

Summary
The latest advances in next-generation sequencing studies and transcriptomic profiling over the past decade have highlighted a surprising frequency of genes regulated by RNA processing mechanisms in the immune system. In particular, two control steps in mRNA maturation, namely alternative splicing and alternative polyadenylation, are now recognized to occur in the vast majority of human genes. Both have the potential to alter the identity of the encoded protein, as well as control protein abundance or even protein localization or association with other factors. In this review we will provide a summary of the general mechanisms by which alternative splicing (AS) and alternative polyadenylation (APA) occur, their regulation within cells of the immune system, and their impact on immunobiology. In particular, we will focus on how control of apoptosis by AS and APA is used to tune cell fate during an immune response.
Keywords: Alternative Splicing, Alternative Polyadenylation, Apoptosis, T cell activation
The central dogma of biology states that DNA is transcribed into RNA, and RNA is translated into protein. At the RNA level, there are a multitude of processing events involved in the maturation of a nascent transcript (pre-mRNA) to a final protein-encoding messenger mRNA, each of which has the potential to expand the protein repertoire to ultimately impact cell activity1–5. Alternative splicing (AS) occurs during RNA processing and has the potential of altering protein identity and expression and therefore cellular function6–9 (Fig. 1,2). Similarly, alternative polyadenylation (APA) impacts the length of the final mRNA, with the potential to truncate the encoded protein or alter the presence of regulatory elements in the 3’ untranslated region (3’ UTR) to control protein expression10,11 (Fig. 2). In recent years there has been a growing body of literature revealing the breadth to which both AS and APA are controlled in the immune system, an increased understanding of the mechanisms driving these processes, and a recognition of the extent to which AS and APA impact the response of lymphocytes to immune challenge (see12,13 and references below). In this review, we summarize the general mechanisms driving AS and APA and provide both classic and recent examples of how AS and APA are regulated in immune cells. Moreover, as a model for the functional consequence of AS and APA in immunology, we focus on AS and APA events in genes encoding apoptotic signaling proteins. Immune cells are especially interesting in the context of apoptosis as dysregulated apoptosis in immune cells plays a large role in autoimmunity, where prolonged and unregulated stimulation has damaging effects to the host14,15. Thus, the dynamic AS and APA changes that occur in apoptotic related genes in activated immune cells, combined with the reliance on apoptosis to downregulate the activity of immune cells following immune perturbance, provides an important framework for understanding the critical nature of mRNA processing events such as AS and APA in tuning the reactivity and homeostasis of the immune system.
Figure 1. An illustration of alternative splicing and polyadenylation patterns.
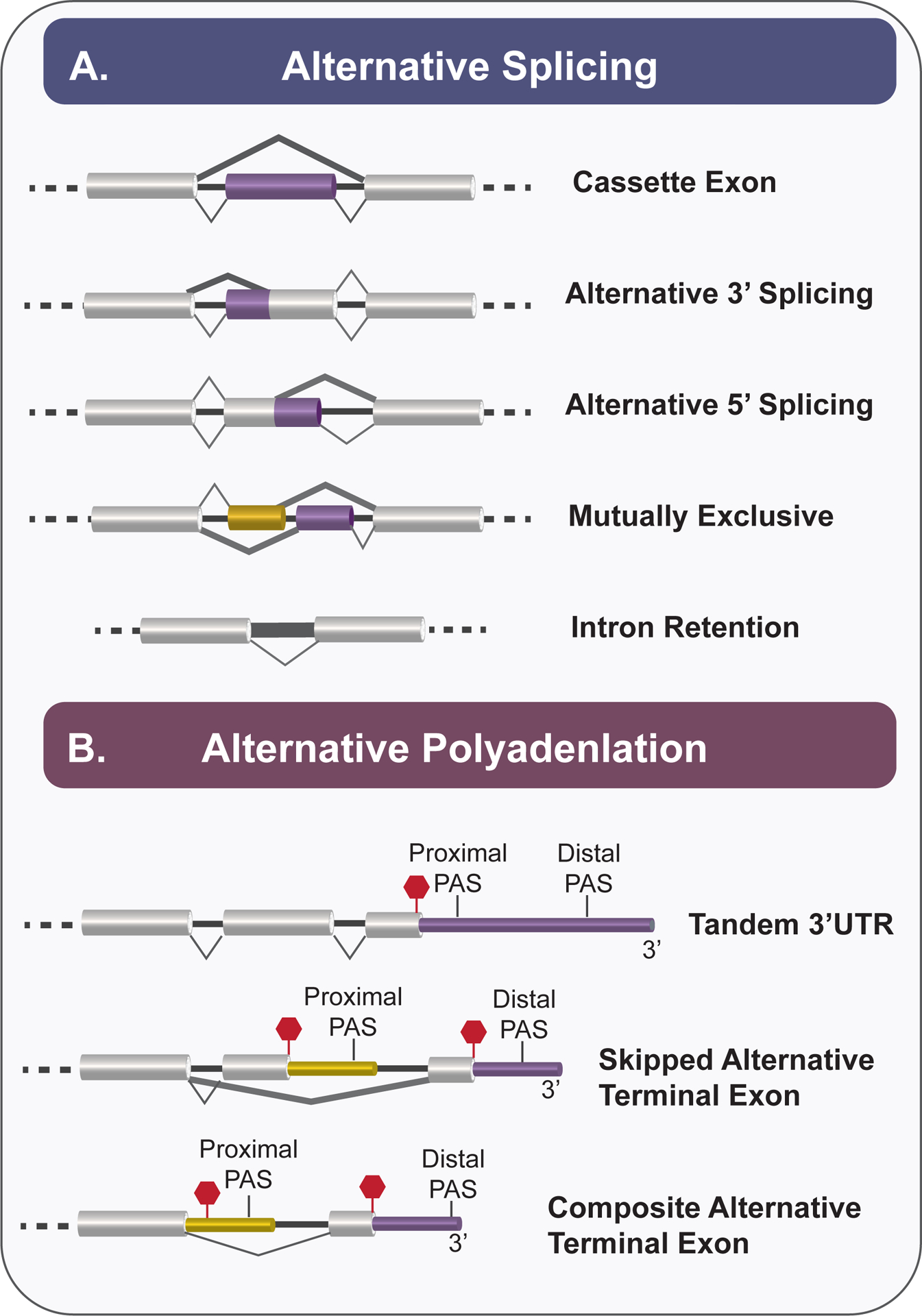
A. Schematic of alternative splicing patterns including: cassette exon, alternative 5’ or 3’ splice site selection, mutually exclusive exons, and intron retention. Constitutive exons are depicted in grey and alternatively splicing exons are depicted in purple or yellow. Thick lines represent alternatives to the canonical (thin line) splicing patterns. B. Examples of alternative polyadenylation patterns include tandem 3’UTR, skipped alternative terminal exon, and composite alternative terminal exon. While the proximal and distal PAS are in proximity to each other within tandem 3’UTRs, proximal PAS may be located within upstream sequences and its inclusion may be subjected to regulation like alternative splicing. Thick lines represent alternatives to the canonical (thin line) splicing patterns. Red hexagon represents translation stop codon. Purple and yellow regions indicate 3’UTRs.
Figure 2. Constitutive splicing and polyadenylation are regulated by RBPs that give rise to alternative splicing and alternative polyadenylation products.
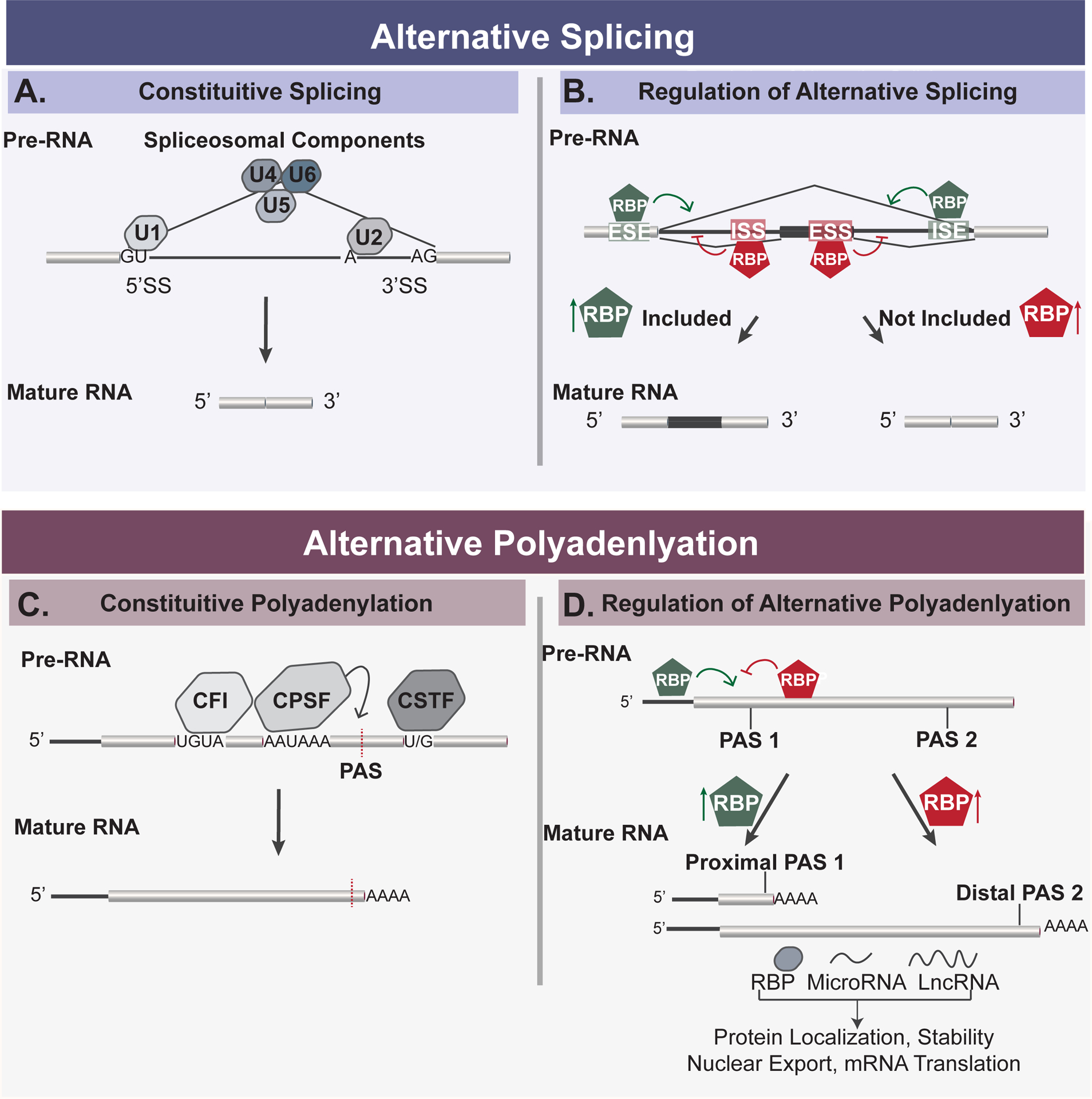
A. Diagram of constitutive splicing. Spliceosomal components, or snRNPs are depicted as U1–U6 and bind to 5’ and 3’ splice sites. snRNPs are essential for constitutive splicing of introns of pre-RNA to produce mature RNA. B. The regulation of alternative splicing is influenced by RNA binding proteins (RBPs) that may either enhance (green hexagon) or repress (red hexagon) exon inclusion. RBPs enhance splicing by binding to exon splicing enhancer (ESE) sequences or intron splicing enhancer (ISE) sequences, or repress splicing by binding to exon splicing silencer (ESS) sequences or intron splicing silencer (ISS) sequences. Due to the combinatorial regulation of RBPs, exon inclusion is mediated by the increased or decreased presence of RBPs, as depicted by arrows. C. Cleavage at the polyadenylation site (PAS) of pre-RNA is mediated by a large multi-component complex containing a core CPSF, CFI, CstF subcomplex. D. The regulation of alternative polyadenylation is regulated by the activity of RBPs that may either enhance (green hexagon) or repress (red hexagon) the usage of PAS sites.
Alternative splicing
Alternative splicing (AS) is a co-transcriptional process that occurs during RNA processing and gives rise to unique RNA transcripts in the cell6,16. The most common types of AS patterns include whole exons that are selectively included or removed from the mature RNA transcript, otherwise known as cassette exons17 (Fig. 1). Other known patterns of AS events are described in Figure 1, all of which alter the identity of the resulting mRNA. Remarkably, genome-wide studies suggests that approximately 95% of human genes undergo AS to yield at least one, and often several, alternative isoforms17,18. Because AS increases the number of unique mRNA transcripts in a cell, AS increases proteome diversity2,5,6. Protein isoforms that arise from alternative splicing have been shown to exhibit changes in enzymatic activity, localization, or stability, and thus to impact cell activity6,7. While the impact of every alternative splicing event in each gene has yet to be characterized, there are numerous studies that exemplify the necessity of regulated AS events for the maintenance of homeostatic cell activity and demonstrate how the dysregulation of AS can lead to disease19–21. In addition, beyond single examples of AS events impacting cell activity, concerted changes in AS have been observed in genome-wide studies and shown to function cooperatively to govern global cell states and tissue maintenance22–25. Therefore, understanding how AS is regulated and coordinated in cells is critical to understanding many physiologic responses, including the response of immune cells to pathogen challenges.
Mechanisms of Alternative Splicing
The spliceosome is a large macromolecular complex that is essential for constitutive splicing of RNA and can be regulated to give rise to AS products16,26. The spliceosome is made up of several small nuclear ribonucleoparticles (U1, U2, U4–U6 snRNPs), each of which is comprised of a small RNA and multiple proteins (Fig. 2a). These snRNPs sequentially bind to the 5’ and 3’ splice sites and branch point sequences on pre-mRNA to direct and catalyze the cleavage of introns and the ligation of exons during constitutive splicing6,16,26. The location and efficiency of splicing carried out by the spliceosome can be regulated by a variety of factors including splice site strength, transcription elongation rates, single nucleotide polymorphisms (SNPs), and RNA binding protein (RBP) activity6,16,26. In this review, we focus mostly on the impact of RBPs on alternative splicing, as RBPs most often regulate condition-specific splicing changes in cells by binding to intronic or exon sequences near splice sites on an immature RNA transcript to block or enhance spliceosomal association16 (Fig. 2b). Sequences that recruit RBPs to enhance splicing are called exonic splicing enhancers (ESE) and intronic splicing enhancers (ISE), while sequences that inhibit splicing are called exonic splicing silencers (ESS) and intronic splicing silencers (ISS)16. Notably, the same sequence motif can function as any of these activities, as the definition is based on the location of the sequence and the ultimate impact on splicing, which results from both the RBP that is recruited and the context of the surrounding sequences (see below). Changes in expression, post-translational modifications, and/or localization of RBPs between cellular states alter the balance of activities on these enhancer and silencer sequences to ultimately alter AS patterns16,27 (Fig. 2b).
RBP Regulation of Alternative Splicing
Splicing regulatory RBPs in humans can largely be divided into three main classes – SR proteins, hnRNP proteins, and hnRNP-like proteins (Fig. 3). Each class of proteins typically contain RNA binding motifs and one or more functional domains that mediate protein-protein interactions. SR proteins are defined by containing at least one RRM (RNA Recognition Motif)-type RNA binding domain as well as a domain comprised of serine-arginine (SR) dipeptides that facilitates homotypic interactions6,28, while hnRNP and hnRNP-like proteins are more heterogeneous in their domain composition and can contain both hnRNPK-homology (KH) (not shown) or arginine-glycine (RGG) RNA binding motifs, as well as RRM motifs. Traditional hnRNP proteins are defined based on their initial co-purification and discovery and are typically more abundant and/or ubiquitously expressed than hnRNP-like proteins, which are RBPs that have hnRNP-like domains but have been isolated independently29,30. All of these three classes of RBPs typically bind to short sequence motifs with a range of promiscuity and redundancy6,31,32. This allows transcripts to simultaneous ensure the ability to bind a necessary subset of RBPs while still adhering to coding or other regulatory constraints.
