

Abstract
Screening already approved drugs for activity against a novel pathogen can be an important part of global rapid-response strategies in pandemics. Such high-throughput repurposing screens have already identified several existing drugs with potential to combat SARS-CoV-2. However, moving these hits forward for possible development into drugs specifically against this pathogen requires unambiguous identification of their corresponding targets, something the high-throughput screens are not typically designed to reveal. We present here a new computational inverse-docking protocol that uses all-atom protein structures and a combination of docking methods to rank-order targets for each of several existing drugs for which a plurality of recent high-throughput screens detected anti-SARS-CoV-2 activity. We demonstrate validation of this method with known drug-target pairs, including both non-antiviral and antiviral compounds. We subjected 152 distinct drugs potentially suitable for repurposing to the inverse docking procedure. The most common preferential targets were the human enzymes TMPRSS2 and PIKfyve, followed by the viral enzymes Helicase and PLpro. All compounds that selected TMPRSS2 are known serine protease inhibitors, and those that selected PIKfyve are known tyrosine kinase inhibitors. Detailed structural analysis of the docking poses revealed important insights into why these selections arose, and could potentially lead to more rational design of new drugs against these targets.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10822-021-00432-3.
Keywords: SARS-CoV-2, Inverse docking, High-throughput, Repurposing, TMPRSS2, PIKfyve
Introduction
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the etiological agent of the COVID-19 pandemic. Since the first cases reported in December 2019, this virus has spread over 223 countries with more than 111 million positive confirmed cases and almost 2.5 million deaths counted up to February 2021 [1]. By the end of 2020, several vaccines have been approved for human immunization against the virus. However, there is a constant need to discover antiviral agents for the treatment of SARS-CoV-2, in order to help control the possibility of new outbreaks, especially from several mutant strains of the virus [2, 3].
The process of de novo drug design against a novel pathogen can require many years of effort, which in the case of the COVID-19 pandemic is a luxury that the world cannot afford. In the light of this difficulty, drug repurposing presents a very practical strategy for drug development against SARS-CoV-2 [4–6]. Drug repurposing is based on the use of already approved drugs for the treatment of other diseases, in this case COVID-19, bypassing the clinical trials and regulatory stages associated with the development of a new drug. The reduction in the number of required steps for approval potentially reduces the time required for the drug to reach the market [4–6]. As a consequence, many repurposed drugs are currently in clinical trials, although there is not yet any approved antiviral medication against SARS-CoV-2.
SARS-CoV-2 is a single-stranded positive-sense RNA virus that belongs to the family of betacoronaviruses. Its genome encodes four structural proteins: envelope (E), nucleocapsid (N), membrane (M) and spike (S) proteins [7], and 16 non-structural proteins, such as the main protease (3CLpro), Papain like protease (PLpro), polimerase (RdRp), helicase, and others [8]. It has already been suggested that the non-structural proteins 3CLpro, PLpro, RdRp and Helicase enzymes are viable antiviral drug targets [8]. During the COVID-19 pandemic a large number of research groups dedicated time and effort to resolve the structures of almost all the previously mentioned viral proteins. For instance, the RCSB Protein Data Bank currently houses approximately 1000 published SARS-CoV-2 related proteins [9, 10]. SARS-CoV-2 proteins whose structures remain unresolved have been the object of structure-predictions made using a variety of computational approaches (Swissmodel [11, 12], Zhang Lab [13, 14], and Alphafold [15]). These predicted structures could become quite important if experimental difficulties slow the release of validated X-ray or cryo-EM-derived structures. Besides the studies on viral proteins, researchers have also focused on key human proteins, identifying several drugs that have anti-SARS-CoV-2 activity by inhibiting host enzymes essential for the SARS-CoV-2 life cycle [16, 17].
Several groups around the world have been combining drug repurposing strategies with high-throughput screening (HTS) in order to find already approved drugs that show in vitro activity against SARS-CoV-2. The antiviral activity in these HTS studies can be measured against an individual known SARS-CoV-2 target, such as some viral enzymes [18, 19], or against cultured viral-infected cells [20–22]. In these later publications, promising compounds showing high antiviral activity on the HTS assay were identified. One disadvantage in these studies, unlike the target-based assays mentioned prior, is that the molecular targets of the tested drugs remain unknown.
The combination of this limitation of the cell-based HTS assays with the accumulated knowledge of all the resolved and predicted SARS-CoV-2 and human related structures provides strong motivation for in silico repurposing campaigns aimed at identifying potential targets. Identifying the viral or human target for any anti-SARS-CoV-2 repurposed drug would be beneficial for focused structural and mutagenic studies as well as optimization.
A powerful in silico methodology to conquer this challenge is inverse docking (INDO), which aims to find the best target(s) for a given drug within a large collection of biologically relevant macromolecular targets [23–26], using molecular docking. Given that molecular docking methods are not conceived to identify potential target(s) for a given ligand, INDO protocols are still relatively immature, and new strategies for improve true positive ligand–protein pairs continue to be reported [24, 26–28].
Here we report an improvement to INDO that aims specifically to identify the potential targets of repurposed drugs with experimentally proved activity against SARS-CoV-2. This strategy is distinct from theoretical drug database screening [29–32] because we restrict our pool of drugs to those already shown by consensus of two or more experimental HTS studies to have anti-SARS-CoV-2 activity. From a joint dataset built from three independent HTS assays that measure inhibition of infection of cultured cells [20–22], we built a ligand set from the top 25% most active compounds in two out of the three studies, comprising 158 distinct compounds. Our target set consists of a total of 18 SARS-CoV-2 proteins and 6 human proteins. Our INDO protocol uses a novel combination of several docking scores which yields increased accuracy in identifying the true positive ligand-protein pairs in a validation set.
In the remainder of this work, we first present our INDO protocol applied over a test set of know protein–ligand complexes in order to find the best scoring function combination. Then, a list of the ligands and targets considered, are indicated. Finally, the INDO study results are analyzed, which in summary show that the preferential targets were human and viral enzymes. Further analysis over every one of this preferential targets were developed in detail, indicating the most suitable repurposed drug for every target, and revealing the molecular interaction patterns present on the binding sites. To the best of our knowledge, this is the first virtual HTS INDO study developed to connect highly active anti-SARS-CoV-2 drugs, gathered from HTS assays, with their respective potential viral and human molecular targets.
Methods
Inverse docking using multiple scoring functions
In the context of ligand-protein interaction prediction, inverse docking (INDO) is a procedure which aims to predict, within a predefined set of proteins, the best target(s) for a given ligand using docking calculations [23–26]. For a successful INDO campaign the most important steps include (i) selection of docking software package(s), (ii) ligand and protein structure preparation, and (iii) proper analysis of the results. In this work, our strategy was first to evaluate different programs and analysis options using a well-defined test set, and then proceed with the identification of potential targets for those molecules found to be active from experimental HTS.
