

Summary
Numerous DNA double-strand breaks (DSBs) arise during meiosis to initiate homologous recombination. These DSBs are usually repaired faithfully, but here we uncover a distinct type of mutational event in which deletions form via joining of ends from two closely spaced DSBs (double cuts) within a single hotspot or at adjacent hotspots on the same or different chromatids. Deletions occur in normal meiosis but are much more frequent when DSB formation is dysregulated in the absence of the ATM kinase. Events between chromosome homologs point to multi-chromatid damage and aborted gap repair. Some deletions contain DNA from other hotspots, indicating that double cutting at distant sites creates substrates for insertional mutagenesis. End joining at double cuts can also yield tandem duplications or extrachromosomal circles. Our findings highlight the importance of DSB regulation and reveal a previously hidden potential for meiotic mutagenesis that is likely to affect human health and genome evolution.
eTOC blurb
Germ cells damage their DNA in meiosis to initiate genetic exchange, but end joining at closely spaced DNA double-strand breaks leads to de novo deletions and duplications, revealing the mutagenic potential of the meiotic genome.
Graphical Abstract
Introduction
Meiotic recombination is essential for faithful genome transmission in most sexually reproducing organisms (Hunter, 2015). Recombination is initiated by the programmed formation of DNA double-strand breaks (DSBs) by the SPO11 protein (Keeney, 2001). SPO11 preferentially cuts within narrow genomic regions called hotspots (Petes, 2001; Tock and Henderson, 2018). Of all DSB hotspots (~15,000 defined in mouse), only a tiny fraction (~250–350) experiences a DSB in any single meiosis (Cole et al., 2012; Lange et al., 2016; Li et al., 2019). DSBs are also nonrandomly dispersed along chromosomes to ensure homolog pairing and recombination (Keeney et al., 2014). This quantitative and spatial regulation of hotspot usage involves hierarchical and combinatorial levels of control (Pan et al., 2011; Lange et al., 2016; Yamada et al., 2017), with the ATM kinase playing a central role (Cooper et al., 2016; Lukaszewicz et al., 2018).
ATM, known for its role in the DNA damage response in mitotic cells (Shiloh, 2006), is activated by meiotic DSBs to limit DSB numbers and to control hotspot usage genome-wide (Lange et al., 2011; Lange et al., 2016). Control of DSB numbers by ATM and its orthologs is conserved, although ATM-deficient mice have the largest reported increase in DSB levels (~10 fold) (Checchi et al., 2014; Lukaszewicz et al., 2018; Tian and Loidl, 2018; Kurzbauer et al., 2021). Control of genome-wide DSB distributions is also conserved (Mohibullah and Keeney, 2017). In yeasts, Tel1 controls DSB formation locally by suppressing double cutting, that is, simultaneous DSB formation at adjacent hotspots (Garcia et al., 2015; Cooper et al., 2016; Fowler et al., 2018). This suppression is strongest at closely spaced hotspots (<10 kb), such that in the absence of Tel1, double cutting at hotspots separated by ~2 kb is more frequent than expected by chance and ~10-fold higher than in wild type. The loss of this Tel1-mediated DSB suppression is thought to contribute to the global increase in DSBs (Garcia et al., 2015; Mohibullah and Keeney, 2017). In mouse, double cutting at adjacent hotspots has not been demonstrated, but potentially may be even more likely in the absence of ATM, considering the extremely elevated DSB numbers. Accordingly, we previously suggested that ATM may inhibit multiple nearby DSBs (Lange et al., 2011; Lange et al., 2016).
Normally, most meiotic DSBs are repaired by recombination between homologous chromosomes. While nonhomologous end-joining (NHEJ) is a robust mitotic DSB repair pathway in mice (Chang et al., 2017), it is thought to be suppressed in meiosis (Kim et al., 2016). However, NHEJ can occur in mouse spermatocytes when DSBs are induced by irradiation (Ahmed et al., 2010; Enguita-Marruedo et al., 2019), and in C. elegans in mutants with compromised DSB processing e.g., (Lemmens et al., 2013; Yin and Smolikove, 2013; Girard et al., 2018) or recombination e.g., (Martin et al., 2005; Macaisne et al., 2018).
The consequences of deregulated meiotic DSB formation are not fully understood. We hypothesized that, in the absence of ATM, formation of DSBs that are too numerous or too closely spaced interferes with the meiotic recombination program, eliciting the use of other repair pathways. We reasoned that mutagenic NHEJ in particular might become more prevalent because resection defects, which impair homologous recombination in mitotic cells (Chen et al., 2017), are found in the absence of ATM in meiotic cells (Joshi et al., 2015; Mimitou et al., 2017; Cannavo et al., 2018; Paiano et al., 2020; Yamada et al., 2020). Moreover, the loss of ATM may cause DSBs to form at similar locations on multiple chromatids — due to the increase in DSB numbers and/or the loss of spatial control — and thus promote NHEJ by damaging the templates needed for recombinational repair.
We tested this hypothesis by asking whether multiple DSBs form at adjacent hotspots in the absence of ATM in mouse spermatocytes and whether they are repaired aberrantly to delete or duplicate the intervening sequences. Further, we asked whether multiple DSBs leading to deletions can occur within an individual hotspot. Our experiments uncovered DSB (mis)repair products, providing insight into the consequences of loss of meiotic DSB control.
Results and Discussion
Hidden potential of the meiotic genome to form deletions
We first catalogued genomic locations at risk for deletion by identifying pairs of closely spaced hotspots in nucleotide-resolution DSB maps obtained by sequencing SPO11-bound oligonucleotides (oligos), a by-product of DSB formation (Lange et al., 2016). We found that numerous DSB hotspots are separated by a short genomic distance (Figure 1A). For example, ~6% of 13,436 autosomal SPO11-oligo hotspots in wild-type mice are located within 5 kb of another hotspot, and this percentage nearly doubles (~10%) in Atm−/− mice, which have more hotspots called (20,417) (Figure S1A). Most hotspots in Atm−/− mice are shared with those in wild type, although a significant fraction are newly called because the few SPO11 oligos that cluster at these sites in wild type are increased in the mutant (Lange et al., 2016) (Figure S1A). Newly called hotspots often emerge near shared hotspots, resulting in an increased number of closely-spaced hotspots in Atm−/− (Figure S1B). Moreover, in addition to the number, the strength of the shared hotspot pairs also disproportionately increases in Atm−/− (Figure S1C), at least in part due to the large increase in DSBs at weaker hotspots (Lange et al., 2016). Thus, while adjacent DSBs may normally be suppressed, this hidden potential for DSBs at adjacent hotspots may be revealed in Atm−/−.
Figure 1. Meiotic deletions at adjacent DSB hotspots.

(A) Distribution of distances separating adjacent SPO11-oligo hotspots on autosomes. The hotspot calling algorithm (Lange et al., 2016) merges hotspots separated by edge-to-edge distances of <1 kb. Hotspot pairs separated by >250 kb are not shown. See also Figure S1.
(B) Strategy to detect deletions between adjacent hotspots. Top panel, SPO11-oligo maps for the hotspot pair on Chr1 (RPM, reads per million). Genomic coordinates (MM10 assembly) are shown below the graphs. The estimated fold increase in absolute SPO11-oligo frequency (RPM × 11.3) is indicated for each hotspot. Vertical pink lines indicate hotspot centers. Lower panel, scheme of the PCR assay, with restriction enzymes and primers indicated (see STAR Methods). The Atp1b1 exon is removed in deletions.
(C) Detection of deletions by agarose gel electrophoresis. Representative gels for the Chr1 hotspot pair are shown.
(D) Deletion frequencies. Three to seven mice of each genotype were analyzed, listed in Table S1. P values are from the Chi-square test.
To test for deletions, we selected DSB hotspot pairs that are shared between wild type and mutant and that contain strong hotspots to increase the chance of simultaneous DSB formation. The chosen hotspot pairs on Chr1 and Chr19 are separated by <2 kb, with hotspots that individually rank in the top quartile of all hotspots in both wild type and Atm−/− (Figures 1B, S1D, and S2A). We tested for deletions by performing nested PCR on testis DNA. For both pairs, concurrent DSB formation at the two hotspot centers with precise joining of the DNA ends would give rise to 1.8-kb deletions (Figures 1B and S2A); in practice, deletions would vary in size depending on the location of SPO11 cleavage in any particular cell. To suppress amplification of parental DNA, genomic DNA was digested with restriction enzymes prior to amplification in some experiments. Nested PCR was performed with input testis DNA equivalent to ~8,000–16,000 haploid genomes per well for a total of ~10–15 million haploid genomes per genotype (Table S1).
Smaller PCR products indicative of deletions were observed in Atm−/− (Figures 1C and S2B). In adult Atm−/− mice, the frequencies of deletions at the Chr1 and Chr19 hotspot pairs were 9.14 × 10−6 and 4.25 × 10−6, respectively, per haploid genome (Figure 1D; Table S1). Deletions were also detected in ATM-proficient cells but were rare. At the Chr19 hotspot pair, the frequencies were 0.15 × 10−6 and 0.31 × 10−6 in wild type and Atm+/−, respectively, reflecting a total of just six events, while no deletions were observed in Spo11−/−. For the Chr1 hotspot pair, only two deletions were observed in ATM-proficient cells, although one was also observed in Spo11−/−. No deletions were detected in somatic Atm−/− controls at the Chr1 hotspot pair (liver, frequency ≤0.10 × 10−6, Table S1), indicating that the events observed in testis DNA are meiosis-specific.
Similar results were obtained in juvenile testes at 15 days post partum (Table S1), when wild-type and mutant testes have a similar distribution of prophase I stages. This finding indicates that the increase in deletion frequency is not due to the different cellular composition of mutant testes caused by prophase I arrest of Atm−/− spermatocytes (Xu et al., 1996; Barlow et al., 1998).
We also tested for deletions in DNA from sperm from Atm+/− and Atm+/+ mice. We captured 7 deletions in Atm+/− at the Chr19 hotspot pair (1.09 × 10−6 per haploid genome), although none from Atm+/+ (≤0.31 × 10−6; Table S1). Thus, meiotic deletions can be transmitted into sperm.
It should be noted that testis DNA is derived from germ cells at different stages of development as well as from some somatic cells, such that the germline deletion frequencies we report for testes are underestimated for all genotypes. Even so, meiotic recombination at a well-studied hotspot that has a similar strength as the individual hotspots examined here occurs at a substantially higher frequency (two to three orders of magnitude: 5.8 × 10−3 noncrossovers per sperm genome (Cole et al., 2010)) than the deletion frequency in Atm−/−. However, taking into account the large number of hotspots located close to one another, the potential to form deletions in meiosis is substantial. For example, considering only hotspot pairs separated by 1–5 kb, we estimated that ~1% of Atm−/− spermatocytes may carry a deletion (Table S3).
Double cutting in the absence of ATM
To map deletion breakpoints, we sequenced deletion products from Atm−/− adults and juveniles: 134 from Chr1 and 58 from Chr19 (Figures 2A and S2C; Table S2). The distribution of deletion breakpoints resembled the distribution of SPO11 oligos, with breakpoints clustered around hotspot centers (Figures 2B, 2C, S2D, and S2E). Smoothed breakpoint and SPO11-oligo profiles displayed similar shapes, except that breakpoint profiles were slightly wider with more prominent smaller peaks (Figures 2D and S2F). We defined the breakpoint center within each hotspot as the position of the smoothed peak in the breakpoint density, in the same manner as hotspot centers were defined (Lange et al., 2016). Breakpoint centers closely matched each hotspot centers within 13 to 39 bp. These findings strongly indicate that deletion formation is preceded by concurrent SPO11 cleavage at the two hotspots, providing evidence for double cutting at adjacent DSB hotspots in mouse.
Figure 2. Deletion breakpoints at the Chr1 hotspot pair.

(A) Deleted DNA sequences at the Chr1 hotspot pair. Deletions (black bars) were combined from adult and juvenile testes (dpp, days post partum). Red asterisks indicate breakpoints in Atm+/− shared with Atm−/−.
(B) Arc diagram of deletions in Atm−/− at the Chr1 hotspot pair. Arcs link the breakpoints of each deletion (134 deletions total). Both deletion breakpoints often map close to the hotspot centers. See also Figure S3A and S3B.
(C) Individual breakpoints cluster around the hotspot centers but are also observed to spread.
(D) Smoothed breakpoint and SPO11-oligo profiles display similar shapes (201-bp Hann filter for smoothing). Offsets between centers of hotspots and breakpoint distributions are indicated.
The median deletion lengths were similar to the distances between the DSB hotspot centers (Chr1: 1954 bp vs 1825 bp; Chr19: 1892 bp vs 1763 bp), as expected from SPO11 cleavage of both hotspots and joining of unprocessed or minimally processed DNA ends. Accordingly, many deletions had breakpoints within the narrow (251 bp) central zones around the hotspot centers that contained the majority (>60%) of SPO11 oligos: Nearly half of the deletions had one breakpoint within the central zone of one hotspot (Chr1, 41%; Chr19, 45%) and about a fifth had both breakpoints within the central zone (Chr1, 21%; Chr19, 19%) (Figure S3A). While the number of deletions with both breakpoints within the zone was lower than predicted from the SPO11-oligo counts (Chr1, 65%; Chr19, 48%; Figure S3B), it was notable that breakpoints outside the zone were not evenly distributed. Instead, some clusters could be observed that overlapped with, but were more prominent than, SPO11-oligo clusters (Figures 2B–D and S2D–F). The overlap of breakpoints and SPO11 oligos implies that DSB ends are usually joined close to the initial SPO11 cleavage sites, whereas the wider distribution of deletion breakpoints may reflect preferential deletion formation for DSBs further from the hotspot center, DNA end processing prior to joining, or multiple DSBs within the same hotspot (see below).
In principle, deletions could arise from single DSBs at one hotspot that are asymmetrically resected. However, this seems highly unlikely, given that both breakpoints usually map in the vicinity of separate hotspots, even with the omission of the restriction digest step (Figure S2G). Moreover, end-joining at single, unprocessed DSBs is predicted to be error-free due to the presence of 2-bp 5′ overhangs after SPO11 cleavage (Yamada et al., 2020; Hatkevich et al., 2021). Although our assay is not designed to detect small deletions at a single hotspot, we address this question below.
