

Abstract
The high accuracy of some CASP14 models at the domain level prompted a more detailed evaluation of structure predictions on whole targets. For the first time in CASP, we evaluated accuracy of difficult domain assembly in models submitted for multidomain targets where the community predicted individual evaluation units with greater accuracy than full-length targets. Ten proteins with domain interactions that did not show evidence of conformational change and were not involved in significant oligomeric contacts were chosen as targets for the domain interaction assessment. Groups were ranked using complementary interaction scores (F1, QS-score and Jaccard coefficient) and their predictions were evaluated for their ability to correctly model inter-domain interfaces and overall protein folds. Target performance was broadly grouped into two clusters. The first consisted primarily of targets containing two evaluation units (EU) wherein predictors more broadly predicted domain positioning and interfacial contacts correctly. The other consisted of complex two- and three-EU targets where few predictors performed well. The highest ranked predictor, AlphaFold2, produced high-accuracy models on eight out of ten targets. Their interdomain scores on three of these targets were significantly higher than all other groups and were responsible for their overall outperformance in the category. We further highlight the performance of AlphaFold2 and the next best group, BAKER-experimental on several interesting targets.
Keywords: CASP14, classification, fold space, protein structure, protein domains, sequence homologs multidomain proteins, structure prediction, protein-protein interactions
1. Introduction
The Critical Assessment of Structure Prediction (CASP) is a collective experiment to determine the current accuracy of protein structure prediction methods1, 2. Predictors are provided with protein sequences and given a limited timeframe within which to submit structure predictions. The submitted models are independently assessed. For the assessment, multidomain protein targets, comprising two or more related evolutionary related structural domains3, can be divided into smaller evaluation units (EUs) roughly corresponding to these domains4. These divisions disentangle the difficulty of individual domain topology prediction from prediction of overall domain positioning. The definition of EUs is based on two factors: the collective performance of the community on any given multidomain target and the availability of multidomain templates at the time of the experiment5. The division of a target into domains is a complex process that is aided by the Evolutionary Classification of protein Domains (ECOD), which plays a pivotal role 6, 7 in determining the complexity of templates available at the time of the CASP experiment.
The CASP14 interdomain assessment was prompted by experience from previous CASP experiments. During CASP108, four targets (T0663, T0690, T0713, and T0734) were both split into EUs and analyzed as full-length FM targets. Consideration of full-length multi-EU targets also appeared in CASP13 as an analysis of 3 FM targets (T0984, T1000, and T1002) with domain interactions (targets annotated as “FM_special”)4. Accurate prediction of the full-length multidomain targets remains a frontier for many methods, as it is complicated by both difficult informatic and biophysical considerations, such as the identification and assembly of multiple templates and the potential complication of multimeric contacts, biological or otherwise. Notably, CASP14 is the first experiment in which these multidomain targets have been collected and assessed as a category focused on domain interactions, rather than being analyzed as a component of tertiary structure assessments. Of the 67 total tertiary structure prediction targets considered for assessment in CASP14, twenty were split into EUs based on the performance of the prediction community (classification paper this issue). Ten of these were selected for the interdomain assessment category. Here we describe the assessment of the domain interaction prediction category and delineate the scores and methods that were used to determine the final overall ranking.
2. Methods
2.1. Full-length targets for interdomain assessment
CASP targets are split into EUs, when necessary, during their classification4, 5. This division enables analysis of model accuracy within domain boundaries or for combinations of domains that are straightforward to model. The accuracy of interdomain contacts and relative domain orientation are usually not assessed as the accuracy of full-length models on multi-EU targets has typically been low. However, in CASP14 several full-length models were very accurate. This performance prompted us to more closely examine domain interactions for certain full-length targets.
For the interdomain assessment, we selected ten out of twenty CASP14 targets with difficult domain organization. These targets were split into multiple EUs based on the relative performance of the prediction community on the full-length target [Kinch et al, Domain classification paper, this issue, PROT-00132] (Table 1) as compared to individual EUs. Relative performance is assessed by analysis of ‘Grishin plots’, comparisons of the GDT_TS of predictions of the full-length target compared to weighted sums of the GDT_TS of individual EUs (Fig 1A). Targets are split when the performance on the individual EUs is better than the performance on the whole (indicated by an increased linear regression slope fit to the data). The other ten were excluded because of 1) conformational changes in the available templates or 2) excessive oligomeric contacts or 3) lack of interaction between the EUs in subunits of chains that assemble into larger complexes due to long linkers or other unstructured interdomain regions. T1044 was excluded from the official ranking because only 17 groups submitted predictions on the full-length target.
