

Abstract
An evolutionary-based definition and classification of target evaluation units (EUs) is presented for the 14th round of the Critical Assessment of Structure Prediction (CASP14). CASP14 targets included 84 experimental models submitted by various structure groups (designated T1024-T1101). Targets were split into EUs based on the domain organization of available templates and performance of server groups. Several targets required splitting (19 out of 25 multidomain targets) due in part to observed conformation changes. All in all, 96 CASP14 EUs were defined and assigned to tertiary structure assessment categories (Topology based FM or High Accuracy based TBM-easy and TBM-hard) considering their evolutionary relationship to existing ECOD fold space: 24 family level, 50 distant homologs (H-group), 12 analogs (X-group), and 10 new folds. Principal component analysis and heatmap visualization of sequence and structure similarity to known templates, as well as performance of servers highlighted trends in CASP14 target difficulty. The assigned evolutionary levels (i.e. H-groups) and assessment classes (i.e. FM) displayed overlapping clusters of EUs. Many viral targets diverged considerably from their template homologs and thus were more difficult for prediction than other homology-related targets. On the other hand, some targets did not have sequence-identifiable templates, but were predicted better than expected due to relatively simple arrangements of secondary structure elements. An apparent improvement in overall server performance in CASP14 further complicated traditional classification, which ultimately assigned EUs into high accuracy modeling (27 TBM-easy and 31 TBM-hard), topology (23 FM) or both (15 FM/TBM).
Keywords: CASP14, evolutionary structure classification, protein structure, fold space, protein domains, sequence homologs, structure analogs, structure prediction, template-based modeling, high accuracy modeling evaluation, free modeling, topology evaluation
1. INTRODUCTION
The Critical Assessment of Protein Structure Prediction (CASP) was envisioned as a large-scale experiment to establish current state-of-the-art methods in predicting protein structure from sequence1. In the experiment, prediction groups are provided the amino acid sequences of target structures whose experimental coordinates are not yet public. Independent assessment teams then evaluate models provided by the prediction groups by comparing them to the experimental structures. The tertiary structure prediction assessors in CASP14 concentrated on two broad categories of targets: high accuracy modeling and topology. The high accuracy modeling category, which was formerly known as template-based modeling or TBM, requires structure models of sufficient accuracy to evaluate the detailed placement of all atoms with respect to the target. The topology category, which was formerly free modeling or FM, addresses the more general placement of secondary structure elements (SSEs) in prediction models of lower quality. Some targets consist of multiple domains with different level of similarity to existing proteins. As such, CASP targets require 1) splitting into Evaluation Units (EUs) and 2) classification into the assessment categories for tertiary structure prediction evaluation.
Traditionally, assessment of CASP targets has been based on domains defined in the context of existing folds that can serve as structure templates for modeling 2–4. Furthermore, the evolutionary relationships between target domains and templates have influenced modeling difficulty5,6. As such, knowledge of sequence-structure relationships catalogued in the Evolutionary Classification of Protein Domains database (ECOD7) provides a solid basis for defining and classifying target domains. Although definitions can vary, domains essentially represent compact structural units that fold independently and act as building blocks for evolution8–10. Domains can be mobile, and their relative orientation in multidomain structures can differ. Thus, accurate assessment of model quality by many of the rigid protein structure comparison methods used in CASP automated evaluation11 has frequently required multidomain targets to be split into their constituent subunits and treated as independent EUs12. However, the recent progress in CASP performance, the availability of superposition independent scores (e.g. LDDT13, CAD-score14 or SphereGrinder15), and the increasing availability of multidomain templates is changing the requirements for splitting targets into EUs16–18.
The experimental structure community contributed 84 targets for assessment in CASP14, which were designated as T1024-T1101. Some of the targets belonged to multimeric complex structures and were indicated with a subunit suffix (i.e. “s1” in T1066s1). Fourteen of the targets were cancelled for various reasons: one was an unverified structure (T1098), one had a template with 100% sequence identity (T1072s2), nine had no structure provided in time for the evaluation (T1051, T1059, T1063, T1066s1 and s2, T1069s1 and s2, T1071, T1075) and 3 were only a single helix (T1048, T1062 and T1072s1). Two targets (T1077 and T1088) were assessed separately alongside the NMR-assisted predictions. Four targets were easier than others for tertiary structure prediction and thus were a-priori designated as “server only”. This report describes domain-based splitting of the remaining targets into EUs, and classification of those EUs into assessment classes for evaluation by high accuracy modeling (TBM-easy and TBM-hard) and topology (FM). A few EUs were assigned to both assessment categories (FM/TBM overlap).
2. METHODS
2.1. Definition of Evaluation Units
The Prediction Center preprocessed coordinate files obtained from experimentalists using similar methods as in previous rounds11. For certain NMR targets having loose ensembles of models (i.e. T1027 and T1029), regions with high flexibility (Cα-Cα deviation of more than 3.5Å between the same residues in different models) were excluded from the target. Target domains were defined initially by the Prediction Center using DomainParser219 and Ddomain20 packages. Automatic domains parsed by these programs were inspected manually, considering several criteria for establishing boundaries. The criteria included globular compactness of secondary structure elements, existence of internal duplications, maintenance of sequence continuity, and establishment of sequence-structure relationships to known folds (from HHpred21 alignments and LGA22 superpositions provided by the Prediction Center).
We evaluated the suitability of defined domains for the purpose of assessment using submitted models. Grishin plots12 provided comparisons of model performance (measured by GDT_TS23) for individual and combined domains. Domains were merged if performance on their combined subunits was comparable to individual subunits and if templates exist with similar domain compositions. Domains were split into separate EUs if performance on individual domains exceeded that on combined domains. For some templates with non-trivial domain organization, the process of defining domains and testing for splits was iteratively repeated with alternately defined boundaries. Decisions to split targets into EUs were also influenced by the existence of conformational changes. When multiple homologous templates existed for a target (see Evolutionary section below), the potential for conformational change was assessed using pairwise superpositions of all related templates (DaliLite Server24) and by examining corresponding literature. Those targets with alternate domain conformations were split into individual EUs. One large target (T1044) was pre-split into smaller targets (T1031, T1033, T1035, T1037, T1039, T1040, T1041, T1042, and T1043) prior to the modeling exercise. The pre-evaluation splits of T1044 were based on the same criteria as listed above, as well as suggestions from the experimentalist.
2.2. Determining Evolutionary Relationships of Targets to Known Templates
Target relationships to known structures were assigned using similar concepts as in the classification hierarchy of the Evolutionary Classification of structure Domains (ECOD) database 7,25,26. We assigned each target domain to its evolutionary position in known structure space (prior to the target’s release date) according to ECOD level (summarized in Table 1). For family level assignments, target sequences were submitted as queries to NCBI CD-Search27 against the conserved domain database28 with default parameters. We consider templates to be related by family level if their sequence identifies the same top family as the template sequence. For both complete and EU-split target sequences, HMM profiles were built using HHblits29 (2 iterations, E-value=1E-3) against UniRef30 database30 (version 2020.06). The resulting profiles were used to identify structure templates (PDB70 profiles updated weekly from http://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/) using HHsearch runs at the Prediction Center on the next day after the target closing date. . Additionally, a modified MSA generation method using PSI-BLAST against the nr70 was used to build search profiles for queries against the PDB70 database on the HHpred server31 (various dates, depending on the target closing date). When the topology of any of the identified sequence-based templates matched that of the target structure (matching topology is consistent with known ECOD relationships7,25,26, and is not in the same family) and the sequence-based and structure-based alignments matched, we consider these templates as homologous (H-group level relationship). To aid in homology and topology level assignments, top structure-based templates from the PDB were identified using structure coordinates of complete targets and split EUs using LGA22, Dalilite24, and RUPEE32. Structure-based templates without significant sequence relationship or other evidence for homology were considered as topology level matches (ECOD X-group). Targets lacking reasonable topology relationships to known structures were considered as new.
Table 1.
