

Abstract
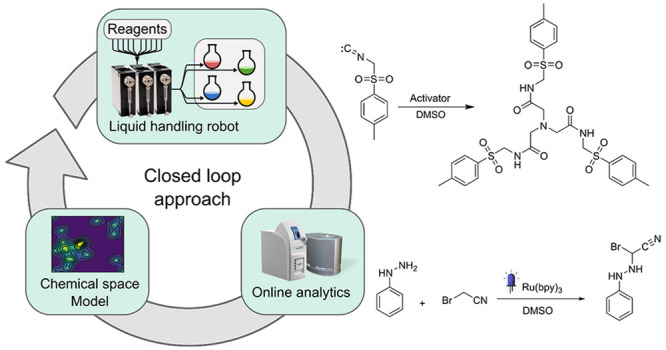
We present a robotic chemical discovery system capable of navigating a chemical space based on a learned general association between molecular structures and reactivity, while incorporating a neural network model that can process data from online analytics and assess reactivity without knowing the identity of the reagents. Working in conjunction with this learned knowledge, our robotic platform is able to autonomously explore a large number of potential reactions and assess the reactivity of mixtures, including unknown chemical spaces, regardless of the identity of the starting materials. Through the system, we identified a range of chemical reactions and products, some of which were well-known, some new but predictable from known pathways, and some unpredictable reactions that yielded new molecules. The validation of the system was done within a budget of 15 inputs combined in 1018 reactions, further analysis of which allowed us to discover not only a new photochemical reaction but also a new reactivity mode for a well-known reagent (p-toluenesulfonylmethyl isocyanide, TosMIC). This involved the reaction of 6 equiv of TosMIC in a “multistep, single-substrate” cascade reaction yielding a trimeric product in high yield (47% unoptimized) with the formation of five new C–C bonds involving sp–sp2 and sp–sp3 carbon centers. An analysis reveals that this transformation is intrinsically unpredictable, demonstrating the possibility of a reactivity-first robotic discovery of unknown reaction methodologies without requiring human input.
Short abstract
A robotic discovery system driven by machine learning is able to navigate a chemical space and successfully discover two unpredictable reactions.
Introduction
Many discoveries in the chemistry laboratory are the result of chance observations, and it is hard to know ahead of time where a new reaction or molecule will be found.1 We can explore chemical space mathematically using rule-based generation methods2 or by mapping chemical reaction databases,3,4 but much of the search is done through traditional approaches using cheminformatics,5,6 artificial intelligence,7−10 or computation.11,12 Reactivity-first approaches13,14 have only recently been tentatively explored, but the vast majority of organic synthesis is target-oriented,15 which means that the discovery of new reactions is a chance event or results from the need to access a new transformation.16 The development of new transformations and methodologies17 however is a complex problem requiring a high degree of expert knowledge.18,19 Furthermore, the current approaches to reaction or method discovery are generally constrained to known heuristics, and the discovery of novel reactions is rare. The search for unexpected results can be accelerated with automated systems, and in the past decade high-throughput experimentation20 has shown its potential in speeding up reaction preparation and analysis (typically applied in reaction optimization and combinatorial chemistry).21−23 However, an increase of reaction throughput does not automatically lead to the serendipitous discovery of entirely new transformations while, on the other hand, the discovery of new reaction pathways from first principles (i.e., in silico, based on quantum mechanics) is hard due to both the combinatorial explosion of possible reaction pathways and the computational cost of accurate modeling of the energy hypersurface. To overcome these limitations, an increasing number of approaches are starting to involve a feedback loop from the online analytics and a decision-making algorithm to perform only a fraction of the possible combinations, considered interesting.22 In such a “closed-loop”24−27 approach, the system automatically explores a chemical space in a trial-and-error fashion mimicking a human experimenter. The system requires three main parts: a chemical robot to perform and analyze the reactions, a program for interpretation of analytical data, and an algorithm that correlates the outcome of the reaction with the input and process parameters. This last part closes the loop by suggesting the predicted optimal parameters for the next reactions (Figure 1).
Figure 1.
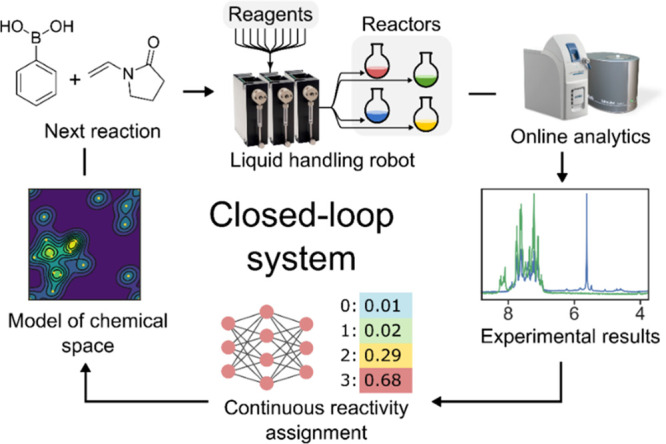
Closed-loop framework for chemical space exploration. A liquid handling robot performs an experiment and collects NMR and MS spectra. These data are processed to assess reactivity and create a model of the chemical space that is queried to formulate the next experiment to be performed.
