 68: 110–118, 1994. doi: 10.1007/s002040050043. [DOI] [PubMed] [Google Scholar]
- 20.Esterline RL, Ray SD, Ji S. Reversible and irreversible inhibition of hepatic mitochondrial respiration by acetaminophen and its toxic metabolite, N-acetyl-p-benzoquinoneimine (NAPQI). Biochem Pharmacol 38: 2387–2390, 1989. doi: 10.1016/0006-2952(89)90481-4. [DOI] [PubMed] [Google Scholar]
- 21.Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol 93: 378–387, 1988. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- 22.Ramsay RR, Rashed MS, Nelson SD. In vitro effects of acetaminophen metabolites and analogs on the respiration of mouse liver mitochondria. Arch Biochem Biophys 273: 449–457, 1989. doi: 10.1016/0003-9861(89)90504-3. [DOI] [PubMed] [Google Scholar]
- 23.Lee KK, Imaizumi N, Chamberland SR, Alder NN, Boelsterli UA. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology 61: 326–336, 2015. doi: 10.1002/hep.27385. [DOI] [PubMed] [Google Scholar]
- 24.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51: 246–254, 2010. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol 32: 391–411, 2002. doi: 10.1080/20024091064273. [DOI] [PubMed] [Google Scholar]
- 26.Baudouin SV, Howdle P, O'Grady JG, Webster NR. Acute lung injury in fulminant hepatic failure following paracetamol poisoning. Thorax 50: 399–402, 1995. doi: 10.1136/thx.50.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akashi S, Tominaga M, Naitou K, Fujisawa N, Nakahara Y, Hiura K, Hayashi S. [Two cases of acetaminophen-induced pneumonitis]. Nihon Kyobu Shikkan Gakkai Zasshi 35: 974–979, 1997. [PubMed] [Google Scholar]
- 28.Price LM, Poklis A, Johnson DE. Fatal acetaminophen poisoning with evidence of subendocardial necrosis of the heart. J Forensic Sci 36: 930–935, 1991. [PubMed] [Google Scholar]
- 29.Bartolone JB, Beierschmitt WP, Birge RB, Hart SG, Wyand S, Cohen SD, Ea K. Selective acetaminophen metabolite binding to hepatic and extrahepatic proteins: an in vivo and in vitro analysis. Toxicol Appl Pharmacol 99: 240–249, 1989. doi: 10.1016/0041-008x(89)90006-9. [DOI] [PubMed] [Google Scholar]
- 30.Breen K, Wandscheer JC, Peignoux M, Pessayre D. In situ formation of the acetaminophen metabolite covalently bound in kidney and lung. Supportive evidence provided by total hepatectomy. Biochem Pharmacol 31: 115–116, 1982. doi: 10.1016/0006-2952(82)90247-7. [DOI] [PubMed] [Google Scholar]
- 31.Bulera SJ, Cohen SD, Khairallah EA. Acetaminophen-arylated proteins are detected in hepatic subcellular fractions and numerous extra-hepatic tissues in CD-1 and C57B1/6J mice. Toxicology 109: 85–99, 1996. doi: 10.1016/0300-483x(96)03309-4. [DOI] [PubMed] [Google Scholar]
- 32.Gu J, Cui H, Behr M, Zhang L, Zhang QY, Yang W, Hinson JA, Ding X. In vivo mechanisms of tissue-selective drug toxicity: effects of liver-specific knockout of the NADPH-cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol Pharmacol 67: 623–630, 2005. doi: 10.1124/mol.104.007898. [DOI] [PubMed] [Google Scholar]
- 33.Micheli L, Cerretani D, Fiaschi AI, Giorgi G, Romeo MR, Runci FM. Effect of acetaminophen on glutathione levels in rat testis and lung. Environ Health Perspect 102, Suppl 9: 63–64, 1994. doi: 10.1289/ehp.94102s963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schønberg SA, Skorpen F. Paracetamol counteracts docosahexaenoic acid-induced growth inhibition of A-427 lung carcinoma cells and enhances tumor cell proliferation in vitro. Anticancer Res 17: 2443–2448, 1997. [PubMed] [Google Scholar]
