

Abstract
The inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) by organophosphates (OPs) as nerve agents and pesticides compromises normal cholinergic nerve signal transduction in the peripheral and central nervous systems (CNS) leading to cholinergic crisis. The treatment comprises an antimuscarinic drug and an oxime reactivator of the inhibited enzyme. Oximes in use have quaternary nitrogens, and therefore poorly cross the brain–blood barrier. In this work, we synthesized novel uncharged thienostilbene oximes by the Wittig reaction, converted to aldehydes by Vilsmeier formylation, and transformed to the corresponding uncharged oximes in very high yields. Eight trans,anti- and trans,syn-isomers of oximes were tested as reactivators of nerve-agent-inhibited AChE and BChE. Four derivatives reactivated cyclosarin-inhibited BChE up to 70% in two hours of reactivation, and docking studies confirmed their productive interactions with the active site of cyclosarin-inhibited BChE. Based on the moderate binding affinity of both AChE and BChE for all selected oximes, and in silico evaluated ADME properties regarding lipophilicity and CNS activity, these compounds present a new class of oximes with the potential for further development of CNS-active therapeutics in OP poisoning.
Keywords: AChE, BChE, reactivation, heterostilbenes, spectroscopy, docking
1. Introduction
Oximes, unlike the symptomatic drugs currently used as inhibitors of acetylcholinesterase (AChE) in the treatment of neurological disorders such as Alzheimer’s and Parkinson’s disease, have an important role for the regeneration of the cholinergic system in cases of poisoning with organophosphate compounds (OPs). OPs present a large group of chemicals used as pesticides, flame retardants, etc., and misused as nerve agents. Their non-selective species toxicity can cause severe biological effects to humans [1,2]. The related enzyme butyrylcholinesterase (BChE) is also involved in the regulation of the cholinergic system as a backup to AChE in hydrolyzing the neurotransmitter acetylcholine (ACh) in neurological disorders [3]. BChE’s role also seems to be the protection of synaptic AChE because it reacts with a variety of drugs, xenobiotics as well as with OPs, but its inhibition is safe for human health [4,5]. Emergency treatment for AChE inhibited with OPs is based on oxime reactivators that regenerate the enzyme through the nucleophilic displacement of the OP moiety conjugated at the catalytic center serine. Oxime therapy comprises anticonvulsants and antimuscarinics to manage symptoms prompted by poisoning manifested as salivation, tremors, respiratory paralysis, and in severe exposures, death [6,7]. Unfortunately, the current standard therapy in OP poisoning is still based on a small number of quaternary pyridinium compounds, 2-PAM, obidoxime, or HI-6, which are not universal reactivators due to the structural variability of OPs and their structural impotence in passing the brain–blood barrier (BBB) [7,8,9,10].
To improve the central nervous system (CNS) activity of oximes, our recent research was directed towards oximes with increased lipophilicity, uncharged oximes, and zwitterionic oximes with a tertiary nitrogen atom [9,10]. Even though uncharged oximes were usually proven to be much better in BBB penetration due to increased lipophilicity [11,12], their downfall is the low affinity as a consequence of the absence of a positive charge for productive interactions with the OP-inhibited enzyme [13,14]. This is where the concept of a peripheral site ligand (PSL) was introduced into the oxime structure as compensation for the positively charged nitrogen in order to preserve, if not increase, the affinity for the enzyme [9,10,15,16]. The introduction of the PSL strategy, which involves the coupling of uncharged pyridine oximes with different PSLs via alkyl chains, has opened up many options for new oxime structures.
In this paper, we describe the synthesis of a new uncharged thienostilbene oxime whose design was based on our long-term experience in the preparation of heterostilbenes with thiophene and furan nuclei [17]. Moreover, a study on (thiophen-2-yl)-aldoximes reported that the sulfur atom, due to its potential for polarization, helps to stabilize the negative charge of its anionic form, similarly to the pyridinium ring of the standard oxime 2-PAM [18]. Therefore, we present here the preparation of a five-membered heterocyclic thiophene nucleus connected by a double bond to the benzene ring with para-position substitution that could be accommodated at the peripheral site and improve binding affinity. It is worth mentioning that the double-bond introduction between two (hetero)aryl groups eliminates the possibility of a pronounced conformational change. Nevertheless, a new oxime group was obtained by an efficient and cost-effective reaction pathway involving the Wittig reaction, Vilsmeier formylation, and the formation of two configurationally stable oxime isomers. We evaluated the kinetic interaction of each isomer with both AChE and BChE, together with their reactivation potential in sarin, cyclosarin, tabun, and VX poisoning. An in silico analysis of basic physicochemical characteristics was performed to estimate the ability of novel oximes in BBB crossing, as well as to assess their potential for further biological testing.
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterization of New Thienostilbene Oximes 13–18
The preparation of the desired thienostilbene oximes 13–18 was achieved according to the three-step reaction path shown in Scheme 1. It began by forming a double bond using the Wittig reaction to yield the starting compounds 1–6 as mixtures of cis- and trans-isomers (48–61%). In the second step, Vilsmeier formylation provided the corresponding mixtures of aldehydes 7–12 (24–47%) and in the third step, formyl derivatives were transformed to oximes 13–18 (57–88%) as the targeted structures. Mixtures of the geometrical isomers of oximes 13–18 were separated to obtain pure compounds by repeated-column and thin-layer chromatography.
Scheme 1.

The three-step reaction pathway for obtaining heterostilbene oxime derivatives 13–18. The yields are expressed for the third reaction step.
The Wittig reaction was performed using commercially available thiophene-2-carbaldehyde, and selected phosphonium salts prepared in our laboratory. The obtained thienostilbenes 1–6 represent the basic building blocks subjected to the Vilsmeier formylation reaction. In this reaction, POCl3 and DMF initially formed a chloriminium ion, a Vilsmeier reagent, which then reacted with the heterostilbene molecule, and an iminium ion was formed, which hydrolyzed to the final aldehydes 7–12. We noticed that the trans-isomers of 1–6 reacted more successfully in the formylation carried out for the partially unreacted cis-isomers, and thus the proportion of trans-isomers of aldehydes 7–12 was significantly higher (18–30%) in comparison to those for the cis-isomers (1–19%). The reaction of converting the synthesized aldehydes 7–12 into the corresponding oximes 13–18 involved the use of NH2OH × HCl and a mixture of ethanol and water as solvents and was based on the reaction described in the literature on simpler heteroaromatic systems [18]. According to 1H NMR spectroscopic analyses, trans-isomers are more present in the reaction mixtures of oxime derivatives 13–18 (Figure 1). In the case of oximes 13–15 and 18 possessing CH3, OCH3, and Cl groups as substituents, respectively, trans,anti-isomers dominate. Oximes 16 and 17 with electron-withdrawing substituents CN and NO2, respectively, have the most trans,syn-isomers in the reaction mixture, while in the case of the N(CH3)2 substituent, the ratio of syn- and anti-isomers was 2:1, with the presence of cis,syn-18 at the same abundance as that of trans,syn-18 (See Experimental Section) [19].
Figure 1.

Ratio of geometrical isomers of the synthesized thienostilbene oximes 13–18.
After successive column and thin-layer chromatography, the trans,anti- and trans, syn-isomers of the individual oxime derivatives 13–18 were isolated at a sufficient quantity to test them as potential cholinesterase reactivators (see Section 2.3). During the experiment, it was found that the anti-isomers of oximes are more polar and less soluble than those of syn-isomers, and due to the difference in their Rf values, they are relatively easily isolated. The similarity of the Rf values of the cis- and trans-isomers of the syn-oximes influenced the lower success of their separation. All of the isolated isomers of oxime 13–18 were fully spectroscopically characterized (See Figure 2 and Figure 3, Experimental Section and Supplementary Materials).
Figure 2.

Downfield region of the 1H NMR spectra (CDCl3) of the mixture of cis-3 and trans-3 (a), mixture of cis-9 and trans-9 (b), and oxime trans,syn-15 (c).
Figure 3.

Downfield region of the 1H NMR spectra (CDCl3+CD3OD) of the oximes trans,anti-13 (a), trans,anti-14 (b), and trans,anti-16 (c).
Additionally, from the reaction mixtures after the Wittig and Vilsmeier reaction, cis- and trans-isomers of thienostilbene 1–6 as well as the cis- and trans-isomers of the formylation products 7–12 were spectroscopically characterized and given for analysis by HRMS (See Experimental Section and Supplementary Materials).
2.2. In Silico Prediction of ADME Properties of Thienostilbene Oximes 13–18
The structure–property relationships [20,21] with predictive models for calculated compounds 13–18 were also analyzed by ACD/Percepta (ver. 14.2.0; Build 2977; ACD/Laboratories, Toronto, ON, Canada), which does not consider the geometrical isomerization. Physico-chemical properties were calculated, specifically the logP values, solubility, and permeability across biological membranes (Table 1). From a medicinal chemistry perspective, the design of drugs capable of penetrating the blood–brain barrier (BBB) and effecting the desired biological response is a formidable challenge. Moderately lipophilic drugs cross the BBB by passive diffusion, and the hydrogen bonding properties of drugs can significantly influence their CNS profiles. Polar molecules are generally poor CNS agents unless they undergo active transport across the CNS. Size, ionization properties, and molecular flexibility are other factors observed to influence the transport of an organic compound [22]. Lipophilicity is one of the most important physical properties that can affect the potency, distribution, and elimination of a drug in the body [23].
Table 1.