Although closed-loop approaches have proved effective in reaction optimization, their application toward the discovery of new reactions remains underexplored. This is because assessing the reactivity of an unknown reaction with unpredictable products is harder than using metrics meant for optimization of a known target compound, such as yield or selectivity. For autonomous reactivity-first discovery to become feasible, both the analytical method as well as reactivity detection algorithm need to be general-purpose and robust. Proton NMR spectroscopy is applicable to a wide range of samples, and inexpensive benchtop instruments amenable to online analysis are available. Still, there are no general-purpose automated algorithms to detect reactivity solely relying on the NMR spectra of the starting materials and that of the reaction mixture. Many reactions are known to lead to subtle changes in the spectrum. In contrast, many trivial phenomena, e.g., proton exchange, can manifest as in visually distinct spectra, requiring an expert to discern whether or not something significant has occurred. In response to this challenge, we devised a convolutional neural network called Reactify that can automatically assess the reactivity of NMR spectra that it has never seen before, having captured the expertise of the human chemist during training. While binary reactive/nonreactive classification of reactions has led to progress,14,28 we hypothesized that a continuous measure would allow the system to abstract the notion of reactivity, rather than restrict it to a given fixed set of reagents. The Reactify network’s ability to assign reactivity values to “unseen” reaction mixtures was then coupled with a closed-loop system aimed at exploring the reactivity of an experimental space. This system comprises a liquid handling platform to prepare and analyze the reactions, Reactify to assign reactivities to the resulting spectral data, and a structural reactivity prediction model to generalize the observed reactivities to the unexplored parts of the chemical space.
Results and Discussion
The Chemical Robot
To emulate a human chemist, experiments were performed in conventional round-bottom flasks that were automatically cleaned after each reaction by flushing them with clean solvent. Starting materials were stored as 1 M stock solutions in dimethyl sulfoxide (DMSO), and the platform used 30 syringe pumps with integrated valves to mix them into six parallel reactors at room temperature. The chemical space was expanded with the addition of either a Lewis acid (24) or a base (23) in order to change the chemical environment. As a further expansion, three of the reactors were also equipped with visible-light light emitting diodes (LEDs) to promote photochemical reactions. During the exploration of chemical space 2 (Figure 3c), the reactions performed in these reactors were prepared by adding 2.5 mol % of a known photocatalyst, associated with the LED wavelength: 2,4,6-triphenylpyrylium tetrafluoroborate (PC1, 405 nm), tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate (PC2, 450 nm), and rose bengal (PC3, 565 nm). After 3 h, the mixtures were analyzed automatically with a benchtop NMR and MS. The software managed the preparation and analysis of the reactions. It was designed to run them in parallel by shifting each experiment starting time to efficiently share the online analytics and cleaning cycles. These physical operations (analysis–cleaning–reaction preparation) lasted around 40 min; hence, having 6 parallel reactors, the individual reaction time is calculated as (6 × 40) – 40 = 200 min. Through this optimized schedule, it was therefore possible to perform up to 36 reactions per day, each with a reaction time of 3.3 h giving a total of over 100 reaction hours per day (Figure 2).
Figure 3.

CNN assessment of reactivity in two different chemical spaces. (a) Structure of the neural network used to assign the reactivity to the NMR spectra. Data of the mixture and the sum of starting materials are used as input. The network is trained using 440 reactions from a chemical space (b) and tested on 1018 reactions performed from combinations of 15 different molecules (c). (d) The accuracy on the test set plotted as a confusion matrix shows that the network successfully learned to generalize the reactivity beyond reagents in the training set. (e) All data were manually classified into four classes.
Figure 2.

Liquid handling platform. (a) Schematic of the platform. A series of reagents are added by dedicated pumps to a mixer flask. A pump expanded with two extra valves (obtained by removing the syringe from a normal pump) is used to transfer the reaction mixture in one of the six reactors1−6 (red); another pump with the same setup (blue) is used to connect the reactors with the benchtop NMR. Finally, a third expanded pump is used for an in-line dilution prior to injection in the MS (green). (b) Picture of the platform. The pumps are visible on the shelves, on two lines. At the bottom, there is the NMR instrument equipped with a flow probe. The MS is on the left and the reactors in the center, while the reagents, the solvent drum, and the waste container are on the left. (c) Six parallel reactions were started with a time-offset to allow the platform to continuously perform physical operations.
The Reactify Neural Network
Initially, we randomly sampled a chemical space made of six simple molecules (chemical space 1, Figure 3b) mixed in binary and tertiary random combinations. The reaction parameters involved different reagent ratios, different temperatures, and the presence of a base (DBN, 7) yielding 440 reactions. We used the 1H NMR spectra collected during this exercise to train a convolutional neural network (CNN, Figure 3a) called Reactify that mimics the reactivity assignments made by a human experimenter. To do so, the reactions were manually scored by an expert organic chemist using four classes of reactivity (0, nonreactive; 3, very reactive) describing the difference between the reaction mixture and the superimposed 1H NMR spectra of the starting materials. High values were assigned to mixtures with several new peaks and the disappearance of the starting material signals. Experiments showing little or chemically insignificant spectral changes were assigned a low reactivity class (Figure 3e).