Notably, the frequency of deletions in Atm−/− at both hotspot pairs is ~100 times lower than the frequency of double cutting estimated from SPO11-oligo read counts with the assumption of independent cutting (Table S3). Thus, it seems likely that only a subset of double cuts undergoes end joining, while the others are correctly repaired, misrepaired by other mechanisms, or not repaired at all, given that Atm−/− spermatocytes accumulate unrepaired DSBs (Barchi et al., 2005).
In wild type and Atm+/−, several deletions had breakpoints that were identical to those observed in Atm−/−, while the one deletion obtained from Spo11−/− had both breakpoints comparatively far from the hotspot centers (Figures 2A and S2C). However, most of the deletion breakpoints in wild type and Atm+/− were not within the narrow central zone (251 bp) of SPO11 oligos around the hotspot centers. For example, 12 deletions (86%) at the Chr19 hotspot pair had both breakpoints outside this zone. Although more spread out, breakpoints still appeared to often cluster where SPO11 oligos clustered (Figure S3C). These observations suggest that in the presence of ATM, deletions may be more prone to occur for DSBs formed outside of hotspot centers or the DSBs involved in the deletion may be more processed.
Double cutting can involve one or multiple chromatids leading to distinct mutational outcomes
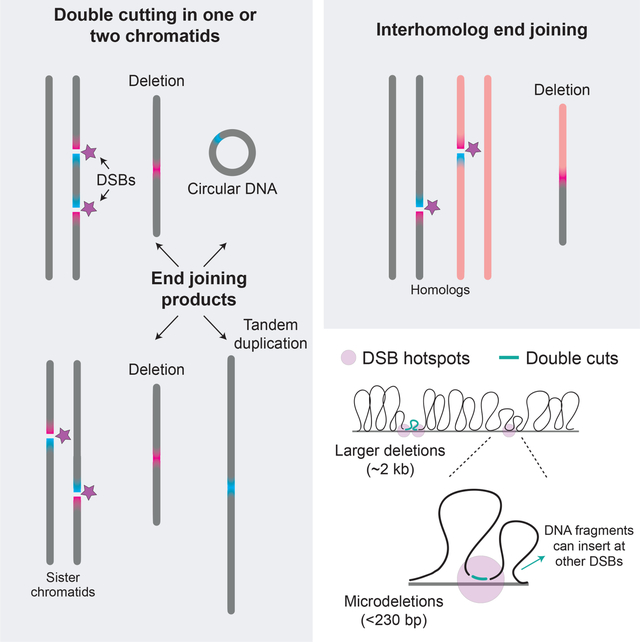
The meiotic deletions we observe could arise from two DSBs on a single chromatid or DSBs on two chromatids. Moreover, double cutting produces four DNA ends, with the potential to give rise to other mutational outcomes. In particular, DSBs on two chromatids can generate tandem duplications as well as deletions, while two DSBs on one chromatid can release a DNA fragment with the potential to circularize (Figure 3A). Finally, DSBs on homologous chromosomes may provoke inter-homolog end-joining. We investigated each of these possibilities.
Figure 3. Tandem duplications and circular DNA from double cutting.

(A) Predicted end-joining products from double cutting in one or two chromatids. Lighter grey indicates DNA segments between two DSBs at adjacent hotspots. Inverse PCR detects both tandem duplications and circles.
(B) Inverse PCR assay at the Chr1 hotspot pair. A 1.3-kb PCR product would arise from a tandem duplication or circular DNA from end joining of double cuts at the two hotspot centers. Lower panel, inverse PCR products detected in Atm−/− testis DNA.
(C) Arc diagrams of breakpoints for duplications/circles at the Chr1 (42 events) and Chr19 (22 events) hotspot pairs in Atm−/−.
(D) Strategy to detect only circular DNA products by inverse PCR. ExoV is used to degrade linear DNA containing tandem duplications. ScaI produces entry sites for ExoV. See also Figure S4B.
(E) Arc diagrams of duplications/circles (27 events) and circles (13 events) at the Chr1 hotspot pair, from Atm−/− testis DNA treated without and with ExoV, respectively. Equal numbers of haploid genomes were tested (Figure S4C).
(F) Distinct breakpoint profiles for duplications/circles and deletions at the Chr1 and Chr19 hotspot pairs in Atm−/−. Arc diagrams of duplications/circles (red arcs) and deletions (blue arcs) are overlaid. Below the arc diagrams are smoothed breakpoint and SPO11-oligo profiles (201-bp Hann filter). Duplication/circle profiles are normalized to the number of deletions. Percentages indicate the fraction of breakpoints mapping to the left or right of the hotspot centers. See also Figure S4D.
Tandem duplications and circular DNA.
We used outward-directed (inverse) PCR, which would give a product from either a tandem duplication or circular DNA (duplication/circle) arising from double cutting (Figure 3A). In Atm−/− testis DNA, duplication/circle products were readily detected (Figure 3B) at a frequency of 2.17 × 10−5 per haploid genome for the Chr1 hotspot pair (42 events) and 2.20 × 10−5 for the Chr19 pair (22 events) (Figure S4A). (We excluded a fraction of products that we were unable to fully sequence due to the presence of palindromes at the junctions, the origin of which is unclear; see Figure S4A.) Both hotspot pairs showed similar breakpoint distributions, with some breakpoints clustering at the most prominent SPO11-oligo peaks while others clustered between the two hotspots (Figure 3C) (see below).
Next, we aimed to distinguish circular DNA junctions from those of duplications to determine if both events occur. To this end, genomic DNA was incubated with E. coli exonuclease V (also known as RecBCD) prior to inverse PCR to specifically degrade linear DNA containing duplications while leaving circles intact (Figures 3D and S4B). We cleaved DNA with a restriction enzyme with sites flanking the hotspot pair to further ensure ExoV degradation, which was confirmed with a control PCR (Figure S4B). Using this approach at the Chr1 hotspot pair, we recovered 13 products from reactions with ExoV, which are predicted to arise only from circles, and a significantly larger number (27) without ExoV (P = 0.0269, Chi-square test; Figures 3E and S4C). This result provides evidence that tandem duplications and circles both occur in Atm−/− testis DNA, and in roughly similar numbers. A few presumptive tandem duplications were also detected in Atm+/+ sperm DNA (Figure S4C), although further investigation is required.
The circle and duplication/circle breakpoint distributions were similar, indicating that end joining positions are not dramatically different within the hotspot pair despite involving DSBs on different numbers of chromatids (Figures 3C and 3E). Distances between the breakpoints, which indicate the duplication length or the circle size, were likewise similar (Chr1: circles, 1267 bp median; duplications/circles, 1287 bp median). In contrast, these distances were significantly smaller than those of deletions (1954 bp median; P < 0.0001, unpaired Mann-Whitney test). Similar findings were obtained for the Chr19 hotspot pair (median deletion size was 1892 bp, compared with 1348 bp for duplications/circles; P < 0.0001, Mann-Whitney test; Figure 3C). This difference was because duplication/circle breakpoints mapped between hotspot centers (64–87%), whereas the majority of deletion breakpoints mapped outside (53–76%, Figure 3F). These shifts were highly significant (Figure S4D) and are consistent with multiple DSBs within hotspots and/or joining of partially resected DSB ends, because both scenarios would yield larger deletions and smaller duplications and circles (Figure S4E).
Interestingly, for duplications/circles only one of the two major breakpoint peaks in each pair was close to the hotspot center. The other peak was shifted ~500 bp inward (Chr1 left hotspot, Chr19 right hotspot, Figure 3F). This shift may be the result of frequent double cuts at these two hotspots, with one cut at the hotspot center and the other ~500 bp away. Supporting this, a small cluster of SPO11 oligos is observed at this position in the Chr19 right hotspot (Figure 3F) and is also seen in the Chr1 left hotspot when SPO11 oligos mapping to repeats are included (Figure S4F). Deletions that would be predicted to arise from double cuts at the Chr1 left hotspot were confirmed using flanking primers (14 events in Atm−/−, 1.75 × 10−6; two events in Atm+/−, 0.25 × 10−6), with the right breakpoints frequently mapping to the shifted peak (Figure S4G). Left breakpoints mapped predominantly outside the hotspot center, implying that double cuts can be more distant than expected or that more than two cuts can form.
Overall, our results provide evidence for multiple mutational outcomes arising from double cutting and end joining in the absence of ATM. Moreover, as circles and tandem duplications are not predicted to arise from a single DSB, their occurrence provides strong evidence for double cutting at two DSB hotspots on either one or two chromatids.
Joining between homologous chromosomes.
To investigate whether deletions involve one or both homologs, we tested the Chr1 hotspot pair in B6 × DBA/2J (DBA) F1 hybrid males, which have a high polymorphism density across this region (Figure 4). Primers were shifted to amplify a larger fragment to aid in identifying the parental chromosome(s) involved in the deletion.
Figure 4. Meiotic deletions within and between chromosomes.

Deletions at the Chr1 hotspot pair in Atm−/− B6 × DBA F1 hybrid males. Nested PCR was performed with the indicated primers (arrows) to identify polymorphisms within the deletion products derived from B6 (red) or DBA (blue). Three deletions were uninformative because the involved chromosome(s) is uncertain. For PRDM9 motif see Figure S5A.
We recovered 37 deletions from Atm−/− testis DNA for a frequency of 4.20 × 10−6 per haploid genome, but none in the presence of ATM (<0.16 × 10−6) (Table S1). The distributions of deletion sizes (median 2046 bp) and breakpoints were broadly similar to those from B6 inbred mice (Figures 4 and S5B). When we analyzed polymorphisms, 30 of the 34 informative deletions (88%) appeared to have originated within one chromosome: 10 on B6 and 20 on DBA (Fig. 4). The higher frequency of deletions on the DBA chromosome might be associated with a polymorphism in the PRDM9 binding site at the left hotspot (Figures 4 and S5A), which could positively affect DSB formation. Thus, the preponderance of deletions arise within one chromosome, involving either one chromatid or sister chromatids.
Strikingly, however, we also captured four deletions that contained sequences from both homologs (Figure 4). In one, the left hotspot from DBA was joined to the right hotspot from B6 (DBA-Δ-B6). In the other three more complex events, a haplotype switch occurred adjacent to one of the deletion breakpoints (DBA-B6-Δ-B6; DBA-B6-Δ-DBA, and B6-DBA-Δ-?, where the question mark indicates that the genotype was uninformative at the right end). In these haplotype switches, 6–8 polymorphisms were incorporated, encompassing up to 500 bp (Figure 4).
The recovery of deletions involving interhomolog interactions shows that DSBs can occur at nearby locations on both homologs in the same cell, at least in Atm−/−. Thus, although homolog synapsis is greatly impaired in the absence of ATM (Xu et al., 1996; Barlow et al., 1998), homolog interactions still occur. Cutting of both homologs at the same hotspot has been inferred from patterns of strand exchange in yeast tetrads (Zhang et al., 2011; Anderson et al., 2015; Marsolier-Kergoat et al., 2018).
Further, the three complex interhomolog events suggest that three or more DSBs had occurred on the two homologs. One possible mechanism for the haplotype switch at a deletion breakpoint is offset DSB formation on both homologs within the same hotspot, with attempted gap repair that is aborted by the DSB in the template, followed by joining to a DSB at the other hotspot (Figure S5C). Alternatively, double cutting within one hotspot on one homolog might release a DNA fragment that then inserts at the DSB in the other homolog (Figure S5D). Irrespective of the precise mechanism, however, these results indicate that loss of ATM can cause damage to multiple chromatids.
Sites of recurrent deletion breakpoints in the absence of ATM
Most deletions between hotspots from Atm−/− mice were unique: 87% (161/185) of events on Chr1 were observed only once, as were 97% (56/58) for Chr19 (Table S2). Microhomologies were present at the breakpoints in 75% of all deletions, but they were generally short, with a median of 1 bp for both the Chr1 and Chr19 deletions (Figure 5A). This frequency is larger than the 44% expected by chance (Figure 5A; STAR Methods). Thus, substantial microhomology is not required for joining, but short overlaps may be favored.
Figure 5. Microhomology at deletion breakpoints in Atm−/−.

(A) Distribution of microhomology lengths observed at the deletion breakpoints for Chr1 and Chr19 hotspot pairs. Microhomology expected by chance was calculated as previously described assuming an unbiased DNA sequence composition (Roth et al., 1985). “Random” indicates microhomology at junctions artificially generated by shifting the original, “Observed” breakpoints by 10 bp outward; this randomization controls for local biases in DNA sequence composition. See STAR Methods for details. Only deletions without insertions or other DNA-end modifications are included (Table S2).
(B) Recurrent deletion within the Chr1 hotspot pair at an imperfect repeat. Twelve simple deletions in Atm−/− map to this repeat at one of the three internal identical repeats of 5, 8, and 6 bp (violet arcs). Four other deletions with inverted insertions also involve this imperfect repeat as well as a 13-bp inverted microhomology at the hotspot center, with the insertions indicated above the deletions (orange arcs). See also Table S4 and Figure S6E.
The 24 recurring deletions on Chr1 had longer microhomologies with a median of 6 bp (Table S4). Twelve of the recurring deletions arose within a 22/23-bp imperfect repeat shared between the two hotspots, and eight of these involved an 8-bp microhomology within the repeat (Figure 5B; Table S4). Thus, the presence and position of short sequence repeats likely influences deletion outcomes.
The presence or absence of microhomologies within end-joining products may be indicative of the end joining pathway used and/or processing at DNA ends. For example, SPO11-bound DSB ends normally require endonucleolytic processing on one strand by MRE11 to release SPO11 oligos (Keeney et al., 1997; Neale et al., 2005; Lange et al., 2011). This may disfavor canonical nonhomologous end-joining and promote alternative pathways that utilize microhomology (Sfeir and Symington, 2015). However, as some DSBs remain unresected in the absence of ATM/Tel1 (Joshi et al., 2015; Mimitou et al., 2017; Cannavo et al., 2018; Paiano et al., 2020; Yamada et al., 2020), deletions may also arise from joining at or very close to SPO11 cleavage positions, which would involve alternative SPO11 removal mechanisms, such as TDP2 (Alvarez-Quilon et al., 2014; Johnson et al., 2019; Paiano et al., 2020). Our findings are thus consistent with multiple end-joining mechanisms being employed to repair adjacent DSBs in the absence of ATM.
