

Abstract
This report describes the tertiary structure prediction assessment of difficult modeling targets in the 14th round of the Critical Assessment of Structure Prediction (CASP14). We implemented an official ranking scheme that used the same scores as the previous CASP topology-based assessment, but combined these scores with one that emphasized physically realistic models. The top performing AlphaFold2 group outperformed the rest of the prediction community on all but two of the difficult targets considered in this assessment. They provided high quality models for most of the targets (86% over GDT_TS 70), including larger targets above 150 residues, and they correctly predicted the topology of almost all the rest. AlphaFold2 performance was followed by two manual Baker methods, a Feig method that refined Zhang-server models, two notable automated Zhang server methods (QUARK and Zhang-server), and a Zhang manual group. Despite the remarkable progress in protein structure prediction of difficult targets, both the prediction community and AlphaFold2, to a lesser extent, faced challenges with flexible regions and obligate oligomeric assemblies. The official ranking of top-performing methods was supported by performance generated PCA and heatmap clusters that gave insight into target difficulties and the most successful state-of-the-art structure prediction methodologies.
Keywords: CASP14, protein structure prediction, topology structure modeling evaluation, homology modeling, machine learning, free modeling, structural bioinformatics
1. INTRODUCTION
The Critical Assessment of Structure Prediction (CASP) provides a critical evaluation of state‐of‐the‐art methods in predicting protein structure from sequence1. CASP provides amino acid sequences corresponding to target structures withheld from the public, and prediction groups submit models for evaluation in various tracks. The previous round of CASP described dramatic progress in structure modeling of the most difficult targets evaluated by the topology assessment, historically known as ‘ab-initio’ or free modeling (FM)2. Prediction of inter-residue distances using deep learning led to substantial improvement in FM model accuracy for monomeric proteins of large families (although the number of required sequences fell considerably)3. For the first time in CASP history, the global topologies for all difficult EUs were roughly captured by at least one model, and near atomic resolution (GDT_TS > 80) was achieved for a few small targets4.
This report describes our topology assessment of the most difficult target evaluation units (EUs) from CASP14. The assessment category included 23 difficult EUs (FM class) and 15 borderline EUs (FM/TBM) that were also evaluated by the high accuracy modeling assessment (TBM assessment, this issue). Many of the EUs in the topology assessment category (48%) had homologous templates that were distantly related to the target. Despite the homologous relationship exemplified by some target EUs to known templates, their structures deviated substantially from one another and their sequences were unrecognizable. The remaining targets had similar topological arrangements of SSEs (26%) or were new folds (26%) (Target Classification, Kinch et al, this issue).
Using similar criteria as in CASP13 to rank predictors in the tertiary structure prediction topology category, we found the AlphaFold2 group outperformed the rest by a large margin and provided remarkably accurate models (29 first models > 80 GDT_TS and 14 first models > 90 GDT_TS). The Baker, Baker-experimental, FEIG-R2, QUARK, Zhang-Server, and Zhang groups followed in rank with scores that were less discriminating but ranked consistently at the top using different evaluation methods. Impressively, the automated server models produced by QUARK and Zhang-Server ranked among these top experimental groups. In fact, CASP14 servers provided topologically correct models that outperformed the top known templates for most target EUs in the assessment and outperformed all CASP13 groups (Progress Paper, this issue). Manual inspection of models highlighted difficulties with multichain assemblies and flexible regions. Finally, we evaluated the current state-of-the-art in protein structure prediction using heatmap visualization and clustering of methodological features provided in CASP14 abstracts and compared these to a baseline server method from CASP13.
2. METHODS
2.1. Scores to identify top models and rank prediction methods
To evaluate performance on topology (FM) targets in CASP14 we considered several sets of scores before selecting a final formula for official ranking of methods. The final ranking was based on models designated as “1” for all groups on “all group” targets encompassing the 23 difficult FM and 15 borderline FM/TBM classes of targets. The model scores were combined by the Prediction Center using SumZ>−2 as previously described with the formula 1*GDT_TS +_1*QCS + 0.1* MolProbity5,6. This ranking scheme was compared to similar schemes where we varied different individual components: replace FM and FM/TBM targets with FM-only targets, use best models instead of first models, and replace chosen scores with FM formula from CASP13, or TBM formula for CASP13. Top prediction model performance trends for individual targets were assessed using Global Distance Test (GDT_TS7), a measure of structure similarity developed to compare prediction models to their target structures with known sequence correspondence. The GDT_TS score quantifies the largest set of residues that can be superposed under specific distance cutoffs (1, 2, 4, and 8 Å) and is expressed as a percentage from 0 to 100. Model to target similarity was compared with template to target similarity using scores from Local-Global Alignment (LGA_S8), which ignores the sequence relationship between target and model for structure superpositions.
Model-to-target Cα-Cα distances on sequence-based superpositions provided by the Prediction Center were used to calculate the local accuracy score corresponding to the performance on various regions in target structures. The average modeling quality of a region or a collection of positions (such as those with regular secondary structures defined by STRIDE9) was calculated using the Cα-Cα distances of the top 50 models ranked by GDT-TS, and they were averaged for each position individually. These residue position averages of Cα-Cα distances were then averaged over the region or the collection of positions to produce the local accuracy score.
2.2. Clustering of prediction method performance on hard targets
To help evaluate the current state-of-the-art in protein structure prediction, we clustered method performance across all FM and FM/TBM targets using principal component analysis (PCA) and heatmaps provided by the ClustVis web tool10. Our methods evaluation was restricted to the top 50 groups ranked by average GDT_TS (filtering out groups that did not provide models for at least 36 out of 38 of the targets). Two groups were excluded that did not provide abstracts and one baseline server (Baker-Robetta) was added that did not change from the previous CASP13. The Prediction Center provided a number of metrics to rate model accuracy and evaluate performance6. We tested many of these scores (some converted* to a 0 to 100 scale) alone and in combination, including GDT_TS, GDT_HA, LDDT, LGA_S, QCS, CAD-aa*, and TMscore. However, we ultimately chose to cluster using a single score (GDT_TS) for ease of heatmap visualization.
For PCA clustering we used a group feature that separated the top performing AlphaFold2 group (Rank1) from the manual groups (manual), the automated server groups (server), and the baseline Baker-Robetta server group (baseline). The GDT_TS scores for all FM and FM/TBM targets were row centered without scaling, and SVD with imputation was used to calculate principal components and to fill in 3 missing target values (T1061-D2, T1080-D1, and T1082-D1) from two groups. The first two principal components that explained that most variance in the group data (N=52 groups) were plotted, and ellipses were drawn around the manual and server group sets so that, with 95% probability, a new observation from the same group would fall inside the ellipse. Many of the CASP14 predictors submitted models as multiple groups. To help distinguish the different methods provided by the same group, we assigned a predictor name feature for each of the groups (using “single” for the remaining groups with only one method and “multiple” for the remaining groups with multiple methods). The assigned names are indicated by different markers in the PCA plot.
In addition to the group and name features used for PCA, we assigned various additional methods related features to the groups for visualization using GDT-TS score heatmap clusters of all FM and FM/TBM targets. From our interpretation of methods described in provided abstracts, we assigned the following yes/no features to the groups: metagenomics, templates, deep learning, attention networks, and server models. Targets were assigned features that were considered in CASP14 classification (Kinch classification paper, this issue); including ECOD levels (New, X-group, or H-group), assessment class (FM or FM/TBM), and taxonomy (Bacteria, fungi or virus). We also included structure determination method (X-ray, NMR, EM, or EM and X-ray) as a target feature. No scaling was applied to rows and imputation was applied to replace missing values. Both rows (38 targets) and columns (52 groups) were clustered using Euclidean distances with Ward linkage and ordered by higher mean value first. GDT_TS scores were colored using a diverging Red (high) to blue (low) heatmap coloring scheme. Targets were split into the top 4 clusters and groups were split into the top 8 clusters by visual inspection of performance.
3. RESULTS
The CASP14 assessment of tertiary structure prediction in the topology category was guided almost entirely by automated scoring used in previous rounds of CASP, and ranks were supported by their consistency using different parameters. Due to the high quality of submitted models for most target EUs, minimal manual inspection was required for their evaluation. The GDT_TS score of top models compared to their target structures was used as a guide to assess models’ overall topology (correct if GDT_TS >45), to identify and discuss those targets that were the most difficult, and to group methods for evaluation of key features that contributed to performance.
