

Abstract
Spike glycoprotein of SARS-CoV-2 is mainly responsible for the recognition and membrane fusion within the host and this protein has an ability to mutate. Hence, T cell and B cell epitopes were derived from the spike glycoprotein sequence of wild SARS-CoV-2. The proposed T cell and B cell epitopes were found to be antigenic and conserved in the sequence of SARS-CoV-2 mutant (B.1.1.7). Thus, the proposed epitopes are effective against SARS-CoV-2 and its B.1.1.7 mutant. MHC-I that best interacts with the proposed T cell epitopes were found, using immune epitope database. Molecular docking and molecular dynamic simulations were done for ensuring a good binding between the proposed MHC-I and T cell epitopes. The finally proposed T cell epitope was found to be antigenic, non-allergenic, non-toxic and stable. Further, the finally proposed B cell epitopes were also found to be antigenic. The population conservation analysis has ensured the presence of MHC-I molecule (respective to the finally proposed T cell) in human population of most affected countries with SARS-CoV-2. Thus the proposed T and B cell epitope could be effective in designing an epitope-based vaccine, which is effective on SARS-CoV-2 and its B.1.1.7mutant.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13721-021-00348-w.
Keywords: SARS-CoV-2, Epitope-based vaccine, T cell epitope, B cell epitope, Spike glycoprotein, Population coverage
Introduction
Coronaviruses belong to the family Coronaviridae. This family is divided into 4 genera, based on genomic structure and phylogenetic relationship as follows: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus (Cui et al. 2019). Betacoronavirus like Severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) infect humans. Some patients in December 2019 were found with pneumonia-like symptoms in Wuhan, China. The reason for infection was found to be a beta coronavirus. This novel virus has never been reported before and thus world health organization (WHO) named it as severe acute respiratory syndrome coronavirus (SARS-CoV-2) (Hui et al. 2020; Zhu et al. 2020). World health organization (WHO) has officially given the name COVID19 for the infection caused by SARS-CoV-2 and declared it as a pandemic on 11 March 2020 (World Health Organization 2020a, b). Globally, SARS-CoV-2 infection has caused millions of deaths and mutation in the virus has made the situation uncontrollable. As of 30 July 2021, 197 million cases and 4 million deaths have been reported globally (Worldometer 2021).
SARS-CoV-2 is a betacoronavirus that has a positive-stranded ribonucleic acid (RNA) (Guo et al. 2020). Its 3′ end contains genome that codes for the following four structural proteins: spike (S) protein, nucleocapsid (N) protein, membrane (M) protein and envelope (E) protein (Phan 2020). N protein codes for the RNA genome of SARS-CoV-2. S, M and E protein is present in the viral envelop of SARS-CoV-2. S protein is divided into two subunits. S1 subunit contains receptor binding domain, which is important for binding with the host receptor and S2 subunit is important for membrane fusion (Babcock et al. 2004). Coronavirus infection is initiated by the interaction of S protein and the host cellular membrane (Schoeman and Fielding 2019). S protein is a very important protein, because it is responsible for receptor binding, membrane fusion, internalization of the virus and initiating the infection inside the host. The genomic sequence of SARS-CoV-2 was first released by the Chinese scientists (Chen et al. 2020).
In a recent study, it has been reported that patients with B.1.1.7 mutation of SARS-CoV-2 get more severely infected, as compared to patients infected with SARS-CoV-2 (Giles et al. 2021). Higher transmission rate of SARS-CoV-2 B.1.1.7 mutant has been reported in 94 countries (Collier et al. 2021). It was first identified in United Kingdom (UK). This mutant contains a variation in S protein, which is mainly responsible for host recognition and its fusion (Planas et al. 2021).
Vaccine development plays an important role in elimination of the spreading virus (Patra et al. 2020). Historically, vaccination has saved lives of large human population from many pathogenic viruses. As compared to the traditional vaccination methods like live, killed and attenuated vaccines, epitope-based vaccine provide a much more rational method of vaccination. The reason could be the presence of specific component, derived from the same pathogen that could elicit an immune response inside the host (Twiddy et al. 2003).Vaccination is the most effective way to prevent this disease, but mutations in SARS-CoV-2 could make these currently available vaccines ineffective. Further, most of the vaccine against SARS-CoV-2 utilize S protein for developing an immunogenic response in the host, but this S proteins are found to be mutated in B.1.1.7 SARS-CoV-2 mutants (Shen et al. 2021). Hence, mutation in SARS-CoV-2 virus genome could be a challenge for available drugs and vaccines. So, it becomes important to design a vaccine, which is effective againstSARS-CoV-2 mutant. The proposed T and B cell epitopes of this study could be helpful for designing of epitope-based vaccine against SARS-CoV-2 and its mutant (B.1.1.7).
Methods
The following flowchart describes the method as seen in Fig. 1.
Fig. 1.

