

Abstract
The trRosetta structure prediction method employs deep learning to generate predicted residue‐residue distance and orientation distributions from which 3D models are built. We sought to improve the method by incorporating as inputs (in addition to sequence information) both language model embeddings and template information weighted by sequence similarity to the target. We also developed a refinement pipeline that recombines models generated by template‐free and template utilizing versions of trRosetta guided by the DeepAccNet accuracy predictor. Both benchmark tests and CASP results show that the new pipeline is a considerable improvement over the original trRosetta, and it is faster and requires less computing resources, completing the entire modeling process in a median < 3 h in CASP14. Our human group improved results with this pipeline primarily by identifying additional homologous sequences for input into the network. We also used the DeepAccNet accuracy predictor to guide Rosetta high‐resolution refinement for submissions in the regular and refinement categories; although performance was quite good on a CASP relative scale, the overall improvements were rather modest in part due to missing inter‐domain or inter‐chain contacts.
Keywords: deep learning, metagenomes, protein structure prediction, refinement, Rosetta
1. INTRODUCTION
Recent work 1 , 2 , 3 , 4 has shown that predicted distances and orientations from deep‐learning networks such as AlphaFold and trRosetta coupled with gradient descent minimization can lead to more accurate 3‐D protein models than previous approaches. We sought to improve the accuracy of such deep learning approaches by incorporating template information and model accuracy estimation methods. We developed a structure prediction pipeline with the following features: (a) joint usage of MSA and template information for the trRosetta distance and orientation predictions 2 ; (b) recombination and rescoring of models made with and without template information guided by the DeepAccNet accuracy predictor; (c) simultaneous modeling of all domains in multi‐domain proteins; and (d) full automation of all modeling stages. Here we describe the performance of this pipeline in CASP14, and also the performance of a full atom refinement protocol with DeepAccNet guided sampling.
2. MATERIALS AND METHODS
2.1. Searching for sequence and structure homologs
We generated six different multiple sequence alignments by multiple‐round iterative HHblits 5 searches against uniclust30 (UniRef30_2020_01) database 6 with gradually relaxed e‐value cutoffs ranging from 1e‐80 up to 1e‐1. In the course of iterations, we picked five alignments corresponding to e‐values of 1e‐80, 1e‐40, 1e‐10, 1e‐3, 1e‐1 followed by filtering at 95% sequence identity and no coverage cutoffs. For the sixth alignment, we selected the one with the lowest e‐value which met one of the two criteria: at least 2,000 sequences with 75% coverage or 5,000 sequences with 50% coverage (both at 90% sequence identity cutoff) were collected. This last alignment was also used to perform template searches against the PDB100 database with hhsearch 5 for the template‐based branch of the structure prediction pipeline.
2.2. Manual MSA curation for human submissions
For targets in which the automated procedure described above resulted in shallow alignments, we performed additional sequence searches against metagenomic and metatranscriptomic datasets provided by JGI 7 ; in the case of viral and phage targets, we also used IMG/VR v2 database storing genomes of cultivated and uncultivated viruses. 8 We converted one of the automatically generated MSAs (usually MSA #6) to an HMM profile and used hmmsearch 9 to collect sequence homologs in the extended sequence database. We then clustered the full‐length sequence hits at 30% identity cutoff using mmseqs2, 10 realigned sequences within each cluster by ClustalW, 11 and used these alignments to build a custom hhblits database specific to the target. This database was then used in conjunction with uniclust30 to generate MSAs for human submissions; e‐value cutoffs were manually tuned on a per‐target basis to balance MSA depth, diversity, and coverage of the target sequence.
2.3. Predicting inter‐residue geometries with trRosetta
We developed two neural networks to predict inter‐residue distance and orientation restraints for subsequent 3D model reconstruction via gradient descent. First, we updated the previously described MSA‐based trRosetta network 2 by incorporating as additional input features TAPE language model embeddings of the target sequence 12 and residue‐residue sequence separation. We also increased the number of filters in the bottom layers of the network from 64 to 128 and decreased the bin size for the predicted angular coordinates from 15° to 10°. Second, we developed a network that utilizes template information in addition to the MSA‐based and TAPE features. The top 25 hhsearch hits were converted into 2D network inputs by extracting pairwise distances and orientations from the template structure for the matched positions only. These were complemented by hhsearch positional similarity and confidence scores (both are 1D) provided by hhsearch, which were tiled along horizontal and vertical axes of the 2D inputs. Features for all unmatched positions were set to zero. Templates were first processed independently by one round of 2D convolutions and then merged together into a single 2D feature matrix using a pixel‐wise attention mechanism. This processed feature matrix was then concatenated with the MSA and TAPE features as in the MSA‐based network described above; the architecture of the upstream part of the network was kept the same (Figure 2(A)). Template‐based trRosetta was first used in Farrell et al 13 in the context of cryo‐EM structure reconstruction.
2.4. Training trRosetta networks
To train the new networks, we compiled an extensive training set based on the entire PDB as of 02/17/2020 and uniclust30 sequence database (version UniRef30_2020_01). We used all non NMR structures with better than 3.5Å resolution; in cases where the same protein was solved multiple times, as well as when there were multiple copies of the same protein in an asymmetric unit we retained all of the conformations to account for potential uncertainties in the structure; in total 208,659 protein chains were selected. All the unique sequences from the selected set of protein chains were then clustered at 30% sequence identity cutoff using mmseqs2 resulting in 22,922 clusters and including 73,193 unique sequences. For each sequence, we collected an MSA using the same procedure as outlined in “Searching for sequence and structure homologs” subsection (only MSA #6 was used here) and identified top 500 templates by running hhsearch against PDB100—the latter was used to train the template‐based variant of trRosetta. Every training epoch, we cycled through all sequence clusters by picking a random sequence member from each cluster. For each selected sequence, a subsampled MSA and a randomly picked protein conformation (in cases where there were multiple) comprise one training example; for the template‐based trRosetta, up to 10 randomly selected templates were also used. In this way, each cluster and each sequence are presented to the network somewhat differently at each training epoch.