Docking software and methodology
The docking calculations were performed with a beta version of Vinardo [33] and Ledock [34]. Vinardo has a scoring function based on Vina [35]. The new beta version is being developed by one of the present authors and was successfully used in the D3R grand challenge 4 competition (see Quiroga/Villarreal submission in Tables 2 and 3 of Parks et al. [36]). Ledock is commercial software with free access for academic use and which has shown good performance in docking benchmarks [37]. We also applied the recently developed coarse grained Korp-PL [38] scoring function to re-score the 10 best poses generated with Vinardo. All programs were used with default options, except the search space, which was adapted to every protein.
In those cases where a ligand is present in a crystallographic target structure, the docking box was defined by extending 8 Å from the ligand’s maximum and minimum x, y, and z coordinates. For the other cases, the docking box was defined to cover the active site of the enzyme or, if the goal was to test the drug action on protein–protein interactions, the interface between protomers. In the Supplementary Materials, Table S6 shows details about the docking boxes and their intent, together with some illustrative figures. All the structures used here are available in PDB format in our repository [39].
Analysis
The docking calculations produced optimal poses of each ligand in every target, together with its score. Generally, any inverse docking protocol uses this set of information to decide which is the preferred target for a given ligand from among all tested targets. A basic approach could be to simply rank the targets for each ligand according to the scoring function in use, with the highest affinity at the top of the list. However, this algorithm does not work well in practice, mainly due to biases in the scoring functions arising from the limited datasets against which they themselves were developed. There are different strategies to overcome this problem, and we follow the approach of Kim et al. [23]. They proposed to normalize the docking scores using a Z-score approach before each ligand selects its preferred targets. An additional way to overcome scoring-function bias is to combine results of multiple distinct scoring functions. Because there are several ways to combine scores, we evaluated both exponential consensus average ranking [40] and a simple arithmetic mean of Z-scores before the selection step. The latter approach was found to be superior in our test set and then was used for the rest of the analysis of the INDO results. When several structures for a given ligand and for a given protein were used, only the combination that produced the best score were considered for this analysis.
Test set
In order to validate the INDO methodology we prepared a control test which consisted of 209 crystallographic protein–ligand complexes taken from the PDBBIND database [41]. PDBBIND is a curated database where every complex is annotated with the experimentally measured binding affinities and UniProt ID, among other useful information. Beginning with PDBBIND’s refined 2018 set, representing more than 4000 complexes, we applied several filters to build a subset more manageable in size and that better matches the properties of the set of drugs selected from the experimental HTS. Molecules in this test set were limited to a molecular weight between 200 to 700 Da, with 3 to 7 rotatable bonds, and experimentally measured affinity of 1 μM or lower. To eliminate redundancy we clustered molecules using a binning clustering method, with a Tanimoto coefficient of 0.6, as implemented in Chemmine tools [42]. Finally, we deleted all but one replica of repeated receptors based on UniProt IDs.
Ligand preparation
Up to five structures per ligand were prepared using Gypsum-DL [43], from the SMILEs in the ChEMBL database. Using the available options of Gypsum-DL, ring conformations were explored for all molecules with aliphatic rings, while stereochemistry was explored only for those molecules without explicit prescription in the corresponding SMILE. Protonation states were explored in the 7.0 + − 1 pH range, and were manually adjusted when deemed necessary. Analysis of the molecular weight (MW) distribution showed that most molecules were between 130 to 750 Da. Five molecules with higher MW and one with a lower MW were not considered.
Protein targets
As previously mentioned, the SARS-CoV-2 RNA genome encodes for at least 20 structural and non-structural proteins. Any and all of these can be used as potential targets for the INDO study, but according to their roles in the viral life cycle and also on the availability of a three-dimensional structure (experimental or by structural modeling), 18 of them were selected for this work. In order to permit the selection of human proteins as targets in the INDO study, several were selected because of their important role on the early stages of SARS-CoV-2 infection. One of the most important is the angiotensin-converting enzyme II (ACE2) receptor, which is responsible for viral recognition and entry into the host cell. After ACE2 recognition, SARS-CoV-2 can enter the host cell following either endosomal or nonendosomal pathways, depending on the infected cell-type. In both pathways, several human enzymes (TMPRSS2, Furin, PIKfyve, etc.) play critical roles in viral entry [16, 17]. Table S1 list the proteins targets used.
For almost all selected targets it was possible to obtain an experimentally solved three-dimensional structure. The exceptions were Nsp6, ExoN, M protein and TMPRSS2, which were obtained from different modeling sources (see Table S6 for more details). In most cases, at least two different structures for every target were used in order to have a wider representation of the conformations of the receptor selected in the INDO study. The exceptions were ORF3a, ORF7a, PIKfyve, and the M protein, for which we were able to either retrieve from the PDB database, or model, only one structure. For the four targets without experimental structure available, we first looked for already modeled structures by the AlphaFold project [15] or by the Zhang lab [13], and then modeled the remaining using the Swiss-Model server [11, 12]. Two modeled structures were obtained from the Alphafold project (Nsp6 and M protein) [15]. The structures of ExoN were obtained from the Swiss-Model server [11, 12] and Zhang lab [13]. The structure retrieved from the Swiss-Model server was based on the structure of SARS-CoV-1 ExoN (PDB accession code 5C8S), with a sequence similarity of 95% with respect to SARS-CoV-2 ExoN, while the model from the Zhang Lab was constructed with I-TASSER [14]. The structures for the soluble domain of TMPRSS2 were modeled with the Swiss-Model server. The templates used were Hepsin serine protease (PDB accession code 5CE1), with a 33% of sequence similarity, and Plasma kallikrein (PDB code 6O1G) with a 44% similarity.
For every viral and human target the docking region was selected based on the previous knowledge of importance on the biological function for the particular target. For enzymes (PLpro, 3CLpro, etc.), the selection was based on the position of the catalytic site and also on the known position of the binding site for the natural substrate. For those targets for which two distinct bindings sites are known, both were included as potential sites in the study (RdRp, Hel, ExoN, 2-O-MT, etc.). In the case of the non-enzyme targets, usually a protein–protein interface was selected as docking site, because this inhibition of interaction with other protein can prevent an important biological function on the viral life cycle. This is the case for several non-structural proteins (such as Nsp7, Nsp8 and Nsp10) that participate in viral replication by binding to functional enzymes. For modeled proteins the position of the ligand present in the template used to generate the structure was taken as reference to construct the docking box. In the case of the M protein, for which no relevant information was available, the docking was set to explore the entire cytosolic domain. See Table S1 and Table S6 for more details.