Shows the flowchart of the work done
S glycoprotein sequence retrieval
S glycoprotein FASTA sequences of wild SARS-CoV-2 (PDB ID: 6XM4 and 6VXX) and B.1.1.7 SARS-CoV-2 mutant (PDB ID: 7LWS and 7LVS) were retrieved from Protein data bank Research Collaboratory for Structural Bioinformatics (PDB RCSB).
T cell epitope prediction
NetCTL1.2 (Larsen et al. 2007) found T cell epitopes, which were derived from the S glycoprotein sequence of wild SARS-CoV-2. These T cell epitopes are effective in eliciting an immune response in the host. This prediction method includes Major histocompatibility complex-I (MHC-I) binding prediction, along with transporter associated with antigen processing (TAP) transport efficiency and proteasomal C-terminal (CT) cleavage prediction. This type of epitope prediction is done taking into consideration of 12 MHC supertypes. MHC class I binding and proteasomal CT cleavage is done by using artificial neural network (ANN) method, whereas TAP transport efficiency is detected by using weight matrix. The following parameters were used in the analysis: threshold—0.5 (for maintain the specificity—0.94 and sensitivity—0.89), Supertype A1, Weight on CT and TAP transport efficiencies of 0.15 and 0.05, respectively.
Further, the immune epitope database (IEDB) analysis tool (Buus et al. 2003) predicted MHC-I, which have a chance to bind with the T cell epitopes. MHC-I molecule binding was calculated using Stabilized matrix-based method (SMM). Allele selection was done and the lengths of the epitopes were set to be 9.0, for proceeding with the binding analysis. This tool finally produced scores for the proteosomal processing, MHC-I binding, TAP transport and an overall score, which indicate the peptides intrinsic potential of a T cell epitopes. Similarly, T cell epitopes and their respective MHC-II molecules were derived from the IEDB analysis tool.
Physiochemical analysis of T cell epitopes
T cell epitope predictions that are effective in eliciting an immune response and are safe for the host, can save a lot of wet lab efforts. The following are the physiochemical properties that must be present for an effective, safe and stable T cell epitope: antigenicity, non-allergenicity, non-toxicity and stability. Based on these properties, 4 T cell epitopes were selected from the 23 T cell epitopes selected from the IEDB.
Antigenicity determination of T cell epitopes
Antigenicity determination predicts the epitopes’ capabilityfor eliciting an immune response inside the host. VaxiJen v2.0 (Doytchinova and Flower 2007) predicts the protective antigens and vaccine subunits. This tool uses a novel alignment-free method of antigen prediction. Most of the other antigen prediction software uses alignment-based protein sequence, but the problem with such methods is two proteins with a dissimilar sequence may have same structure and respective function. This method of antigen prediction allows classification of antigen based on the physicochemical properties of the protein.
Allergenicity determination of T cell epitopes
Allergenicity determination predicts the epitopes capability of producing any kind of allergic reactions or hypersensitivity. AllerTOP v. 2.0 (Dimitrov et al. 2014) defines whether epitope can be allergen or not. This tool uses auto cross covariance transformation method, which is applied for structure–activity relationship study of peptides.
Toxicity determination of the T cell epitopes
ToxinPred online server (Gupta et al. 2013) predicts the toxicity of the epitope. Toxicity prediction is done in order to check any kind of cross-reactivity or tolerance of the epitope inside the host. In this Swiss-Prot based (SVM) prediction method was used with an E-value cut-off value of 10 for the motif-based method. SVM threshold was set as 0.0.
Stability prediction of the T cell epitopes
ProtParam online server (Garg et al. 2016) computes the physical and chemical parameters that are stored in the SWISS-Prot or TrEMBL, like the stability of the epitope. ProtParam tool computes molecular weight, pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY). Here, we were mainly interested in computing the stability, which was revealed by instability index generated by ProtParam tool (Gasteiger et al. 2005).
Population coverage analysis of T cell epitopes
Population coverage analysis was done to check the presence of the MHC-I molecule that binds to the proposed T cell epitopes in the human population of the SARS-CoV-2 affected countries. Because if these MHC-I molecules are present in the human population of the affected countries, then only they will bind to the T cell epitope for eliciting an immune response in the host against SARS-CoV-2. IDEB analysis tool was used for this population coverage analysis.
3D structure modeling of MHC-I and T cell epitopes
As the 3D structure of the T cell epitopes and the MHC-I were not present in any database, they were 3D modeled. The T cell epitopes and MHC-I 3D structures were predicted and modeled using PEP-FOLDD v3.5 (Maupetit et al. 2009) and SWISS-MODELonline server (Waterhouse et al. 2018), respectively.
Molecular docking analysis of T cell epitopes
Molecular docking analysis of T cell epitopes were done in order to check the binding affinities between the chosen T cell epitopes and their corresponding MHC-I molecules that were derived from the immune databases. Autodock flexible receptor (ADFR) v4.2 tool (Ravindranath et al. 2015) was used to do molecular docking for knowing the receptor- ligand interactions and binding affinities.