Table 1 –
CASP14 multiple EU targets selected and rejected for interdomain assessment
| Target | EUs | #Groups | Selected | Templates1 | Rejection Reason |
|---|---|---|---|---|---|
| T1024 | 2 | 113 | No | D0–5gxb/1pvf | Conformational change in templates |
| T1100 | 2 | 108 | No | D0–6yue/4cq4 | Conformational change in templates |
| T1092 | 2 | 109 | No | D1–6gbj_D D2–6p1k_J | No interaction between domains3 |
| T1096 | 2 | 104 | No | D1–3les D2–3t5v_B | No interaction between domains3 |
| T1047s2 | 3 | 117 | No | D0–3cr8 | Mainly oligomeric2 |
| T1050 | 3 | 126 | No | D0–4a2l/4a2m | Conformational change in template |
| T1093 | 3 | 106 | No | D1–4l35 D2–4ylo_D D3–3hgb_A | No interaction between domains3 |
| T1070 | 4 | 116 | No | D0– 5iv7 | Mainly oligomeric2 |
| T1091 | 4 | 104 | No | D0–6m3y/6m48 | Conformational change in template |
| T1030 | 2 | 111 | Yes | D1–2dgj D2–6o8b | |
| T1044 | 9 | 17 | Yes | N/A | Excluded during analysis for low model submission rate |
| T1053 | 2 | 108 | Yes | D1–3akk D2–1yo7 | |
| T1058 | 2 | 110 | Yes | D1–6g94 D2–4exr | |
| T1086 | 2 | 109 | Yes | D1–5a7d D2–5i9e | |
| T1094 | 2 | 107 | Yes | D1–6edt_C D2–4a3k_B | |
| T1101 | 2 | 105 | Yes | D1–6qey D2–1vdx | |
| T1038 | 2 | 105 | Yes | D1–3i48 D2–6hg9_B | |
| T1052 | 3 | 126 | Yes | D1–6f7k D2-Unk D31n06 | |
| T1061 | 3 | 109 | Yes | D1-Unk D2-Unk D3Unk | |
| T1085 | 3 | 106 | Yes | D1–6ipe D2–6qk8 D32wh0_B |
Templates were identified by LGA searches of model structure against the PDB. Top ranked structures (with chain when necessary) are identified by EU per full-length target. In some cases no template could be found by LGA, this is signified by ‘Unk’. All LGA results are available at http://predictioncenter.org
Targets whose templates suggested that oligomeric interactions were the major contribution to multidomain organization were not included
Targets with no interaction between EUs were individual subunits from a multisubunit complex whose domain interactions were dictated by the complex.
Figure 1. Grishin plots and domain architecture for CASP14 Interdomain targets.

A) The Grishin plots of interdomain targets compare the weighted sum of GDT_TS of constituent EUs vs the GDT_TS of the full-length target. Non-linearity of scatter plots was indicative of targets where some predictors determined correct domain arrangements and interactions B) The 10 CASP14 interdomain targets colored by EU. The target set consisted of 7 double EU targets (T1030, T1038, T1053, T1058, T1086, T1094, and T1101) and 3 triple (T1052, T1061, T1085) EU targets.
All CASP14 interdomain targets are composed of either two or three EUs (Fig. 1B). There were no known covering templates for the selected interdomain targets when compared against the PDB at the time of the experiment using LGA CA-CA deviations 9. EUs containing multiple domains did so due to the presence of a covering template. Where a target has been divided into multiple EUs, those EUs receive a suffix denoting their position (e.g., D1, D2, or D3).
T1030, T1085, and T1086 are elongated targets composed of short helical repeats. Notably, these targets could be formed with a near-native domain interface but also with significant conformational differences distant from the interface. This property guided our decision to not include scores which emphasized topology in the official ranking. T1030-D1 had a template structure in a bacterial adhesin protein (PDB: 2DGJ) whereas T1030-D2 had a nearby structural template in a threonine protein kinase TBK1 (PDB: 6OB8). There was no covering template for full-length T1030.
T1038 is a viral glycoprotein composed of two EUs deposited as a dimer (PDB: 6ya2) and was assessed as a multimer in that category {multimer assessment this issue}.
T1052 and T1061 were defined as three EUs each, but both had EUs with multidomain templates available. T1061 also had a small deteriorated domain that was not included in any EU but remained in the full-length interdomain target. T052 and T061 were both deposited as trimers and were assessed as in the assembly category {multimer assessment this issue, PROT-00145}.
T1053 is a Legionella effector with an N-terminal protein kinase-like domain followed by a unique C-terminal helical bundle domain that is not typically associated with protein kinases.
T1058 has a unique “ABABA” EU definition that arose from a duplication of a unit with a soluble “B” domain inserted in the middle of a transmembrane helix “A” domain.
T1094 has a complex multidomain organization, where T1094-D2 is inserted into T1094-D1. T1094-D1 contains an α + β domain with an additional deteriorated all-β subdomain, giving the D1/D2 interface a characteristic “knob in socket” appearance.
T1101 is a two-domain RNAse where both domains have known templates but the overall architecture combining the two domains is exclusive to the target.
2.2. Evaluation scores for ranking
We evaluated the performance of predictors by comparison of QS, F1 and Jaccard scores from the QS10 and Iface-check11 programs. These programs had been previously used to evaluate oligomer prediction in CASP1211 and CASP1312. These scores are publicly available from the Prediction Center website13.