Insertion of hotspot-derived fragments
Unexpectedly, we observed insertions in 11% of deletions from Atm−/− mice (27/243; two deletions with two inserts, for 29 total inserts; Table S2). All but one of the inserted DNA fragments mapped to chromosome positions that showed SPO11 activity, either the assayed hotspot pairs themselves or other hotspots. The insert that mapped to a sequence with no SPO11 oligos contained a CT-rich microsatellite repeat and shared significant microhomology with both deletion breakpoints (12 and 9 bp) (Table S5), suggesting a possible DNA synthesis-based insertion mechanism.
Ectopic insertions from other hotspots.
Fifteen inserts of 15–349 bp mapped to other chromosome positions, either at hotspots or at SPO11-oligo clusters that were below the hotspot-calling threshold (Figures 6A–C and S6A–C; Table S5). Microhomologies at both sides of inserts were often short (0 to 2 bp), similar to deletions without insertions (Figure 6D; Table S5).
Figure 6. Insertions derived from DSB hotspots.

(A–D) Ectopic insertions from other hotspots. A, Example of a deletion on Chr1 from Atm−/− containing a 79-bp insertion derived from a hotspot on Chr7. B, Inserts from other hotspots map close to their respective hotspot centers. Three inserts map to SPO11-oligo clusters rather than called hotspots (asterisks). Inserts 1 and 3 from the non-PAR ChrY map to multiple hotspots. See also Figures S6A, S6B and Table S5. C, Non-PAR ChrY hotspot to which inserts 1 and 2 can be mapped. Some SPO11 oligos can be uniquely mapped to this hotspot, but, as it is one copy of a family of repeats, numerous other SPO11 oligos can be mapped to both this and other copies of the repeat (“Multi”). D, Microhomologies at insert breakpoints are often short (median, 1 bp).
(E–H) Inverted insertions derived locally from the hotspot pair. E, Deletions with inverted insertions at the Chr19 hotspot pair. Inserts are derived in inverted orientation from the left or the right hotspot. For the Chr1 hotspot pair, see Figure S6E. F, Example of an inverted insertion (top) and possible mechanisms (bottom). The right insertion breakpoint shares a 5-bp inverted microhomology with the left deletion breakpoint, while the left insertion breakpoint and the right deletion breakpoint do not share microhomology. Potential models explaining the inversion mechanism involve double cutting at the left hotspot or overhang foldback. G, Microhomologies at breakpoints for insertions derived from the Chr1 and Chr19 hotspot pairs. Inserts mapping to the left hotspot in a pair have long microhomologies present at their right breakpoints. Inserts mapping to the right hotspot mirror this pattern. See also Table S6. H, Inserts from the Chr1 and Chr19 hotspot pairs map close to hotspot centers.
Most of these inserts could be uniquely mapped, but three were derived from the non-pseudoautosomal region (non-PAR) of the Y chromosome. The mouse non-PAR Y is highly ampliconic (Soh et al., 2014), so these inserts cannot be assigned to a single location. Hotspots have been called at some of these locations based on the assignment of unique SPO11 oligos, although most SPO11 oligos map to multiple locations that are highly similar (Figures 6B, 6C, S6B and S6C; Table S5). Among all chromosomes, the non-PAR Y experiences the largest increase in SPO11-oligo density in the absence of ATM (~30 fold) (Lange et al., 2016).
Inserts came from sites at or close to hotspot/cluster centers: nearly half had an end within 50 bp (Figure 6B). Their sizes were small (median 106 bp) even though larger inserts would have been detectable (Figures S6C and S6D). We envision that these inserts derive from DNA fragments released by SPO11 double cutting; we infer that they were not circularized because they are not permuted. It seems remarkable that inserts can sometimes originate from weak SPO11-oligo clusters, as such weak sites would be unlikely to experience two DSBs in the same cell if each DSB occurred independently at a population-average frequency. However, these findings are readily explained by the recent observation of concerted SPO11 double cleavage within hotspots (especially in Atm−/−) (Johnson et al., 2021; Prieler et al., 2021). The ectopic insertion of SPO11-generated fragments into other SPO11-generated DSB locations represents a distinct pathway for meiosis-specific, nonreciprocal genome modification by translocation of sequences.
Inverted insertions derived locally from the hotspot pair.
Twelve deletions had insertions of 16 to 709 bp that mapped to one of the hotspots but were reinserted in opposite orientation (inverted insertions; Figures 6E, S6C and S6E), including one with an inverted insertion from each hotspot in the pair. Reconstructing the insertion breakpoints illuminated potential inversion mechanisms. Specifically, in each case, one insertion breakpoint shared an inverted microhomology with one deletion breakpoint (median 5 bp) (Figures 6F and 6G). The other insertion breakpoint, which was at or near this deletion breakpoint, had no or little microhomology with the other deletion breakpoint (median 0 bp). Inserts from the left and right hotspots mirrored each other in the sense that those from the left hotspot had the longer microhomology at the right breakpoint, whereas those from the right hotspot had the longer microhomology at the left breakpoint (Figures 6E, 6G and S6E; Table S6). Insert sequences clustered around hotspot centers (Figure 6H), indicating that they originated from DSB formation.
We propose two models to explain these events (Figure 6F). In one, DSBs occur at or near the two inverted microhomologies, creating a small double-cut fragment within one hotspot, and a third DSB in the other hotspot generates a larger fragment destined for deletion. One end of the small fragment remains unprocessed, preventing normal recombination at this hotspot, while both ends with the inverted microhomology are at least partially resected, allowing strand annealing and fragment inversion. The unprocessed end of the now inverted fragment joins to the third DSB at the other hotspot. This is an attractive model because it incorporates defects seen or inferred in Atm−/− mice, namely, impaired DSB processing (Paiano et al., 2020; Yamada et al., 2020) and closely spaced DSBs ((Johnson et al., 2021); this study). In the second model, a DSB occurs next to just one of the inverted microhomologies and resection exposes the second microhomology. A foldback structure formed by annealing the inverted microhomologies is further processed by a structure-specific nuclease, generating a 5′ overhang that is filled in prior to joining the DSB end at the other hotspot. Foldback structures may be common in Atm−/−, because the excess of DSBs may exhaust the pool of RPA (Toledo et al., 2013), which normally prevents formation of secondary structures (Chen et al., 2013).
Interestingly, four inverted insertions from the right hotspot at the Chr1 hotspot pair involved the previously identified recurrent deletion site (Figures 5B and S6E). In this case, 13 bp overlapping the recurrent deletion shares inverted microhomology with the most prominent DSB peak. Thus, this genomic location often engages in inappropriate repair (9% of all Chr1 deletions), demonstrating that DSB formation at or nearby such sequences may cause (mis)repair products to arise.
Microdeletions within a single hotspot
If more than one DSB can form inside the same hotspot, and not just at adjacent hotspots, we predicted that small deletions might also arise within single hotspots. The presumed origin of insertions from hotspot fragments implies that closely spaced DSBs can indeed occur within individual hotspots on one chromatid. We applied deep sequencing to test for such microdeletions. We selected a hotspot on Chr1 ranked among the five strongest in both Atm−/− and wild type. The SPO11-oligo profile for this hotspot displays a major central peak, a secondary peak, and several minor peaks, with SPO11 oligos spreading up to ~500 bp on both sides of the central peak, especially to the right (Figure 7A). We designed four overlapping amplicons, ~580 bp each, to scan ~900 bp of the hotspot. This strategy would recover deletions around the major peak as well as those with one or both breakpoints away from the major peak, as often seen for inserts (Figure 7B). Multiple, independent PCRs were carried out, each with input testis DNA equivalent to ~16,000 haploid genomes, for a total of ~10 million per amplicon. From pools of amplified DNA we sequenced ~3.5–5 million molecules per amplicon.
Figure 7. Microdeletions within a single DSB hotspot.

(A) Detection of microdeletions at a strong DSB hotspot on Chr1 using amplicon deep sequencing. The hotspot displays a major SPO11-oligo peak, a secondary peak, and several minor peaks, as shown in the smoothed (21-bp Hann filter) and unsmoothed profiles. Four overlapping amplicons were deep sequenced to identify microdeletions. Arc diagrams of independent microdeletions found in each amplicon for the indicated genotypes are shown. For microdeletion frequencies, see Table S7.
(B, C) Microdeletions from Atm−/− in the region overlapping in amplicons 1, 2, and 3. B, Arc diagram of independent microdeletions. C, Distributions of the left and right breakpoints relative to the SPO11-oligo distribution. Both unsmoothed and smoothed profiles are shown.
(D, E) Microdeletion length distributions in the overlapping region. D, Density and scatter plots of microdeletion lengths (>10 bp). E, Arc diagrams of microdeletions stratified by length. Microdeletions >160 bp are not shown, as their location may be more restricted by the size of the assessed region. See also Figures S7E and S7F for larger microdeletion distribution.
We identified reads containing microdeletions ranging from 11 to 229 bp (Figure 7A). (Reads with ≤10-bp deletions were not considered; Figure S7A). In Atm−/−, 216 reads with microdeletions were detected from the four amplicons, for a total frequency of 1.58 × 10−5, significantly greater than in Spo11−/− (0.19 × 10−5), wild type (0.54 × 10−5), and Atm+/− (0.23 × 10−5) (p<0.0001, Chi square test) (Table S7). For some amplicons, reads from wild type and Atm+/− were also more frequent than in Spo11−/− (Table S7). If we disregard duplicate reads for a specific microdeletion from a given amplicon, we conservatively estimate that we obtained 149 independent microdeletions in total from Atm−/− (Figure 7A). This is likely an underestimate since some microdeletions were found in multiple amplicons (Figure S7B).
Microdeletion breakpoints frequently clustered at SPO11-oligo sites in Atm−/− (Figure 7A). Prominent clusters were observed at the major SPO11-oligo peak (amplicons 1 to 3) and at the secondary SPO11-oligo peak (amplicons 1 to 4), indicating that microdeletions arose from SPO11-generated DSBs. This was even more evident when we pooled the 82 independent microdeletions from amplicons 1, 2, and 3 for which both breakpoints mapped within the 304-bp overlapping region (i.e., between the forward primer of amplicon 3 and the reverse primer of amplicon 1) (Figure 7B). It was visually striking that many microdeletions had both breakpoints within these two SPO11-oligo peaks (17/82; 21%, 3 of which were recurring; Figure S7B), implying that they originated from double cutting. In the smoothed breakpoint profiles, the most prominent left breakpoint peak matched the major SPO11-oligo peak, while the most prominent right breakpoint peak matched the secondary SPO11-oligo peak (Figure 7C). These breakpoint peaks became even more prominent when all 116 reads in this overlap region were considered, something not seen with random sampling of reads (Figure S7C). Thus, at least some of the duplicated reads likely represent independent microdeletions.
Microdeletions in the overlap region displayed a distinct length distribution (Figure 7D), with two prominent microdeletion classes of ~30 bp and ~60 bp. The ~60-bp microdeletions predominantly mapped between the major SPO11-oligo peak and the secondary SPO11-oligo peak (71%, 17/24 from 49–70 bp; Figure 7E), which are separated by 55 bp (Figure 7C); reads within this class were often duplicated (Figure S7D). A subset of the ~30-bp microdeletions (22%, 4/18 from 27–42 bp; Figure 7E) mapped between a minor SPO11-oligo peak and the secondary SPO11-oligo peak 34 bp away (Figure 7C). These results imply that hotspot substructure predicts double cutting and, hence, microdeletion locations. Conversely, double cutting will affect SPO11-oligo distribution within a hotspot when SPO11 oligos predominantly arise from double cutting.
In addition to microdeletions between two SPO11-oligo peaks, ~30-bp (56%, 10/18) and ~60-bp (21%, 5/24) microdeletions often had only one breakpoint at a SPO11-oligo peak (Figure 7E). Assuming they also arose from two SPO11 breaks, the distinct size classes may reflect a more general underlying mechanism for double cutting, such that the length of double-cut fragments may be constrained. In that case, it remains possible that some of the ~60-bp microdeletions may have arisen from two double cuts (triple cutting), releasing two ~30-bp fragments, rather than a single double cut, although this may be less likely given that very few of the ~30-bp microdeletions have left breakpoints within the major SPO11-oligo peak like the ~60-bp microdeletions.
Microdeletions, especially the ~60-bp class, frequently removed the PRDM9 binding site (Figure 7E). PRDM9 binding could potentially affect the spacing of double cuts. However, as similar microdeletion lengths are observed outside the PRDM9 motif, another or additional factors must exist that lead to this size class.
Interestingly, the major SPO11-oligo peak and the adjacent minor peak are separated by 21 bp (Figure 7C), but the few small microdeletions (11–24 bp) did not map between these peaks (Figures 7D and 7E), implying that two SPO11-induced DSBs may be constrained to longer distances. Such a constraint implies that microdeletions arise from two DSBs in one chromatid (gaps), rather than from two single DSBs forming independently on two chromatids.
A less prominent microdeletion class of ~140 bp was also observed (129–153 bp, Figure 7D). In several of these longer microdeletions, the left breakpoints clustered at the major SPO11-oligo peak, and the right breakpoints were at sites with fewer SPO11 oligos (46%, 6/13; Figure 7E). These longer microdeletions may also arise from double cutting. Long SPO11 oligos are observed in Atm−/− testes (Lange et al., 2011), but are underrepresented in SPO11-oligo maps because of size selection during library preparation (Thacker et al., 2014; Lange et al., 2016). Even longer microdeletions were also observed that peaked at ~180 bp and ~230 bp (Figure 7D), especially when considering all amplicons where microdeletions were less size-constrained (Figures S7E and S7F). Some of these long microdeletions may have arisen from multiple double-cut fragments that generated long gaps. However, it is notable that the median insert size from other hotspots in the paired hotspot deletions was quite long (~100 bp), with one insert extending 349 bp, suggesting that long double-cut fragments can be generated.
In Atm+/+ and Atm+/−, microdeletions did not display obvious spatial patterns (Figures 7A and 7E). Because the frequency of microdeletions in the presence of ATM is only slightly higher in some amplicons than in Spo11−/−, we consider that a portion of these events arose from PCR errors or other genomic damage, as in Spo11−/−. Strikingly, however, a ~30-bp (25–43-bp) microdeletion class was also apparent, but not in the Spo11−/− control, although the prominent ~60-bp microdeletion class found in Atm−/− was not observed (Figures 7D and S7E). Microdeletions from the ~30-bp class did not map within SPO11-oligo peaks, although some breakpoints clustered (Fig. 7E). Possibly, in the presence of ATM, microdeletions are more likely to occur at DSBs formed outside of SPO11-oligo peaks. In both the presence and absence of ATM, microhomology at breakpoints was generally short (median 1 bp) (Figure S7G), similar to deletions between hotspot pairs.