In the important case of the SARS-CoV-2 spike (S) protein complex, we searched for potential ligand binding sites at interprotomer interfaces, the rationale being that binding in such a location could either anchor the trimer in the closed conformation or prevent post-activation separation of protomers. First we analyzed the 10 μs molecular dynamics simulation trajectories of the glycosylated S trimer in the closed state from Shaw Research [44]. From these trajectories we calculated the atomic density of the highly flexible glycans, which revealed that almost all interprotomer interfaces at the complex surface are within reach of a glycan (Fig. S7). This suggested that the interprotomer interfacial sites on the exterior of S are not good candidates for high affinity binding sites due to glycan competition. We therefore elected to search for druggable sites in the interior of the S protein. We performed exploratory docking assays using probe molecules taken from a set of 300 randomly selected FDA approved drugs with molecular weight from 400 to 600 Da and with a logP from 0 to 6. Structures and properties of the FDA molecules were obtained from the SuperDrug2 database [45]. After exhaustive docking in the interior space of the S protein, clustering analysis of the center of mass positions of the docked molecules revealed six clear sites with extensive interchain contacts and with good predicted affinity for the FDA approved drugs (Figs. S8 and S9). Three of these sites are equivalent due to the symmetry of the trimer, leaving four distinct internal sites. Details of the locations of these sites are given in Table S6. Besides these interior sites, we also performed the INDO procedure on the RBD domain, which is occluded from water and glycans in the closed state, but fully exposed in the open state. In this case we used the structure co-crystallized with the ACE2 receptor and explored independently the S and the ACE2 sides of complex [46]. This is a critical point, since if we had explored the actual RBD:ACE2 complex, the search could produce enhancers of the interaction rather than inhibitors. Taking into consideration all the mentioned binding sites, a total of 65 protein structures representing 35 distinct sites were selected for the INDO study (Table S6).
Results and discussion
Test set
The INDO validation test consisted of docking of every ligand against every target in the test set, resulting in a total of 43,681 (209 × 209) docking calculations with each of the three docking programs. Then based on the individual or composite Z-scores (see Analysis in Methods section), every ligand produced an ordered list of its preferred targets. From these lists, the fraction of ligands which select the correct crystallographic target as its first option (“top-1”) or in general within the first N targets (“top-N”) was calculated. The results are shown in Fig. S1. Clearly, the combined use of Ledock, Korp-PL, and Vinardo scoring functions results in a better recovery of the correct protein–ligand complex compared to use of any one individually. The combined use of the scoring functions recovered 66%, 75%, 81% and 88% in the top-1, top-3, top-5, and top-10 respectively. We further characterized the discrimination performance of our procedure by plotting the ROC curves (Fig. S2). Two extreme cases were analyzed, first considering all the predictions, and second considering only top-1 predictions. When using all the predictions the AUC is 0.95, while for top-1 only predictions the AUC is 0.85. However reassuring these high AUC values are, they may also reflect the fact that some molecules may only be good binders for certain target proteins. The major differences between true positive rate (TPR) and false positive rate (FPR) are obtained at the average Z-scores of − 0.90 and − 1.50 for each case. These threshold values are useful references and were used to guide analysis of the following results of INDO.
The highly active repurposed drug (HARD) list
In the work of Heiser et al. [20], the anti-SARS-CoV-2 activity of 1670 drugs was studied via HTS, including FDA-approved drugs, EMA-approved drugs, and compounds in late stage clinical trials. In the HTS made by Touret et al. [21], 1520 off-patent drugs from the prestwick chemical library, most of them approved by FDA, EMA and other agencies, were tested. Finally, the HTS by Ellinger et al. [22] use 5632 compounds including 3488 compounds that are marketed or have been tested in human clinical trials. All three HTS studies were conducted using different assays techniques in order to measured the anti-SARS-CoV-2 activity for every repurposed drug.
Our HARD list is constructed of those compounds that were among the top 25% most active compounds in at least two of the three studies mentioned above. This information was obtained from the ChEMBL database version 27 [47, 48]. To make the activity entrees of different assays compatible, we associate those drugs that share the same parent compound according to the molecule-hierarchy table of the database. In this way, we avoid distinguishing different salts of the same drug. ChEMBL IDs of the resulting list can be found in our repository [39], together with the set of scripts and SQLite queries used to process the ChEMBL database. The preferred compound names of the resulting list is shown in the supporting information (Table S2).
This list comprises 158 drugs. After applying the MW filter mentioned in the Methods section, we obtain a total of 152 compounds. This HARD list was submitted to the INDO procedure in order to identify the most likely potential SARS-CoV-2-related target for every compound.
Control compounds (CC)
In order to further test the INDO procedure, 14 known inhibitors of different targets included in this study were added as control compounds (CC). The targets of these CC are five human (Cathepsin L, PIKfyve, TMPRSS2, Trypsin and Furin) and four viral (3CLpro, PLpro, E protein and 2-O-MT) proteins. These CC, described in Table S3, which are known to inhibit the activity of one or two specific targets, were combined with the 152 compounds from the HARD list and all were analyzed in a single INDO experiment. Table S3 and Fig. 1 displays the top-5 target selection for every CC after the INDO procedure. The number of CC compounds that select its correct target in top-1, top-3 and top-5 position are 3, 10 and 12, which translates to 21%, 71% and 85% correct predictions respectively. This values are only slightly lower than the one obtained in the Test set, but this reduction is somewhat expected, as in this case the receptor is not always co-crystallized with the ligand as they are in the test cases, and also some CC’s showed reported inhibition for several selected targets, as is the case for several inhibitors of serine-proteases, discussed on the next section. These confounding factors were mitigated in the design of the Test set by clustering the ligands and selecting the receptors by UniProt ID. Notably, in the case of CC with two known targets, like FOY251 and Diminazene, the INDO procedure was also able to pick both targets in the top-3 (Fig. 1). These results clearly show the robustness of the INDO procedure in finding the correct SARS-CoV-2 protein/enzyme target. In addition to this reassuring finding, the CCs also serve as a reference to analyze the interactions of the predicted binders of the HARD list. In this way, it is possible to advance in the chemical rationale behind the target selection and not to rely in the calculated Z-scores alone. A detailed discussion of the targets detected by INDO for both CC and HARD is given in next section.
Fig. 1.

Target identification for control compounds. Green arrows indicate that the corresponding target is obtained among the top-5 of each compound. Orange dashed lines indicate those compounds for which the corresponding target was not found in the top-5. Line thickness shows a fine partition of the prediction ranking as indicated in the legend
Inverse docking (INDO)
Preferential targets
Based on the results for the test set, it is clear that lower average Z-scores correlate with higher probability of correctly assigning the target for a given compound (Fig. S2). In order to give an overview of the preferred targets for the drugs in the HARD list, we analyzed which targets were selected as top-1 for every compound, with an average Z-score of − 1.0 or lower. With this threshold value we expected to include all true positive results. In total we found 52 repurposed drugs, and we show how they are distributed across targets in Fig. 2 and, with further details, in Table S4. The most preferred targets are enzymes, with the human enzymes TMPRSS2 and PIKfyve being the most populated with 7 and 6 drugs, respectively. These targets were followed by three viral targets, two enzymes and one structural protein: the catalytic site of Helicase, the S protein at the internal site S4 and PLpro, with 5, 5 and 4 compounds respectively. Notable absences on this list are the non structural (NSP) viral proteins, the ACE2 receptor and the RBD of the S protein. This analysis clearly shows that the human enzymes included in this INDO procedure were highly selected as top-1 targets, over all the viral targets included in the same study, possibly due to a critical role of those enzymes in the viral cycle of SARS-CoV-2, but also possibly due to the fact that the original targets for these drugs were also human enzymes of similar functions to those found here.
Fig. 2.