Molecular dynamics simulation of T cell epitope and MHC-I complex
To mimic the physiological state of the T cell epitope and MHC-I complex, which has the highest affinity for each other were selected on the basis of docking results. This was done to check the atomic stability of the complex as a whole, using GROMACS v 2018.1 (Abraham et al. 2015) was used for doing molecular dynamic simulation. The simulation was run for 50 ns. The whole system containing the complex was made electrostatistically neutral by adding counter ions. The complex was solvated within TIP3P water box and minimized using steepest descent method. Then, the complex was equilibrated with constant pressure and temperature (NPT) and steady volume and temperature (NVT). Then, to evaluate the structural and binding feature of the complex, root mean square deviation (RMSD) and root mean square fluctuations (RMSF) were calculated.
Presence of T and B cell epitope in SARS-CoV-2 and its B.1.1.7 mutant
Multiple sequence alignment was done using ClustalW (Larkin et al. 2007) for finding the presence of proposed T and B cell epitopes in S glycoprotein sequence of SARS-CoV-2 (PDB ID: 6XM4 and 6VXX) and its B.1.1.7 mutant (PDB ID: 7LWS and 7LVS).
B cell epitope prediction
B cell epitopes interact with the B lymphocyte for eliciting immune response (Nair et al. 2002). Hydrophilicity, surface exposed, turns, accessibility, flexibility, antigenic propensity and polarity are the parameters of a protein that could be linked with the presence of continuous epitopes. These result in evolution of methods that use some predefined empirical rules, following which some protein sequence features can predict continuous epitopes. These predictions are based on propensity scale, which assigns a value to every amino acid. IEDB tool was used for identifying B cell epitopes and its antigenicity using different predefined methods. These B cell epitopes were derived from the S glycoprotein of wild SARS-CoV-2.
Results
T cell epitope prediction
T cells destroy the virus-infected cells if virus has already infected the host (Ledford 2021).This previous infection could be the result of virus causing infection or vaccination (Palm and Henry 2019). T cell present inside the host recognizes epitope, which are a part of viral protein to fight infection, effectively (da Silva and Hughes 1998). Proteasome complex cleaves the peptide bonds and converts these proteins into small peptides. These small peptide molecules get associated with class-I MHC molecules and are further presented to T helper cells. These helper T cells are responsible for initiating the production of antibodies and killer T cells, which finally destroys virus infected cells (Ledford 2021).
The binding predictions of MHC-I and processing were done from IDEB tool that generates proteasomal CT processing, TAP transport, MHC-I and processing score. The overall score predicts the peptides potential to be a T cell epitope as shown in Table S1. From the S glycoprotein region of wild SARS-CoV-2, potential T cell epitopes were predicted using NetCTL server in a preselected environment. 23 T cell epitopes were found and used for the further analysis as shown in Table S2. MHC-I binding predictions resulted in a range of MHC-I alleles that interact with selected T cell epitopes. The MHC-I that were having highest binding affinity were chosen for further analysis. Further, T cell epitope prediction along with the MHC-II molecules was done using IEDB database analysis tool. This can be observed from table S5.
Physiochemical analysis of T cell epitopes
For an epitope to be effective and safe for the host, it must be antigenic, non-allergenic, non-toxic and stable. On the basis of these parameters four epitopes were selected for further analysis (LDSKVGGNY, SKVGGNYNY, NDLCFTNVY and GQTGKIADY), as shown in Table S3.
Population coverage analysis of T cell epitopes
The population coverage analysis of the proposed epitopes is shown in Table S4. T cell recognizes a complex of MHC and pathogen-derived epitope. This means that the particular MHC needs to be present in the individual so that it can binds to a particular T cell pathogen-derived epitope for eliciting an immune response. This is known as MHC restriction of T cell response. As MHC molecules are polymorphic and different human leukocyte antigen (HLA) alleles are present in human population. If peptides are selected that binds with HLA with a high affinity and that HLA is present in target human population, then epitope-based vaccine could be more effective. Therefore, careful considerations must be taken care so that the vaccine is not ethnically biased. For the issue discussed above, IEDB population coverage analysis helps in calculating the fraction of individuals that contains the predicted MHC.
3D structure prediction of T cell epitopes
3D structure modeling of T cell epitopes was done using homology modeling approach using SWISS MODEL web server that is fully automated and freely available. Further, MHC-I were modeled using a de-novo approach, which predicts 3D structure of protein from its linear amino acid sequence.
Molecular docking analysis of T cell epitopes with their respective MHC-I molecules
ADFR tool was used to find the binding affinities between the T cell epitope and MHC-I, which are having a lower value of IC50 (as predicted from NetCTL server). GQTGKIADY T cell epitope binds with HLA-C*03:03 most strongly as described in Table 1, because it is having the highest binding affinity. Figure 2 shows the complex of GQTGKIADY T cell epitope and HLA-C*03:03.
Table 1.