The QS(best) score (QSb) reflects the number of correctly predicted contacts in an interface as a fraction of the total number of predicted contacts.
The F1 score (a.k.a. interface contact similarity score) is another measure estimating accuracy of predicted interface contacts in terms of the harmonic mean of the precision and recall.
The Jaccard coefficient (JC, a.k.a. interface patch similarity score) measures the similarity between domain interface patches in the model and the target as the ratio between the number of interface residues common to both structures and the number of residues in the union of model and target domain-domain interfaces.
2.3. Overall Group Ranking
The group ranking is computed using a sum of Z-scores measures discussed in 2.2. First, raw scores are converted into Z-scores based on per-target distributions of model_1 (i.e. the model designated by the prediction group as their top model). scores. We chose to base our calculation on models designated as first (model_1) scores to show preference for the groups who managed to submit their best model as the first. Then, Z-scores from the first round are adjusted. All models with Z-score<−2 (i.e. outlier models scoring two standard deviations below the mean) are removed from the model set, and Z-scores are recalculated from the distribution of scores for the remaining models. All models that scored below the average (Z < 0) in both calculation rounds are assigned Z=0; the remaining models retain their second-round Z-scores. This adjustment prevents models with very low scores from obscuring distinctions among top-scoring methods and is typically implemented in CASP rankings 12. A group’s overall ranking is defined by the sum of its QS-, F1- and Jaccard-based Z-scores for all targets.
Our mandate was to assess interface modeling, not overall structural modeling, and thus structural similarity scores such as GDT_TS and LDDT were not included in our final ranking. We did, however, calculate the overall ranking of interdomain targets by these structural superposition scores as a comparison. The same method for ranking groups by summed Z-scores was used over an equally weighted combination of GDT_TS14 and LDDT15.
2.4. Heatmap and principal components analysis
All measures provided by the prediction center for domain interaction assessment (F1, JC, and QSb from the official ranking as well as QS-score(global), precision, and recall) were combined into a single performance score for use in heatmaps and principal components analysis (PCA). Precision and recall were largely redundant with F1. QS-score(global) refers to the QS score calculated over all interface residues, whereas QSb refers to the best observed QS score among target interfaces. In a set largely made up of two-EU targets, the two QS scores are redundant and QSb was chosen for the overall ranking. These six scores represent the full set of publicly available scores that were used during assessment for exploratory analysis. A reduced set of scores used for the overall ranking was chosen to reduce redundancy and provide the most concise overall measure. To obtain a single score, Z-scores were calculated across groups for each measure on a particular target. The performance score represents the sum of Z-scores for each measure. Performance scores for each target (columns) and group (rows) are colored from high (red) through medium (yellow) to low (blue) performance (Fig. 4). Heatmaps were clustered by hierarchical clustering, where linkage was determined using Ward’s method on Euclidean distances16. The R ‵pheatmap‵ library was used to generate plots and do exploratory analysis17, 18. PCA of the individual scores for each group was calculated using nonlinear iterative partial least squares (NIPALS) to impute missing data19. Groups were required to submit predictions from at least 9 targets to be considered. Ellipses denoting the joint 95% confidence region were calculated using the ggplot2 library20 using an assumption of a multivariate t-distribution.
Figure 4. Heatmap of Z-score sums of interdomain scores.

Z-scores for 6 interdomain scores were summed for each group (row) and target (column). Groups are identified by their CASP14 group accession identifier. Groups are annotated by category: Top1 (purple), Top5 (cyan), TopServer (magenta). Other groups are identified by either manual (salmon) or server (green). Targets are annotated by two (green) or three (blue) domains. Rows and columns are cluster by hierarchal clustering using Euclidean distances and Ward linkage. Notable divergence in performance between targets with two and three evaluation units.
3. Results and Discussion
3.1. Performance of prediction methods on domain interfaces
The official domain interface ranking scheme (cumulative Z-score as described in 2.3) shows one group, AlphaFold2 {AlphaFold2 ref this issue} (Top1), performs especially well in this category (Fig. 2A). The other methods in the Top5 are BAKER-experimental {BAKER ref this issue}, MULTICOM21, BAKER {BAKER ref this issue}, and ProQ3D22. The top server-based method (TopS) by this ranking is BAKER-ROSETTASERVER {BAKER ref this issue}. The average interface contact similarity score for first models from the top-performing AlphaFold2 group on all interdomain targets (F1 78.95, JC 0.78) is much higher than that of the next best BAKER-experimental group (F1 48.36, JC 0.52) or the top Baker server (F1 43.8, JC 0.51). On the other hand, the averages (F1 17.7, JC 0.26) for models from a baseline server (Baker-ROBETTASERVER), whose method did not change from the previous CASP13 round, are much worse than these top performing groups. Performance rankings for top groups were mostly unaltered by the choice to first rank models (i.e., the model chosen by the group as the best) vs the best model as scored by our scheme (Fig S1). However, these rankings and average scores only signify relative performance of the groups across all targets.