ADME properties of the oxime derivatives 13–18.
| Compound | logP a | Solubility b [mg/mL] | Permeability c [10−6 cm/s] |
PPB d | CNS e |
|---|---|---|---|---|---|
| 13 | 3.95 | 0.004 | 23.6 | 83% | −1.86 |
| 14 | 3.70 | 0.009 | 23.7 | 77% | −1.77 |
| 15 | 4.27 | 0.002 | 23.3 | 90% | −2.05 |
| 16 | 3.32 | 0.009 | 23.4 | 77% | −1.82 |
| 17 | 3.58 | 0.01 | 23.6 | 81% | −1.92 |
| 18 | 3.94 | 0.01 | 23.6 | 83% | −1.86 |
a lipophilicity logP (<−2.0 very hydrophilic, −2.0–(−1.0) hydrophilic, −1.0–4.2 optimal, 4.2–5.0 lipophilic); b solubility (<0.01 highly insoluble, 0.01–0.10 insoluble, >0.10 soluble); c Caco permeability (<1.0 × 10−6 poorly permeable, 1.0–7.0 × 10−6 moderately permeable, >7.0 × 10−6 highly permeable); d PPB—plasma-protein binding (10–40% weakly bound, 40–80% moderately, 80–90% strongly bound, >90% extensively bound); e CNS < −3.5 non-penetrant, −3.50–(−3.0) weak penetrant, >−3.0 penetrant); (ACD/Percepta (ver. 14.2.0; Build 2977; ACD/Laboratories, Toronto, ON, Canada).
In general, the solubility of selected oximes 13–18 was low. Nitro derivative 17 and dimethylamino derivative 18 showed the highest solubility (0.01 mg/mL). All of the compounds showed an optimal lipophilicity value, meaning they should readily penetrate the BBB, since the majority of CNS drugs have logP within the range of 3.5 to 4.5 (ACD/Percepta (ver. 14.2.0; Build 2977; ACD/Laboratories, Toronto, ON, Canada). Moreover, estimations of the CNS factor displayed their high penetrant ability. Plasma protein binding (PPB) results showed high bioavailability and a long biological half-life because the bound portion may act as a reservoir from which the drug is slowly released in the unbound form to maintain equilibrium. Nevertheless, based on these in silico ADME analyses, chloro derivative 15 seems to be the one with the least-desired physico-chemical properties for a CNS active drug, yet the solubility of all thienostilbenes require improvement in the future.
2.3. In Vitro Evaluation of Thienostilbene Oximes 13–18 in Reactions with Cholinesterases
Eight thienostilbene oxime derivatives 13–18 were tested as reversible inhibitors of AChE and BChE, and dissociation constants are given in Table 2. Generally, all of the oximes were moderate reversible inhibitors of both enzymes with Ki constants in the micromolar range. All of the oximes, except trans,anti-16, were more potent inhibitors of AChE than BChE. Out of all of the tested oximes, BChE also exhibited higher potency for cis,syn-18, which has nitrogen at the substituent, as well as for trans,anti-16. In the case of AChE, it seems that the CN substituent affected the binding affinity, as Ki for trans,anti-16 is up to 10-fold higher than for the most potent inhibitor of AChE trans,syn-15. In addition, inhibition of AChE was uncompetitive for all compounds, except for trans,syn-15, while all of the compounds exhibited competitive binding for BChE.
Table 2.
Dissociation constants, Ki (μM) ± SE, for reversible inhibition of human acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) with new oximes at 25 °C.
| Thienostilbene Oximes | AChE | BChE |
|---|---|---|
| trans,syn-13 | 46 ± 7 | 355 ± 26 |
| trans,anti-13 | 41 ± 4 | 322 ± 38 |
| trans,syn-14 | 67 ± 12 | 271 ± 24 |
| trans,anti-14 | 54 ± 7 | 172 ± 14 |
| trans,syn-15 | 52 ± 12 | 573 ± 35 |
| trans,anti-15 | 76 ± 21 | 482 ± 45 |
| trans,anti-16 | 158 ± 40 | 44 ± 9 |
| cis,syn-18 | 39 ± 11 | 89 ± 16 |
The reactivation of human AChE and BChE inhibited with nerve agents (sarin, cyclosarin, VX, and tabun) was screened with one concentration of thienostilbene oxime derivatives (0.1 mM). In the case of AChE inhibited with any of the four nerve agents, no reactivation higher than 20% was observed within 5 h, which does not present an improvement over TMB-4, 2-PAM, HI-6, and obidoxime in the case of AChE inhibited with tabun, VX, sarin, and cyclosarin, respectively. Unfortunately, reactivation could not be tested with a higher concentration due to poor solubility and solvent DMSO, which affects AChE activity. In the case of BChE, only the cyclosarin-inhibited BChE was prone to reactivation, and Figure 4 shows the percentage of maximal reactivation and the observed reactivation rate with 0.1 mM oximes. The fastest reactivation was observed with trans,anti-15 and trans,anti-14, while with their trans,syn-counterparts follow. These four derivatives reactivated cyclosarin-inhibited BChE up to 70% in two hours of reactivation. Although the maximal percentage of reactivation was similar to the standard oxime HI-6, the observed reactivation rate was about four-fold slower than that obtained for HI-6. BChE inhibited by other nerve agents was resistant to reactivation with these compounds.
Figure 4.

Reactivation of cyclosarin-inhibited human BChE with eight thienostilbene oxime derivatives and standard oxime HI-6 (0.1 mM) given as observed reactivation constant (kobs) and maximal reactivation in 22 h.
Nevertheless, oximes with 4-methoxyphenyl and 4-chlorophenyl substituents 14 and 15 seem to be successful in the reactivation of cyclosarin-inhibited human BChE. These findings are in accordance with our recent studies with quinuclidine-3 oximes from which the most potent reactivators of cyclosarin-inhibited BChE were bromo- or chlorobenzyl oximes [24].
2.4. Docking of Potential Reactivators
To gain insight into the structure of the enzyme–inhibitor complex after the oxime approaches the active site, we performed docking of thienostilbene oximes into inhibited cholinesterase. According to our experimental results, the significant reactivation ability of tested thienostilbene oximes is found only for cyclosarin-inhibited BChE; therefore, we conducted docking of the compounds of interest into the active site of BChE inhibited by cyclosarin. It should be mentioned that, unlike the cyclosarin-inhibited AChE, the crystal structure of cyclosarin-inhibited BChE is not available in the literature. To solve this obstacle, we tried to use the conformation of cyclosarin bound to AChE (from PDB structure 3ZLU [25]) and incorporate it within the active site of BChE. However, because of the differences between active sites of BChE and AChE, specifically, due to the positioning of His438 in BChE, this approach resulted in sterically unfavorable interactions (Figure S1). Cuya et al. performed a molecular dynamics study [26] to obtain the most realistic inhibitor conformation, but they also used cyclosarin-inhibited AChE. Therefore, in our work, the experimentally obtained crystal structure of BChE inhibited by tabun is utilized, replacing the dimethylamino and ethoxy groups of tabun with methyl and cyclohexyloxy groups of cyclosarin, respectively. The orientation of replaced substituents in the resulting cyclosarin conformation was as similar to the structure in AChE as possible, given the steric limitations in the active site of BChE (Figure S2).
As the compounds with the best reactivating capability, trans,anti-14 and trans,anti-15 were chosen for docking into the active site of cyclosarin-inhibited BChE. For the sake of comparison, we also performed docking of the poor reactivator cis,syn-18. The structures of the active site of cyclosarin-inhibited BChE with docked thienostilbene oximes are shown in Figure 5. For each of these three oximes, all conformations obtained by docking into the inhibited active site are superimposed, so the most abundant orientation of the reactivator is clearly depicted.
Figure 5.

Structures of the active site of cyclosarin-inhibited BChE with docked thienostilbene oximes: trans,anti-14, trans,anti-15, and cis,syn-18. Cyclosarin (presented using the ball and stick model) is covalently bound to Ser198.
Molecules 14 and 15 take various conformations, but it is visible from the presentations in Figure 5 that, in most cases, the oxime group is oriented toward His438. In contrast, for the molecule cis,syn-18, its oxime group is always oriented in the opposite direction. The orientation of the oxime group is relevant for the reactivation, as well as its proximity to His438 and the phosphorus atom of cyclosarin, because the first step of reactivation is a nucleophilic attack of the oxygen of the oxime group to phosphorus. Since the oximes in this study entered the active site in neutral form, the first step of efficient reactivation should be preceded by the total (or at least partial) deprotonation of the oxime group. This deprotonation is possible only if the proton of the oxime is in a sterically favorable position to the unprotonated nitrogen atom of His438. Although neutral histidine is a weak base and thus unable to deprotonate the oxime group, here it is placed close to Glu325, which accepts the proton of the epsilon nitrogen and thus facilitates proton transfer from the oxime to the delta nitrogen of histidine. Figure 5 shows that none of the conformations obtained by docking of cis,syn-18 are not in a favorable position to proceed with reactivation. In contrast, for 14 and 15, most conformers are placed so as to enable reactivation. In Figure 6, the most favorable conformations of docked molecules 14 and 15 are presented, along with some geometry parameters.
Figure 6.

The most favorable conformation of trans,anti-14 and trans,anti-15 docked into the active site of cyclosarin-inhibited BChE.
Figure 6 shows that the structures of the complexes formed between trans,anti-14 and trans,anti-15 with the active site of cyclosarin-inhibited BChE are favorable for the reactivation of the enzyme. As already mentioned, the thienostilbene oximes studied here are not in ionic form; this is important because neutral oximes have an improved ability to cross the blood–brain barrier compared to charged ones. However, to be able to break the covalent P-O bond between the inhibitor and catalytic Ser198, the oxime should be totally or partially deprotonated by histidine, such as in the semi-empirical calculations of BChE reactivation reported recently [27]. Therefore, the proximity of the oxime to histidine along with the position of the oxime group, which provides the absence of steric hindrance for the oxygen attack of phosphorus, are crucial for efficient reactivation.
In the static structure obtained by docking, the distance between the covalently bound inhibitor and oxygen of the neutral oxime is too big to enable the reaction to start immediately (the nucleophilic attack of oxygen to phosphorus). However, the favorable orientation of oxime and its proximity to His438 make this process feasible. To additionally scrutinize this conclusion, we performed scanning of the Potential Energy Surface (PES), utilizing quantum chemistry calculations (see details in section Materials and Methods) on a small-model system consisting of His448, Glu325, and Ser198 with covalently bound cyclosarin and thienostilbene oxime trans,anti-15. Scanning the PES involved crude geometry optimization for the set of structures obtained by an incremental (0.1 Å) decrease in the distance between the oxygen of oxime and phosphorus, d(OOxime-P). The starting value of the distance was 8 Å. The system’s total energy also decreased until the oxygen approached phosphorus at d = 4.3 Å (Figure S3 and Table S1). Further shortening the d(OOxime-P) did not cause significant energy change until the oxygen was 3 Å from phosphorus. It may be concluded that the approximate equilibria distance d(OOxime-P) before the start of reaction between oxime and cyclosarin is between 4 and 3 Å. It is important to emphasize that this structure may be achieved spontaneously (the process being exothermic, and more relevant, without energy barriers), starting from the first geometry obtained by docking.