Figure 3. RNA binding proteins characterized to regulate alternative splicing and alternative polyadenylation.
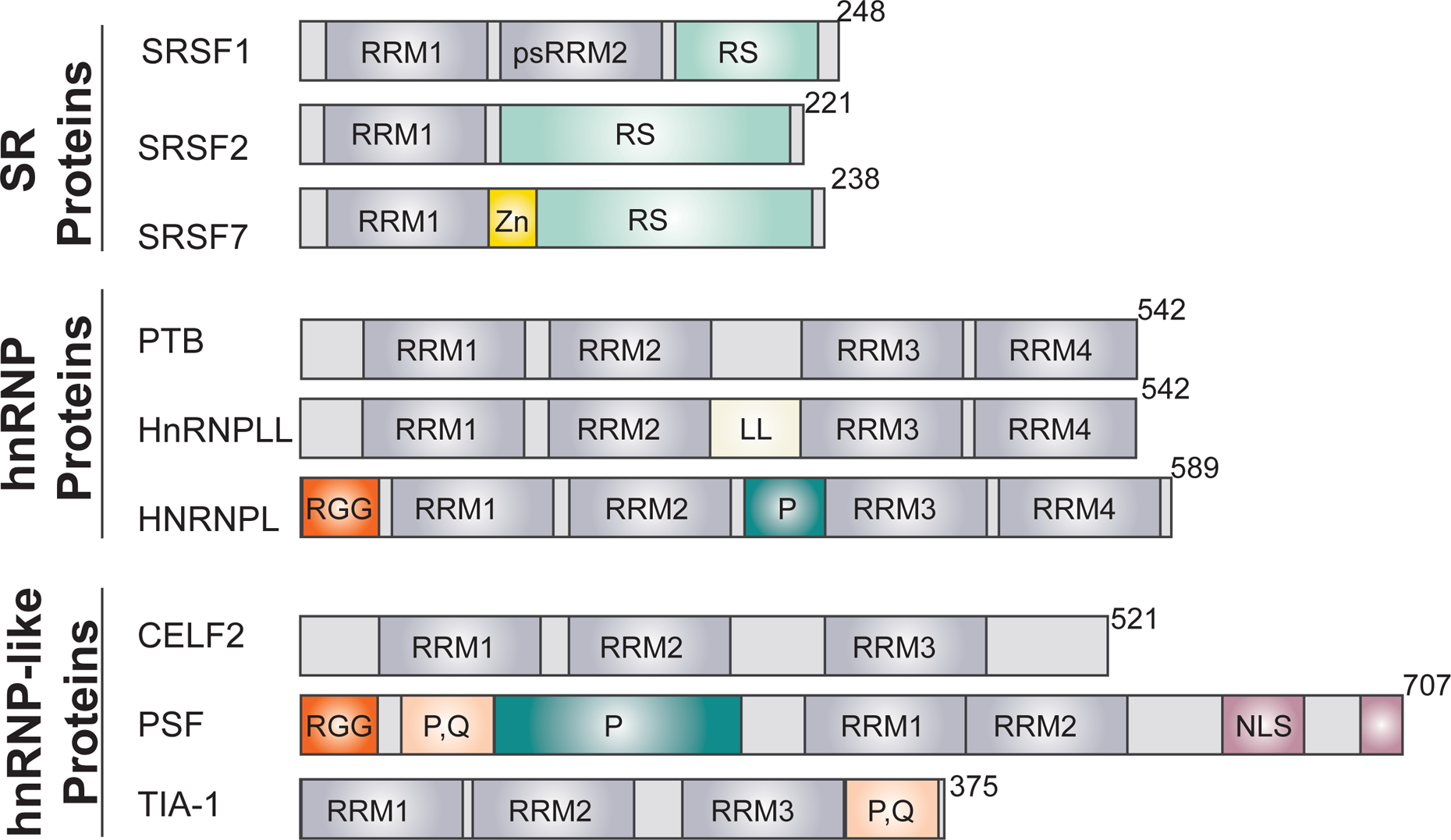
RRM, RNA recognition motif (a classic 2 alpha helix, 4 beta-strand fold); psRRM2, pseudo-RRM (similar fold to an RRM but lacking key residues); P, proline-rich region; RGG, arginine/glycine/glycine repeat region; P, Q, proline/glutamine rich sequence; RS, arginine/serine dipeptide repeat sequence; Zn, zinc finger; NLS, nuclear localization sequence; LL, unique L-like linker sequence.
Importantly, the RBPs discussed above function in a highly combinatorial manner such that even modest changes in the abundance or activity of individual RBPs or core spliceosomal proteins can result in large splicing changes in particular transcripts6,16. Moreover, each gene is regulated by a discrete subset of RBPs, with each RBP typically regulating several hundred splicing events6,16. Overall, this combinatorial pattern of RBP association results in a complex network of coregulation, in which any change in RBP expression or activity can potentially impact the splicing of some or many transcripts (e.g. see Fig. 4 below). Consistently, signal- or developmental-responsive changes in AS are typically mediated by changes in RBP abundance due to either regulated transcription or mRNA stability of the messages encoding RBPs, or changes in activity or localization mediated by post-translational modification of the protein itself (e.g. see reviews27,33). Examples of such regulation of AS via RBPs in activated immune cells are detailed later in this review.
Figure 4. Classic examples of AS in the immune response.
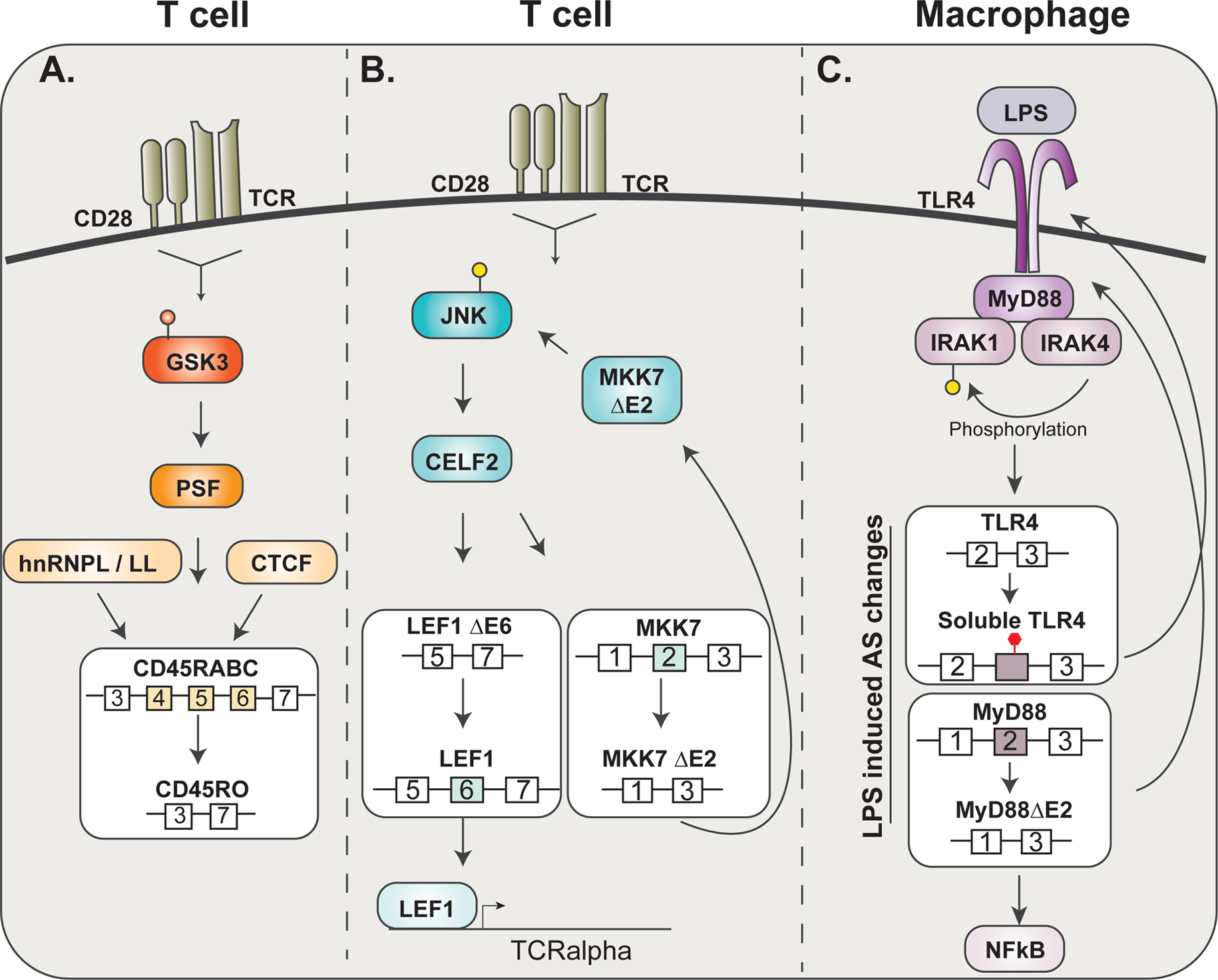
A. Upon T cell activation, GSK3 inactivation leads to subsequent PSF activity to then influence CD45RABC splicing to CD45RO along with hnRNPL/LL and CTCF. B. Upon T cell activation, the activation of JNK kinase signaling regulates CELF2 activity to influence the splicing of LEF1 and MKK7. A feed-forward loop is established as MKK7 ΔE2 phosphorylates JNK to then further regulate CELF2 activity. C. LPS incudes macrophage activation through TLR4 signaling. TLR4 recruits MyD88 which further recruits IRAK4 (only the long form of MyD88) and IRAK1. IRAK1 is phosphorylated by IRAK4, when co-recruited, to promote NFkB signaling.
Alternative Polyadenylation
Beyond AS regulation, protein expression in cells is also controlled at the RNA-level through the presence or absence of regulatory sequences located in the 5’ and 3’ untranslated regions (UTRs) of mRNA10,34. In particular, 3’ UTRs are the primary site of miRNA association and function10,11 as well as containing binding sites for proteins that regulate translation, mRNA stability, and subcellular localization of mRNA10,11,35. Indeed, even protein phosphorylation state, protein complex assembly, and overall protein activity has been shown to be influenced by the presence of regulatory elements in 3’UTRs36,37.
Importantly, the length of the 3’UTR of a given gene is not static, but rather can vary in a tissue and condition-specific manner through the process of alternative polyadenylation (Fig. 1, 2c,d). Polyadenylation itself, which occurs on virtually all mRNAs, as well as some non-coding RNAs, involves the cleavage of the nascent RNA during transcription, followed by the subsequent addition of ~200 adenosine bases to the exposed 3’ hydroxyl end38. Alternative polyadenylation (APA) refers to instances in which cleavage, and subsequent polyadenylation, occurs at different positions on a transcript in a condition or cell-type specific manner11 (Fig. 1). APA results in the truncation of the open reading frame if cleavage occurs prior to the final coding exons (e.g. either version of the alternate terminal exon, Fig, 1). On the other hand, when both or all of the cleavage events are downstream of the stop codon, APA results in 3’UTRs of distinct lengths and sequence (Fig. 1, Tandem 3’UTR). Since 3’UTR identity determines the ultimate expression and regulation of the upstream open reading frame, as described above, APA after the stop codon can have a profound effect on protein expression or function10,11,35,39. Notably, recent transcriptome-wide studies have observed APA in over half of human protein-coding genes11,40. Therefore, regulation of APA provides an additional prevalent level of RNA-based control of cellular activity.
Mechanisms and Regulation of Alternative Polyadenylation
Cleavage and polyadenylation is mediated by a large multi-component complex consisting of a core CPSF (Cleavage and Polyadenylation Stimulation Factor) subcomplex as well as additional enhancer subcomplexes CFI (cleavage factor I), CFII (cleavage factor II) and CstF (cleavage stimulatory factor), each of which are comprised of 2–6 proteins38 (Fig. 2c). These four subcomplexes (aka the Cleavage and Polyadenylation Factors or CPAFs) associate in a cooperative manner with genomic-encoded sequence motifs comprising a hexameric AAUAAA and upstream and downstream U/G-rich sequences (collectively known as the Polyadenylation Sequence or PAS). The AAUAAA hexamer is the primary binding site for the CPSF complex and directs the location of cleavage, whereas CFI binds to an upstream UGUA and CstF associates with a U/G-rich element downstream of the core hexamer38 (Fig. 2c). Importantly, since each of these interactions is relatively weak, the cooperative assembly of CPSF with CFI and CstF helps ensure efficiency of binding and accurate positioning of the overall complex; the strengthening of one interaction can compensate for suboptimal (aka “weak”) interactions at other regions11. Indeed, much regulation of APA is thought to be controlled by the expression level of various CPAFs, in which increases in abundance of proteins such as the RNA-binding component of CstF (CstF64) increases the use of weak APA site through promoting occupancy of low affinity sites through mass action (41,42 and see also below).
Additionally, the efficiency of CPAF complex recruitment to specific locations on the pre-mRNA substrate can also be controlled by RBPs in a manner analogous that the regulation of AS (Fig. 2d). Several SR, hnRNP and hnRNP-like RBPs have been shown to promote recruitment of CPAFs to weak PAS sites or to compete with complexes such as CFI or CstF for their cognate binding sites43–45. As described above for the regulation of splicing, changes in the expression or activity of RBPs in response to environmental or developmental cues can redirect the activity of the CPAFs on individual substrate pre-mRNAs to alter 3’UTR identity of specific genes. Other recent reviews provide an excellent comprehensive discussion of the mechanisms and regulation of APA11,38, while below we focus on specific examples of APA regulation in the immune system.
AS and APA in the Immune System
Much early work on gene expression in the immune system made use of microarrays and the use of exon junction or isoform-specific probes to infer splicing or polyadenylation changes allowed several early studies demonstrating widespread AS changes across the transcriptome during activation of human CD4+ and CD8+ T cells46 and APA changes upon activation of T cells, B cells and monocytes39. However, the microarray platform was never optimized to detect APA or AS47,48. Therefore, most early predictions of AS or APA in the immune system were incomplete and not well validated experimentally12. In the past few years, however, there has been a tremendous increase in our appreciation for AS and APA throughout biology due to advances in next-generation sequencing such as increasing the number and length of sequencing reads obtained by short-read RNA-Seq, the emergence of long-read RNA-Seq, and the development of robust methods to quantify AS and APA from both such RNA-Seq data42,49,50.
One of the first studies to apply RNA-Seq data for AS prediction in the immune system was from our own lab, studying activation of primary and cultured human T cells51. This work resulted in the identification of ~200 genes that exhibited robust, validated AS changed upon activation of Jurkat cells with PMA and primary CD4+ T cells with PHA51. Since then, many groups have used RNA-Seq data to gain a genome-wide view of AS and/or APA in various primary immune cell types (e.g. see52–61 and Table 1 for several examples). Together these studies have confirmed the broad impact AS and APA have on gene expression during the innate and adaptive immune responses. In addition, the deposition in public repositories of RNA-Seq data from these and other studies is a tremendous benefit for future discovery, as this enables investigators to directly compare transcriptomes from a wide range of conditions to seek further insight. For example, RNA-Seq data collected for gene expression studies by one group, may be used to investigate AS or APA by others (e.g.52). It is anticipated, therefore, that our appreciation for the depth of regulation of immune cell function by AS and APA will grow tremendously in the near future. Already there are many clear examples of how AS and APA impact immune responses. Below we discuss some of the best studied instances of AS and APA in immune cells and what is known about the regulators and mechanisms driving AS and APA in T cell, B cells and macrophages.