Evolutionary Assignment of CASP13 Targets among Existing Folds
| Target EU | Taxonomy | Class | ECOD Architecture | ECOD Assignment (X-group/H-group) | Level |
|---|---|---|---|---|---|
| T1024-D1 | Bacteria | TBM-easy | alpha complex | Major facilitator superfamily (MFS) transporter | H-group |
| T1024-D2 | Bacteria | TBM-easy | alpha complex | Major facilitator superfamily (MFS) transporter | H-group |
| T1025 | Bacteria | TBM-easy | a/b 3-layered sandwich | Rossmann-related | Family |
| T1026 | Virus | TBM-hard | beta sandwiches | Nucleoplasmin-like/VP (viral coat and capsid proteins) | H-group |
| T1027 | Eukaryota | FM | alpha arrays | na | New |
| T1028 | Bacteria | TBM-easy | a+b complex | C-type lectin-like | Family |
| T1029 | Bacteria | FM | a+b two layers | Type III secretory system chaperone-like | New |
| T1030-D1 | Bacteria | TBM-hard | alpha bundles | Bacterial immunoglobulin/albumin-binding domains | H-group |
| T1030-D2 | Bacteria | TBM-hard | alpha bundles | Bacterial immunoglobulin/albumin-binding domains | H-group |
| T1031 | Virus | FM | a+b two layers | dsRBD-like | X-group |
| T1032 | Eukaryota | TBM-hard | a+b duplicates/obligate | Smc hinge domain | Family |
| T1033 | Virus | FM | alpha arrays | Enhancer of polycomb-like protein 1 (Epl1) N-domain | X-group |
| T1034 | Eukaryota | TBM-easy | beta complex | Hedgehog/intein | Family |
| T1035 | Virus | FM/TBM | alpha arrays | na | New |
| beta complex | Viral glycoprotein ectodomain-like | ||||
| T1036s1 | Virus | TBM-easy | beta barrels | first barrel domain in viral glycoproteins | Family |
| beta barrels | Glycoprot B PH2 | ||||
| T1037 | Virus | FM | a+b complex | na | New |
| T1038-D1 | Virus | FM | a+b complex | na | New |
| T1038-D2 | Virus | FM/TBM | beta sandwiches | Immunoglobulin-related | H-group |
| T1039 | Virus | FM | alpha bundles | Fatty acid responsive transcription factor FadR, C-domain | X-group |
| T1040 | Virus | FM | alpha arrays | na | New |
| T1041 | Virus | FM | alpha complex | RNAi polymerase helical domain | H-group |
| T1042 | Virus | FM | alpha arrays | na | New |
| T1043 | Virus | FM | beta barrels | cradle loop barrel | X-group |
| T1045s1 | Eukaryota | TBM-easy | a+b two layers | UBC-like | Family |
| T1045s2 | Eukaryota | TBM-hard | a/b 3-layered sandwich | HAD domain-related | H-group |
| T1046s1 | Virus | FM/TBM | alpha bundles | Sigma2 domain-like | X-group |
| T1046s2 | Virus | TBM-hard | a+b three layers | Sensor domain | H-group |
| T1047s1 | Bacteria | FM | beta barrels | secretin domain | H-group |
| T1047s2-D1 | Bacteria | FM/TBM | beta barrels | RIFT-related | H-group |
| T1047s2-D2 | Bacteria | TBM-hard | a+b two layers | Ring-building motif II in type III secretion system | H-group |
| T1047s2-D3 | Bacteria | FM/TBM | a+b complex | na | New |
| T1049 | Bacteria | FM | beta sandwiches | Immunoglobulin-like beta-sandwich | X-group |
| T1050-D1 | Bacteria | TBM-easy | beta duplicates/obligate | beta-propeller | Family |
| T1050-D2 | Bacteria | TBM-easy | beta duplicates/obligate | beta-propeller | Family |
| T1050-D3 | Bacteria | TBM-easy | beta sandwiches | Immunoglobulin-related | Family |
| T1052-D1 | Virus | TBM-easy | beta duplicates/obligate | Pectin lyase-like | Family |
| beta sandwiches | Domain in virus attachment proteins | ||||
| T1052-D2 | Virus | TBM-easy | a/b 3-layered sandwich | SGNH hydrolase | H-group |
| T1052-D3 | Virus | FM/TBM | beta barrels | RIFT-related | H-group |
| T1053-D1 | Bacteria | FM/TBM | a+b complex | Protein kinase/SAICAR synthase/ATP-grasp | H-group |
| T1053-D2 | Bacteria | FM/TBM | alpha bundles | Bromodomain-like | X-group |
| T1054 | Bacteria | TBM-hard | a+b two layers | amino-terminal domain of OmpATb | H-group |
| T1055 | Virus | FM/TBM | alpha arrays | Sec63 N-terminal subdomain-like | H-group |
| alpha array | LEM/SAP HeH motif | H-group | |||
| T1056 | Virus | TBM-hard | a+b complex | alpha/beta-Hammerhead/Barrel-sandwich hybrid | Family |
| T1057 | Bacteria | TBM-easy | a/b 3-layered sandwich | Rossmann-related | Family |
| T1058-D1 | Bacteria | FM/TBM | alpha bundles | Transmembrane heme-binding four-helical bundle | H-group |
| T1058-D2 | Bacteria | TBM-hard | a+b two layers | Cystatin/monellin | H-group |
| T1060s2 | Virus | TBM-hard | beta barrels | RIFT-related | H-group |
| beta barrels | RIFT-related | ||||
| T1060s3 | Virus | TBM-hard | beta sandwiches | N-terminal Ig-like domain in baseplate protein ORF48 | H-group |
| beta barrels | RIFT-related | ||||
| T1061-D1 | Virus | FM/TBM | a+b complex | N0 domain in phage tail proteins and secretins | H-group |
| beta barrels | RIFT-related | ||||
| a+b complex | C-terminal insertion domain in phage tail proteins | ||||
| T1061-D2 | Virus | FM | beta sandwiches | jelly-roll | X-group |
| T1061-D3 | Virus | TBM-easy | beta sandwiches | Immunoglobulin-related | H-group |
| T1064 | Virus | FM | beta sandwiches | Immunoglobulin-related | H-group |
| T1065s1 | Bacteria | TBM-hard | a+b two layers | RelE-like | H-group |
| T1065s2 | Bacteria | FM/TBM | a+b two layers | na | New |
| T1067 | Bacteria | TBM-hard | beta complex | L,D-transpeptidase catalytic domain-like | Family |
| T1068-D1 | Eukaryota | TBM-hard | alpha arrays | Thymine dioxygenase JBP1 DNA-binding domain | H-group |
| T1070-D1 | Virus | FM | beta duplicates/obligate | Phage tail fiber protein trimerization domain | H-group |
| T1070-D2 | Virus | TBM-easy | beta sandwiches | gp9 N-terminal domain-related | H-group |
| T1070-D3 | Virus | TBM-hard | beta sandwiches | Agglutinin HPA-like | H-group |
| T1070-D4 | Virus | TBM-easy | beta sandwiches | Agglutinin HPA-like | H-group |
| T1073 | Bacteria | TBM-easy | alpha arrays | HTH | H-group |
| T1074 | Bacteria | FM | beta barrels | Lipocalins/Streptavidin | X-group |
| T1076 | Bacteria | TBM-easy | a/b 3-layered sandwich | Thiamin diphosphate-binding fold (THDP-binding) | |
| a/b 3-layered sandwich | Rossmann-related | Family | |||
| a/b 3-layered sandwich | Thiamin diphosphate-binding fold (THDP-binding) | ||||
| T1078 | Eukaryota | TBM-hard | beta barrels | Allergen Alt a 1 | H-group |
| T1079 | Bacteria | TBM-hard | a+b complex | Lysozyme-like | Family |
| alpha arrays | PGBD-like | ||||
| beta complex | L,D-transpeptidase catalytic domain-like | ||||
| T1080 | Bacteria | FM/TBM | beta duplicates/obligate | Phage tail fiber protein trimerization domain | H-group |
| T1082 | Virus | FM/TBM | alpha arrays | PABC(PABP) domain | H-group |
| T1083 | Bacteria | TBM-hard | alpha duplicates/obligate | Ferritin/Heme oxygenase/4-helical cytokines | X-group |
| T1084 | Bacteria | TBM-hard | alpha duplicates/obligate | Ferritin/Heme oxygenase/4-helical cytokines | X-group |
| T1085-D1 | Bacteria | TBM-hard | alpha superhelices | ARM repeat | H-group |
| T1085-D2 | Bacteria | FM/TBM | alpha superhelices | ARM repeat | H-group |
| T1085-D3 | Bacteria | TBM-hard | alpha superhelices | ARM repeat | H-group |
| T1086-D1 | Bacteria | TBM-easy | alpha superhelices | ARM repeat | Family |
| T1086-D2 | Bacteria | TBM-hard | alpha superhelices | ARM repeat | H-group |
| T1087 | Bacteria | TBM-hard | alpha duplicates/obligate | Ferritin/Heme oxygenase/4-helical cytokines | X-group |
| T1089 | Bacteria | TBM-easy | beta duplicates/obligate | beta-propeller | Family |
| T1090 | Eukaryota | FM | beta complex | ETN0001 domain-like | H-group |
| T1091-D1 | Bacteria | TBM-easy | beta sandwiches | Immunoglobulin-related | H-group |
| T1091-D2 | Bacteria | TBM-easy | beta sandwiches | Immunoglobulin-related | H-group |
| T1091-D3 | Bacteria | TBM-easy | beta sandwiches | Immunoglobulin-related | Family |
| T1091-D4 | Bacteria | TBM-easy | beta sandwiches | Immunoglobulin-related | Family |
| T1092-D1 | Virus | TBM-hard | a+b complex | N-terminal domain in RNA-polymerase beta-prime subunit | H-group |
| T1092-D2 | Virus | TBM-easy | beta barrels | RIFT-related | Family |
| T1093-D1 | Virus | FM | a+b complex | 1st helical domain in RNA-polymerase beta-prime subunit | H-group |
| T1093-D2 | Virus | TBM-hard | a+b complex | 2nd helical domain in RNA-polymerase beta-prime subunit | |
| a+b two layers | MoeA-I/ODC-C/Reverse ferredoxin-like domain in RNA-pol | H-group | |||
| T1093-D3 | Virus | FM | a+b complex | alpha/beta-Hammerhead/Barrel-sandwich hybrid | H-group |
| T1094-D1 | Virus | TBM-hard | a+b complex | N-domain in beta subunit of DNA dependent RNA-pol | Family |
| T1094-D2 | Virus | FM | a+b complex | insert domain in beta subunit of DNA dependent RNA-pol | H-group |
| beta barrels | RIFT-related | ||||
| T1095 | Virus | TBM-hard | a+b complex | alpha/beta-Hammerhead/Barrel-sandwich hybrid | |
| a+b complex | alpha/beta-Hammerhead/Barrel-sandwich hybrid | ||||
| a+b complex | C-domain in beta subunit of DNA dependent RNA-pol | Family | |||
| T1096-D1 | Virus | FM | alpha complex | Sigma2 domain of RNA polymerase sigma factors | H-group |
| T1096-D2 | Virus | FM | alpha complex | na | New |
| T1099 | Virus | TBM-hard | alpha arrays | Hepatitis B viral capsid (hbcag) | Family |
| T1100-D1 | Archaea | TBM-hard | extended segments | NarQ transmembrane domain | H-group |
| alpha duplicates/obligate | HAMP domain | ||||
| T1100-D2 | Archaea | TBM-hard | a+b three layers | sensor domains | H-group |
| T1101-D1 | Eukaryota | TBM-easy | a+b two layers | KH-domains | H-group |
| T1101-D2 | Eukaryota | TBM-easy | beta barrels | LigT-related | Family |
Table 1. CASP14 target EU information. Taxonomic group, assessment class, and ECOD hierarchy: ECOD Architecture retains similar secondary structure compositions and geometric shapes, ECOD name assignment to X-groups that display similar topology but lack justification for homology, H-groups with homologous folds, or “na” for new folds, with hierarchy level in last column.