Our assumption that SPO11 double cutting occurs within individual hotspots, preceding microdeletion formation, is consistent with recent findings from S. cerevisiae in which double-cut fragments were observed (Johnson et al., 2021; Prieler et al., 2021). Their minimum size was determined to be ~30 bp but longer fragments in increments of ~10 bp were abundant as well. The authors suggest models in which Spo11 is constrained to make the simultaneous cleavages. The recent finding of condensates of the yeast DSB-formation machinery suggests a mechanism whereby centers of DSB activity form which could provide such a platform (Claeys Bouuaert et al., 2021). Our single hotspot data fit well with both of these concepts. Further, potential SPO11 double-cut fragments have been computationally identified within mouse hotspots (Johnson et al., 2021) from SPO11-oligo libraries (Lange et al., 2016). Assuming a similar lower size limit in mouse as in yeast, double-cut fragments are predicted to peak at ~32 bp and extend to ≤60 bp in Atm−/−, near the size limit of the SPO11-oligo library preparation (Lange et al., 2016). Our analysis of microdeletions is in line with this finding, given a prominent microdeletion class of ~30 bp, but also suggests that SPO11-oligo double-cut fragments extend much longer, i.e., from ≥60 bp to at least ~230 bp, congruent with the observation of long SPO11 oligos in Atm−/− (Lange et al., 2011).
Conclusions
Our study uncovers an unexpected pathway for germline mutagenesis in mice involving nonhomologous end-joining at SPO11 double cuts and leading to various mutational outcomes. Although pronounced in the absence of ATM, these de novo mutations also occur at low frequency in the presence of ATM. Deletions and tandem duplications result in copy number variations (CNVs), predicting a role in genome evolution (Collins et al., 2020). Moreover, since about 50% of hotspots are in genic regions (Smagulova et al., 2016), SPO11-mediated events could disrupt or otherwise modify gene function. Indeed, in both of our hotspot pairs, deletions and duplications remove and duplicate exons, respectively. The occurrence of events involving two chromosomes also predicts gross rearrangements. Thus, our findings reveal a previously hidden potential in meiosis for mutagenesis and the disruption of genome integrity. Furthermore, double-cut fragments appear to be released from hotspots and inserted at other genomic sites undergoing DSBs, also with mutagenic consequences. Because DNA gaps are thought to initiate a subset of meiotic recombination events as part of the normal meiotic program (Johnson et al., 2021; Prieler et al., 2021), this raises the question as to how re-insertion of released double-cut fragments is usually suppressed. We speculate that circularization of these fragments could be a mechanism to suppress reintegration into the genome.
Microdeletions resulted in PRDM9 motif deletion in 38% of events in the absence of ATM and 11% in the presence of ATM. By eliminating PRDM9 motifs, such deletions are predicted to shape the recombination landscape, thus influencing genome evolution (Cole et al., 2014; Baker et al., 2015) and potentially contributing to speciation (Davies et al., 2016; Smagulova et al., 2016). Because PRDM9 drives recombination away from promoters (Brick et al., 2012), deletions may be better tolerated in PRDM9-dependent meiotic recombination, including in humans, and in turn have a greater potential impact on human genome evolution. This prediction is supported by recent findings of an enrichment of structural variants (insertions and deletions ≥50 bp) at human DSB hotspots (Beyter et al., 2021).
Limitations of the study
Although short double-cut fragments are observed at single hotspots (Johnson et al., 2021), consistent with our observations of microdeletions, our physical evidence for the existence of large double-cut fragments between hotspots relies on the detection of circular DNA. The longevity of linear fragments and their conversion to circles remains to be addressed.
We showed that nonhomologous end-joining at double cuts yields mutational outcomes, however genetic studies are needed to determine which end joining pathway(s) is responsible. The methods we developed will enable such experiments in various knockout mice in the future.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maria Jasin (m-jasin@ski.mskcc.org).
Materials availability
Mouse strains associated with this study are available at the Jackson Laboratory. Other materials will be provided upon request to the lead contact.
Data and code availability
Original data including gel pictures, Sanger sequencing, all data processing spreadsheets and R scripts are available from the lead contact upon request. Raw and processed amplicon deep sequencing data files are deposited at GEO. This paper also analyzes existing SPO11-oligo data that are publicly available. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Taq DNA polymerase | Thermo Fisher Scientific | Cat# EP0406 |
| Pfu Turbo DNA polymerase | Agilent | Cat# 600254 |
| Non-acetylated ultra-pure bovine serum albumin (BSA) | Invitrogen | Cat# AM2616 |
| dNTPs, PCR-grade | Roche | Cat# 3622614001 |
| S1 Nuclease | Thermo Fisher Scientific | Cat# EN0321 |
| Restriction enzymes (Listed in Method Details) | New England Biolabs | N/A |
| Exo V (RecBCD) | New England Biolabs | Cat# M0345S |
| Thermolabile ExoI | New England Biolabs | Cat# M0568S |
| Proteinase K, PCR-grade | Roche | Cat# 03115828001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Invitrogen PureLink Quick Gel Extraction Kit | Invitrogen | Cat# 210012 |
| PureLink PCR Purification Kit | Invitrogen | Cat# K310002 |
|
| ||
| Deposited data | ||
|
| ||
| Amplicon sequencing data | This paper | GEO:GSE182210 |
| SPO11-oligo data | Lange et al., 2016 | GEO:GSE84689 |
| Mouse genome assembly GRCm38/mm10 | N/A | http://genome.ucsc.edu |
| Mouse polymorphism database | N/A | https://www.sanger.ac.uk/data/mouse-genomes-project/ |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: B6.129S6-Atmtm1Awb/J | The Jackson Laboratory | RRID:IMSR_JAX:008536 |
| Mouse: DBA/2J mice for backcrossing Atm+/− mice | The Jackson Laboratory | RRID:IMSR_JAX:000671 |
| Mouse: Spo11−/− | Baudat et al., 2000 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers (Listed in Method Details) | Integrated DNA Technologies | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R (versions 3.4.4 to 3.6.3) | N/A | http://www.r-project.org |
| GraphPad Prism 8 | N/A | https://www.graphpad.com/scientific-software/prism/ |
| ApE | N/A | https://jorgensen.biology.utah.edu/wayned/ape/ |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Mice were purchased from The Jackson Laboratory where indicated. Atm+/− males on the C57BL/6J (B6) background (B6.129S6-Atmtm1Awb/J; stock #008536) were crossed with B6 females (stock #000664) to establish a breeding colony and to obtain experimental animals. The Atm mutation was previously described (Barlow et al., 1996). Atm+/− B6 mice were crossed with Atm+/− DBA/2J (DBA) mice to get F1 hybrid males. Atm+/− mice on the DBA background were obtained by six backcrosses of Atm+/− mice that were partially on an A/J background with DBA inbred mice (stock #000671). The previously described Spo11 mutation (Baudat et al., 2000) was maintained on the B6 background. Animals were fed regular rodent chow with continuous access to food and water. All experiments were conducted according to US Office of Laboratory Animal Welfare regulatory standards and were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee.
METHOD DETAILS
Isolation of testis and sperm DNA
DNA was isolated from testicular single-cell suspensions to enrich for germ cells. Testis dissociation was based on protocols previously described (Cole et al., 2014; Gaysinskaya et al., 2014) with some modifications. Testes from juvenile 15-day-old and adult 2- to 5-month-old mice were decapsulated and seminiferous tubules incubated at 33 °C in 10 ml Gey’s Balanced Salt Solution (GBSS) (Sigma) with 0.5 mg/ml collagenase (Sigma) and 2 μg/ml DNase I (Roche) for 15–20 min, shaking at 500 rpm. After 10 min and at the end of the incubation the tubules were gently pipetted up and down using a transfer pipette. The tubules were washed twice with 10 ml GBSS and digested with 0.5 mg/ml trypsin (Sigma) in 10 ml GBSS, supplemented with 2 μg/ml DNase I, for 25–40 min at 33 °C, shaking at 500 rpm. Pipetting up and down was performed at the start, midpoint and end of the incubation. Trypsin was inhibited by adding 10% fetal calf serum. Cell suspension was obtained by repeated pipetting for 2 min and filtration through a 70-μm cell strainer (Thermo Fisher Scientific). All of the mutant cell suspension and one-third to one-half of the wild-type cell suspension was transferred to a 15 ml tube and centrifuged for 3 min at 1550 × g. The cell pellet was resuspended by tapping the tube, followed by the addition of 600 μl lysis buffer (200 mM NaCl, 100 mM Tris-Cl pH 7.5, 5 mM EDTA, 0.5% SDS, 0.5 mg/ml Proteinase K). The lysate was transferred to a 1.5 ml Eppendorf tube and incubated overnight at 55 °C. Genomic DNA was isolated using phenol-chloroform extraction and alcohol precipitation, with the extraction step repeated twice. DNA was dissolved in 200–400 μl H2O, incubated for 1 h at 55 °C and overnight at 4 °C. DNA concentration was quantified by NanoDrop (Thermo Fisher Scientific). DNA was diluted to ~200 ng/μl and incubated for another 2–3 h at 37 °C to reduce the viscosity of the DNA solution. DNA concentration was measured again three times before freezing. DNA samples were stored at −20 °C.
Sperm DNA was isolated as previously described (Peterson et al., 2020). Liver DNA (somatic control) was extracted (Cole and Jasin, 2011) from the same Atm−/− mouse as testis DNA.
Detection of deletions at hotspot pairs by nested PCR
Deletions were detected by nested PCR using primers flanking the two hotspots of a pair (Figures 1B and S2A). Prior to PCR, genomic DNA was digested with restriction enzymes to suppress parental DNA amplification, although this step can be omitted (Figure S2G). Restriction digestions were performed in a 60 μl volume with 1200 ng of DNA and 4–4.5 μl restriction enzyme(s) (Chr1 hotspot pair: NsiI, NcoI, and BglII or NcoI alone; Chr19 hotspot pair: BamHI or KpnI; New England Biolabs). Reactions were incubated overnight at 37 °C followed by an additional 3 h of incubation with a 1.5–2 μl of fresh restriction enzyme(s), then placed on ice before PCR. In the majority of PCRs, 2.5 μl of the digestion reaction was used per PCR, equivalent to 50 ng of genomic DNA, with this DNA concentration being optimal for the estimation of deletion frequencies; 1.25 μl and 5 μl, equivalent to 25 ng and 100 ng, respectively, were also tested. In the experiments with no digestion, 50 ng of DNA template was used per PCR in most cases.
Nested PCR was based on the previously described protocol (Cole and Jasin, 2011) with some adjustments. A first round of PCR was performed using the following primers: Chr1_DH_FW1: 5′-GGTCAGGAGGATGGGGAG-3′ and Chr1_DH_RV1: 5′-CCAAGAAGGTGCTTATCGTCC-3′ for the Chr1 hotspot pair, Chr19_DH_FW2: 5′-TGGAATGTAGCCTCTGGCAG-3′ and Chr19_DH_RV2: 5′-GTGCTGTCAGAAGCCGTGG-3′ for the Chr19 hotspot pair, Chr1_DH_DB_FW1: 5′-CCTGGAGACCAAAGCCAC-3′ and Chr1_DH_RV1: 5′-CCAAGAAGGTGCTTATCGTCC-3′ for the Chr1 hotspot pair in B6/DBA F1 hybrids. A second round of PCR was performed using the following primers: Chr1_DH_FW2: 5′-GCCTTGGCTCAAAGTGTCTG-3′ and Chr1_DH_RV2: 5′-TCCCTTCTGCCATCCTTCTG-3′ for the Chr1 hotspot pair, new Chr19_DH_FW2: 5′-CCCAAAGTTGTCCTTGCTTCC-3′ and new Chr19_DH_RV2: 5′-TGGTTTCCAGAGGAGAGGAC-3′ for the Chr19 hotspot pair, Chr1_DH_DB_FW2: 5′-GGCATTGCTTCATTCATGGTCTG-3′ and Chr1_DH_RV2–3: 5′-CATTGTCATCTTGAGCACTCCTC-3′ for the Chr1 hotspot pair in B6/DBA F1 hybrids. The annealing temperature was optimized for each primer pair by gradient PCR with testis genomic DNA of known high quality. DNA samples from different testes were also tested for the appearance of a strong parental PCR band using the primary PCR primers.
For nested PCR, each primary PCR was performed in a 50 μl volume containing: 1x Jeffreys buffer (10x: 450 mM Tris-Cl pH 8.8, 110 mM (NH4)2SO4, 45 mM MgCl2, 67 mM 2-mercaptoethanol, 44 μM EDTA, 10 mM each dNTP (Roche), 1.13 mg/ml ultra-pure (non-acetylated) BSA (Invitrogen), 12.5 mM Tris base), 0.2 μM primers, 0.03 U/μl Taq DNA polymerase (Thermo Fisher Scientific) and 0.006 U/μl Pfu Turbo DNA polymerase (Agilent). The primary PCR product (0.6 μl) was treated with S1 nuclease (Thermo Fisher Scientific) at room temperature for 30 min, in a 5 μl volume containing: 0.5x S1 buffer, 0.2 U/μl S1 nuclease and 5 ng/μl sonicated salmon sperm DNA. The reaction was diluted with 45 μl of dilution buffer (10 mM Tris-Cl pH 7.5 and 5 ng/μl sonicated salmon sperm DNA). 4 μl of the diluted S1 reaction was seeded into the secondary PCR, which was performed in a 20 μl volume containing the same components as the primary PCR. Deletion products were identified by electrophoresis on a 1% agarose gel (Figures 1C and S2B). PCR products smaller than the parental size were excised, purified (Invitrogen PureLink™ Quick Gel Extraction Kit), and Sanger sequenced using primers from the second round of PCR and in some cases additional primers. Diluted S1 reactions were stored at −20 °C and used again for secondary PCRs if more PCR product was needed for sequencing.
PCR conditions were tested for each hotspot pair. The best result, that is, the appearance of more intense, smaller PCR products, was obtained when the extension time was shortened (<60 s/kb) in the primary PCR to inhibit amplification of the longer parental PCR product. The extension time in the secondary PCR was slightly shorter.