The investigation performed in this section does not aim to answer how different substituents at the phenyl unit of thienostilbene oximes affect their reactivating ability. However, the presented results indicate that trans,anti-thienostilbene oximes have a conformational advantage compared to their cis,syn-counterparts, thus rationalizing the experimental findings shown in Figure 4.
3. Materials and Methods
3.1. General
Petroleum ether (PE), bp 40–60 °C, was used. Solvents were purified by distillation. Column chromatography was carried out on columns with silica gel (Fluka 0.063–0.2 nm and Fluka 60 Å, technical grade). TLC was carried out using plates coated with silica gel (0.2 mm, Kiselgel 60 F254). Organic layers were routinely dried with anhydrous MgSO4 and evaporated using a rotary evaporator. 1H and 13C NMR spectra were recorded on a spectrometer at 600 MHz. All NMR spectra were measured in CDCl3 using tetramethylsilane as a reference. The following abbreviations were used in the NMR spectra: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), and m (multiplet). For the full characterization of targeted oximes, the additional techniques 2D-CH correlation (HSQC) and 2D-HH-COSY were used. Mass spectra were obtained on a UPLC-MS system. Melting points were obtained using a microscope-equipped apparatus and have not been corrected. HRMS analyses were carried out on a mass spectrometer (MALDI TOF/TOF analyzer), equipped with an Nd:YAG laser operating at 355 nm with a firing rate of 200 Hz in the positive (H+) or negative (-H) ion reflector mode. The initial phosphonium salts were prepared in the laboratory from the corresponding bromides, while the other starting compounds used were purchased chemicals.
3.2. Synthesis of Thienostilbenes 1–6
Thienostilbenes 1–6 were prepared by the Wittig reaction. The corresponding phosphonium salt (11 mmol) was dissolved in 130 mL of EtOH (dried on 4 Å sieves) in a three-necked flask. The appropriate aldehyde (10 mmol) was added. The content of the flask was mixed on a magnetic stirrer and purged with nitrogen. The reaction mixture was heated to 40 °C for 30 min. The NaOEt obtained from Na (11 mmol) reacted with dry EtOH (10 mL) and was added dropwise to the flask. Upon completion of the dropwise addition, the system was closed, and the reaction mixture was stirred on a magnetic stirrer overnight at room temperature. EtOH was applied to a rotary evaporator under reduced pressure. Extraction was carried out in a system of toluene and water, after which the organic layer was dried over MgSO4, filtered, and the toluene was evaporated. The dry reaction mixture was purified by column chromatography on silica gel using a PE/E or PE/DCM variable polarity eluent. The column chromatography yielded mixtures of cis- and trans-isomers of the desired products 1–6, which were used as reactants in further Vilsmeier formylation reactions. The spectroscopic data for the geometrical isomers of compounds 1–6 were obtained from the 1H NMR spectra of the mixtures.
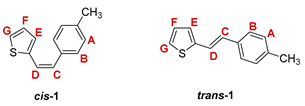
cis-2-(4-methylstyryl)thiophene (cis-1) [28]: 404 mg (18.4%); Rf (petroleum ether) = 0.71; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.14–7.21 (m, 4H, HA, HB), 7.09 (d, 1H, J = 4.7 Hz, HG), 6.98 (d, 1H, J = 3.6 Hz, HE), 6.89 (dd, 1H, J = 4.7; 3.6 Hz, HF), 6.66 (d, 1H, J = 12.1 Hz, HC/D), 6.54 (d, 1H, J = 12.1 Hz, HC/D), 2.37 (s, 3H, -CH3);
trans-2-(4-methylstyryl)thiophene (trans-1) [28,29]: 810 mg (36.8%); Rf (petroleum ether) = 0.57; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.37 (d, 2H, J = 7.8 Hz, HA/B), 7.26 (d, 1H, J = 5.2 Hz, HG), 7.21-7.14 (m, 3H, HC/D, HA/B), 7.05 (d, 1H, J = 3.6 Hz, HE), 7.00 (dd, 1H, J = 5.2; 3.6 Hz, HF), 6.91 (d, 1H, J = 16.0 Hz, HC/D), 2.36 (s, 3H, -CH3);
MS (ESI) m/z (%, fragment): 201 (100, M+H+); HRMS (m/z) for C13H12S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 201.0659, [M+H]+measured = 201.0660.
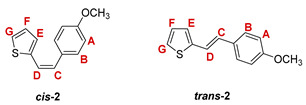
cis-2-(4-methoxystyryl)thiophene (cis-2) [30]: 403 mg (17.0%); Rf (petroleum ether/diethyl ether (1%)) = 0.30; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.29 (d, 2H, J = 8.7 Hz, HA/B), 7.09 (d, 1H, J = 4.9 Hz, HG), 6.98 (dd, 1H, J = 4.9; 3.6 Hz, HF), 6.98 (d, 1H, J = 3.6 Hz, HE), 6.87 (d, 2H, J = 8.7 Hz, HA/B), 6.63 (d, 1H, J = 11.8 Hz, HC/D), 6.51 (d, 1H, J = 11.8 Hz, HC/D), 3.83 (s, 3H, -OCH3);
trans-2-(4-methoxystyryl)thiophene (trans-2) [29]: 803 mg (33.8%); Rf (petroleum ether/diethyl ether (1%)) = 0.23; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.40 (d, 2H, J = 8.4 Hz, HA/B), 7.15 (d, 1H, J = 4.9 Hz, HG), 7.10 (d, 1H, J = 15.9 Hz, HC/D), 7.02 (d, 1H, J = 3.6 Hz, HE), 7.01 (d, 1H, J = 15.9 Hz, HC/D), 6.89 (d, 2H, J = 8.4 Hz, HA/B), 6.88 (dd, 1H, J = 4.9; 3.6 Hz, HF), 3.83 (s, 3H, -OCH3);
MS (ESI) m/z (%, fragment): 217 (100, M+H+); HRMS (m/z) for C13H12OS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 217.0609, [M+H]+measured = 217.0605.
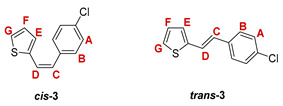
cis-2-(4-chlorostyryl)thiophene (cis-3): 591 mg (24.4%); Rf (petroleum ether) = 0.75; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.33-7.28 (m, 4H, HA/B), 7.12 (d, 1H, J = 5.0 Hz, HG), 6.97 (d, 1H, J = 3.6 Hz, HE), 6.90 (dd, 1H, J = 5.0; 3.6 Hz, HF), 6.71 (d, 1H, J = 11.8 Hz, HC/D), 6.50 (d, 1H, J = 11.8 Hz, HC/D);
trans-2-(4-chlorostyryl)thiophene (trans-3) [29]: 629 mg (26.0%); Rf (petroleum ether) = 0.65; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.39 (d, 2H, J = 8.5 Hz, HA/B), 7.33-7.28 (m, 2H, HA/B), 7.21 (d, 1H, J = 5.0 Hz, HG), 7.20 (d, 1H, J = 16.2 Hz, HC/D), 7.08 (d, 1H, J = 3.4 Hz, HE), 7.01 (dd, 1H, J = 5.0; 3.4 Hz, HF), 6.87 (d, 1H, J = 16.2 Hz, HC/D);
MS (ESI) m/z (%, fragment): 221/223 (80, M+H+), 186 (100); HRMS (m/z) for C12H9ClS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 221.0112, [M+H]+measured = 221.0114.
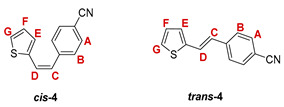
cis-4-(2-(thiophen-2-yl)vinyl)benzonitrile (cis-4): 831 mg (35.8%); Rf (petroleum ether/diethyl ether (30%)) = 0.52; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.63 (d, 2H, J = 8.0 Hz, HA/B), 7.47 (d, 2H, J = 8.0 Hz, HA/B), 7.14 (d, 1H, J = 4.9 Hz, HG), 6.98 (d, 1H, J = 3.6 Hz, HE), 6.92 (dd, 1H, J = 4.9; 3.6 Hz, HF), 6.79 (d, 1H, J = 12.0 Hz, HC/D), 6.52 (d, 1H, J = 12.0 Hz, HC/D);
trans-4-(2-(thiophen-2-yl)vinyl)benzonitrile (trans-4) [29]: 585 mg (25.2%); Rf (petroleum ether/diethyl ether (30%)) = 0.43; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.62 (d, 2H, J = 8.3 Hz, HA/B), 7.53 (d, 2H, J = 8.3 Hz, HA/B), 7.34 (d, 1H, J = 16.2 Hz, HC/D), 7.28 (d, 1H, J = 5.1 Hz, HG), 7.15 (d, 1H, J = 3.5 Hz, HE), 7.04 (dd, 1H, J = 5.1; 3.5 Hz, HF), 6.90 (d, 1H, J = 16.2 Hz, HC/D);
MS (ESI) m/z (%, fragment): 212 (100, M+H+); HRMS (m/z) for C13H9NS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 212.0458, [M+H]+measured = 212.0456.