- 35.Hart SG, Cartun RW, Wyand DS, Khairallah EA, Cohen SD. Immunohistochemical localization of acetaminophen in target tissues of the CD-1 mouse: correspondence of covalent binding with toxicity. Fundam Appl Toxicol 24: 260–274, 1995. doi: 10.1006/faat.1995.1029. [DOI] [PubMed] [Google Scholar]
- 36.Neff SB, Neff TA, Kunkel SL, Hogaboam CM. Alterations in cytokine/chemokine expression during organ-to-organ communication established via acetaminophen-induced toxicity. Exp Mol Pathol 75: 187–193, 2003. doi: 10.1016/s0014-4800(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 37.Placke ME, Wyand DS, Cohen SD. Extrahepatic lesions induced by acetaminophen in the mouse. Toxicol Pathol 15: 381–387, 1987. doi: 10.1177/019262338701500401. [DOI] [PubMed] [Google Scholar]
- 38.Nassini R, Materazzi S, Andrè E, Sartiani L, Aldini G, Trevisani M, Carnini C, Massi D, Pedretti P, Carini M, Cerbai E, Preti D, Villetti G, Civelli M, Trevisan G, Azzari C, Stokesberry S, Sadofsky L, McGarvey L, Patacchini R, Geppetti P. Acetaminophen, via its reactive metabolite N-acetyl-p-benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. FASEB J 24: 4904–4916, 2010. doi: 10.1096/fj.10-162438. [DOI] [PubMed] [Google Scholar]
- 39.Jeffery EH, Haschek WM. Protection by dimethylsulfoxide against acetaminophen-induced hepatic, but not respiratory toxicity in the mouse. Toxicol Appl Pharmacol 93: 452–461, 1988. doi: 10.1016/0041-008x(88)90048-8. [DOI] [PubMed] [Google Scholar]
- 40.Sandoval J, Orlicky DJ, Allawzi A, Butler B, Ju C, Phan CT, Toston R, De Dios R, Nguyen L, McKenna S, Nozik-Grayck E, Wright CJ. Toxic acetaminophen exposure induces distal lung ER stress, proinflammatory signaling, and emphysematous changes in the adult murine lung. Oxid Med Cell Longev 2019: 7595126, 2019. doi: 10.1155/2019/7595126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kennon-McGill S, McGill MR. Extrahepatic toxicity of acetaminophen: critical evaluation of the evidence and proposed mechanisms. J Clin Transl Res 3: 297–310, 2018. doi: 10.18053/jctres.03.201703.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin-Murphy BV, Kominsky DJ, Orlicky DJ, Donohue TM Jr, Ju C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology 57: 1575–1584, 2013. doi: 10.1002/hep.26134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherlock LG, Sjostrom K, Sian L, Delaney C, Tipple TE, Krebs NF, Nozik-Grayck E, Wright CJ. Hepatic-specific decrease in the expression of selenoenzymes and factors essential for selenium processing after endotoxemia. Front Immunol 11: 595282, 2020. doi: 10.3389/fimmu.2020.595282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michaelis KA, Agboke F, Liu T, Han K, Muthu M, Galambos C, Yang G, Dennery PA, Wright CJ. IκBβ-mediated NF-κB activation confers protection against hyperoxic lung injury. Am J Respir Cell Mol Biol 50: 429–438, 2014. doi: 10.1165/rcmb.2013-0303OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobrinskikh E, Al-Juboori SI, Oria M, Reisz JA, Zheng C, Peiro JL, Marwan AI. Heterogeneous response in rabbit fetal diaphragmatic hernia lungs after tracheal occlusion. J Surg Res 250: 23–38, 2020. doi: 10.1016/j.jss.2019.12.025. [DOI] [PubMed] [Google Scholar]