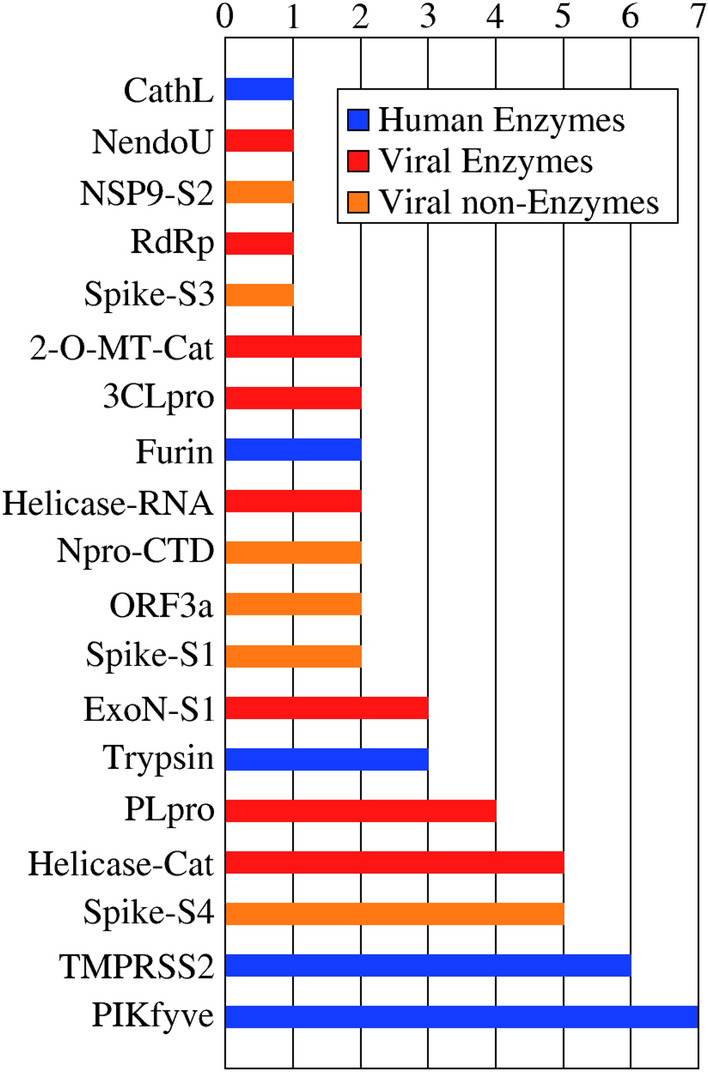
Preferred target distribution. Top-1 predictions with a average Z-score
Analysis of selected targets
In this section, some of the preferred targets have been selected in order to thoroughly analyze the repurposed drugs from the HARD list that showed potential as inhibitors. We based this selection on a combination of Z-scores values and a predicted mode of interaction resembling the pharmacophoric interaction of known inhibitors used in the Control Compounds section. All the gather information is resumed on Fig. 3 and Table S5.
Host targets
Serine proteases
The infection of SARS-CoV-2 to the human cell depends on the ACE2 receptor and a diverse set of host proteases [16, 49, 50]. These proteases prime the S protein of SARS-CoV-2 and are responsible for efficient fusion with the host lipid membrane to deliver the viral genetic material [16]. Among the most important proteases, three serine proteases, transmembrane protease/serine 2 (TMPRSS2), furin, and trypsin [16] recognize a similar protease cleavage site pattern (Arginine-Serine, R-S) present on the S1/S2 and S2 sites of the SARS-CoV-2 S protein [16]. In the case of furin, the S1/S2 site also contains the furin multibasic cleavage motif (RRAR) [7, 51]. Similar protease cleavage patterns indicated similar catalytic binding sites for the three serine proteases, composed of a catalytic triad (Ser-His-Asp) and a pocket containing an Asp residue, responsible for the substrate Arg interaction recognition [7]. Comparable catalytic binding sites suggest the possibility that one compound can inhibit the biological function of more than one of these enzymes. This is the case for two of the selected CC, Diminazene (CC7) [7] and the Camostat derivative, FOY251 (CC6) [52, 53], that have been reported to have inhibitory activity against furin/trypsin and TMPRSS2/trypsin, respectively (Table S3).
After INDO, Diminazene’s top-3 target matches represented the three serine proteases with a high average Z-score (between − 2.01 and − 2.62, Table S3). Previous work showed that Diminazene displays inhibitory activity against furin and trypsin [7, 54]. This experimental evidence agrees with the present INDO analysis, where these targets represent the second and third position among Diminazene’s selections. Diminazene’s top-selected target is TMPRSS2, making this serine protease a very reasonable preferred target for Diminazene. From the HARD list, two compounds share the same top-3 potential target patterns: Nafamostat and Hydroxystilbamidine (Fig. 3). Nafamostat displayed the mentioned top-3 sequence with a high average Z-score (between − 1.99 to − 2.70, shown in Table S5). Previous work showed that this drug inhibits trypsin and TMPRSS2 [53], which is consistent with our results. A superposition of the lower energy interaction conformation of Diminazene and Nafamostat in the active binding site of furin showed that both drugs display identical interaction pattern with residues of the active site (Figs. 4a, S3a and S3b), producing hydrogen bonds and ionic interactions in three separated pockets: (1) the Asp pocket with Asp306, (2) the catalytic triad site with Ser368 and (3) the opposite side with Asp191 or Asn192. This common interaction motif and the high average Z-score strongly suggest that furin is also a preferred target for Nafamostat.
Fig. 3.

Selected cases for target identification obtained in this work and discussed in the text. Line thickness shows the predicted ranking as indicated in the legend. The colors of the arrows allows to visualize the different discussion sections in the text. Target identification for the 152 drugs of the HARD list is available in our repository [39]
Fig. 4.

3D superposition of: a Diminazene (orange), Nafamostat (yellow) and Hydroxystilbamidine (green) on the catalytic site of Furin; b Apilimod (orange), Pexidartinib (yellow) and Vatalanib (green) on the active site of PIKfyve; c GRL-0617 (orange), Clebopride (yellow) and Mosapride (green) on the S3 and S4 PLpro subsites; d Sofalcone (orange), Bumetanide (yellow) and Stepronin (green) on the Helicase NTPase binding site. Hydrogen bond interaction represented as black dashed lines
Hydroxystilbamidine also displayed the same top-3 targets as did Diminazene and Nafamostat (Figs. 1 and 3). Hydroxystilbamidine, selected furin as preferred target on the third position and with a lower average Z-score than Diminazene and Nafamostat. Previous work showed that Hydroxystilbamidine does not inhibit furin [7], and consistent with this fact, our docking calculations showed that Hydroxystilbamidine only displays hydrogen bond interactions with two of the previously mentioned pockets: (1) the Asp pocket with Asp306 and (2) the opposite side pocket with Asp191 (Fig. S3c). We therefore attribute Hydroxystilbamidine’s lack of furin activity to the absence of a third hydrogen bond interaction with residues from the the catalytic triad site (Ser368). According to the present analysis, only trypsin and TMPRSS2 were selected as preferred targets for Hydroxystilbamidine.