3.1. Consistent Ranking of Top Prediction Groups using Different Criteria to Evaluate Performance
The official CASP14 ranking was based primarily on two complementary scores (GDT_TS and QCS) that have been used to guide evaluation of the topology category for the past several rounds4,5,11. The ranking formula also included an aggregated score (MolProbity) of prediction model quality that combined the clash, rotamer outlier, and Ramachandran favored components of experimental structure model evaluation12. The official rank formula used Z-score sums over the FM and FM/TBM targets for first models (Figure 1A). Notably, the Z-score sum for AlphaFold2 outranked the sums for all remaining groups by a wide margin, and the average Z-score for AlphaFold2 (2.51) was more than twice the next best group average (Baker, 1.02). The top ranked groups by the official ranking scheme were ordered AlphaFold2, Baker, Baker-experimental, FEIG-R2, QUARK, Zhang-server and Zhang. Notably, two automated servers (QUARK and Zhang-server) were among the top performing groups.
Figure 1. Ranks of Tertiary Structure Prediction Groups.
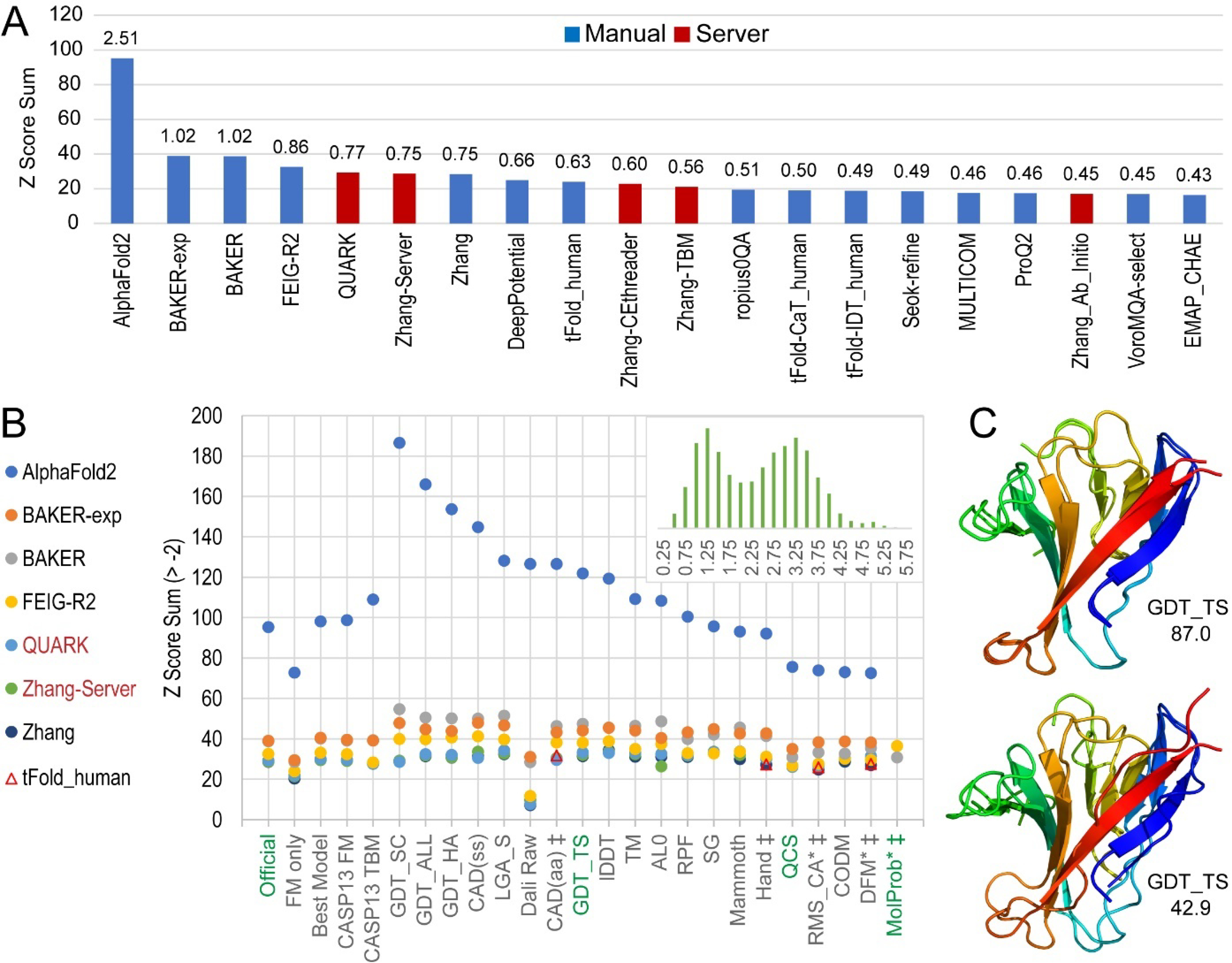
A) Column graph represents official ranks of the top 20 groups by their Z-score sum (> −2.0) over all FM and FM/TBM target EUs for weighted chosen scores on first models (weights: 1*GDT_TS +_1*QCS + 0.1* MolProbity). Groups are labeled and colored according to Manual (blue) and Server (red) types. The average chosen score over all FM and FM/TBM targets is indicated above the bar. B) Scatter of Z Score Sums (>2) from the official CASP14 ranking scheme for the top 7 consistently ranked groups (labeled to the side, circle markers) according to a majority of the scores (labeled on the X axis, official component scores colored green). Alternate group(s) (open triangle markers) rank among the top 7 for a few individual scores (marked by a double dagger). For each scoring strategy, the label is named by the component of the official ranking scheme that is replaced. For example, the ‘FM only’ score ignores the FM/TBM targets, and the ‘CASP13 FM’ score replaces the weighted score. Scores calculated with inverted raw scores are indicated with an asterisk. Inset histogram depicts MolProbity score distribution for first models. C) AlphaFold2 top first model is superimposed with T1064 (GDT_TS 87.0, upper panel) and significantly outperforms the next best first model from Xianmingpan superimposed with T1064 (GDT_TS 42.9, lower panel). All models are colored in rainbow from N-terminal (blue) to C-terminal (red).
We tested various permutations of the official scoring scheme to evaluate the consistency of the top ranked groups (Figure 1B). The top seven groups were ranked in the same order when we narrowed the targets to FM-only, which excluded FM/TBM target EUs but used the same official scoring scheme, or if we replaced our score weights with the official FM scoring scheme from CASP13 (1*GDT_TS+1*QCS). When the ranks were evaluated using the best models instead of first models, the last two positions (rank 6 and rank 7) were inverted. Alternately, the CASP13 TBM scoring scheme, (GDT_HA + (SG + lDDT + CAD)/3 + ASE), inverted positions 2 and 3 and positions 5 and 6, but the same seven groups were at the top. This high accuracy scoring scheme widened the distribution of the Z-score sums for the top-performing groups, suggesting that the quality of AlphaFold2 models became increasingly distinct from the rest using scores that favored higher quality models with correct overall backbone folds.
In addition to these permutations of the official ranking scheme, we also performed ranking using select individual scores provided by the Prediction Center. The same seven groups were ranked at the top for 15 out of 20 individual scores, which represented 80% of all permuted scoring schemes (20 out of 25 total scores). Among those scores with altered top rankings, two that were developed to assess CASP10 FM target topology (DFM and Hand13) as well as root-mean-square deviation calculated on Cα atoms of sequence based superposition (RMS_CA) swapped the rank 7 Zhang group with the group tFold_human. Similarly, two tFold groups (tFold_human and tFold-CaT_human) broke into the top ranks (position 5 and 6, replacing QUARK and Zhang-server) using the Contact Area Difference score for all atoms (CAD-aa14), which evaluates contacts without rigid-body superposition. While this change in ranks for CAD-aa might have suggested an improved performance for tFOLD using measures that are less sensitive to conformation change, their average performance for CAD-aa on all FM and FM/TBM target EUs was only marginally better than the servers they replaced. Furthermore, similar methods that measured contacts without rigid body superposition (like CAD-ss, LDDT, and SphereGrinder) ranked the same top seven groups as the official scoring scheme ranks.
Interestingly, the highest Z-score sum for AlphaFold2 among all single score metrics was for a GDT-style score that measures the correct placement of side chains (186.4, GDC_SC15). In fact, components from two scores produced by the Prediction Center separated side chains from other atoms. Each side chain component shifted the AlphaFold2 Z-score sum higher, with CAD-ss increasing the sum by 14.5% over CAD-aa and GDC_SC increasing the sum by 12.4% over GDC_ALL. Because these sensitive side chain measures required models with approximately correct backbone folds, the increased performance by AlphaFold2 with respect to the remaining groups might have reflected their prediction models having more accurate sidechain positions, but could have also reflected improved backbone positions. This AlphaFold2 outperformance trend was recapitulated in other scores used for evaluating high accuracy models where the Baker group also consistently outperformed Baker-experimental. Measures that have traditionally evaluated model topology swapped the two Baker groups (Figure 1B).