Figure 2. Performance on interdomain target assessment.
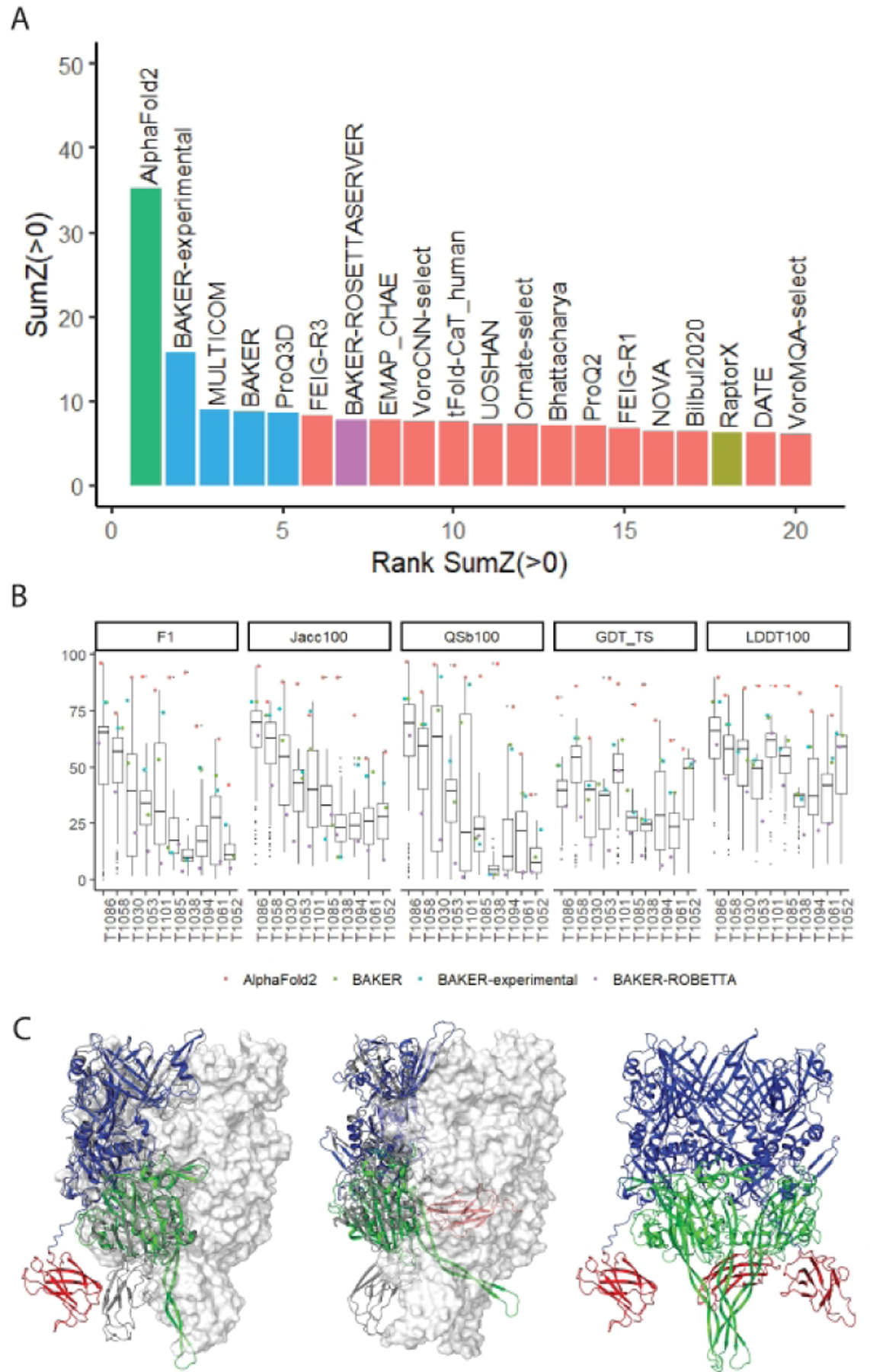
A) Participants in the interdomain category were ranked by the sum of the Z-scores for F1, Jaccard coefficient, and QS(best) domain interaction score distributions over the first submitted model for each group. Groups were categorized by the top group (cyan), top 5 groups (blue), and top server (magenta). For the sake of clarity, only the top 20 groups ranked by this measure are shown here. Additionally, servers not in these three groups were identified as manual (salmon) or server (olive) prediction methods. B) Boxplots were generated using the ‵ggplot2‵ R library. The order of the targets was determined manually. The line represents the median of the distribution. The box is drawn from the 1st (25%) to the 3rd (75%) quartile. The interquartile range (IQR) is defined as the distance between the 1st and the 3rd quartile. Whiskers extend from the box to the highest observation or no more than 1.5 * IQR above the 3rd quartile, and to the lowest observation or no less than 1.5 * IQR below the first quartile. Outliers that fall outside the bounds of the whiskers are plotted as individual points. Observations from AlphaFold2, BAKER, BAKER-experimental, and BAKER-ROBETTA (i.e. the baseline server) were plotted against the boxplots for comparison. C) Difficult multidomain trimeric assembly for T1061 (target monomer in grey cartoon, with two additional chains in white surface). Superimposed models (AlphaFold2 left and BAKER center) are colored by domains: D1 (blue), D2 (green), and D3 (red). AlphaFold2 model assembles into a trimer, with conformation change of the C-terminal domains (right).