Camostat belongs to the same family as Nafamostat and it inhibits trypsin and TMPRSS2 [53]. Our INDO analysis showed these enzymes as the top-2 selected targets (Fig. 3), with average Z-score of − 2.02 and − 2.09, respectively (Table S5), supporting the present analysis as an adequate tool to find serine-protease inhibitors.
Finally, Fig. 3 showed that Mosapride, Apixaban and Clebopride also displayed trypsin and TMPRSS2 in their top-3 for preferred targets, with high average Z-scores between − 1.09 and − 1.75 (Tables S5). Considering that these average Z-score values are similar to those from known trypsin and TMPRSS2 inhibitors (FOY251, Table S3 and Camostat, Table S5) [52, 53], we suggest that these three drugs are potential inhibitors of the above-mentioned serine proteases.
PI-3P-5-kinase (PIKfyve)
Binding of a virus to specific host cell receptor triggers membrane fusion, which can occur directly at the plasma membrane or following endocytic uptake [55]. Viruses that require endocytic uptake can use different initial trafficking routes [55]. One endolysosomal system used by SARS-CoV-2 is conducted through phosphoinositides, where the phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) is particularly important for endosome homeostasis [55, 56]. PI(3,5)P2 is produced by PI-3P-5-kinase (PIKfyve) through the phosphorylation of the D-5 position in phosphatidylinositol-3-phosphate (PI3P) [55–57]. It has been reported that inhibitors of PIKfyve can inhibit infection by several viruses, including Ebola and Lassa [58, 59]. Among these inhibitors, Apilimod was reported to inhibit viral entry of MERS-CoV, SARS-CoV and SARS-CoV-2 [55]. This agrees with our INDO study, where Apilimod displays PIKfyve as the top-1 preferred target (Fig. 1) with a very high average Z-score of − 2.21 (Table S3). This supports the value of the INDO procedure as a tool in the search for PIKfyve inhibitors.
In a recent publication reporting the structure of the PIKfyve lipid kinase complex, Lys1877 was confirmed as catalytically essential in the enzyme active site [57]. A detailed observation of the active site shows that it is formed mostly by hydrophobic residues, such as Ile54, Lys56 (Lys1877), Phe69, Ala90, Met116, Phe120, Leu199, Tyr216 and Ile217. The lowest energy interaction conformation of Apilimod on the active site of PIKfyve (Figs. 4b and S4a) shows mainly hydrophobic interactions with these residues, separated into two different binding pockets. Apilimod’s 3-methylphenyl moiety is placed in binding pocket 1, lined by Phe66, Phe69, Ala90, Ile92, Leu114, Tyr216 and Ile217 present on one side of the active site. Apilimod’s pyridine ring occupies pocket 2 on the other side of the PIKfyve binding site, formed by Ile54, Leu119, Phe120 and Leu199, and also displays a hydrogen bond interaction with Leu119. To the best of our knowledge, this is the first study to postulate a structural model of a putative inhibitor such as Apilimod bound in the PIKfyve active site.
Several HARD have selected PIKfyve as a top-1 preferred target (Fig. 3), with average Z-scores between − 1.08 and − 1.94 (Table S5). These compounds belong to the pharmacological group of tyrosine kinase inhibitors, developed for a variety of clinical purposes. Several previous publications have reported that approved drugs from the family of tyrosine kinase inhibitors, in particular Imatinib, Dasatinib and Saracatinib, can block coronavirus infection (SARS-CoV and MERS-CoV) at early stages of the viral life cycle [17, 60–62]. This activity has been attributed to the inhibition of various tyrosine kinases, such as Abelson tyrosine kinase 2 (Abl2) [61, 62] and the Src-family of tyrosine kinases (SFKs) [60], but the reported results were not conclusive on whether these enzymes are the actual targets for these inhibitors in the context of their anti-coronavirus activity.
Figures 4b and S4, displayed that this family of compounds exhibited high similar interaction pattern to the reference compound Apilimod. Figure S4 shows that Pexidartinib and Vatalanib place the halogenated ring, present in every structure, in pocket 1, and the heteroaromatic ring displaying the previously-mentioned hydrogen bond interaction with Leu119 and the hydrophobic interactions, in pocket 2.
Considering that PIKfyve is a lipid kinase, it is reasonable to speculate that it shares a similar active domain with the family of tyrosine kinase enzymes. It has been reported that active domains in tyrosine kinases showed the presence of an ATP binding pocket, where a Lys residue (Lys52) is responsible for holding the ATP alpha and beta-phosphates in position for the phosphorylation catalysis procedure. As was previously mentioned, PIKfyve has Lys1877 in the active site, which is critical kinase activity. It stands to reason, therefore, that drugs that compete for ATP binding can be active against both tyrosine kinases and PIKfyve. The gathered information previously reported, added to the INDO study performed in this work, suggests that tyrosine kinase inhibitors show activity against coronaviruses, including SARS-CoV-2, potentially through the inhibition of the PIKfyve enzyme during the early stages of the viral life cycle.
Viral targets
Papain-like protease (PLpro)
SARS-CoV-2 PLpro is a cysteine protease which is responsible for the early cleavage of the viral polypeptide [63] during virus maturation. PLpro has also been reported to suppress host innate immune responses through the reversal of certain post-translational protein modifications [64, 65]. Ratia et al. developed a series of naphthalene derivatives as SARS-CoV-1 PLpro inhibitors, among which the compound GRL-0617 displayed the highest activity against SARS-CoV-1 PLpro [63]. Recently, Freitas et al. tested several of those naphthalene derivatives against SARS-CoV-2 PLpro [66]. As with SARS-CoV-1, GRL-0617 displayed the highest activity in inhibiting PLpro as well as SARS-CoV-2 replication [66]. In Table S3 (CC10) we show that our INDO protocol indeed selects SARS-CoV-2 PLpro as the top-1 target for GRL-0617.
Crystallographic structures of SARS-CoV-1 and SARS-CoV-2 PLpro-GRL-0617 complexes have been reported [63, 67]. In both cases, the inhibitor occupies the same position away from the catalytic active site and instead bound to the S3 and S4 PLpro subsites, because despite the differences between SARS-CoV-1 and SARS-CoV-2 PLpro, the residues lining the active site and the nearby S3 and S4 cavities are identical [63, 66, 67]. The interaction between GRL-0617 and PLpro is stabilized mainly through a pair of hydrogen bonds and a series of hydrophobic interactions [63, 66, 67]. The present INDO study successfully recapitulates the crystallographic interaction pattern of the PLpro-GRL-0617 complex. The amide moiety of GRL-0617 displays two hydrogen bond interactions with residues Asp165 and Gln270. The rest of the inhibitor structure, including the naphthyl and the substituted benzyl rings, shows hydrophobic interactions with non-polar residues lining the mentioned sites: Leu163, Gly164, Pro249, Tyr265, Try269 and Tyr274 (Figs. 4c and S5a) [63, 66, 67].