Primary PCR conditions were denaturation at 96 °C for 2 min, followed by 37 cycles of amplification as follows: denaturation at 96 °C for 20 s, annealing at 59 °C for 30 s, extension at 65 °C for 2 min for the Chr1 and Chr19 hotspot pairs (3.6-kb parental products), and 2 min 30 s for the Chr1 hotspot pair in B6/DBA hybrids (4 kb-parental product). Secondary PCR conditions where the same, but with different extension times: 3 min for the Chr1 and Chr19 hotspot pairs (3.4-kb parental products) and 3 min 30 s for the Chr1 hotspot pair in B6/DBA hybrids (3.9-kb parental products).
For detection of smaller deletions at the Chr1 left hotspot, genomic DNA was digested with NsiI, and nested PCR was performed with the forward primers used to detect deletions in B6/DBA F1 hybrids (see above), and the reverse primers used to detect duplications/circles (see below). Primary and secondary PCR conditions were the same: denaturation at 96 °C for 20 s, annealing at 61 °C for 30 s, extension at 65 °C for 1 min 20 s (2.1-kb parental products).
Sequenced PCR products were analyzed using ApE software (https://jorgensen.biology.utah.edu/wayned/ape/) to identify deletions, microhomologies, and polymorphisms in DNA sequences. The probability of microhomology at a junction of a given length (x) occurring by chance can be calculated for a random DNA sequence using the equation P(x) = (x + 1)(1/4)x(3/4)2 (Roth et al., 1985). The probability of fortuitously observing microhomology of 1 bp or greater is thus 1 – P(0) = 0.4375. As a separate test to account for the nonrandom sequence composition around the assayed genomic sites, we shifted observed deletion breakpoints by 10 bp outward. This exercise produced similar microhomology profiles to those expected by chance for unbiased sequence composition, for both hotspot pairs (Figure 5A). Deletion breakpoint profiling was performed using R versions 3.4.4 to 3.6.3 (http://www.r-project.org). Graphing was performed in R and GraphPad Prism version 8.
Detection of tandem duplications and circular DNA by nested inverse PCR
Primary and secondary PCR reaction protocols, including S1 digest, were the same as in the nested PCR protocol detecting deletions. For the primary PCR, the following primers were used: Chr1_Inv_FW1: 5′-CACTCATTCCCACTTGGCTC-3′ and Chr1_Inv_RV4: 5′-GGACGCATGATAAGCAAGGAG-3′ for the Chr1 hotspot pair, Chr19_Inv_FW3: 5′-TTCTGAGAGAGTCTTGCATTGG-3′ and Chr19_Inv_RV1: 5′-CCTGTACTGTTCATTCGCCATC-3′ for the Chr19 hotspot pair. For the secondary PCR, the following primers were used: Chr1_Inv_FW2: 5′-CAATCCGATGGCATTTAGATTAGG-3′ and Chr1_Inv_RV5: 5′-GGAGAAATGAGTTAATGAAATAGCC-3′ for the Chr1 hotspot pair, new Chr19_Inv_FW4: 5′-CTCAGTGCTGGAATTACAAGAG-3′ and Chr19_Inv_RV2: 5′-CACTAAACCGGAGTTCGCTG-3′ for the Chr19 hotspot pair. Inverse PCR does not amplify parental DNA. Therefore, we paired each primer with another primer to test for the appearance of the parental PCR product and to optimize the annealing temperature by gradient PCR. These control primers were the same primers we used to detect deletions.
Primary and secondary inverse PCR conditions were the same: 96 °C for 2 min followed by 30 cycles at 96 °C for 20 s, 61 °C for 30 s and 65 °C for 3–4 min. For both hotspot pairs, if double cutting occurs at hotspot centers, the expected PCR product is 1.4 kb and 1.3 kb for the primary and secondary PCR, respectively. In our experiments, the median size of secondary PCR products was 702 bp for Chr1 and 844 bp for Chr19. However, the more frequent appearance of smaller PCR products was not due to the extension time.
Inverse PCR product sequencing and further analysis were the same as for deletions.
ExoV digestion prior to inverse PCR to detect circular DNA
ExoV (RecBCD; New England Biolabs) was used to degrade linear DNA but not circular DNA. To ensure DNA degradation at the Chr1 hotspot pair we digested genomic DNA with the ScaI restriction enzyme (Figures 3D and S4B). Restriction digestions were performed overnight at 37 °C in a 60 μl volume with 1200 ng of DNA, 4 μl ScaI (New England Biolabs), followed by 3 h of incubation with a fresh portion, 2 μl, of the enzyme. Digestions were inactivated at 80 °C for 20 min, and placed on ice. Next, reactions were split into two and half was treated with ExoV in a 60 μl final volume containing: 1x CutSmart buffer, 1mM ATP and 0.2 U/μl ExoV (or H2O). Reactions were incubated at 37 °C for 1–2 h and inactivated at 70 °C for 30 min, then placed on ice. To remove short oligonucleotides generated by ExoV, 3 μl of Thermolabile ExoI (New England Biolabs) was added to each reaction and samples were incubated for 15 min at 37 °C, 5 min at 80 °C, then placed on ice.
For each inverse PCR, 2.5 μl of the digestion reaction (+/− ExoV) was used, equivalent to 25 ng of pre-digestion genomic DNA. In parallel, a control PCR was performed to test ExoV efficiency. To this end, an aliquot of the digestion reaction was diluted (1:10, 1:100, and 1:1000), and used in nested PCR with control primers (reverse inverse primers plus forward primers used to detect deletions in B6 × DBA F1 hybrids), amplifying a 2.1-kb PCR product (Figure S4B). PCR conditions for inverse PCR and control PCR were the same as described above.
Detection of microdeletions at a single hotspot by amplicon deep sequencing
Four ~580-bp overlapping amplicons of the hotspot (Figure 7A) were amplified using the following primer pairs: amplicon 1, Chr1_MiSeq_FW1: 5′-TCATGTCCGGTTTGGATGCC-3′ and Chr1_MiSeq_RV1: 5′-CGAATCAAAGACTCCAGAAGAGG-3′; amplicon 2, Chr1_MiSeq_FW2: 5′-AGTAGCAAGAAGGGCTGGAG-3′ and Chr1_MiSeq_RV2: 5′-AGAGACAGATGTGGAGGCTAAC-3′; amplicon 3, Chr1_MiSeq_FW4: 5′-CTGAATCTCTCCTGGTGTGTC-3′ and Chr1_MiSeq_RV4: 5′-AGGACATTGAATGTTTTTATGGTTC-3′; amplicon 4, Chr1_MiSeq_FW5: 5′-AAACAGAAGAAGCAGTAATGCC-3′ and Chr1_MiSeq_RV5: 5′-GTGTGCAACTGGGTATATTTCAG-3′. For each primer pair the annealing temperature was tested by gradient PCR with testis genomic DNA of known high quality. To minimize PCR bias and jackpot amplification of single deletion events, the number of amplification cycles was determined by when a faint PCR product was first visible, which was 22 cycles for all samples. For each amplicon, PCR conditions were the same: 96 °C for 2 min followed by 22 cycles at 96 °C for 20 s, 59 °C for 30 s and 65 °C for 35 s. PCR was performed in a 50 μl total containing 1.5 U Taq DNA polymerase (Thermo Fisher Scientific) and 0.3 U Pfu Turbo DNA polymerase (Agilent) in the Jeffreys buffer described above for nested PCR with 50 ng of testis genomic DNA. Testis DNA was derived from 3–5 different mice for each genotype and amplified independently.
PCRs were carried out in 96-well plates to generate a large number of independently derived PCR products for sequencing. For each amplicon, 576 PCRs were performed with a total of 28.8 μg genomic DNA, equivalent to 9.6 million haploid genomes. PCRs were pooled, and a 1200-μl aliquot was purified and concentrated on spin columns (Invitrogen PureLink™ PCR Purification Kit) as follows: For each 100 μl PCR aliquot, 400 μl binding buffer from the kit was added, and then the sample was applied to the column and centrifuged. This step was repeated 6 times in each of two columns before washing and elution with 37 μl elution buffer. Eluted DNA was combined from the two columns and a 15-μl aliquot was run on a 2% agarose gel to estimate DNA concentration, which was ~2 ng/μl. The remaining ~60 μl was submitted to the Integrated Genomics Operation at MSK for library preparation and deep sequencing. The library was prepared by ligation of indexed adapter sequences using the KAPA HyperPrep Kit (Roche). Sequencing was performed using Illumina MiSeq (2 × 300-bp paired-end reads). In a single run, libraries from amplicons 1–4 were mixed with 5% PhiX DNA control library (Illumina) and sequenced. Because MiSeq can deliver 15–20 million reads per lane, in our experiment, 3.6–4.8 million reads per amplicon were estimated, which was about 2 times lower than the 9.6 million input DNA templates. With that calculation, not all deletion events were recovered, but most recovered deletions should have yielded only a single read.
We performed 4 MiSeq runs with amplicons from different genotypes (Figure S7A). FASTQ sequencing files were analyzed using CRISPResso2 (Pinello et al., 2016; Clement et al., 2019) by the Bioinformatics Core Facility at MSK. Reads were aligned to the reference amplicon sequence and 2.9–5.3 million paired-end reads were mapped per amplicon. Alignments matching the target sequence for the first 20 and last 20 bases and with one deletion event were filtered. Reads with >1 bp deletion and with no mutations in the sequence were analyzed in R and ApE. Breakpoint distribution analyses and random sampling (Figure S7C) were performed in R. Graphs were plotted using R and GraphPad Prism.
SPO11-oligo maps
SPO11-oligo maps were generated previously (GEO accession number GSE84689) (Lange et al., 2016). Mice for the Atm−/− data set “Atm null 1”, which was used in all the analyses, were on a mixed B6 and 129/Sv strain background. Two Atm+/+ data sets are available, “B6”, from mice on a pure B6 background, and “Atm wt”, from the same colony as Atm−/− mice. Hotspots selected in this study were called in the “B6” data set (Figures 1B, S1D and S2A), while analysis of hotspot distributions was done with the “Atm wt” data set (Figures 1A and S1A–C). The “B6” data set was also used to calculate double cutting frequency per meiosis (Table S3), and check if SPO11 oligos map to the genomic locations from which inserts derived (Figures S6A and S6B).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using R and GraphPad Prism. Statistical parameters and tests are reported in the figures and corresponding figure legends. In cases where outliers were removed for plotting purposes, the removed data points were included in statistical analyses.
Supplementary Material
Figure S1. Higher potential in Atm−/− for DSB formation at adjacent hotspots, related to Figure 1.
Analyses were performed for autosomes.
(A) Hotspot pairs separated by 1–5 kb are observed more frequently in Atm−/− than in Atm+/+. Data are derived from SPO11-oligo maps (Lange et al., 2016). “Shared” hotspots are those called in both Atm−/− and Atm+/+. “New” hotspots are those called only in Atm−/−, although corresponding SPO11-oligo clusters are typically present in the Atm+/+ map (Lange et al., 2016). Hotspot pairs in Atm−/− are separated into classes according to whether each hotspot is shared or new.
(B) Distribution of distances separating hotspots in Atm−/− hotspot pairs, shown up to 250 kb. The proportion of each hotspot pair class from panel A is indicated. The increase in the number of hotspot pairs in Atm−/− separated by short distances (Figure 1A) is primarily the result of the emergence of new hotspots close to shared hotspots.
(C) Strength of individual hotspots in hotspot pairs separated by 1–5 kb. Hotspot pair strength, the median of total RPM (reads per million) for the two hotspots in a pair, is indicated. Note that the RPM values do not account for the 11.3-fold increase in SPO11 oligos in Atm−/− (Lange et al., 2011). Outliers with >300 RPM (4 in Atm−/−; 18 in Atm+/+) are not shown.
(D) Hotspot strength for the individual hotspots in the Chr1 and Chr19 hotspot pairs. RPM values in Atm−/− are multiplied by 11.3, the average increase in whole-testis SPO11-oligo levels in Atm−/− mice. Hotspot rank is the rank of the individual hotspot within all hotspots for Atm+/+ or Atm−/−.
Figure S2. Deletions at the Chr19 hotspot pair and deletions at both hotspot pairs from experiments without prior genomic DNA digest, related to Figure 2.
(A) SPO11-oligo maps in wild type and Atm−/− at the Chr19 hotspot pair, with the scheme of the nested PCR assay below. The fold increase in the SPO11 oligos (RPM × 11.3) is indicated for each hotspot. Prior to amplification, genomic DNA is optionally digested with restriction enzymes to reduce amplification of parental products. The Pip5k1b exon located at the left hotspot is removed in many of the deletions.
(B) Representative gel of PCR products in Atm−/−. ~16,000 haploid genomes were seeded per PCR. Asterisks indicate deletions.
(C–F) Analysis of deletions at the Chr19 hotspot pair. C, Deleted DNA sequences (black bars) from adult and juvenile testes, and sperm. Deletions detected in Atm+/+ and Atm+/− often had one breakpoint that was also found for a deletion in Atm−/− (red asterisks). See also Figure S3C. D, Arc diagram of deletions in Atm−/− (58 deletions total). See also Figures S3A and S3B. E, Distribution of individual breakpoints. F, Smoothed breakpoint and SPO11-oligo profiles (201-bp Hann filter for smoothing). Offsets between centers of hotspots and breakpoint distributions are indicated.
(G) Distribution of deletions in Atm−/− at the Chr1 and Chr19 hotspot pairs from experiments without prior genomic DNA digest. Deletion breakpoints clustered around hotspot centers. A few deletion products maintaining at least one restriction site were captured (green arcs). The majority of events would be detected if genomic DNA was digested.
Figure S5. Deletions in Atm−/− B6 × DBA F1 hybrid mice with models for haplotype switching, related to Figure 4.
(A) Predicted PRDM9 recognition sequences at the left hotspot in B6 and DBA, aligned to the PRDM9 motif from SPO11-oligo analysis (Lange et al., 2016).
(B) Arc diagram of deletions from the Chr1 hotspot pair (37 deletions total). Two interhomolog deletions are indicated (pink arcs).