Table 1:
Selected RNA-Seq based studies of splicing and APA in Immune Cells.
| Study | Organism/Cell Type | Stimulus | Analysis method | Findings | Reference | |
|---|---|---|---|---|---|---|
| Alternative Splicing | Radens et al. RNA 2020 | Human Naïve CD4+ T cells | Anti-CD3/CD28 | MAJIQ | Identified hundreds of splicing changes and compared these to polarized Th cells | 1 |
| Liu et al., NAR 2018 | Human M1 and M2 Macrophages | GM-CSF and M-CSF | rMATs, MISO, SpliceTrap | 1933 and 1262 events in 1327 and 903 genes are specific to GM- and M-CSF conditions | 2 | |
| Lin et al., 2016 | Human Macrophages | Monocyte Differentiation | PennDiff, DEXseq, and IUTA | Detected 233 M1-induced AS events | 3 | |
| Kalam et al., PLoS Pathogens 2017 | Human Macrophages | Mycobacterium tuberculosis | rMats | 1057 and 583 splicing events were found unique to H37Ra and H37Rv Mtb strains by 48hrs respectively | 4 | |
| Pai et al., PLoS Genetics 2016 | Human Macrophages | Bacterial infection | MISO | Identified hundreds of retained introns and alternative exons | 5 | |
| West et al., Cell Reports 2019 | Mouse RAW 264.7 Macrophages | Salmonella Infection with hnRNP M KD | MAJIQ | 94 LSVs in uninfected versus hnRNP M KD macrophages and 67 LSVs in Salmonella-infected versus hnRNP M KD macrophages | 6 | |
| Janssen et al., G3 2020 | Mouse Avelolar Macrophages | LPS | DEXseq | 1,000 genes with altered isoform usage on Day 3 vs. baseline (Day 0) upon LPS challenge | 7 | |
| Alternative Polyadenylation | Kalam et al., PLoS Pathogens 2017 | Human Macrophages | Mycobacterium tuberculosis | DaPars | increase in the uses of distal poly-A sites and increased expression of transcripts with longer 3’-UTR | 4 |
| Gruber et al., Nat Comm 2014 | Mouse T cell | Anti-CD3/CD28/IL2 | 3’end seq | 3,116 genes undergo alternative polyadenylation (APA) at tandem poly(A) sites | 8 | |
| Beisang et al., Gene 2014 | Human T cells | Anti- CD3/CD28 | 3’end seq | 2352 3′UTRs length changed over time following T cell activation and 203 of these transcripts were CELF1 targets | 9 | |
| Singh et al., Nat Comm 2018 | Human T cells, B cells | NA | 3’end seq | Intronic polyadenylation isoforms are widely expressed in immune cells, differentially used during B-cell development or in different cellular environments, and can generate truncated proteins lacking C-terminal functional domains | 10 | |
| Pai et al., PLoS Genetics 2016 | Human Macrophages | Bacterial infection | MISO | Identified hundreds of genes with shortened 3’ UTRs upon infection | 5 |
Prototypical regulators and regulation of AS in Immune Cells
Two classic examples of AS regulated by immune activation, discovered more than 20 years ago, includes the regulated splicing of CD45 (PTPRC) upon T cell activation62–64 and the alternative splicing of TLR4 signaling proteins in LPS-treated macrophages65,66 (Fig. 4). Both examples display induced changes in isoform expression, limiting the extent of cellular signaling in response to subsequent stimuli, thereby preventing prolonged activation and, in turn, promoting recovery of the immune system to a basal state. Consistently, defects in CD45 and MyD88 AS have been linked to hyperactivity of immune cells and the development of autoimmune diseases or cancer67–70. More recently, the functional impact of many additional activation-induced AS events in lymphocytes and macrophages have been studied in detail. Since several reviews have previously described many of these cases in depth12,13, we focus below on only a few of the most extensively studied examples of AS to provide an overview of the functional impact of AS in the immune system and highlight what is known about the mechanisms, signaling pathways and RBPs connecting splicing to immune challenge.
CD45 and the global impact of hnRNP L and LL in T cell biology
CD45 is a transmembrane protein tyrosine phosphatase that is expressed on the cell surface of nucleated hematopoetic cells71. The intracellular phosphatase activity of CD45 plays a critical role in antigen signaling in lymphocytes by promoting signaling cascades triggered upon engagement of the T cell receptor (TCR) or B cell receptor (BCR). By contrast, the extracellular domain regulates CD45 phosphatase activity. As early as the late 1980s it was recognized that three exons (exons 4–6, also termed A-C in the older literature) within the gene encoding CD45 (now called PTPRC) undergo alternative splicing72. Exons 4–6 encode a portion of the extracellular domain of CD45, thus their differential inclusion impacts the ability of antibodies to interact with CD45 on the cell surface71. In particular, exclusion of exon 4–6 results in a smaller isoform that specifically interacts with CD45RO antibodies, while isoforms encoded by mRNAs that include exons 4, 5 or 6 react with antibodies CD45RA, RB and RC respectively71,73. The ability to detect different isoforms of CD45 by flow cytometry led to the recognition that isoform expression is a useful marker of cell identity and state. Namely B cell express an isoform (B220, also called RABC) that is recognized by the RA, RB and RC antibodies (includes all of exons 4–6), while naïve T cells predominantly express an isoform reactive to RA and RB (primarily includes exons 4&5)71,73. Perhaps most importantly for both immunology and understanding splicing regulation, T cells switch from the RA to the RO isoform upon antigen stimulation due to repression of exon 4 (and 5 and 6 to a lesser extent) inclusion upon activation71,74. The smaller (RO) isoform lacks bulky glycosylation and sialyation groups on the extracellular side and thus is more prone to homodimerization than the larger RA isoforms75. As dimerization causes steric hindrance of the intracellular phosphatase domain, the switch from RA to RO upon activation decreases phosphatase activity, effectively downregulating subsequent antigen signaling75–77. Consistently, several studies have suggested that the switch in CD45 expression from RA to RO is critical to maintain T cell homeostasis, and removing this “break” leads to immune hyperactivity77 (and see discussion of C77G SNP below).
The CD45 RA to RO switch has been used extensively by immunologists to track T cell activation, but also provides an excellent model for mechanistic studies of activation-induced splicing regulation. Indeed, several groups, including our own, have examined CD45 AS as a model for identifying RBPs that regulate splicing in immune cells and in response to antigen stimulation (Fig. 4). Each of the three variable exons of CD45 contain a conserved Activation Responsive Sequence (ARS) that represses inclusion (i.e. functions as an inducible Exonic Splicing Silencer (ESS))78,79. In naïve T cells, the ARS is primarily bound by hnRNP L, which exerts a weak level of repression79,80. However, upon T cell activation, two additional proteins – the hnRNP L homologue hnRNP L-like (hnRNP LL) and PSF (aka SFPQ) – also are recruited to the ARS elements and provide much greater repressive activity, thus promoting skipping of exons 4–6 and production of the RO isoform81–84. The signal-responsive recruitment of hnRNP LL and PSF are due to distinct regulatory mechanisms. HnRNP LL is poorly expressed in naïve T cells and undergoes significant transcriptional upregulation in response to antigen stimulation82–84. By contrast, PSF is hyperphosphorylated in naïve T cells by the kinase GSK3 and bound to a partner protein, TRAP150, that blocks access of RNA to the RNA-binding domain (RRM) of PSF85,86. Upon activation, GSK3 activity is downregulated and PSF thus loses phosphorylation on key residues that are critical for interact with TRAP150 – thereby allowing association between PSF and RNA85. The functional significance of the association of hnRNP L/LL/PSF complex with RNA is highlighted by the presence of a SNP within the ARS sequence of exon 4 (C77G) that results in aberrantly high levels of CD45RA, and corresponding low expression of CD45 RO87–89. This SNP weakens the overall association of proteins with the ARS and, consistent with the above functional consequence of the RA to RO switch, correlates with susceptibility to autoimmune diseases67,90–92.
Finally, it is worth noting that in addition to hnRNP L, LL and PSF, CD45 exon 5 is also regulated by the presence of CTCF bound to DNA93 (Fig. 4). CTCF is a DNA-binding protein that regulates genome architecture, functioning as both an insulator between genes and a regulator of transcriptional pausing within genes93–95. In the CD45 gene, binding of CTCF to the DNA encoding exon 5 results in pausing of RNA Polymerase II (Pol II) during transcription, which in turn promotes assembly of the splicing machinery on the weak splice sites of exon 5, thereby increasing inclusion of exon 5 in the final mRNA93. Such activity of CTCF in regulating splicing is consistent with a general mechanistic interplay between transcription and splicing96–98. In particular, numerous studies have demonstrated that the speed of Pol II elongation can directly impact AS patterns by influencing competition between splice sites or splicing regulatory elements97. In the case of CD45, the impact of CTCF in controlling Pol II elongation, and thus exon 5 inclusion, is regulated by both the expression level of CTCF in B cells and by methylation of its DNA target in T cells93. In particular, activated and memory T cells have increased DNA methylation as compared to naïve T cells and this increased methylation reduces binding of CTCF resulting in the observed loss of exon 5 inclusion upon T cell activation and in the transition from naïve to memory T cell state93,99. In thinking about this mechanism, it is worth noting that the transcription elongation factor ELL2 has also been implicated as a general regulator of splicing in B cells, although the specificity of this effect is not fully understood100.
In sum, the regulated splicing of CD45 provides both an excellent example of the functional relevance of AS in lymphocytes and also has led to the identification and characterization of many factors involved in tuning splicing in a condition-specific manner in T cells. In particular, the induced expression of hnRNP LL and regulated activity of PSF that is observed upon T cell activation provide prototypical examples of how RBPs can be regulated in a signal-dependent manner27, while regulation of CTCF binding via DNA methylation represents a general model for how epigenetic marks may influence splicing101.
PSF has a complex and poorly identified RNA recognition sequence, and depletion of PSF has not yet been linked to widespread splicing changes in lymphocytes86,102. Similarly, although genome-wide studies have identified a number of regulated splicing events in T cells that are directly bound and regulated by CTCF, the functional consequence of these remain unexplored99. By contrast to PSF and CTCF, hnRNP L and hnRNP LL both clearly play a major role in shaping immune function. These homologues are both highly expressed in activated T cells, bind efficiently to CA-rich elements in RNA, and have been shown to widely regulate splicing in T cells82–84,103–106. Analysis of in vivo binding of hnRNP L in primary human CD4+ cells shows enriched binding to genes involved in Wnt and TCR signaling104, and knock-out of hnRNP L in developing thymocytes results in increased Lck activity, a defect in transition to CD4+CD8+ double positive thymocytes and lack of migration of single positive cells to the periphery103. Similarly, several lines of evidence suggest that hnRNP LL has widespread impact on AS and function in both B and T cells. hnRNP LL increases upon differentiation to memory T cells and plasma cells from naïve T and B cells respectively and regulates the splicing of many genes involved in immune function and the differentiated state82–84,105,106. Moreover, mice with a hypermorphic allele of hnRNP LL exhibit a decrease in circulating naïve and memory CD4+ T cells due to lack of persistence of these cells in the periphery84. Given that hnRNP LL is highly enriched in lymphoid cells over many other tissues, this protein may be uniquely evolved to regulate splicing during the adaptive immune response.
MKK7, LEF1 and regulation by CELF2
Another RBP that is highly regulated in lymphocytes is the hnRNP-like CUG-binding protein and ELAV-Like Factor (CELF2, Fig. 3). During thymocyte development and in response to T cell activation, CELF2 is regulated at the level of transcription, by mRNA stability and the level of splicing, to alter the expression of active protein107,108. Transcription of CELF2 is induced by NFkB, which binds near the transcription start site of CELF2 in TNF or PMA-activated cells107. By contrast, the stability of the CELF2 mRNA is promoted in a JNK-dependent manner107,109. Together these signaling events result in an increase in CELF2 mRNA and protein.
As its name implies, CELF2 binds to CUG and UG-rich repeats in RNA110. When this binding occurs in or around exons in a nascent transcript, the result is often CELF2-dependent changes in splicing110,111. Indeed, both targeted and whole-transcriptome RNA-Seq studies have identified hundreds of genes that are regulated in a CELF2-dependent manner in cultured Jurkat T cells, many of which overlap with JNK-regulated AS events in primary CD4+ cells and/or AS events that are regulated upon the DN to DP transition in thymic development107,109,111,112. Two of the best characterized of these CELF2-dependent splicing events, in terms of both mechanism and functional impact, are observed in the genes encoding the LEF1 transcription factor and the MAP kinase, MKK7 (Fig. 4).
In the case of LEF1, CELF2 binds to a regulatory element downstream of variable exon 6 to promote its inclusion108,111. Inclusion of exon 6, in turn, adds 28 amino acids to a regulatory domain of LEF1 which promotes its ability to activate transcription from the TCRalpha promoter108. Thus, increased expression of CELF2 during thymocyte development is a likely driver of TCRalpha expression as cells progress from the DN to DP state107, although this has not been directly proven in vivo.
In contrast to its activity in promoting exon inclusion in LEF1, CELF2 inhibits inclusion of exon 2 in the gene encoding the JNK kinase MKK7109. Specifically, binding of CELF2 immediately upstream of MKK7 exon 2 blocks spliceosome assembly at this exon and promotes exon skipping111. Exon 2 encodes 16 amino acids that disrupt the docking domain through which MKK7 interacts with its substrate JNK, therefore CELF2-induced skipping of exon 2 enhances MKK7 phosphorylation of JNK and subsequent JNK signaling109. As mentioned above, expression of CELF2 itself is enhanced by JNK signaling, thus the regulation of MKK7 by CELF2 provides a feedforward regulatory loop to augment JNK signaling, and subsequent cytokine release, in response to T cell activation109. Together, LEF1 and MKK7 provide two examples in which CELF2-dependent splicing clearly tunes the activity of T cells to promote optimal development and responsiveness of the immune response. Moreover, the differential activity of CELF2 on LEF1 and MKK7 underscore a common rule in splicing regulation, namely, RBPs can have both positive and negative impacts on exon inclusion depending on the location and context of their binding site16,111. Although the mechanistic basis of such differential activity has not been fully characterized, one explanation is that when a RBP binding site directly overlaps a splice site, competitive binding leads to exon skipping; while binding of an RBP adjacent to a site required for spliceosome assembly can promote splicing through cooperative protein-protein interactions16,111.
Regulation of AS in innate immunity – TLR4 signaling and hnRNP M
As alluded to above, the innate immune response is also tuned by alternative splicing. This is best demonstrated in the TLR4 signaling pathway (Fig. 4). Toll-like receptor 4 (TLR4) is a transmembrane protein, one of several pattern recognition receptors (PRRs) that recognize features of microbial infection, and is found to be critical for tailoring the immune activation response of APCs including dendritic cells and macrophages113. When bound by an agonist such as lipopolysaccharide (LPS), TLR4 nucleates an intracellular complex via MyD88 that recruits several IL-1 receptor-associated kinases (IRAK) including IRAK4 and IRAK113,114. When co-associated with MyD88, IRAK4 phosphorylates IRAK1, which in turn propagates signaling leading to activation of NF-kB114. Notably, TLR4, MyD88 and IRAK1 all undergo AS in response to LPS stimulation in a manner that attenuates further signaling13. In the case of TLR4, LPS stimulation of monocytes or macrophages leads to inclusion of a so-called “poison” exon (i.e. one that includes a stop codon), prior to the transmembrane domain, such that the resultant encoded protein is secreted and competitively binds to soluble LPS, thus limiting engagement of cell-bound receptors65,115.
Similarly, in response to LPS stimulation exon 2 of the gene encoding MyD88 is skipped, resulting in an in-frame deletion, removing the portion of MyD88 that is necessary for IRAK4 association. The resulting smaller MyD88 isoform (MyD88s), functions as a dominant negative of TLR4 signaling as incomplete complex assembly on TLR4 no longer leads to IRKA1 phosphorylation66,116. Finally, IRAK1 also exists as multiple isoforms through AS, which differ in their ability to interact with and activate downstream effectors in inflammatory signaling117–119. Surprisingly, despite the clear functional significance of the LPS-mediated AS of TRL4, MyD88 and IRAK-1, little is known about the mechanisms through which this splicing regulation is conferred. Although changes in the expression of core splicing components have been shown to alter the balance of some of these splicing choices69,120,121, much work remains to uncover the signaling pathways and RBPs that tune LPS sensitivity of macrophages via AS of these key genes.