While we chose a combined score for our official ranking, the GDT_TS score alone provided better separation of the top groups and represented a middle ground between topology-level assessment scores like QCS and scores that require accurate models like GDC_SC. Using this GDT_TS measure as a guide to compare the AlphaFold2 model1 performance to the next best model, AlphaFold2 significantly outperformed the runner-up on the SARS-CoV-2 ORF8 accessory protein target T1064 (Figure 1C, top panel, GDT_TS 87.0). The AlphaFold2 model superimposed with 1.4 Å RMSD over 96.7% of the target. Only a few residues at the C-terminus, in two adjacent loops, and in the boundary of a flexible loop (that was excluded from the EU) deviated by over 1 Å. In the experimental crystal structure of T1064, the conformation of the excluded flexible loop appeared to be determined by crystal packing. Residues from the C-terminus and two adjacent loops that were modeled with less accuracy by AlphaFold2 were in a homodimer interface. These observations suggest the AlphaFold2 model approached a theoretical upper boundary of model accuracy. Exceeding the score of such a boundary would require considering crystal interactions. The next best model from the Xianmingpan group roughly predicted most of the overall topology of the target (Figure 1C, lower panel, GDT_TS 42.9). However, this model failed to position the N-terminal strand correctly (although it was close to the C-terminal strand where it should interact), and the overall accuracy of the fold was worse (RMSD 3.1 Å over 81.5% of the target).
3.2. AlphaFold2 Provided High Quality Models for Most Difficult Targets
AlphaFold2 models designated as first achieved impressive scores for target EUs in the difficult FM and FM/TBM categories (38 targets): with 33 higher than 70 GDT_TS, 29 higher than 80 GDT_TS, and 14 higher than 90 GDT_TS (Figure 2A). While AlphaFold2 consistently outperformed on almost all target EUs (average GDT_TS 84.6), several other groups approached their model quality on individual targets (~40% of targets had first models within 15% of the AlphaFold2 GDT_TS score). For example, a first model for T1033 from the Baker group (GDT_TS 75.5) achieved 2.1 Å RMSD over 99% of the 100-residue long target structure (Figure 2B). While this performance score approached 15% of that achieved by AlphaFold2, it far exceeded the average server performance (GDT_TS 33.1) on this difficult target. Another impressive first model was produced by the top performing server on target T1082 (QUARK, GDT_TS 72.67). The model provided the correct overall topology of the target and achieved a 2.7 Å RMSD over 75% of the structure (Figure 2C). Top first models from the rest of the prediction community (i.e., excluding those from AlphaFold2) achieved good average overall performance on all FM and FM/TBM targets of GDT_TS 66.5. This average performance improved on the quality of top models reported for CASP13 (GDT_TS 62)4. The performance of CASP14 servers are discussed in the Server performance section below.
Figure 2. Topology Assessment of Tertiary Structure Predictions.
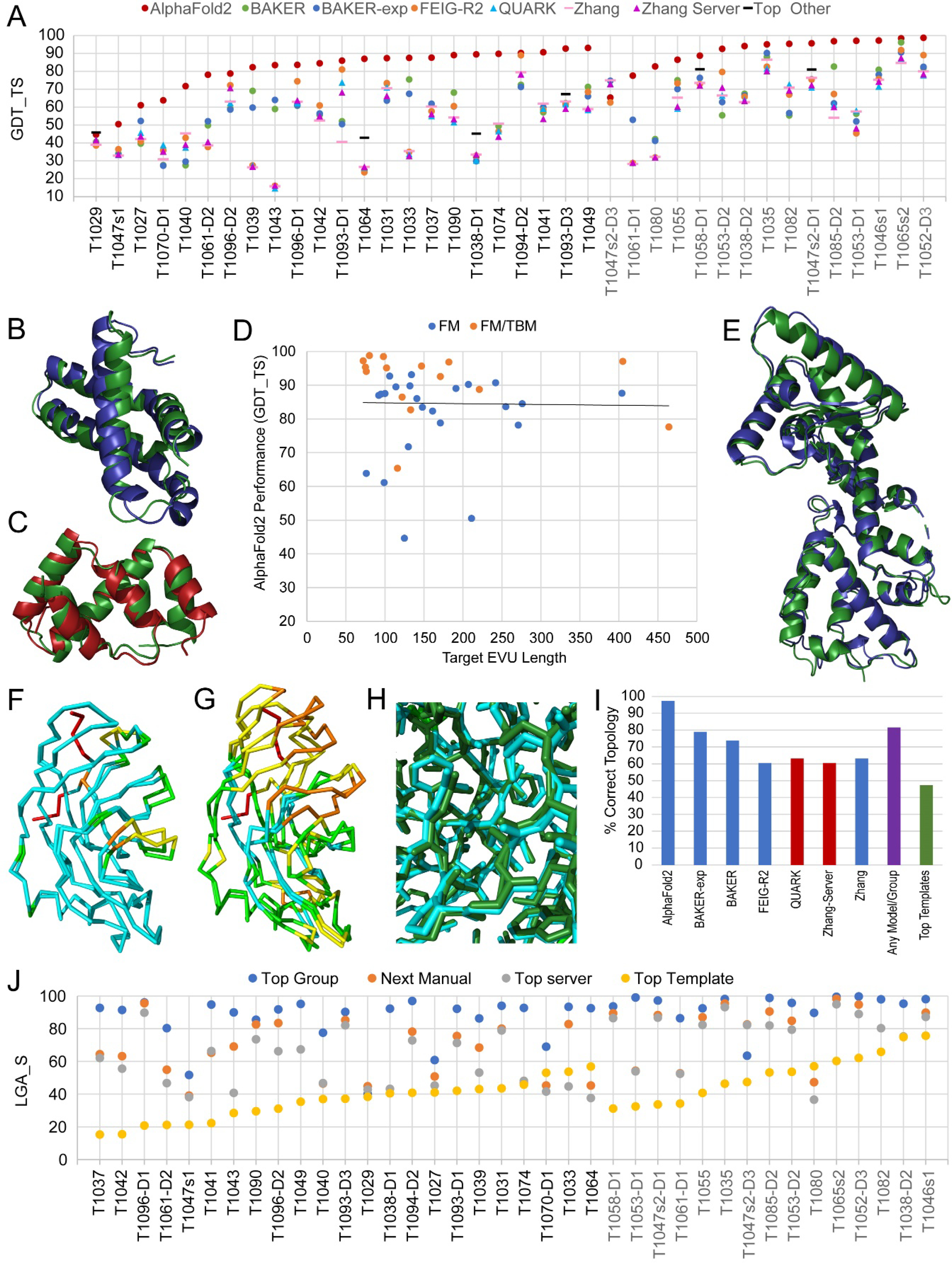
A) Scatter plot of GDT_TS scores for first models of top performing groups (labels in top legend; Top other is any group that performs 5% better that the listed groups). Servers are indicated by triangles. FM (black labels) and FM/TBM (gray labels) target EUs are each sorted by the GDT_TS scores for AlphaFold2. B) Excluding AlphaFold2 models, the next best performing manual group Baker outperforms on T1033 (green) with their superimposed first model (blue, GDT_TS 75.5. lower panel), C) while the best performing server QUARK outperforms on T1082 (green) with their superimposed first model (red, GDT_TS 72.67, upper panel). D) Scatterplot depicts lack of correlation for AlphaFold2 model GDT_TS scores on FM (orange circles) and FM/TBM (blue circles) targets with their size in residue length. The black line is a linear fit to the datapoints. E) Relatively large (276 residues) two-domain target T1042 (green) superimposed with AlphaFold2 model (blue, GDT_TS 70.97). F) Target 1049 superimposed with AlphaFold2 first model colored by local accuracy: 0–1 Å (cyan), 1–2 Å (green), 2–4 Å (yellow), 4–8 A (orange), and >8 (red). G) Target 1049 superimposed with tFold-IDT server first model colored as in F. H) Target1049 (dak green) superimposed with AlphaFold2 first model colored as in F with sidechains in stick. I) Column graph represents the percentage of FM and FM/TBM targets having prediction models with the correct topology (estimated by model GDT_TS scores > 45) selecting among first models for either the top performing groups (labeled and colored as above), among best models provided by any group other than AlphaFold2 (any model/group), or counting Top templates assigned as homologs as a reference. J) Scatter of LGA_S scores for each FM (label in black) and FM/TBM (label in gray) target, with each ordered by the top template score (yellow circles). AlphaFold2 first model (Top group, blue circles) performance is compared to the next best manual group (orange circles) and the top server (gray circles).