Protein chains over 300 residues in length were cropped during training to fit into GPU memory. In addition to the continuous crops used to train the original trRosetta network in Yang et al, 2 we also explored discontinuous crops in which two randomly selected non‐intersecting sequence fragments along with the corresponding intra‐ and inter‐fragment portions of the network inputs and outputs were used during training. This cropping strategy better handles interactions between residues distant along the sequence in long multi‐domain proteins.
2.5. Recombining trRosetta predictions from different MSAs
Each of the six MSAs generated as described above for the query sequence was used as input to both the MSA only and the MSA plus template variants of the trRosetta network. Following Anishchenko et al, 14 we estimated the quality of these predictions on a per residue pair basis by calculating the KL‐divergence (D KL,ijk , indices i,j define a residue pair and index k enumerates MSAs which were used to get the predictions) of the predicted distance and orientation distributions from the background (higher D KL values correspond to more peaked distributions and hence more confident predictions). We then merged predictions, separately for each type of the network, by calculating the weighted sum of the predicted distributions where the weights are softmaxed D KL,ijk values along the last k dimension.
2.6. Recreating 3D structure from network predictions
For each residue pair, the predicted distance and orientation distributions were converted into restraints following the reference state correction step 2 , 15 and were then used to generate protein structures using trRosetta folding protocol based on restrained minimization. For this CASP14, we introduced a few tweaks to the protocol to improve its convergence and increase the model quality as measured by the MolProbity score. 16 First, we added a Savitzky‐Golay filter 17 to smoothen the trRosetta‐derived restraints before feeding them into Rosetta as cubic spline functions. We also implemented the automatic detection of disulfides to favor bond formation between closely located cysteines that could form an S‐S bond. Additionally, during energy minimization in the centroid mode, the weight of the repulsive van der Waals energy term was ramped up from 3.0 to 10.0, while the restraint weights for distances and orientations were ramped down from 3.0 to 1.0 and from 1.0 to 0.5, respectively. To further improve stereochemistry and local quality of the final models, two‐step full atom relaxation 18 was introduced. In the first step, the full‐atom model was relaxed in torsion space with elevated restraint weights (3.0 and 1.0 for distances and angles, respectively) as well as Rosetta full‐atom scores to add side chains while keeping overall backbone structures similar to the input centroid level structures satisfying given restraints well. In the second step, the full‐atom model was further relaxed with much weaker pairwise distance restraints (weight: 0.1) in torsion angle space followed by relaxation in the Cartesian space. For each of the merged predictions from two different trRosetta networks with different MSAs, 45 protein models were generated using this improved folding protocol with a various subset of restraints.
2.7. Model accuracy estimation using DeepAccNet
A deep learning framework called DeepAccNet was developed for model accuracy estimations. 19 DeepAccNet estimates per‐residue accuracy in l‐DDT and residue‐residue distance signed error (represented as histograms of residue‐pair distance errors, or estogram in short) in protein models and can be used to guide Rosetta protein structure refinement. Two variants of DeepAccNet were tested in CASP14; DeepAccNet‐MSA incorporates trRosetta distance predictions as an additional input feature to the network to improve prediction accuracy; DeepAccNet‐cen represents protein models in a coarse‐grained level and can be enumerated faster by an order of magnitude than the regular DeepAccNet.
2.8. Model recombination by trRefine and scoring
To recombine the two sets of models from two trRosetta networks (one with and the other without template information), we developed a new network, called trRefine, which takes the outputs of the two trRosetta networks as well as generated model structures and their predicted distance errors as inputs and generates the refined predictions for residue‐residue geometries (Figure 1(B)). Among the total 90 models from both trRosetta network predictions, the top 10 scored models were selected based on REF2015 energy function. 20 The inter‐residue Cβ–Cβ distance errors (Cα for GLY) were estimated by DeepAccNet‐MSA, and they were combined with corresponding model conformations represented in pairwise distance and orientation maps. The combined 2D features for each model conformation were independently processed by a single 2D residual convolution block and then merged into a single 2D feature matrix using pixel‐wise attention. The outputs from two trRosetta networks were also processed by a single 2D residual convolution block and were merged into a single feature matrix. The processed structural features and predicted inter‐residue geometry features were concatenated together and processed further by 2D residual convolution networks to predict refined inter‐residue geometries. This trRefine network was trained on decoy structures generated by two trRosetta networks for 7,307 protein chains used to train DeepAccNet. 19 For data augmentation, three subsampled MSAs were generated with various depths (i.e., number of sequences in MSA), and for each subsampled MSA, 15 models were generated for each of trRosetta networks. The distance errors in trRosetta models were estimated by DeepAccNet‐MSA. During the training, one of the subsampled MSAs was randomly selected, and the corresponding trRosetta outputs and structures with predicted distance errors were used to optimize the trRefine network.
FIGURE 1.