Although gene-specific regulators of TLR4, MyD88 and IRAK1 splicing have not yet been identified, one RBP that has been conclusively linked with AS and TLR4 signaling is hnRNP M53. HnRNP M is a repressor of splicing of weak introns in IL-6 and other genes involved in the innate immune response, reducing the expression of fully processed transcripts. Consistently, hnRNP M attenuates innate immune signaling as demonstrated by the fact that macrophages depleted of hnRNP M exhibit higher expression of pro-inflammatory cytokines, such as IL-6, upon LPS activation and reduced infection with virus or Salmonella53. In other words, knockdown of hnRNP M leads to hyperactivation of macrophages in response to viral and bacterial pathogens. This repressive activity of hnRNP M is partially countered by TLR4 signaling, which phosphorylates hnRNP M, thereby reducing its association with IL-6 and other target genes and resulting in an increase in IL-6 expression53. In sum, while much more remains to be discovered regarding the molecular mechanisms of AS during the innate immune response, it is clear that regulation of splicing plays an essential role in shaping the response of the innate immune system to viral and bacterial pathogens.
Prototypical regulators and regulation of APA Regulation in Immune Cells
Compared to AS, our understanding of the mechanisms and impact of APA regulation in immune cells, and across all of biology, is still relatively limited. AS has been studied in more depth and detail than APA perhaps due to the notion that changes in AS more frequently have a predictable consequence on protein expression than changes in APA. Nevertheless, the transcriptome-wide regulation of APA in human cells, particularly the global trends displayed in anti-CD3/CD28 activated CD4+ T cells described above (39 and Table 1), highlights the need to investigate the regulation of APA in the immune system. Indeed, already there are a few well documented examples of APA playing a robust role in the behavior of the immune system, and control of APA in immune cells has been observed to be driven both by changes in the expression of core CPAFs as well as via the activity of specific RBPs. Below, we review several of the best characterized examples of the mechanism and impact of APA in immune cells to both provide an overview of what is known and to motivate further studies.
Ig heavy chain and regulation by altering abundance of a core CPAF: CstF64
One of the first examples of regulated APA in human cells was identified through the study of the switch from membrane-bound to secreted forms of immunoglobin heavy chain (IgH) that occurs for at least the mu isotype of IgH during B cell activation. In 1980, the groups of Baltimore, Hood and Wall simultaneously reported that the membrane-bound form of IgH expressed in pre-B and primary B cells, and the secreted form of IgH that predominates in plasma cells, are generated by differential processing of the 3’ end of the gene encoding the Ig heavy chain122–124. The gene encoding IgH has two alternative PAS sites125. The first is after the exons encoding the constant region of IgH (PASs), while the second is after two additional exons that encode the transmembrane domain (PASm) (Fig. 5). In primary B cells the constant-region exons are spliced to the transmembrane exons. By contrast, upon differentiation to plasma cells the IgH message is cleaved at the first PAS site immediately after the constant-region exons. This prevents the transmembrane exons from being transcribed or spliced to the rest of the message, such that the encoded protein lacks a membrane-anchoring domain and thus is secreted into the extracellular environment125.
Figure 5. Classic examples of APA in the immune response.
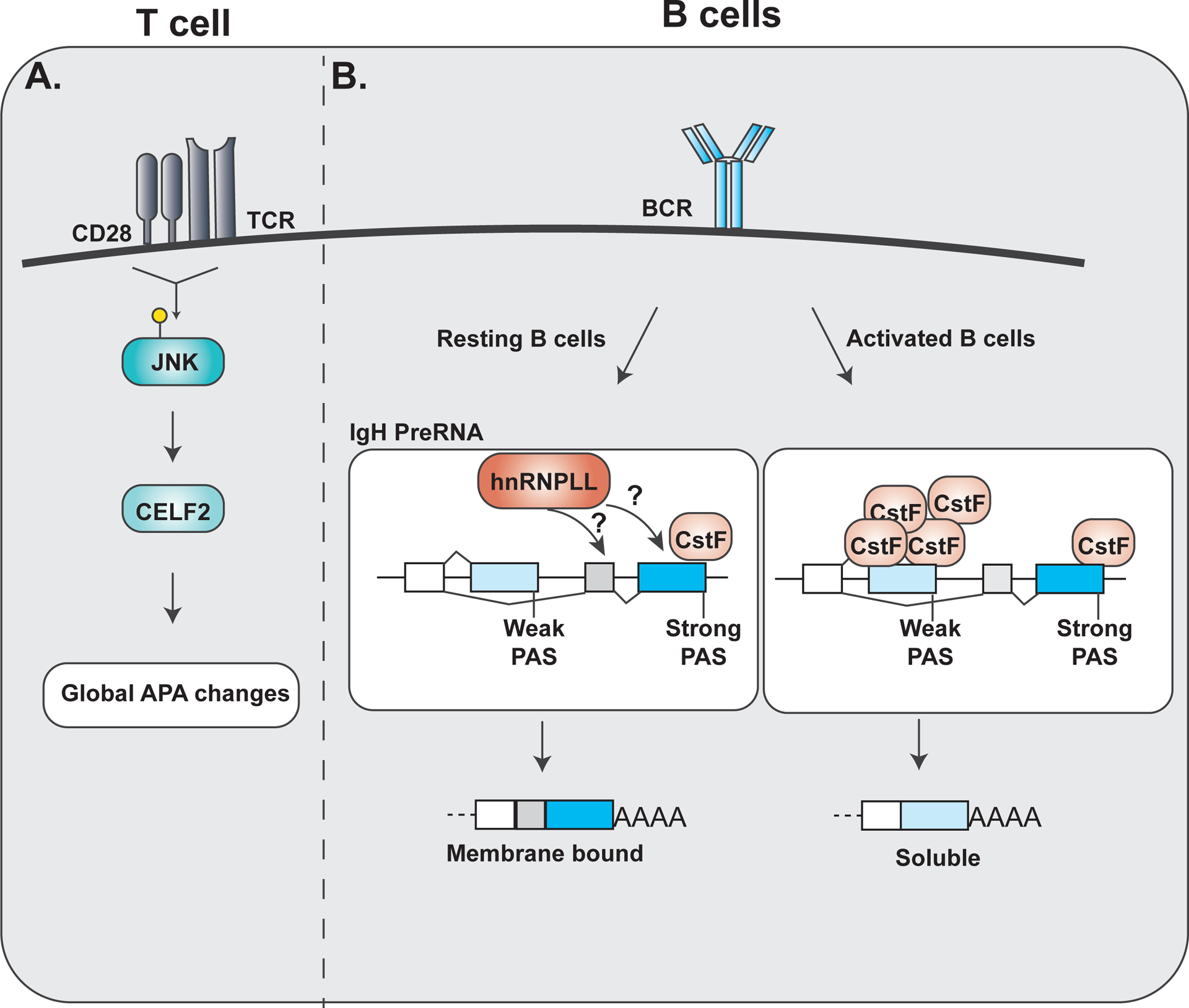
A. Upon T cell stimulation phosphorylated JNK activates CELF2 activity to mediate global APA changes within cells. B. Within resting B cells, low levels of CstF binding to the strong distal Pas site and leads to increased production of membrane bound immunoglobulin heavy chain (IgH). hnRNPLL activity is also attributed to the selection of the strong PAS site selection in resting B cells. Upon B cell activation, increased expression of CstF promotes usage of the weaker proximal PAS site and increased production of soluble IgH.
As described above, PAS sequences and their surrounding contexts can differ in the affinity with which they recruit core components of the cleavage and polyadenylation machinery (see Fig. 2c, d). In particular, the sequence that recruits the protein CstF64 is highly degenerate, thus CstF64 exhibits a wide range of binding affinities to these target sites. The CstF64 binding site adjacent to the PASm in IgH is a high affinity “strong” site126; thus even at low cellular concentrations of CstF64 this protein binds to this distal site in IgH to promote recruitment of the CPSF complex and cleavage of the transcript. By contrast, the CstF64 binding site at the proximal PASs has lower affinity for CstF64126. Therefore, CstF64 does not nucleate the CFAP complex at this more proximal PAS site unless the abundance of CstF64 is high. Primary B cells have a relatively low concentration of CstF64, consistent with the skipping of the proximal PAS site, and use of the distal site, in IgH under these conditions126. However, upon B cell differentiation, such as triggered by LPS stimulation, the expression of CstF64 is dramatically upregulated126, resulting in “filling” of the proximal site by CstF64 and cleavage at this site before the distal site is even transcribed. Thus, regulation of IgH is a clear example of the functional implications of changing the expression level of core CFAP factors, as discussed in the previous section. Given the central role of CstF64 in APA, it is almost certain that the change in CstF64 expression during B cell differentiation drives APA of other physiologically-relevant targets. Notably, CstF64 expression is also higher in effector versus naïve T cells, where this difference has been implicated in at least one study to drive altered isoform expression of the NF-AT transcription factor127. Additionally, LPS-stimulated macrophages exhibit increased CstF64, which has been attributed to changes in both abundance and APA of downstream genes128. Therefore, CstF64 regulation is foundational in shaping 3’ UTR identity, gene expression and cellular function not just in B cells, but throughout both the adaptive and innate immune system.
CELF2 – a RBP regulator of APA as well as AS in T cells
Just as CELF2 binding to target RNAs can alter binding of splicing factors to their cognate sequences, binding of CELF2 near PAS sites also can sterically hinder binding of CPAFs. Notably, the optimal binding site for CELF2 (UG-repeats110) is similar to that of CFIm25 (UGUA129) or CstF64 (U/UG-rich sequences130). Therefore, one can readily appreciate that subtle difference in RNA sequence, or in the relative concentration of CELF2 versus CFIm25 or CstF64, can result in the preferential binding of CELF2 over the CPAFs. Indeed, competitive binding between CELF2 and CFIm25 and CstF64 has recently been demonstrated in biochemical assays and has been shown in cells to alter APA43. Given that CELF2 is regulated by JNK signaling in activated T cells, it is likely that CELF2 may drive much of the APA that has previously been observed upon antigen stimulation of CD4+ primary T cells. Indeed, CELF2 regulates a wide program of activation-induced APA in the Jurkat model T cell line43. However, the functional consequence of CELF2-mediated APA regulation on immune cell function remains relatively poorly studied and merits much further attention. Interestingly, one study recently suggested that hnRNP LL might also be a specific regulator of APA, as depletion of hnRNP LL increases the ratio of the above-described soluble versus membrane-bound IgH131; however, this effect appears to be at the level of splicing and is inconsistent with the increase in both hnRNP LL expression and soluble IgH observed in the transition from B cells to plasma cells106.
Regulation of Apoptosis in T cells via AS and APA
Apoptosis
A newly emerging example of the functional impact of AS and APA in the immune system is in regard to apoptosis. Apoptosis is a tightly regulated form of controlled cell death that occurs in response to both extracellular stimuli and intracellular stress132,133. Hallmarks of apoptotic cells include cellular shrinkage, phosphatidylserine exposure on the extracellular membrane, DNA fragmentation, and cellular blebbing132. In the immune system, apoptosis plays a critical role in maintaining homeostasis and limiting responsiveness of immune cells. For example, one of the outcomes of T cell engagement with antigen is Activation-Induced Cell Death (AICD), where cells are eliminated after multiple rounds of stimulation through the triggering of apoptosis134,135. Such AICD is critical to control the degree and duration of the T cell response to enable a return to homeostasis and prevent hyper-reactivity134. In addition, T cells undergo apoptosis in the presence of a weak stimulation, or in the absence of co-stimulation, to prevent spurious immune responses136,137. Consistently, defects in apoptosis regulation in immune cells has been linked to autoimmune disease138–140.
Both the cell-extrinsic or intrinsic triggering of apoptosis activates signaling cascades that involve extensive cross-talk between two major protein families: Caspases and Bcl-2 proteins141–144 (Fig. 6). Caspases are cysteine proteases that are categorized into two different groups. Initiator caspases (Casp-8, −9, −10) function at the apex of apoptotic signaling to link apoptotic triggers to downstream signaling events144. The initiator caspases exist initially as pro-form monomers and are activated through their dimerization to cleave target proteins. Executioner caspases (Casp-3, −6, −7), on the other hand, exist as inactive caspase dimers and are activated through cleavage by initiator caspases to promote cellular responses144. By contrast to the proteolytic activity of caspases, BCL2 family members control mitochondrial membrane permeabilization and can either promote or inhibit apoptotic signaling133,143. There are approximately 20 Bcl-2 family members, characterized by their relatively conserved Bcl-2 homology (BH) domains that promote protein interaction and dimerization. Pro-apoptotic BCL2 family proteins include Bax, Bak, Bid and Bim; while Bcl-xL, Bcl-w, MCL1 and Bcl2 are examples of family members that inhibit apoptosis133,143.
Figure 6. Signaling regulators of the apoptosis signaling pathway are regulated at the level of alternative spicing.
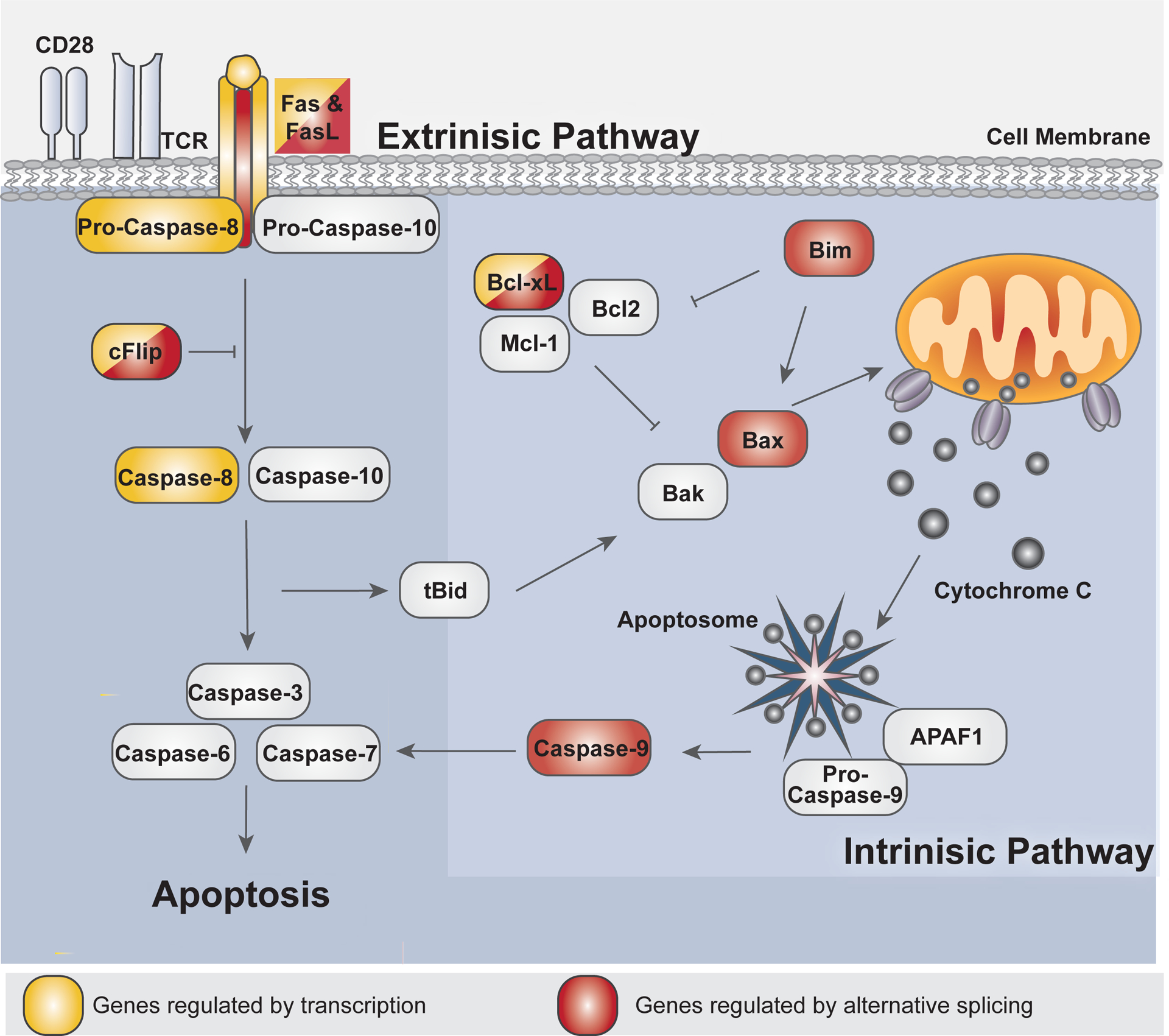
The extrinsic pathway and intrinsic signaling pathway both converge to activate effector caspase activity (Caspase-3,−6–7) to promote apoptosis within the cell. The extrinsic pathway events are activated upon Fas/Fas ligand signaling. Caspase-8/10 are subsequently activated and then cleaves and activates downstream effector caspases. Caspase-8/10 activation may be blocked by cFLIP activity. The cleavage of tBid by Caspase-8/10 then promotes intrinsic pathway signaling. The intrinsic pathway includes pro-apoptotic and anti-apoptotic BCL2 family protein members to regulate mitochondrial outer-membrane depolarization (MOMP) and the release cytochrome C into the cytoplasm. Upon MOMP, activated caspase-9 later cleaves and activates effector caspases. Fas152, FasL152, and Caspase-8132, labeled in yellow, are characterized to be regulated at the level of transcription in T cell. Casp9227, Bax109, and Bim188, labeled in red, are characterized to be regulated at the level of AS. cFLIP194,229 and Bcl-x151 are labeled in red and yellow are characterized to be regulated at both the level of transcription and AS.