- 46.Dobrinskikh E, Al-Juboori SI, Shabeka U, Reisz JA, Zheng C, Marwan AI. Heterogeneous pulmonary response after tracheal occlusion: clues to fetal lung growth. J Surg Res 239: 242–252, 2019. doi: 10.1016/j.jss.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 47.Marwan AI, Shabeka U, Reisz JA, Zheng C, Serkova NJ, Dobrinskikh E. Unique heterogeneous topological pattern of the metabolic landscape in rabbit fetal lungs following tracheal occlusion. Fetal Diagn Ther 45: 145–154, 2019. doi: 10.1159/000487752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallrabe H, Svindrych Z, Alam SR, Siller KH, Wang T, Kashatus D, Hu S, Periasamy A. Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci Rep 8: 79, 2018. doi: 10.1038/s41598-017-18634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 16: 1227–1236, 2002. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 50.Arnaiz SL, Llesuy S, Cutrín JC, Boveris A. Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med 19: 303–310, 1995. doi: 10.1016/0891-5849(95)00023-q. [DOI] [PubMed] [Google Scholar]
- 51.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem 273: 17940–17953, 1998. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 52.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos 31: 1499–1506, 2003. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 53.Yan MZ, Huo YZ, Yin ST, Hu HB. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol 17: 274–283, 2018. doi: 10.1016/j.redox.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu Y, Zhang C, Chen YH, Wang H, Zhang ZH, Chen X, Xu DX. Immature mice are more susceptible than adult mice to acetaminophen-induced acute liver injury. Sci Rep 7: 42736, 2017. doi: 10.1038/srep42736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ginsberg GL, Placke ME, Wyand DS, Cohen SD. Protection against acetaminophen-induced hepatotoxicity by prior treatment with fenitrothion. Toxicol Appl Pharmacol 66: 383–399, 1982. doi: 10.1016/0041-008x(82)90305-2. [DOI] [PubMed] [Google Scholar]
- 56.Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci 65: 135–150, 2002. doi: 10.1093/toxsci/65.1.135. [DOI] [PubMed] [Google Scholar]
- 57.Muhammad-Azam F, Nur-Fazila SH, Ain-Fatin R, Mustapha Noordin M, Yimer N. Histopathological changes of acetaminophen-induced liver injury and subsequent liver regeneration in BALB/C and ICR mice. Vet World 12: 1682–1688, 2019. doi: 10.14202/vetworld.2019.1682-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeong TB, Kim JH, Kim SH, Lee S, Son SW, Lim Y, Cho JY, Hwang DY, Kim KS, Kwak JH, Ys J. Comparison of toxic responses to acetaminophen challenge in ICR mice originating from different sources. Lab Anim Res 35: 16, 2019. doi: 10.1186/s42826-019-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamamoto H, Fujii K, Hayakawa T. Inhibitory effect of cold stress against acetaminophen-induced hepatic injury in B6C3F1 and ICR mice. Toxicol Lett 81: 125–130, 1995. doi: 10.1016/0378-4274(95)03424-2. [DOI] [PubMed] [Google Scholar]
- 60.Nagy G, Kardon T, Wunderlich L, Szarka A, Kiss A, Schaff Z, Bánhegyi G, Mandl J. Acetaminophen induces ER dependent signaling in mouse liver. Arch Biochem Biophys 459: 273–279, 2007. doi: 10.1016/j.abb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 61.Dobrinskikh E, Sherlock LG, Orlicky DJ, Zheng L, De Dios R, Balasubramaniyan D, Sizemore T, Butler B, Wright CJ. The developing murine lung is susceptible to acetaminophen toxicity. Am J Physiol Lung Cell Mol Physiol 320: L969–L978, 2021. doi: 10.1152/ajplung.00072.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masubuchi Y, Nakayama J, Watanabe Y. Sex difference in susceptibility to acetaminophen hepatotoxicity is reversed by buthionine sulfoximine. Toxicology 287: 54–60, 2011. doi: 10.1016/j.tox.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 63.Goldring CE, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP, McMahon M, Hayes JD, Itoh K, Yamamoto M, Bk P. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology 39: 1267–1276, 2004. doi: 10.1002/hep.20183. [DOI] [PubMed] [Google Scholar]