(C, D). Models for haplotype switching in deletions. B, Attempted double-strand gap repair. Examples of one intrachromosomal (top) and one interhomolog (bottom) deletion with haplotype switches. In both cases, two DSBs produce a double-stranded gap between the two hotspots, releasing a double-cut fragment (lighter blue or red double line). One DSB end at the gap invades the homolog and is extended by DNA synthesis, but a DSB in the repair template leads to aborted synthesis, such that this end is ejected and joins the other end of the gap (intrachromosomal end joining) or the end of a DSB formed at the other hotspot in the homolog (interhomolog end joining). In the case of interhomolog deletions, it is also possible that only one DSB is formed on the chromosome initiating strand invasion. Thus, in both examples at least three DSBs occur, indicating extensive damage to multiple chromatids at adjacent hotspots when ATM is absent. The substantial template damage in this scenario may favor nonhomologous end-joining mechanisms. C, Precise insertion due to homologous annealing. An example of an intrachromosomal deletion with a haplotype switch. At a minimum four DSBs are formed, involving double cutting within one hotspot on one chromosome and at both hotspots on the other chromosome. A released double-cut fragment from one hotspot is precisely inserted at the double-stranded gap in the homolog through annealing of homologous sequences exposed by resection. Importantly this scenario depends on the relative position of the two DSBs in chromosomes. If the relative position were switched (bottom right), resection would not expose homologous sequences. Nonhomologous insertion is not compatible because it would produce a small deletion (bottom right) or a small duplication (bottom left). As we do not observe these types of repair products in our deletions, we consider the attempted double-strand gap repair scenario more likely.
Figure S3. Distribution of deletions for the Chr1 and Chr19 hotspot pairs, related to Figure 2.
(A) Deletion arcs from Atm−/− are segregated according to the positions of breakpoints relative to 251-bp zones around each hotspot center (yellow). The percentage of SPO11 oligos is the number of SPO11 oligos within each 251-bp zone divided by the total number of SPO11 oligos mapping between the left or right primer (arrows) and the midpoint between hotspot centers. Deletions were grouped according to whether both breakpoints (violet arcs), one breakpoint (red and blue arcs), or neither breakpoint (gray arcs) mapped within the 251-bp zones. The percentage of deletions within each group is indicated.
(B) Predicted and observed percentage of deletions with both breakpoints, one breakpoint, or neither breakpoint within 251 bp around hotspot centers. The predicted distribution of deletions is equivalent to the expected distribution of double cuts calculated from SPO11-oligo counts (RPMs) at the hotspot pairs.
(C) Deletions detected in the presence of ATM in testes and sperm at the Chr19 hotspot pair. Note that 12 of 14 deletions mapped outside of the 251-bp zone around the hotspot centers, which is in contrast to deletions observed in Atm−/−. The blue asterisk indicates a breakpoint cluster overlapping a SPO11-oligo cluster. See also Figure S2C.
Figure S4. Extended analysis of tandem duplications and circular DNA, related to Figure 3.
(A) Frequencies of duplications/circles from the inverse PCR experiments. Sequencing was used to verify duplication/circle products. The few products that could not be fully sequenced were excluded from the frequency calculation. Partial sequences indicated the presence of palindromes of ≥7–30-bp that would impede sequencing because of hairpin formation. These products were found in all genotypes, indicating likely PCR errors or SPO11-independent events. One mouse of each genotype was analyzed for both hotspot pairs. P values are from the Chi-square test.
(B) ExoV experiment to detect circular DNA at the Chr1 hotspot pair, related to Figures 3D and 3E. Left, SPO11-oligo map in Atm−/− and the location of nested primer pairs (arrows) used in inverse PCR (light gray) and in control PCR (purple and light gray). Restriction sites for ScaI, which was used to digest genomic DNA prior to ExoV treatment, flank the hotspot pair. Middle, representative gel of inverse PCR products obtained from reactions either digested with ExoV to detect circles or not digested with ExoV to detect duplications/circles. Note that ExoV treatment appears to remove products that form hairpins. Right, corresponding gel of control PCR to assess ExoV efficiency, showing PCR products from the primary (PCR 1) and secondary (PCR 2) PCRs. The estimated number of pre-digestion haploid genomes per PCR is shown. No products were amplified in PCRs from reactions treated with ExoV.
(C) Frequencies of duplications/circles and circles from the ExoV experiments at the Chr1 hotspot pair. For Atm−/−, testis DNA from two mice was analyzed. No products were observed in the small-scale experiment using Atm+/− testis DNA. In the larger experiment with Atm+/− and Atm+/+ sperm from one mouse per genotype, three duplication/circle products were detected, but no circles. This would be consistent with the idea that circle DNA is not transmitted into sperm; however, their low frequency and the more frequent appearance of PCR products with hairpins in –ExoV experiments obscures our evaluation of these genotypes. P value is from the Chi-square test.
(D) Distribution of breakpoint distances from the hotspot centers for deletions and duplications/circles, related to Figure 3F. Scatter plots of breakpoint positions at the left and right hotspots in the Chr1 and Chr19 hotspot pairs. P values are from an unpaired Mann-Whitney test.
(E) Multiple double cuts should shift breakpoint junctions for deletions versus tandem duplications and circles. Left, double cutting at two DSB hotspots (one DSB per hotspot) is expected to lead to similarly sized deletions, tandem duplications, and circles. Right, multiple double cuts involving additional DSBs at each hotspot will lead to shorter tandem duplications and circles, but larger deletions.
(F) Imputed multi-mapping SPO11 oligos at the Chr1 hotspot pair (Lange et al., 2016). A SPO11-oligo cluster is evident at ~ 500 bp from the left hotspot center (red asterisk), which is not observed in the unique SPO11-oligo map (Figure 3F). Duplication/circle breakpoints and smaller deletion breakpoints, shown in G, frequently map at and close to this SPO11-oligo cluster.
(G) Deletions detected within the Chr1 left hotspot in Atm−/− (14 events) and Atm+/− (2 events). Nested PCR was carried out with primers flanking the hotspot (same primers used in the control PCR in B), with genomic DNA digested with NsiI; eight of 16 deletion products contained the restriction site, indicating incomplete digest. Note that the right breakpoints map to similar positions as duplication/circle breakpoints, and the left breakpoints map to similar positions to the left deletion breakpoints that are more spread out. This supports the idea that multiple double cuts (E) can yield distinct breakpoint profiles for duplications/circles and deletions (Figure 3F).
Figure S7. Extended analysis of microdeletions at a single hotspot, related to Figure 7.
(A–C) Evaluation of reads with microdeletions from deep sequencing. A, Frequency of reads that had microdeletions of the indicated lengths. Four overlapping amplicons across the hotspot (1–4) were deep sequenced using MiSeq. For each amplicon, 576 separate PCRs were carried out with a total input of 9.6 million haploid genome equivalents, using a limited number of amplification cycles (see Methods). The PCR products for each amplicon were then pooled for sequencing. Four MiSeq runs were performed (I–IV) with the indicated amplicons and genotypes. For clarity, sequencing results are grouped by run (Top) or genotype (Bottom). At top, note that reads with small deletions (2–4 bp) were frequently observed in all amplicons for each genotype, including Spo11−/−, and that their frequency varied between sequencing runs, indicating a significant batch effect. Short deletions (e.g., 2 bp) are known to arise from PCR and sequencing errors (Schirmer et al., 2015), and we saw that they became progressively less abundant in all genotypes up to 10 bp. Excluded from the 4-bp deletions is a product that was represented by >1000 reads in amplicons 1–3. By contrast, reads with deletions >10 bp were more frequent in Atm−/− than in the other genotypes. At bottom, note that reads with 11–25-bp microdeletions were more frequent in Atm−/−. Therefore, 11 bp was used as the lower cutoff to define a microdeletion for subsequent analyses. Our sequencing design (pooling of many low-cycle PCRs) is expected to minimize PCR jackpots and reduce instances where PCR duplicates from the same input DNA molecule are sequenced. However, two amplicons in Atm+/+ showed multiple reads for one microdeletion each (amplicon 1: 26-bp microdeletion, 14 reads; amplicon 2: 31-bp microdeletion, 22 reads; Table S7), which therefore may be PCR duplicates. Thus, in some subsequent analyses we applied a stringent definition of independent events by including only one read for a given deletion product from a given amplicon. B, Recurring microdeletions within the hotspot. Top, smoothed (21-bp Hann filter) and unsmoothed SPO11-oligo maps at the Chr1 hotspot in Atm−/−. Bottom, recurring microdeletions. These identical microdeletions (black bars) were found in independent PCRs for different amplicons. i) In Atm−/−, amplicons that yielded the recurring microdeletions are indicated. For example, the microdeletion with the AG microhomology arose in amplicons 3 and 4. Note that one or both breakpoints for each of the recurring microdeletions occurred at SPO11-oligo peaks (dashed vertical lines). ii) In Atm+/− and Atm+/+, some microdeletions were identical to those found in Atm−/−, in some cases in multiple amplicons. For example, the microdeletion with the GGAAGGAG microhomology was recovered in all three genotypes, but in different amplicons (Atm−/−, 1; Atm+/−, 2; Atm+/+, 4). iii) Some of the microdeletions found in Spo11−/− recurred in other genotypes. Note that these map to the same region of the hotspot away from the center. C, Many of the duplicated reads in Atm−/− amplicons likely represent independent microdeletion events rather than PCR duplicates. For each amplicon, we randomly sampled with replacement from a set of reads representing all of the unique microdeletions observed. The sampled group was equal in number to the duplicated reads actually obtained for that amplicon. Then, the sampled reads with microdeletions mapping to the region where amplicons 1, 2, and 3 overlap were combined, and their breakpoint distribution was compared with the distribution of the observed duplicate reads. Smoothed breakpoint profiles (21-bp Hann filter) of independently obtained reads, as in Figure 7C, and of all reads are also shown. In the breakpoint profiles of the observed duplicates, the left breakpoints were particularly enriched in a peak that matched the central peak of SPO11 oligos, and the right breakpoints were enriched at a position that matched the secondary peak of SPO11 oligos. These patterns were not recapitulated by random sampling: both the left and right breakpoints were more widely dispersed without such clear enrichment coincident with the SPO11-oligo peaks. Because the observed duplicates were more likely than the random sample to have breakpoints aligned with the strongly favored locations of DSBs, these findings are consistent with the conclusion that most of the duplicated reads represent independent microdeletion events.
(D) Arc diagrams of microdeletions in duplicated reads stratified by length from Atm−/−, as in Figure 7C. Microdeletions >160 bp are not shown, as their location may be more constrained by restricting analysis to the region where the amplicons overlap.
(E) Microdeletion length distribution for all amplicons by genotype for independent reads. Density plots are shown (>10 bp); densities in Atm+/−, Atm+/+, and Spo11−/− are normalized to Atm−/− to reflect read numbers in each genotype. As in the analysis of microdeletions in the overlap region of amplicons 1, 2, and 3 (Figure 7D), distinct size classes were observed in the presence and absence of ATM, but not in Spo11−/−. Larger microdeletion size classes (~140 bp, ~180 bp, ~230 bp) were well defined when all amplicons are considered. A smaller size class (~15 bp) was more prominent outside of the overlap region.
(F) Arc diagrams of the ~140-bp, ~180-bp, and ~230-bp microdeletions observed for Atm−/−.
(G) Microhomology lengths at microdeletion breakpoints in the presence and absence of ATM. Analyses were performed for independent microdeletions in the overlapping region between amplicons 1, 2, and 3.
Figure S6. Characteristics of insertions in Atm−/−, related to Figure 6.
(A) Derivation of inserts mapping to DSB hotspots/clusters on other chromosomes. The genomic locations of inserts (blue bars) are mapped relative to SPO11 oligos for Atm−/− and Atm+/+. We indicated whether these locations were called as hotspots in one or both genotypes or instead contain SPO11-oligo clusters. Inserts mapping to the non-PAR ChrY are shown in B. One insert mapping to a microsatellite repeat is not shown. See also Table S5.
(B) Inserts from the non-PAR ChrY map to multiple DSB hotspots. Three inserts identified in Atm−/− deletions map to non-PAR ChrY repeats (Table S5), some of which have called hotspots. SPO11-oligo profiles are shown only for those repeats containing hotspots in Atm−/−, Atm+/+, or both, with the position of the inserts (blue bars) for each hotspot indicated. Inserts 1 and 2 are part of the same repeat family, and in one case they map to a single hotspot. Inserts 1 and 3 map to three and two hotspots, respectively. SPO11 oligos map uniquely to these hotspots (“Unique”) or also map to other copies of the repeats (“Multi”). It remains possible that other repeat locations to which SPO11 oligos do not map uniquely also contain hotspots. Given that all insert-matching repeats (67 total) contain an intact PRDM9 motif, it is possible that DSBs are formed in many such repeats. The locations of multi-mapping SPO11 oligos were imputed, meaning they were distributed among mapped positions proportionally to local densities of uniquely mapping SPO11 oligos (Lange et al., 2016).
(C) Insert length distribution. Median insert lengths are indicated. See also Tables S5 and S6.
(D) No correlation (Pearson’s r) between insert and deletion lengths for inserts from other chromosomes. Note: Deletions of median size (1943 bp) with ~400 bp insertions should be readily detected (~1900 bp PCR), as should somewhat longer insertions.
(E) Deletions with inverted insertions at the Chr1 hotspot pair in B6 and B6 × DBA mice. Asterisks denote deletions where one insert breakpoint had the same microhomology as the site with recurring deletions (Figure 5B). Note that for the two deletions on the DBA chromosome, the inserts also derived from the DBA chromosome.
Highlights.