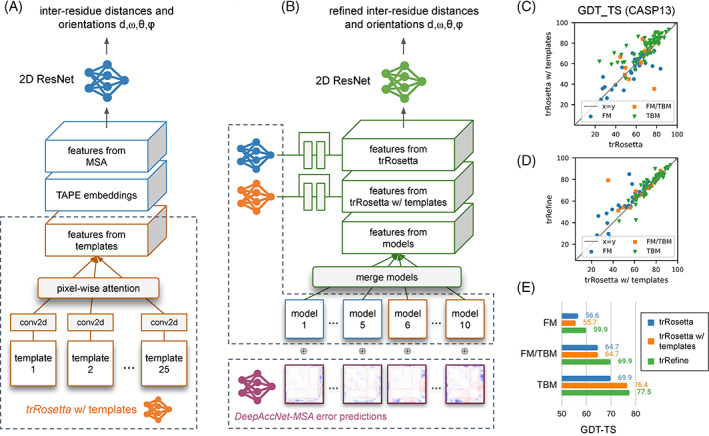
Deep neural networks for protein structure prediction, model recombination and rescoring. (A) Revised trRosetta network incorporating TAPE language model and homologous template derived features. (B) trRefine network utilizing DeepAccNet guided model recombination (see Methods for details). (C)–(E) Benchmarking of the newly developed networks on CASP13 targets. MSAs for CASP13 targets were taken from Yang et al 2 ; hhsearch templates sharing more than 30% sequence identity to the target were excluded. (E) Average GDT‐TS scores for each target difficulty category
Based on trRefine predictions, the new pool of structure models was generated by the trRosetta folding protocol. The trRefine‐derived models were re‐scored using DeepAccNet‐MSA, and among the top 10 scored models, three models were picked for submissions 1–3 after clustering. Submissions 4 and 5 were the top models from the MSA‐only and template‐based trRosetta, respectively.
2.9. Human intervention
We sought to test two human interventions for the human category submissions. At the first stage, MSAs different from that used by the server (details in the previous “Manual MSA curation for human submissions” section) were used to build ensembles of new model structures, and the model with the best match to the predicted distances was chosen for the starting point of refinement. When more than one MSA led to comparable distance predictions, all those were subject to refinement and ranked by DeepAccNet‐MSA at the end. Automated refinement (see below) was applied to every domain <300 aa that was not intertwined or engaged in inter‐domain contacts.
2.10. Model accuracy estimation guided refinement protocol
We experimented with deep learning‐based model refinement in CASP14 in both regular (TS) and refinement (TR) categories. This was an advance over our refinement protocol in the last CASP where no deep learning component was utilized. 21 In this CASP, we tested two refinement protocols integrating variants of DeepAccNet. The standard protocol integrates DeepAccNet‐MSA into our standard Rosetta refinement protocol, 19 and resulting models were submitted for the group “BAKER.” The experimental protocol uses DeepAccNet‐cen directly in the Rosetta Monte Carlo (MC) search algorithm, and resulting models were submitted for the group “BAKER‐experimental.”
The standard protocol was used (a) for the final stage refinement of trRefine models in human regular category predictions, as described previously, and (b) for refinement category predictions. DeepAccNet‐MSA was incorporated into every iteration in the refinement protocol at three levels. Estograms were converted to residue‐residue interaction potentials which were added to the Rosetta energy function as restraints to guide Rosetta sampling. Second, the per‐residue l‐DDT predictions were used to decide which regions to intensively sample or to recombine with other models. Third, global l‐DDT prediction was used as the objective function during the selection stages of the evolutionary algorithm and to control the model diversity in the pool during iteration. More details of the protocol can be found in Hiranuma et al. 19 , 22
The experimental protocol was designed to facilitate more frequent communication between the deep neural network and Rosetta modeling components within the conformation search stage. In the basic sampling unit, MC search using fragment insertion and/or partial chunk rigid‐body movements was guided by using the inverse of l‐DDT (−l‐DDT) predicted by DeepAccNet‐cen as score with temperature factor kT = 0.01. Two‐hundred independent 5000 step MC trajectories were carried out from the initial model at the first iteration, and then 40 MC trajectories (with the same 5000 steps) were run for each of the five structures selected after all‐atom relaxation and DeepAccNet‐MSA evaluation using a simple evolutionary algorithm. This “coarse‐grained sampling stage” was iterated five times. Twenty models selected by DeepAccNet‐MSA from the entire iterations were subjected to 10 iterations of all‐atom refinement protocol, 22 and the top 5 models selected by DeepAccNet‐MSA at the end were submitted.
3. RESULTS
3.1. Updates to trRosetta and development of the trRefine network
We sought to improve the trRosetta network by incorporating additional features beyond raw multiple sequence alignments. Recent studies have shown that language models trained in a self‐supervised way on the massive body of protein sequence data produce learned representations capturing the fundamental properties of proteins like secondary structure, inter‐residue contacts, biological activity, and others. 12 , 23 , 24 We trained a version of the trRosetta network that uses TAPE language model embeddings 12 as additional input features (Figure 1(A)), using an updated training set that employs different MSA variants at each training epoch (see Methods). When benchmarked on CASP13 targets, this updated network results in much better models for the FM category, improving the average TM‐score from 51.9 (reported in Yang et al 2 ) to 63.7 on the same set of MSAs.
Despite good performance on hard protein targets, the quality of sequence‐based trRosetta models in the homology modeling regime was not high. To tackle this issue, we developed a modified version of trRosetta (referred to as template‐based trRosetta, see Methods) in which MSA‐based and TAPE input features were complemented by structural information derived from top‐scoring homologs identified by hhsearch (Figure 1(A)). As expected, the biggest benefit of using template‐based trRosetta compared to the sequence only method is in the TBM category: the GDT‐TS score improves from 69.9 to 76.4 for the networks with and without templates, respectively (Figure 1(C),(E)).