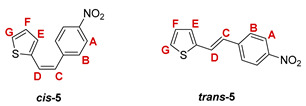
cis-2-(4-nitrostyryl)thiophene (cis-5): 582 mg (22.9%); Rf (petroleum ether/diethyl ether (15%)) = 0.63; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.19 (d, 2H, J = 8.4 Hz, HA/B), 7.53 (d, 2H, J = 8.4 Hz, HA/B), 7.17 (d, 1H, J = 5.2 Hz, HG), 6.99 (d, 1H, J = 3.5 Hz, HE), 6.99 (dd, 1H, J = 5.2; 3.5 Hz, HF), 6.83 (d, 1H, J = 12.0 Hz, HC/D), 6.55 (d, 1H, J = 12.0 Hz, HC/D);
trans-2-(4-nitrostyryl)thiophene (trans-5) [29]: 630 mg (24.8%); Rf (petroleum ether/diethyl ether (15%)) = 0.51; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.21 (d, 2H, J = 8.6 Hz, HA/B), 7.58 (d, 2H, J = 8.6 Hz, HA/B), 7.39 (d, 1H, J = 16.1 Hz, HC/D), 7.30 (d, 1H, J = 5.2 Hz, HG), 7.18 (d, 1H, J = 3.7 Hz, HE), 7.05 (dd, 1H, J = 5.2; 3.7 Hz, HF), 6.95 (d, 1H, J = 16.1 Hz, HC/D);
MS (ESI) m/z (%, fragment): 232 (100, M+H+); HRMS (m/z) for C12H9NO2S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 232.0356, [M+H]+measured = 232.0354.
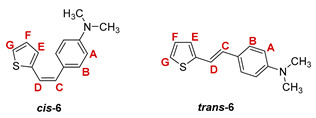
cis-N,N-dimethyl-4-(2-(thiophen-2-yl)vinyl)aniline (cis-6) [31]: 524 mg (20.8%); Rf (petroleum ether/diethyl ether (30%)) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.28 (d, 2H, J = 8.7 Hz, HA/B), 7.10 (d, 1H, J = 5.2 Hz, HG), 7.02 (d, 1H, J = 3.6 Hz, HE), 6.91 (dd, 1H, J = 5.2; 3.6 Hz, HF), 6.69 (d, 2H, J = 8.7 Hz, HA/B), 6.54 (d, 1H, J = 11.9 Hz, HC/D), 6.49 (d, 1H, J = 11.9 Hz, HC/D), 2.98 (s, 6H, -N(CH3)2);
trans-N,N-dimethyl-4-(2-(thiophen-2-yl)vinyl)aniline (trans-6) [31]: 630 mg (25.0%); Rf (petroleum ether/diethyl ether (30%)) = 0.47; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.37 (d, 2H, J = 8.7 Hz, HA/B), 7.12 (d, 1H, J = 4.7 Hz, HG), 7.05 (d, 1H, J = 16.0 Hz, HC/D), 6.99 (d, 1H, J = 3.6 Hz, HE), 6.98 (dd, 1H, J = 4.7; 3.6 Hz, HF), 6.88 (d, 1H, J = 16.0 Hz, HC/D), 6.71 (d, 2H, J = 8.7 Hz, HA/B), 2.99 (s, 6H, -N(CH3)2);
MS (ESI) m/z (%, fragment): 230 (100, M+H+); HRMS (m/z) for C14H15NS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 230.0929, [M+H]+measured = 230.0925.
3.3. Synthesis of Aldehydes 7–12
The obtained thienostilbenes, products of Wittig reactions 1–6, were subjected to Vilsmeier formylation. The selected thienostilbenes, as a mixture of isomers, were dissolved in 2 mL of DMF and stirred for 10 min at 10 °C. This temperature was achieved using a water bath with a few ice cubes and monitored with a thermometer. The weighed amount of POCl3 was slowly added dropwise. After 30 min, the water bath was removed, and the reaction mixture was allowed to stir at room temperature. In the reactions to prepare compounds 7–9, the reaction mixture was stirred at room temperature for 48 h. The reaction mixture for preparing compounds 10 and 11 was heated to 80 °C, and the reaction mixture for preparing compound 12 to 60 °C. Heating was continued for 3 h, after which time the reaction mixture was left to stir at room temperature for 48 h. Upon completion of the reaction, the reaction mixture was neutralized using 10% NaOH solution. When neutralization was achieved, extraction was carried using diethyl ether and water. The combined organic layer was washed with water and dried over MgSO4, filtered, and the diethyl ether was evaporated on a rotary evaporator. The dry reaction mixture was purified by column chromatography on silica gel using a PE/E or PE/DCM variable polarity eluent. In the first fractions, the unreacted substrates 1–6 were isolated (mostly as cis-isomers), while in the last fractions, the desired formyl derivatives 7–12 were obtained (mostly as trans-isomers). They were further used in the preparation reactions for oximes 13–18. The spectroscopic data for the geometrical isomers were obtained from the 1H NMR spectra of the mixtures of compounds 7–12.
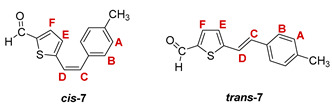
cis-5-(4-methylstyryl)thiophene-2-carbaldehyde (cis-7): 167 mg (12.1%); Rf (petroleum ether/diethyl ether (10%)) = 0.41; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.79 (s, 1H, -CHO), 7.36 (d, 2H, J = 8.1 Hz, HA/B), 7.18 (d, 2H, J = 8.1 Hz, HA/B), 7.18-7.12 (m, 2H, HE, HF), 6.65 (d, 1H, J = 11.7 Hz, HC/D), 6.54 (d, 1H, J = 11.7 Hz, HC/D), 2.36 (s, 3H, -CH3);
trans-5-(4-methylstyryl)thiophene-2-carbaldehyde (trans-7): 335 mg (24.2%); Rf (petroleum ether/diethyl ether (10%)) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.85 (s, 1H, -CHO), 7.66 (d, 1H, J = 3.8 Hz, HE/F), 7.40 (d, 2H, J = 8.9 Hz, HA/B), 7.14 (d, 1H, J = 16.3 Hz, HC/D), 7.15 (d, 2H, J = 8.9 Hz, HA/B), 7.13 (d, 1H, J = 3.8 Hz, HE/F), 6.90 (d, 1H, J = 16.3 Hz, HC/D), 2.37 (s, 3H, -CH3);
MS (ESI) m/z (%, fragment): 229 (100, M+H+); HRMS (m/z) for C14H12OS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 229.0609, [M+H]+measured = 229.0608.
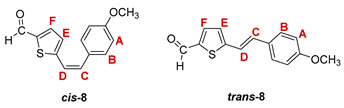
cis-5-(4-methoxystyryl)thiophene-2-carbaldehyde (cis-8): 66 mg (4.8%); Rf (petroleum ether/diethyl ether (10%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.79 (s, 1H, -CHO), 7.55 (d, 1H, J = 3.9 Hz, HE/F), 7.45 (d, 2H, J = 8.8 Hz, HA/B), 7.04 (d, 1H, J = 3.9 Hz, HE/F), 6.87 (d, 2H, J = 8.8 Hz, HA/B), 6.75 (d, 1H, J = 11.9 Hz, HC/D), 6.61 (d, 1H, J = 11.9 Hz, HC/D), 3.82 (s, 3H, -OCH3);
trans-5-(4-methoxystyryl)thiophene-2-carbaldehyde (trans-8) [32]: 261 mg (19.1%); Rf (petroleum ether/diethyl ether (10%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.83 (s, 1H, -CHO), 7.65 (d, 1H, J = 3.9 Hz, HE/F), 7.45 (d, 2H, J = 8.8 Hz, HA/B), 7.11 (d, 1H, J = 16.1 Hz, HC/D), 7.10 (d, 1H, J = 3.9 Hz, HE/F), 7.07 (d, 1H, J = 16.1 Hz, HC/D), 6.91 (d, 2H, J = 8.8 Hz, HA/B), 3.84 (s, 3H, -OCH3);
MS (ESI) m/z (%, fragment): 245 (100, M+H+), 121 (40); HRMS (m/z) for C14H12O2S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 245.0458, [M+H]+measured = 245.0456.
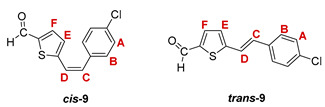
cis-5-(4-chlorostyryl)thiophene-2-carbaldehyde (cis-9): 74 mg (5.4%); Rf (petroleum ether/diethyl ether (30%)) = 0.56; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.80 (s, 1H, -CHO), 7.56 (d, 1H, J = 3.9 Hz, HE/F), 7.34 (d, 2H, J = 8.2 Hz, HA/B), 7.26 (d, 2H, J = 8.2 Hz, HA/B), 7.02 (d, 1H, J = 3.9 Hz, HE/F), 6.74 (d, 1H, J = 12.1 Hz, HC/D), 6.70 (d, 1H, J = 12.1 Hz, HC/D);
trans-5-(4-chlorostyryl)thiophene-2-carbaldehyde (trans-9) [32]: 370 mg (26.9%); Rf (petroleum ether/diethyl ether (30%)) = 0.44; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.87 (s, 1H, -CHO), 7.67 (d, 1H, J = 3.9 Hz, HE/F), 7.43 (d, 2H, J = 8.4 Hz, HA/B), 7.35 (d, 2H, J = 8.4 Hz, HA/B), 7.19 (d, 1H, J = 16.1 Hz, HC/D), 7.16 (d, 1H, J = 3.9 Hz, HE/F), 7.09 (d, 1H, J = 16.1 Hz, HC/D);
MS (ESI) m/z (%, fragment): 249/251 (100, M+H+), 121 (30); HRMS (m/z) for C13H9ClOS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 249.0061, [M+H]+measured = 249.0063.
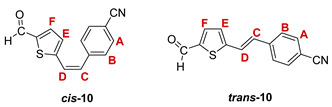
cis-4-(2-(5-formylthiophen-2-yl)vinyl)benzonitrile (cis-10): obtained only in traces; Rf (petroleum ether/DCM (50%)) = 0,17;
trans-4-(2-(5-formylthiophen-2-yl)vinyl)benzonitrile (trans-10): 472 mg (29.4%); Rf (petroleum ether/DCM (50%)) = 0.16; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.90 (s, 1H, -CHO), 7.70 (d, 1H, J = 3.9 Hz, HE/F), 7.67 (d, 2H, J = 8.3 Hz, HA/B), 7.59 (d, 2H, J = 8.3 Hz, HA/B), 7.32 (d, 1H, J = 16.1 Hz, HC/D), 7.23 (d, 1H, J = 3.9 Hz, HE/F), 7.12 (d, 1H, J = 16,1 Hz, HC/D);
MS (ESI) m/z (%, fragment): 240 (100, M+H+); HRMS (m/z) for C14H9NOS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 240.1331, [M+H]+measured = 240.1335.