Shows the binding affinities between the 4 T cell epitopes and the MHC-I that have lowest value of IC50
| S. no | T cell epitope | MHC-I (having the lowest value of IC50) | Binding affinity (kcal/mol) | Status |
|---|---|---|---|---|
| 1 | LDSKVGGNY | HLA-C*12:03 | − 0.3 | Rejected |
| 2 | SKVGGNYNY | HLA-B*15:02 | 0.7 | Rejected |
| 3 | NDLCFTNVY | HLA-C*12:03 | 3.5 | Rejected |
| 4 | GQTGKIADY | HLA-C*03:03 | − 0.7 | Accepted |
Fig. 2.
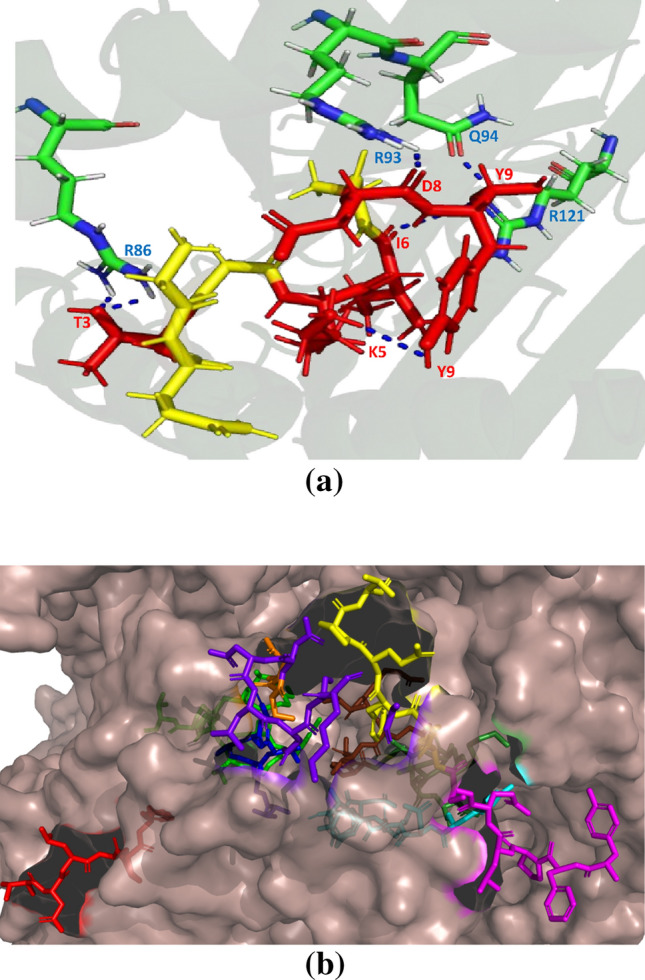
Representation of a hydrogen bond interaction between GQTGKIADT (red colour shows the interacting residues and yellow colour shows the non-interacting residues of the T cell epitope) and HLA-C*03:03. b Position of T cell epitope GQTGKIADT (forest green) and B cell epitope TNLCPFG (red), TFKCYGVSPT (green), TGCVIA (blue), CYFPLQSY (magentas), ADYNYKLPDD (cyan), NSNNLD (purple blue), YGFQPT (yellow) and VRQIAPGTGKID (chocolate red) on spike protein of SARS-CoV-2 (PDB ID: 6VXX)
R86, R93, Q94 and R121 interact with GQTGKIADT T cell epitope on S protein of SARS-CoV-2. These hydrogen bonding (H-bonding) interactions show the reason for the stability of GQTGKIADT and HLA-C*03:03 complex. This can be observed from Fig. 2a. Further, Fig. 2b shows that GQTGKIADT T cell epitope is present on the surface of S protein and hence accessible to MHC-I molecule.
Molecular dynamic (MD) simulation of T cell epitope and MHC-I complex
MD simulations were applied for the modeled structure of the finally selected T cell epitope (GQTGKIADT) and their respective MHC-I (HLA-C*03:03) complex. The selection was made by considering the docking score. This was done in order to understand the stability and dynamics of the complex for 50 ns in GROMACS. The system was solvated in TIP3P salvation box and CHARMM36 all atom force field.
Root mean square deviation (RMSD) was done in order to calculate the average distance of the backbone C-alpha (Cα) atom of the superimposed frames. RMSD quantifies the structural stability of a protein complex (Khan et al. 2021). An initial change can be observed between 0 and 10 ns. After that, another change is observed between 30 and 40 ns, after which the system gets quite stable. As observed from the Fig. 3a, the complex of T cell epitope GQTGKIADT and HLA-C*03:03 is stable after 40 ns. Next, Root mean square fluctuation (RMSF) was applied to the system trajectories as observed in Fig. 3b. RMSF calculates the average residual mobility of complex residues from its mean position. Minor fluctuations can be observed, which mean that the complex is stable, except between 30 and 40 ns.
Fig. 3.