- 64.Williams DP, Garcia-Allan C, Hanton G, LeNet JL, Provost JP, Brain P, Walsh R, Johnston GI, Smith DA, Bk P. Time course toxicogenomic profiles in CD-1 mice after nontoxic and nonlethal hepatotoxic paracetamol administration. Chem Res Toxicol 17: 1551–1561, 2004. doi: 10.1021/tx049846x. [DOI] [PubMed] [Google Scholar]
- 65.Newsome PN, Henderson NC, Nelson LJ, Dabos C, Filippi C, Bellamy C, Howie F, Clutton RE, King T, Lee A, Hayes PC, Plevris JN. Development of an invasively monitored porcine model of acetaminophen-induced acute liver failure. BMC Gastroenterol 10: 34, 2010. doi: 10.1186/1471-230X-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petersen P, Vilstrup H. Relation between liver function and hepatocyte ultrastructure in a case of paracetamol intoxication. Digestion 19: 415–419, 1979. doi: 10.1159/000198403. [DOI] [PubMed] [Google Scholar]
- 67.Andersson BS, Rundgren M, Nelson SD, Harder S. N-acetyl-p-benzoquinone imine-induced changes in the energy metabolism in hepatocytes. Chem Biol Interact 75: 201–211, 1990. doi: 10.1016/0009-2797(90)90118-7. [DOI] [PubMed] [Google Scholar]
- 68.Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol 91: 761–773, 2017. doi: 10.1007/s00204-016-1692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marwan AI, Shabeka U, Dobrinskikh E. Suggested mechanisms of tracheal occlusion mediated accelerated fetal lung growth: a case for heterogeneous topological zones. Front Pediatr 5: 295, 2017. doi: 10.3389/fped.2017.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 43: 149–173, 2003. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 71.Hukkanen J, Pelkonen O, Raunio H. Expression of xenobiotic-metabolizing enzymes in human pulmonary tissue: possible role in susceptibility for ILD. Eur Respir J Suppl 32: 122s–126s, 2001. [PubMed] [Google Scholar]
- 72.Crawford EL, Weaver DA, DeMuth JP, Jackson CM, Khuder SA, Frampton MW, Utell MJ, Thilly WG, Willey JC. Measurement of cytochrome P450 2A6 and 2E1 gene expression in primary human bronchial epithelial cells. Carcinogenesis 19: 1867–1871, 1998. doi: 10.1093/carcin/19.10.1867. [DOI] [PubMed] [Google Scholar]
- 73.Botto F, Seree E, el Khyari S, de Sousa G, Massacrier A, Placidi M, Cau P, Pellet W, Rahmani R, Barra Y. Tissue-specific expression and methylation of the human CYP2E1 gene. Biochem Pharmacol 48: 1095–1103, 1994. doi: 10.1016/0006-2952(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 74.Hukkanen J, Hakkola J, Anttila S, Piipari R, Karjalainen A, Pelkonen O, Raunio H. Detection of mRNA encoding xenobiotic-metabolizing cytochrome P450s in human bronchoalveolar macrophages and peripheral blood lymphocytes. Mol Carcinog 20: 224–230, 1997. doi: 10.1002/(sici)1098-2744(199710)20:2<224::aid-mc9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 75.Forkert PG. CYP2E1 is preferentially expressed in Clara cells of murine lung: localization by in situ hybridization and immunohistochemical methods. Am J Respir Cell Mol Biol 12: 589–596, 1995. doi: 10.1165/ajrcmb.12.6.7766423. [DOI] [PubMed] [Google Scholar]
- 76.Bao Y, Wang P, Shao X, Zhu J, Xiao J, Shi J, Zhang L, Zhu HJ, Ma X, Manautou JE, Zhong XB. Acetaminophen-induced liver injury alters expression and activities of cytochrome P450 enzymes in an age-dependent manner in mouse liver. Drug Metab Dispos 48: 326–336, 2020. doi: 10.1124/dmd.119.089557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44: 88–106, 2012. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.