To better understand group performance on individual targets, we examined per target group score distributions highlighting those from the top groups and the baseline server (Fig. 2B). The box plot distributions for the three domain interface scores (F1, JC, and QSb) distinguish five relatively easy targets (T1086, T1053, T1030, T1053 and T1101), where the average group performance is roughly better than the average performance on the rest of the targets. First models from AlphaFold2 and Baker-EXPERIMENTAL outperform the rest of the groups on all five ‘easy’ targets (points are outside the box). Among these targets, the outperformance of the top groups is less for the transmembrane protein T1058, whose domain interactions could be determined by their partitioning into soluble and membrane regions.
The first models for AlphaFold2 also outperform on the remaining difficult interdomain targets (T1085, T1038, T1094, T1061, and T1052). For three of these (T1085, T1038, and T1094), their performance extends beyond the 1.5 times the interquartile range of the scores (i.e. beyond the ‘whisker’) and likely contributes to their outperformance in the domain interaction rankings. One of these difficult targets represents an elongated ARM repeat fold that was separated into 3 domains (T1085, structure not yet published). AlphaFold2 predicts the relative orientation and interface of all three domains, while the rest of the predictions do not. The other two targets are discussed below as examples (Specific prediction highlights section).
The domain interfaces for two of the most difficult targets in the assessment (T1061 and T1052) were challenging for the entire prediction community, although first models from AlphaFold2, BAKER and BAKER-experimental outranked the rest using domain interface evaluation measures (Fig. 2B, F1, JC, and QSb). While each of these difficult interdomain targets included three defined EUs, their domain count according to ECOD was higher: T1061 had six domains and T1052 had four domains. When collections of domains had a covering template and were predicted with high GDT_TS, they were not split into multiple EUs. Some EUs are made up of multiple domains. Additionally, each of these multidomain targets assembled into trimers. Both the AlphaFold2 and BAKER predictions for T1061 correctly place the first two EUs relative to each other. However, they each incorrectly place the C-terminal domain, which has a relatively small interaction surface, in different orientations. Impressively, the AlphaFold2 model is consistent with the trimeric assembly and could potentially indicate an alternate orientation of the C-terminal domains (Fig. 2C).
3.2. Performance of prediction methods on full-length targets
The interdomain analysis and official ranking (Fig. 2A) specifically focused on the reproduction of interfacial contacts in the interdomain targets. The selected targets (Fig. 1A) generally have predictions where some members of the community show similar prediction accuracy (by GDT_TS) of the whole target compared to the weighted sum of individual EUs. In general, interfacial contact scores are meaningful when each of the interacting domains are predicted correctly. The final two measures depicted in Fig. 2B represent a rigid body, superposition based measure (GDT_TS9, 14) and superposition free measure (LDDT15) that evaluate all contacts in the structure and capture the overall performance of predictions on full-length targets. As expected, the rigid body superposition score distributions (Fig. 2B, GDT_TS) for all full-length targets are lower than the superposition free distributions that are less sensitive to global domain movements (Fig. 2B, LDDT). The AlphaFold2 model LDDT scores were higher relative to the GDT_TS scores for the two most difficult multidomain targets that adopted trimeric assemblies (T1061 and T1052, discussed in the section above) as well as for T1030, T1085, and T1086.
Ranks of CASP14 group performance on full-length interdomain targets using GDT_TS and LDDT reaffirmed separation of AlphaFold2 from the remainder of predictor groups (Fig. 3A). Three other members of the Top5 (BAKER, BAKER-experimental, and ProQ-3D) remain among the top 5 ranked groups (Fig. 3B). MULTICOM was in the Top5 by domain interaction scores but ranked 18 by structural scores, whereas tfold-CaT human ranked 4 by structural scores (and ranked 10 by interaction scores). Caution must be used evaluating relative rankings by Z-score sum when the absolute difference in Z-scores is small.
Figure 3. Overall ranking by superposition scores for CASP14 interdomain targets.

A) Interdomain targets were scored by equally weighted structural superposition scores (GDT-TS and LDDT). Rankings largely recapitulated those seen by interaction scores. B) Comparison of stack rankings for structural superposition Z-scores and domain interaction Z-scores (RSumZ). C) T1030 target (left) compared to T1030 superimposed with AlphaFold2 model 1 demonstrates how in some models with a well-formed interface structural superposition can still deviate due to global deformation.