The extrinsic pathway of apoptosis is initiated by the activation of a death receptor on the cell surface, such as Fas (CD95), TRAIL receptors, or TNF receptors141,145. Upon oligomerization of death receptors, pro-caspase 8 and FADD are recruited to the intracellular domain of the death receptor to promote dimerization and activation of pro-caspase 8145. In addition to activating the executioner caspases, activated Casp-8 also cleaves the BCL2 family member, Bid, to produce the cleavage product tBid133. tBid links the extrinsic and intrinsic pathways together146. The intrinsic apoptotic pathway consists of signaling events amongst numerous Bcl-2 family members that culminate in the breakdown of the outer mitochondrial membrane, known as mitochondrial outer membrane permeabilization or MOMP133,147. Not only can the intrinsic pathway be activated by the events downstream of the extrinsic pathway, but the intrinsic pathway can also be directly activated by the presence of reactive oxygen species (ROS), nutrient deprivation, cellular stress, and by many other means141,142. MOMP leads to the subsequent release of cytochrome c from the mitochondria147. Once released, cytosolic cytochrome c nucleates the oligomerization of APAF-1 and Casp-9 into a complex named the apoptosome144. The apoptosome is essential for the activation of the enzymatic activity of the initiator Casp-9. Once activated, Casp-9 cleaves the above-mentioned effector caspases to promote apoptosis, in a manner analogous to the extrinsic pathway133,144.
Alternative splicing has been well documented to generate pro- or anti-apoptotic forms of Caspases, Bcl-2 family members or other apoptotic signaling proteins, to impact cellular viability148–150 (Fig. 6). For example, the alternative splicing of Bcl-x, was one of the earliest examples of physiologic relevance of AS in humans, as altering the ratio between the long anti-apoptotic (Bcl-xL) and short pro-apoptotic (Bcl-xS) directly impacts cell survival150,151. More recently, APA of several apoptosis related genes has also been described (see below). Given the critical role of apoptosis in shaping the reactivity of immune system, understanding the role of AS and APA in setting the threshold of sensitivity for apoptosis is essential for appreciating the impact of AS and APA in immune responses. Below we discuss recent evidence for how apoptotic-related AS and APA is controlled in T cells to shape immune response and homeostasis. We also highlight critical questions that remain to be answered in hopes of motivating further study.
Regulation of Apoptosis in the Immune System by Alternative Splicing
FAS
Perhaps the most fully characterized example of how AS is used to tune apoptosis in the immune system is with regards to the extrinsic apoptosis receptor Fas (also known as CD95 or APO-1) (Fig. 6). Fas belongs to the tumor necrosis factor receptor family (TNF-R)145 and is the key trigger for the above-described AICD in activated T cells152. Upon activation, both Fas and its ligand, FasL, are upregulated in T cells152. Engagement of Fas by FasL results in trimerization of Fas on the cell surface and recruitment and activation of Casp-8/10142. Interestingly, at the lower concentration of Fas and FasL found in naïve T cells, the Fas/FasL interaction may provide a co-stimulatory effect to boost early T cell activity and proliferation153,154.
Fas is regulated not only by changes in gene expression but also at the level of AS (Fig. 7). Exclusion of exon 6 from the final mRNA transcript produces a soluble Fas protein that lacks the transmembrane region to anchor the receptor to the extracellular membrane155–158. As for TLR4 discussed above, the soluble Fas receptor competes with membrane-bound Fas for FasL ligation, thereby limiting Fas signaling to the cell155,158. Activation of T cells promotes exon 6 inclusion, favoring the pro-apoptotic, membrane bound form of Fas required for AICD157,159. However, defects in this switch have been commonly observed and linked to a variety of diseases, including autoimmune phenotypes such as lupus (SLE), autoimmune lymphoproliferative syndrome (ALPS) and leukemias, all of which exhibit increased skipping of Fas exon 6 consistent with this form decreasing apoptotic signaling155,159–161.
Figure 7. Canonical apoptosis signaling mediators are regulated at the level of AS in T cells.
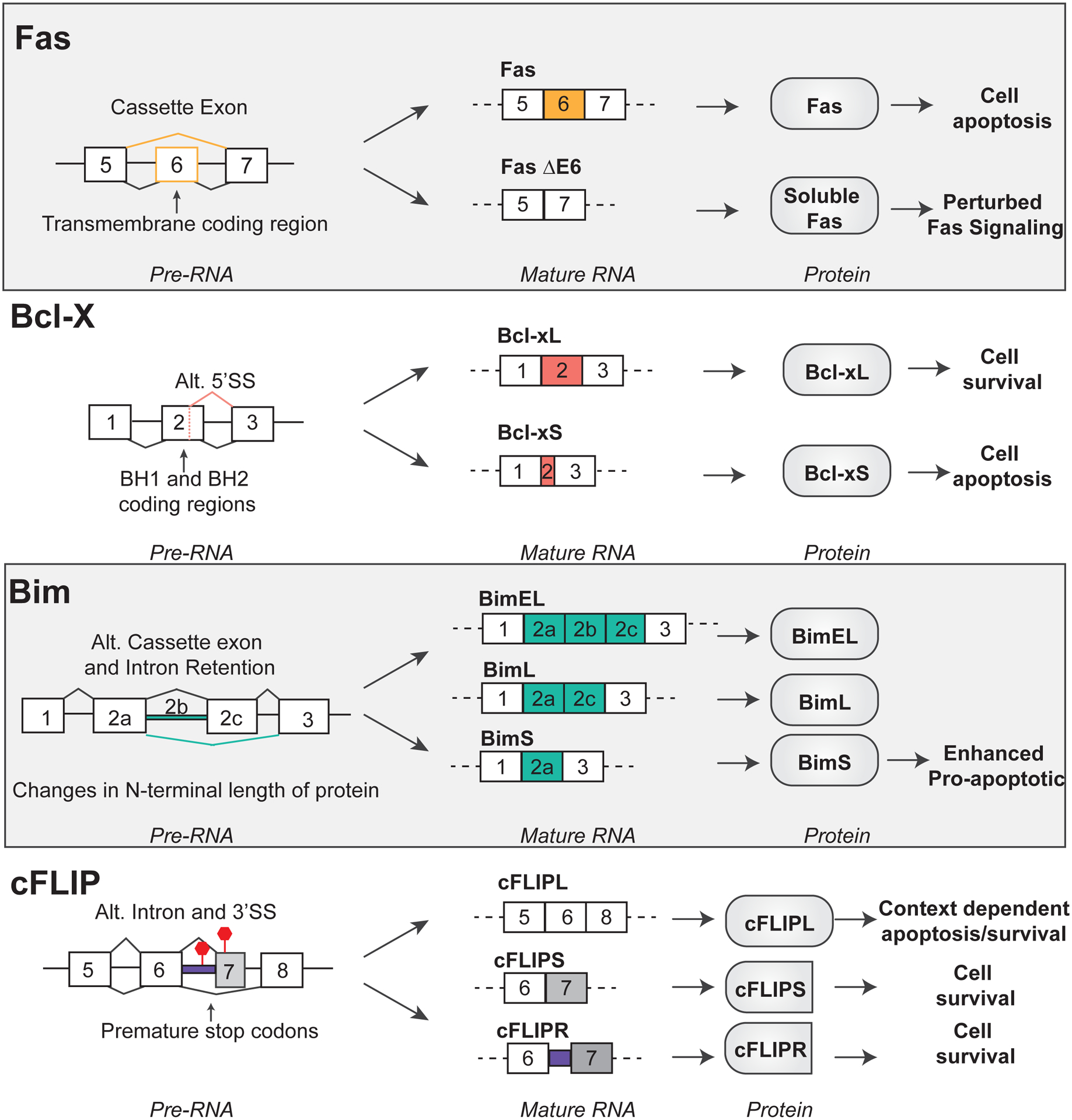
Fas, Bcl-x, Bim, and cFLIP are regulated at the level of alternative splicing. Diagrams depict the pattern of alternative splicing at the pre-RNA and mature RNA level. The impact on protein and cell physiology are illustrated for each alternative splicing event.
The regulation of Fas exon 6 inclusion has been attributed to hundreds of genes and a variety of signaling pathways162. Most directly, TIA-1 and PTB bind immediately around exon 6 and act in competition with each other, with TIA-1 binding downstream of Fas exon 6 to promote its inclusion, while PTB binds upstream and represses use of exon 6163,164. T cell activation increases both the expression and phosphorylation of TIA-1 to promote exon 6 inclusion, while IL-7 has been shown to decrease PTB via the activity of the miRNA, mir-124, also leading to increased exon 6 inclusion165–167. Interestingly, caspase cleavage of core splicing factors such as U2AF65 further influences the balance of Fas exon 6 inclusion to create a feedback loop that tunes Fas expression during apoptosis168. Moreover, some correlations been made between dysregulated RBPs and dysregulated Fas splicing, such as the finding of increased expression of the splicing protein SPF45 in glioblastoma, which drives increased exon 6 skipping and is a potential therapeutic target for glioma169,170. However, further insight into the regulation of Fas AS in disease is certain to yield valuable information regarding the treatment of immunologic pathologies.
Bcl-x
As mentioned above, Bcl-x was among the first genes shown to encode pro- and anti-apoptotic isoforms through alternative splicing150,151. Bcl-x is a transmembrane protein that lies in the outer mitochondria membrane to regulate MOMP events within the intrinsic apoptotic signaling cascade171 (Fig. 6). Two well characterized AS isoforms of Bcl-x exist: the anti-apoptotic longer isoform, Bcl-xL, and the pro-apoptotic shorter isoform Bcl-xS due to an alternative 5’ splice site within exon 2 (Fig. 7). Both isoforms are in-frame and produce stable proteins: Bcl-xL is 233 amino acids long while Bcl-xS lacks 63 amino acids, resulting in the removal of BH1 and BH2 domains from the protein. Bcl-xL prevents apoptosis by binding to Bax and tBid via its BH1 and BH2 domains, thereby preventing Bax and tBid from inducing MOMP150,171–173. Bcl-xS, on the other hand, not only lacks the ability to interact with and suppress Bax and tBid, but also can dimerize with Bcl-xL thereby functioning as a dominant-negative to the anti-apoptotic activity of the long isoform150,174. Consistently, overexpression of Bcl-xL, or knocking down Bcl-xS, increases cellular survival; while overexpressing Bcl-xS, or repressing Bcl-xL, increases the cellular apoptosis171,175.
As described for Fas, the mechanisms of AS of Bcl-x have also been extensively studied. A host of factors, including SR proteins SRSF 2,3,7,9 and 10 and hnRNPs F/H, A1, K and PTB, all regulate the relative use of the competing alternative 5’ splice sites in Bcl-x exon 2 to tune the balance of expression of the short and long isoforms (recently reviewed in150). However, most of the studies regarding Bcl-x AS have been done in the context of Hela and 293 cells, or in response to DNA damage, rather than in cells of the immune system.
In T cells, Bcl-xL expression plays a critical role in survival during T cell development in the thymus and also effector responses in the periphery176,177. Naïve peripheral T cells express both Bcl-x isoforms at low levels. T cell activation results in an increased expression of both isoforms, but favors expression of Bcl-xL as a mechanism to promote the survival and function of effector T cells, particularly with CD28 co-stimulation177. For the most part, the induced expression of Bcl-xL upon T cell activation is due to transcription, with apparent little impact on AS177. Notably however, two identified regulators of Bcl-x AS, SF3B1(SAP155) and Sam68, are regulated respectively by PI3K and Fyn, two kinases activated by antigen engagement of T cells178–180. Therefore, it seems plausible that splicing of Bcl-x, in addition to transcription, maybe regulated in at least some stages of T cell activation. Moreover, as clearly shown in B cells, dysregulation of Bcl-x splicing often is a driver of cellular transformation and cancer181. An interesting question warranting further study, therefore, is whether dysregulation of Bcl-x is observed in other diseases of the immune system such as immunodeficiencies or autoimmunity.
Bim
Another Bcl-2 family member that is regulated at the level of alternative splicing is Bim (Fig. 7). Bim is a pro-apoptotic BH3-only protein that interacts with other Bcl2 family proteins such as MCL1, Bcl-xL, and BCL2 to promote apoptotic signaling133 (Fig. 6). Numerous lines of evidence demonstrate a critical role for Bim activity in regulating T cell development, survival, and function135,182. For example, knock-out of Bim in mice results in a severe block of T cell development in the thymus183, while in peripheral T cells, the absence of Bim reduces rates of AICD induced in both in vivo infection and in vitro stimulation184,185. Similarly, in human T cells knock-down of Bim by siRNA also protects cells from AICD185.