To calculate the distribution of CASP targets among ECOD architectures, observed frequencies for CASP13 and CASP14 target domains were divided by expected frequencies based on the ECOD domain counts (database version 277). ECOD domains were made non-redundant at the family level by keeping a single representative from each assigned family ID (14,251 total non-redundant domains). Expected frequencies were calculated by dividing the non-redundant domain counts in each architecture by the total number of non-redundant domains, and observed frequencies were calculated by dividing the number of target domains in each architecture by the total number of target domains in CASP13 and CASP14.
2.3. Combining Prediction Center Metrics to Classify Targets
The Prediction Center provides a number of metrics that can be used to help classify templates11. We chose the same metrics as in previous rounds of CASP5,16 to assign CASP14 target EUs to evaluation categories. The sequence-structure relationship of EUs to known folds (measured by the average of HHscore and LGA_S of targets to top templates) was compared to the top20 server performance (average server model 1 GDT_TS) in a scatter plot. The HHscore (product of the alignment coverage of the query and the HHsearch probability5) was calculated for all top-ranked templates identified by any of the search methods described above. We considered the template with the highest HHscore (among all methods) as the top sequence-based template for classification and combined this score with the top-scoring structure template (by LGA_S22), regardless of the template’s relationship to the target.
For non-trivial assignments, we clustered EUs using principal component analysis and heatmaps from the ClustVis web tool33 using the three measures from the scatterplot (HHscore, LGA_S, and top 20 server performance) and a number of additional measures described below that were developed to assess target difficulty. To avoid scaling the measures, we chose scores that could be calculated as percentages (from 0 to 100), which match the scaling of the classification plot metrics. For those servers that declared “PARENT” templates for some targets and no templates for others, we calculated the percentage of declared templates (%parentTBM) for each target by summing up the number of all models with a declared template and dividing by the total number of models for all selected server groups. The number of effective sequences for each target (Neff%Max) was calculated based on the entropy of the MSA used for HHpred searches (Neff header in HHpred results files). To calculate the percentage, we divided the Neff by the theoretical maximum entropy of 2031. Finally, we included two scores from Dalilite: coverage (DaliCvg) calculated as the length of the alignment divided by the number of target residues and the DaliZ score (Dali%Max) expressed as a percentage of the maximum (from DaliZ target self-score). Data was preprocessed using the Singular Value Decomposition (SVD) with imputation option in ClustVis33 to replace missing values with no scaling. Rows were centered for Principal Component Analysis (PCA) and prediction ellipses were drawn around the clustered features that represent 0.95 probability a new observation from the same group will fall inside. Heatmap rows were clustered using correlation distance and Ward linkage, and columns were clustered using Euclidean distance and Ward linkage. To help classify target EUs near difficulty category boundaries in the traditional classification plot, we considered the following features in PCA and heatmap: preliminary classification based on the traditional plot (FM, FM/TBM, TBM-hard, and TBM-easy), evolutionary relationship to known structures (Family, H-group, X-group, and New) and taxonomy (Virus, Archaea, Bacteria, and Eukaryota).
3. RESULTS AND DISCUSSION
3.1. Domain-Based Definition of Evaluation Units
For domain-based establishment of EUs in CASP14, we considered similar criteria for domain definition as in previous rounds of CASP (i.e. domain parser results, internal duplications, sequence continuity, and sequence/structure relationships). For defined multidomain targets, the decision to split into EUs was based on performance comparisons in Grishin plots as well as on the presence of flexible templates with alternate conformations. Domains in previous CASPs often crossed assessment categories2,5,6,12,16 due to evolutionary mechanisms that favor their recombination into new functional units 10. Such domain mobility posed a problem for classification of CASP14 targets into evaluation categories. For example, the V. cholera effector MavQ target (T1053) included two domains with different evolutionary relationships (Figure 1A). Sequence-based templates existed for the N-terminal protein kinase-like domain. Yet, the C-terminal helical bundle was unique, with templates that were related only by structure. Accordingly, the Grishin plot (Figure 1B) supported splitting the target domains into independent EUs (T1053-D1 and T1053-D2). In contrast, the first two domains from the Salmonella phage epsilon15 tail spike protein (Figure 1C, T1052) existed in the same orientation in sequence-related templates (for example the tails pike from Salmonella phage Det7, 6f7d), and the Grishin plot supported merging the two domains into a single EU (Figure 1D). Thus, for this target the domain count and the EU count differed.
Figure 1. Domain Based Definition of Evaluation Units.
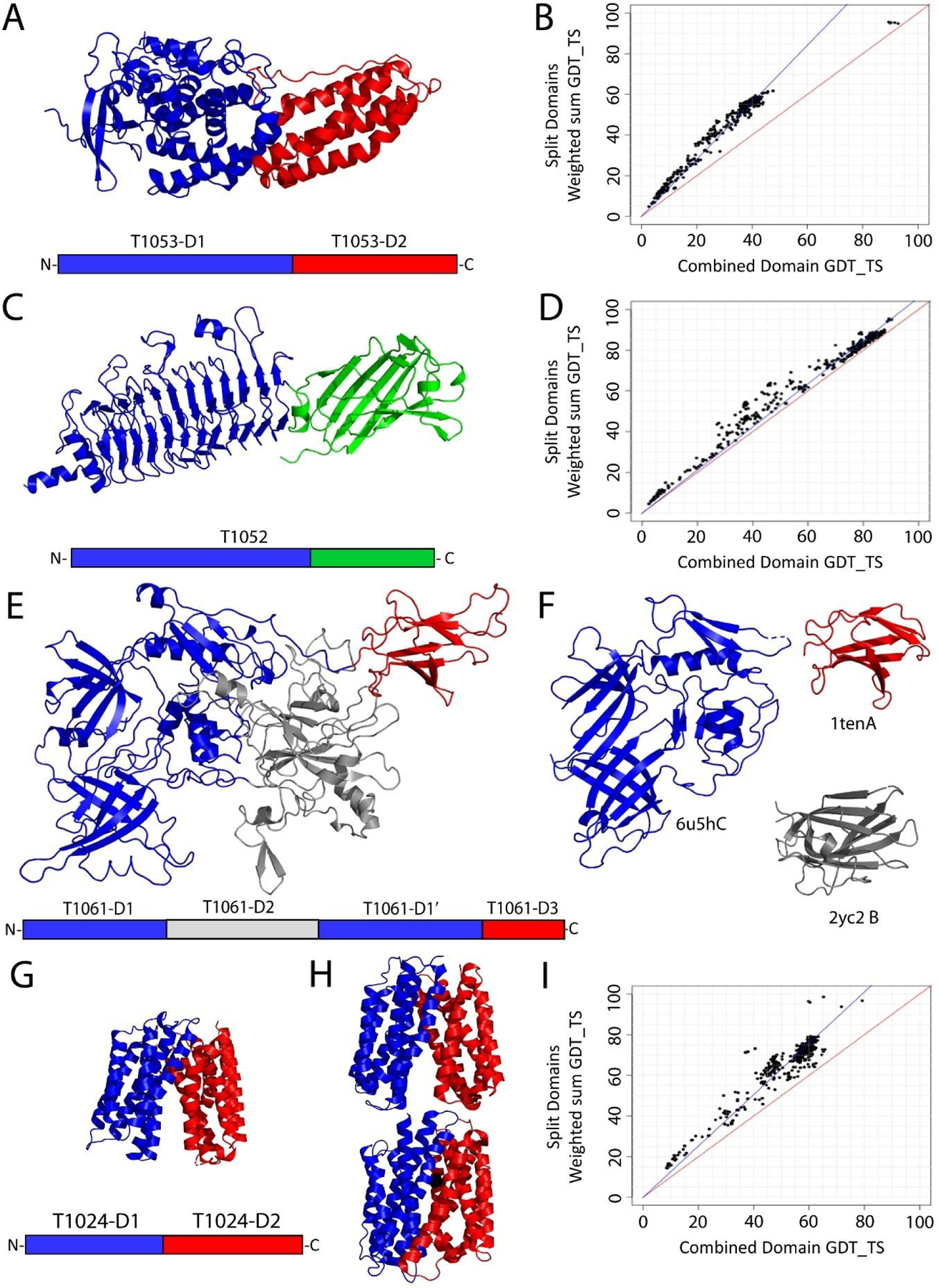
A) Simple domain organization for Target T1053 (primary sequence schematic below), which has an N-terminal protein kinase-like domain (blue) and a unique C-terminal helical bundle (red). B) T1053 Grishin plot demonstrates higher modeling accuracy of individual domains (Y-axis) when compared to whole target (X-axis), and thus suggests splitting domains. C) Target T1052-D1 includes two domains (schematic below). D) T1052-D1 Grishin plot suggests merging domains (the data regression line running close to the diagonal). E) Complex domain organization of T1061 (schematic below): T1061-D1 (blue) has a unique insert (T1061-D2, gray) followed by T1061-D3 (red). F) T1061 sequence templates: left Pyocin R2 (blue), upper right Fibronectin type III (red). Lower right: structure template (gray). G) The MFS transporter T1024 has 12 TMH formed by an internal duplication of two 6TMH domains (colored blue and red, primary sequence schematic below). H) MFS templates adopt outward-facing (3wdo, upper panel) and inward-facing (4j05, lower panel) conformations by changing the relative orientation of the two domains. I) Shifted clouds of combined domain performance in the T1024 Grishin plot reflect model choice of alternate conformations.