Top performing models for difficult FM and FM/TBM targets from the previous round of CASP achieved “near atomistic” resolution for small targets (ranging in size from 41 to 154 residues in CASP13)4. Yet for this round of CASP, difficult targets of all sizes obtained comparable high-quality predictions. In fact, AlphaFold2 model performance displayed no correlation with size (Figure 2D). Four of the five targets where their models failed to achieve high quality (GDT_TS>70) were small: T1070-D1 (76 residues), T1027 (99 residues), T1047s2-D3 (116 residues), and T1029 (125 residues). Out of the fifteen larger targets above 150 residues in length, AlphaFold2 successfully predicted 14 to GDT_TS 70, and 12 to GDT_TS 80. One such prediction was for a 276-residue protein fragment from a phage DNA-dependent RNA polymerase (T1042, Figure 2E). The AlphaFold2 model achieved a GDT_TS of 84.5, which represented 1.6 Å RMSD over 97.8% of the structure. For another target (T1049) where the first AlphaFold2 model performed the best among all FM targets (Figure 2F), the prediction achieved high quality (GDT_TS 93.1), while the score for the next best tFOLD-IDT server model (GDT_TS 71.3) reflected a more typical topology-level prediction for difficult targets (Figure 2G). At the high-quality level achieved by AlphaFold2 on this target, the model correctly placed most sidechains (Figure 2H).
Although this assessment considered topology-level performance, the high quality of AlphaFold2 models highlighted in Figure 2 prompted more comprehensive evaluation of their side chain placement. Several FM (T1049, T1074, T1090, and T1064) and FM/TBM targets (T1065s2, T1046s1, and T1082) with resolution better than 2A (and one with 2.02A) were chosen to compare the performance of AlphaFold2 with the next best method using measures for side chain evaluation (GDC_SC and CAD-ss). Table 1 summarizes the performance on side chain placement. Similar to the increased outperformance of AlphaFold2 according to Z-score sums for these side chain evaluation scores (Figure 1B), their average scores on high resolution targets (62.6 for GDT_SC and for 0.68 CAD-ss) significantly outperformed average for the next best groups (29.8 for GDT_SC and 0.4 for CAD-ss). The placement of sidechains in the first AlphaFold2 model for T1049 model illustrated in figure 2H ranked second among these, with their lowest performance on T1074 (GDT_SC 56.97) predicting a majority of the sidechains correctly.
Table 1.
Side chain placement comparisons
| CASP14 Target | AlphaFold2 | Next Best Group (ranked by GDC_SC) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| EU | Res. (Å) | class | GDT_TS | GDC_SC | CAD-ss | Group | GDT_TS | GDC_SC | CAD-ss |
| T1065s2 | 1.59 | FM/TBM | 98.47 | 71.47 | 0.71 | BAKER | 96.17 | 59.98 | 0.63 |
| T1046s1 | 1.65 | FM/TBM | 97.22 | 60.6 | 0.67 | BAKER | 80.9 | 30.29 | 0.44 |
| T1082 | 1.52 | FM/TBM | 95.33 | 64.02 | 0.68 | MESHI | 70 | 26.19 | 0.29 |
| T1049 | 1.75 | FM | 93.1 | 65.06 | 0.68 | BAKER | 71.27 | 34.45 | 0.52 |
| T1074 | 1.5 | FM | 89.77 | 56.97 | 0.66 | laufer_ros | 60.61 | 16.68 | 0.36 |
| T1090 | 1.77 | FM | 89.02 | 60.23 | 0.68 | BAKER | 68.12 | 29.52 | 0.45 |
| T1064 | 2.04 | FM | 86.96 | 59.91 | 0.66 | PerezLab_Gators | 33.15 | 11.34 | 0.11 |
3.3. Models Predicted Correct Topologies and Outperformed Templates
The outstanding quality of CASP14 models on difficult targets allowed us to evaluate the topology-level performance of the prediction community using a score cutoff (GDT_TS > 50) as an estimation of correct fold. Several predictions were close to but did not achieve the topology cutoff. Manual inspection of the models was consistent with the chosen boundary. For example, the next best model for T1064 (Figure 1C, lower panel) achieved a GDT_TS just lower than 45 and failed to place the N-terminal strand. Borderline models from top groups (between 45 and 50 GDT_TS) tended to distort the overall topology in a subjectively acceptable way. For example, T1074 adopted an unusual lipocalin-like fold with a flattened barrel. Models from QUARK (GDT_TS 46.6) and Baker-Rosettaserver (GDT_TS 47.7) each correctly predicted the 8-stranded lipocalin-like β-meander but failed to close the barrel at one of the unusually flattened edges of the target.
Using the accepted score cutoff of GDT_TS 50 as a gauge of generally correct topology, all top performing groups performed reasonably well on CASP14 target EUs (Figure 2I). AlphaFold2 achieved the correct topology for almost all targets, while the Baker groups predicted the correct topology for almost 80% (Baker-experimental) and 74% (Baker) of the targets. The top performing server groups were around 63% (QUARK) and 61% (Zhang-server) of targets predicted with correct topology, while the combined set of models produced by groups other than AlphaFold2 achieved the correct topology for over 81% of the targets. Overall, the ability of the prediction community to establish the topology of difficult targets exceeded the percentage of targets with homologous known folds (Figure 2B, green bar), highlighting the utility of de novo prediction in CASP14.
As indicated in Figure 2I, almost half of the difficult targets in CASP14 had distantly related homologous templates (Kinch classification, this issue). However, top-scoring LGA_S templates among known folds for these difficult targets were often sets of SSEs from unrelated folds that displayed higher scores than the template homologs. As such, the top templates rarely achieved a similarity score above 50 LGA_S (Figure 2J, yellow circles). Exceptions with higher template homolog similarity scores (LGA_S >50) included several domains from viral targets with fast evolving sequence. Two of the viral targets were phage proteins (T1070-D1 and T1080) with top template homologs represented by phage tail fiber protein trimerization domains (4uxg_B, LGA_S 53.1 and 4uw8_C, LGA_S 57.1, respectively). Another two viral targets from SARS-CoV2 ORF8 protein (T1064) and tomato spotted wilt tospovirus glycoprotein precursor domain (T1038-D2) adopted immunoglobulin-related folds similar to their top template domains in the RL42 T cell receptor β chain (3skn_H, LGA_S 56.9) and the Interleukin-17 receptor C (6hg9_B, LGA_S 74.8) that function in host immunity and may have been acquired by the virus.
Regardless of target/template evolutionary relationships, prediction models from AlphaFold2 displayed much higher similarity to the target than the top template for all difficult targets (Figure 2J, blue circles). On average, the AlphaFold2 model scores improved on the top template scores by over 145%, with the highest improvement of over 500% for the target T1037, where the AlphaFold2 model achieved 92.7 LGA_S (the top target was 15.3 LGA_S). This model covered 99.2% of the large 404 residue-long target with 1.6 Å RMSD. Top models from the rest of the prediction community exceeded the similarity of top templates for 35 of the 38 difficult targets. They improved over the top templates by just over 95% on average, with the highest improvement (358%, from 20.8 to 83.4 LGA_S) by the FEIG-R2 group on T1096-D1. The server performance compared to top templates was also impressive. Servers provided better models than the top templates for 34 out of the 38 difficult targets, with the best (330% improvement to 66.2 LGA_S) from the Zhang-server on T1096-D1.
3.4. Server Performance on Difficult Targets
Two automated servers from the Zhang group (QUARK and Zhang-server) were among the top performing groups for difficult CASP14 targets. The Zhang group also submitted automated server models for Zhang-CEthreader, Zhang-TBM, and Zhang_Ab_Initio methods. The next best performing tFold-CaT server from Tencent AI Lab was accompanied by another two servers: tFold and tFold-IDT. The Baker group provided Baker-Rosettaserver, as well as the Robetta server from the previous round of CASP whose method was not modified so that we used it as a baseline for performance comparisons. Finally, the Yang group provided 3 server methods (Yang_server, Yang_FM and Yang_TBM) and the Cheng group provided 4 server methods (MULTICOM-Hybrid, MULTICOM-Dist, MULTICOM-Deep, and MULTICOM-Construct). The GDT_TS score distributions over all combined FM and FM/TBM targets for the Zhang servers that performed among the top groups were compared to distributions for the top-performing method from each of these multi-groups (Figure 3A). Each of these had a bimodal score distribution with a break around the cutoff chosen as indicating correct overall topology (45 GDT_TS). These distributions highlighted the ability of these server methods to provide models with correct topologies for some portion of difficult targets. The top performing QUARK and Zhang-servers distinguished themselves from the remaining servers by having the largest number of models scoring in the high GDT_TS peak. While the Baker-Rosettaserver provided one of the best-scoring models above 90 GDT_TS, first models for a majority of the targets fell in the lower-scoring peak. Similar trends in peak distributions existed for the remaining servers (Figure 3A).