T1030 adopts an elongated structure consisting of repeating helical units. The relatively poor performance of the AlphaFold2 (and other group) models using GDT_TS with respect to LDDT results from deviations at the ends of the elongated structure, despite correct prediction of the native interface (Fig. 3C). The overall GDT_TS (scaled 0–100) of the first AlphaFold2 model was 63.0 whereas the respective scores for D1 and D2 were 75.3 and 89.5. The LDDT scores for the same models (scaled to 0–100) were 85.0 for the full-length prediction, and 87.0 and 82.0 for the D1 and D2, respectively. T1086 and T1085 provide similar examples of elongated helical ARM repeats whose relative orientations can deviate at the ends of the repeating units. T1086 had a similar correct prediction of the native interface, but T1085 had the additional complexity of having three domains (discussed in the section above).
Overall, the Pearson correlation coefficient considering the difference between Z-score ranks or Z-scores is similar, 0.92 and 0.94 respectively (Fig. S2). This analysis principally re-iterates the observation that the majority of the CASP community still benefits from analysis of some special cases of split domains rather than full-length targets.
3.3. Heatmaps and PCA of interface scores support rankings and highlight target difficulty
To visualize the performance of groups on individual interdomain targets, heatmap clustering and PCA of domain interface scores (see Methods) were performed to reveal general trends within the category (Fig. 4). Heatmap scores highlight AlphaFold2 outperformance on nearly all targets, with this top group clustering independently from the rest. The top server (BAKER-ROSETTASERVER) is present in a cluster that contains the remaining top-performing groups and forms an outgroup to AlphaFold2. Two and three-EU targets separate in the heatmap clustering, with the exception of T1038 (discussed in Specific prediction highlights section below) and T1094 (discussed above, includes 4 domains in a trimeric assembly). T1094 possesses a relatively large interdomain interface (1127 Å2) and was one of the more complex prediction targets among the two-EU targets. Overall, the heatmap analysis reveals that there is a large cluster of groups/methods (row clusters 1–3 from the top, Fig. 4) that predict simple two-EU targets with generally correct domain positioning.
Finally, we performed principal components analysis (PCA) upon the domain interaction score to cluster prediction methods by their performance. The PCA plot re-iterated that AlphaFold2 was distinct from all other methods, server and manual (Fig. 5A). The combination of the heatmap and the PCA suggest that AlphaFold2 had significantly distinct performance on T1038, T1094, and T1053. We recalculated the PCA without AlphaFold2 in order to get a clearer separation among the remaining groups (Fig. 5B). This recalculation established that BAKER-experimental and BAKER were clearly distinct from the remaining server and manual groups. However, the performance of the remaining groups, which included the top server, was difficult to distinguish.
Figure 5. Principal components analysis (PCA) of interdomain assessment scores.

Six interdomain contact scores (F1, Precision, Recall, Jaccard Coefficient, Q(global), Q(best) were reduced to sums of Z-scores. A) These Z-scores were then analyzed by non-iterative partial least squares (NIPALS) PCA to evaluate comparative performance. AlphaFold2 (A, gray) clearly demonstrated significantly different performance than members of the Top5 (orange), the top server (light blue), and the clusters of manual (green ellipse/dots) and server (yellow ellipse/methods). B) The method was repeated with AlphaFold2 removed in order to better visualize the spread between the remaining methods.
3.4. Specific prediction highlights in difficult interdomain targets
Here we discuss the interdomain contact results for three targets that potentially illustrate the difference in performance between the top two methods: T1038, T1094, and T1053. Notably, AlphaFold2 outperformed on each of these targets with respect to the rest of the prediction community. Although T1044 was excluded from our analysis due to a lack of participation in its prediction, we include this difficult target as an example to highlight an impressive model from the Baker-experimental group.
T1038 is Tomato spotted wilt topsovirus glycoprotein contains two beta-sandwich domains (Figure 6A, left). Both a monomer and a dimer were present in the asymmetric unit of the deposited structure, with the interchain interface (944 Å2) of the dimer having a similar area as the interdomain interface (733 Å2). The monomer exhibits a disorder to order loop transition upon dimerization. Of the submitted models, only AlphaFold2 (Fig. 6A, middle) correctly positioned the two domains and predicted a structure that would accommodate the dimeric interaction (F1 score, 92.2). The dimer interaction surface from T1038 is contributed by two interaction loops and one β-strand edge from D1 (residues 76–86, 94–102, and 115–122, respectively, gray in Figure 6A), with part of the surface also forming the domain interaction. The flexibility of the loops contributing to this homodimeric interaction may have led to the difficulty observed for most groups in predicting its conformation. For comparison, the second-place overall group (BAKER-experimental, Fig. 6A, right, whose model ranked 80th for this target by F1) predicted this region as three beta strands and failed to assemble the domains in the correct orientation (F1 score 8.8).
Figure 6. Interdomain prediction examples from top ranked predictors.