Bim is expressed as 3 major AS isoforms that arise due to alternative splicing. The three major isoforms exist due to the selective inclusion of exons 2a and 2c, and the intronic region between these two exons, which is also referred to as exon 2b (Fig. 7). BimEL is the longest isoform with the inclusion of exons 2a, 2b, 2c. BimL includes exons 2a and 2c, while BimS includes only exon 2186. While the functional differences between these isoforms has not been clearly documented, at least one study suggests that BimS has the greater pro-apoptotic activity than the longer isoforms187. Consistent with this notion, and the role of Bim in AICD, stimulation of CD4+ T cells results in a reduction in BimEL and an increase in BimS188. However, despite the clear AS changes in Bim between naïve and activated T cells, the mechanisms and RBPs involved in this regulation are entirely unknown. Moreover, given that most studies with Bim have focused on only one isoform, it would be of interest to directly compare the roles of the alternate isoforms in more cell types and conditions through manipulation of isoform expression by CRISPR or treatment with antisense oligonucleotides. Additionally, biochemical studies are needed to better understand if BimS, BimL or BimEL exhibit differential binding partners with other BCL2 family interaction members. Clearly the study of Bim is ripe for uncovering additional mechanisms and functional implications of AS in the immune system.
cFLIP
A final example of regulation of apoptosis by AS with relevance to the immune system is cFLIP (cellular FLICE-like inhibitory protein). cFLIP, encoded by CFLAR, is a negative regulator of the extrinsic pathway signaling events activated by death-receptor signaling (Fig. 6). cFLIP is structurally related to caspase-8, but lacks proteolytic activity; therefore, the competitive binding and heterodimerization of cFLIP to caspase-8 inhibits caspase-8 cleavage and subsequent activation189,190. Three major AS isoforms of cFLIP are expressed at the protein level in T cells including: cFLIPL, cFLIPS, cFLIPR (Fig. 7). All major cFLIP isoforms contain two DED domains, which allows for the heterodimerization with caspase-8189. The longest isoform, cFLIPL, also contains the catalytically inactive caspase-like domain. By contrast, cFLIPR and cFLIPS are truncated prior to the caspase-like domain due to inclusion of intron 6 or poison exon 7, respectively, both of which introduce a premature stop codon189. All isoforms maintain the capability to be a negative regulator of caspase-8 and −10 activation due to the DED domains; however, cFLIPL function is more complex, as under certain experimental conditions and concentrations it also can enhance Casp-8 activity via its caspase-like domain191,192. The cFLIP isoforms also appear to differ in their effects on T cell differentiation, in that siRNA knockdown of cFLIPL increased IFNgamma secretion in Th1 cells and IL4 production in Th2 cells, while siRNA knockdown of cFLIPS decreased IL4 production and GATA3 expression of Th2 cells193.
cFLIP is yet another example where isoform levels are differentially regulated upon T cell activation. Specfically, cFLIPS, but not cFLIPL, is upregulated in primary T cells activated with either PHA or anti-CD3 antibody and upon restimulation of effector T cells with anti-CD3 and anti-CD28 antibodies194–196. cFLIPS is also selectively upregulated by activated T cells polarized to the Th2 subset compared to Th1 and Th0 (unpolarized) cells, in a manner which is dependent on STAT6 signaling193. NFAT, p38alpha, and DHX32 have all been implicated to be specific inducers of cFLIPS expression and activity in T cells, although it is not clear if this is due to splicing or to selective stabilization of the cFLIPS protein191,194,197,198. Although the mechanisms by which RBP activity may regulate AS changes in cFLIP are not well characterized in T cells, a few studies have begun to characterize the role of RBP activity mediating cFLIP AS in other cell types. The knockdown of RNA binding proteins hnRNPA2 and hnRNPB1, and the combined deletion of RBM5,6, and 10, have both been demonstrated to increase the inclusion of exon 7 in glioblastoma cancer cells and HeLa cells, respectively, to promote expression of cFLIPS199,200.
One strong piece of evidence in favor of functional relevance of cFLIP AS is the discovery of a SNP that has been implicated to regulate inclusion levels of cFLIP isoforms. The Shwerk group discovered a SNP in the 3’ splice site of intron 6 (rs10190751A) that is characterized to enhance levels of cFLIPR compared to cFLIPS in humans201. Comparison of the frequency of rs10190751A between a population of healthy cells and primary follicular lymphoma samples revealed a significant association with the SNP in follicular lymphoma samples compared to control201, consistent with cFLIPS having a stronger anti-apoptotic impact than other isoforms195,196. The molecular mechanism by which rs10190751A increases cFLIPS is yet to be determined, although this likely involves changes in the binding of splicesomal components or trans-acting factors to regulate the abundance of cFLIPS. Additionally, the phenotypic consequences of having the SNP allele in a cancer context remains an important unanswered question.
Regulation of Apoptosis in the Immune System by Alternative Polyadenylation
CD5
Although, to date, there is little evidence for APA regulation of caspases, Bcl-2 proteins or other core apoptotic machinery, the expression of several regulators of apoptosis is controlled through APA. One example of this in T cells is the gene encoding the CD5 type-1 transmembrane glycoprotein (Fig. 8). CD5 is a regulator of TCR signaling whose expression is tightly controlled and regulated during T cell development and activation202,203. Upon T cell activation, tyrosine residues in intracellular domain of CD5 become phosphorylated, forming a docking site for multiple signal transduction proteins including PI3K, rasGAP and CK2204. One major consequence of CD5 activity is the regulation of activation induced cell death (AICD)202,203,205–208. CK2 is a pro-survival kinase and the CD5/CK2 signaling axis promotes cell survival in thymocytes208. Consistently, depletion of CD5/CK2 in mice reduces their susceptibility to experimental autoimmune encephalomyelitis (EAE) through increased apoptosis and accelerated AICD205. Separate from CDK2 signaling, CD5 also protects tumor-specific T cells from AICD by regulating the expression of FasL and thus activation of Caspase-8209.
Figure 8. Genes characterized to regulate apoptosis in T cells are regulated at the level of APA.

A. Three polyadenylation sites are utilized in the 3’UTR region of CD5. Proximal PAS usage (PAS 1, PAS2) are regulated by miR-204 and PTB activity. Proximal PAS usage correlates with increased protein output, as opposed to distal PAS usage. B. The presence of a SNP (rs6598) within the hexamer sequence of Gimap5 increases the usage of the distal PAS, compared to WT allele.
Although there have been limited studies detailing the mechanisms controlling CD5 expression, recently a group demonstrated that in T cells CD5 expression is regulated at the RNA level by APA210. Three polyadenylation sites are utilized in the 3’UTR region of CD5 (pa1, pa2, pa3) in resting Jurkat and primary T cells (Fig. 8). Upon T cell activation, there is a preference for the proximal PAS sites210; consequently, the length of the CD5 3’UTR becomes shorter. Use of luciferase reporter assays demonstrated that use of the most proximal PAS site of CD5 results in increased protein production as compared to the distal PAS sites210. The 3’ UTR sequences between the CD5 promixal and distal PAS sites contains a miR-204 binding site. Mutation of the miR-204 binding site in the longer 3’UTR phenocopies use of the proximal PAS in the luciferase assay210, demonstrating that the switch from distal to proximal PAS promotes CD5 expression by evading miRNA-mediated repression. Although the mechanism driving CD5 APA is not conclusively proven, biochemical studies suggest that binding of the hnRNP protein PTB near the CD5 proximal PAS promotes its use210. Obvious remaining questions of interest are whether there is a correlation between PTB and miR-204 levels and CD5 expression in the thymocytes and peripheral T cells, and if the perturbance of PTB and miR-204 have effects on T cell effector functions through CD5 regulation. Moreover, given that both the shortening of the 3’UTR of CD5 and the result of removal of a miRNA binding site, are consistent with the global studies on APA during T cell activation39, it will be interesting to determine if PTB has a more widespread impact of APA in T cells beyond CD5.
GIMAP5
GIMAP, GTPase immune-associated protein, is a family of small putative GTPases also referred to as immune-associated nucleotide-binding proteins (IANs)211,212. There are 7 GIMAP proteins encoded in the human genome, several of which play a role in T cell activity211,212. Notably, GIMAP5 not only plays an important anti-apoptotic role in T cells, but dysregulation of APA of this gene leads to autoimmune disease211,213 (Fig. 8).
The BB (Biobreeding) rat was the first model system to suggest GIMAP5 activity on apoptosis in T cells. The BB rat is prone to spontaneously acquire type-1 diabetes and also develops life-long lymphopenia. At least the lymphopenia in the BB rat is due to a frame-shift mutation of GIMAP5 that produces a truncated protein product214–216. The expression of this truncated GIMAP5 protein results in increased apoptosis rates and a shortened lifespan of T cells in the BB rat model, with a corresponding 10-fold reduction in CD4+ and CD8+ T cells216–218. Recent studies have associated this phenotype with impaired calcium signaling and loss of mitochondrial integrity in T cells, therefore insinuating a role of the intrinsic apoptosis pathway in mediating the increased apoptosis rates219–221. Parsing out the role of GIMAP5 in human T cells have led to contradicting results as one study found overexpressing this protein leads to apoptosis in Jurkat T cells and in PHA/IL2 activated primary human T cells grown without apoptotic inducers222, while another study showed that overexpression of GIMAP5 leads to increased resistance to apoptosis in Jurkat T cells induced with gamma-radiation and okadaic acid223. Overall, these studies suggest that GIMAP5 is regulating T cell apoptosis; however, more work is necessary to detail the exact role and context of GIMAP5 function.
Interestingly, GIMAP5 is regulated through APA in at least some autoimmune diseases. A SNP in the 3’UTR region of GIMAP5 is enriched in a subset of SLE patients compared to healthy controls and is also correlated with the increased presence of IA-2 autoantibodies in patients with type-1 diabetes224,225. The SNP (rs6598) converts the canonical hexamer sequence utilized by polyadenylation machinery (AATAAA) to the sequence, AATAGA (Fig. 8). The presence of the AATAGA hexamer sequence is correlated with a longer 3’UTR when comparing individuals that is homozygous for the allele against heterozygous and WT controls225. This SNP confirms an association between dysregulated GIMAP5 APA and disease correlation; however, it remains unknown how the change in 3’UTR alters GIMAP5 expression or function. Importantly, regardless of mechanism, this GIMAP5 SNP highlights the need to investigate sequence variations outside of coding sequences as potential drivers of disease and suggests the potential for additional unanticipated regulation of APA impacting apoptosis, as well as other activities of immune cells.
Conclusions and Future Directions
The regulation of gene expression in immune cells is, by necessity, highly complex to enable accurate and robust response to the many different challenges faced by innate and adaptive immune systems. Here we lay out existing evidence for a broad impact by both regulated splicing (AS) and 3’ end processing (APA) in shaping the function of immune cells and the immune response. We further describe some of the regulatory proteins and mechanisms that have been most widely implicated in the control of AS and APA in lymphocytes and macrophages. Most importantly, we highlight the many questions that remain unanswered with regards to AS and APA in immunity. In particular, we focus on the regulation of apoptosis during the immune response as a field ripe for further discovery. Indeed, beyond the gene regulation examples described above, RNA-Seq studies have already suggested AS changes in Bax and Casp-8 in T cells; and APA of cFLIP109,226; although none of these have been validated nor has the functional impact been explored. In addition, there are AS isoforms of other apoptosis signaling proteins that have been well studied in other cell types, but have yet to be investigated in immune cells, such as Casp-9227. In sum, the continued focus on transcriptomics in immune cells is predicted to yield many additional examples of how apoptosis and other critical features of immune regulation are controlled by AS and APA. Finally, given the proven ability to regulate isoform expression for therapeutic value228, further discovery of AS and APA in the immune system is also critical as it is predicted to suggest novel therapeutic avenues for the treatment of immunologic disease.
Acknowledgements
The authors acknowledge the support of NIH grants R35 GM118048 and GM118048-S1 (KWL) and F31 GM140978 (DB)
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152(6):1252–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol. 2016;23(12):1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sterne-Weiler T, Martinez-Nunez RT, Howard JM, et al. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013;23(10):1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Floor SN, Doudna JA. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agosto LM, Gazzara MR, Radens CM, et al. Deep profiling and custom databases improve detection of proteoforms generated by alternative splicing. Genome Res. 2019;29(12):2046–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ule J, Blencowe BJ. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol Cell. 2019;76(2):329–345. [DOI] [PubMed] [Google Scholar]
- 7.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiszbein A, Kornblihtt AR. Alternative splicing switches: Important players in cell differentiation. Bioessays. 2017;39(6). [DOI] [PubMed] [Google Scholar]
- 9.Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12(10):715–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayr C. Regulation by 3’-Untranslated Regions. Annu Rev Genet. 2017;51:171–194. [DOI] [PubMed] [Google Scholar]
- 11.Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18(1):18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev. 2013;253(1):216–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carpenter S, Ricci EP, Mercier BC, Moore MJ, Fitzgerald KA. Post-transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol. 2014;14(6):361–376. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev. 2003;193:70–81. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Xu X, Liu Y. Activation-induced cell death in T cells and autoimmunity. Cell Mol Immunol. 2004;1(3):186–192. [PubMed] [Google Scholar]
- 16.Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15(10):689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–1415. [DOI] [PubMed] [Google Scholar]
- 19.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136(4):777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montes M, Sanford BL, Comiskey DF, Chandler DS. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. 2019;35(1):68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Wu D, Zhang H, et al. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci Rep. 2018;8(1):10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbosa-Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338(6114):1587–1593. [DOI] [PubMed] [Google Scholar]
- 23.Ule J, Ule A, Spencer J, et al. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37(8):844–852. [DOI] [PubMed] [Google Scholar]
- 24.Lee JA, Damianov A, Lin CH, et al. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron. 2016;89(1):113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Licatalosi DD, Yano M, Fak JJ, et al. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 2012;26(14):1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci. 2012;37(5):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heyd F, Lynch KW. DEGRADE, MOVE, REGROUP: signaling control of splicing proteins. Trends Biochem Sci. 2011;36(8):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu X-D. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 29.Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 1988;2(2):215–227. [DOI] [PubMed] [Google Scholar]
- 30.Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19(5):327–341. [DOI] [PubMed] [Google Scholar]
- 31.Ray D, Kazan H, Cook KB, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499(7457):172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dominguez D, Freese P, Alexis MS, et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell. 2018;70(5):854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giudice J, Cooper TA. RNA-binding proteins in heart development. Adv Exp Med Biol. 2014;825:389–429. [DOI] [PubMed] [Google Scholar]