Ideally, a single network (i.e., template‐based trRosetta) should be sufficient to capture all the relevant information from both input sequences and template structures. In practice, we found it challenging to balance the two sources of information within one network, and there were a small number of targets for which the sequence‐based variant of trRosetta gave better models (Figure 1(C), points below the diagonal). To mitigate this, we developed a separate neural network called trRefine (Figure 1(B), see Methods) to recombine predictions from the two trRosetta networks guided by DeepAccNet‐MSA and generate refined predictions for inter‐residue geometries; a new pool of structure models was then generated by the trRosetta folding protocol and these were evaluated with DeepAccNet‐MSA. The joint use of model recombination and rescoring increased the overall accuracy across all CASP13 targets by additional +2.3 GDT‐TS score units over template‐based trRosetta when averaged across all difficulty categories (Figure 1(D),(E)); the improvement mostly comes from recombination of predictions from the sequence‐ and template‐based variants of the trRosetta network for multi‐domain targets when the two networks disagree in their predictions for different domains.
3.2. Fully automated structure prediction pipeline
We incorporated these ideas into a fully automated protein structure prediction pipeline outlined in Figure 2(A), which was used in CASP14 both by our BAKER‐ROSETTASERVER group, and with several interventions described below, our human group (BAKER). The pipeline starts with collecting multiple sequence alignments and structure templates for the target sequence that are then passed through the two variants of the trRosetta network to predict inter‐residue distances and orientations. Inter‐residue geometry restraints derived from the network predictions are used to fold structures by direct minimization and relaxation in Rosetta yielding two sets of structure models. The two pools of models are rescored and recombined using DeepAccNet‐MSA and trRefine networks, and the refined inter‐residue geometry restraints are used to guide a second round of minimization‐based folding from which final models are selected by DeepAccNet‐MSA.
FIGURE 2.
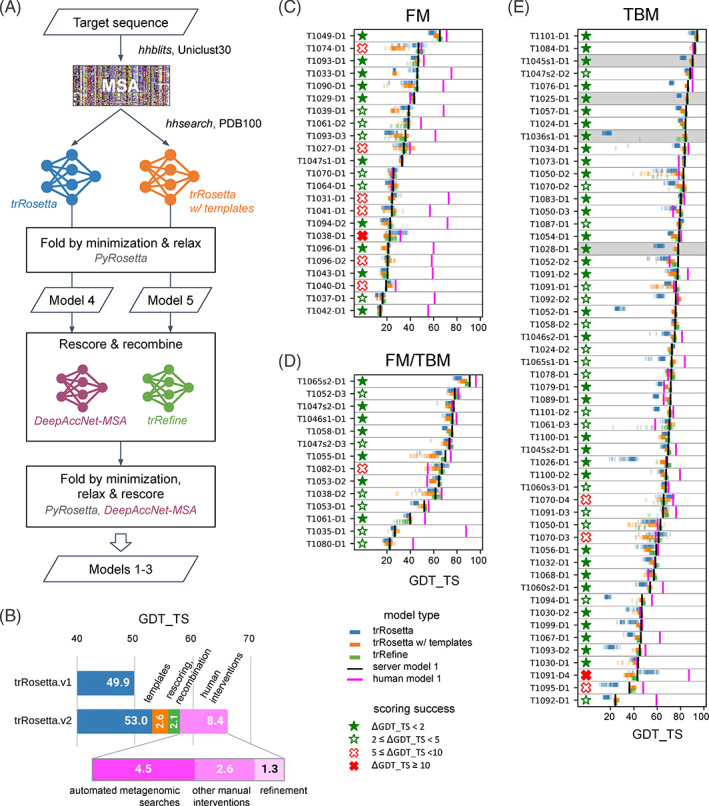
Contributions to CASP14 structure prediction accuracy. (A) Fully automated structure prediction pipeline. (B) Contribution of different factors to the overall performance on CASP14 targets; trRosetta.v1 is the original network from Yang et al, 2 and trRosetta.v2 incorporates the TAPE embeddings and was trained on the new training set. (C)–(E) Per‐target analysis of models generated by the pipeline for FM, FM/TBM and TBM targets respectively. Blue, orange and green dots indicate models produced by trRosetta, trRosetta with templates, and trRefine networks respectively. Black vertical lines represent server model 1, while magenta lines correspond to model 1 of the human submissions. Stars and crosses on the left of panels (C)–(E) visualize scoring success by measuring the difference in the GDT‐TS score between the submitted server model 1 and the best model generated by the pipeline
Since for a given sequence, different MSAs lead to network predictions with different accuracies, and the deepest alignment does not necessarily result in the best prediction accuracy, 2 , 25 we chose to generate six alignments using different inclusion cutoffs and then recombined network predictions from all these alignments (see Methods). Using updated sequence and structure databases for training and incorporating the TAPE embeddings resulted in more accurate models (ΔGDT‐TS = +3.1 on CASP14 targets) compared to the original trRosetta when run on the alignments generated by BAKER‐ROSETTASERVER (Figure 2(B)). Incorporation of template information yielded ΔGDT‐TS = +2.6 improvement over the sequence‐only network (Figure 2(B)), and model recombination and rescoring with trRefine and DeepAccNet‐MSA, an additional ΔGDT‐TS = +2.1. For 55% (or 85%) of domains the automatically selected model 1 was within 2 (or 5) GDT‐TS score units from the best model sampled throughout the whole modeling protocol (Figure 2(C)–(E), closed and open green stars).