Figure 3. Server Performance.
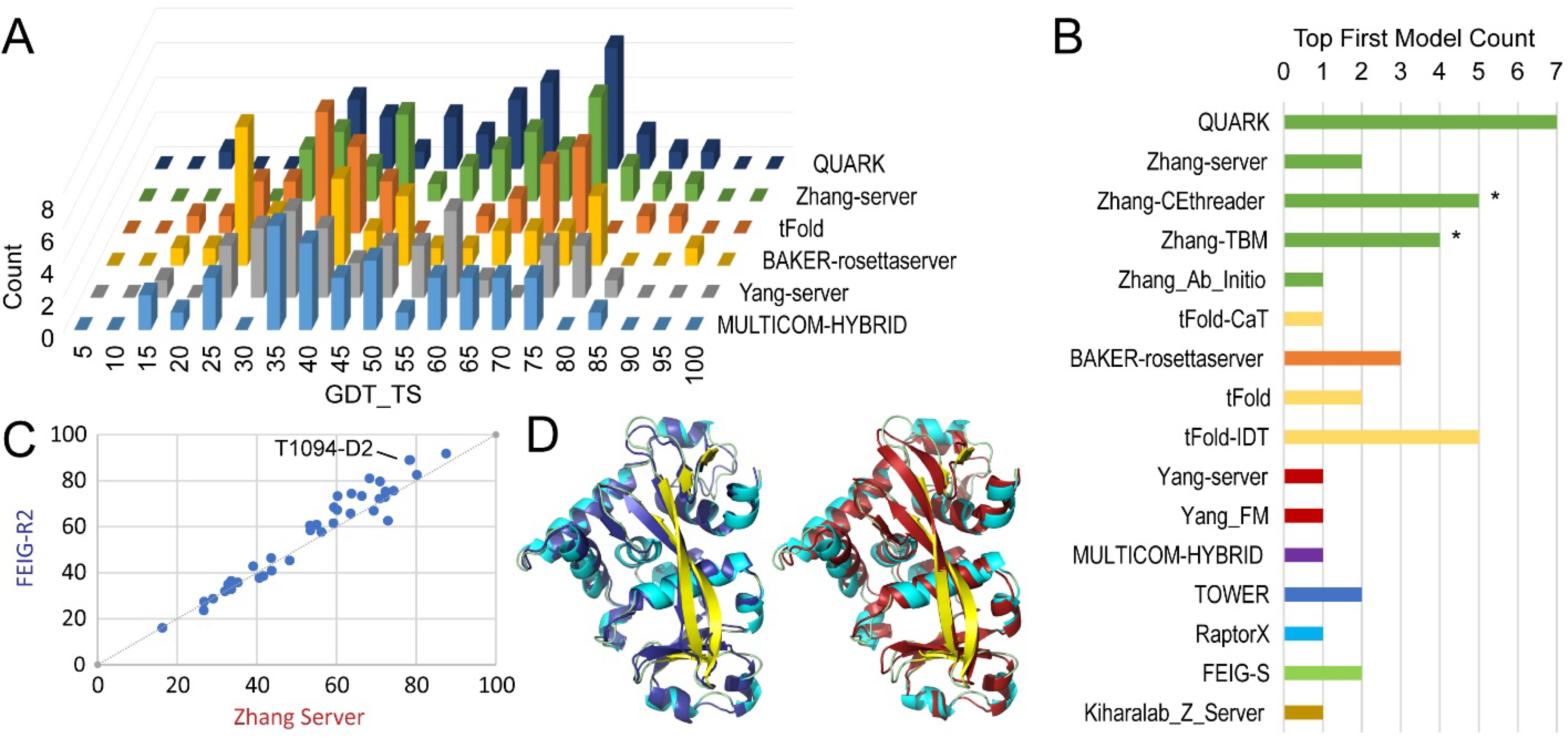
A) Histogram of GDT_TS scores (X-axis) across first models for all FM and FM/TBM target EUs for each server (Z-axis, labeled on the right). B) Bar graph represents count of top first models among server predictions for all FM and FM/TBM target EUs. Bars are colored according to independent predictor groups, who could register multiple methods and an asterisk ‘*’ marks a pair of models tied for first. C) GDT_TS score of Zhang-server models (X-axis) above GDT_TS 50 were improved by FEIG-R2 (Y-axis) refinement. An example target EU with improved model quality is labeled. D) T1094-D2 is colored according to the SSE: strand (yellow) and helix (cyan) and superimposed with the FEIG-R2 model in blue (left panel) or with Zhang-server model in red (right panel).
While the shifts in score distributions towards the higher peak are dictated by the overall server performance, many of the methods provided correct fold predictions for some of the targets. To better understand the ability of servers to provide high quality models, we counted the number of rank1 (by GDT_TS among servers only) first models for each server method (Figure 3B). Using the scores for the server rank1 models, completely automated predictions achieved the correct topology (GDT_TS >50) for 25 of 38 (66%) targets, with an average GDT_TS score of 61 for the top ranked models. While the QUARK and Zhang-server were both among the top groups overall, the QUARK server provided seven top-scoring models, the most among all servers, followed by Zhang-CEthreader and tFOLD-IDT at five models each. If each multi-method group could combine their methods by selecting top models, the Zhang servers would outperform with a count of 18, followed by tFOLD with a count of 8.
Interestingly, one of the top-performing manual groups (FEIG-R2) refined Zhang-server models. Comparison of the refined FEIG-R2 models to the initial Zhang-server models (Figure 3C) highlighted the ability to improve models with correct overall topology (GDT_TS above 50). The FEIG refinement improved 21 out of 23 Zhang-server models that started above 50 GDT_TS with an average improvement of 5 GDT_TS. As an example of improved server predictions by the FEIG refinement, the FEIG-R2 model for T1094-D2 superimposes better with the target (GDT_TS 89.0, 1.2 Å RMSD over the whole structure, Figure 3D left) than the Zhang-server model does (GDT_TS 78.3, 1.8 Å RMSD over the whole structure, Figure 3D right). Refinement of this target approached the performance of AlphaFold2 (within 1.2 GDT_TS), improved the backbone positions of SSEs by approximately 1 Å and improved loops by 2 Å in local accuracy of model-target Cα to Cα distances calculated from a sequence dependent-superposition. This model improvement was also reflected in the MolProbity scores, where the average overall GDT_TS performance on all difficult targets (measured by Z-score sums) correlates (R2 0.7) with the MolProbity scores averaged over all difficult targets for each of the Zhang manual and server groups and FEIG-R2 (data not shown). In this correlation plot, the refinement method achieved the highest Z-score sum average and the lowest MolProbity score, followed by QUARK, Zhang-server, and Zhang. Thus, despite the overall impressive performance of the Zhang group automated servers, room for improvement exists in refinement and selection of server models among all the group’s prediction methods.
3.5. Prediction Difficulty Remains for Flexible Regions and Non-Globular Assemblies
AlphaFold2 models did not rank first for only two of the difficult CASP14 targets (T1029 and T0147s2-D3). Each one of these represented a broad category of target types that remained difficult for the current state-of-the-art in protein structure prediction: flexibility and obligate oligomeric assembly. T1029 was one of two NMR structures with generally poor-quality models. The best performing model for T1029 (Figure 4A left) was from the kiharalab-Z-server. The outperformance of this model compared to the prediction from AlphaFold2 (rank 3 in figure 4A right) was due to its overall better placement of the β-sheet with respect to two N-terminal α-helices. None of the top-performing first models positioned the N-terminal end, the C-terminal end, or the C-terminal α-helix (red positions in Figure 4A right). The other difficult NMR target (T1027) was represented by a loose ensemble that required removal of several flexible regions for assessment (kinch classification paper, this issue). The relatively low top20 average server performance (38.8 GDT_TS) on this target signified the difficult nature of flexible regions to predict an assess in CASP.
Figure 4. Difficult Targets and Local quality assessment.