T1038 (A), T1053 (B), T1094 (C) have performance differences between AlphaFold2 and BAKER-experimental. Structures in A-C are colored by domain (D1, blue and D2, red). Interacting residues (within 4 angstrom) from the target structure are depicted as dots (colored light blue from D1 and pink from D2) in the target structure and in the models for the corresponding residues. Model structures were aligned to the target (left), except for the BAKER-experimental model for T10838, which is shown as a convenient orientation to see the predicted domain interaction. An interdomain (F1) and structural superposition (GDT_TS) score is depicted for each example. Residues from an oligomeric interaction surface for T1038 (gray cartoon) partially overlaps with the domain interaction surface (gray dots). T1053 interdomain interface was formed from multiple high contact order patches complicating the prediction. The large interdomain interface in T1094 was partly due to a pseudodomain (cyan) contained with the T1094-D1 (blue) evaluation unit. T1044 (D, colored by EU) from BAKER-experimental had excellent JC and F1 scores, 56.0 and 62.0, respectively. Structures of T1044 (left) and the AlphaFold2 model (right) are shown side by side in one orientation to the left, then with a 90° rotation on the right.
T1053 is a two-domain protein from Legionella that was classified as two EUs, with the N-terminal EU representing a protein kinase followed by a C-terminal helical bundle (Fig. 6B, left). Notably, the EU boundary was within a kinked helix extended from the kinase C-lobe. The interaction region between D1 and D2 is principally helical, with D1 and D2 contributing three (331–341, 355–367, and 394–406) and four (407–412, 444–456, 500–513, and 558–576) distinct helical regions to the domain interface, respectively. AlphaFold2 correctly predicted the position of both the loop 444–456 and the two N-terminal short helices in residues 558–576 (Fig. 6B, middle). Although BAKER-experimental (Fig. 6B, right) largely predicted the topology of the protein correctly and the relative placement of the two domains, the differing placement of the loop and the prediction of the 588–576 region as a single out-of-position helix likely led to the difference in interdomain scores between the top two groups (F1 90.1 and 48.9, respectively).
T1094 was evaluated as a two-EU target, but notably one of the EUs contained a small pseudo-domain (defined by a combination of the sequence that borders the D2 insertion and the C-terminus) that contributed to its large interface surface area (Fig. 6C, left). This protein had potential templates in ECOD in both the “N-terminal domain in beta subunit of DNA dependent RNA-polymerase“ and “insertion domain in beta subunit of DNA dependent RNA-polymerase”. This target was difficult for most groups. Models from both AlphaFold2 and BAKER-experimental correctly predicted the overall domain arrangement, which is reflected in their F1 scores (68.3 and 50.0 respectively). The domain interface of T1094 consists of two faces: one composed of two helices of D1 and a helix and loop of D2, and a second composed of a series of small loops from D1 and a small beta-sheet from D2. The D1-D2 helical interface is packed more tightly in the AlphaFold2 model (Fig. 6C, middle) than in the target, whereas in the BAKER-experimental (Figure 6C, right), the overall separation in this face of the interaction is more similar to the target. Conversely, the first model for AlphaFold2 extends the interface in the sheet/loop face of the interface beyond that which was presented in the target model. T1094 is an excellent example of how two groups can predict correct topology and positioning of domains in a multidomain target but still get very different scores.
T1044 represents a 2166 residue-long phage polymerase that was split into nine different targets prior to releasing the sequence to predictors (Kinch classification, this issue, PROT-00132). The full-length target also includes domains from the active site that were excluded from the tertiary structure prediction category (Figure 6D, left two structures rotated 180° about the Y axis). The top prediction models for this target were all by the BAKER-experimental group, with the first model scoring 56.0 for F1 and 0.62 for JC (Fig. 6D, right two structures rotated 180° about the Y axis). This prediction was also scored well by structural superposition scores achieving a 49.03 GDT_TS and 57.0 LDDT scores. Notably, the domains are positioned correctly relative to each other, with those closer to the surface being more distorted than the ones in the center.
4. Conclusions
Assessment of domain interactions in 10 CASP14 targets composed of difficult multiple EUs highlighted the clear outperformance of AlphaFold2 on most targets, followed by the method of BAKER-experimental, which could be distinguished from the remaining groups by PCA. One server (BAKER-ROSETTASERVER) was among a cluster of the top-performing methods (Fig. 4), but their performance was not easily distinguished from one another (Fig. 5B). This top cluster generally performed better than most groups on four out of 5 targets we designated as ‘easy’ for most of the groups assessed herein. However, for two difficult targets (T1052 and T1061), neither the models from AlphaFold2 nor the models from the rest of the prediction community succeeded in correctly predicting the domain organization for the entire multidomain target (Figure 2B and Figure 4). These two targets included more domains (six and four) than the rest of the targets, and both assembled into trimers. Collectively, these three targets were also assessed as multimers {multimer assessment this issue}. While we excluded targets whose structures rely on assembly for folding, these two retained domain interactions that appeared to be distinct from assembly. Whether or not the poor performance reflected domain flexibility or the requirement for assembly remains a question that will likely present itself again in future CASPs. Future domain interaction assessments will likely require more careful consideration of the oligomeric interaction surface. It also may require careful enumeration of which components of an interaction surface are supplied by individual EUs or domains. Given the conceptual similarity of scoring and evaluation of domain interactions in multidomain structures, multimer interactions in homo-oligomer assemblies and subunit interactions in protein complexes, the assessments might benefit from being combined in future CASP experiments.