3.3. Modeling multi‐domain targets
Considering targets as a whole during modeling and not splitting them into domains allowed in many cases for accurate recapitulation of inter‐domain interactions: our automated server was scored #1 by the assessors among all the servers or #7 overall with Z‐score = 7.84; the next best server is RaptorX with Z‐score = 6.32 and ranked #18 overall. Modeling results for 3‐domain target T1052 (Figure 3(A)–(C)) exemplify how the input sequence and structural information was recombined by trRosetta to yield high‐quality predictions for all three domains. As shown in Figure 3(A), multiple templates were selected for both domains D1 and D2; D1 could not have been accurately modeled without templates (ΔGDT‐TS with the MSA‐based trRosetta model is +48.1), while MSA was the only source of information for domain D3. The attention mechanism utilized to merge the signal from multiple templates into a single feature matrix makes it possible to track which templates were selected for each residue pair within the target (Figure 3(B)). For D2, none of the selected templates were particularly close to the target: GDT‐TS to the closest template structure was 52. However, by recombining multiple templates along with the sequence‐derived signal, template‐based trRosetta yielded a model with GDT‐TS = 70.1 (for comparison, MSA‐based trRosetta gave GDT‐TS = 66.2, both picked by Rosetta energy 20 ); model quality was improved further by trRefine and DeepAccNet‐MSA networks increasing GDT‐TS to 78.1. The final BAKER‐ROSETTASERVER model 1 for T1052‐D2 was the most accurate among all servers with ΔGDT‐TS = +4.1 from the next best server model.
FIGURE 3.
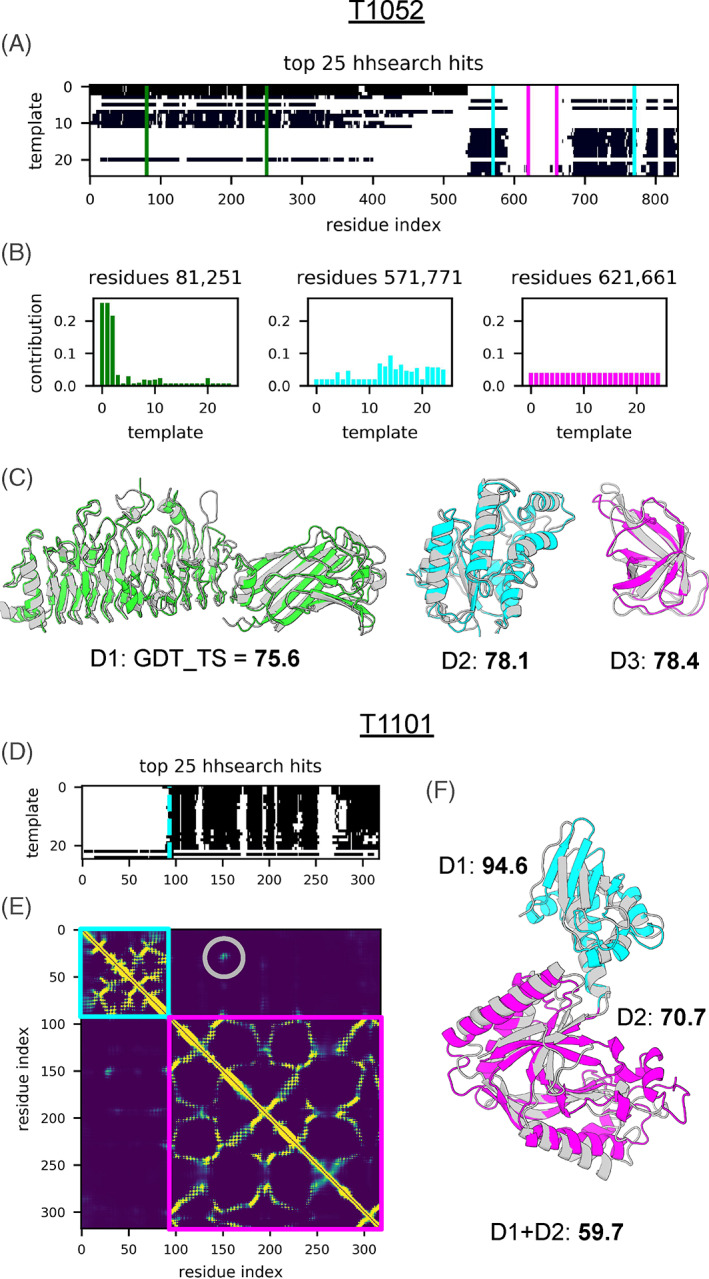
Recombination of input sequence and structural information by the automated trRosetta pipeline. Panels (A) and (D) show coverage of targets T1052 and T1101 respectively by the top 25 templates identified by hhsearch. (B) Examples of pixel‐wise attentions assigned by the network to the templates for the three residue pairs each belonging to a different domain of the 3‐domain target T1052: pairs 81–251, 252–570, 571–771 from domains D1, D2, D3 are marked in green, cyan, and magenta respectively. Panels (C) and (F) show experimental structures (gray) of targets T1052 and T1101 overlaid with the BAKER‐ROSETTASERVER model 1 of the respective domains. GDT‐TS scores are indicated for each domain for both targets, and for all of T1101
Despite good quality models for individual domains, the automated pipeline was not able to recapitulate domain‐domain interactions of full length T1052 due to lack of signal from both MSA and templates. Unlike T1052, MSA provided sufficient signal to predict a patch of inter‐domain contacts in the case of T1101 (Figure 3(D)–(F)) so that the domain‐domain arrangement was also recapitulated; BAKER‐ROSETTASERVER full length model 1 was the most accurate among all servers with the full‐length GDT‐TS = 59.7 (next best full‐length GDT‐TS = 57.0 is from another trRosetta‐based server Yang_FM); individual domains D1 and D2 from this target were also ranked #1 among servers.