Defining the domain boundaries was difficult for a multidomain target (T1061) representing E. coil phage tail (Figure 1E). Automatic domain parsing programs were inconsistent, and their results produced sequence discontinuous domains that tended not to reflect evolution. Fortunately, sequence-related templates of pyocin R2 (6u5hC), which consisted of four domains in ECOD (Figure 1F, left), and a tenascin fibronectin type III domain (Figure 1F, upper right), helped define a sequence insert that was unique to the target (Figure1E, gray). The unique insert identified templates with low structure scores (Figure 1F lower left, top LGA_S 21.2 to 2yc2B) due to numerous SSE decorations and deteriorated edge strands of its core jelly-roll fold. The decorated domain was inserted in between two intimately associated RIFT-related barrels (the second T1061-D1’ included two additional continuous domains), which resulted in a complex target domain organization (Figure 1E, lower schematic). We chose to merge the four domains into a single EU (T1061-D1), which shifted the target difficulty as measured by performance. The top performance on independent domains corresponding to the first RIFT-related domain (max GDT_TS 88.0) and the remaining domains (T1061-D1’, max GDT_TS 89.6) was better than on the sequence discontinuous T1061-D1 EU (max GDT_TS 77.1).
Some CASP14 targets exemplified proteins that can adopt multiple conformations, such as the major facilitator superfamily (MFS) target T1024 (Figure 1G). MFS transporters move substrates across membranes using an alternating access model where the protein adopts inward and outward-facing conformations34. The conformational changes occur primarily between two duplicated domains of six core transmembrane helices (TMH), and templates existed for both the inward and outward-facing states (Figure 1H). Accurate modeling of the conformation tends to be difficult for such targets without additional information about ligands, chemical modifications, detergent composition for solubilization, or other criteria that stabilize one conformation over the other. As such, we split CASP14 targets into independent EUs when template homologs existed with alternate domain conformations. The performance scatter in the Grishin plot for T1024 (Figure 1I) highlighted two clouds, with one of the clouds shifted towards better performance on the merged domains. These two clouds roughly corresponded to the two conformation states of the target.
One unique component of CASP14 was a requirement for a large target structure to be pre-split into component domains prior to its release to predictors (due to an expectation of difficulty in acquiring targets during a global pandemic and large size of the target). The 2166-residue phage DNA-dependent RNA polymerase structure (T1044) was split into sequence fragments corresponding to 12 domains, nine of which were released for prediction as separate targets (T1031, T1033, T1035, T1037, T1039, T1040, T1041, T1042, and T1043). While similar fragments of larger proteins have often been used for X-ray crystallography and NMR, their successful structure determination would presumably require the fragments to fold into stable units. Unfortunately, the domains defined a priori for T1044 do not necessarily correspond to stable folding units that can exist independently. Although similar considerations were used to pre-define domains for T1044 as they were for the remaining targets, the boundaries were not based on performance and could not be iteratively adjusted.
In summary, CASP14 consisted of a total of 84 targets. This number included both the whole multidomain target (T1044) and its nine pre-evaluation split domains (listed above). Of the remaining targets, 33 were considered as single domains and 25 were multidomain, ranging anywhere from 2 to 6 domains. Most of the multidomain targets were split into their component domains (15 targets were split into 37 EUs), while a smaller number were considered as single EUs that merged all the domains (6 targets). The four remaining multidomain targets had a combination of both combined and split domains, with a total of 17 domains that were split into 11 EUs. Given the number of multidomain targets with performance plots that suggested splitting, CASP14 introduced a new category of assessment to evaluate predictor performance on domain interactions ([Inter-domain assessment, Schaeffer et al, this issue]). This new assessment evaluated inter-domain interactions in a similar manner as the evaluation of multimeric targets. The chosen multidomain target subset excluded those targets that 1) were split due to conformation changes, 2) lacked interactions or 3) had interactions dictated by their oligomeric state.
3.2. Evolutionary relationships of targets to known folds
An essential component of defining target EUs and classifying them into assessment categories was understanding their relationship to fold space, as the nature of these relationships (in terms of sequence/structure similarity) has tended to correlate with target difficulty5,16. The distribution of CASP14 EUs among evolutionary levels (summarized in Table 1) changed from that of CASP13 (figure 2A). In CASP13 the most populated evolutionary group represented family-level similarity and included almost half of the dataset (46%), while in CASP14 this group included only quarter of targets, whereas the most populated group represented distantly related homology (H-group, 52% of the targets). This shift toward more distant template relationships forecasted an increase in CASP14 prediction difficulty. This difficulty was further elevated by a more challenging taxonomic distribution of CASP14 targets (Figure 2B), where a notable share of entries was of viral origin (44%). Given the evolutionary pressures of viral proteins to adapt to their hosts35, their classification in fold space as well as their structure prediction tends to be challenging. When compared to the family-level domain space of ECOD, complex structure architectures as well as duplicates/obligate multimers, which tended to be more difficult, were overrepresented in CASP14 (Figure 2C). On the contrary, architectures with more regular SSEs, such as a+b two layers or a/b barrels were under-represented or missing altogether. This representation is skewed when compared to the somewhat more regularly distributed CASP13 target domains.
Figure 2. Target Relationships to Known Folds.