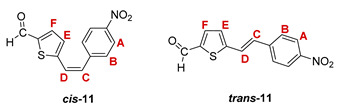
cis-5-(4-nitrostyryl)thiophene-2-carbaldehyde (cis-11): 122 mg (9.0%); Rf (petroleum ether/diethyl ether (50%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.82 (s, 1H, -CHO), 8.23 (d, 2H, J = 8.7 Hz, HA/B), 7.60 (d, 2H, J = 8.7 Hz, HA/B), 7.57 (d, 1H, J = 3.9 Hz, HE/F), 7.03 (d, 1H, J = 3.9 Hz, HE/F), 6.85 (d, 1H, J = 12.2 Hz, HC/D), 6.80 (d, 1H, J = 12.2 Hz, HC/D);
trans-5-(4-nitrostyryl)thiophene-2-carbaldehyde (trans-11) [33]: 244 mg (18.0%); Rf (petroleter/diethyl ether (50%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.91 (s, 1H, -CHO), 8.25 (d, 2H, J = 8.6 Hz, HA/B), 7.71 (d, 1H, J = 4.0 Hz, HE/F), 7.65 (d, 2H, J = 8.6 Hz, HA/B), 7.37 (d, 1H, J = 16.2 Hz, HC/D), 7.26 (d, 1H, J = 4.0 Hz, HE/F), 7.18 (d, 1H, J = 16.2 Hz, HC/D);
MS (ESI) m/z (%, fragment): 260 (100, M+H+); HRMS (m/z) for C13H9NO3S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 260.0303, [M+H]+measured = 260.0306.
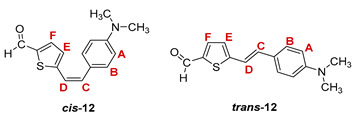
cis-5-(4-(dimethylamino)styryl)thiophene-2-carbaldehyde (cis-12): 247 mg (19.1%); Rf (DCM) = 0.51; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.63 (s, 1H, -CHO), 7.02 (d, 1H, J = 3.4 Hz, HE/F), 6.70 (d, 1H, J = 3.4 Hz, HE/F), 6.68 (d, 1H, J = 11.8 Hz, HC/D), 6.66 (d, 2H, J = 8.7 Hz, HA/B), 6.55 (d, 2H, J = 8.7 Hz, HA/B), 6.49 (d, 1H, J = 11.8 Hz, HC/D), 2.98 (s, 6H, -N(CH3)2);
trans-5-(4-(dimethylamino)styryl)thiophene-2-carbaldehyde (trans-12) [34]: 227 mg (27.5%); Rf (DCM) = 0.44; 1H NMR (CDCl3, 600 MHz) δ/ppm: 9.80 (s, 1H, -CHO), 7.56 (d, 1H, J = 4.0 Hz, HE/F), 7.25 (d, 2H, J = 8.7 Hz, HA/B), 7.23 (d, 2H, J = 8.7 Hz, HA/B), 7.10 (d, 1H, J = 16.5 Hz, HC/D), 7.09 (d, 1H, J = 4.0 Hz, HE/F), 7.00 (d, 1H, J = 16.5 Hz, HC/D), 2.99 (s, 6H, -N(CH3)2);
MS (ESI) m/z (%, fragment): 258 (100, M+H+); HRMS (m/z) for C15H15NOS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 258.0874, [M+H]+measured = 258.0879.
3.4. Synthesis of Oximes 13–18
The obtained mixture of cis- and trans-isomers of aldehydes, products of Vilsmeier formylations 7–12, were converted into corresponding oximes according to the literature [25]. Crystals of NH2OH × HCl were dissolved in a prepared mixture of 10 mL of EtOH and 3 mL of distilled water. After a homogeneous solution was obtained, the corresponding prepared heterostilbene aldehyde derivative 7–12 was added. The reaction mixture was stirred at room temperature for 24 h. When the reaction was completed, the solvent was evaporated on a rotavapor under reduced pressure. The reaction mixture was purified by repeated column chromatography on silica gel using PE/DCM and DCM/methanol variable polarity eluents. The desired oxime derivatives 13–18 as pure geometrical isomers were thus isolated. The spectroscopic data and yields of pure isolated isomers of compounds 13–18 are given below.
5-(4-methylstyryl)thiophene-2-carbaldehyde oxime. Yield 465 mg (87%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is cis,syn-: trans,syn-: trans,anti- = 1:3:15;
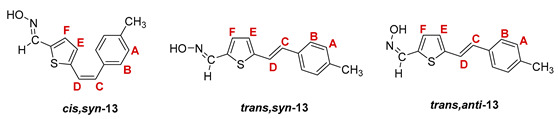
cis,syn-5-(4-methylstyryl)thiophene-2-carbaldehyde oxime (cis,syn-13): 5 mg; Rf (petroleum ether/diethyl ether (30%)) = 0.33; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.14 (s, 1H, -CH = NOH), 7.25 (d, 2H, J = 7.6 Hz, HA/B), 7.15 (d, 2H, J = 7.6 Hz, HA/B), 6.98 (d, 1H, J = 3.6 Hz, HE/F), 6.89 (d, 1H, J = 3.6 Hz, HE/F), 6.61 (d, 1H, J = 12.3 Hz, HC/D), 6.58 (d, 1H, J = 12.3 Hz, HC/D), 2.17 (s, 3H, -CH3);
trans,syn-5-(4-methylstyryl)thiophene-2-carbaldehyde oxime (trans,syn-13): 55 mg; Rf (petroleum ether/diethyl ether (30%)) = 0.34; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (s, 1H, -CH = NOH), 7.47 (bs, 1H, = N-OH), 7.36 (d, 2H, J = 7.9 Hz, HA/B), 7.16 (d, 2H, J = 7.9 Hz, HA/B), 7.12 (d, 1H, J = 15.9 Hz, HC/D), 7.08 (d, 1H, J = 3.5 Hz, HE/F), 6.96 (d, 1H, J = 3.5 Hz, HE/F), 6.94 (d, 1H, J = 15.9 Hz, HC/D), 2.36 (s, 3H, -CH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 145.2 (d, -CH = NOH), 138.1 (s), 133.8 (s), 133.6 (s), 130.5 (d, CE/F), 130.2 (d, CC/D), 129.5 (2d, CA/B), 126.5 (s), 126.4 (2d, CA/B), 125.9 (d, CE/F), 120.4 (d, CC/D), 21.4 (q, -CH3);
trans,anti-5-(4-methylstyryl)thiophene-2-carbaldehyde oxime (trans,anti-13): 67 mg; Rf (petroleum ether/diethyl ether (30%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.64 (s, 1H, -CH = NOH), 7.38 (d, 2H, J = 8.1 Hz, HA/B), 7.17 (d, 1H, J = 16.0 Hz, HC/D), 7.17 (d, 2H, J = 8.1 Hz, HA/B), 7.16 (d, 1H, J = 3.5 Hz, HE/F), 7.04 (d, 1H, J = 16.0 Hz, HC/D), 7.03 (d, 1H, J = 3.5 Hz, HE/F), 2.36 (s, 3H, -CH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 141.8 (d, -CH = NOH), 138.1 (s), 133.8 (s), 132.6 (d), 130.2 (d, CC/D), 129.5 (2d, CA/B), 128.9 (s), 126.5 (2d, CA/B), 126.5 (s), 124.8 (s), 120.4 (d, CC/D), 21.4 (q, -CH3);
MS (ESI) m/z (%, fragment): 244 (100, M+H+); HRMS (m/z) for C14H13NOS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 244.0718, [M+H]+measured = 244.0716.
5-(4-methoxystyryl)thiophene-2-carbaldehyde oxime. Yield 288 mg (83%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is cis,syn-:trans,syn-:trans,anti- = 1:6:20;
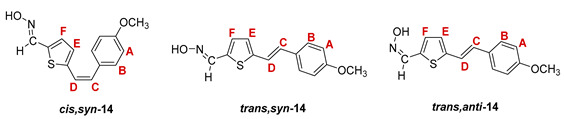
cis,syn-5-(4-methoxystyryl)thiophene-2-carbaldehyde oxime (cis,syn-14): 5 mg; Rf (DCM) = 0.38; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.15 (s, 1H, -CH = NOH), 7.29 (d, 2H, J = 8.6 Hz, HA/B), 7.10 (bs, 1H, -CH = N-OH), 6.98 (d, 1H, J = 3,3 Hz, HE/F), 6.90 (d, 1H, J = 3,3 Hz, HE/F), 6.88 (d, 2H, J = 8.6 Hz, HA/B), 6.58 (d, 1H, J = 12.0 Hz, HC/D), 6.55 (d, 1H, J = 12.0 Hz, HC/D), 3.83 (s, 3H, -OCH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.7 (d, -CH = NOH), 134.0 (s), 132.7 (s), 129.5 (2d), 128.8 (d), 128.7 (s), 128.5 (s), 127.3 (2d), 124.9 (d), 118.8 (d), 113.7 (d), 54.7 (q);
trans,syn-5-(4-methoxystyryl)thiophene-2-carbaldehyde oxime (trans,syn-14): 68 mg; Rf (DCM) = 0.38; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (s, 1H, -CH = NOH), 7.54 (bs, 1H, = N-OH), 7.41 (d, 2H, J = 8.6 Hz, HA/B), 7.07 (d, 1H, J = 3.8 Hz, HE/F), 7.04 (d, 1H, J = 16.1 Hz, HC/D), 6.94 (d, 1H, J = 3.8 Hz, HE/F), 6.93 (d, 1H, J = 16.1 Hz, HC/D), 6.89 (d, 2H, J = 8.6 Hz, HA/B), 3.83 (s, 3H, -OCH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 145.2 (d, -CH = NOH), 133.3 (s), 130.7 (s), 130.4 (d, CE/F), 129.9 (d, CC/D), 129.4 (s), 127.8 (2d, CA/B), 125.4 (d, CE/F), 122.0 (s), 119.3 (d, CC/D), 114.3 (2d, CA/B), 55.2 (q, -OCH3);
trans,anti-5-(4-methoxystyryl)thiophene-2-carbaldehyde oxime (trans,anti-14): 93 mg; Rf (DCM) = 0.19; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.64 (s, 1H, -CH = NOH), 7.42 (d, 2H, J = 8.6 Hz, HA/B), 7.26 (d, 1H, J = 4.0 Hz, HE/F), 7.09 (d, 1H, J = 16.2 Hz, HC/D), 7.02 (d, 1H, J = 16.0 Hz, HC/D), 7.01 (d, 1H, J = 4.0 Hz, HE/F), 6.90 (d, 2H, J = 8.6 Hz, HA/B), 4.62 (bs, 1H, -CH = NOH), 3.83 (s, 3H, -OCH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 141.2 (d, -CH = NOH), 133.3 (s), 132.6 (s), 132.1 (d, CE/F), 129.4 (d, CC/D), 128.9 (s), 128.7 (s), 127.4 (2d, CA/B), 124.0 (d, CE/F), 118.8 (d, CC/D), 113.8 (2d, CA/B), 54.9 (q, -OCH3);
MS (ESI) m/z (%, fragment): 260 (100, M+H+); HRMS (m/z) for C14H13NO2S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 260.0667, [M+H]+measured = 260.0666.