The Reactify reactivity assignment model was designed purposely without any information about the chemical structures of the materials and trained to detect reactive reactions by correlating the raw spectroscopic data to the values assigned by a chemist. The network was built using a combination of convolutional and dense layers, taking as input a pair of NMR spectra corresponding to the mixture before and after the reaction, the former estimated by superimposing the spectra of the starting materials. The output was designed to vary continuously between 0 and 1 and trained using the reactivity classes described above normalized to 1 (0 = 0, 1 = 0.33, 2 = 0.66, 3 = 1). Since the four classes are not evenly distributed in the chemical space—unreactive combinations (reactivity = 0) make up 45% of the space—we implemented a weighted loss function based on the relative abundance of each class in order to emphasize the correct prediction of the less abundant reactive examples. Following the training on data from 440 initial reactions (chemical space 1), the model’s performance was evaluated on 1018 reactions between 15 different starting materials (chemical space 2, Figure 3c). The results are shown in Figure 3d, where the confusion matrix compares the class assignments made by the neural network (predicted) versus manual assignment (true values). The network predicted the correct reactivity class 56% of the time with the top-2 accuracy of 89% (the random baseline being 25% and 50% for the top-2). The encouraging results for an unseen set of molecules suggest that the CNN was capable of generalizing a notion of reactivity in NMR spectra independent of the reagents used.
Algorithm for the Exploration of the Chemical Space
To close the loop and drive the exploration of the chemical space, we needed a representation of chemical space that could accommodate many different classes of molecules and correlate the presence of various structural motifs with the observed reactivity. To this end, we used the junction tree variational autoencoder algorithm29 to translate the molecular structure of the reagents into fixed-length fingerprint vectors. The autoencoder combines a tree-structured scaffold generated over chemical substructures with a graph message passing network. The resulting 56-dimensional vectors are then used as input to the reactivity estimation neural network we developed (Figure 4a). In order to keep the model consistent with binary and ternary combinations in the case of a two-component reaction, a vector of 56 zeroes was submitted as the third vector. The idea behind the algorithm is to train the model on a small fraction of the chemical space, explored at random, and use the knowledge acquired to predict the reactivity of the remaining possible combinations. The reactant combinations would then be sorted by predicted reactivity and the best candidates reacted in the platform. After each reaction the model is retrained using the newly obtained reactivity information. By guiding the robot with such a reactivity-driven algorithm it will be possible to perform the reactive combinations first, meaning that only a fraction of the chemical space will need to be explored. Furthermore, thanks to the structural fingerprint, the encoding network is able to abstract the reactivity from the identity of the molecules involved. It is therefore possible to update and use the model on any organic reaction involving three reagents, meaning that this method is easily scalable to vast chemical spaces with a large number of starting materials. We imagine that by training the model on bigger data sets the scope of the predictions will also expand.
Figure 4.
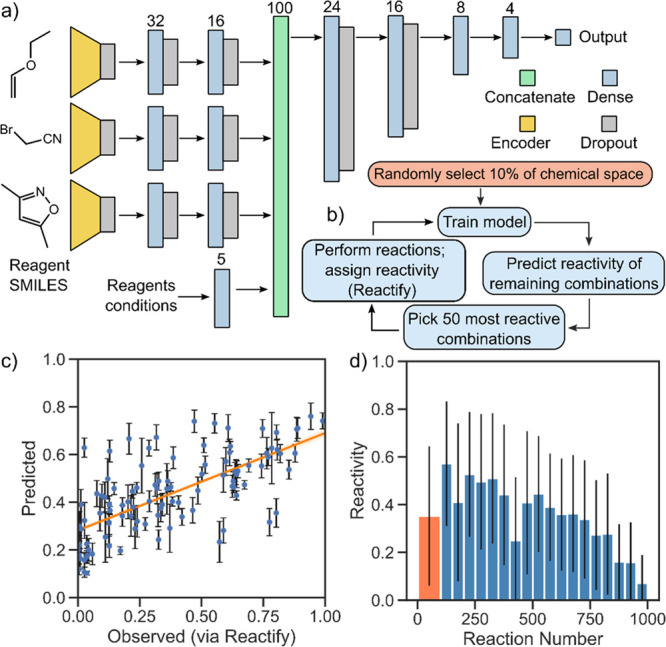
Aspects of the autonomous chemical space exploration algorithm. (a) Structure of the neural network used for reactivity prediction. A junction-tree neural network encodes the molecular structure of each reactant into a 56 dimensional vector. (b) Scheme of the algorithm used to simulate the chemical space exploration. (c) Correlation of predicted versus observed reactivity (assigned via Reactify) for the test set. The correlation is demonstrated by fitting a linear regression model with the shaded area representing the 99% confidence interval obtained using bootstrap. Prediction uncertainties (calculated as standard deviations) are shown within error bars (see Section S3.1 for connection between uncertainty and error lower bound). (d) Results of the chemical space exploration simulation. After the initial selection of 100 random reactions (orange), the algorithm starts to create a model that correlates parameters to reactivity. By prioritizing combinations that are predicted to be reactive, the space is explored in a more efficient way. The error bars show standard deviation.
We validated the reactivity estimation neural network on the data acquired from the 1018 random combinations of chemical space 2 and tested the network’s ability to predict the reactivity of the reagent combinations. To do so, NMR spectra resulting from these combinations were first assessed for reactivity using the Reactify convolutional neural network. We then trained the reactivity prediction neural network to connect the reagent structural fingerprints to the reactivity outcome, using 90% of these reactivity assignments for training. The results (Figure 4c) show a mean squared error in the prediction of 0.035 for the test set (reactivity values normalized to 1, see the SI for alternative metrics and training scenarios).