The INDO procedure on the HARD list shows numerous compounds beyond GRL-0617 with PLpro as the top-1 preferred target (Table S5). A closer look at the interaction patterns of these compounds with PLpro showed that none were able to participate in the previously mentioned hydrogen bond interactions with both Asp165 and Gln270 residues. From Fig. 3, Mesopride and Clebopride display PLpro as a potential target on their top-3 position. The interaction patterns of the mentioned compounds with the residues of PLpro showed a similar interaction pattern as GRL-0617, especially because of the presence of an amide group on their structures, allowing hydrogen bond interactions with residues Asp165 and Gln270 (Figs. 4c, S5b and S5c). The similarities between these two compounds, regarding both the high average Z-score and the interaction with residues from S3 and S4 PLpro subsites with the PLpro inhibitor GRL-0617, explain why Mosepride and Clebopride also claim PLpro as their potential target.
Helicase
Helicase, together with the RNA-dependent RNA polymerase (RdRp), form the main components of the replication-transcription complex (RTC) responsible for viral genome replication and transcription of coronaviruses like SARS-CoV-2 [8, 68, 69]. Previous reports revealed that Helicase exhibits multiple enzymatic activities, including hydrolysis of NTPs and dNTPs, unwinding of DNA and RNA duplexes and RNA 5′-triphosphatase activity [8, 69]. This enzyme is one of the most evolutionarily conserved proteins in coronaviruses and therefore an important target for drug development [8, 68]. It had been reported that small molecules can inhibit the NTPase activity of helicase through interference with ATP binding [8, 68]. This NTPase domain is positioned in a cleft between domains 1A and 2B, lining by the residues Lys288, Ser289, Asp374, Glu375, Gln404 and Arg567 [8, 68].
The present INDO study showed various HARD with helicase as their most preferred target option (Fig. 3). All the mentioned compounds present a carboxylic acid on their structures. From this list of compounds, Sofalcone, Bumetanide and Stepronin displayed the most relevant interaction pattern with residues from the helicase NTPase binding domain (Figs. 4d and S6). These compounds placed their acid groups on the phosphate binding position from the natural NTPs substrates [68], producing extensive hydrogen bond interactions with residues Gly285, Gly287, Lys288, Gln404, Arg443 and Arg567. This interaction pattern resembles that of natural substrates across the NTPase binding site residues and the elevated average Z-score obtained on the INDO suggest Sofalcone, Bumetanide and Stepronin as promising potential inhibitors of helicase.
S protein
SARS-CoV-2 S protein plays a key role in the early stage of viral infection, with the S1 domain responsible for the molecular recognition to ACE2 and the S2 domain mediating the membrane fusion. Due to this important role in the viral cycle, we analyzed several docking sites in the interior of S protein as previously described in the Methods section. The S protein has no known catalytic function, but in order to interact with the ACE2 receptor a conformational change from the closed to the open state must occur. The interior docking sites found in the closed state (See Methods section and Figs. S7, S8 and S9) have extensive interprotomer contacts and therefore ligands at those sites may stabilize the closed state or prevent post-activation separation of protomers. We also explored the spike RBD and the ACE2, in this cases searching for molecules that may impair its interaction. As was already mentioned, our results showed that no compound from the HARD list selected either the spike RBD or the ACE2 in their respective top-3 choices. This result indicated that these interfaces are not druggable sites for these compounds.
However, from the four distinct internal sites analyzed (Figs. S29 to S32), internal site 4 is preferred, both in terms of number of drugs and in Z-scores values (Fig. 2, Table S5). This site is located at the apex of the trimer in the closed state, and it permits access to residues of the RBD that form the interaction interface with ACE2. It is reasonable to speculate that drugs at this position may prevent the opening of the RBD, resulting in inhibition of SARS-CoV-2 entry. Ponatinib and Silodosin select the internal site 4 as their top-1 choice with average Z-scores of − 2.20 and − 1.57, respectively (Fig. 3 and Table S5). Figure S10 schematically shows this interaction.
Conclusion
In this work, an inverse docking procedure was performed on 152 approved drugs experimentally determined to be highly active against SARS-CoV-2 (HARD) in three independent in vitro HTS assays. Our objective was to identify potential human and viral targets for these drugs. Our INDO approach was first validated on a test set of 209 protein–ligand crystallographic complexes not involving SARS-CoV-2 targets. This validation showed that the combined use of three different scoring functions (Ledock, Korp-PL and Vinardo) resulted in accurate recovery of the correct protein–ligand complex compared to use of any one scoring function individually. When applied to known inhibitors of different SARS-CoV-2 targets this INDO methodology successfully identified 10 out of 14 in their top-3 preferences, showing good accuracy in identifying the correct SARS-CoV-2 protein/enzyme target.
Through this work we have relied mostly on experimental evidence to define the docking search space. This approach has the advantage of concentrating the computational efforts on known interaction sites. Alternatively, it is possible to use blind docking techniques to reveal new interaction sites that have not yet been experimentally tested. This would open up the possibility of discovering new druggable binding sites.
The analysis of the INDO results of the 152 potentially repurposable drugs showed that the preferential targets were the human enzymes TMPRSS2 and PIKfyve, followed by the viral enzymes Helicase and PLpro. This observation is in line with the fact that enzymes are more druggable targets that any other non-structural or structural SARS-CoV-2 protein.
A closer analysis over the preferential targets showed that the three human serine proteases included in this work were selected as top-3 position targets for Nafamostat, showing the same preference as one of the control compounds, Diminazene. The fact that these drugs are proven inhibitors of different serine-proteases included in this work supports the present analysis as a powerful tool to find serine-protease inhibitors. Conversely, we found Hydroxystilbamidine and Camostat as inhibitors of only human Trypsin and TMPRSS2, because they were not able to perform the required molecular interaction on the active site of furin.
The INDO procedure selected PIKfyve as the top-1 target for five drugs from the HARD list. These compounds belong to the family of tyrosine-kinase inhibitors and they were able to establish the same molecular interaction as did Apilimod, a well-known PIKfyve inhibitor, on the active site of the enzyme. This information suggests that a tyrosine-kinase inhibitor can show anti SARS-CoV-2 activity by inhibiting the human PIKfyve enzyme.
The observations over the two preferential viral targets showed that PLpro select this target as top-1 for the GRL-0617, the control compound reported as specific inhibitor of this viral enzyme. Also the docked pose reproduce the crystallographic interaction pattern observed in the PLpro:GRL-0617 complex. These pharmacophoric interactions were also observed by two drugs from the HARD list: Mosapride and Clebopride, which display PLpro as their top-3 target choices. For helicase, the present study found several compounds selected this enzyme as their top-1 target, acting on the catalytic site of the enzyme. All such drugs share a carboxylic acid moiety on their structures, possibly playing the role of the phosphate group present on the natural NTPs substrates.