5-(4-chlorostyryl)thiophene-2-carbaldehyde oxime. Yield 363 mg (77%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is cis,syn-:trans,syn-:trans,anti- = 1:5:13;
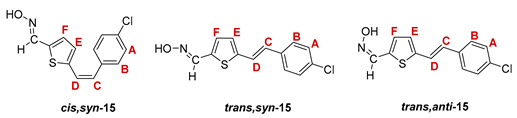
cis,syn-5-(4-chlorostyryl)thiophene-2-carbaldehyde oxime (cis,syn-15): 9 mg; Rf (DCM/methanol (1%)) = 0.58; 1H NMR (CDCl3, 600 MHz) δ/ppm (data from the mixture with trans,syn-15): 8.15 (s, 1H, -CH = NOH), 7.32 (d, 2H, J = 8.5 Hz, HA/B), 7.29 (d, 2H, J = 8.5 Hz, HA/B), 7.14 (d, 1H, J = 3.4 Hz, HE/F), 6.89 (d, 1H, J = 3.4 Hz, HE/F), 6.64 (d, 1H, J = 12.1 Hz, HC/D), 6.56 (d, 1H, J = 12.1 Hz, HC/D);
trans,syn-5-(4-chlorostyryl)thiophene-2-carbaldehyde oxime (trans,syn-15): 56 mg; Rf (DCM/methanol (1%)) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (s, 1H, -CH = NOH), 7.39 (d, 2H, J = 8.5 Hz, HA/B), 7.31 (d, 2H, J = 8.5 Hz, HA/B), 7.14 (bs, 1H, = N-OH), 7.14 (d, 1H, J = 16.2 Hz, HC/D), 7.09 (d, 1H, J = 3.7 Hz, HE/F), 6.99 (d, 1H, J = 3.7 Hz, HE/F), 6.91 (d, 1H, J = 16.2 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 145.2 (d, -CH = NOH), 144.4 (s), 135.1 (s), 134.2 (s), 133.6 (s), 130.4 (d, CE/F), 129.0 (2d, CA/B), 128.7 (d, CC/D), 127.6 (2d, CA/B), 126.5 (d, CE/F), 121.9 (d, CC/D);
trans,anti-5-(4-chlorostyryl)thiophene-2-carbaldehyde oxime (trans,anti-15): 94 mg; Rf (DCM/methanol (1%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.66 (s, 1H, -CH = NOH), 7.40 (d, 2H, J = 8.5 Hz, HA/B), 7.32 (d, 2H, J = 8.5 Hz, HA/B), 7.27 (d, 1H, J = 4.0 Hz, HE/F), 7.19 (d, 1H, J = 16.1 Hz, HC/D), 7.06 (d, 1H, J = 4.0 Hz, HE/F), 7.00 (d, 1H, J = 16.1 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 141.6 (d, -CH = NOH), 135.1 (s), 133.7 (s), 132.5 (d, CE/F), 130.1 (s), 129.0 (2d, CA/B), 128.8 (s), 128.7 (d, CC/D), 127.7 (2d, CA/B), 125.5 (d, CE/F), 122.0 (d, CC/D);
MS (ESI) m/z (%, fragment): 264/266 (100, M+H+), 121 (35); HRMS (m/z) for C13H10ClNOS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 264.0172, [M+H]+measured = 264.0171.
4-(2-(5-((hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile. Yield 366 mg (73%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is cis,syn-:trans,syn-:trans,anti- = 1:30:10;
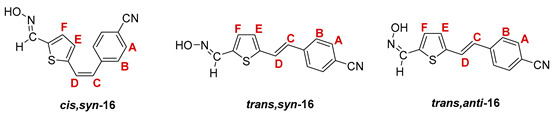
cis,syn-4-(2-(5-((hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile (cis,syn-16): 3 mg; Rf (DCM/methanol (2.5%)) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm (data from the mixture with trans,syn-16): 8.14 (s, 1H, -CH = NOH), 7.64 (d, 2H, J = 8.1 Hz, HA/B), 7.47 (d, 2H, J = 8.1 Hz, HA/B), 7.35 (s, 1H, -CH = NOH), 6.99 (d, 1H, J = 3.7 Hz, HE/F), 6.89 (d, 1H, J = 3.7 Hz, HE/F), 6.73 (d, 1H, J = 12.1 Hz, HC/D), 6.58 (d, 1H, J = 12.1 Hz, HC/D);
trans,syn-4-(2-(5-((hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile (trans,syn-16): 83 mg; Rf (DCM/methanol (2.5%)) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.23 (s, 1H, -CH = NOH), 7.62 (d, 2H, J = 8.4 Hz, HA/B), 7.53 (d, 2H, J = 8.4 Hz, HA/B), 7.43 (s, 1H, -CH = NOH), 7.27 (d, 1H, J = 16.1 Hz, HC/D), 7.11 (d, 1H, J = 3.6 Hz, HE/F), 7.06 (d, 1H, J = 3.6 Hz, HE/F), 6.93 (d, 1H, J = 16.1 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.9 (d, -CH = NOH), 143.5 (s), 141.1 (s), 135.4 (s), 132.6 (2d, CA/B), 132.5 (s), 130.4 (d, CE/F), 127.9 (d, CC/D), 127.8 (d, CE/F), 126.8 (2d, CA/B), 124.8 (d, CC/D), 118.9 (s, -CN);
trans,anti-4-(2-(5-((hydroxyimino)methyl)thiophen-2-yl)vinyl)benzonitrile (trans,anti-16): 28 mg; Rf (DCM/methanol (2.5%)) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.68 (s, 1H, -CH = NOH), 7.63 (d, 2H, J = 8.2 Hz, HA/B), 7.55 (d, 2H, J = 8.2 Hz, HA/B), 7.33 (d, 1H, J = 16.2 Hz, HC/D), 7.29 (d, 1H, J = 4.0 Hz, HE/F), 7.13 (d, 1H, J = 4.0 Hz, HE/F), 7.03 (d, 1H, J = 16.2 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 141.4 (d, -CH = NOH), 141.1 (s), 132.7 (s), 132.6 (2d, CA/B), 132.5 (d, CE/F), 129.6 (s), 127.8 (d, CC/D), 127.2 (s), 126.8 (2d, CA/B), 126.7 (d, CE/F), 124.9 (d, CC/D), 118.9 (s, -CN);
MS (ESI) m/z (%, fragment): 255 (100, M+H+); HRMS (m/z) for C14H10N2OS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 255.0514, [M+H]+measured = 255.0512.
5-(4-nitrostyryl)thiophene-2-carbaldehyde oxime. Yield 220 mg (57%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is trans,syn-:cis,anti-:trans,anti- = 8:1:5;
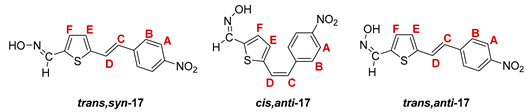
trans,syn-5-(4-nitrostyryl)thiophene-2-carbaldehyde oxime (trans,syn-17): 37 mg; Rf (DCM/methanol (1%)) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.24 (s, 1H, -CH = NOH), 8.21 (d, 2H, J = 8.8 Hz, HA/B), 7.59 (d, 2H, J = 8.8 Hz, HA/B), 7.33 (d, 1H, J = 16.2 Hz, HC/D), 7.32 (bs, 1H, -CH = NOH), 7.13 (d, 1H, J = 3.7 Hz, HE/F), 7.10 (d, 1H, J = 3.7 Hz, HE/F), 6.98 (d, 1H, J = 16.2 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.4 (s), 141.5 (s), 133.7 (s), 132.5 (d), 128.2 (d), 127.3 (d), 126.9 (2d), 126.8 (s), 125.8 (d), 124.2 (2d), (1 signal is missing);
cis,anti-5-(4-nitrostyryl)thiophene-2-carbaldehyde oxime (cis,anti-17): 6 mg; Rf (DCM/methanol (1%)) = 0,45; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.20 (d, 2H, J = 8.5 Hz, HA/B), 7.58 (s, 1H, -CH = NOH), 7.54 (d, 2H, J = 8.5 Hz, HA/B), 7.19 (d, 1H, J = 4.0 Hz, HE/F), 6.94 (d, 1H, J = 4.0 Hz, HE/F), 6.81 (d, 1H, J = 12.1 Hz, HC/D), 6.65 (d, 1H, J = 12.1 Hz, HC/D);
trans,anti-5-(4-nitrostyryl)thiophene-2-carbaldehyde oxime (trans,anti-17): 16 mg; Rf (DCM/methanol (1%)) = 0.43; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.22 (d, 2H, J = 8.6 Hz, HA/B), 7.68 (s, 1H, -CH = NOH), 7.60 (d, 2H, J = 8.6 Hz, HA/B), 7.38 (d, 1H, J = 16.2 Hz, HC/D), 7.30 (d, 1H, J = 3.9 Hz, HE/F), 7.16 (d, 1H, J = 3.9 Hz, HE/F), 7.08 (d, 1H, J = 16.2 Hz, HC/D); MS (ESI) m/z (%, fragment): 275 (100, M+H+); HRMS (m/z) for C13H10N2O3S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 275.0412, [M+H]+measured = 275.0413.