In a different experiment aimed at simulating exploration, we trained the model on a random batch comprising 10% of the data set (ca. 100 reactions) and predicted the reactivity values of the remaining ca. 900 combinations. We then simulated performing the most reactive combinations by revealing the outcome of 50 combinations predicted to have the highest reactivity. The model was retrained on the expanded data set of 150 reactions, and the process was repeated until all of the 1018 reactions were explored. The results of the simulation are shown in Figure 4d. The initial random batch of experiments had an average reactivity of 0.40 ± 0.29. Following training, the first generation of 50 reactions suggested by the model gave an average reactivity of 0.60 ± 0.23. Over time, the algorithm is trained on more data, but at the same time, the reactive combinations are taken out of the data set, leaving only the unreactive ones, as evidenced by the decline in reactivity of subsequent generations (Figure 4d). This simulated exploration experiment has also been repeated 100 times with different initial states in order to measure the efficiency of the algorithm in finding the unknown reactions presented in the following section. The reactions reported in Figure 5d,e were found, respectively, after 2.6 and after 6.0 iterations out of 19 (random baseline would be 9.5 iterations).
Figure 5.

Five reactions showing high reactivity have been found and characterized. Reactions a and b have been previously reported in the literature30−32 with the exact same product. Reaction c is known in the literature but has never been used to make 27.32 Reactions d and e are unreported in the literature.
Discovery of a Multistep-One-Substrate Reaction Cascade
Based on the data obtained from the benchtop instruments, a small selection of five combinations were randomly selected from a pool of reactions showing high reactivity. These five reactions were repeated in the platform and the products manually isolated (Figure 5). Two of these are known molecules already reported in the literature30−32 and one a new product for which the generic procedure is known,33 and the last two are “novel” as they lead to unknown molecules through unknown procedures, making them genuine discoveries. Reaction a is a C–H functionalization made through photoredox catalysis. It has already been described in 201230 where the authors used the same building blocks, [Ir(ppy)2(dtbbpy)]PF6 as photocatalyst, 2 equiv of Na2HPO4, and acetonitrile as the solvent. Reaction b has been reported as a method for the synthesis of N-aryl-C,C-dimethoxycarbonylnitrones31 and originally involved sodium hydroxide and THF as the solvent. The formation of molecule 26 following reaction b is known.31 Reaction c is a hydroamidation33 promoted by diethyl Br-malonate, and the product is unreported in the literature.
Reaction d is a photochemical reaction involving the addition of phenylhydrazine and bromoacetonitrile in the presence of tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate and 450 nm irradiation. In this reaction, a new C–N bond is formed while the bromide, usually a leaving group, is kept in its place. It is unreported in the literature. Reaction e was discovered during the analysis of the mixture of p-toluenesulfonylmethyl isocyanide (TosMIC) and diethyl bromomalonate. An X-ray analysis of the isolated product confirmed that the trimeric product 29 was formed. The molecular structure of the product showed an unusual increase in complexity and a nontrivial mechanism of formation taking in consideration the three central methylene carbons. Interestingly, the XRD showed a tubular supramolecular assembly composed of six molecules packed as a ring and multiple rings stacked together leaving void space with an average diameter of 12 Å (Figure 6a) (details in Section S6).
Figure 6.

Reaction of diethyl bromomalonate and TosMIC discovered with the automated platform. (a) General scheme of the reaction. 6 equiv of isocyanide is consumed in the presence of an activator, water, and DMSO 29; on the right-hand side are the X-ray structure of 29 and its tube-shaped supramolecular structure. (b) Analogous products obtained with variations of the isocyanide. (c) The reaction has been carried out using variations of diethyl bromomalonate, yielding the same product. (d) By performing the reaction in the presence of an amine, we observed variations of the product, suggesting the mechanism reported in the next figure.
To explore this transformation further, we decided to perform the reaction with a range of isocyanides to elucidate a possible mechanism. From the seven isocyanides (Section S3.6) tested, a similar product was isolated in the reaction with the 1H-benzotriazol-1-ylmethyl isocyanide 30 while traces of the reaction with (trimethylsilyl)methyl isocyanide 31 were detected by LC-MS (Figure 6b). Variations of diethyl bromomalonate have also been explored, finding working alternatives in several similar molecules including trifluoroacetic acid (TFA, 38) (Figure 6c). All of these variations yielded the same product, suggesting that the second reagent is not directly involved in the product formation but rather acts as some kind of a promoter; this is because the reaction does not give the product in any detectable amount in the absence of it. In order to confirm the involvement of the hypothesized intermediates presented in the mechanism below, the reaction has also been carried out in the presence of various amines (Figure 6d), and the presence of the relative products 39 and 43 has been established by LC-MS in all cases. These correspond to an asymmetric version of the product 29, where one or two branches have been replaced with the amine R-group. The possibility of tuning the branches in this way gave us precious information about the mechanism and increased the flexibility and the possible applications of this reaction.