Overall, to the best of our knowledge, this is the first INDO study that suggests potential viral and/or human targets for a list of HARD with anti-SARS-CoV-2 activity. The results presented in this work contribute to the characterization of drugs with potential for directly repurposing against SARS-CoV-2 and to the further development of novel compounds with anti-SARS-CoV-2 activity. When combined with high-throughput screening and structure-based rational drug design, the INDO protocol demonstrated here should therefore contribute substantially to the fight against SARS-CoV-2 and other diseases.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thanks to Marcelo Puiatti, Rodrigo Quiroga and Alfredo Quevedo for contributions in the initial stages of this project. This work used computational resources from CCAD, Universidad Nacional de Córdoba (https://ccad.unc.edu.ar), which are part of SNCAD, MinCyT, República Argentina. Special thanks to CCAD for give us a priority and exclusive access to the computer resources as part of the global efforts to fight against Covid-19 pandemic.
Author contributions
SRR, SAP and MAV designed the study, performed the experiments and analyzed the data. CAF made contribution to the discussions during the work. All authors prepared the manuscript and provided feedback on the paper
Funding
This work was supported by CONICET-Consejo Nacional de Investigaciones Científicas y Tecnológicas, http://www.conicet.gov.ar; Secyt-UNC.Secretaría de Ciencia y Técnica de la Universidad Nacional de Córdoba (http://www.unc.edu.ar/investigacion). S. A. Paz acknowledges the financial support from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT-FONCyT Grant PICT-2017-0621). C. F. Abrams acknowledges support from the National Institutes of Health (AI150471 and GM100472).
Data availibility
Protein structures, ligand SMILES and SQL scripts used in this work are available in our public GitHub repository at https://github.com/alexispaz/HTIDocking-Cov2. The scripts extract data from the ChEMBL database version 27, downloaded from http://www.ebi.ac.uk/chembl. Detailed description of the docking boxes are in the supplementary materials file. Gypsum-dl version 1.1.7 was downloaded from https://durrantlab.pitt.edu/gypsum-dl/. The latest free academic version of Ledock (v1.0) was downloaded from Lephar web site at http://www.lephar.com/download.htm. KORP-PL version 0.1.1 was used as downloaded from the author’s page at https://team.inria.fr/nano-d/software/korp-pl. Our beta version of Vinardo is available upon request.
Declarations
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World Health Organization. https://covid19.who.int/
- 2.Korber B, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, Owen CJ, Pang J, Tan CC, Boshier FA, Ortiz AT, Balloux F. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020;83:104351. doi: 10.1016/j.meegid.2020.104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohamed K, Yazdanpanah N, Saghazadeh A, Rezaei N. Computational drug discovery and repurposing for the treatment of COVID-19: a systematic review. Bioorg Chem. 2021;106:104490. doi: 10.1016/j.bioorg.2020.104490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahamad S, Branch S, Harrelson S, Hussain MK, Saquib M, Khan S. Primed for global coronavirus pandemic: emerging research and clinical outcome. Eur J Med Chem. 2021;209:112862. doi: 10.1016/j.ejmech.2020.112862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batalha PN, Forezi LS, Lima CG, Pauli FP, Boechat FC, de Souza MCB, Cunha AC, Ferreira VF, da Silva FdC. Repurposing for the treatment of COVID-19: pharmacological aspects and synthetic approaches. Bioorg Chem. 2021;106:104490. doi: 10.1016/j.bioorg.2020.104488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C, et al. Furin: a potential therapeutic target for COVID-19. iScience. 2020;23:101642. doi: 10.1016/j.isci.2020.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohaim MA, El Naggar RF, Clayton E, Munir M. Structural and functional insights into non-structural proteins of coronaviruses. Microb Pathog. 2021;150:104641. doi: 10.1016/j.micpath.2020.104641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Protein Data Bank. https://www.rcsb.org/
- 10.Berman HM. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.SWISS-MODEL. https://swissmodel.expasy.org/
- 12.Bienert S, Waterhouse A, de Beer TAP, Tauriello G, Studer G, Bordoli L, Schwede T. The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res. 2017;45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Lab. https://zhanglab.ccmb.med.umich.edu/COVID-19/
- 14.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alphafold. https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19
- 16.Luan B, Huynh T, Cheng X, Lan G, Wang H-R. Targeting proteases for treating COVID-19. J Proteome Res. 2020;19:4316–4326. doi: 10.1021/acs.jproteome.0c00430. [DOI] [PubMed] [Google Scholar]
- 17.Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fakhar Z, Khan S, AlOmar SY, Alkhuriji A, Ahmad A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci Rep. 2021;11:234. doi: 10.1038/s41598-020-79918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghahremanpour MM, Tirado-Rives J, Deshmukh M, Ippolito JA, Zhang C-H, Cabeza de Vaca I, Liosi M-E, Anderson KS, Jorgensen WL. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heiser K, McLean PF, Davis CT, Fogelson B, Gordon HB, Jacobson P, Hurst B, Miller B, Alfa RW, Earnshaw BA, Victors ML, Chong YT, Haque IS, Low AS, Gibson CC. Identification of potential treatments for COVID-19 through artificial intelligence-enabled phenomic analysis of human cells infected with SARS-CoV-2. bioRxiv. 2020;53:1689–1699. [Google Scholar]
- 21.Touret F, Gilles M, Barral K, Nougairède A, van Helden J, Decroly E, de Lamballerie X, Coutard B. In vitro screening of a fda approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci Rep. 2020;10:13093. doi: 10.1038/s41598-020-70143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellinger B, Bojkova D, Zaliani A, Cinatl J, Claussen C, Westhaus S, Reinshagen J, Kuzikov, M, Wolf M, Geisslinger G, Gribbon P, Ciesek S (2020) Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. PREPRINT available at Research Square. Version 1, 1–19
- 23.Kim SS, Aprahamian ML, Lindert S. Improving inverse docking target identification with Z-score selection. Chem Biol Drug Des. 2019;93:1105–1116. doi: 10.1111/cbdd.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee A, Lee K, Kim D. Using reverse docking for target identification and its applications for drug discovery. Expert Opin Drug Discov. 2016;11:707–722. doi: 10.1080/17460441.2016.1190706. [DOI] [PubMed] [Google Scholar]
- 25.Kharkar PS, Warrier S, Gaud RS. Reverse docking: a powerful tool for drug repositioning and drug rescue. Future Med Chem. 2014;6:333–342. doi: 10.4155/fmc.13.207. [DOI] [PubMed] [Google Scholar]