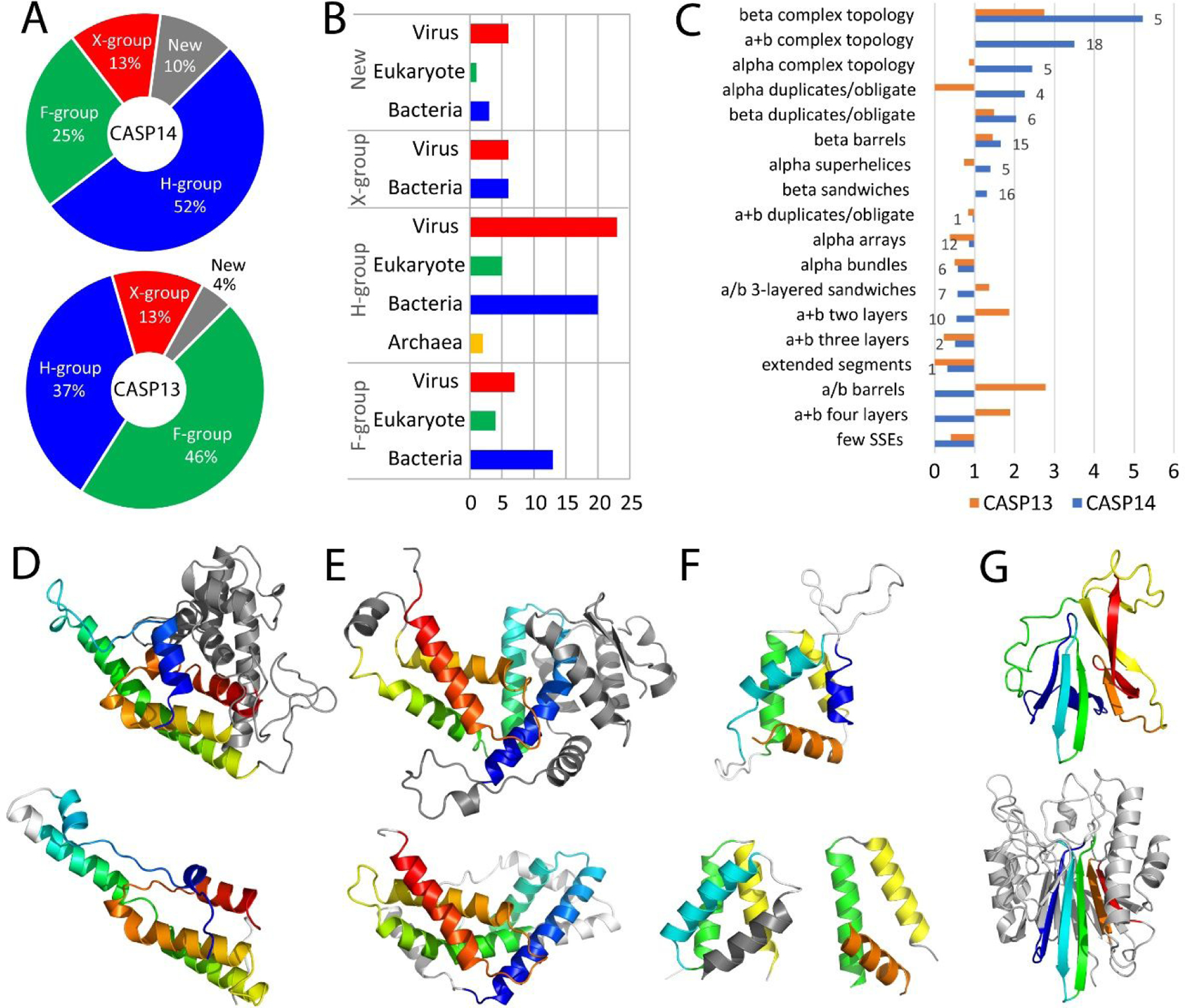
A) Pie chart distribution of CASP14 EUs (upper) among ECOD evolutionary categories is compared to CASP13 (lower). B) Bar chart depicts counts of CASP14 EUs in taxonomic groups split into ECOD evolutionary categories. C) Observed ECOD architecture (labeled) frequencies of target domains from CASP14 (blue) and CASP13 (orange) are overrepresented (above 1) and underrepresented (below 1) as compared to expected frequencies of all ECOD family-level domains. The number of observed CASP14 domains are indicated to the outside of the bar. D) CrAss-like phage polymerase domain (T1041, upper) includes large extension (gray) to the core set of SSEs (colored in rainbow from N- to C- terminus) that are present in the closest homolog from RNAi polymerase (PDB: 2j7nA, lower, white inserts). E) Bacillus phage polymerase subunit domain (T1096-D1, upper) is compared to top structure template homolog (PDB:3les, lower), colored as above. F) New fold in CrAss-like phage polymerase helical array domain (T1035, upper), with SSEs colored in rainbow, can be assembled from analogous SSE arrangements in : H2-H4 in repetitive alpha hairpin of unknown function (PDB: 6e4j, lower left) and H1-H3 in a helical subdomain from gamma-tubulin complex component 5 (PDB: 6l81, lower right). G) New fold in a+b complex topology domain from viral glycoprotein precursor (T1038-D1, upper), with SSEs colored in rainbow, is compared to an unrelated a+b four layers domain from top the structure template (PDB: 3i48, lower), with corresponding SSEs colored similarly.
CASP14 targets were assigned at the family level when they belonged to the same sequence group (defined by the conserved domain database28) as their template (24 EUs). Due to an increased similarity between family level target/template structures (average LGA_S 72.2), many of the multidomain targets were merged into single EUs (T1036s1, T1052, T1076, T1079, T1095 and T1099) at this level. However, some were split into their component domains due to the existence of templates with alternate conformations. The Bacteroides ovatus response regulator (T1050) was assigned to the periplasmic ligand-binding sensor domain superfamily (COG3292). Hybrid two-component system sensor templates belonging to this superfamily adopted alternate conformations in the apo and ligand-bound state (PDB: 4a2m and 4a2l 36). The N-terminal beta-propeller of one template rotated with respect to the adjacent beta-propeller and immunoglobulin-related domains of the other. Thus, high accuracy prediction of domain interactions for this target would require knowledge of the ligand, and the model quality in Grishin plots reflected the choice of template conformations (data not shown). Templates for the pilus tip adhesin PitA (T1091) also exhibited a conformation change, and the target was split into its four immunoglobulin-related domains. Two of the four domains were recognized by sequence at the family level, and the others presumably evolved beyond sequence recognition (H-level). Several other family-level multidomain targets were split when their component domains belonged to different evolutionary categories (T1086, T1091, T1092, T1094 and T1101), while the domains from a thermophilic worm structure (T1101) represented a novel combination of a eukaryotic KH domain (H-group) and a LigT-related domain (pfam10469) with a potential conformation change (noted by the experimentalist).
CASP14 targets that were distantly related to their template homologs (H-group) tended to diverge significantly from their templates (average LGA_S 57.4). For example, the top-scoring structure template for a CrAss-like phage DNA-dependent RNA polymerase domain (T1041, defined as a pre-evaluation split) was from a distantly related homolog, RNAi polymerase from Neurospora crassa (Figure 2D). The T1041 domain included a relatively large helical extension not found in the RNAi polymerase or other existing folds. The extension, along with other diverging α-helices, resulted in a low structure similarity score for the target when compared to the top template (LGA_S 25.03). Similarly, a domain (T1096-D1) from one subunit of the bacillus phage AR9 DNA-directed RNA polymerase had a top structure template from a distantly related homolog, RNA polymerase sigma factor SigA from Thermus aquaticus (Figure 2E). The T1096-D1 target had a relatively large N-terminal extension and diverging α-helical orientations, which resulted in the lowest structure score (LGA_S 20.84) among the H-group EUs. Examples like these included novel SSE decorations that might be more difficult to model as a significant portion of their structure. Almost half of the H-group targets (23 EUs), like these two examples, were viral proteins that tended to evolve rapidly.
The X-group targets (12EUs, Figure 2A) were related to existing templates by topology, and lacked any other justification typically used to infer homology7. Most of the X-group target EUs adopted folds with common topologies that could have arisen multiple times in evolution: such as immunoglobulin-like β-sandwich, bromodomain-like helix bundle, or RIFT-related barrel. These targets lacked sequence evidence for homology and their structures had relatively low scores to top templates (average LGA_S 41.5). The remaining X-group target EUs exhibited simple topologies with few SSEs and high structure scores to fragments of unrelated targets (average LGA_S 82.1). For example, the anti-parallel helix pairs of T1083, T1084, and T1087 had varying degrees of histidine-rich sequence that resembled a DELD family protein Dld1 (PDB: 5los, HHprob 65 to T1087) with a monomeric histidine zipper of anti-parallel coiled-coils37. However, the targets lacked the DELD motif and formed four-helix bundles from dimers of antiparallel helix hairpins. This arrangement prompted their placement in the alpha duplicates/obligate multimer architecture of ECOD, and the topology of dimers from T1087 and T1083 (T1084 was a mirror image) resembled that found in the ferritin/heme oxygenase (FHO) X-group of ECOD (where we placed the targets). The top structure templates for each of the three target hairpins are analogous helix pairs from unrelated bacterial hemolysins (LGA_S range from 84.8 to 92.9). Similarly, the simple three-helix bundle from T1046s1 identified a top partial structure template from an α-helical array (LGA_S 74.6 to 5utgA).
Finally, CASP14 targets included several new folds (10EUs, Figure2A). Four of these were from the pre-evaluation defined domains of CrAss-like phage DNA-dependent RNA polymerase (T1035, T1037, T1040, and T1042), which has since been described as having much of its structure in a “new region of fold space”38. While the overall SSE topology was unique in targets assigned to the new fold category, most could be assembled from analogous SSE arrangements existing in the PDB. The phage polymerase domain T1035 adopted an array of five α-helices with mainly perpendicular orientations (Figure 2F). While no existing template included the same topology, the array could be assembled from multiple analogous structures. The T1035 helix H2-H4 could be represented by three helices in a repetitive α-hairpin from uncharacterized protein PF2048.1 (PDB: 6e4jA1), and the helix H3-H5 could be represented by three helices from gamma-tubulin complex component 5 (PDB: 6l81A). Another new fold from the N-terminal domain in tomato spotted wilt tospovirus glycoprotein precursor (T1038-D1) adopted an a+b complex topology architecture (Figure 2G). The top structure template (3i48, LGA_S 40.5) belonged to a different a+b four layers architecture from Staphylococcus aureus beta toxin and is unrelated. The template SSEs from a central β-sheet in one of the layers corresponded to a β-sheet in the target formed by a three-stranded β-meander and an interacting β-hairpin. This target could also be separated into SSE arrangements existing in known structures.
3.3. Target Difficulty Highlighted by PCA and Heatmap Clustering of EUs
In the previous round of CASP, the top-performing state-of-the-art methods utilized deep learning techniques for predicting protein structures17,18,39. Such knowledge-based prediction methods relied on information gleaned from sequence and structure databases. Thus, combining scores that emulated the evolutionary relationships of targets to known folds helped to establish their difficulty. To achieve this task in CASP14, we chose several scores that represented sequence, structure, and performance components of the target EUs (see methods for score description). The scores were clustered as multivariate data, and principal component analysis was used to visualize the variance of target features, such as assigned ECOD classification level (Figure 3A) and potential tertiary structure prediction category (Figure 3B).
Figure 3. Target Feature Clustering Informs Classification.