To understand the formation of the core of the molecule, we prepared two isotopically enriched versions of the TosMIC substrate, labeling the isocyanide carbon and the methylene carbon. To confirm the source of the three oxygen atoms found in the product, we performed the reaction in both synthesized 18O–DMSO and anhydrous DMSO with small amounts of H218O. These experiments revealed that all of the three central methylene carbons come from the CH2 carbons of the TosMIC, while the oxygen atoms come from DMSO (Figure 7b). This means that the product is obtained using at least 6 equiv of isocyanide and that DMSO is also taking part in the reaction; in fact, the reaction has been attempted in DMF and MeCN without success (Section S4.4). While testing the reaction in different conditions, we also noticed its poor reproducibility, with significant yield fluctuations even under apparently identical conditions. Upon further investigation, it was found that the amount of trace water present in the solvent has a marked effect on the reaction profile. By testing different amounts of water, we found that the reaction does not yield any product under strictly anhydrous conditions and in the presence of more than 2 equiv of water. The best yields were obtained with 0.4 equiv (Section S4.3). The reaction kinetics have been investigated with on-line HPLC, showing the formation of an intermediate after 2 h that eventually disappears after 34 h with the simultaneous formation of product 29 (Section S4.2). An MS analysis of the corresponding peak showed a mass consistent with compound 55: the two-branch imine analogous to the product 29. The chromatogram also showed the presence of high amounts of the single-branched amine 52. This was in accordance with the data reported in Figure 7d and supported the hypothesis of a mechanism involving the formation of a central amine group that undergoes two identical semireactions to build the other two branches. Given this information, we propose the mechanism reported in Figure 7a. The role of diethyl bromomalonate (and the other activators) is to promote the oxidation of the isocyanide group to isocyanate.34 The formation of the central methylene carbons can be explained with the formation of amine 50 and following elimination of the sulfonate to yield imine 51.3551 is then reduced36 to form the single-branched amine 52 and attacked by it to form 53, which undergoes an elimination of ammonia followed by a reduction to produce 55.37
Figure 7.

Reaction mechanism and cheminformatic analysis. (a) Scheme of the proposed mechanism. Two of the intermediates have been found by HPLC-MS analysis while the isocyanate group, CO2, DMS, and ammonium have been detected at IR and NMR, respectively. (b) The isotopic labeling of carbon atoms C1 and C2 of TosMIC and the DMSO oxygen helped determine the source of core atoms in the product molecule. (c) Comparison of the structural change between reagents and products among known reactions of TosMIC (red line indicates discovered reaction). (d) Estimation of reaction unpredictability by estimating the size of the relevant reaction network. A full simulation could only be carried out for the first eight steps of the simulation as the combinatorial explosion produces a vastly greater number of molecules than could be feasibly analyzed.
The mechanism is then repeated with the consumption of another equivalent of 51 to yield the final product 29. In addition, this mechanistic hypothesis is supported by an on-line IR experiment showing the presence of an isocyanate group (47 and 49) and CO2 as well as the observation of dimethyl sulfide and ammonium signals in the NMR analysis of the mixture (Sections S4.6 and S4.7). To compare this new reaction with known transformations in the literature, we compiled a set of 1656 known reactions found in the Reaxys database38 involving TosMIC. The RDkit39 python library was used to extract a fingerprint difference between starting materials and products, indicating the extent to which each reaction transforms the structure of its input reagents.
To compare fingerprint differences among reactions, the l2-norm of the fingerprint-difference vectors was calculated. Low values of the l2-norm indicate a high structural similarity between reagents and products while high values correspond to complex reactions involving several transformations. The results are shown in Figure 7c, where the l2-norm values of literature reactions are grouped into bins on a logarithmic x-axis. The l2-norm of the reaction discovered is indicated with the red line, signaling an unusual degree of structural change compared to known reactions. To gauge the serendipitous nature of our discovery quantitatively, we carried out a simulation to assess the size of the chemical reaction network. This network was generated by repeatedly applying a set of common reaction templates, including the ones invoked in our proposed mechanism, to generate an expanding pool of chemicals. Comparing the total size of the resulting network to the subset leading to the product gives an indication of the unpredictability for the particular product obtained. The results are shown in Figure 7d, where blue lines indicate the reaction network relevant to product formation. The overall network is vastly larger in size than the subset potentially leading to a product (1010 chemicals vs 105 chemicals), indicating that the observed pathway is highly improbable to predict a priori.
Conclusions
In summary, we showed that closed-loop approaches combining automatic reaction execution and reactivity assessment using machine learning can play a crucial role in the discovery of novel reactions in unexplored parts of chemical space. Our neural network model can abstract the reactivity from the identity of the reagents, and we expect that this type of algorithm will also progressively improve in accuracy when presented with reactivity data for subsequent chemical spaces. The continuous reactivities provided by the CNN are correlated with reagent structural features, showing that it is possible to explore chemical space intelligently and discover unpredictable reactions, thanks to the unbiased nature of our system. Our results demonstrate the possibility of NMR-driven universal reactivity detection as a key enabler of autonomous discovery in a closed loop. Within this framework, we also show the potential of reactivity-first chemical space search and its suitability to the discovery of novel molecules and mechanisms.