Some targets with predictions having correct domain positioning and interdomain contacts scored poorly according to the overall fold scores. Among these, significant deviations existed in structure prediction distant from the scored interfaces, especially in non-globular targets (e.g. elongated ARM repeats). This observation was exemplified by predictions of the helical bundle T1030, which had a simple interface between EUs and could score well by F1 and JC, while scoring poorly by GDT_TS (but not LDDT) when the angle of the bundle or interacting helices were slightly skewed from native. In hindsight, the lower performance by the prediction community on these examples of elongated structures was not caused by difficulty in predicting domain interaction but instead by the choice of using rigid body GDT_TS assessment scores to determine multidomain splits during classification {classification paper, this issue, PROT-00132 }. The elongated structures were each solved by X-ray crystallography and exhibit extensive interactions from crystal contacts that are generally not considered in CASP assessment. Future assessments, both for domain interactions and tertiary structure predictions, might need to consider such higher order chain assemblies from crystal packing. Interestingly, such cases do not necessarily capture biologically relevant conformations, and models from current state of the art protein structure prediction methods may or may not capture relevant conformations. Finally, our selection of only the most difficult domain interaction targets limited our conclusions about the performance of protein structure prediction methods on domain assemblies. The limited dataset precluded statistically relevant performance comparisons, and future domain interaction assessments would benefit from evaluating all domains instead of a select few difficult ones.
Supplementary Material
Acknowledgements
The study is supported in part by the grants from the National Institutes of Health (GM127390 to NVG and GM100482 to AK) and the Welch Foundation (I-1505 to NVG). We would like to thank the CASP organizers for their continued dedication to this experiment, the structural biologists who contributed data, and the predictors whose participation is vital to this experiment.
References
- 1.Kryshtafovych A., et al. , Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins, 2019. 87(12): p. 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moult J., et al. , A large-scale experiment to assess protein structure prediction methods. Proteins, 1995. 23(3): p. ii–v. [DOI] [PubMed] [Google Scholar]
- 3.Yang S and Bourne PE, The evolutionary history of protein domains viewed by species phylogeny. PLoS One, 2009. 4(12): p. e8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinch LN, et al. , CASP13 target classification into tertiary structure prediction categories. Proteins, 2019. 87(12): p. 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinch LN, et al. , CASP9 target classification. Proteins, 2011. 79 Suppl 10: p. 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng H., et al. , ECOD: an evolutionary classification of protein domains. PLoS Comput Biol, 2014. 10(12): p. e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaeffer RD, et al. , ECOD: identification of distant homology among multidomain and transmembrane domain proteins. BMC Mol Cell Biol, 2019. 20(1): p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor TJ, et al. , Definition and classification of evaluation units for CASP10. Proteins, 2014. 82 Suppl 2: p. 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zemla A., LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res, 2003. 31(13): p. 3370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertoni M., et al. , Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep, 2017. 7(1): p. 10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lafita A., et al. , Assessment of protein assembly prediction in CASP12. Proteins, 2018. 86 Suppl 1: p. 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzenko D., et al. , Assessment of protein assembly prediction in CASP13. Proteins, 2019. 87(12): p. 1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kryshtafovych A, Monastyrskyy B, and Fidelis K., CASP11 statistics and the prediction center evaluation system. Proteins, 2016. 84 Suppl 1: p. 15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zemla A., et al. , Processing and analysis of CASP3 protein structure predictions. Proteins, 1999. Suppl 3: p. 22–9. [DOI] [PubMed]
- 15.Mariani V., et al. , lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics, 2013. 29(21): p. 2722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murtagh F and Legendre P., Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? Journal of Classification, 2014. 31(3): p. 274–295. [Google Scholar]
- 17.Kolde R, pheatmap: Pretty Heatmaps. 2019.
- 18.Jolliffe I, Principal Component Analysis. 2006: Springer; New York. 488. [Google Scholar]
- 19.Wold H, Estimation of principal components and related models by iterative least squares, in Multivariate Analysis, Krishnaiah PR, Editor. 1966, Academic Press: New York. [Google Scholar]
- 20.Wickham H, ggplot2: elegant graphics for data analysis (use R!). Springer, New York, doi, 2009. 10: p. 978–0. [Google Scholar]
- 21.Hou J., et al. , The MULTICOM Protein Structure Prediction Server Empowered by Deep Learning and Contact Distance Prediction. Methods Mol Biol, 2020. 2165: p. 13–26. [DOI] [PubMed] [Google Scholar]
- 22.Uziela K., et al. , ProQ3D: improved model quality assessments using deep learning. Bioinformatics, 2017. 33(10): p. 1578–1580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.