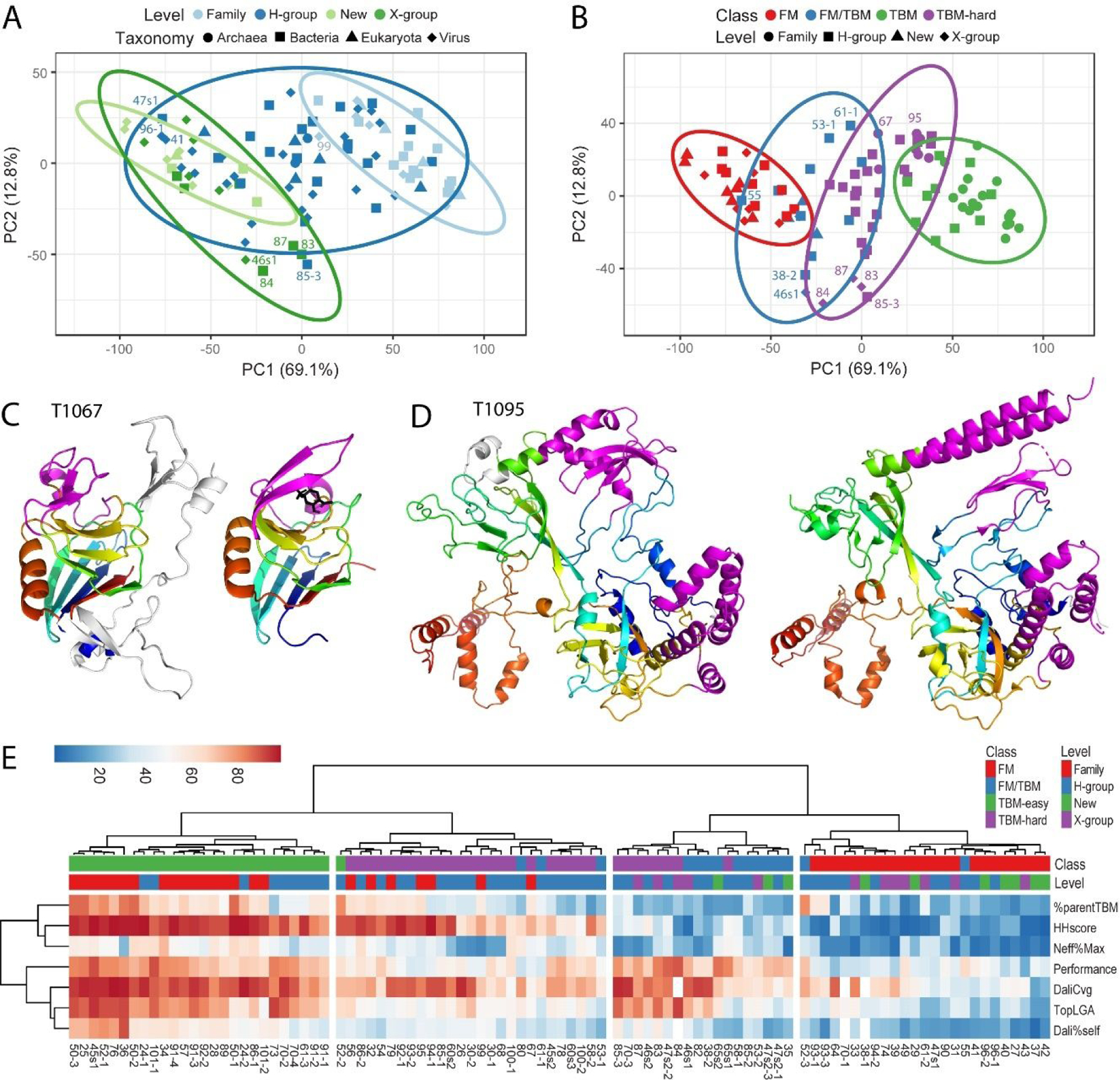
A) PCA clusters of ECOD classification level: Family (light blue), H-group (blue), X-group (green), and New (light green); symbols represent taxonomy: Archaea (circle), Bacteria (square), Eukaryota (triangle) and Virus (diamond) are based on seven scores: sequence relationship to template (HHscore), predictor declaration of parent template (%parentTBM), effective sequences (Neff%Max), performance of top 20 servers (performance), and structure relationship to template using LGA_S (TopLGA), DaliLite Z-scores (Dali%self), and DaliLite coverage (DaliCvg). Ellipses represent 95% confidence level. Some targets discussed in the text are labeled, omitting “T10” or “T1” prefix for brevity. B) PCA clusters of tertiary structure prediction class: TBM-easy (green), TBM-hard (purple), FM/TBM (blue), and FM (red); symbols represent level: Family (circle), H-group (square), X-group (diamond) and New (triangle) use the same scores and are labeled similarly. C) YkuD-like superfamily member T1067 (left) has insertions (white) with respect to the core fold (rainbow) present in the YkuD-like template (PDB: 6fj1C, right). Each have modifications (magenta) surrounding the presumed active site (black stick, right). D) Four domain target T1095 (left) with a single EU has alternate conformations of SSEs (magenta) with respect to the sequence-related template (PDB: 6j9fC, right) with the same fold (rainbow cartoon). E) Heatmap depicts score range (0–100) using a diverging color scheme from blue to red. Scores (rows) are clustered using correlation distance and Ward linkage and Target EUs (columns) are clustered using Euclidean distance and Ward linkage. Target EU features of ECOD level and tertiary prediction class are colored in the columns above the heatmap according to the legend.
In terms of ECOD classification level (Figure 3A), target EUs that belonged to the same family as their template structures separated from new folds and analogous folds (X-group). The X-group EUs mainly overlapped with the New EUs, except for four targets with simple SSE arrangements (T1083, T1084, and T1087 with helix hairpins and T1046s1 with a small 3-helix bundle). CASP14 EUs with distant homology to their templates (H-group) overlapped with both the family-level and the new/X-group level targets. The viral targets (diamonds in the figure) tended to shift to the left in the principal component representing most of the variance in the data (PC1, 69.1%). For example, the family-level viral target T1099 fell outside the confidence ellipse (95%) due to a unique insertion not found in the template. Similarly, while the viral targets T1096-D1 and T1041 (from Figure 2D and E) were homologous to their templates, their large insertions shifted their positions towards the X-group/New clusters. The bacterial target T1047s1 was also shifted towards the X-group/new EUs. T1047s1 adopted an unusual elongated fold that was largely based on oligomeric interactions. The distribution of EUs according to ECOD hierarchy suggested that a distinction should be made between easy (mainly family-level, TBM-easy) and hard (mainly H-group level, TBM-hard) EUs in high accuracy evaluation of template-based models.
As scores were selected to discriminate the tertiary structure prediction class, this feature (Figure 3B) displayed less overlap than the ECOD level feature (Figure 3A). The TBM-easy and FM classes formed tighter clusters than the others and were well-separated in PC1. The wider distribution of the difficult to categorize TBM-hard and FM/TBM classes along the second component (PC2, 12.8%) was dictated by the X-group EUs with simple SSE arrangements. These simple SSE EUs overlapped with TBM-hard despite lacking significant sequence similarity to their templates. On the opposite side of the PC2 variance, the TBM-hard target structures like T1067 or T1095 diverged from their sequence-related templates. The family-level target T1067 belonged to the YkuD-like superfamily of transpeptidases/ carboxypeptidases together with its top template (6fj1C, LGA_S 50.65). T1067 had insertions with respect to the shared core fold, and the structure surrounding the active site diverged between the target and the template (Figure 3C, magenta). Target T1095 encompassed four domains that were treated as a single EU and showed alternate orientations of interacting SSEs with respect to the sequence-related template (Figure 3D, magenta). While T1095 clustered near the TBM-easy targets, the alternate SSE interactions preclude the target (and others near it) from being easy.
A heatmap visualization of the same data clustered the target EUs generally by class (Figure 3E, columns), and the scores by type (Figure 3E, rows). The three sequence-related scores (%parentTBM, HHscore, and Neff%Max) separated from the three structure-related scores (Top LGA_S, Dali%Self and DaliCvg), while performance branched from the structure scores. TBM-easy and FM targets formed independent clusters on opposite sides of the tree, with a few exceptions (T1052-D2, T1052-D3, and T1055). T1052 had a RIFT-related domain inserted into the middle of a SGNH hydrolase. This difficult domain organization caused T1052-D2 to cluster with more difficult TBM-hard targets and T1052-D3 to cluster with more difficult FM targets. We ultimately chose to keep T1052-D2 together with the TBM easy targets due to the high sequence scores, while we chose to keep T1052-D3 as FM/TBM due to the relatively good performance. The other exception, T1055, included a HEH motif identified by sequence merged with a more distant Sec63 N-like domain lacking a sequence relationship. While the larger Sec63 N-like domain dictated its clustering with the FM targets, we included T1055 in FM/TBM due to the relatively good performance and the presence of the HEH.