Germline mutagenesis at mouse DSB hotspots arises via double cutting and end joining
Double cutting within single DSB hotspots can initiate microdeletions
Double-cut fragments can circularize or insert into other DSB locations
Mutational events are more frequent when DSB control is affected in ATM mutant mice
Acknowledgments
We thank Matthew Neale and Franz Klein for sharing unpublished results; members of the Jasin and Keeney labs for assistance and discussions, in particular, Shintaro Yamada; and MSK core facilities, in particular the Integrated Genomics Operation (Cassidy Cobbs, Neeman Mohibullah, Agnès Viale) and the Bioinformatics Core (Nicholas Socci), which are supported by NIH grant P30 CA008748. This work was supported by March of Dimes grant 1-FY17-799 (A.L.), American Cancer Society fellowship PF-12-157-01-DMC (J.L.), and NIH grants R35 GM118092 (S.K.), RM1HG009490, and R35 GM118175 (M.J.).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed EA, Philippens ME, Kal HB, de Rooij DG, and de Boer P (2010). Genetic probing of homologous recombination and non-homologous end joining during meiotic prophase in irradiated mouse spermatocytes. Mutat Res 688, 12–18. [DOI] [PubMed] [Google Scholar]
- Alvarez-Quilon A, Serrano-Benitez A, Lieberman JA, Quintero C, Sanchez-Gutierrez D, Escudero LM, and Cortes-Ledesma F (2014). ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat Commun 5, 3347, doi: 10.1038/ncomms4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Oke A, Yam P, Zhuge T, and Fung JC (2015). Reduced Crossover Interference and Increased ZMM-Independent Recombination in the Absence of Tel1/ATM. PLoS Genet 11, e1005478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Kajita S, Walker M, Saxl RL, Raghupathy N, Choi K, Petkov PM, and Paigen K (2015). PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet 11, e1004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi M, Mahadevaiah S, Di Giacomo M, Baudat F, de Rooij DG, Burgoyne PS, Jasin M, and Keeney S (2005). Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol 25, 7203–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, and Wynshaw-Boris A (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–171. [DOI] [PubMed] [Google Scholar]
- Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, and Wynshaw-Boris A (1998). Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development 125, 4007–4017. [DOI] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, and Keeney S (2000). Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6, 989–998. [DOI] [PubMed] [Google Scholar]
- Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, Atlason BA, Kristmundsdottir S, Mehringer S, Hardarson MT, et al. (2021). Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 53, 779–786. [DOI] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, and Petukhova GV (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Johnson D, Andres SN, Kissling VM, Reinert JK, Garcia V, Erie DA, Hess D, Thoma NH, Enchev RI, et al. (2018). Regulatory control of DNA end resection by Sae2 phosphorylation. Nat Commun 9, 4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, and Lieber MR (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checchi PM, Lawrence KS, Van MV, Larson BJ, and Engebrecht J (2014). Pseudosynapsis and decreased stringency of meiotic repair pathway choice on the hemizygous sex chromosome of Caenorhabditis elegans males. Genetics 197, 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Kass EM, Yen WF, Ludwig T, Moynahan ME, Chaudhuri J, and Jasin M (2017). ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proc Natl Acad Sci U S A 114, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lisby M, and Symington LS (2013). RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell 50, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys Bouuaert C, Pu S, Wang J, Oger C, Daccache D, Xie W, Patel DJ, and Keeney S (2021). DNA-driven condensation assembles the meiotic DNA break machinery. Nature 592, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Baudat F, Grey C, Keeney S, de Massy B, and Jasin M (2014). Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat Genet 46, 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, and Jasin M (2011). Isolation of meiotic recombinants from mouse sperm. Methods Mol Biol 745, 251–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Kauppi L, Lange J, Roig I, Wang R, Keeney S, and Jasin M (2012). Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat Cell Biol 14, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Keeney S, and Jasin M (2010). Comprehensive, fine-scale dissection of homologous recombination outcomes at a hot spot in mouse meiosis. Mol Cell 39, 700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alfoldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TJ, Garcia V, and Neale MJ (2016). Meiotic DSB patterning: A multifaceted process. Cell Cycle 15, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. (2016). Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enguita-Marruedo A, Martin-Ruiz M, Garcia E, Gil-Fernandez A, Parra MT, Viera A, Rufas JS, and Page J (2019). Transition from a meiotic to a somatic-like DNA damage response during the pachytene stage in mouse meiosis. PLoS Genet 15, e1007439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler KR, Hyppa RW, Cromie GA, and Smith GR (2018). Physical basis for long-distance communication along meiotic chromosomes. Proc Natl Acad Sci U S A 115, E9333–E9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Gray S, Allison RM, Cooper TJ, and Neale MJ (2015). Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaysinskaya V, Soh IY, van der Heijden GW, and Bortvin A (2014). Optimized flow cytometry isolation of murine spermatocytes. Cytometry A 85, 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard C, Roelens B, Zawadzki KA, and Villeneuve AM (2018). Interdependent and separable functions of Caenorhabditis elegans MRN-C complex members couple formation and repair of meiotic DSBs. Proc Natl Acad Sci U S A 115, E4443–E4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatkevich T, Miller DE, Turcotte CA, Miller MC, and Sekelsky J (2021). A pathway for error-free non-homologous end joining of resected meiotic double-strand breaks. Nucleic Acids Res 49, 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N (2015). Meiotic Recombination: The Essence of Heredity. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Allison RM, Cannavo E, Cejka P, and Neale MJ (2019). Removal of Spo11 from meiotic DNA breaks in vitro but not in vivo by Tyrosyl DNA Phosphodiesterase 2. bioRxiv doi: 10.1101/527333. [DOI] [Google Scholar]
- Johnson D, Crawford M, Cooper T, Claeys Bouuaert C, Keeney S, Llorente B, Garcia V, and Neale MJ (2021). Concerted cutting by Spo11 illuminates meiotic DNA break mechanics. Nature 594, 572–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Brown MS, Bishop DK, and Borner GV (2015). Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol Cell 57, 797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S (2001). Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol 52, 1–53. [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, and Kleckner N (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. [DOI] [PubMed] [Google Scholar]
- Keeney S, Lange J, and Mohibullah N (2014). Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 48, 187–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Peterson SE, Jasin M, and Keeney S (2016). Mechanisms of germ line genome instability. Semin Cell Dev Biol 54, 177–187. [DOI] [PubMed] [Google Scholar]
- Kurzbauer MT, Janisiw MP, Paulin LF, Prusen Mota I, Tomanov K, Krsicka O, Haeseler AV, Schubert V, and Schlogelhofer P (2021). ATM controls meiotic DNA double-strand break formation and recombination and affects synaptonemal complex organization in plants. Plant Cell 33, 1633–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Pan J, Cole F, Thelen MP, Jasin M, and Keeney S (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Yamada S, Tischfield SE, Pan J, Kim S, Zhu X, Socci ND, Jasin M, and Keeney S (2016). The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 167, 695–708 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens BB, Johnson NM, and Tijsterman M (2013). COM-1 promotes homologous recombination during Caenorhabditis elegans meiosis by antagonizing Ku-mediated non-homologous end joining. PLoS Genet 9, e1003276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Bitoun E, Altemose N, Davies RW, Davies B, and Myers SR (2019). A high-resolution map of non-crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat Commun 10, 3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Lange J, Keeney S, and Jasin M (2018). Control of meiotic double-strand-break formation by ATM: local and global views. Cell Cycle 17, 1155–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaisne N, Kessler Z, and Yanowitz JL (2018). Meiotic Double-Strand Break Proteins Influence Repair Pathway Utilization. Genetics 210, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsolier-Kergoat MC, Khan MM, Schott J, Zhu X, and Llorente B (2018). Mechanistic View and Genetic Control of DNA Recombination during Meiosis. Mol Cell 70, 9–20 e26. [DOI] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, and Boulton SJ (2005). RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 25, 3127–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Yamada S, and Keeney S (2017). A global view of meiotic double-strand break end resection. Science 355, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohibullah N, and Keeney S (2017). Numerical and spatial patterning of yeast meiotic DNA breaks by Tel1. Genome Res 27, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Pan J, and Keeney S (2005). Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiano J, Wu W, Yamada S, Sciascia N, Callen E, Paola Cotrim A, Deshpande RA, Maman Y, Day A, Paull TT, and Nussenzweig A (2020). ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis. Nat Commun 11, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. (2011). A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SE, Keeney S, and Jasin M (2020). Mechanistic Insight into Crossing over during Mouse Meiosis. Mol Cell 78, 1252–1263 e1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD (2001). Meiotic recombination hot spots and cold spots. Nat Rev Genet 2, 360–369. [DOI] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieler S, Chen D, Huang L, Mayrhofer E, Zsoter S, Vesely M, Mbogning J, and Klein F (2021). Spo11 generates gaps through concerted cuts at sites of topological stress. Nature 594, 577–582. [DOI] [PubMed] [Google Scholar]
- Roth DB, Porter TN, and Wilson JH (1985). Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol 5, 2599–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, Ijaz UZ, D’Amore R, Hall N, Sloan WT, and Quince C (2015). Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res 43, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, and Symington LS (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends in biochemical sciences 40, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y (2006). The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 31, 402–410. [DOI] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero RD, and Petukhova GV (2016). The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh YQ, Alfoldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, Fulton RS, Kremitzki C, Koutseva N, Mueller JL, et al. (2014). Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 159, 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D, Mohibullah N, Zhu X, and Keeney S (2014). Homologue engagement controls meiotic DNA break number and distribution. Nature 510, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, and Loidl J (2018). A chromatin-associated protein required for inducing and limiting meiotic DNA double-strand break formation. Nucleic Acids Res 46, 11822–11834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tock AJ, and Henderson IR (2018). Hotspots for Initiation of Meiotic Recombination. Front Genet 9, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, and Baltimore D (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10, 2411–2422. [DOI] [PubMed] [Google Scholar]
- Yamada S, Hinch AG, Kamido H, Zhang Y, Edelmann W, and Keeney S (2020). Molecular structures and mechanisms of DNA break processing in mouse meiosis. Genes Dev 10.1101/gad.336032.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Kim S, Tischfield SE, Jasin M, Lange J, and Keeney S (2017). Genomic and chromatin features shaping meiotic double-strand break formation and repair in mice. Cell Cycle 16, 1870–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, and Smolikove S (2013). Impaired resection of meiotic double-strand breaks channels repair to nonhomologous end joining in Caenorhabditis elegans. Mol Cell Biol 33, 2732–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kim KP, Kleckner NE, and Storlazzi A (2011). Meiotic double-strand breaks occur once per pair of (sister) chromatids and, via Mec1/ATR and Tel1/ATM, once per quartet of chromatids. Proc Natl Acad Sci U S A 108, 20036–20041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Higher potential in Atm−/− for DSB formation at adjacent hotspots, related to Figure 1.
Analyses were performed for autosomes.
(A) Hotspot pairs separated by 1–5 kb are observed more frequently in Atm−/− than in Atm+/+. Data are derived from SPO11-oligo maps (Lange et al., 2016). “Shared” hotspots are those called in both Atm−/− and Atm+/+. “New” hotspots are those called only in Atm−/−, although corresponding SPO11-oligo clusters are typically present in the Atm+/+ map (Lange et al., 2016). Hotspot pairs in Atm−/− are separated into classes according to whether each hotspot is shared or new.
(B) Distribution of distances separating hotspots in Atm−/− hotspot pairs, shown up to 250 kb. The proportion of each hotspot pair class from panel A is indicated. The increase in the number of hotspot pairs in Atm−/− separated by short distances (Figure 1A) is primarily the result of the emergence of new hotspots close to shared hotspots.
(C) Strength of individual hotspots in hotspot pairs separated by 1–5 kb. Hotspot pair strength, the median of total RPM (reads per million) for the two hotspots in a pair, is indicated. Note that the RPM values do not account for the 11.3-fold increase in SPO11 oligos in Atm−/− (Lange et al., 2011). Outliers with >300 RPM (4 in Atm−/−; 18 in Atm+/+) are not shown.
(D) Hotspot strength for the individual hotspots in the Chr1 and Chr19 hotspot pairs. RPM values in Atm−/− are multiplied by 11.3, the average increase in whole-testis SPO11-oligo levels in Atm−/− mice. Hotspot rank is the rank of the individual hotspot within all hotspots for Atm+/+ or Atm−/−.
Figure S2. Deletions at the Chr19 hotspot pair and deletions at both hotspot pairs from experiments without prior genomic DNA digest, related to Figure 2.
(A) SPO11-oligo maps in wild type and Atm−/− at the Chr19 hotspot pair, with the scheme of the nested PCR assay below. The fold increase in the SPO11 oligos (RPM × 11.3) is indicated for each hotspot. Prior to amplification, genomic DNA is optionally digested with restriction enzymes to reduce amplification of parental products. The Pip5k1b exon located at the left hotspot is removed in many of the deletions.
(B) Representative gel of PCR products in Atm−/−. ~16,000 haploid genomes were seeded per PCR. Asterisks indicate deletions.
(C–F) Analysis of deletions at the Chr19 hotspot pair. C, Deleted DNA sequences (black bars) from adult and juvenile testes, and sperm. Deletions detected in Atm+/+ and Atm+/− often had one breakpoint that was also found for a deletion in Atm−/− (red asterisks). See also Figure S3C. D, Arc diagram of deletions in Atm−/− (58 deletions total). See also Figures S3A and S3B. E, Distribution of individual breakpoints. F, Smoothed breakpoint and SPO11-oligo profiles (201-bp Hann filter for smoothing). Offsets between centers of hotspots and breakpoint distributions are indicated.
(G) Distribution of deletions in Atm−/− at the Chr1 and Chr19 hotspot pairs from experiments without prior genomic DNA digest. Deletion breakpoints clustered around hotspot centers. A few deletion products maintaining at least one restriction site were captured (green arcs). The majority of events would be detected if genomic DNA was digested.
Figure S5. Deletions in Atm−/− B6 × DBA F1 hybrid mice with models for haplotype switching, related to Figure 4.
(A) Predicted PRDM9 recognition sequences at the left hotspot in B6 and DBA, aligned to the PRDM9 motif from SPO11-oligo analysis (Lange et al., 2016).
(B) Arc diagram of deletions from the Chr1 hotspot pair (37 deletions total). Two interhomolog deletions are indicated (pink arcs).
(C, D). Models for haplotype switching in deletions. B, Attempted double-strand gap repair. Examples of one intrachromosomal (top) and one interhomolog (bottom) deletion with haplotype switches. In both cases, two DSBs produce a double-stranded gap between the two hotspots, releasing a double-cut fragment (lighter blue or red double line). One DSB end at the gap invades the homolog and is extended by DNA synthesis, but a DSB in the repair template leads to aborted synthesis, such that this end is ejected and joins the other end of the gap (intrachromosomal end joining) or the end of a DSB formed at the other hotspot in the homolog (interhomolog end joining). In the case of interhomolog deletions, it is also possible that only one DSB is formed on the chromosome initiating strand invasion. Thus, in both examples at least three DSBs occur, indicating extensive damage to multiple chromatids at adjacent hotspots when ATM is absent. The substantial template damage in this scenario may favor nonhomologous end-joining mechanisms. C, Precise insertion due to homologous annealing. An example of an intrachromosomal deletion with a haplotype switch. At a minimum four DSBs are formed, involving double cutting within one hotspot on one chromosome and at both hotspots on the other chromosome. A released double-cut fragment from one hotspot is precisely inserted at the double-stranded gap in the homolog through annealing of homologous sequences exposed by resection. Importantly this scenario depends on the relative position of the two DSBs in chromosomes. If the relative position were switched (bottom right), resection would not expose homologous sequences. Nonhomologous insertion is not compatible because it would produce a small deletion (bottom right) or a small duplication (bottom left). As we do not observe these types of repair products in our deletions, we consider the attempted double-strand gap repair scenario more likely.