According to the official rankings, BAKER‐ROSETTASERVER was the best automated server for FM/TBM and TBM targets with Z‐score = 63.4 (combined Z‐scores > 0.0 according to assessors' formula) vs Z‐score = 49.7 of the next‐best Zhang‐Server. The relatively poor performance on the free modeling targets compared to other groups likely reflects the limited sequence information utilized (uniclust30 was the only sequence database used (UniRef30_2020_01)). Incorporating the BFD database 26 , 27 into the pipeline and re‐running it increased the average GDT‐TS score over all targets by +4.5 (“automated metagenomic searches” bar on Figure 2(B) and a scatter plot comparison in Figure 4(B)); the biggest improvements are for the FM targets with average ΔGDT‐TS = +11.0.
FIGURE 4.
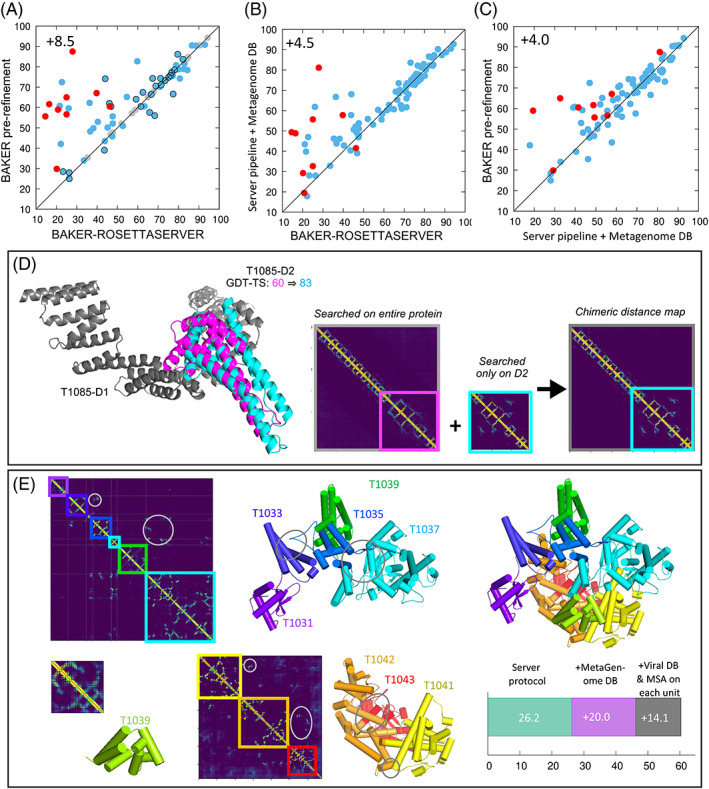
Improvements brought by human interventions. (A)–(C) Overall model quality in GDT‐TS by automated prediction pipeline (x‐axis) and (A) by modeling after human sequence search (y‐axis) or (B) by the same pipeline but with the metagenome database ran after CASP14 (y‐axis). (C) Comparison of Y‐axis values in panels (A) and (B) shows that expert sequence augmentation produced modest improvements for all but the hardest targets once sequence database effects are controlled for. Targets part of the viral RNA polymerase (PDB ID 6vr4) are highlighted by red dots. (D) T1085, an example when sequence search by domains helped. (E) Viral RNA polymerase (target numbers T1031,33,35,37,39,40,41,42,43) modeled as three sets of domains (modeling units). Inter‐domain contacts are highlighted by gray circles. Models for the units are overlaid on the crystal structure on the right. The improvements for this target from additional sequence search and per‐domain MSA generation are indicated in the bottom right corner
3.4. Impact of MSA curation
The human group utilized the same automated pipeline but achieved better performance primarily through manual MSA curation. As we were not aware until after CASP14 of the improvement in performance simply by including the BFD database in the automated pipeline, custom target‐specific hhblits databases were generated using metagenome information from the JGI (see Methods). In Figure 4(A), the model quality before the human sequence search (i.e., server models) and after the search are compared. The net improvement brought about by the manual MSA curation and, in a few cases described below, by adjusting the query sequence boundaries was 6.8 units in GDT‐TS (“automated metagenomic searches” + “other manual interventions” bars on Figure 2(B) and a scatter plot comparison in Figure 4(A)). The improvements were largest for multi‐domain modeling problems. For example, T1085 (Figure 4(D)), which was modeled as a whole unit by the server but had a sequence coverage issue at its D2, was split into two modeling units for sequence search and was rebuilt by “hybridizing” distance maps from two domain to improve the GDT‐TS of D2 by 23 units. Modeling of 8 targets from an 8‐domain viral RNA polymerase (PDB ID 6vr4) also benefited from additional sequence search (Figure 4(E)) against metagenomic and viral sequence databases that generated MSAs yielding more accurate intra and interdomain distance and orientation predictions; based on the predicted inter‐domain contacts, the 8 targets were grouped into 3 modeling units and the resulting models were considerably better than those of the automated pipeline (red points in Figure 4(A)).
3.5. Deep learning guided refinement
We have tested two refinement protocols in CASP14 that utilize the DeepAccNet deep neural network to guide sampling. The “standard” protocol used DeepAccNet‐msa only on the resulting models from Rosetta fragment‐assembly Monte Carlo (MC) search, while the “experimental” protocol used DeepAccNet‐cen also inside the MC search; details can be found in Methods. Both protocols were tested in the TR category; the standard protocol was also applied to trRefine models for our human group “BAKER” submissions in the TS category.
The net improvements by the standard protocol on 52 TS domains and 44 TR domains, and by the experimental protocol on 44 TR domains are reported in Figure 5(A). The average improvement added by the standard refinement protocol on TS domains was 4.4 GDT‐HA units (2.3 in GDT‐TS; the overall contribution is +1.3 in GDT‐TS (Figure 2) because only 55% of targets were subjected to refinement). The net improvement on regular TS targets was larger and more consistent than for TR targets (average ΔGDT‐HA = +2.0). Still, the results on the TR targets were ranked first in the refinement category by assessors.