5-(4-dimethylaminostyryl)thiophene-2-carbaldehyde oxime. Yield 436 mg (88%); according to 1H NMR spectroscopy the ratio of isomers in the mixture is cis,syn-:trans,syn-:trans,anti- = 1:1:1;
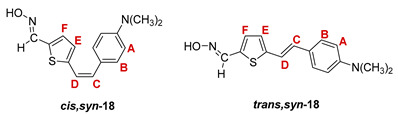
cys,syn-5-(4-dimethylaminostyryl)thiophene-2-carbaldehyde oxime (cys,syn-18): 75 mg; Rf (DCM/methanol (1%)) = 0.21; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.42 (s, 1H, -CH = NOH), 7.34 (d, 2H, J = 8.8 Hz, HA/B), 7.31 (broad s, 1H, -CH = NOH), 7.10 (d, 1H, J = 3.9 Hz, HE/F), 6.99 (d, 2H, J = 8.8 Hz, HA/B), 6.90 (dd, 1H, J = 3.9; 1.9 Hz, HE/F), 6.65 (d, 1H, J = 11.9 Hz, HC/D), 6.48 (d, 1H, J = 11.9 Hz, HC/D), 2.77 (s, 6H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 149.7 (s), 148.7 (d), 132.9 (s), 130.4 (d), 127.6 (2d), 127.6 (2d), 125.9 (d), 122.4 (d), 117.8 (d), 116.9 (s), 108.8 (s), 44.6 (2q);
trans,syn-5-(4-dimethylaminostyryl)thiophene-2-carbaldehyde oxime (trans,syn-18) (it was not possible to isolate this isomer without the traces of cis,syn-18): Rf (DCM/methanol (1%)) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.43 (s, 1H, -CH = NOH), 7.79 (broad s, 1H, -CH = NOH), 7.42 (dd, 1H, J = 8.5; 2.0 Hz, HA/B), 7.17 (d, 1H, J = 4.1 Hz, HE/F), 7.15 (d, 1H, J = 15.6 Hz, HC/D), 7.07-6.97 (m, 4H), 6.88 (d, 1H, J = 15.6 Hz, HC/D), 2.78 (s, 6H);
MS (ESI) m/z (%, fragment): 273 (100, M+H+); HRMS (m/z) for C15H16N2OS (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 273.0553, [M+H]+measured = 273.0557.
3.5. Chemicals for Enzyme Assays
Recombinant human AChE was kindly donated by Dr Zoran Radić (Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, La Jolla, CA, USA), while purified BChE isolated from human plasma was prepared and generously gifted by Dr Florain Nachon (Institut de Recherche Biomédicale des Armées, Bretigny-sur-Orge, France). Enzymes were diluted in 1% of BSA buffer and stored at 4 °C as work solutions. OP compounds sarin, cyclosarin, VX, and tabun were purchased from the NC Laboratory (Spiez, Switzerland). OP stock solutions (5000 μg/mL) were made in isopropyl alcohol, and further dilutions were made in water just before use. The newly synthesized oximes were prepared in a DMSO solvent at a concentration of 100 mM and stored at 4 °C. Other reagents and solvents used in cholinesterase activity measurement were purchased from Sigma-Aldrich (St. Louis, MO, USA).
3.6. Cholinesterase Activity Measurements
Reversible inhibition of AChE and BChE with oximes (50–200 µM) was evaluated in the presence of the substrate concentration range ATCh (0.1–0.7 mM) in 0.1 mM phosphate buffer. Enzyme activity was measured by the Ellman method [35] at 25 °C and 412 nm on a Tecan Infinite M200PRO plate reader (Tecan Austria, GmbH, Salzburg, Austria). Due to the low solubility, a 100 mM stock solution of the tested compounds with the following aliquots was prepared in DMSO, so the same solvent was used in controls as well. The inhibition constants were evaluated as previously described [36].
For the reactivation of OP-inhibited cholinesterases, the enzyme was incubated with 10-fold excess sarin, cyclosarin, VX, or tabun concentrations for about 60 min to achieve a 95−100% inhibition rate. The inhibition mixture was fractionated on a Sephadex G-50 spin column (Roche Diagnostic GmbH, Mannheim, Germany) to remove the unconjugated nerve agent. The reactivation mixture, containing oxime (0.1 mM) in a 0.1 M sodium phosphate buffer pH 7.4, was combined with an inhibited enzyme for the initiation of the reactivation reaction. At specified time intervals, an aliquot was 100-fold diluted in a 0.1 M sodium phosphate buffer pH 7.4 and upon the addition of the substrate ATCh (1 mM) and the reagent DTNB (0.3 mM), residual enzyme activity was measured by the Ellman method [35] at 25 °C and 412 nm on a spectrophotometer (CARY 300, Varian Inc., Mulgrave,Australia). Activity was monitored up to 24 h with consistent correction for the inhibitory effect of oximes and the oxime-induced hydrolysis of ATCh. The first-order reactivation rate constants kobs at given oxime concentrations were determined by non-linear regression as described earlier [37].
3.7. Docking of Thienostilbene Oximes
The model system for the docking study was prepared using the geometry of the active site of butyrylcholinesterase, taken from the PDB structure 3DJY [38], where the inhibitor tabun was replaced by cyclosarin, as described above. The docking of the oximes into the active site of butyrylcholinesterase (15 residues) was performed using the Autodock program suite [39]. The presented results were obtained using the Lamarckian Genetic Algorithm, with a maximum of 2.5 million energy evaluations for docking calculations. The number of requested genetic algorithm dockings was 10, and 10 binding poses for each ligand were obtained, resulting in different structures of the complex between the cyclosarin-inhibited active site and the potential reactivator. Scanning of PES was carried out using the Gaussian09 program package [40], at the B3LYP/6-31G(d) level of theory.
3.8. In Silico Prediction of ADME Properties
In silico predictions of ADME properties were performed with ACD/Percepta (ver. 14.2.0; Build 2977; ACD/Laboratories, Toronto, ON, Canada).
4. Conclusions
This paper presents the design and synthesis of new, uncharged 2-thienostilbene oximes by an economical three-step synthesis using the Wittig reaction, Vilsmeier formylation, and the transformation of aldehydes into oximes in high yields. The expected targeted products were pure cis- and trans-isomers of syn- and anti-oximes containing different substituents bound in the para-position of the benzene ring. Finally, in each oxime formation, although four isomers were expected, cis,syn, trans,syn-, cis,anti-, and trans,anti-, considering the configuration of the carbon–carbon and carbon–nitrogen double bonds, after successive column and thin layer chromatography of the reaction mixtures, only some of isomers 13–18 were successfully isolated. Out of the eight oximes selected for the reactivation of nerve-agent inhibited cholinesterases, trans,anti-15 and trans,anti-14 and their trans,syn-counterparts reactivated cyclosarin-inhibited BChE up to 70%. This is the first study to show the potential of thienostilbene oximes as therapeutics in OP poisoning, and it seems that further design of the compounds e.g., with amide, OH, mono- and dimethylamino groups, or a triazole ring, could provide a new platform for further antidote and scavenger development for exposure to organophosphates.
Acknowledgments
The authors are grateful to Zoran Radić and Florian Nachon for a generous gift of enzymes. The authors thank Makso Herman for language editing. We also gratefully acknowledge the NMR Centre at RBI for recording all the NMR spectra.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ph14111147/s1: Spectra contributions: 1H and 13C NMR spectra of the new investigated compounds, along with the 2D NMR spectra and mass spectra of all new isolated oxime derivatives 13–18; Figure S1: Superposition of the cyclosarin-bound AChE (3ZLU) and BChE (3DJY) with cyclosarin in the same conformation at the active serine; Figure S2: Superposition of cyclosarin-bound AChE (3ZLU) and BChE (3DJY) with cyclosarin bound at the active serine obtained by replacing the dimethylamino and ethoxy groups of tabun with methyl and cyclohexyloxy groups of cyclosarin, respectively; Figure S3: Energy profile for the incremental decrease of the distance between the oxygen of oxime and phosphorus; Table S1: Data obtained by scanning of PES, presented in Figure S3.