The TBM-hard and FM/TBM classes also tended to group separately, with an exception of one subgroup of 7 EUs designated as TBM-hard that clustered with the FM/TBM EUs. This subgroup included the previously discussed X-group targets with simple SSE arrangements (T1083, T1084, and T1087), and exhibited good structure and performance scores with low sequence scores. Interestingly, the additional FM/TBM structures in this group (T1046s2, T1047s2-D2 and T1070-D3 and T1085-D3) were all relatively small domains ( 57 to 141 residues) with common SSE arrangements. They were placed near other small EUs (72–75 residues) in the heatmap tree (T1046s1, T1038-D2 and T1082) and were classified as distant homologs despite low sequence scores. The T1085-D3 ARM repeat and the T1070-D3 agglutinin HPA-like domain diverged from domain duplications in their target structures. The phage holin lysis mediator (T1046s2) had significant structure similarity to profilin-like sensor domains. Finally, the simple SSE arrangement of the bacterial flagellar P-ring protein domain (T1047s2-D2, 83 residues) was assigned as a ring-building motif II domain in type III secretion system (T3SS) based on the evolutionary relationship between the flagellum and the T3SS[36], despite its top template belonging to an alternate ECOD X-group (3mmlH, Glucose permease domain IIB-like). Other single EU exceptions that clustered in the TBM-hard class and were ultimately considered as TBM/FM included a merged four domain EU T1061-D1 (Figure 1E), an EU T1080 whose conformation was dictated by oligomeric state, and an EU (T1053-D1) that had large insertions with respect to the template.
3.4. Performance Improvement Complicated Traditional EU Classification
The correlation of predictor performance with target difficulty as measured by sequence and structure distance to known templates has been used in past CASP rounds to reliably categorize target EUs into high accuracy modeling (formerly TBM) and topology (formerly FM) evaluation categories. For CASP14, a broadened scatter of target EU data suggested the boundaries between these categories have become increasingly blurred (Figure 4A). Several factors could have theoretically contributed to this lack of correspondence. To gain an understanding of the factors that contributed to the broadened scatter in CASP14, we split the traditional performance plot into its sequence (Figure 4B, left panel) and structure (Figure 4B, right panel) components, and we compared the scatter from each component to data from CASP13 (Figure 4C, left and right, respectively).
Figure 4. Relationship of Target EU Difficulty with Performance.
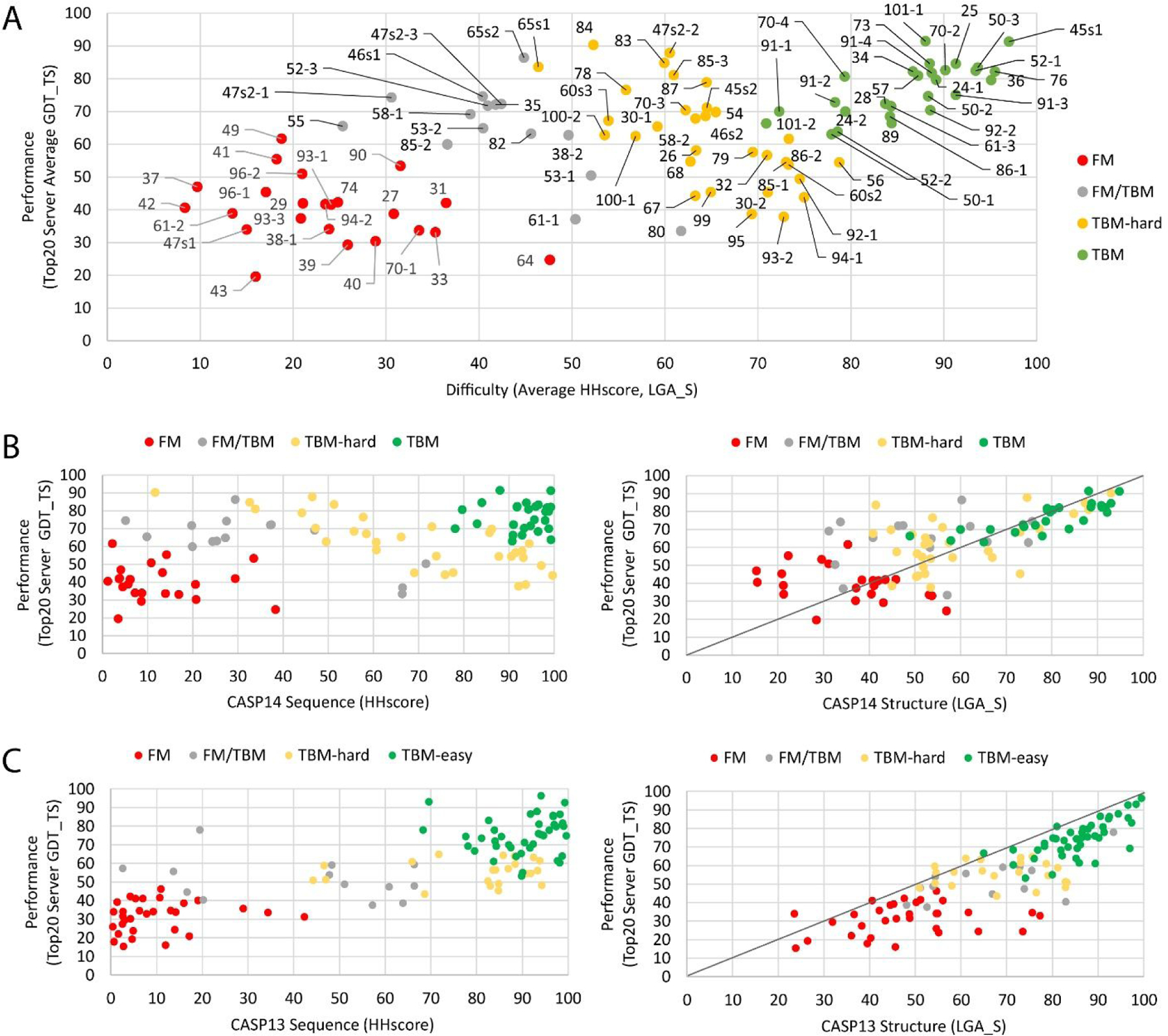
A) CASP14 Target difficulty (X-axis) represented by the average of scores for the top template by sequence (HHscore ) and by structure (LGA_S) correlates with performance (Average GDT_TS scores for top20 server first models, Y axis) with a broad scatter. Target EUs are labeled, omitting the “T10” or “T1” prefix in the target number for brevity, and are colored according to four evaluation categories: high accuracy modeling (TBM-easy), difficult high accuracy modeling (TBM-hard), topology (FM), and a questionable overlapping set (FM/TBM). B) CASP14 target difficulty separated into the sequence (left) and structure (right) components was compared to C) CASP13 target difficulty, colored as above. A line was drawn through normal (0,0 to 100,100) in the structure component plots (right).
For the CASP14 sequence component, an increasing number of target EUs found themselves between homologs on the right (above ~60 HHscore) and analogs on the left (below ~20 HHscore) when compared to CASP13 (Figure 4B and C, left panels). The tendency to keep multidomain targets whole, which could lower the HHscore coverage, as well as the over-abundance of viral targets (whose sequences diverge rapidly) shifted the sequence component of many EU homologs towards lower scores. On the other hand, many prediction methods incorporated large metagenomic sequence datasets such as BFD40 or MGnify41 into their multiple sequence alignments, while our assignment of template difficulty was limited to publicly available sequences. This lack of information could have led to an overestimation of difficulty at the sequence level for some other target EUs, although it provided the basis for a fair comparison of target difficulty with CASP13. Though an a-posteriori analysis showed that larger databases did not help find substantially more evolutionary related sequences in cases where searches versus public databases (e.g., Uniref30) returned an insignificant number of hits.
The structure component scatter of CASP14 EU data shifted notably towards higher average performance levels when compared to CASP13 (Figure 4B and C, right panels). While the average performance of the top20 servers rarely passed above the diagonal for target EUs in CASP13, half of the CASP14 target EUs mapped higher than that. This skew towards higher performance highlighted a potential breakthrough in CASP14 for many server prediction methods that do not rely on manual intervention for operation. Some of the server groups maintain publicly available webservers like I-Tasser42, trRosetta 43, MULTICOM44, Psipred45 or RaptorX46, highlighting the availability of state-of-the-art structure prediction methods for the community. Different components of CASP14 server performance were evaluated in the high accuracy modeling (M. Hartmann, A. Lupas et al, this issue) and topology (L. Kinch, N. Grishin et al, this issue) tertiary structure prediction evaluations.
CONCLUSIONS
Over the course of CASP assessments, the requirement for splitting targets into EUs has gradually changed. In the earlier rounds of CASP where most of the predictions had low scores and there were relatively fewer templates in the PDB, targets were routinely split into their component domains2,4,47. Some of these earlier splits were required for evaluation purposes. Rigid body structure comparison methods developed for the earliest CASP assessments have since been expanded with additional scores like LDDT13, CAD-score14 or SphereGrinder15 that could evaluate whole structure models when their domain orientations do not exactly follow that in targets. During CASP14 almost 175,000 structures existed in the PDB, representing a significant increase from the first CASP (~2800 structures). This increase in experimentally determined structures has expanded the known fold space, providing novel arrangements of SSEs that may be useful for prediction methods. Similarly, progress in experimental structure determination methods like cryo-EM has led to an ever-increasing number of larger and more complete protein structures. In fact, the growing number of existing multidomain templates precluded the requirement for some domain splits (e.g. T1052 in Figure 1C). The increasing PDB structure count has also provided paradigms for conformation change in superfamilies, as observed in several CASP14 targets, including T1024 (Figure 1G), T1050, and T1091, among others. For high accuracy modeling of these targets, information about ligands or other criteria would need to be considered and may need to become a component of future CASP experiments. Given the lack of such information in CASP14, targets with indications of conformational change (seen as performance clouds in the Grishin plots, where overall performance improved on individual domains) were split. While overall performance improved after these straightforward domain splits, this strategy did not necessarily account for all conformation change of such structures.