Materials and Methods
General Experimental Remarks
Chemicals and solvents were supplied by Fisher Chemicals, Sigma-Aldrich, Lancaster Chemicals Ltd., and Tokyo chemicals industry, used as received. Deuterated solvents were obtained from Goss Scientific Instruments Ltd. and Cambridge Isotope Laboratories Inc. All commercial starting materials were used as supplied, without further purification. Off-line NMR data were recorded on a Bruker Advance 600 MHz or a Bruker Advance 400 MHz instrument, in deuterated solvent, at T = 298 K, using TMS as the scale reference. Chemical shifts are reported using the δ-scale, referenced to the residual solvent protons in the deuterated solvent for 1H and 13C NMR (i.e., 1H, δ (CDCl3) = 7.26; 13C, δ (CDCl3) = 77.16). All chemical shifts are given in ppm, and all coupling constants (J) are given in Hz (J) as absolute values. Characterization of spin multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublet, dt = double triplet, dq = double quartet, and ddt = double doublet of triplets. Chromatographic separation of the reaction mixture was achieved with a reverse phase column by Agilent (Poroshell 120 HPH C18, 3.0 × 100 mm, 2.7 μm) on a Thermo Fisher UltiMate 3000 HPLC instrument. The MS apparatus was a Bruker MaXis Impact instrument, acquisition range at 50–2000 m/z.
Liquid Handling Platform
The control over the fluids was performed using C3000 model TriContinent pumps (Tricontinent Ltd., Auburn, CA). They were equipped with distribution (3-way) and 90°/120° (2-way) valves. 5 mL syringes (TriContinent) were used for all functions except the pumps connected to the MS instrument and the photocatalysts which used a 0.5 mL syringe. The pumps were connected to the computer and each other by a daisy chain with an RS232 serial communication cable and DA-15 connectors. The liquid connectivity was assured using PTFE tubing (1/16” 1.6 mm OD × 0.8 mm ID) cut to the desired length and PEEK/PTFE flangeless fittings. To perform a reaction, the robot mixed 2 mL of the selected starting materials (from 1 M stock solutions) into the mixer flask and then moved the mixture into one of the six round-bottom flasks (25 mL). They were placed on top of two hot plates for magnetic stirring, and three of them were irradiated with a visible-light LED (Thorlabs). After 3 h, the mixture was analyzed, and the flask was washed with 5 mL of DMSO three times. The software to control the platform was written in Python and was optimized to continuously run six reactions in parallel.
Benchtop NMR Spectroscopy
The online NMR spectra were recorded using a Spinsolve benchtop NMR from Magritek (60 MHz). Shimming was performed before each experiment directly on the sample. The instrument was equipped with a flow cell to allow online analysis. The cell was designed to go through the instrument, and its location placed the thicker part (5 mm diameter) at the center of the magnets. Both the inlet and outlet were connected to normal PTFE tubing with screw caps (Figure S1). The flow cell allowed automatic reaction monitoring in real time by pumping 3 mL of solution from the reaction mixture. The instrument was controlled with Python through a TCP connection with the API exposed by Spinsolve software.
Benchtop MS Spectroscopy
The spectra were recorded using an Advion Expression CMS equipped with an ESI (electrospray ionization) module. The mass spectrometer was controlled using a Python library created to wrap around the binary libraries supplied by Advion. Before injection, the mixture was diluted by taking 0.1 mL of reaction mixture into the syringe and adding with 0.4 mL of acetonitrile. 0.4 mL of the diluted solution was pumped into waste and the process repeated five times to obtain a 10–4 M solution. After each injection, the instrument was cleaned by flushing it with acetonitrile and a water/acetonitrile 1:1 solution.
Automatic Reactivity Assignment
NMR data were checked manually, and a reactivity value was assigned between 0 and 3. The mixture spectrum was compared with the superimposition of the starting materials’ spectra, and the criteria for the assignment are the appearance of new peaks, their intensity, peaks shifting, and reagent peaks disappearing. Although there were borderline cases between two values, some general guidelines were followed: (a) absolutely no difference or a slight shift = 0, (b) one peak appearing or a big shift, medium intensities = 1, (c) two or three peaks appearing in high intensity = 2, and (d) more than three peaks appearing with a high intensity = 3. Before the training of the neural network, the NMR spectra were resampled to rescale them from 4878 to 271 points. They were then normalized to 1, and the solvent peak was removed by cutting the spectrum at 3 ppm. In order to avoid overfitting, a random scaling (y-axis) and shifting (x-axis) were applied on both the mixture spectrum and the reagent superimposition during training. A 2 × 271 matrix obtained by the processed spectra of the mixture and the superimposition of the starting materials was used as input for the neural network. Details about the neural network architecture can be found in Section S2.3. The network was trained on 440 reactions obtained from combinations of chemical space 1 (Figure 3b) and used to assess its accuracy on 1018 reactions from chemical space 2 (Figure 3c).
Reactivity Predictions and Automatic Space Exploration
The reactivity data assigned by the neural network were correlated with the starting materials represented as 56 dimensional fingerprint vectors. The fingerprints were calculated from the reagents SMILES using the encoding part of the junction tree autoencoder developed by Jin et al.29 The other reaction conditions (presence of acid, base, and photocatalyst) were fed into the model as a one-hot encoded vector. The software for training and testing the neural network was written using the Tensorflow library for Python. The network was used to run a simulation of the chemical space exploration where data from the full data set were progressively (in batches of 50 reactions) accessed following the reactivity predictions generated by the linear regressor. The simulation was implemented in Python.