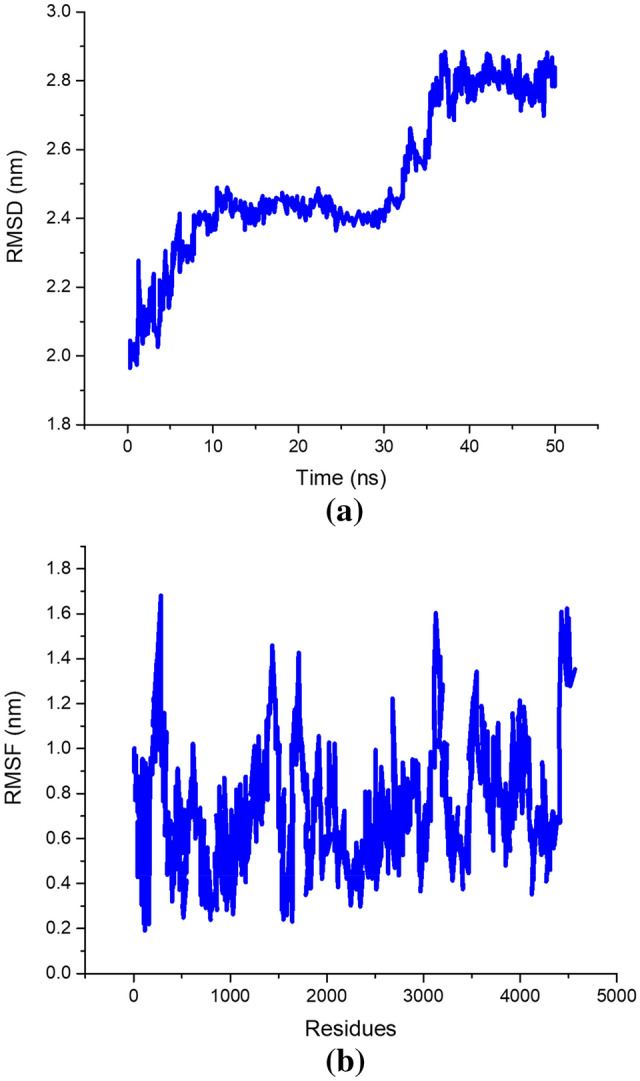
a RMSD and b RMSF plot of T cell epitope GQTGKIADT and HLA-C*03:03 complex
Presence of T cell epitope in SARS-CoV-2 mutant
Further, the presence of T cell epitope GQTGKIADT was also found in SARS-CoV-2 B.1.1.7 mutant, as shown in Fig. 4. This means that the proposed T cell epitope GQTGKIADT can be used in designing an epitope-based vaccine against SARS-CoV-2 and its mutant (B.1.1.7).
Fig. 4.

T cell epitope conserved in spike glycoprotein of SARS-CoV-2 (PDB ID: 6VXX and 6XM4) and its mutant (PDB ID: 7LWS and 7LWV)
B cell epitope identification
When an antigenic molecule encounters a B cell, it proliferates into antibody-producing effector cells. Further, these effector cells produce antibodies, which are helpful in fighting against infection (Alberts et al. 2002). The epitopes are the part of antigen that are responsible for evoking the immunogenic response in the host cell. A different analysis method was used from IEDB tool to identify B cell epitope. This tool uses amino acid scale-based method.
Prediction method of Kolaskar and Tongaonkar (KT) antigenicity
The determination of antigenicity was as per the physicochemical properties of the amino acids. The average antigenic propensity of the S glycoprotein from wild SARS-CoV-2 was 1.043, with a maximum value (1.214) and minimum value (0.907). The antigenic determination threshold was 1.043 (> 1.00 are potential antigenic determinants). The resultant six epitopes were satisfying the threshold and so they have the ability for expressing B cell response. Results are summarized and shown in Fig. 5 and Table 2.
Fig. 5.

Kolaskar and Tongaonkar antigenicity analysis of the conserved region of spike glycoprotein of SARS-CoV-2
Table 2.
B cell epitopes that have the potential to elicit an immune response, as predicted from Kolaskar and Tongaonkar antigenicity analysis of the spike glycoprotein of wild SARS-CoV-2
| S. no | Start | End | Peptide | Length | Antigenic score |
|---|---|---|---|---|---|
| 1 | 4 | 10 | TNLCPFG | 7 | 1.1812 |
| 2 | 47 | 56 | TFKCYGVSPT | 10 | 1.5059 |
| 3 | 101 | 106 | TGCVIA | 6 | 0.4716 |
| 4 | 159 | 166 | CYFPLQSY | 8 | 0.9394 |
| 5 | 176 | 198 | YQPYRVVVLSFELLHAPATVCGP | 23 | 0.4697 |
Prediction method of Emini surface accessibility
For being a potential B cell epitope, it must be accessible to the surface. Therefore, this method is used to predict the peptide surface accessibility. The average value of peptide antigenic propensity was 1.00, with a maximum value (4.805) and minimum value (0.073). The antigenic determination threshold was 1.00. The region between 90 and 100 amino acid residues was found to be more accessible in the conserved S glycoprotein of SARS-CoV-2 as shown in Fig. 6 and Table 3.
Fig. 6.

Emini surface accessibility prediction of the spike glycoprotein derived from wild SARS-CoV-2
Table 3.