The progress in prediction method performance over time has also contributed to a changing requirement for splitting targets into domains. In fact, the substantial progress in state-of-the-art deep learning methods from the previous CASP1318 called for a suggestion to cease splitting targets into EUs for the topology category39. At the same time, an inadvertent omission of splitting a target with a conformational change led to a noted performance outlier in the CASP13 high accuracy modeling category17. For this round of CASP, sets of high-performing models existed that did not follow the general trend of the remaining predictions that performed better on split domains (see Figure 1B). Such remarkable performance skewed the Grishin plots for many multidomain CASP14 targets, and we tended to merge domains for this round that would previously have been split (i.e. novel insertions or extensions to known folds in Figure 2D&E). This tendency to keep multidomain targets as single EUs shifted the overall model accuracy lower and increased the target difficulty for CASP14 (shifts to the left in Figure 4B with respect to Figure 4C). In future rounds of CASP, the requirements for splitting targets into EUs are likely to follow the same progressive dichotomy.
Maintaining similar target evaluation strategies across CASP rounds allows a fair comparison from one round to the next and provides a consistent sense of target difficulty. However, given the current advancements of structure prediction methods, the increased scatter observed for CASP14 EU data in the traditional classification plot (Figure 4A) will likely continue in the future. With metagenomics advancements in the age of next-generation sequencing, both the size and diversity of sequence datasets are increasing rapidly48–50. This data explosion requires advanced bioinformatics tools and databases, some of which are utilized in structure prediction methods40,41 but not in evaluation of target difficulty. Including these tools in future CASP target classification strategies would probably provide a better sense of target difficulty. Similarly, advances in structure determination methods like cryo-EM are providing structures that are not limited by their ability to crystalize. Categories of proteins like those that span the membrane, exist as dynamic macromolecular complexes or form fibrous assemblies are beginning to dominate newly released structures. Thus, a more complete picture of structure space is emerging that includes non-domain sequence not easily classified in traditional evolutionary terms. Given these technology advancements, the ability of state-of-the-art deep learning methods to detect increasingly distant relationships between sequence and structure datasets will likely exceed the ability of experts to classify structures using traditional evolutionary concepts. As such, placing future CASP targets into evaluation categories should shift towards a performance-centric strategy where high accuracy modeling assessment simply applies to targets with high performance, while topology assessment applies to low performance targets.
ACKNOWLEDGMENTS
We thank the CASP organizers for their invitation for us to participate in CASP14, the protein crystallographers, NMR spectroscopists and cryo-EM scientists that contributed targets and fellow assessors for helpful discussion about the targets and their classification. This research was supported by the US National Institute of General Medical Sciences (NIGMS/NIH) grants R01GM100482 (AK) and R35GM127390 (NVG) and the Welch foundation grant I-1505 (NVG).
REFERENCES
- 1.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995;23(3):ii–v. [DOI] [PubMed] [Google Scholar]
- 2.Kinch LN, Qi Y, Hubbard TJ, Grishin NV. CASP5 target classification. Proteins 2003;53 Suppl 6:340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murzin A, Hubbard TJ. Prediction targets of CASP4. Proteins 2001;Suppl 5:8–12. [DOI] [PubMed]
- 4.Murzin AG. Structure classification-based assessment of CASP3 predictions for the fold recognition targets. Proteins 1999;Suppl 3:88–103. [DOI] [PubMed]
- 5.Kinch LN, Kryshtafovych A, Monastyrskyy B, Grishin NV. CASP13 target classification into tertiary structure prediction categories. Proteins 2019;87(12):1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinch LN, Li W, Schaeffer RD, et al. CASP 11 target classification. Proteins 2016;84 Suppl 1:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng H, Schaeffer RD, Liao Y, et al. ECOD: an evolutionary classification of protein domains. PLoS Comput Biol 2014;10(12):e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holland TA, Veretnik S, Shindyalov IN, Bourne PE. Partitioning protein structures into domains: why is it so difficult? J Mol Biol 2006;361(3):562–590. [DOI] [PubMed] [Google Scholar]
- 9.Majumdar I, Kinch LN, Grishin NV. A database of domain definitions for proteins with complex interdomain geometry. PLoS One 2009;4(4):e5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol 2004;14(2):208–216. [DOI] [PubMed] [Google Scholar]
- 11.Kryshtafovych A, Monastyrskyy B, Fidelis K. CASP11 statistics and the prediction center evaluation system. Proteins 2016;84 Suppl 1:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinch LN, Shi S, Cheng H, et al. CASP9 target classification. Proteins 2011;79 Suppl 10:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013;29(21):2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olechnovic K, Kulberkyte E, Venclovas C. CAD-score: a new contact area difference-based function for evaluation of protein structural models. Proteins 2013;81(1):149–162. [DOI] [PubMed] [Google Scholar]
- 15.Lukasiak P, Antczak M, Ratajczak T, Blazewicz J. SphereGrinder - reference structure-based tool for quality assessment of protein structural models. Paper presented at: Proceedings of the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2015; Washington, DC, USA. [Google Scholar]
- 16.Abriata LA, Kinch LN, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. Definition and classification of evaluation units for tertiary structure prediction in CASP12 facilitated through semi-automated metrics. Proteins 2018;86 Suppl 1:16–26. [DOI] [PubMed] [Google Scholar]
- 17.Croll TI, Sammito MD, Kryshtafovych A, Read RJ. Evaluation of template-based modeling in CASP13. Proteins 2019;87(12):1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019;87(12):1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo JT, Xu D, Kim D, Xu Y. Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res 2003;31(3):944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Xue B, Zhou Y. DDOMAIN: Dividing structures into domains using a normalized domain-domain interaction profile. Protein Sci 2007;16(5):947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005;33(Web Server issue):W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res 2003;31(13):3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. Proteins 1999;Suppl 3:22–29. [DOI] [PubMed]
- 24.Holm L DALI and the persistence of protein shape. Protein Sci 2020;29(1):128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaeffer RD, Kinch L, Medvedev KE, Pei J, Cheng H, Grishin N. ECOD: identification of distant homology among multidomain and transmembrane domain proteins. BMC Mol Cell Biol 2019;20(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer RD, Liao Y, Grishin NV. Searching ECOD for Homologous Domains by Sequence and Structure. Curr Protoc Bioinformatics 2018;61(1):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 2004;32(Web Server issue):W327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu S, Wang J, Chitsaz F, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 2020;48(D1):D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 2011;9(2):173–175. [DOI] [PubMed] [Google Scholar]
- 30.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, UniProt C. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015;31(6):926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabler F, Nam SZ, Till S, et al. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr Protoc Bioinformatics 2020;72(1):e108. [DOI] [PubMed] [Google Scholar]
- 32.Ayoub R, Lee Y. RUPEE: A fast and accurate purely geometric protein structure search. PLoS One 2019;14(3):e0213712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 2015;43(W1):W566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang XC, Zhao Y, Heng J, Jiang D. Energy coupling mechanisms of MFS transporters. Protein Sci 2015;24(10):1560–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daugherty MD, Malik HS. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 2012;46:677–700. [DOI] [PubMed] [Google Scholar]
- 36.Lowe EC, Basle A, Czjzek M, Firbank SJ, Bolam DN. A scissor blade-like closing mechanism implicated in transmembrane signaling in a Bacteroides hybrid two-component system. Proc Natl Acad Sci U S A 2012;109(19):7298–7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nostadt R, Hilbert M, Nizam S, et al. A secreted fungal histidine- and alanine-rich protein regulates metal ion homeostasis and oxidative stress. New Phytol 2020;227(4):1174–1188. [DOI] [PubMed] [Google Scholar]
- 38.Drobysheva AV, Panafidina SA, Kolesnik MV, et al. Structure and function of virion RNA polymerase of a crAss-like phage. Nature 2021;589(7841):306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abriata LA, Tamo GE, Dal Peraro M. A further leap of improvement in tertiary structure prediction in CASP13 prompts new routes for future assessments. Proteins 2019;87(12):1100–1112. [DOI] [PubMed] [Google Scholar]
- 40.Steinegger M, Mirdita M, Soding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods 2019;16(7):603–606. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell AL, Almeida A, Beracochea M, et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res 2020;48(D1):D570–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods 2015;12(1):7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci U S A 2020;117(3):1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou J, Wu T, Cao R, Cheng J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. Proteins 2019;87(12):1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 2019;47(W1):W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jing X, Zeng H, Wang S, Xu J. A Web-Based Protocol for Interprotein Contact Prediction by Deep Learning. Methods Mol Biol 2020;2074:67–80. [DOI] [PubMed] [Google Scholar]
- 47.Sippl MJ, Lackner P, Domingues FS, et al. Assessment of the CASP4 fold recognition category. Proteins 2001;Suppl 5:55–67. [DOI] [PubMed]
- 48.Kim M, Lee KH, Yoon SW, Kim BS, Chun J, Yi H. Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics Inform 2013;11(3):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koonin EV, Makarova KS, Wolf YI. Evolution of Microbial Genomics: Conceptual Shifts over a Quarter Century. Trends Microbiol 2021. [DOI] [PMC free article] [PubMed]
- 50.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010;11(1):31–46. [DOI] [PubMed] [Google Scholar]