General Procedure for Synthesis of Products 25, 26, 27, 28, and 29
Diethyl 2-bromomalonate (2 mmol, 0.41 mL), p-toluenesulfonylmethyl isocyanide (2 mmol, 0.39 g), and water (0.8 mmol, 15 μL) are mixed in 4 mL of anhydrous DMSO and stirred for 24 h at 30 °C. The reaction mixture is diluted with water (20:1) and extracted with ethyl acetate. The organic phase is separated and washed with brine. Mg2SO4 is then added to the reaction mixture, and after filtration, the solvent is removed under reduced pressure. Products 27 and 29 precipitated as white solids during the evaporation of ethyl acetate; they are isolated by filtration and washed with ethyl acetate. Products 25, 26, and 28 were purified with a chromatographic column.
Code availability
Online code repositories are provided for training and testing the Reactify neural network (https://github.com/croningp/Reactify); reactivity-first chemical space exploration, cheminformatic estimation of the novelty and predictability of the trimer 29 discovery (https://github.com/croningp/Rx1st); and the junction tree variational autoencoder for chemical structures (https://github.com/croningp/JTNN-VAE). The code is provided under the MIT license. Relevant data sets have been deposited online at https://zenodo.org/record/4670997.
Safety Statement
No unexpected or unusually high safety hazards were encountered in the work reported.
Acknowledgments
The authors gratefully acknowledge financial support from the EPSRC (Grants EP/S030603/1, EP/S019472/1, EP/S017046/1, EP/L015668/1, EP/L023652/1) and the ERC (project 670467 SMART-POM). J. M. Granda acknowledges financial support from the Polish Ministry of Science and Higher Education grant 1295/MOB/IV/2015/0. We would also like to thank Prof. Bartosz Grzybowski and his team for comments on the reaction mechanism involving TOSMIC.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00435.
Hardware specifications, Reactify neural network, reactivity prediction network, reaction discovered and variations, study of reaction mechanism, cheminformatics simulation, other reactions discovered and rediscovered, crystal structure details, and 1H and 13C NMR spectra (PDF)
Author Contributions
L. Cronin conceived the concept, the abstraction, algorithm, and the project and coordinated the efforts of the research team. D. Caramelli built and coded the platform with help from A. B. Henson, developed the algorithms for data analysis and manually characterized the reactions with help from J. M. Granda, and gathered data for the proposed mechanism with help from D. Cambie. S. H. M. Mehr and J. M. Granda did the cheminformatics analysis. L. Cronin, D. Caramelli, J. M. Granda, and S. H. M. Mehr wrote the manuscript with input from all of the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Grzybowski B. A.; Bishop K. J. M.; Kowalczyk B.; Wilmer C. E. The ‘wired’ universe of organic chemistry. Nat. Chem. 2009, 1, 31–36. 10.1038/nchem.136. [DOI] [PubMed] [Google Scholar]
- Oeschger R.; et al. Diverse functionalization of strong alkyl C – H bonds by undirected borylation. Science 2020, 368, 736–741. 10.1126/science.aba6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond J. L.; Ruddigkeit L.; Blum L.; van Deursen R. The enumeration of chemical space. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 717–733. 10.1002/wcms.1104. [DOI] [Google Scholar]
- Schwaller P.; Probst D.; Vaucher A. C.; et al. Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach Intell 2021, 3, 144–152. 10.1038/s42256-020-00284-w. [DOI] [Google Scholar]
- Herges R. Reaction planning: prediction of new organic reactions. J. Chem. Inf. Comput. Sci. 1990, 30, 377–383. 10.1021/ci00068a006. [DOI] [Google Scholar]
- Suleimanov Y. V.; Green W. H. Automated Discovery of Elementary Chemical Reaction Steps Using Freezing String and Berny Optimization Methods. J. Chem. Theory Comput. 2015, 11, 4248–4259. 10.1021/acs.jctc.5b00407. [DOI] [PubMed] [Google Scholar]
- Segler M. H. S.; Kogej T.; Tyrchan C.; Waller M. P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin I. I.; Madzhidov T. I.; Antipin I. S.; Varnek A. A. Artificial intelligence in synthetic chemistry: achievements and prospects. Russ. Chem. Rev. 2017, 86, 1127–1156. 10.1070/RCR4746. [DOI] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. Machine Learning in Computer-Aided Synthesis Planning. Acc. Chem. Res. 2018, 51, 1281–1289. 10.1021/acs.accounts.8b00087. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Vaucher A. C.; Laino T.; Reymond J. L. Prediction of chemical reaction yields using deep learning. Mach. Learn.: Sci. Technol. 2021, 2, 015016. 10.1088/2632-2153/abc81d. [DOI] [Google Scholar]
- Katsila T.; Spyroulias G. A.; Patrinos G. P.; Matsoukas M. T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. 10.1016/j.csbj.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor D. P.; et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 2018, 3, 5–20. 10.1038/s41578-018-0005-z. [DOI] [Google Scholar]
- Gromski P. S.; Henson A. B.; Granda J. M.; Cronin L. How to explore chemical space using algorithms and automation. Nat. Rev. Chem., 2019, 3, 119–128. 10.1038/s41570-018-0066-y. [DOI] [Google Scholar]