Author Contributions
Conceptualization, I.Š. and Z.K.; methodology, M.M. and T.Č.; formal analysis, Ž.M. and K.L.; investigation, M.M., T.Č., D.B., I.P., and A.R.; resources, I.Š. and Z.K.; writing—original draft preparation, I.Š., Z.K., T.Č., and D.B.; writing—review and editing, M.M., T.Č., D.B., A.R., Z.K., and I.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC was funded by the Croatian Science Foundation (project IP-2018-01-7683). The University of Zagreb’s short term scientific support (2020) under the project Preparation of new annelated heteropolycyclic systems is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dolgin E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med. 2013;19:1194. doi: 10.1038/nm1013-1194. [DOI] [PubMed] [Google Scholar]
- 2.Steindl D., Boehmerle W., Körner R., Praeger D., Haug M., Nee J., Schreiber A., Scheibe F., Demin K., Jacoby P., et al. Novichok nerve agent poisoning. Lancet. 2021;397:249–252. doi: 10.1016/S0140-6736(20)32644-1. [DOI] [PubMed] [Google Scholar]
- 3.Darvesh S., Hopkins A.D., Geula C. Neurobiology of Butyrylcholinesterase. Nat. Rev. Neurosci. 2003;4:131–138. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 4.Giacobini E. In: Butyrylcholinesterase: Its Function and Inhibitors. 1st ed. Martin D., editor. Taylor & Francis Group plc; London, UK: 2003. [Google Scholar]
- 5.Čadež T., Kovarik Z. Advancements in recombinant technology for production of butyrylcholinesterase, a bioscavenger of nerve agent. Period. Biol. 2020;121–122:55–63. doi: 10.18054/pb.v121-122i1-2.10867. [DOI] [Google Scholar]
- 6.Sidell F.R., Borak J. Chemical warfare agents: II. nerve agents. Ann. Emerg. Med. 1992;21:865–871. doi: 10.1016/S0196-0644(05)81036-4. [DOI] [PubMed] [Google Scholar]
- 7.Timperley C.M., Abdollahi M., Al-Amri A.S., Baulig A., Benachour D., Borrett V., Cariño F.A., Geist M., Gonzalez D., Kane W., et al. Advice on assistance and protection by the Scientific Advisory Board of the Organisation for the Prohibition of Chemical Weapons: Part 2. On preventing and treating health effects from acute, prolonged, and repeated nerve agent exposure, and the identific. Toxicology. 2019;413:13–23. doi: 10.1016/j.tox.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Timperley C.M., Forman J.E., Abdollahi M., Al-Amri A.S., Baulig A., Benachour D., Borrett V., Cariño F.A., Geist M., Gonzalez D., et al. Advice on assistance and protection provided by the Scientific Advisory Board of the Organisation for the Prohibition of Chemical Weapons: Part 1. On medical care and treatment of injuries from nerve agents. Toxicology. 2019;415:56–69. doi: 10.1016/j.tox.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Maček Hrvat N., Kovarik Z. Counteracting poisoning with chemical warfare nerve agents. Arh. Hig. Rada Toksikol. 2021;71:266–284. doi: 10.2478/aiht-2020-71-3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zorbaz T., Kovarik Z. Neuropharmacology: Oxime antidotes for organophosphate pesticide and nerve agent poisoning. Period. Biol. 2020;121–122:35–54. doi: 10.18054/pb.v121-122i1-2.10623. [DOI] [Google Scholar]
- 11.Zorbaz T., Mišetić P., Probst N., Žunec S., Zandona A., Mendaš G., Micek V., MačEk Hrvat N., Katalinić M., Braïki A., et al. Pharmacokinetic evaluation of brain penetrating morpholine-3-hydroxy-2-pyridine oxime as an antidote for nerve agent poisoning. ACS Chem. Neurosci. 2020;11:1072–1084. doi: 10.1021/acschemneuro.0c00032. [DOI] [PubMed] [Google Scholar]
- 12.Sit R.K., Kovarik Z., Maček Hrvat N., Žunec S., Green C., Fokin V.V., Sharpless K.B., Radic Z., Taylor P. Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther. 2018;367:363–372. doi: 10.1124/jpet.118.249383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuca K., Jun D., Musilek K. Structural requirements of acetylcholinesterase reactivators. Mini Rev. Med. Chem. 2006;6:269–277. doi: 10.2174/138955706776073510. [DOI] [PubMed] [Google Scholar]
- 14.Norrrahim M.N.F., Ahmad Shah N.A., Jamal S.H., Yunus W.M.Z.W., Ernest V.F.K.V., Kasim N.A.M. Nanocellulose-based filters as novel barrier systems for chemical warfare agents. Solid State Phenom. 2021;317:180–186. doi: 10.4028/www.scientific.net/SSP.317.180. [DOI] [Google Scholar]
- 15.de Koning M.C., van Grol M., Noort D. Peripheral site ligand conjugation to a non-quaternary oxime enhances reactivation of nerve agent-inhibited human acetylcholinesterase. Toxicol. Lett. 2011;206:54–59. doi: 10.1016/j.toxlet.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Kliachyna M., Santoni G., Nussbaum V., Renou J., Sanson B., Colletier J.P., Arboléas M., Loiodice M., Weik M., Jean L., et al. Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- Aldoxime and -Amidoxime hybrids as efficient uncharged reactivators of nerve agent-Inhibited human acetylcholinesterase. Eur. J. Med. Chem. 2014;78:455–467. doi: 10.1016/j.ejmech.2014.03.044. [DOI] [PubMed] [Google Scholar]
- 17.Šagud I., Škorić I., Šindler-Kulyk M. Excited state transformations of heterostilbenes: Pathways to polycyclic skeleta. Comptes Rendus. Chim. 2018;21:1043–1052. doi: 10.1016/j.crci.2018.09.008. [DOI] [Google Scholar]
- 18.Ribeiro T.S., Prates A., Alves S.R., Oliveira-Silva J.J., Riehl C.A.S., Figueroa-Villar J.D. The effect of neutral oximes on the reactivation of human acetylcholinesterase inhibited with paraoxon. J. Braz. Chem. Soc. 2012;23:1216–1225. doi: 10.1590/S0103-50532012000700004. [DOI] [Google Scholar]
- 19.Porcheddu A., Giacomelli G. Synthesis of Oximes and Hydroxamic Acids. In: Liebman J.F., Rappoport Z., editors. The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. John Wiley & Sons; New York, NY, USA: 2009. pp. 163–231. [Google Scholar]
- 20.Alavijeh M.S., Chishty M., Qaiser M.Z., Palmer A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRX. 2005;2:554–571. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Testa B., Crivori P., Reist M., Carrupt P.A. The influence of lipophylicity on the pharmacokinetic behavior of drugs: Concepts and examples. Perspect. Drug Discov. Des. 2000;19:179–211. doi: 10.1023/A:1008741731244. [DOI] [Google Scholar]
- 22.Pajouhesh H., Lenz G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stocks M. The small molecule drug discovery process-from target selection to candidate selection. In: Jefferis R., Roberts S., Ganellin R., editors. Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies. The University of Nottingham; England, UK: 2013. pp. 81–126. [DOI] [Google Scholar]
- 24.Zandona A., Katalinić M., Šinko G., Radman Kastelic A., Primožič I., Kovarik Z. Targeting organophosphorus compounds poisoning by novel quinuclidine-3 oximes: Development of butyrylcholinesterase-based bioscavengers. Arch. Toxicol. 2020;94:3157–3171. doi: 10.1007/s00204-020-02811-5. [DOI] [PubMed] [Google Scholar]
- 25.Artursson E., Andersson P.O., Akfur C., Linusson A., Borjegren S., Ekstrom F. Catalytic-site conformational equilibrium in nerve-agent adducts of acetylcholinesterase; possible implications for the HI-6 antidote substrate specificity. Biochem. Pharmacol. 2013;85:1389–1397. doi: 10.1016/j.bcp.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Cuya T., da Silva Gonçalves A., da Silva J.A.V., Ramalho T.C., Kuca C.C.K., França T.C.C. The role of the oximes HI-6 and HS-6 inside human acetylcholinesterase inhibited with nerve agents: A computational study. J. Biomol. Struct. Dyn. 2018;36:3444–3452. doi: 10.1080/07391102.2017.1389307. [DOI] [PubMed] [Google Scholar]
- 27.Maraković N., Knežević A., Rončević I., Brazzolotto X., Kovarik Z., Šinko G. Enantioseparation, in vitro testing, and structural characterization of triple-binding reactivators of organophosphate-inhibited cholinesterases. Biochem. J. 2020;477:2771–2790. doi: 10.1042/BCJ20200192. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S., Pandey A.K., Singh R., Singh K.N. Decarboxylative arylation of α,β-unsaturated carboxylic acids using aryl trazenes by copper/ionic liquid combination in PEG-400. Eur. J. Org. Chem. 2018;2018:5942–5949. doi: 10.1002/ejoc.201800930. [DOI] [Google Scholar]
- 29.Qu J., Cao C.T., Cao C. Determining the excited-state substituent constants of furyl and thienyl groups. J. Phys. Org. Chem. 2018;31:3799. doi: 10.1002/poc.3799. [DOI] [Google Scholar]
- 30.Lee S.K., Min H.Y., Huh K.S., Kim E.Y., Lee E., Song S., Kim S. Styrylheterocycles: A novel class of inhibitors on lipopolysaccharide-induced nitric oxide production. Bioorganic Med. Chem. Lett. 2003;13:3689–3692. doi: 10.1016/j.bmcl.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Jin J., Li X., Zhang J., Zhao P., Tian H. Rational design of double-check mercury ion chemosensors based on photochromic compounds. Isr. J. Chem. 2013;53:288–293. doi: 10.1002/ijch.201200102. [DOI] [Google Scholar]
- 32.Jones G., Stanforth S.P. The Vilsmeier reaction of fully conjugated carbocycles and heterocycles. Org. React. 1997;49:1–330. [Google Scholar]
- 33.Yoshimura S., Takahashi S., Kawamata A., Kikugawa K., Suehiro H., Aoki A. Synthesis of α-substituted alkanoic acids and inhibition of platelet aggregation. Chem. Pharm. Bull. 1978;26:685–702. doi: 10.1248/cpb.26.685. [DOI] [PubMed] [Google Scholar]
- 34.Jiang X., Yang X., Zhao C., Jin K., Sun L. Electrogenerated chemiluminiscence of a series of donor-acceptor molecules and X-ray crystallographic evidence for the reaction mechanisms. J. Phys. Chem. C. 2007;111:9595–9602. doi: 10.1021/jp071015n. [DOI] [Google Scholar]
- 35.Ellman G.L., Courtney K.D., Andres V., Featherstone J., Featherstone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 36.Kovarik Z., Ciban N., Radić Z., Simeon-Rudolf V., Taylor P. Active site mutant acetylcholinesterase interactions with 2-PAM, HI-6, and DDVP. Biochem. Biophys. Res. Commun. 2006;342:973–978. doi: 10.1016/j.bbrc.2006.02.056. [DOI] [PubMed] [Google Scholar]
- 37.Maček Hrvat N., Zorbaz T., Šinko G., Kovarik Z. The estimation of oxime efficiency is affected by the experimental design of phosphylated acetylcholinesterase reactivation. Toxicol. Lett. 2018;293:222–228. doi: 10.1016/j.toxlet.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 38.Carletti E., Li H., Li B., Ekstrom F., Nicolet Y., Loiodice M., Gillon E., Froment M.T., Lockridge O., Schopfer L.M., et al. Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation. J. Am. Chem. Soc. 2008;130:16011–16020. doi: 10.1021/ja804941z. [DOI] [PubMed] [Google Scholar]
- 39.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., et al. Gaussian 09; Revision D.01. Gaussian, Inc.; Wallingford, CT, USA: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and Supplementary Material.