- 26.Wang F, Wu F-X, Li C-Z, Jia C-Y, Su S-W, Hao G-F, Yang G-F. ACID: a free tool for drug repurposing using consensus inverse docking strategy. J Cheminform. 2019;11:73. doi: 10.1186/s13321-019-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Zhi D. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins. 2001;43:217–226. doi: 10.1002/1097-0134(20010501)43:2<217::AID-PROT1032>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 28.Wang J-C, Chu P-Y, Chen C-M, Lin J-H. idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res. 2012;40:W393–W399. doi: 10.1093/nar/gks496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferraz WR, Gomes RA, S Novaes AL, Goulart Trossini GH. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med Chem. 2020;12:1815–1828. doi: 10.4155/fmc-2020-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jade D, Ayyamperumal S, Tallapaneni V, Joghee Nanjan CM, Barge S, Mohan S, Nanjan MJ. Virtual high throughput screening: potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur J Pharmacol. 2021;901:174082. doi: 10.1016/j.ejphar.2021.174082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barge S, Jade D, Gosavi G, Talukdar NC, Borah J. In-silico screening for identification of potential inhibitors against SARS-CoV-2 transmembrane serine protease 2 (TMPRSS2) Eur J Pharm Sci. 2021;162:105820. doi: 10.1016/j.ejps.2021.105820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quiroga R, Villarreal MA. Vinardo: a scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS ONE. 2016;11:e0155183. doi: 10.1371/journal.pone.0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Huang D. Hydrogen bonding penalty upon ligand binding. PLoS ONE. 2011;6:e19923. doi: 10.1371/journal.pone.0019923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parks CD, Gaieb Z, Chiu M, Yang H, Shao C, Walters WP, Jansen JM, McGaughey G, Lewis RA, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK. D3R grand challenge 4: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des. 2020;34:99–119. doi: 10.1007/s10822-020-00289-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Sun H, Yao X, Li D, Xu L, Li Y, Tian S, Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys. 2016;18:12964–12975. doi: 10.1039/C6CP01555G. [DOI] [PubMed] [Google Scholar]
- 38.Kadukova M, Machado KdS, Chacón P, Grudinin S. KORP-PL: a coarse-grained knowledge-based scoring function for protein-ligand interactions. Bioinformatics. 2021;37:943–950. doi: 10.1093/bioinformatics/btaa748. [DOI] [PubMed] [Google Scholar]
- 39.Ribone SP, Paz SA, Abrams CF, Villarreal M (2021) High-throughput inverse docking of SARS-Cov-2. 10.5281/zenodo.5557825. https://github.com/alexispaz/HTIDocking-Cov2
- 40.Palacio-Rodríguez K, Lans I, Cavasotto CN, Cossio P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci Rep. 2019;9:5142. doi: 10.1038/s41598-019-41594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47:2977–2980. doi: 10.1021/jm030580l. [DOI] [PubMed] [Google Scholar]
- 42.Backman TWH, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011;39:W486–W491. doi: 10.1093/nar/gkr320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ropp PJ, Spiegel JO, Walker JL, Green H, Morales GA, Milliken KA, Ringe JJ, Durrant JD. Gypsum-DL: an open-source program for preparing small-molecule libraries for structure-based virtual screening. J Cheminform. 2019;11:34. doi: 10.1186/s13321-019-0358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw DE. http://www.deshawresearch.com/resources_sarscov2.html
- 45.Siramshetty VB, Eckert OA, Gohlke B-O, Goede A, Chen Q, Devarakonda P, Preissner S, Preissner R. SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res. 2018;46:D1137–D1143. doi: 10.1093/nar/gkx1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 47.Mendez D, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47:D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.EMBL-EBI (2020) CHEMBL Database Release 27
- 49.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.M P, Reddy GJ, Hema K, Dodoala S, Koganti B. Unravelling high-affinity binding compounds towards transmembrane protease serine 2 enzyme in treating SARS-CoV-2 infection using molecular modelling and docking studies. Eur J Pharmacol. 2021;890:173688. doi: 10.1016/j.ejphar.2020.173688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffmann M, et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv. 2021;65:103255. doi: 10.1016/j.ebiom.2021.103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shrimp JH, Kales SC, Sanderson PE, Simeonov A, Shen M, Hall MD. An enzymatic TMPRSS2 assay for assessment of clinical candidates and discovery of inhibitors as potential treatment of COVID-19. ACS Pharmacol Transl Sci. 2020;3:997–1007. doi: 10.1021/acsptsci.0c00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kayode O, Huang Z, Soares AS, Caulfield TR, Dong Z, Bode AM, Radisky ES. Small molecule inhibitors of mesotrypsin from a structure-based docking screen. PLoS ONE. 2017;12:e0176694. doi: 10.1371/journal.pone.0176694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang Y-L, Chou Y-Y, Rothlauf PW, Liu Z, Soh TK, Cureton D, Case JB, Chen RE, Diamond MS, Whelan SPJ, Kirchhausen T. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci. 2020;117:20803–20813. doi: 10.1073/pnas.2007837117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ou X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lees JA, Li P, Kumar N, Weisman LS, Reinisch KM. Insights into lysosomal PI(3,5)P2 homeostasis from a structural-biochemical analysis of the PIKfyve lipid kinase complex. Mol Cell. 2020;80:736–743.e4. doi: 10.1016/j.molcel.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson EA, et al. The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. PLoS Negl Trop Dis. 2017;11:e0005540. doi: 10.1371/journal.pntd.0005540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hulseberg CE, Fénéant L, Szymańska-de Wijs KM, Kessler NP, Nelson EA, Shoemaker CJ, Schmaljohn CS, Polyak SJ, White JM. Arbidol and other low-molecular-weight drugs that inhibit Lassa and Ebola viruses. J Virol. 2019;93:e02185. doi: 10.1128/JVI.02185-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin J, Jung E, Kim M, Baric R, Go Y. Saracatinib inhibits middle east respiratory syndrome-coronavirus replication in vitro. Viruses. 2018;10:283. doi: 10.3390/v10060283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coleman CM, Sisk JM, Mingo RM, Nelson EA, White JM, Frieman MB. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and middle east respiratory syndrome coronavirus fusion. J Virol. 2016;90:8924–8933. doi: 10.1128/JVI.01429-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sisk JM, Frieman MB, Machamer CE. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J Gen Virol. 2018;99:619–630. doi: 10.1099/jgv.0.001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng CTK, Baker SC, Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freitas BT, Durie IA, Murray J, Longo JE, Miller HC, Crich D, Hogan RJ, Tripp RA, Pegan SD. Characterization and noncovalent inhibition of the Deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 67.Gao X, Qin B, Chen P, Zhu K, Hou P, Wojdyla JA, Wang M, Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm Sin B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hao W, Wojdyla JA, Zhao R, Han R, Das R, Zlatev I, Manoharan M, Wang M, Cui S. Crystal structure of middle east respiratory syndrome coronavirus helicase. PLOS Pathog. 2017;13:e1006474. doi: 10.1371/journal.ppat.1006474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen J, Malone B, Llewellyn E, Grasso M, Shelton PM, Olinares PDB, Maruthi K, Eng ET, Vatandaslar H, Chait BT, Kapoor TM, Darst SA, Campbell EA. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020;182:1560–1573.e13. doi: 10.1016/j.cell.2020.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein structures, ligand SMILES and SQL scripts used in this work are available in our public GitHub repository at https://github.com/alexispaz/HTIDocking-Cov2. The scripts extract data from the ChEMBL database version 27, downloaded from http://www.ebi.ac.uk/chembl. Detailed description of the docking boxes are in the supplementary materials file. Gypsum-dl version 1.1.7 was downloaded from https://durrantlab.pitt.edu/gypsum-dl/. The latest free academic version of Ledock (v1.0) was downloaded from Lephar web site at http://www.lephar.com/download.htm. KORP-PL version 0.1.1 was used as downloaded from the author’s page at https://team.inria.fr/nano-d/software/korp-pl. Our beta version of Vinardo is available upon request.