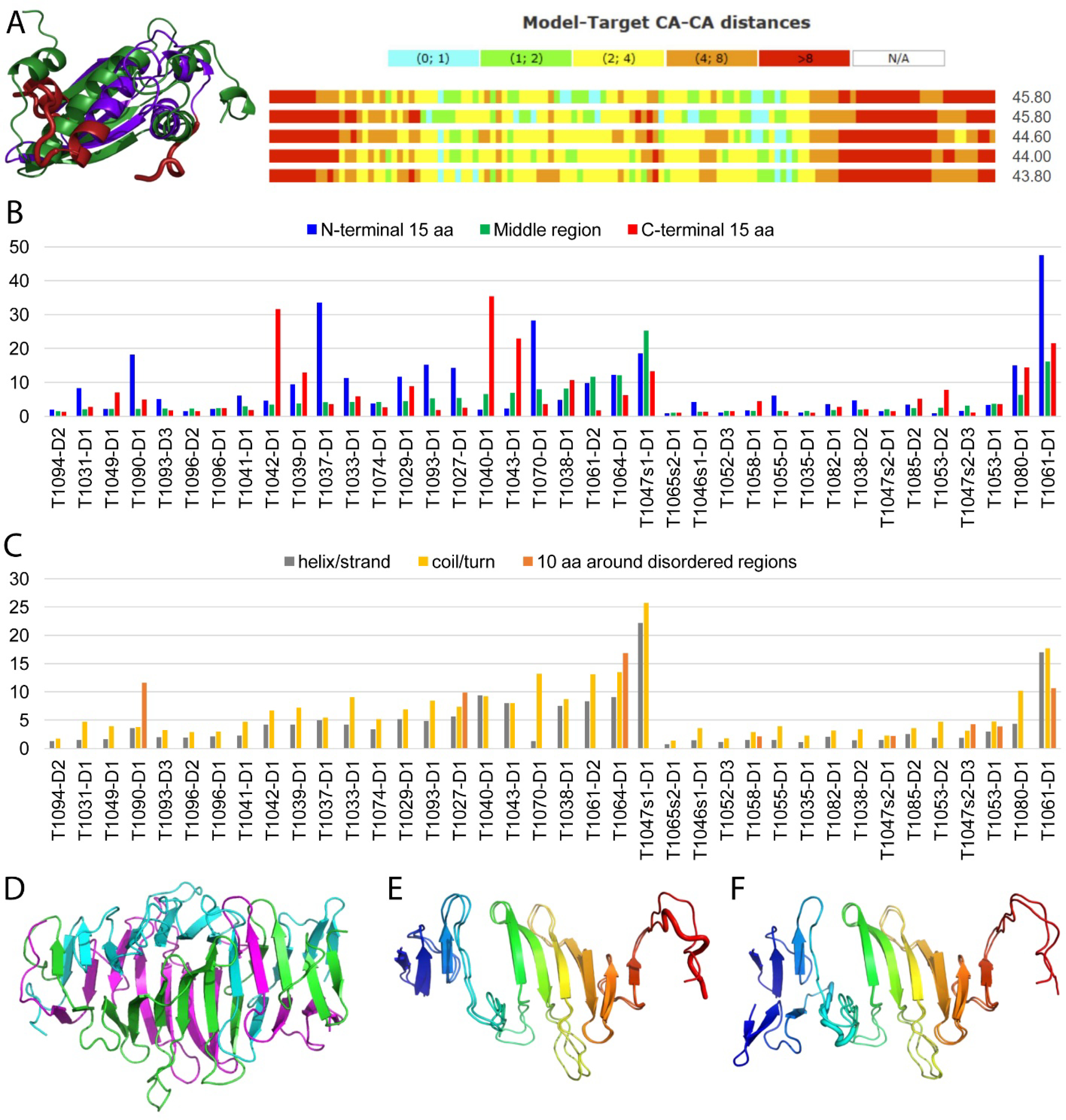
A) The top model for T1029 from Kiharalab_Z_Server (GDT_TS 45.8, slightly outperformed AlphaFold2) is superimposed with the target on the left. Modeled residues within 8A are colored blue, with the rest colored red. Local T1029 accuracy (measured by model-target Cα-Cα distance) of top five methods ranked by GDT-TS scores of first models (right panel, with GDT-TS scores shown to the right). B) Local quality scores (see Materials and methods) for the N-terminal region (N-terminal 15aa), the C-terminal region (C-terminal 15aa), and the rest of the protein (Middle region) for each FM (left) and FM/TBM targets (right). Targets from each class are ranked according to the local quality scores of the Middle region. C). Local quality scores for regions with regular secondary structures (helix/strand), loops (coil/turn) and regions around disordered or inserted segments (10aa around disordered regions) are depicted for each target and sorted as in panel B. D) Oligomeric assembly of T1080 trimer, with chains colored in cyan, magneta, and green. E) Superposition of T1080 monomer with AlphaFold2 first model is colored in rainbow from the N- to C- terminus. An incorrect turn is in thick ribbon. F) Superposition of T1080 monomer with AlphaFold2 model4, with incorrect turn (thick ribbon) positioning the small N-terminal domain in the register of another monomer from the assembly.
To examine the difficulty of CASP14 structure prediction methods on flexible regions, we manually inspected local accuracy plots and model-target superpositions, finding common difficulties in target regions at the N- and C-terminal ends, in loops, and in regions surrounding disordered segments or inserted domains. To illustrate these observations, first, we zoomed in on 15 termini residues in the models. The modeling quality of these regions was quantified by average residue Cα-Cα distances in global sequence dependent LGA model-target structural alignments for the top 50 models (Figure 4B). In 15 out of 23 FM targets and in 6 out of 15 FM/TBM targets, the N-terminal region was predicted with less local accuracy (higher Cα to Cα distances). For example, the N-terminus of T1090-D1 included a helical extension that formed crystal packing contacts and was connected to the rest of the fold by a disordered loop. This region in T1090-D1 was not predicted by any of the groups, including AlphaFold2. Similarly, the N-terminus of T1037 formed an elongated β-hairpin that was connected to the rest of the structure by a domain insertion. First models from AlphaFold2 and Baker-experimental were the only two that positioned the hairpin correctly within the context of the rest of the target. For the C-terminal region, 10 out of 23 FM and 6 out of 15 FM/TBM targets had less accurate predictions on average. T1042-D1 also included a C-terminal helical region that was connected to the rest of the target by an inserted domain. AlphaFold2 was the only group to correctly position all but the C-terminal loop of this target region.
We also examined the performance differences between the residues in helices or strands and the residues in loops. The average Cα-Cα distances were calculated for these regions and then compared to those surrounding disordered segments or domain insertions (Figure 4C). The average local accuracy for the top 50 predictions on loops was worse than on helices/strands for all but one of the FM and FM/TBM targets. The T1040-D1 fragment from a larger structure was the single exception to this observation due to the presence of an extended C-terminal, mainly helical segment that did not make any local contacts with the rest of the domain in the target, but instead interacted with other domains from the larger structure. AlphaFold2 was the only group to position this extended segment correctly (representing one third of the target) relative to the rest of the structure. One outlier target (T1070-D1) included predictions with much lower performance on loops with respect to other SSEs. The N-terminal portion of this target, which encompassed over half of its total length, formed structurally defined β-strands only in the context of a trimeric assembly and was difficult for all groups to predict. AlphaFold2 correctly predicted the relative placement of the C-terminal half of this N-terminal segment (~20 residues), while the rest of the groups missed the entire segment (~45 residues).
In addition to flexibility, prediction difficulty on another category of CASP14 targets was represented by relatively poor AlphaFold2 performance on T0147s2-D3 from an unusual elongated heterodimeric ring assembly (structure not yet published, see figure 2A for GDT_TS scores). T1047 adopted a much higher order elongated heterodimeric complex assembly (from T1047s1 and T1047s2). T1047s1 was one of the most difficult targets for the structure prediction community (Average Top20 GDT_TS 33.9), with the first model from AlphaFold2 adopting the correct topology (GDT_TS 50.5), but incorrectly predicting the position of a swapped N-terminal loop that helps form the assembly. Comparing the AlphaFold2 model to a target with a “non-swapped” N-terminal loop reshuffled the score (GDT_TS 59.7), since the prediction did not manage to correctly distinguish the inter-chain from the intra-chain distances.
CASP14 targets also included structures formed by non-globular assemblies of obligate interactions (T1070 and T1080). These targets adopted phage tail fiber protein trimerization domain folds with interdigitated β-strands forming long triangular sheets in the trimer (Figure 4D). For such targets distinguishing between inter- and intra-residue distances was a challenge for top-performing distance-based deep learning methods. Many phage tail trimerization structures existed among known folds that could have served as templates or for training (top template LGA_S 57.1 for T1080). AlphaFold2 models for T1080 were the only predictions that improved this top template (Figure 2J). Their first model achieved the correct overall topology (Figure 4E), placing a turn between two single interdigitating β-strands, followed by a β-meander (GDT_TS 82.7). Yet the turn that placed the final interdigitating β-strand was incorrect for the trimer. On the other hand, their model 3 correctly placed the final β-strand (Figure 4F), but the turn in between the first two interdigitating strands was incorrect (GDT_TS 67.9), shifting the register of the N-terminus to the sheet from another chain.