B cell epitopes that are accessible at the surface, as predicted from Emini surface accessibility analysis of the spike glycoprotein of wild SARS-CoV-2
| S. no | Start | End | Peptide | Length | Antigenic score |
|---|---|---|---|---|---|
| 1 | 90 | 99 | ADYNYKLPDD | 10 | 0.6956 |
| 2 | 108 | 113 | NSNNLD | 6 | 1.1859 |
| 3 | 126 | 139 | LFRKSNLKP FERDI | 14 | 0.3610 |
| 4 | 165 | 171 | YGFQPT | 6 | 1.6231 |
Prediction method of Karplus and Schulz (KS) flexibility
Experimentally, the antigenicity is correlated with its peptide flexibility (Rose et al. 1985). Therefore, this method was implemented to investigate the flexibility of the peptide. The average value of peptide antigenic propensity has been found to be 0.989, with a maximum value (1.112) and minimum value (0.896). The antigenic determination threshold was found to be 0.989. The region from 80 to 88 amino-acid was found to be the most flexible as shown in Fig. 7.
Fig. 7.

Karplus and Schulz flexibility prediction of the spike glycoprotein derived from wild SARS-CoV-2
Prediction method of Bepipred linear epitope
This method uses hiddenmarkov model (HMM) method (Most suitable method for linearB cell epitope prediction). The average antigenic propensity of the peptide was 0.075, with a maximum value (1.896) and minimum value (0.021). The antigenic determination threshold was 0.350. Peptide sequence from 165 to 178 is capable of induction of the desired immune response from the B cellepitope. The result is shown in Fig. 8 and Table 4.
Fig. 8.

Bepipred linear epitope prediction of the spike glycoprotein derived from wild SARS-CoV-2
Table 4.
B cell epitopes that are capable of producing a desired immune response, as predicted from Bepipred linear epitope analysis of the spike glycoprotein of wild SARS-CoV-2
| S. no | Start | End | Peptide | Length | Antigenic score |
|---|---|---|---|---|---|
| 1 | 78 | 91 | VRQIAPGQTGKIAD | 14 | 1.2606 |
| 2 | 110 | 118 | NNLDSKVGG | 9 | 0.8904 |
| 3 | 144 | 154 | YQAGSTPCNGV | 11 | 0.0881 |
| 4 | 166 | 177 | YGFQPTNGVGYQ | 12 | 0.7136 |
Prediction method of Chow and Fasman (CF) beta-turn
Often, the beta turns are hydrophilic and accessible. These two properties are of the antigenic region of a protein (Marshall 2004) and so this method was used. The average value of antigenic propensity for peptide was 1.044, with a maximum value (1.397) and minimum value (0.694). The antigenic determination threshold was 1.044. The region of the peptide from 141 to 179 was considered as a beta turn region as shown in Fig. 9.
Fig. 9.

Chou and Fasman beta-turn prediction of the spike glycoprotein derived from wild SARS-CoV-2
Presence of B cell epitope in SARS-CoV-2 mutant
TNLCPFG, TFKCYGVSPT, TGCVIA, CYFPLQSY, ADYNKLPDD, NSNNLD, YGFQPT, VRQIAPGQTGKIAD, NNLDSKVGG and YQAGSTPCNGV are B cell epitopes that are effective against SARS-CoV-2 and its mutant (B.1.1.7). Conserved B cell epitopes TNLCPFG, TFKCYGVSPT, TGCVIA and CYFPLQSY are shown in Fig. 10a, ADYNKLPDD, NSNNLD and YGFQPT are shown in Fig. 10b, VRQIAPGQTGKIAD, NNLDSKVGG and YQAGSTPCNGV are shown in Fig. 10c.
Fig. 10.

B cell epitope conserved in spike glycoprotein of SARS-CoV-2 (PDB ID: 6VXX and 6XM4) and its mutant (PDB ID: 7LWS and 7LWV) derived from a Kolaskar and Tongaonkar antigenicity analysis. b Emini surface accessibility analysis. c Bepipred linear epitope analysis
Discussion
Advancement in computational biology and sequence-based technology has led to development of huge database, which could be used in the treatment of infection. Therefore, an effort has been made in this paper to find a novel T cell epitope and some B cell epitopes, which show antigenic response against SARS-CoV-2 and its mutant B.1.1.7.This study is an in silico-based study and the data have been extracted from the various immune databases, but such a type of study has previously been validated with wet-lab results (Shrestha and Diamond 2004). So, the proposed B and T cell epitope could also be effective in eliciting an immune response against SARS-CoV-2 and its mutant (B.1.1.7).