FIGURE 5.
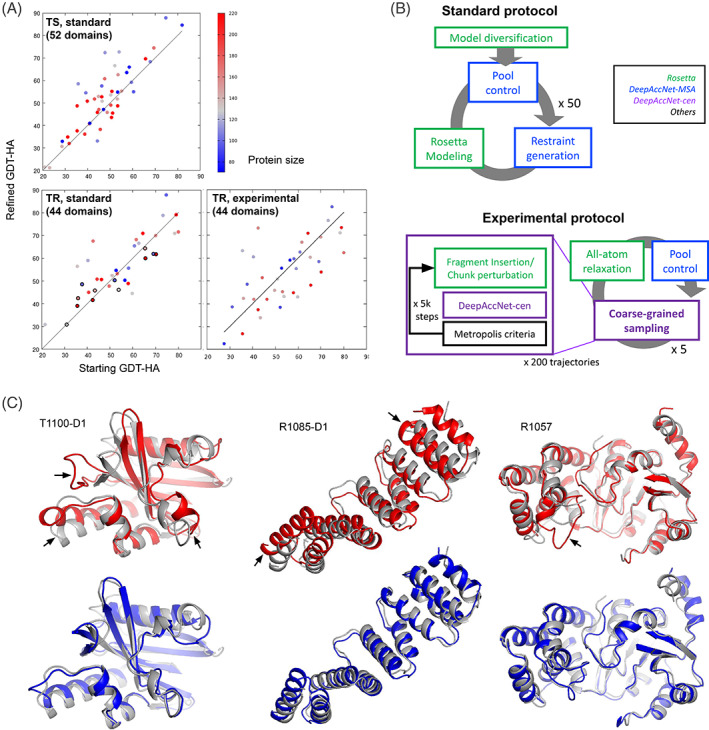
Refinement performance. (A) Scatter plots comparing starting model (x‐axis) vs refined model (y‐axis) quality for TS category predictions (top), TR BAKER predictions (bottom left), and TR BAKER‐experimental predictions (bottom right). (B) Schematic descriptions of refinement protocols. (C) Refinement success on relatively large proteins. Native structures, starting models, and final refined models are shown in gray, red, and blue, respectively. Improvements in GDT‐HA are +19.6, +25.0, and +6.5 for T1100‐D1 (166aas), R1085‐D1 (160aas), and R1057 (241aas), respectively
The primary advance compared to previous CASPs was refinement of larger proteins. Because of the very large search space, refinement methods have typically not been successful at improving models with more than 150 residues. We overcame this limitation in CASP14 by using DeepAccNet; residue pair distance restraints derived from the network focused sampling in the correct region of conformational space. Four of 9 non‐AlphaFold2 models bigger than >150 residues in the TR category were improved by more than 5 GDT‐HA units with the standard protocol (this was best performance on these targets according to the assessor).
The somewhat better performance in refining TS compared to TR models likely reflects differences in how the protocol was applied. During TS modeling, we attempted to include the larger protein context (neighboring domains, etc.) whenever possible, while for TR modeling, the provided starting model was refined in isolation in a fully automated manner. This difference is exemplified by modeling results for T1035, which was released both for TS and TR categories (Figure S1, part of full protein shown in Figure 4(C)). Modeling this domain alone in the TR category resulted in model degradation (ΔGDT‐HA = −7), which contrasts with our TS submission (ΔGDT‐HA = +1) refined along with neighboring domains.
The improvements brought about by refinement, while among the best in the refinement category in CASP14, were somewhat smaller in CASP14 than in recent benchmark studies on more controlled datasets. 19 Missing inter‐chain or domain contacts in the current refinement protocol can lead to failures because the physically based Rosetta energy function 20 requires the larger protein context for accurate energy evaluation (as illustrated by several of the AlphaFold2 predictions, deep learning methods can in some cases implicitly account for “missing” contacts). Also, for more complex protein topologies, the more intensive use of DeepAccNet to guide sampling in the experimental protocol led to structures with predicted accuracies quite a bit higher than the actual accuracies. This highlights the challenges of direct optimization of quantities computed by deep learning networks with millions of parameters—the models can be very good at prediction generally, but because of the large numbers of parameters and search space, direct optimization is very prone to false minima. Reducing these false minima through adversarial training 28 and including the Rosetta energy along with the predicted accuracy in the Monte Carlo search could help alleviate this problem.
4. DISCUSSION
We developed an accurate, fast, and fully automated protein structure prediction pipeline. Analysis of results revealed that joint use of MSAs and templates within a single network helped in building more accurate structure models compared to an MSA‐only network. Additional improvement resulted from recombination and rescoring with the networks, trRefine and DeepAccNet‐MSA. Still further improvements followed from manual curation of MSAs and the use of extended sequence databases, as was demonstrated by our human BAKER group. We have also been benchmarking our CASP14 automated structure prediction pipeline in CAMEO, 29 and it has been consistently ranked first over the last 6 months. Our combination of the Rosetta physically based model with DeepAccNet to guide sampling showed promise in CASP14, but more consistent results will likely require inclusion of all interacting domains in the modeling process, and reduction or avoidance of false optima in the accuracy predictor.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26194.
Supporting information
Figure S1 Refinement failures due to missing inter‐domain contacts. Native, starting, and refined models are shown in gray, red, and blue, respectively. Other domains having direct interactions but not included in the refinement scenario are shown in ivory color in right panels.