- 34.Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science. 2016;352(6292):1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litterman AJ, Kageyama R, Le Tonqueze O, et al. A massively parallel 3’ UTR reporter assay reveals relationships between nucleotide content, sequence conservation, and mRNA destabilization. Genome Res. 2019;29(6):896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma W, Mayr C. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3’UTR-Mediated Protein-Protein Interactions. Cell. 2018;175(6):1492–1506 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berkovits BD, Mayr C. Alternative 3’ UTRs act as scaffolds to regulate membrane protein localization. Nature. 2015;522(7556):363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29(9):889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008;320(5883):1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY, Ji Z, Tian B. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3’-end of genes. Nucleic Acids Res. 2008;36(17):5581–5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayr C, Bartel DP. Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia Z, Donehower LA, Cooper TA, et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat Commun. 2014;5:5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatrikhi R, Mallory MJ, Gazzara MR, et al. RNA Binding Protein CELF2 Regulates Signal-Induced Alternative Polyadenylation by Competing with Enhancers of the Polyadenylation Machinery. Cell Rep. 2019;28(11):2795–2806 e2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nazim M, Masuda A, Rahman MA, et al. Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res. 2017;45(3):1455–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi M, Zhang H, Wu X, et al. ALYREF mainly binds to the 5’ and the 3’ regions of the mRNA in vivo. Nucleic Acids Res. 2017;45(16):9640–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T-cell activation. RNA. 2007;13:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blencowe BJ, Ahmad S, Lee LJ. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 2009;23(12):1379–1386. [DOI] [PubMed] [Google Scholar]
- 48.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaquero-Garcia J, Barrera A, Gazzara MR, et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife. 2016;5:e11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park JW, Tokheim C, Shen S, Xing Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods Mol Biol. 2013;1038:171–179. [DOI] [PubMed] [Google Scholar]
- 51.Martinez NM, Pan Q, Cole BS, et al. Alternative splicing networks regulated by signaling in human T cells. RNA. 2012;18(5):1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Radens CM, Blake D, Jewell P, Barash Y, Lynch KW. Meta-analysis of transcriptomic variation in T-cell populations reveals both variable and consistent signatures of gene expression and splicing. RNA. 2020;26(10):1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West KO, Scott HM, Torres-Odio S, West AP, Patrick KL, Watson RO. The Splicing Factor hnRNP M Is a Critical Regulator of Innate Immune Gene Expression in Macrophages. Cell Rep. 2019;29(6):1594–1609 e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janssen WJ, Danhorn T, Harris C, et al. Inflammation-Induced Alternative Pre-mRNA Splicing in Mouse Alveolar Macrophages. G3 (Bethesda). 2020;10(2):555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H, Lorenzini PA, Zhang F, et al. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res. 2018;46(12):6069–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin J, Hu Y, Nunez S, et al. Transcriptome-Wide Analysis Reveals Modulation of Human Macrophage Inflammatory Phenotype Through Alternative Splicing. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(7):1434–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalam H, Fontana MF, Kumar D. Alternate splicing of transcripts shape macrophage response to Mycobacterium tuberculosis infection. PLoS Pathog. 2017;13(3):e1006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pai AA, Baharian G, Page Sabourin A, et al. Widespread Shortening of 3’ Untranslated Regions and Increased Exon Inclusion Are Evolutionarily Conserved Features of Innate Immune Responses to Infection. PLoS Genet. 2016;12(9):e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gruber AR, Martin G, Müller P, et al. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nature Communications. 2014;5(1):5465. [DOI] [PubMed] [Google Scholar]
- 60.Beisang D, Reilly C, Bohjanen PR. Alternative polyadenylation regulates CELF1/CUGBP1 target transcripts following T cell activation. Gene. 2014;550(1):93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh I, Lee SH, Sperling AS, et al. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat Commun. 2018;9(1):1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Birkeland ML, Johnson P, Trowbridge IS, Pure E. Changes in CD45 isoform expression accompany antigen-induced murine T cell activation. Proc Natl Acad Sci USA. 1989;86:6734–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deans DM, Boyd AW, Pilarski LM. Transitions from high to low molecular weight isoforms of CD45 (T200) involve rapid activation of alternate mRNA splicing and slow turnover of surface CD45R. J Immunol. 1989;143:1233–1238. [PubMed] [Google Scholar]
- 64.Saga Y, Lee JS, Saraiya C, Boyse EA. Regulation of alternative splicing in the generation of isoforms of the mouse Ly-5 (CD45) glycoprotein. Proc Natl Acad Sci. 1990;87:3728–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iwami KI, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J Immunol. 2000;165(12):6682–6686. [DOI] [PubMed] [Google Scholar]
- 66.Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of Interleukin-1- and Lipopolysaccharide-Induced NF-kappaB Activation by Alternative Splicing of MyD88. Curr Biol. 2002;12(6):467–471. [DOI] [PubMed] [Google Scholar]
- 67.Jacobsen M, Schweer D, Ziegler A, et al. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genet. 2000;26:495–499. [DOI] [PubMed] [Google Scholar]
- 68.Tchilian EZ, Beverley PC. CD45 in memory and disease. Arch Immunol Ther Exp (Warsz). 2002;50(2):85–93. [PubMed] [Google Scholar]
- 69.Blumhagen RZ, Hedin BR, Malcolm KC, et al. Alternative pre-mRNA splicing of Toll-like receptor signaling components in peripheral blood mononuclear cells from patients with ARDS. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L930–l939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108–117. [DOI] [PubMed] [Google Scholar]
- 71.Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. [DOI] [PubMed] [Google Scholar]
- 72.Streuli M, Hall LR, Saga Y, Schlossman SF, Saito H. Differential usage of three exons generates at least five different mRNAs encoding human leukocyte common antigens. J Exp Med. 1987;166:1548–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trowbridge IS, Thomas ML. CD45: An emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol. 1994;12:85–116. [DOI] [PubMed] [Google Scholar]
- 74.Lynch KW, Weiss A. A model system for the activation-induced alternative-splicing of CD45 implicates protein kinase C and Ras. Mol Cell Biol. 2000;20:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu Z, Weiss A. Negative regulation of CD45 by differential homodimerization of the alternatively spliced isoforms. Nat Immunol. 2002;3(8):764–771. [DOI] [PubMed] [Google Scholar]
- 76.Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 1998;279:88–91. [DOI] [PubMed] [Google Scholar]
- 77.Majeti R, Xu Z, Parslow TG, et al. An Inactivation Point Mutation in the Inhibitory Wedge of CD45 Causes Lymphoproliferation and Autoimmunity. Cell. 2000;103:1059–1070. [DOI] [PubMed] [Google Scholar]
- 78.Rothrock C, Cannon B, Hahm B, Lynch KW. A conserved signal-responsive sequence mediates activation-induced alternative splicing of CD45. Mol Cell. 2003;12(5):1317–1324. [DOI] [PubMed] [Google Scholar]
- 79.Tong A, Nguyen J, Lynch KW. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J Biol Chem. 2005;280(46):38297–38304. [DOI] [PubMed] [Google Scholar]
- 80.Rothrock CR, House AE, Lynch KW. HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 2005;24:2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Melton AA, Jackson J, Wang J, Lynch KW. Combinatorial control of signal-induced exon repression by hnRNP L and PSF. Mol Cell Biol. 2007;27(19):6972–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Topp JD, Jackson J, Melton AA, Lynch KW. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. Rna. 2008;14(10):2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science. 2008;321(5889):686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu Z, Jia X, de la Cruz L, et al. Memory T cell RNA rearrangement programmed by heterogeneous nuclear ribonucleoprotein hnRNPLL. Immunity. 2008;29(6):863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heyd F, Lynch KW. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell. 2010;40(1):126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yarosh CA, Tapescu I, Thompson MG, et al. TRAP150 interacts with the RNA-binding domain of PSF and antagonizes splicing of numerous PSF-target genes in T cells. Nucleic Acids Res. 2015;43(18):9006–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lynch KW, Weiss A. A CD45 polymorphism associated with Muliple Sclerosis disrupts an exonic splicng silencer. J Biol Chem. 2001;276:24341–24347. [DOI] [PubMed] [Google Scholar]
- 88.Zilch CF, Walker AM, Timon M, Goff LK, Wallace DL, Beverley PCL. A point mutation within CD45 exon A is the cause of variant CD45RA splicing in humans. Eur J Immunol. 1998;28:22–29. [DOI] [PubMed] [Google Scholar]
- 89.Tchilian EZ, Wallace DL, Imami N, et al. The exon A (C77G) mutation is a common cause of abnormal CD45 splicing in humans. J Immunol. 2001;166(10):6144–6148. [DOI] [PubMed] [Google Scholar]
- 90.Tchilian EZ, Wallace DL, Dawes R, et al. A point mutation in CD45 may be associated with an increased risk of HIV-1 infection. Aids. 2001;15(14):1892–1894. [DOI] [PubMed] [Google Scholar]
- 91.Dawes R, Hennig B, Irving W, et al. Altered CD45 expression in C77G carriers influences immune function and outcome of hepatitis C infection. Journal of medical genetics. 2006;43(8):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Motta-Mena LB, Smith SA, Mallory MJ, Jackson J, Wang J, Lynch KW. A disease-associated polymorphism alters splicing of the human CD45 phosphatase gene by disrupting combinatorial repression by heterogeneous nuclear ribonucleoproteins (hnRNPs). J Biol Chem. 2011;286(22):20043–20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479(7371):74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xiang JF, Corces VG. Regulation of 3D chromatin organization by CTCF. Curr Opin Genet Dev. 2021;67:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singh P, Lee DH, Szabo PE. More than insulator: multiple roles of CTCF at the H19-Igf2 imprinted domain. Front Genet. 2012;3:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neugebauer KM. Nascent RNA and the Coordination of Splicing with Transcription. Cold Spring Harb Perspect Biol. 2019;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Giono LE, Kornblihtt AR. Linking transcription, RNA polymerase II elongation and alternative splicing. Biochem J. 2020;477(16):3091–3104. [DOI] [PubMed] [Google Scholar]
- 98.Tellier M, Maudlin I, Murphy S. Transcription and splicing: A two-way street. Wiley Interdiscip Rev RNA. 2020;11(5):e1593. [DOI] [PubMed] [Google Scholar]
- 99.Marina RJ, Sturgill D, Bailly MA, et al. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. EMBO J. 2016;35(3):335–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nelson AM, Carew NT, Smith SM, Milcarek C. RNA Splicing in the Transition from B Cells to Antibody-Secreting Cells: The Influences of ELL2, Small Nuclear RNA, and Endoplasmic Reticulum Stress. J Immunol. 2018;201(10):3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marina RJ, Oberdoerffer S. Epigenomics meets splicing through the TETs and CTCF. Cell Cycle. 2016;15(11):1397–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yarosh CA, Iacona JR, Lutz CS, Lynch KW. PSF: nuclear busy-body or nuclear facilitator? Wiley Interdiscip Rev RNA. 2015;6(4):351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gaudreau MC, Heyd F, Bastien R, Wilhelm B, Moroy T. Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. J Immunol. 2012;188(11):5377–5388. [DOI] [PubMed] [Google Scholar]
- 104.Shankarling G, Cole BS, Mallory MJ, Lynch KW. Transcriptome-wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Mol Cell Biol. 2014;34(1):71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu Z, Yates AL, Hoyne GF, Goodnow CC. Consequences of increased CD45RA and RC isoforms for TCR signaling and peripheral T cell deficiency resulting from heterogeneous nuclear ribonucleoprotein L-like mutation. J Immunol. 2010;185(1):231–238. [DOI] [PubMed] [Google Scholar]
- 106.Benson MJ, Aijo T, Chang X, et al. Heterogeneous nuclear ribonucleoprotein L-like (hnRNPLL) and elongation factor, RNA polymerase II, 2 (ELL2) are regulators of mRNA processing in plasma cells. Proc Natl Acad Sci U S A. 2012;109(40):16252–16257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mallory MJ, Allon SJ, Qiu J, et al. Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. Proc Natl Acad Sci U S A. 2015;112(17):E2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mallory MJ, Jackson J, Weber B, Chi A, Heyd F, Lynch KW. Signal- and development-dependent alternative splicing of LEF1 in T cells is controlled by CELF2. Mol Cell Biol. 2011;31(11):2184–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martinez NM, Agosto L, Qiu J, et al. Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev. 2015;29(19):2054–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dasgupta T, Ladd AN. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2012;3(1):104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ajith S, Gazzara MR, Cole BS, et al. Position-dependent activity of CELF2 in the regulation of splicing and implications for signal-responsive regulation in T cells. RNA Biol. 2016;13(6):569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gazzara MR, Mallory MJ, Roytenberg R, et al. Ancient antagonism between CELF and RBFOX families tunes mRNA splicing outcomes. Genome Res. 2017;27(8):1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol. 2014;5(461). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell. 2020;180(6):1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jaresova I, Rozkova D, Spisek R, Janda A, Brazova J, Sediva A. Kinetics of Toll-like receptor-4 splice variants expression in lipopolysaccharide-stimulated antigen presenting cells of healthy donors and patients with cystic fibrosis. Microbes Infect. 2007;9(11):1359–1367. [DOI] [PubMed] [Google Scholar]
- 116.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197(2):263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jensen LE, Whitehead AS. IRAK1b, a novel alternative splice variant of interleukin-1 receptor-associated kinase (IRAK), mediates interleukin-1 signaling and has prolonged stability. J Biol Chem. 2001;276(31):29037–29044. [DOI] [PubMed] [Google Scholar]
- 118.Rao N, Nguyen S, Ngo K, Fung-Leung WP. A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in Toll/IL-1R-induced inflammatory signaling. Mol Cell Biol. 2005;25(15):6521–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yanagisawa K, Tago K, Hayakawa M, Ohki M, Iwahana H, Tominaga S. A novel splice variant of mouse interleukin-1-receptor-associated kinase-1 (IRAK-1) activates nuclear factor-kappaB (NF-kappaB) and c-Jun N-terminal kinase (JNK). Biochem J. 2003;370(Pt 1):159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Arras L, Alper S. Limiting of the innate immune response by SF3A-dependent control of MyD88 alternative mRNA splicing. PLoS Genet. 2013;9(10):e1003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.O’Connor BP, Danhorn T, De Arras L, et al. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet. 2015;11(2):e1004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Early P, Rogers J, Davis M, et al. Two mRNAs can be produced from a single immunoglobulin mu gene by alternative RNA processing pathways. Cell. 1980;20(2):313–319. [DOI] [PubMed] [Google Scholar]
- 123.Rogers J, Early P, Carter C, et al. Two mRNAs with different 3’ ends encode membrane-bound and secreted forms of immunoglobulin mu chain. Cell. 1980;20(2):303–312. [DOI] [PubMed] [Google Scholar]
- 124.Alt FW, Bothwell AL, Knapp M, et al. Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3’ ends. Cell. 1980;20(2):293–301. [DOI] [PubMed] [Google Scholar]
- 125.Peterson ML. Mechanisms controlling production of membrane and secreted immunoglobulin during B cell development. Immunol Res. 2007;37(1):33–46. [DOI] [PubMed] [Google Scholar]
- 126.Takagaki Y, Seipelt RL, Peterson ML, Manley JL. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell. 1996;87(5):941–952. [DOI] [PubMed] [Google Scholar]
- 127.Chuvpilo S, Zimmer M, Kerstan A, et al. Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity. 1999;10(2):261–269. [DOI] [PubMed] [Google Scholar]
- 128.Shell SA, Hesse C, Morris SM Jr., Milcarek C. Elevated levels of the 64-kDa cleavage stimulatory factor (CstF-64) in lipopolysaccharide-stimulated macrophages influence gene expression and induce alternative poly(A) site selection. J Biol Chem. 2005;280(48):39950–39961. [DOI] [PubMed] [Google Scholar]
- 129.Yang Q, Gilmartin GM, Doublié S. Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3’ processing. Proc Natl Acad Sci U S A. 2010;107(22):10062–10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yao C, Biesinger J, Wan J, et al. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci U S A. 2012;109(46):18773–18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peng Y, Yuan J, Zhang Z, Chang X. Cytoplasmic poly(A)-binding protein 1 (PABPC1) interacts with the RNA-binding protein hnRNPLL and thereby regulates immunoglobulin secretion in plasma cells. J Biol Chem. 2017;292(29):12285–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicologic Pathology. 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20(3):175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7(7):532–542. [DOI] [PubMed] [Google Scholar]
- 135.Zhan Y, Carrington EM, Zhang Y, Heinzel S, Lew AM. Life and Death of Activated T Cells: How Are They Different from Naïve T Cells? Front Immunol. 2017;8:1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lechner O, Lauber J, Franzke A, Sarukhan A, von Boehmer H, Buer J. Fingerprints of anergic T cells. Curr Biol. 2001;11(8):587–595. [DOI] [PubMed] [Google Scholar]
- 137.Fathman CG, Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol. 2007;7(8):599–609. [DOI] [PubMed] [Google Scholar]