SARS-CoV-2 is a virus containing RNA as a genetic material and so gets mutated more frequently (Manzin et al. 1998). The genome size is 29,700 bases. This genome contains 14 open reading frames (ORFs) and 4 structural protein S, E, M and N. Interaction between SARS-CoV-2 S protein and ACE (angiotensin-converting enzyme) results in internalization and fusion within the host (Joshi et al. 2020a, b).This initiates the multiplication of SARS-CoV-2 within the host cells and causes respiratory damages (Joshi et al. 2020a, b). S protein can induce a faster and long-lived mucosal immune response, as compared to the other proteins of the virus (Khan et al. 2014). Hence S glycoprotein could be a suitable target for combating SARS-CoV-2 infection. But mutations mostly occur at the outer membrane S glycoprotein and increase the sustainability of the virus by escaping the humoral and cell-mediated immune response (Ma et al. 2014).
SARS-CoV-2 infection is spreading very fast, resulting in deaths of millions and economic losses due to lockdown (Khan et al. 2021).Vaccines are one of the most suited ways of combating such infections. But mutation in the virus could make these vaccines ineffective (Akhtar et al. 2020). So, one should proceed with designing of vaccines, which are effective against both wild and mutated species of the virus. In this direction, we have derived 23 T cell epitopes from the S glycoprotein of wild SARS-CoV-2. MHC-I that best interacts with these 23 T cell epitopes have been found. Further, from the 23 T cell epitopes 4 T cell epitopes were found to be antigenic, non-allergenic, non-toxic and stable. Molecular docking studies were done in order to ensure a good binding of the MHC-I molecule with their respective T cell epitopes. Finally, it was revealed that T cell epitope (GQTGKIADY) binds with HLA-C*03:03, with a high binding affinity and is found to be stable in molecular dynamic simulations after 40 ns. The population conservation analysis has ensured the presence of HLA-C*03:03 in the human population of the most affected countries with SARS-CoV-2. Further, some T cell epitope and its antigenic scores were also calculated along with their respective MHC-II molecules.
Finally, a novel T cell epitope GQTGKIADY was found that is antigenic, non-allergenic, non-toxic and stable. It binds with C*03:03 with good affinity and is found in the human population of countries affected by SARS-CoV-2 like China, India, Russia, United kingdom, United states and Italy. Further, it was found that GQTGKIADY is conserved in S glycoprotein of wild and B.1.1.7 mutant of SARS-CoV-2. Hence, GQTGKIADY is a novel epitope, which could be utilized for development of epitope-based vaccine for SARS-CoV-2 and its B.1.1.7 mutant. Further, some B cell epitopes were found to be antigenic and conserved in SARS-CoV-2 and its B.1.1.7 mutant like TNLCPFG, TFKCYGVSPT, TGCVIA, CYFPLQSY, ADYNKLPDD, NSNNLD, YGFQPT, VRQIAPGQTGKIAD, NNLDSKVGG and YQAGSTPCNGV. These epitopes can also help in developing immunogenic response against SARS-CoV-2 and its B.1.1.7 mutant. This can lead to development of epitope-based vaccine, which is effective against both wild and B.1.1.7 mutated SARS-CoV-2.
Conclusion
A novel T cell epitope GQTGKIADY was found in this study, which is antigenic, non-allergen, non-toxic and stable. GQTGKIADY epitope binds well with HLA-C*03:03, as revealed by molecular docking and molecular dynamic studies. HLA-C*03:03 was found in the human population of China, India, Russia, United kingdom, United states and Italy, and hence its effective binding with GQTGKIADY epitope could result in eliciting good immunogenic response in human population of these countries. Further, some B cell epitopes were found to be antigenic. These T and B cell epitopes were derived from wild SARS-CoV-2 and found to be conserved in B.1.1.7 mutant of SARS-CoV-2. These novel T and B cell epitopes can be used for designing of epitope-based vaccine against SARS-CoV-2 and its B.1.1.7 mutant. The limitation of this work is that the proposed epitopes are not effective against all the SARS-CoV-2 mutants.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are thankful to Indian Institute of Information Technology, Allahabad for providing necessary facilities and infrastructure, required for the completion of the work.
Author contributions
All authors contribute equally.
Funding
No funding.
Availability of data
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Code availability
Not applicable.
Declarations
Conflict of interest
There is no conflict of interest between the authors.
Ethical approval
Not applicable.
Consent for publication
All the authors have read and approved the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vidhu Agarwal, Email: mail2vidhu.alld@gmail.com.
Akhilesh Tiwari, Email: atiwari@iiita.ac.in.
Pritish Varadwaj, Email: pritish@iiita.ac.in.