- Granda J. M.; Donina L.; Dragone V.; Long D. L.; Cronin L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 2018, 559 (7714), 377–381. 10.1038/s41586-018-0307-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke M. D.; Lalic G. Teaching target-oriented and diversity-oriented organic synthesis at Harvard University. Chem. Biol. 2002, 9, 535–541. 10.1016/S1074-5521(02)00143-6. [DOI] [PubMed] [Google Scholar]
- Mcnally A.; Prier C. K.; Macmillan D. W. C. Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334 (6059), 1114. 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben C.; Lapkin A. A. Automatic discovery and optimization of chemical processes. Curr. Opin. Chem. Eng. 2015, 9, 1. 10.1016/j.coche.2015.07.001. [DOI] [Google Scholar]
- Gajewska E. P.; et al. Algorithmic Discovery of Tactical Combinations for Advanced Organic Syntheses. Chem. 2020, 6, 280–293. 10.1016/j.chempr.2019.11.016. [DOI] [Google Scholar]
- Schwaller P.; et al. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. 10.1039/C9SC05704H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennen S. M.; et al. The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org. Process Res. Dev. 2019, 23, 1213–1242. 10.1021/acs.oprd.9b00140. [DOI] [Google Scholar]
- Poscharny K.; et al. Machine assisted reaction optimization: A self-optimizing reactor system for continuous-flow photochemical reactions. Tetrahedron 2018, 74, 3171–3175. 10.1016/j.tet.2018.04.019. [DOI] [Google Scholar]
- Häse F.; Roch L. M.; Kreisbeck C.; Aspuru-Guzik A. Phoenics: A Bayesian Optimizer for Chemistry. ACS Cent. Sci. 2018, 4, 1134–1145. 10.1021/acscentsci.8b00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery J. High-Throughput Discovery of New Chemical Reactions. Science 2011, 333, 1387–1389. 10.1126/science.1210735. [DOI] [PubMed] [Google Scholar]
- Perera D.; et al. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. 10.1126/science.aap9112. [DOI] [PubMed] [Google Scholar]
- Richmond C. J.; et al. A flow-system array for the discovery and scale up of inorganic clusters. Nat. Chem. 2012, 4, 1037–1043. 10.1038/nchem.1489. [DOI] [PubMed] [Google Scholar]
- Schweidtmann A. M.; et al. Machine learning meets continuous flow chemistry: Automated optimization towards the Pareto front of multiple objectives. Chem. Eng. J. 2018, 352, 277–282. 10.1016/j.cej.2018.07.031. [DOI] [Google Scholar]
- Cherkasov N.; Bai Y.; Exposito A. J.; Rebrov E. V. OpenFlowChem – a platform for quick, robust and flexible automation and self-optimization of flow chemistry, React. Chem. Eng. 2018, 3, 769–780. 10.1039/C8RE00046H. [DOI] [Google Scholar]
- Dragone V.; Sans V.; Henson A. B.; Granda J. M.; Cronin L. An autonomous organic reaction search engine for chemical reactivity. Nat. Commun. 2017, 8, 15733. 10.1038/ncomms15733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Barzilay R.; Jaakkola T.. Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv, 2019. https://arxiv.org/pdf/1802.04364.pdf (accessed March 30, 2021).
- Jiang H.; et al. Direct C-H functionalization of enamides and enecarbamates by using visible-light photoredox catalysis. Chem. - Eur. J. 2012, 18, 15158–15166. 10.1002/chem.201201716. [DOI] [PubMed] [Google Scholar]
- Tomioka Y.; Nagahiro C.; Nomura Y.; Maruoka H. Synthesis and 1,3-Dipolar Cycloaddition Reactions of N-Aryl-C,C-dimethoxycarbonylnitrones. J. Heterocycl. Chem. 2003, 40, 121–127. 10.1002/jhet.5570400116. [DOI] [Google Scholar]
- El Hassan I.; Lauricella R.; Tuccio B. Formation of β-fluorinated aminoxyl radicals from N-arylketonitrones. Mendeleev Commun. 2006, 16, 149–151. 10.1070/MC2006v016n03ABEH002308. [DOI] [Google Scholar]
- Ma W.; et al. Iron-Catalyzed Anti-Markovnikov Hydroamination and Hydroamidation of Allylic Alcohols. J. Am. Chem. Soc. 2019, 141, 13506–13515. 10.1021/jacs.9b05221. [DOI] [PubMed] [Google Scholar]
- Le H. V.; Ganem B. Trifluoroacetic anhydride-catalyzed oxidation of isonitriles by DMSO: A rapid, convenient synthesis of isocyanates. Org. Lett. 2011, 13 (10), 2584–2585. 10.1021/ol200695y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leusen A. M.; Wildeman J.; Oldenziel O. H. Base-Induced Cycloaddition of Sulfonylmethyl Isocyanides to C, N Double Bonds. Synthesis of 1, 5-Disubstituted and 1, 4, 5-Trisubstituted Imidazoles from Aldimines and Imidoyl Chlorides. J. Org. Chem. 1977, 42, 1153–1159. 10.1021/jo00427a012. [DOI] [Google Scholar]
- Gallant A. J.; Patrick B. O.; MacLachlan M. J. Mild and selective reduction of imines: Formation of an unsymmetrical macrocycle. J. Org. Chem. 2004, 69, 8739–8744. 10.1021/jo049197u. [DOI] [PubMed] [Google Scholar]
- Guérin C.; Bellosta V.; Guillamot G.; Cossy J. Mild nonepimerizing N -alkylation of amines by alcohols without transition metals. Org. Lett. 2011, 13 (13), 3534–3537. 10.1021/ol201351a. [DOI] [PubMed] [Google Scholar]
- Reaxys. https://new.reaxys.com/ (accessed March 30, 2021).
- RDKit. www.rdkit.org (accessed March 30, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.