- 138.Lleo A, Selmi C, Invernizzi P, Podda M, Gershwin ME. The consequences of apoptosis in autoimmunity. J Autoimmun. 2008;31(3):257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sankari SL, Babu NA, Rajesh E, Kasthuri M. Apoptosis in immune-mediated diseases. J Pharm Bioallied Sci. 2015;7(Suppl 1):S200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lamhamedi-Cherradi SE, Luan JJ, Eloy L, Fluteau G, Bach JF, Garchon HJ. Resistance of T-cells to apoptosis in autoimmune diabetic (NOD) mice is increased early in life and is associated with dysregulated expression of Bcl-x. Diabetologia. 1998;41(2):178–184. [DOI] [PubMed] [Google Scholar]
- 141.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. [DOI] [PubMed] [Google Scholar]
- 142.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–163. [DOI] [PubMed] [Google Scholar]
- 143.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. [DOI] [PubMed] [Google Scholar]
- 144.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23(6):1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. [DOI] [PubMed] [Google Scholar]
- 147.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8(2):115–128. [DOI] [PubMed] [Google Scholar]
- 148.Paronetto MP, Passacantilli I, Sette C. Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ. 2016;23(12):1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lin JC, Tsao MF, Lin YJ. Differential Impacts of Alternative Splicing Networks on Apoptosis. Int J Mol Sci. 2016;17(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stevens M, Oltean S. Modulation of the Apoptosis Gene Bcl-x Function Through Alternative Splicing. Front Genet. 2019;10:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Boise LH, Gonzalez-Garcia M, Postema CE, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74(4):597–608. [DOI] [PubMed] [Google Scholar]
- 152.Brunner T, Mogil RJ, LaFace D, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373(6513):441–444. [DOI] [PubMed] [Google Scholar]
- 153.Siegel RM, Ka-Ming Chan F, Chun HJ, Lenardo MJ. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nature Immunology. 2000;1(6):469–474. [DOI] [PubMed] [Google Scholar]
- 154.Paulsen M, Valentin S, Mathew B, et al. Modulation of CD4+ T-cell activation by CD95 co-stimulation. Cell Death Differ. 2011;18(4):619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cheng J, Zhou T, Liu C, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263(5154):1759–1762. [DOI] [PubMed] [Google Scholar]
- 156.Cascino I, Fiucci G, Papoff G, Ruberti G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol. 1995;154(6):2706–2713. [PubMed] [Google Scholar]
- 157.Liu C, Cheng J, Mountz JD. Differential expression of human Fas mRNA species upon peripheral blood mononuclear cell activation. Biochem J. 1995;310 ( Pt 3)(Pt 3):957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Papoff G, Cascino I, Eramo A, Starace G, Lynch DH, Ruberti G. An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J Immunol. 1996;156(12):4622–4630. [PubMed] [Google Scholar]
- 159.Roesler J, Izquierdo J-M, Ryser M, et al. Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95ΔTM), is the basis for ALPS Ia in a family with duplicated 3′ splice site AG in CD95 intron 5 on one allele. Blood. 2005;106(5):1652–1659. [DOI] [PubMed] [Google Scholar]
- 160.Cascino I, Papoff G, De Maria R, Testi R, Ruberti G. Fas/Apo-1 (CD95) receptor lacking the intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J Immunol. 1996;156(1):13–17. [PubMed] [Google Scholar]
- 161.Papo T, Parizot C, Ortova M, et al. Apoptosis and expression of soluble Fas mRNA in systemic lupus erythematosus. Lupus. 1998;7(7):455–461. [DOI] [PubMed] [Google Scholar]
- 162.Tejedor JR, Papasaikas P, Valcarcel J. Genome-Wide Identification of Fas/CD95 Alternative Splicing Regulators Reveals Links with Iron Homeostasis. Mol Cell. 2014. [DOI] [PubMed] [Google Scholar]
- 163.Izquierdo JM, Majos N, Bonnal S, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19(4):475–484. [DOI] [PubMed] [Google Scholar]
- 164.Forch P, Puig O, Kedersha N, et al. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol Cell. 2000;6(5):1089–1098. [DOI] [PubMed] [Google Scholar]
- 165.Izquierdo JM, Valcárcel J. Fas-activated Serine/Threonine Kinase (FAST K) Synergizes with TIA-1/TIAR Proteins to Regulate Fas Alternative Splicing. Journal of Biological Chemistry. 2007;282(3):1539–1543. [DOI] [PubMed] [Google Scholar]
- 166.Sánchez-Jiménez C, Izquierdo JM. T-cell intracellular antigens in health and disease. Cell Cycle. 2015;14(13):2033–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yin Y, Zhang S, Luo H, et al. Interleukin 7 up-regulates CD95 protein on CD4+ T cells by affecting mRNA alternative splicing: priming for a synergistic effect on HIV-1 reservoir maintenance. J Biol Chem. 2015;290(1):35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Izquierdo JM. Fas splicing regulation during early apoptosis is linked to caspase-mediated cleavage of U2AF65. Mol Biol Cell. 2008;19(8):3299–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Corsini L, Bonnal S, Basquin J, et al. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat Struct Mol Biol. 2007;14(7):620–629. [DOI] [PubMed] [Google Scholar]
- 170.Lu J, Li Q, Cai L, et al. RBM17 controls apoptosis and proliferation to promote Glioma progression. Biochem Biophys Res Commun. 2018;505(1):20–28. [DOI] [PubMed] [Google Scholar]
- 171.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6(6):e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Edlich F, Banerjee S, Suzuki M, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145(1):104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lindenboim L, Borner C, Stein R. Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ. 2001;8(9):933–942. [DOI] [PubMed] [Google Scholar]
- 175.Trisciuoglio D, Tupone MG, Desideri M, et al. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017;8(12):3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Behrens TW, Mueller DL. Bcl-x and the regulation of survival in the immune system. Immunol Res. 1997;16(2):149–160. [DOI] [PubMed] [Google Scholar]
- 177.Watts TH. Staying Alive: T Cell Costimulation, CD28, and Bcl-xL. The Journal of Immunology. 2010;185(7):3785–3787. [DOI] [PubMed] [Google Scholar]
- 178.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176(7):929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Massiello A, Roesser JR, Chalfant CE. SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5’ splice site selection of Bcl-x pre-mRNA. FASEB J. 2006;20(10):1680–1682. [DOI] [PubMed] [Google Scholar]
- 180.Shultz JC, Vu N, Shultz MD, Mba MU, Shapiro BA, Chalfant CE. The Proto-oncogene PKCiota regulates the alternative splicing of Bcl-x pre-mRNA. Mol Cancer Res. 2012;10(5):660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tessoulin B, Papin A, Gomez-Bougie P, et al. BCL2-Family Dysregulation in B-Cell Malignancies: From Gene Expression Regulation to a Targeted Therapy Biomarker. Front Oncol. 2018;8:645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bouillet P, O’Reilly LA. CD95, BIM and T cell homeostasis. Nature Reviews Immunology. 2009;9(7):514–519. [DOI] [PubMed] [Google Scholar]
- 183.Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. [DOI] [PubMed] [Google Scholar]
- 184.Hildeman DA, Zhu Y, Mitchell TC, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16(6):759–767. [DOI] [PubMed] [Google Scholar]
- 185.Snow AL, Oliveira JB, Zheng L, Dale JK, Fleisher TA, Lenardo MJ. Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct. 2008;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Miyashita T, Shikama Y, Tadokoro K, Yamada M. Molecular cloning and characterization of six novel isoforms of human Bim, a member of the proapoptotic Bcl-2 family. FEBS Lett. 2001;509(1):135–141. [DOI] [PubMed] [Google Scholar]
- 187.O’Connor L, Strasser A, O’Reilly LA, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17(2):384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Trushin SA, Carena AA, Bren GD, et al. SDF-1α degrades whereas glycoprotein 120 upregulates Bcl-2 interacting mediator of death extralong isoform: implications for the development of T cell memory. J Immunol. 2012;189(4):1835–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tsuchiya Y, Nakabayashi O, Nakano H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int J Mol Sci. 2015;16(12):30321–30341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Safa AR. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J Carcinog Mutagen. 2013;Suppl 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol. 2006;6(3):196–204. [DOI] [PubMed] [Google Scholar]
- 192.Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277(1):76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kylaniemi MK, Kaukonen R, Myllyviita J, Rasool O, Lahesmaa R. The regulation and role of c-FLIP in human Th cell differentiation. PLoS One. 2014;9(7):e102022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ueffing N, Schuster M, Keil E, Schulze-Osthoff K, Schmitz I. Up-regulation of c-FLIP short by NFAT contributes to apoptosis resistance of short-term activated T cells. Blood. 2008;112(3):690–698. [DOI] [PubMed] [Google Scholar]
- 195.Kirchhoff S, Muller WW, Krueger A, Schmitz I, Krammer PH. TCR-mediated up-regulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J Immunol. 2000;165(11):6293–6300. [DOI] [PubMed] [Google Scholar]
- 196.Kirchhoff S, Muller WW, Li-Weber M, Krammer PH. Up-regulation of c-FLIPshort and reduction of activation-induced cell death in CD28-costimulated human T cells. Eur J Immunol. 2000;30(10):2765–2774. [DOI] [PubMed] [Google Scholar]
- 197.Tourian L, Jr., Zhao H, Srikant CB. p38alpha, but not p38beta, inhibits the phosphorylation and presence of c-FLIPS in DISC to potentiate Fas-mediated caspase-8 activation and type I apoptotic signaling. J Cell Sci. 2004;117(Pt 26):6459–6471. [DOI] [PubMed] [Google Scholar]
- 198.Alli Z, Chen Y, Abdul Wajid S, Al-Saud B, Abdelhaleem M. A role for DHX32 in regulating T-cell apoptosis. Anticancer Res. 2007;27(1A):373–377. [PubMed] [Google Scholar]
- 199.Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcarcel J. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell. 2008;32(1):81–95. [DOI] [PubMed] [Google Scholar]
- 200.Golan-Gerstl R, Cohen M, Shilo A, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472. [DOI] [PubMed] [Google Scholar]
- 201.Ueffing N, Singh KK, Christians A, et al. A single nucleotide polymorphism determines protein isoform production of the human c-FLIP protein. Blood. 2009;114(3):572–579. [DOI] [PubMed] [Google Scholar]
- 202.Raman C. CD5, an important regulator of lymphocyte selection and immune tolerance. Immunol Res. 2002;26(1–3):255–263. [DOI] [PubMed] [Google Scholar]
- 203.Tabbekh M, Mokrani-Hammani M, Bismuth G, Mami-Chouaib F. T-cell modulatory properties of CD5 and its role in antitumor immune responses. Oncoimmunology. 2013;2(1):e22841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Voisinne G, Gonzalez de Peredo A, Roncagalli R. CD5, an Undercover Regulator of TCR Signaling. Front Immunol. 2018;9(2900). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Axtell RC, Webb MS, Barnum SR, Raman C. Cutting edge: critical role for CD5 in experimental autoimmune encephalomyelitis: inhibition of engagement reverses disease in mice. J Immunol. 2004;173(5):2928–2932. [DOI] [PubMed] [Google Scholar]
- 206.Alotaibi F, Rytelewski M, Figueredo R, et al. CD5 blockade enhances ex vivo CD8(+) T cell activation and tumour cell cytotoxicity. Eur J Immunol. 2020;50(5):695–704. [DOI] [PubMed] [Google Scholar]
- 207.Tabbekh M, Franciszkiewicz K, Haouas H, et al. Rescue of tumor-infiltrating lymphocytes from activation-induced cell death enhances the antitumor CTL response in CD5-deficient mice. J Immunol. 2011;187(1):102–109. [DOI] [PubMed] [Google Scholar]
- 208.Mier-Aguilar CA, Cashman KS, Raman C, Soldevila G. CD5-CK2 Signaling Modulates Erk Activation and Thymocyte Survival. PLoS One. 2016;11(12):e0168155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Friedlein G, El Hage F, Vergnon I, et al. Human CD5 protects circulating tumor antigen-specific CTL from tumor-mediated activation-induced cell death. J Immunol. 2007;178(11):6821–6827. [DOI] [PubMed] [Google Scholar]
- 210.Domingues RG, Lago-Baldaia I, Pereira-Castro I, et al. CD5 expression is regulated during human T-cell activation by alternative polyadenylation, PTBP1, and miR-204. Eur J Immunol. 2016;46(6):1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Filén S, Lahesmaa R. GIMAP Proteins in T-Lymphocytes. J Signal Transduct. 2010;2010:268589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Nitta T, Takahama Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol. 2007;28(2):58–65. [DOI] [PubMed] [Google Scholar]
- 213.Ciucci T, Bosselut R. Gimap and T cells: a matter of life or death. Eur J Immunol. 2014;44(2):348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.MacMurray AJ, Moralejo DH, Kwitek AE, et al. Lymphopenia in the BB Rat Model of Type 1 Diabetes is Due to a Mutation in a Novel Immune-Associated Nucleotide (Ian)-Related Gene. Genome Research. 2002;12(7):1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Michalkiewicz M, Michalkiewicz T, Ettinger RA, et al. Transgenic rescue demonstrates involvement of the Ian5 gene in T cell development in the rat. Physiological Genomics. 2004;19(2):228–232. [DOI] [PubMed] [Google Scholar]
- 216.Ramanathan S, Norwich K, Poussier P. Antigen activation rescues recent thymic emigrants from programmed cell death in the BB rat. J Immunol. 1998;160(12):5757–5764. [PubMed] [Google Scholar]
- 217.Woda BA, Like AA, Padden C, McFadden ML. Deficiency of phenotypic cytotoxic-suppressor T lymphocytes in the BB/W rat. J Immunol. 1986;136(3):856–859. [PubMed] [Google Scholar]
- 218.Lang JA, Kominski D, Bellgrau D, Scheinman RI. Partial activation precedes apoptotic death in T cells harboring an IAN gene mutation. Eur J Immunol. 2004;34(9):2396–2406. [DOI] [PubMed] [Google Scholar]
- 219.Serrano D, Ghobadi F, Boulay G, Ilangumaran S, Lavoie C, Ramanathan S. GTPase of the Immune-Associated Nucleotide Protein 5 Regulates the Lysosomal Calcium Compartment in T Lymphocytes. Front Immunol. 2017;8:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Pandarpurkar M, Wilson-Fritch L, Corvera S, et al. Ian4 is required for mitochondrial integrity and T cell survival. Proc Natl Acad Sci U S A. 2003;100(18):10382–10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Keita M, Leblanc C, Andrews D, Ramanathan S. GIMAP5 regulates mitochondrial integrity from a distinct subcellular compartment. Biochem Biophys Res Commun. 2007;361(2):481–486. [DOI] [PubMed] [Google Scholar]
- 222.Dalberg U, Markholst H, Hornum L. Both Gimap5 and the diabetogenic BBDP allele of Gimap5 induce apoptosis in T cells. Int Immunol. 2007;19(4):447–453. [DOI] [PubMed] [Google Scholar]
- 223.Sandal T, Aumo L, Hedin L, Gjertsen BT, Døskeland SO. Irod/Ian5: an inhibitor of gamma-radiation- and okadaic acid-induced apoptosis. Mol Biol Cell. 2003;14(8):3292–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Shin JH, Janer M, McNeney B, et al. IA-2 autoantibodies in incident type I diabetes patients are associated with a polyadenylation signal polymorphism in GIMAP5. Genes Immun. 2007;8(6):503–512. [DOI] [PubMed] [Google Scholar]
- 225.Hellquist A, Zucchelli M, Kivinen K, et al. The human GIMAP5 gene has a common polyadenylation polymorphism increasing risk to systemic lupus erythematosus. Journal of medical genetics. 2007;44(5):314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Eckhart L, Henry M, Santos-Beneit AM, et al. Alternative splicing of caspase-8 mRNA during differentiation of human leukocytes. Biochem Biophys Res Commun. 2001;289(4):777–781. [DOI] [PubMed] [Google Scholar]
- 227.Shultz JC, Chalfant CE. Caspase 9b: a new target for therapy in non-small-cell lung cancer. Expert Rev Anticancer Ther. 2011;11(4):499–502. [DOI] [PubMed] [Google Scholar]
- 228.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev.24(15):1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 1.Radens CM, Blake D, Jewell P, Barash Y, Lynch KW. Meta-analysis of transcriptomic variation in T-cell populations reveals both variable and consistent signatures of gene expression and splicing. RNA. 2020;26(10):1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H, Lorenzini PA, Zhang F, et al. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res. 2018;46(12):6069–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin J, Hu Y, Nunez S, et al. Transcriptome-Wide Analysis Reveals Modulation of Human Macrophage Inflammatory Phenotype Through Alternative Splicing. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(7):1434–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalam H, Fontana MF, Kumar D. Alternate splicing of transcripts shape macrophage response to Mycobacterium tuberculosis infection. PLoS Pathog. 2017;13(3):e1006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pai AA, Baharian G, Page Sabourin A, et al. Widespread Shortening of 3’ Untranslated Regions and Increased Exon Inclusion Are Evolutionarily Conserved Features of Innate Immune Responses to Infection. PLoS Genet. 2016;12(9):e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West KO, Scott HM, Torres-Odio S, West AP, Patrick KL, Watson RO. The Splicing Factor hnRNP M Is a Critical Regulator of Innate Immune Gene Expression in Macrophages. Cell Rep. 2019;29(6):1594–1609 e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janssen WJ, Danhorn T, Harris C, et al. Inflammation-Induced Alternative Pre-mRNA Splicing in Mouse Alveolar Macrophages. G3 (Bethesda). 2020;10(2):555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruber AR, Martin G, Müller P, et al. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nature Communications. 2014;5(1):5465. [DOI] [PubMed] [Google Scholar]
- 9.Beisang D, Reilly C, Bohjanen PR. Alternative polyadenylation regulates CELF1/CUGBP1 target transcripts following T cell activation. Gene. 2014;550(1):93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh I, Lee SH, Sperling AS, et al. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat Commun. 2018;9(1):1716. [DOI] [PMC free article] [PubMed] [Google Scholar]