3.6. Insights into Prediction Methodology
PCA analysis of GDT-TS scores on all FM and FM/TBM targets was used to visualize the variance of top performing CASP14 groups in terms of their overall performance level and group type (Figure 5A, rank1 group, other top manual or server groups, and a baseline server used in CASP13). The plot of the top two components (PC1 and PC2) explained 40.6% and 13.7% of the variance, respectively. The most striking separation of groups was in PC1 between the top-performing method AlphaFold2 (purple diamond) and other methods, with the baseline control (red filled circle) on the other end of the PC1 axis. This PC1 axis likely explained the overall group GDT_TS performance, while the distribution of groups along PC2 reflected their performance on different targets. AlphaFold2 fell far outside the 95% confidence outline (blue ellipse) of other top performing manual methods. One other manual method that fell just outside the ellipse was Baker, while all the CASP14 server methods clustered within their 95% confidence ellipse (blue).
Figure 5. PCA and Heatmap clusters.
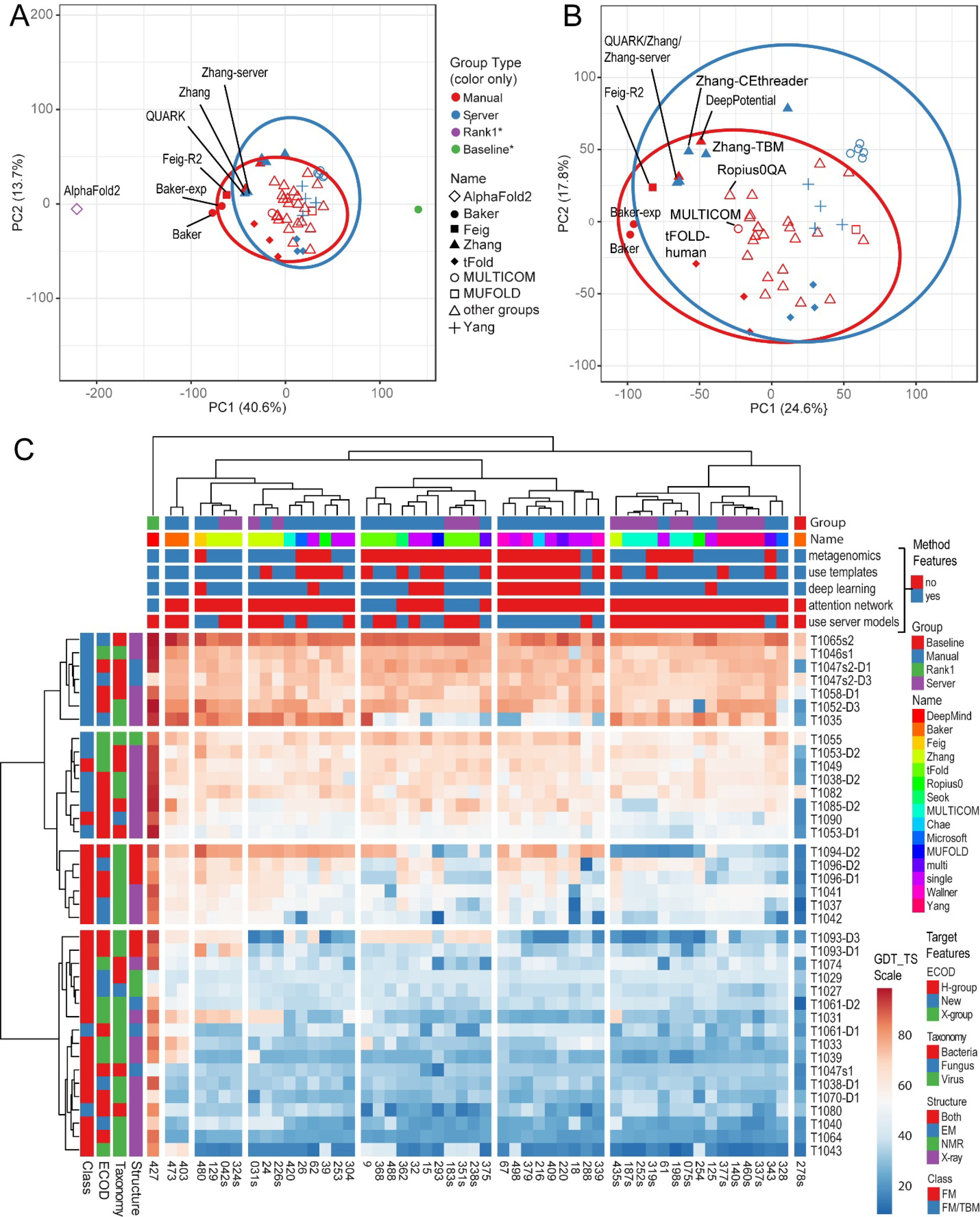
A) Scatter of PC1 and PC2 variance from PCA clustering of methods (represented by markers listed to the right) using GDT_TS performance scores on all FM and FM/TBM targets. The two ellipses represent 95% confidence for manual methods cluster (red) and server method cluster (blue). Top-performing groups are labeled. B) Scatterplot of PC1 and PC2 variance from PCA clustering of methods (markers to left) using GDT_TS and QCS performance scores on all FM and FM/TBM targets excluding AlphaFold2 and the baseline server (markers with * for both). C) GDT_TS Heatmap (from low scores in blue to high scores in red) of methods in the columns (features colored on top, only group numbers provided for brevity) and targets in rows (features on the left, with names labeled to the right). Method features are retrieved from abstracts submitted by participants. Columns and rows are clustered according to Euclidean distance with Ward linkage.
The top-performing manual methods (labeled red points) performed better than the server methods (blue points) as seen in their shift in PC1 away from the baseline server. The two methods from the Baker group exhibited similar performance, while methods from the Zhang group separated into two clusters. The top-performing Zhang-server, QUARK, and Zhang (manual) formed one cluster and the other four methods (Zhang-TBM, Zhang-CEthreader, DeepPotential, and Zhang-AbInitio) formed another cluster that shifted towards the baseline server along PC1 and upwards along PC2, suggesting these methods displayed variable performance on different CASP14 targets. Different methods submitted by the tFold group, the Yang group, and the MULTICOM servers also formed relatively tight clusters that exhibited little variance along PC1. The tFold and Yang group methods tended to distribute more along the PC2 axis that reflected target groups, suggesting that they might benefit from being combined.
The top performing AlphaFold2 group (and the baseline) fell outside the confidence clusters for the other manual and server groups. Excluding these outliers from the performance analysis might better separate the performance of the remaining methods. To assess the topology-level performance of the remaining groups, PCA of GDT_TS and QCS scores was performed (Figure 5B). While the PC1 variance was lower (24.6%), the distribution of group performance was similar, and the components probably reflected GDT_TS/QCS performance (PC1) and target performance (PC2). The manual groups tended to outperform the servers, and the two Baker groups clustered outside the 95% confidence level of the servers.
To better understand the variance in target performance displayed along PC2 as well as the differences in methodologies employed by the CASP14 groups, we clustered the methods and targets by their GDT_TS scores using the heatmap tool in ClustVis10. According to abstracts provided by the predictors, their methods essentially differed in terms of algorithms and data sources. The group methods were classified by these features, including the use of metagenomics sources for additional sequence information, the use of templates for structure information, the general use of deep learning, or the more specific use of attention networks, and the incorporation of server predictions. Figure 5B highlights the methods in columns across the top of the heatmap whose clusters were ordered by mean overall GDT_TS performance. Top-performing standalone methods including AlphaFold2 (cluster 1, numbered from high to low performance), the top two Baker methods (cluster2), and servers from the Zhang group (cluster3) each applied deep learning, used sequence information from metagenomics, and used structure information from available templates. AlphaFold2 prediction quality was distinct from the rest of the methods, with their key development being the use of an attention network for deep learning that directly outputs structure coordinates. Other deep-learning methods predicted contacts and/or distances that were used for subsequent model building and refinement.
Compared to the top-performing standalone methods, one large cluster of manual groups (cluster 6) ranked and refined server models (with one exception). While these groups performed better than a large cluster of groups (cluster 7) that included many of the servers they could have employed, they were generally worse than the top performing servers. The best method in this category of using server models was FEIG-R2 (in cluster3), which was able to improve the models from the Zhang-server (in the same cluster) it used for refinement. The MULTICOM human group (cluster 4), which incorporated all server models in their method, performed much better than their individual servers (cluster 7), highlighting their relative success in choosing among server models. Similarly, the three manual group methods from tFold performed relatively better than their three servers although they all clustered together (cluster 5).