References
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Akhtar N, Joshi A, Singh B, Kaushik V. Immuno-informatics quest against COVID-19/SARS-COV-2: determining putative T-cell epitopes for vaccine prediction. IDDT. 2020 doi: 10.2174/1871526520666200921154149. [DOI] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J et al (2002) Molecular biology of the cell, 4th edition. Garland Science. New York. https://www.ncbi.nlm.nih.gov/books/NBK26884/
- Babcock GJ, Esshaki DJ, Thomas WD, Ambrosino DM. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. JVI. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buus S, Lauemøller SL, Worning P, et al. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62:378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- Chen H-Z, Tang L-L, Yu X-L, et al. Bioinformatics analysis of epitope-based vaccine design against the novel SARS-CoV-2. Infect Dis Poverty. 2020;9:88. doi: 10.1186/s40249-020-00713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier DA, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature. 2021;593:136–141. doi: 10.1038/s41586-021-03412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva J, Hughes AL. Conservation of cytotoxic T lymphocyte (CTL) epitopes as a host strategy to constrain parasite adaptation: evidence from the nef gene of human immunodeficiency virus 1 (HIV-1) Mol Biol Evol. 1998;15:1259–1268. doi: 10.1093/oxfordjournals.molbev.a025854. [DOI] [PubMed] [Google Scholar]
- Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP vol 2—a server for in silico prediction of allergens. J Mol Model. 2014;20:2278. doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg VK, Avashthi H, Tiwari A, et al. MFPPI—multi FASTA ProtParam interface. Bioinformation. 2016;12:74–77. doi: 10.6026/97320630012074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, et al., editors. The proteomics protocols handbook. Totowa: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Giles B, Meredith P, Robson S, et al. The SARS-CoV-2 B.1.1.7 variant and increased clinical severity—the jury is out. Lancet Infect Dis. 2021 doi: 10.1016/S1473-3099(21)00356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Military Med Res. 2020;7:11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8:e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui DS, Azhar IE, Madani TA, et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Sunil Krishnan G, Kaushik V. Molecular docking and simulation investigation: effect of beta-sesquiphellandrene with ionic integration on SARS-CoV2 and SFTS viruses. J Genet Eng Biotechnol. 2020;18:78. doi: 10.1186/s43141-020-00095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Joshi BC, Mannan MA, Kaushik V. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Info Med Unlocked. 2020;19:100338. doi: 10.1016/j.imu.2020.100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MK, Zaman S, Chakraborty S, et al. In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol Immunol. 2014;61:16–22. doi: 10.1016/j.molimm.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Khan RJ, Jha RK, Amera GM, et al. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J Biomol Struc Dynam. 2021;39:2679–2692. doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H. How ‘killer’ T cells could boost COVID immunity in face of new variants. Nature. 2021;590:374–375. doi: 10.1038/d41586-021-00367-7. [DOI] [PubMed] [Google Scholar]
- Ma C, Li Y, Wang L, et al. Intranasal vaccination with recombinant receptor-binding domain of MERS-CoV spike protein induces much stronger local mucosal immune responses than subcutaneous immunization: implication for designing novel mucosal MERS vaccines. Vaccine. 2014;32:2100–2108. doi: 10.1016/j.vaccine.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzin A, Solforosi L, Petrelli E, et al. Evolution of hypervariable region 1 of hepatitis C virus in primary infection. J Virol. 1998;72:6271–6276. doi: 10.1128/JVI.72.7.6271-6276.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SJ. Developing countries face double burden of disease. Bull World Health Organ. 2004;82:556. [PMC free article] [PubMed] [Google Scholar]
- Maupetit J, Derreumaux P, Tuffery P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucl Acids Res. 2009;37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair DT, Singh K, Siddiqui Z, et al. Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J Immunol. 2002;168:2371–2382. doi: 10.4049/jimmunol.168.5.2371. [DOI] [PubMed] [Google Scholar]
- Palm A-KE, Henry C. Remembrance of things past: long-term B cell memory after infection and vaccination. Front Immunol. 2019;10:1787. doi: 10.3389/fimmu.2019.01787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra P, Mondal N, Patra BC, Bhattacharya M. Epitope-based vaccine designing of nocardia asteroides targeting the virulence factor MCE-family protein by immunoinformatics approach. Int J Pept Res Ther. 2020;26:1165–1176. doi: 10.1007/s10989-019-09921-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. Novel coronavirus: from discovery to clinical diagnostics. Inf Genet Evol. 2020;79:104211. doi: 10.1016/j.meegid.2020.104211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D, Bruel T, Grzelak L, et al. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat Med. 2021 doi: 10.1038/s41591-021-01318-5. [DOI] [PubMed] [Google Scholar]
- Ravindranath PA, Forli S, Goodsell DS, et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol. 2015;11:e1004586. doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Glerasch LM, Smith JA (1985) Turns in peptides and proteins. In: Advances in protein chemistry. Elsevier, Hoboken, pp 1–109 [DOI] [PubMed]
- Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Tang H, McDanal C, et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe. 2021;29:529–539.e3. doi: 10.1016/j.chom.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Role of CD8+ T cells in control of west nile virus infection. JVI. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiddy SS, Holmes EC, Rambaut A. Inferring the rate and time-scale of dengue virus evolution. Mol Biol Evol. 2003;20:122–129. doi: 10.1093/molbev/msg010. [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucl Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2020a) Naming the coronavirus disease (COVID-19) and the virus that causes it. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it. Accessed 28 Mar 2020
- World Health Organization (2020b) Statement on the second meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV). https://www.who.int/news-room/detail/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov). Accessed 28 Mar 2020
- Worldometer (2021) COVID-19 coronavirus pandemic. https://www.worldometers.info/coronavirus. Accessed 30 Jul 2021
- Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Not applicable.