Figure S3. Distribution of deletions for the Chr1 and Chr19 hotspot pairs, related to Figure 2.
(A) Deletion arcs from Atm−/− are segregated according to the positions of breakpoints relative to 251-bp zones around each hotspot center (yellow). The percentage of SPO11 oligos is the number of SPO11 oligos within each 251-bp zone divided by the total number of SPO11 oligos mapping between the left or right primer (arrows) and the midpoint between hotspot centers. Deletions were grouped according to whether both breakpoints (violet arcs), one breakpoint (red and blue arcs), or neither breakpoint (gray arcs) mapped within the 251-bp zones. The percentage of deletions within each group is indicated.
(B) Predicted and observed percentage of deletions with both breakpoints, one breakpoint, or neither breakpoint within 251 bp around hotspot centers. The predicted distribution of deletions is equivalent to the expected distribution of double cuts calculated from SPO11-oligo counts (RPMs) at the hotspot pairs.
(C) Deletions detected in the presence of ATM in testes and sperm at the Chr19 hotspot pair. Note that 12 of 14 deletions mapped outside of the 251-bp zone around the hotspot centers, which is in contrast to deletions observed in Atm−/−. The blue asterisk indicates a breakpoint cluster overlapping a SPO11-oligo cluster. See also Figure S2C.
Figure S4. Extended analysis of tandem duplications and circular DNA, related to Figure 3.
(A) Frequencies of duplications/circles from the inverse PCR experiments. Sequencing was used to verify duplication/circle products. The few products that could not be fully sequenced were excluded from the frequency calculation. Partial sequences indicated the presence of palindromes of ≥7–30-bp that would impede sequencing because of hairpin formation. These products were found in all genotypes, indicating likely PCR errors or SPO11-independent events. One mouse of each genotype was analyzed for both hotspot pairs. P values are from the Chi-square test.
(B) ExoV experiment to detect circular DNA at the Chr1 hotspot pair, related to Figures 3D and 3E. Left, SPO11-oligo map in Atm−/− and the location of nested primer pairs (arrows) used in inverse PCR (light gray) and in control PCR (purple and light gray). Restriction sites for ScaI, which was used to digest genomic DNA prior to ExoV treatment, flank the hotspot pair. Middle, representative gel of inverse PCR products obtained from reactions either digested with ExoV to detect circles or not digested with ExoV to detect duplications/circles. Note that ExoV treatment appears to remove products that form hairpins. Right, corresponding gel of control PCR to assess ExoV efficiency, showing PCR products from the primary (PCR 1) and secondary (PCR 2) PCRs. The estimated number of pre-digestion haploid genomes per PCR is shown. No products were amplified in PCRs from reactions treated with ExoV.
(C) Frequencies of duplications/circles and circles from the ExoV experiments at the Chr1 hotspot pair. For Atm−/−, testis DNA from two mice was analyzed. No products were observed in the small-scale experiment using Atm+/− testis DNA. In the larger experiment with Atm+/− and Atm+/+ sperm from one mouse per genotype, three duplication/circle products were detected, but no circles. This would be consistent with the idea that circle DNA is not transmitted into sperm; however, their low frequency and the more frequent appearance of PCR products with hairpins in –ExoV experiments obscures our evaluation of these genotypes. P value is from the Chi-square test.
(D) Distribution of breakpoint distances from the hotspot centers for deletions and duplications/circles, related to Figure 3F. Scatter plots of breakpoint positions at the left and right hotspots in the Chr1 and Chr19 hotspot pairs. P values are from an unpaired Mann-Whitney test.
(E) Multiple double cuts should shift breakpoint junctions for deletions versus tandem duplications and circles. Left, double cutting at two DSB hotspots (one DSB per hotspot) is expected to lead to similarly sized deletions, tandem duplications, and circles. Right, multiple double cuts involving additional DSBs at each hotspot will lead to shorter tandem duplications and circles, but larger deletions.
(F) Imputed multi-mapping SPO11 oligos at the Chr1 hotspot pair (Lange et al., 2016). A SPO11-oligo cluster is evident at ~ 500 bp from the left hotspot center (red asterisk), which is not observed in the unique SPO11-oligo map (Figure 3F). Duplication/circle breakpoints and smaller deletion breakpoints, shown in G, frequently map at and close to this SPO11-oligo cluster.
(G) Deletions detected within the Chr1 left hotspot in Atm−/− (14 events) and Atm+/− (2 events). Nested PCR was carried out with primers flanking the hotspot (same primers used in the control PCR in B), with genomic DNA digested with NsiI; eight of 16 deletion products contained the restriction site, indicating incomplete digest. Note that the right breakpoints map to similar positions as duplication/circle breakpoints, and the left breakpoints map to similar positions to the left deletion breakpoints that are more spread out. This supports the idea that multiple double cuts (E) can yield distinct breakpoint profiles for duplications/circles and deletions (Figure 3F).
Figure S7. Extended analysis of microdeletions at a single hotspot, related to Figure 7.
(A–C) Evaluation of reads with microdeletions from deep sequencing. A, Frequency of reads that had microdeletions of the indicated lengths. Four overlapping amplicons across the hotspot (1–4) were deep sequenced using MiSeq. For each amplicon, 576 separate PCRs were carried out with a total input of 9.6 million haploid genome equivalents, using a limited number of amplification cycles (see Methods). The PCR products for each amplicon were then pooled for sequencing. Four MiSeq runs were performed (I–IV) with the indicated amplicons and genotypes. For clarity, sequencing results are grouped by run (Top) or genotype (Bottom). At top, note that reads with small deletions (2–4 bp) were frequently observed in all amplicons for each genotype, including Spo11−/−, and that their frequency varied between sequencing runs, indicating a significant batch effect. Short deletions (e.g., 2 bp) are known to arise from PCR and sequencing errors (Schirmer et al., 2015), and we saw that they became progressively less abundant in all genotypes up to 10 bp. Excluded from the 4-bp deletions is a product that was represented by >1000 reads in amplicons 1–3. By contrast, reads with deletions >10 bp were more frequent in Atm−/− than in the other genotypes. At bottom, note that reads with 11–25-bp microdeletions were more frequent in Atm−/−. Therefore, 11 bp was used as the lower cutoff to define a microdeletion for subsequent analyses. Our sequencing design (pooling of many low-cycle PCRs) is expected to minimize PCR jackpots and reduce instances where PCR duplicates from the same input DNA molecule are sequenced. However, two amplicons in Atm+/+ showed multiple reads for one microdeletion each (amplicon 1: 26-bp microdeletion, 14 reads; amplicon 2: 31-bp microdeletion, 22 reads; Table S7), which therefore may be PCR duplicates. Thus, in some subsequent analyses we applied a stringent definition of independent events by including only one read for a given deletion product from a given amplicon. B, Recurring microdeletions within the hotspot. Top, smoothed (21-bp Hann filter) and unsmoothed SPO11-oligo maps at the Chr1 hotspot in Atm−/−. Bottom, recurring microdeletions. These identical microdeletions (black bars) were found in independent PCRs for different amplicons. i) In Atm−/−, amplicons that yielded the recurring microdeletions are indicated. For example, the microdeletion with the AG microhomology arose in amplicons 3 and 4. Note that one or both breakpoints for each of the recurring microdeletions occurred at SPO11-oligo peaks (dashed vertical lines). ii) In Atm+/− and Atm+/+, some microdeletions were identical to those found in Atm−/−, in some cases in multiple amplicons. For example, the microdeletion with the GGAAGGAG microhomology was recovered in all three genotypes, but in different amplicons (Atm−/−, 1; Atm+/−, 2; Atm+/+, 4). iii) Some of the microdeletions found in Spo11−/− recurred in other genotypes. Note that these map to the same region of the hotspot away from the center. C, Many of the duplicated reads in Atm−/− amplicons likely represent independent microdeletion events rather than PCR duplicates. For each amplicon, we randomly sampled with replacement from a set of reads representing all of the unique microdeletions observed. The sampled group was equal in number to the duplicated reads actually obtained for that amplicon. Then, the sampled reads with microdeletions mapping to the region where amplicons 1, 2, and 3 overlap were combined, and their breakpoint distribution was compared with the distribution of the observed duplicate reads. Smoothed breakpoint profiles (21-bp Hann filter) of independently obtained reads, as in Figure 7C, and of all reads are also shown. In the breakpoint profiles of the observed duplicates, the left breakpoints were particularly enriched in a peak that matched the central peak of SPO11 oligos, and the right breakpoints were enriched at a position that matched the secondary peak of SPO11 oligos. These patterns were not recapitulated by random sampling: both the left and right breakpoints were more widely dispersed without such clear enrichment coincident with the SPO11-oligo peaks. Because the observed duplicates were more likely than the random sample to have breakpoints aligned with the strongly favored locations of DSBs, these findings are consistent with the conclusion that most of the duplicated reads represent independent microdeletion events.
(D) Arc diagrams of microdeletions in duplicated reads stratified by length from Atm−/−, as in Figure 7C. Microdeletions >160 bp are not shown, as their location may be more constrained by restricting analysis to the region where the amplicons overlap.
(E) Microdeletion length distribution for all amplicons by genotype for independent reads. Density plots are shown (>10 bp); densities in Atm+/−, Atm+/+, and Spo11−/− are normalized to Atm−/− to reflect read numbers in each genotype. As in the analysis of microdeletions in the overlap region of amplicons 1, 2, and 3 (Figure 7D), distinct size classes were observed in the presence and absence of ATM, but not in Spo11−/−. Larger microdeletion size classes (~140 bp, ~180 bp, ~230 bp) were well defined when all amplicons are considered. A smaller size class (~15 bp) was more prominent outside of the overlap region.
(F) Arc diagrams of the ~140-bp, ~180-bp, and ~230-bp microdeletions observed for Atm−/−.
(G) Microhomology lengths at microdeletion breakpoints in the presence and absence of ATM. Analyses were performed for independent microdeletions in the overlapping region between amplicons 1, 2, and 3.
Figure S6. Characteristics of insertions in Atm−/−, related to Figure 6.
(A) Derivation of inserts mapping to DSB hotspots/clusters on other chromosomes. The genomic locations of inserts (blue bars) are mapped relative to SPO11 oligos for Atm−/− and Atm+/+. We indicated whether these locations were called as hotspots in one or both genotypes or instead contain SPO11-oligo clusters. Inserts mapping to the non-PAR ChrY are shown in B. One insert mapping to a microsatellite repeat is not shown. See also Table S5.
(B) Inserts from the non-PAR ChrY map to multiple DSB hotspots. Three inserts identified in Atm−/− deletions map to non-PAR ChrY repeats (Table S5), some of which have called hotspots. SPO11-oligo profiles are shown only for those repeats containing hotspots in Atm−/−, Atm+/+, or both, with the position of the inserts (blue bars) for each hotspot indicated. Inserts 1 and 2 are part of the same repeat family, and in one case they map to a single hotspot. Inserts 1 and 3 map to three and two hotspots, respectively. SPO11 oligos map uniquely to these hotspots (“Unique”) or also map to other copies of the repeats (“Multi”). It remains possible that other repeat locations to which SPO11 oligos do not map uniquely also contain hotspots. Given that all insert-matching repeats (67 total) contain an intact PRDM9 motif, it is possible that DSBs are formed in many such repeats. The locations of multi-mapping SPO11 oligos were imputed, meaning they were distributed among mapped positions proportionally to local densities of uniquely mapping SPO11 oligos (Lange et al., 2016).
(C) Insert length distribution. Median insert lengths are indicated. See also Tables S5 and S6.
(D) No correlation (Pearson’s r) between insert and deletion lengths for inserts from other chromosomes. Note: Deletions of median size (1943 bp) with ~400 bp insertions should be readily detected (~1900 bp PCR), as should somewhat longer insertions.
(E) Deletions with inverted insertions at the Chr1 hotspot pair in B6 and B6 × DBA mice. Asterisks denote deletions where one insert breakpoint had the same microhomology as the site with recurring deletions (Figure 5B). Note that for the two deletions on the DBA chromosome, the inserts also derived from the DBA chromosome.
Data Availability Statement
Original data including gel pictures, Sanger sequencing, all data processing spreadsheets and R scripts are available from the lead contact upon request. Raw and processed amplicon deep sequencing data files are deposited at GEO. This paper also analyzes existing SPO11-oligo data that are publicly available. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Taq DNA polymerase | Thermo Fisher Scientific | Cat# EP0406 |
| Pfu Turbo DNA polymerase | Agilent | Cat# 600254 |
| Non-acetylated ultra-pure bovine serum albumin (BSA) | Invitrogen | Cat# AM2616 |
| dNTPs, PCR-grade | Roche | Cat# 3622614001 |
| S1 Nuclease | Thermo Fisher Scientific | Cat# EN0321 |
| Restriction enzymes (Listed in Method Details) | New England Biolabs | N/A |
| Exo V (RecBCD) | New England Biolabs | Cat# M0345S |
| Thermolabile ExoI | New England Biolabs | Cat# M0568S |
| Proteinase K, PCR-grade | Roche | Cat# 03115828001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Invitrogen PureLink Quick Gel Extraction Kit | Invitrogen | Cat# 210012 |
| PureLink PCR Purification Kit | Invitrogen | Cat# K310002 |
|
| ||
| Deposited data | ||
|
| ||
| Amplicon sequencing data | This paper | GEO:GSE182210 |
| SPO11-oligo data | Lange et al., 2016 | GEO:GSE84689 |
| Mouse genome assembly GRCm38/mm10 | N/A | http://genome.ucsc.edu |
| Mouse polymorphism database | N/A | https://www.sanger.ac.uk/data/mouse-genomes-project/ |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: B6.129S6-Atmtm1Awb/J | The Jackson Laboratory | RRID:IMSR_JAX:008536 |
| Mouse: DBA/2J mice for backcrossing Atm+/− mice | The Jackson Laboratory | RRID:IMSR_JAX:000671 |
| Mouse: Spo11−/− | Baudat et al., 2000 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers (Listed in Method Details) | Integrated DNA Technologies | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R (versions 3.4.4 to 3.6.3) | N/A | http://www.r-project.org |
| GraphPad Prism 8 | N/A | https://www.graphpad.com/scientific-software/prism/ |
| ApE | N/A | https://jorgensen.biology.utah.edu/wayned/ape/ |