The CASP14 targets with four assigned features, including class, ECOD16 level, taxonomy, and structure determination method, were clustered into four major groups based on the GDT_TS heatmap (Figure 5B). The top cluster included only FM/TBM targets whose topologies were predicted by all top50 groups, including half of the targets being predicted by the baseline server. The next cluster was primarily made up of FM/TBM targets, but it also included two that were classified as FM (T1049 and T1090). Most of the top50 methods consistently predicted the correct topology of these targets (GDT_TS scores in shades of red), but with relatively lower GDT_TS scores than in the first target cluster. The third target cluster included FM targets that were predicted with a range of performances across the top50 groups. This group of targets included three fragments (T1037, T1041, and T1042) from the large phage DNA-dependent RNA polymerase, as well as domains from two subunits of another phage polymerase (T1094-D2, T1096-D1, and T1095-D2). These domains tended to distinguish the poor performance of groups from cluster 5 (did not use metagenomic sequences) and cluster 7 (mainly servers) as well as the intermediate performance on some targets for groups in cluster 6 (who chose among server models). The fourth target group was made up of mostly FM targets and proved to be difficult for all methods except for AlphaFold2, with the Baker manual groups predicting the topology for a subset of the targets.
4. CONCLUSIONS
Tertiary structure prediction models produced by AlphaFold2 consistently achieved high quality across all difficult FM and FM/TBM targets in CASP14 (Figure 2A). In fact, they excelled (GDT_TS >70) on over 86% of the difficult targets. For the first time in CASP history, high quality models extended to large structures and the performance of AlphaFold2 did not depend on target size (Figure 2D). In fact, one of the best-scoring models from this group was for an unusual 405 residue long protein kinase-like domain (T1053-D1) from a legionella T4SS effector. While we split this target due to the lack of performance by the rest of the prediction community, the AlphaFold2 model was also high quality (GDT_TS 89.4) for the combined domains (T1053-D12), which represented a much larger target (576 residues). Although their model achieved impressive scores: 1.3 Å RMSD over 90% of the two-domain structure, the active site lacked predicted ADP binding site correlation by FTMap (function assessment paper, this issue), suggesting that implicit treatment of substrates might need to be replaced by explicit modeling of small molecules in future rounds of CASP.
Excluding AlphaFold2, the structure prediction community predicted the correct topology for 81% of the difficult FM and FM/TBM targets. The top performing methods included two from the Baker group, a refinement method from the Feig group (that refined models from one of the top performing servers), two automated servers from the Zhang group (QUARK and Zhang-server), and a manual Zhang group (Figure 1A). Models from these top-performing groups achieved relatively high overall quality (average 66.5 GDT_TS for top first models), predicted the correct topology for most of the targets (Figure 2I), and beat top scoring templates (Figure 2J). While some automated servers performed among the top groups, potential exists for their improvement. The ability of individual groups to submit multiple methods not only provided more opportunities to predict the correct target structures, but also generated competition among the multiple submitted servers from the same groups. If individual groups could successfully select their top server models among predictions from all of their methods, they could improve on the number of rank 1 predictions of difficult targets. Additionally, a manual group used refinement to improve those Zhang-server models that started with the correct fold. Such an improvement suggests the servers could incorporate similar refinement strategies for added performance.
Difficulties for the current state-of-the-art structure prediction methods remain for some multidomain targets (Schaeffer, this issue), non-globular target assemblies, and flexible regions (Figure 4). Assemblies of non-globular protein chains provided a particular challenge for contact distance based deep learning methods. Difficulties on such targets could have arisen from a lack of representation of non-globular domains among existing structures. Examples of extended regions of folds that are stabilized by obligate chain or domain interactions are in their infancy and are now being provided by atomic resolution EM structure determination methods that do not require crystallization and can solve structures for much larger proteins and their complexes. Finally, the symmetry arising from CASP14 target assemblies (in T1070 and T1080 homotrimers and higher order heteromers of T1047) presumably added an additional challenge of distinguishing between inter- and intra-chain distances, whether the assemblies were explicitly considered or implied through deep learning.
Flexible structure targets provided another general difficulty for the prediction community (Figure 4 A–C), although some AlphaFold2 models could improve the termini and loop predictions for select targets. Poor modeling of flexible regions may have resulted from targets adopting multiple conformations that depended on either solution/crystal conditions or interactions with other domains/proteins. Other possible explanations could have reflected technical difficulties for deep learning methods. The amino acid sequences of protein termini are often more divergent than regions in the core, which could lead to missing regions or mis-alignments in the sequence profiles that represent crucial components of many structure modeling methods. We also observed that poor alignment quality tends to occur in loop/turn regions and regions surrounding disordered segments (those that are not observed in X-ray structures or adopt different conformations in NMR structures) or in segments that connect inserted domains (in split target EUs) in contrast to regions adopting regular secondary structures. Such flexibility in protein structure was a challenge for all aspects of the CASP experiment, from classifying target EUs (kinch classification, this issue), to predicting target structures, and assessing the predictions. In fact, the difficulty for some high quality AlphaFold2 models in terms of flexible sequence regions was approaching a theoretical boundary of model accuracy that would require considering chain interactions, such as those formed in crystal packing (i.e. T1064).
The clear state-of-the-art in protein structure prediction methodology for CASP14 was provided by AlphaFold2, who modified their successful contact distance-based deep learning method from CASP13. The AlphaFold2 group replaced their previous convolutional neural network architecture with an attention-based neural network that outputs structure coordinates directly. A few other groups (i.e. tFold) used attention networks, but they did not use metagenomics sequence data, and their neural network output predicted contacts/distances instead of structure coordinates. The rest of the structure prediction community appeared to catch up with the previous performance of AlphaFold2 (AlphaFold from CASP13) after publication of the initial version of their method17. These groups included automated servers that provide publicly available structure prediction methods to the scientific community. If the past is indicative of the future, we might expect to access to open-source protein structure prediction implementations that rival the performance of AlphaFold2 in the future.
ACKNOWLEDGMENTS
We thank the CASP organizers for their invitation for us to participate in CASP14, the protein crystallographers, NMR spectroscopists and cryo-EM scientists that contributed targets and fellow assessors for helpful discussion about topology modeling performance. This research was supported by the US National Institute of General Medical Sciences (NIGMS/NIH) grants R01GM100482 (AK) and R35GM127390 (NVG) and the Welch foundation grant I-1505 (NVG).
REFERENCES
- 1.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins. 1995;23(3):ii–v. [DOI] [PubMed] [Google Scholar]
- 2.Abriata LA, Kinch LN, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. Definition and classification of evaluation units for tertiary structure prediction in CASP12 facilitated through semi-automated metrics. Proteins. 2018;86 Suppl 1:16–26. [DOI] [PubMed] [Google Scholar]
- 3.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins. 2019;87(12):1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abriata LA, Tamo GE, Dal Peraro M. A further leap of improvement in tertiary structure prediction in CASP13 prompts new routes for future assessments. Proteins. 2019;87(12):1100–1112. [DOI] [PubMed] [Google Scholar]
- 5.Kinch LN, Li W, Monastyrskyy B, Kryshtafovych A, Grishin NV. Evaluation of free modeling targets in CASP11 and ROLL. Proteins. 2016;84 Suppl 1:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryshtafovych A, Monastyrskyy B, Fidelis K. CASP11 statistics and the prediction center evaluation system. Proteins. 2016;84 Suppl 1:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. Proteins. 1999;Suppl 3:22–29. [DOI] [PubMed] [Google Scholar]
- 8.Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31(13):3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32(Web Server issue):W500–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abriata LA, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins. 2018;86 Suppl 1:97–112. [DOI] [PubMed] [Google Scholar]
- 12.Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tai CH, Bai H, Taylor TJ, Lee B. Assessment of template-free modeling in CASP10 and ROLL. Proteins. 2014;82 Suppl 2:57–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olechnovic K, Kulberkyte E, Venclovas C. CAD-score: a new contact area difference-based function for evaluation of protein structural models. Proteins. 2013;81(1):149–162. [DOI] [PubMed] [Google Scholar]
- 15.Keedy DA, Williams CJ, Headd JJ, et al. The other 90% of the protein: assessment beyond the Calphas for CASP8 template-based and high-accuracy models. Proteins. 2009;77 Suppl 9:29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng H, Schaeffer RD, Liao Y, et al. ECOD: an evolutionary classification of protein domains. PLoS Comput Biol. 2014;10(12):e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577(7792):706–710. [DOI] [PubMed] [Google Scholar]