ACKNOWLEDGMENTS
This work is supported by National Science Foundation Award # DBI 1937533 (I.A.), Eric and Wendy Schmidt by recommendation of the Schmidt Futures program (H.P.), the Howard Hughes Medical Institute (D.B., D.K., S.M.), NIAID Federal Contract # HHSN272201700059C (M.B.), a gift from Amgen (I.H.), The Open Philanthropy Project Improving Protein Design Fund (J.D.), The Audacious Project at the Institute for Protein Design (D.B.), and a gift from Microsoft (S.M. and M.B.).
Anishchenko I, Baek M, Park H, et al. Protein tertiary structure prediction and refinement using deep learning and Rosetta in CASP14 . Proteins. 2021;89(12):1722-1733. 10.1002/prot.26194
Ivan Anishchenko, Minkyung Baek, and Hahnbeom Park contributed equally to this work.
Funding information Eric and Wendy Schmidt by recommendation of the Schmidt Futures program; gift from Amgen; gift from Microsoft; Howard Hughes Medical Institute; National Science Foundation, Grant/Award Number: DBI 1937533; NIAID Federal Contract, Grant/Award Number: HHSN272201700059C; The Audacious Project at the Institute for Protein Design; The Open Philanthropy Project Improving Protein Design Fund
DATA AVAILABILITY STATEMENT
All the codes and deep learning models will be made available at https://github.com/RosettaCommons/trRosetta2 under the MIT license. Fully automated modeling is accessible through the webserver https://robetta.bakerlab.org/.
REFERENCES
- 1. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577(7792):706‐710. [DOI] [PubMed] [Google Scholar]
- 2. Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci U S A. 2020;117(3):1496‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu J. Distance‐based protein folding powered by deep learning. Proc Natl Acad Sci U S A. 2019;116(34):16856‐16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greener JG, Kandathil SM, Jones DT. Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat Commun. 2019;10(1):3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mirdita M, von den Driesch L, Galiez C, Martin MJ, Söding J, Steinegger M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017;45(D1):D170‐D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen I‐MA, Markowitz VM, Chu K, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45(D1):D507‐D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paez‐Espino D, Chen I‐MA, Palaniappan K, et al. IMG/VR: a database of cultured and uncultured DNA viruses and retroviruses. Nucleic Acids Res. 2017;45(D1):D457‐D465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35(11):1026‐1028. [DOI] [PubMed] [Google Scholar]
- 11. Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947‐2948. [DOI] [PubMed] [Google Scholar]
- 12. Rao R, Bhattacharya N, Thomas N, et al. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst. 2019;32:9689‐9701. [PMC free article] [PubMed] [Google Scholar]
- 13. Farrell DP, Anishchenko I, Shakeel S, et al. Deep learning enables the atomic structure determination of the Fanconi Anemia core complex from cryoEM. IUCrJ. 2020;7(Pt 5):881‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anishchenko I, Chidyausiku TM, Ovchinnikov S, Pellock SJ, Baker D. De novo protein design by deep network hallucination. bioRxiv. 10.1101/2020.07.22.211482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou H, Zhou Y. Distance‐scaled, finite ideal‐gas reference state improves structure‐derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11(11):2714‐2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams CJ, Headd JJ, Moriarty NW, et al. MolProbity: more and better reference data for improved all‐atom structure validation. Protein Sci. 2018;27(1):293‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Savitzky A, Golay MJE. Smoothing and differentiation of data by simplified least squares procedures. Anal Chem. 1964;36(8):1627‐1639. [Google Scholar]
- 18. Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23(1):47‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiranuma N, Park H, Baek M, Anishchanka I, Dauparas J, Baker D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat Commun. 2021;12(1):1340‐1350. 10.1101/2020.07.17.209643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park H, Bradley P, Greisen P Jr, et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J Chem Theory Comput. 2016;12(12):6201‐6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park H, Lee GR, Kim DE, Anishchenko I, Cong Q, Baker D. High‐accuracy refinement using Rosetta in CASP13. Proteins. 2019;87(12):1276‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park H, Ovchinnikov S, Kim DE, DiMaio F, Baker D. Protein homology model refinement by large‐scale energy optimization. Proc Natl Acad Sci U S A. 2018;115(12):3054‐3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A. 2021;118(15):e2016239118. 10.1073/pnas.2016239118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: towards cracking the language of Life's code through self‐supervised deep learning and high performance computing. IEEE Trans Pattern Anal Mach Intell. 2021;14(8):1‐11. 10.1101/2020.07.12.199554 [DOI] [Google Scholar]
- 25. Kandathil SM, Greener JG, Jones DT. Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins. 2019;87(12):1092‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steinegger M, Mirdita M, Söding J. Protein‐level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods. 2019;16(7):603‐606. [DOI] [PubMed] [Google Scholar]
- 27. Steinegger M, Söding J. Clustering huge protein sequence sets in linear time. Nat Commun. 2018;9(1):2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madry A, Makelov A, Schmidt L, Tsipras D, Vladu A. Towards deep learning models resistant to adversarial attacks. Poster presented at: 6th International Conference on Learning Representations, ICLR 2018, Vancouver, BC, Canada, April 30 ‐ May 3, 2018. https://openreview.net/pdf?id=rJzIBfZAb [Google Scholar]
- 29. Haas J, Barbato A, Behringer D, et al. Continuous automated model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins. 2018;86(Suppl 1):387‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Refinement failures due to missing inter‐domain contacts. Native, starting, and refined models are shown in gray, red, and blue, respectively. Other domains having direct interactions but not included in the refinement scenario are shown in ivory color in right panels.
Data Availability Statement
All the codes and deep learning models will be made available at https://github.com/RosettaCommons/trRosetta2 under the MIT license. Fully automated modeling is accessible through the webserver https://robetta.bakerlab.org/.